

Visual Abstract

Keywords: primary hyperoxaluria type 1, RNAi, lumasiran, urinary oxalate, kidney stones, nephrocalcinosis, oxalosis, plasma oxalate, RNAi therapeutics
Abstract
Background and objectives
In the rare disease primary hyperoxaluria type 1, overproduction of oxalate by the liver causes kidney stones, nephrocalcinosis, kidney failure, and systemic oxalosis. Lumasiran, an RNA interference therapeutic, suppresses glycolate oxidase, reducing hepatic oxalate production. The objective of this first-in-human, randomized, placebo-controlled trial was to evaluate the safety, pharmacokinetic, and pharmacodynamic profiles of lumasiran in healthy participants and patients with primary hyperoxaluria type 1.
Design, setting, participants, & measurements
This phase 1/2 study was conducted in two parts. In part A, healthy adults randomized 3:1 received a single subcutaneous dose of lumasiran or placebo in ascending dose groups (0.3–6 mg/kg). In part B, patients with primary hyperoxaluria type 1 randomized 3:1 received up to three doses of lumasiran or placebo in cohorts of 1 or 3 mg/kg monthly or 3 mg/kg quarterly. Patients initially assigned to placebo crossed over to lumasiran on day 85. The primary outcome was incidence of adverse events. Secondary outcomes included pharmacokinetic and pharmacodynamic parameters, including measures of oxalate in patients with primary hyperoxaluria type 1. Data were analyzed using descriptive statistics.
Results
Thirty-two healthy participants and 20 adult and pediatric patients with primary hyperoxaluria type 1 were enrolled. Lumasiran had an acceptable safety profile, with no serious adverse events or study discontinuations attributed to treatment. In part A, increases in mean plasma glycolate concentration, a measure of target engagement, were observed in healthy participants. In part B, patients with primary hyperoxaluria type 1 had a mean maximal reduction from baseline of 75% across dosing cohorts in 24-hour urinary oxalate excretion. All patients achieved urinary oxalate levels ≤1.5 times the upper limit of normal.
Conclusions
Lumasiran had an acceptable safety profile and reduced urinary oxalate excretion in all patients with primary hyperoxaluria type 1 to near-normal levels.
Clinical Trial registry name and registration number:
Study of Lumasiran in Healthy Adults and Patients with Primary Hyperoxaluria Type 1, NCT02706886
Introduction
Primary hyperoxaluria type 1 is a rare autosomal recessive disorder characterized by overproduction of oxalate due to a deficiency of the hepatocyte peroxisomal enzyme alanine-glyoxylate aminotransferase (AGT) (1). Primary hyperoxaluria type 1 is the most severe form of primary hyperoxaluria and accounts for approximately 80% of diagnosed cases of primary hyperoxaluria (2 –4). The diagnosed prevalence of primary hyperoxaluria type 1 ranges from one to three cases per 1,000,000 (5,6). However, a recent analysis suggests that the genetic prevalence could be two to six times greater (7). Moreover, consanguineous unions further increase the prevalence of the disease (8,9). Most patients present in early childhood, and severe cases present with kidney failure in early infancy (7,10).
In primary hyperoxaluria type 1, deficiency of AGT, which converts glyoxylate to glycine, results in glyoxylate oxidation to oxalate (Figure 1). Excess oxalate is excreted by the kidneys where it can form insoluble calcium oxalate crystals, resulting in nephrocalcinosis, recurrent kidney stones, and, ultimately, kidney failure (2,8). As kidney function worsens, oxalate excretion is further compromised, leading to a progressive increase in plasma oxalate (8,11). Deposition of calcium oxalate crystals in other tissues, including bone, blood vessels, heart, eye, and skin, results in a devastating condition recognized clinically as systemic oxalosis.
Figure 1.

Defect in glyoxylate metabolism in hepatocytes of patients with primary hyperoxaluria type 1 and lumasiran therapeutic hypothesis. Patients with primary hyperoxaluria type 1 have reduced or absent alanine-glyoxylate aminotransferase (AGT) function, resulting in increased oxalate and glycolate production by the hepatocyte, both of which are excreted by the kidneys. AGT in the liver peroxisome metabolizes glyoxylate to glycine. When AGT is deficient (black X), glyoxylate cannot be metabolized to glycine. Excess glyoxylate accumulates and is converted to oxalate. Excess hepatic oxalate is then transported to the kidney for excretion in the urine, leading to hyperoxaluria. In the kidneys, excess oxalate combines with calcium, which due to its insolubility, can readily crystallize in the urinary tract and lead to recurrent urolithiasis and nephrocalcinosis, causing progressive kidney damage. As kidney function worsens, there is a reduction in the capacity of the kidneys to clear oxalate from the blood, leading to an increase in plasma oxalate and subsequent deposition of calcium oxalate crystals in tissues and vital organs, including bone, heart, retina, and skin, recognized clinically as systemic oxalosis. Lumasiran targets liver hydroxyacid oxidase 1 mRNA (red X), decreasing production of glycolate oxidase (GO), and hence, reducing hepatic oxalate production and delivery of oxalate to the kidneys for excretion. GR, glyoxylate reductase; LDH, lactate dehydrogenase. Adapted from ref. 27, with permission.
Hyperhydration, high-dose pyridoxine (a coenzyme for AGT), and crystallization inhibitors are recommended in patients with relatively preserved kidney function and may delay disease progression (8,12). However, these treatments are burdensome for patients and caregivers. Patients who progress to kidney failure and have high plasma oxalate levels are often treated with intensive hemodialysis schedules (6 d/wk), which often remain inadequate to offset the production of oxalate (13). The only option for metabolic correction is liver transplantation, and for those with advanced kidney disease, a dual liver-kidney transplantation is required. However, transplantation is associated with significant morbidity and mortality (14 –16) and may not be readily accessible. As such, patients with primary hyperoxaluria type 1 have an urgent need for new treatments that can effectively reduce hepatic oxalate production.
Lumasiran is a subcutaneously administered RNA interference (RNAi) therapeutic previously demonstrated to reduce the hepatic production of oxalate in animal models by degrading glycolate oxidase mRNA (17). Efficient targeting of lumasiran to hepatocytes is achieved via cognate interaction of N-acetyl galactosamine ligand covalently linked to the RNAi and the hepatocyte asialoglycoprotein receptor. Glycolate oxidase catalyzes the conversion of glycolate to glyoxylate; hence, its inhibition reduces hepatic oxalate production by limiting the availability of glyoxylate, the substrate for metabolic conversion to oxalate, while also increasing glycolate levels (17,18) (Figure 1). Rare cases of glycolate oxidase deficiency in humans, with marked and well-tolerated elevations in glycolate levels (19 –21), suggest that suppression of glycolate oxidase may be a safe strategy for the treatment of primary hyperoxaluria type 1 (22).
In this first-in-human phase 1/2 trial, we evaluated the safety, pharmacokinetic, and pharmacodynamic profiles of lumasiran in healthy participants and in patients with primary hyperoxaluria type 1.
Materials and Methods
Study Design and Oversight
This randomized, single-blind, placebo-controlled, phase 1/2 trial was conducted in two parts between March 8, 2016 and January 23, 2019. Part A evaluated single doses in ascending dose groups in healthy adult participants at a single center in the United Kingdom. Part B evaluated multiple doses in ascending dose groups in adult and pediatric patients with primary hyperoxaluria type 1 in a multicenter study conducted at nine clinical sites across five countries (Israel, France, Germany, the United Kingdom, and The Netherlands). The protocol, including a detailed summary of changes, and statistical analysis plan were developed by the sponsor, Alnylam Pharmaceuticals, and are available at ClinicalTrials.gov. Important changes to methods after trial commencement are summarized in Supplemental Table 1. The trial was approved by independent ethics committees/institutional review boards in each country or site, per local regulations. The study was conducted in accordance with Good Clinical Practice guidelines of the International Council for Harmonization and the provisions of the Declaration of Helsinki. All patients or their legal guardians provided written informed consent. A safety review committee evaluated safety prior to all dose escalation decisions and convened monthly for the duration of the study. Stopping rules were on the basis of toxicity using the standard toxicity grading of the Common Terminology Criteria for Adverse Events (CTCAE) to assess adverse events (AEs). The protocol included two sets of rules: one for study cohorts in both parts A and B and one for individual patients in part B. Detailed stopping rules are provided in the protocol.
Participants
Eligibility criteria for part A healthy participants are outlined in Supplemental Material. Eligible participants for part B were patients aged 6–64 years with a diagnosis of primary hyperoxaluria type 1 confirmed by genetic analysis or reduced AGT enzyme activity. Additional inclusion criteria for part B included 24-hour urinary oxalate excretion >0.7 mmol/24 h per 1.73 m2 (upper limit of normal [ULN] is 0.46 mmol/24 h per 1.73 m2) (23) and eGFR>45 ml/min per 1.73 m2 calculated on the basis of the Modification of Diet in Renal Disease formula for adults and the Schwartz bedside formula for children, as detailed in the protocol. These criteria ensured kidney function was sufficient for urinary oxalate excretion to reliably reflect hepatic oxalate production. Full listings of part A and part B eligibility criteria are provided in the protocol.
Randomization and Blinding
During screening, and after eligibility was confirmed for part A and the initial three cohorts in part B, participants were randomly assigned (3:1) to either lumasiran or placebo in each dose-level cohort in a single-blinded manner. All patients in the open-label expansion cohorts in part B received lumasiran and therefore were not blinded. As members of the safety review committee, investigators and sponsor were not blinded for either part to permit ongoing review of safety, tolerability, pharmacokinetics, and pharmacodynamic data. No study participant was a member of more than one cohort. A unique identification number, incorporating the clinical study center number, was assigned sequentially to each study participant. The clinical study center pharmacy staff randomized the participants in accordance to a cohort-specific, computer-generated randomization list generated by the contract research organization biostatistician. Randomized treatment assignment was on the basis of permuted block randomization method with a block size of four with a ratio of 3:1 (lumasiran to placebo). Block size was not known to the investigators. Because lumasiran was potentially visually distinguishable from the placebo, syringes containing dispensed study drug were masked in the pharmacy prior to being transferred to the clinic.
Procedures
In part A, the starting dose was on the basis of preclinical data (17). Four cohorts of eight subjects were randomized 3:1 to receive a single subcutaneous injection of lumasiran at 0.3, 1, 3, or 6 mg/kg body wt (milligrams per kilogram) or sterile normal saline solution (0.9% NaCl) as a placebo (Supplemental Figure 1). Healthy participants were followed for at least 57 days after the single injection. Healthy participants returned to the site on day 85, and then every 28 days until plasma glycolate decreased to 20% above baseline or less than or equal to ULN (≤14 µmol/L). The lowest dose to have a pharmacologic effect in part A, in the opinion of the safety review committee, was selected as the starting dose in part B. Patients with primary hyperoxaluria type 1 were administered up to three doses of lumasiran (1 or 3 mg/kg) at monthly or quarterly intervals (Figure 2). The monthly interval was on the basis of preclinical data, in which a dosing interval of 1 month was required to maintain target suppression. Preliminary data from part A informed the option for quarterly dosing in part B.
Figure 2.
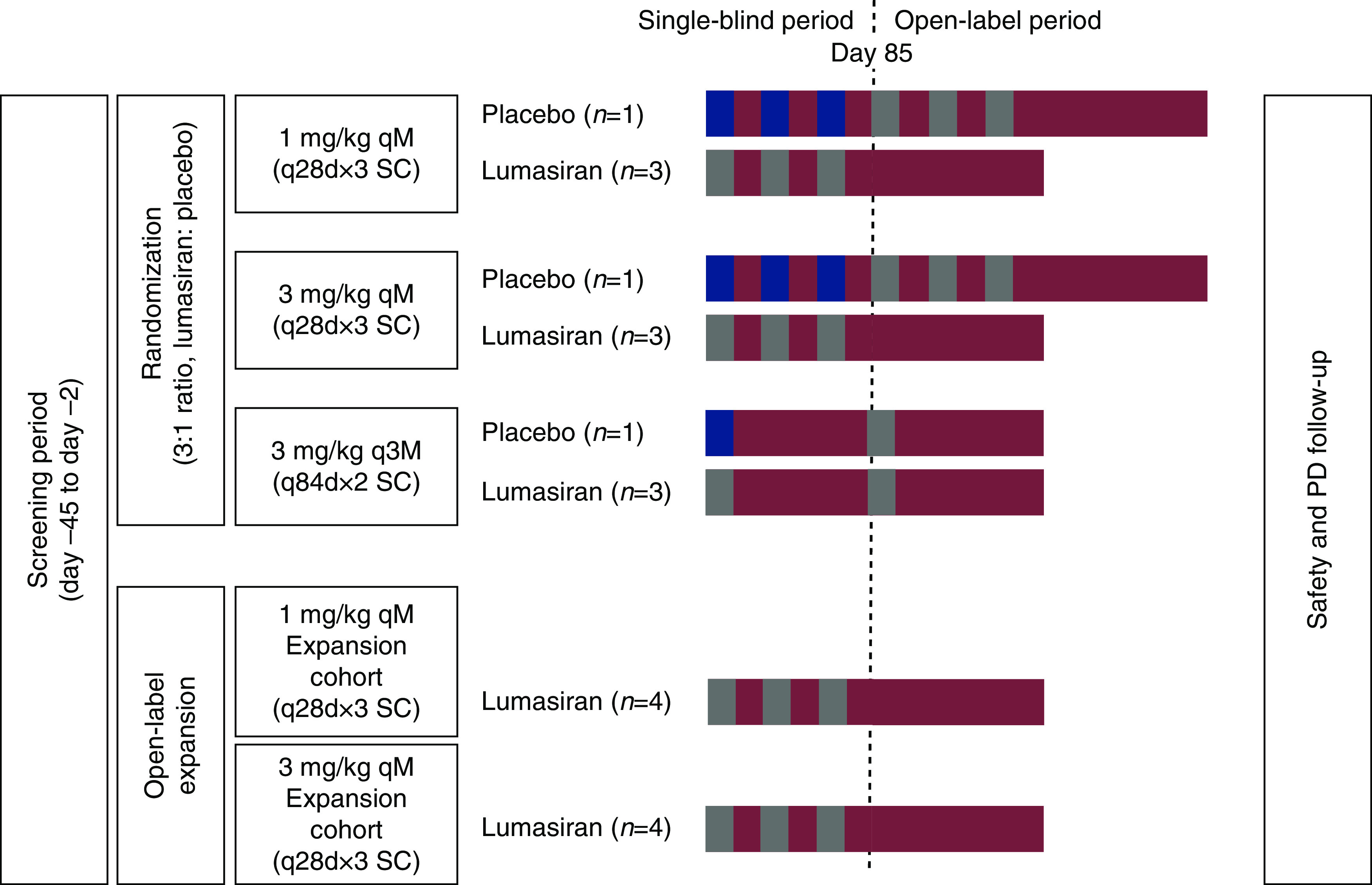
Study design part B (patients with primary hyperoxaluria type 1). In this multiple ascending dose part of the study, patients with primary hyperoxaluria type 1 were screened within 45 days prior to study drug administration. Patients were randomized 3:1 to lumasiran (gray boxes) or placebo (blue boxes) between days –1 and 1 into each dosing cohort (1 mg/kg once per month, 3 mg/kg once per month, or 3 mg/kg once every 3 months). All patients received the first dose of lumasiran or placebo on day 1. Patients who received study drug once per month received the remaining two single-blind doses at days 29 and 57. Patients who received study drug once every 3 months received the remaining dose at day 85. Patients initially randomized to placebo received subsequent dosing of lumasiran on day 85 at the same dose administered to the cohort into which they were initially randomized. The 1- and 3-mg/kg monthly dosing cohorts were each expanded by an additional four patients who received three open-label doses of lumasiran. These patients were not randomized or blinded as each patient received open-label lumasiran starting on day 1. The 1-mg/kg once per month expansion cohort was enrolled first, followed by the 3-mg/kg once per month expansion cohort. During the PD follow-up period, patients were monitored for at least 12 weeks (84 days) following the last dose of study drug. N, number of patients; PD, postdose; q28d, every 28 days; q84d, every 84 days; qM, once monthly; q3M, once quarterly; SC, subcutaneous.
Patients initially assigned to placebo crossed over to open-label lumasiran treatment on day 85 at the same dose as the group to which they had been enrolled. Following the blinded period in part B, two open-label expansion cohorts were initiated. The doses of lumasiran (1.0 mg/kg once monthly or 3.0 mg/kg once monthly) were determined on the basis of review of the accumulated safety, tolerability, and pharmacodynamic data and endorsed by the safety review committee. The duration of follow-up in part B was amended during the study to minimize the duration of exposure to high urinary oxalate levels for patients who wished to continue receiving lumasiran in the separate open-label extension study (NCT03350451; results are not reported here). All patients were followed for at least 12 weeks (Supplemental Table 1). All patients continued their standard of care therapy through the duration of the study, including hyperhydration, pyridoxine, and crystallization inhibitors.
Outcomes
The primary outcome of this study was the incidence of AEs. Safety assessments also included vital signs, 12-lead electrocardiograms, clinical laboratory assessments, physical examinations, and echocardiography (part B only). All AEs were categorized according to the Medical Dictionary for Regulatory Activities, version 16.0 or higher, and were graded using the National Cancer Institute, CTCAE, version 5.0.
Secondary and exploratory outcomes included pharmacokinetic and pharmacodynamic parameters and evaluation of antidrug antibodies. In part A, secondary pharmacodynamic parameters included plasma and spot urinary glycolate levels (healthy participant sample numbers for pharmacodynamic and pharmacokinetic analyses are in Supplemental Material). Part B included additional secondary parameters of 24-hour urinary oxalate excretion, urinary oxalate-creatinine ratio, and calculated creatinine clearance to estimate kidney function. Exploratory outcomes included plasma oxalate concentration in part B patients.
Oxalate was measured using the dual enzymatic modified Trinity kit method in urine and with an ion-chromatographic method in plasma. Urinary and plasma glycolate levels were measured using gas chromatography-mass spectrometry. Urinary creatinine was determined using an enzymatic colorimetric method. Pharmacokinetic measurements of lumasiran in plasma and urine samples were analyzed by liquid chromatography–time-of-flight mass spectrometry, and noncompartmental measurements were calculated using Phoenix WinNonlin, version 7 (Certara). Antidrug antibodies against lumasiran were evaluated using a bispecific ELISA to detect IgG and IgM with a minimal required dilution of 50-fold. Antidrug antibodies were tested on the basis of a tierwise approach to screen, confirm, and titer the response.
Statistical Analyses
Formal power calculations were not conducted for this study where assessment of safety and tolerability was the primary objective. Sample size was determined on the basis of feasibility considerations, including the rarity of the disease. Safety analyses included all participants who received at least one injection of lumasiran or placebo. Pharmacokinetic and pharmacodynamic analyses included all participants with at least one evaluable postdose blood or urine sample collected. Pharmacokinetic analyses included all participants who received at least one dose of lumasiran (24 of the 32 healthy participants in part A and all 20 patients in part B). All 32 healthy participants (part A) and 20 patients with primary hyperoxaluria type 1 (part B) were included in the pharmacodynamic analysis.
Statistical analyses were performed with Statistical Analysis Software (SAS software, version 9.2 or higher; SAS Institute). Descriptive statistics were used to analyze the data. Because there was no intention to apply the results observed in this phase 1/2 study to any other sample of similar patients, no inferential statistics were performed. For continuous variables, mean, SD, and SEM are reported. For categoric and ordinal variables, frequencies and percentages of patients in each category are reported.
Results
Trial Population
Part A enrolled 32 healthy participants, and part B enrolled 20 patients with primary hyperoxaluria type 1 (Supplemental Figure 1). Data from part A are presented from day 1 to day 85 (placebo comparison period). Data from part B are presented in two ways: the placebo comparison period from day 1 to day 85, which compares the first 85 days on study for each patient, and the lumasiran treatment experience from day 1 to day 197, which includes the time after the first lumasiran dose for each patient.
Demographic characteristics were generally balanced among cohorts within part A and part B (Table 1). Baseline clinical characteristics of patients with primary hyperoxaluria type 1, including eGFR and baseline urinary oxalate excretion, were balanced among dosing cohorts (Supplemental Table 2).
Table 1.
Demographic and clinical characteristics of the study participants at baseline
| Characteristic | Part A Healthy Participants | Part B Patients with Primary Hyperoxaluria Type 1 | |||
|---|---|---|---|---|---|
| Lumasiran, n=24 | Placebo, n=8 | Lumasiran, n=17 | Placebo, n=3 | Overall,a n=20 | |
| Age, yr | 29 (6) | 30 (6) | 14 (8) | 21 (19) | 15 (10) |
| Women, n (%) | 11 (46) | 5 (63) | 12 (71) | 1 (33) | 13 (65) |
| Race, n (%) | |||||
| White | 18 (75) | 7 (88) | 13 (77) | 2 (67) | 15 (75) |
| Black | 2 (8) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Asian | 1 (4) | 1 (13) | 3 (18) | 1 (33) | 4 (20) |
| Other | 3 (13) | 0 (0) | 1 (6) | 0 (0) | 1 (5) |
| Body weight, kg | 72 (12) | 67 (14) | 47 (25) | 64 (43) | 50 (27) |
| Height, cm | 172 (9) | 173 (8) | 146 (20) | 158 (30) | 148 (21) |
| BMI, kg/m2 | 24.3 (2.4) | 22.3 (3.0) | 20.7 (5.3) | 23.0 (8.2) | 21.0 (5.6) |
| Age at diagnosis, yr | — | — | 4 (3) | 9 (3) | 4 (4) |
| Genotype, n (%) | |||||
| PR/*b | — | — | 1 (6) | 1 (33) | 2 (10) |
| M/M or M/N | — | — | 9 (53) | 1 (33) | 10 (50) |
| N/N | — | — | 7 (41) | 1 (33) | 8 (40) |
| Pyridoxine use, n (%) | — | — | 10 (59) | 3 (100) | 13 (65) |
| 24-h urine oxalate excretion,c mmol/24 h per 1.73 m2 | — | — | 1.66 (0.64) | 1.96 (0.32) | 1.71 (0.60) |
| 24-h urine oxalate-creatinine ratio,d mg/mg | — | — | 0.17 (0.08) | 0.18 (0.04) | 0.17 (0.07) |
| eGFR,e ml/min per 1.73 m2 | — | — | 82 (21), n=11 | 61 (12), n=2 | 78 (21), n=14 |
| Plasma oxalate,f µmol/L | — | — | 7.9 (4.1) | 15.6 (6.9) | 8.8 (4.8) |
Data are mean (SD), unless otherwise stated. n, number of patients; BMI, body mass index; —, not applicable.
Patients initially randomized to placebo were rebaselined, and their first day of lumasiran administration was set at day 1.
Genotype: M, Missense; N, Nonsense; PR, Pyridoxine-Responsive; *, any genotype of PR, M, or N.
Upper limit of normal =0.46 mmol/24 h per 1.73 m2; 1 mmol/24 h per 1.73 m2=88 mg/24 h per 1.73 m2.
1 mg/mg=1.256 mmol/mmol.
eGFR was calculated on the basis of the Modification of Diet in Renal Disease formula for patients ≥18 years of age and the Schwartz bedside formula for patients <18 years of age.
Upper limit of normal =1.6 µmol/L.
Primary Outcome Measure
The majority of AEs in this trial were mild to moderate in severity and were considered unrelated to the study treatment. No lumasiran-related serious AEs and no AEs leading to death, treatment discontinuation, or withdrawal from the study were reported in part A or part B.
In part A (days 1–85), AEs were reported in 20 (83%) lumasiran-treated and five (63%) placebo-treated healthy participants (Supplemental Table 3). The most common AEs reported in part A in lumasiran-treated healthy participants were nasopharyngitis (29%), headache (21%), and injection site pain (17%). There were no serious AEs, and one (4%) healthy participant who received 0.3 mg/kg lumasiran had a severe AE that was assessed as not related to study drug. In the lumasiran-treated cohort, four (17%) healthy participants had injection site reactions assessed as related to study drug by the investigator; all were mild, transient injection site pain.
During the placebo comparison period of part B (days 1–85), AEs were reported in ten (59%) lumasiran-treated patients and two (67%) placebo-treated patients (Table 2). The most common AEs reported in patients treated with lumasiran were abdominal pain (18%), headache (18%), rhinitis (12%), nephrolithiasis (12%), and cough (12%). Most AEs were mild in severity, with serious AEs reported in one placebo-treated and two (12%) lumasiran-treated patients; none were considered related to treatment (Table 2, Supplemental Table 4). There was one severe AE in the placebo group and no severe AEs in the lumasiran group. Two patients had AEs related to lumasiran, which consisted of mild or moderate transient injection site reactions (Table 2).
Table 2.
Primary outcome measure: Treatment-emergent adverse events in patients with primary hyperoxaluria type 1 during the placebo comparison period (days 1–85)
| Adverse Events, n (%) | Placebo, n=3 | Lumasiran | |||
|---|---|---|---|---|---|
| 1 mg/kg Once Monthly, n=7 | 3 mg/kg Once Monthly, n=7 | 3 mg/kg Once Every 3 mo, n=3 | All Lumasiran, n=17 | ||
| Any adverse event | 2 (67) | 5 (71) | 4 (57) | 1 (33) | 10 (59) |
| Serious adverse eventa | 1 (33) | 0 (0) | 2 (29) | 0 (0) | 2 (12) |
| Severe adverse eventb | 1 (33) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Adverse event leading to treatment discontinuation | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Death | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Injection site reactionsc | 0 (0) | 1 (14) | 1 (14) | 0 (0) | 2 (12) |
n, number of patients.
Patient treated with placebo: nephrolithiasis and pyelonephritis acute. Patients treated with lumasiran: vomiting and nephrolithiasis.
Pyelonephritis acute.
Includes all adverse events mapping to the Medical Dictionary for Regulatory Activities high-level term injection site reactions.
No clinically significant changes in laboratory measures or in findings from physical examinations were noted in either part. Additional details on safety outcomes, including analysis of all 20 patients after receiving lumasiran, are included in Supplemental Material; results were consistent with those in the placebo comparison period (Supplemental Table 5).
Secondary and Exploratory Outcome Measures
Pharmacokinetics.
Pharmacokinetic profiles (Supplemental Figure 2) showed a rapid elimination of lumasiran from the systemic circulation, indicative of rapid hepatic uptake. Urinary excretion is a minor route of elimination for lumasiran, accounting for 7%–26% of drug clearance (additional pharmacokinetic data are summarized in Supplemental Tables 6 and 7). In part B, three positive antidrug antibody measurements were observed in two patients who received 1 mg/kg lumasiran. All measurements were transient low titers (1:50), were not associated with AEs, and had no effect on pharmacokinetic or pharmacodynamic parameters.
Pharmacodynamics.
To provide an informative measure of target engagement, plasma glycolate levels were assessed in part A. Healthy participants received single subcutaneous doses of lumasiran 0.3, 1, 3, or 6 mg/kg with subsequent increases in plasma glycolate at higher levels (Figure 3). Median time to recovery (plasma glycolate <20% above baseline or less than or equal to ULN) after a single dose was 134.5 (range, 57–225) days in the 3-mg/kg cohort and 169 (range, 113–242) days in the 6-mg/kg cohort. Urinary glycolate showed a similar response (Supplemental Figure 3). The lowest dose in part A producing an appreciable pharmacologic effect, in the opinion of the safety review committee, was 1 mg/kg, which was selected as the starting dose in part B.
Figure 3.

Plasma glycolate levels after a single dose of lumasiran in healthy adult participants (part A).
Three lumasiran treatment regimens (1 or 3 mg/kg monthly or 3 mg/kg once every 3 months) were examined in part B in patients with primary hyperoxaluria type 1 (Figure 4A). In all 20 patients after treatment with lumasiran (days 1–197), the mean maximum reduction in 24-hour urinary oxalate excretion levels was 75% (range, 43%–92%) from baseline (1.69 mmol/24 h per 1.73 m2). The reduction observed in 24-hour urinary oxalate excretion was rapid and sustained over time (Figure 4A). Corresponding trends in 24-hour urine oxalate-creatinine ratios were observed (Figure 4B, Supplemental Table 8).
Figure 4.
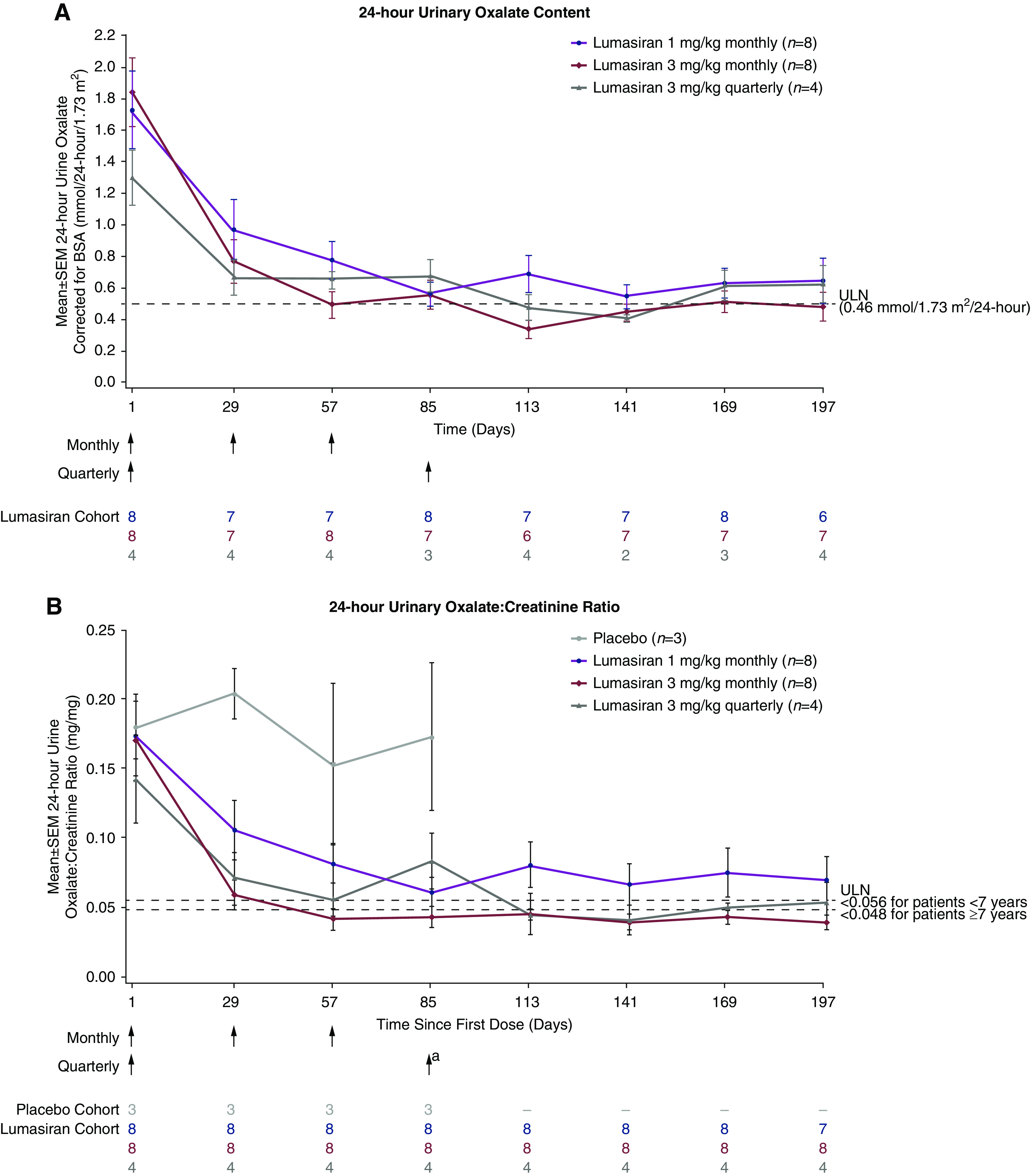
Urinary oxalate assessments after multiple doses of lumasiran in patients with primary hyperoxaluria type 1 (part B). (A) 24-hour urinary oxalate excretion (mmol per 24 hours per 1.73 m2). (B) 24-hour urinary oxalate-creatinine ratio. Values for patients during placebo treatment are only shown in (B) because incomplete collections are excluded from quantification of 24-hour urinary oxalate content, resulting in limited data for this measure. BSA, body surface area; N, number of patients with valid collections at each time point; ULN, upper limit of normal. aPatients initially randomized to placebo received their first dose of lumasiran on day 85. For the purpose of this analysis, they are also included in the lumasiran dosing cohort in which they were randomized, with day 1 relative to first dose of lumasiran; the patient randomized to placebo in 3-mg/kg quarterly dosing received a single dose of lumasiran, designated as day 1.
All 20 lumasiran-treated patients achieved a near-normal 24-hour urinary oxalate excretion of ≤1.5 times ULN (≤0.69 mmol/24 h per 1.73 m2), and 15 patients (75%) were within the normal range (≤0.46 mmol/24 h per 1.73 m2). Of the 12 patients treated with 3 mg/kg once monthly or once every 3 months, 11 (92%) had 24-hour urinary oxalate excretion levels within the normal range.
Following administration of lumasiran, there was an observed reduction in plasma oxalate concentration in a subset of patients with available values (Supplemental Figure 4, Supplemental Table 8), with a mean maximum reduction of 74% (range, 38%–94%). Changes observed in urinary glycolate-creatinine ratios after treatment were as anticipated on the basis of lumasiran’s mechanism of action (Supplemental Table 8). Analyses of plasma glycolate data are not presented for part B of this study due to a laboratory issue (Supplemental Material).
Discussion
In this first-in-human study of lumasiran, we demonstrate the preliminary safety, tolerability, and proof of mechanism in healthy adult participants along with lowering of urinary oxalate in pediatric and adult patients with primary hyperoxaluria type 1. The majority of lumasiran-related AEs consisted of mild or moderate transient injection site reactions, and no serious or severe AEs were considered lumasiran related. There were no clinically significant changes in clinical laboratory measures, including hematology and liver function tests.
Previous identification of a rare human case of glycolate oxidase deficiency, along with other studies, suggested that suppression of this enzyme in the oxalate synthesis pathway could offer a viable strategy for substrate reduction therapy in primary hyperoxaluria type 1 by trapping substrate in a benign form (glycolate) that would not promote kidney injury (19–22,24). Of note, calcium glycolate is approximately 15,000-fold more soluble than calcium oxalate, and there are no known toxicities of increased urinary glycolate excretion. Consequently, lumasiran was developed to test the hypothesis that glycolate oxidase inhibition would starve substrate for oxalate production. Preclinical work using small interfering RNA (siRNA) targeting the mRNA encoding glycolate oxidase confirmed this approach could interrupt oxalate production in a rodent model of primary hyperoxaluria type 1 (17,18) and could provide a potentially transformative therapy for patients with this disease.
On the basis of observations in glycolate oxidase–deficient individuals and results from lumasiran treatment of healthy rodents and primates (17), plasma glycolate measures were predicted to provide an informative measure of target engagement. The responses observed in plasma glycolate concentrations in healthy participants allowed for efficient selection of doses for use in patients. Pharmacokinetic profiles showed that lumasiran is rapidly absorbed and eliminated from plasma upon repeated monthly or quarterly dosing, and the kidney route is a minor pathway of elimination for lumasiran.
All 20 lumasiran-treated patients achieved a normal or near-normal 24-hour urinary oxalate excretion. The potential clinical significance of this result is clear in the context of the natural history of the disease. Among patients with primary hyperoxaluria without kidney failure at baseline, the degree of hyperoxaluria at diagnosis appears to be associated with the subsequent risk of kidney failure (4). As described in a recent publication supported by the Kidney Health Initiative, a substantial reduction in urine oxalate (e.g., near normalization) is expected to predict clinical benefit in patients with primary hyperoxaluria type 1 (25). These findings suggest that the profound lowering of urinary oxalate excretion observed following lumasiran treatment may lead to improvements in clinical outcomes, such as kidney stone events, nephrocalcinosis, and kidney function. The 3-mg/kg once monthly dose regimen appeared to lead to more rapid and higher-magnitude reduction of 24-hour urinary oxalate excretion relative to the 1-mg/kg once monthly and 3-mg/kg once every 3 months regimens. In addition, in patients treated with 3 mg/kg once every 3 months, lumasiran demonstrated a sustained effect on urinary oxalate throughout the 3-month dosing interval. These results are consistent with evidence that chemically stabilized siRNA molecules, such as lumasiran, slowly release the functional siRNA into the RNAi pathway over time (24). Both rapid onset and sustained reduction are important factors in selecting appropriate dosage regimens due to the episodic and unpredictable exacerbations of the disease and the potential acute loss of kidney function in patients with primary hyperoxaluria type 1 (12,26).
Patients enrolled in this trial were required to have an eGFR>45 ml/min per 1.73 m2 so that urinary oxalate excretion would be unimpaired and the effect of lumasiran in suppressing hepatic oxalate production would be accurately reflected in 24-hour urine oxalate excretion measurements. Importantly, these patients had modest elevations in plasma oxalate, which lumasiran treatment appeared to reduce, although available data were limited.
This is the first report of the use of RNAi to successfully lower oxalate in patients with primary hyperoxaluria type 1. Study limitations included its small number of patients, descriptive statistical evaluations, limited duration of dosing, and the inability to present plasma glycolate data for part B of this study due to a laboratory issue. Following study completion, all patients in part B were enrolled in a long-term, open-label extension study for continued dosing (NCT03350451; results will be reported elsewhere).
In conclusion, lumasiran demonstrated an acceptable safety profile in adult and pediatric patients with primary hyperoxaluria type 1. All patients exhibited normal or near-normal levels of urinary oxalate excretion after treatment with lumasiran. These data support the therapeutic hypothesis and the three ongoing phase 3 clinical trials (NCT03681184, NCT03905694, and NCT04152200), which include patients of all ages and at all stages of kidney function. This novel approach has the potential to provide the first nonsurgical therapeutic for all patients with primary hyperoxaluria type 1.
Disclosures
P. Cochat reports employment with Université Claude-Bernard Lyon 1, France, and Hospices Civils de Lyon, France; consultancy fees and invitations to scientific meetings from Alnylam Pharmaceuticals and Dicerna during the conduct of the study; invitations to scientific meetings from Advicenne and Sanofi Aventis outside of the submitted work; receiving honoraria from Alnylam Pharmaceuticals, Dicerna, and Scientific Advisory Board (SAB); and serving as an expert for Advicenne Pharma, principal investigator (PI) and SAB member of Alnylam Pharmaceuticals, PI and SAB member of Dicerna, and editor-in-chief of Elsevier-Masson Eds. G. Deschênes reports employment with Assistance Publique-Hôpitaux de Paris Robert Debré, Paris University; consultancy agreements with Chiesi, Raptor Phamaceuticals, and SANOFI Genzyme; serving as Advicenne biocodex principal investigator, Alexion principal investigator, Alnylam Pharmaceuticals principal investigator, Biocodex principal investigator, Dicerna principal investigator, Oxthera principal investigator, and Roche principal investigator; receiving honoraria from Alnylam Pharmaceuticals, Biocodex, Chiesi, Fresenius, Novartis, and Orphaneurope; and patents and inventions for Institut National de la Santé et de la Recherche Médicale transfert. G. Deschênes also reports consultancy fees from Alnylam Pharmaceuticals, Biocodex, and Dicerna Pharmaceuticals and was a PI for research funded by OxThera during the conduct of the study. D. V. Erbe is employed by, has a pending patent related to the research of, and holds stock in Alnylam Pharmaceuticals. Y. Frishberg reports employment with Shaare Zedek Medical Center, consultancy fees from Alnylam Pharmaceuticals, serving as a member of the Safety Review Committee for Alnylam Pharmaceuticals, and serving as a Data Monitoring Committee member for Alexion Pharmaceuticals. P. P. Garg reports employment with Alnylam Pharmaceuticals, holding stock in Alnylam Pharmaceuticals, and serving as a scientific advisor or membership of SQZ Biotech. J. W. Groothoff reports employment with Amsterdam University Medical Center (Academic Medical Center); consultancy agreements with Alnylam Pharmaceuticals, Dicerna Pharmaceuticals, and UniQure Pharmaceuticals; receiving research funding from Alnylam Pharmaceuticals, Dicerna Pharmaceuticals, and UniQure Pharmaceuticals; and serving as a scientific advisor or member of Alnylam Pharmaceuticals. B. A. Habtemarian reports employment with and holds stock in Alnylam Pharmaceuticals. J. Harambat reports employment with Bordeaux University Hospital, France; consultancy agreements with Alnylam Pharmaceuticals and Chiesi; receiving research funding from Advicenne, Alnylam Pharmaceuticals, and GSK; receiving honoraria from Alnylam Pharmaceuticals and Chiesi; and serving as European Society for Paediatric Nephrology/ European Renal Association - European Dialysis and Transplant Association Chairman, International Pediatric Nephrology Association member, European Society for Paediatric Nephrology member, European Dialysis and Transplant Association member, and on the Pediatric Nephrology editorial board. P. Haslett is a full-time employee of and holds stock in Alnylam Pharmaceuticals, the sponsor of the study reported in the article. S.-A. Hulton reports employment with Birmingham Women’s and Children’s Hospital NHS Foundation Trust; consultancy agreements with Alnylam Pharmaceuticals, Chiesi, and Dicerna; receiving research funding from Alnylam Pharmaceuticals and Dicerna; receiving honoraria from Alnylam Pharmaceuticals, Chiesi, and Dicerna; serving as president of the British Association for Paediatric Nephrology, vice president of the Renal Association, and a scientific advisor or member of the OxalEurope Committee; and speakers bureau for Alnylam Pharmaceuticals, Chiesi, and Dicerna. S.-A. Hulton also reports travel expenses to participate in clinical research meetings and consultancy fees paid to Birmingham Children’s Hospital Renal Research Fund from Alnylam Pharmaceuticals during the conduct of the study. J. C. Lieske reports employment with Mayo Clinic; consultancy agreements with Allena, Alnylam Pharmaceuticals, the American Board of Internal Medicine, Dicerna, Novobiome, Orfan, OxThera, Siemens, and Synlogic; receiving research funding from Allena, Alnylam Pharmaceuticals, Dicerna, OxThera, Retrophin, Siemens, and Synlogic; grants from Alnylam Pharmaceuticals during the conduct of the study; grants from Dicerna, Retrophin, OxThera, and Siemens outside the submitted work; other from Orfan-Bridgebio outside the submitted work; grants and other from Allena outside the submitted work; receiving honoraria from the American Board of Internal Medicine and UpToDate; and serving as a scientific advisor or member of American Board of Internal Medicine, Kidney International, and the Oxalosis & Hyperoxaluria Foundation. U. Lorch reports employment with Richmond Pharmacology Ltd., which is a clinical research organization that provides clinical research services to coauthor colleagues. J. Lu reports employment with and ownership interest in Alnylam Pharmaceuticals. D. Magen reports employment with Rambam Health Care Campus, Haifa, Israel and Technion–Israeli Institute of Technology, Haifa, Israel; consultancy agreements with Alnylam Pharmaceuticals; and receiving research funding and honoraria from Alnylam Pharmaceuticals. T. L. McGregor reports employment with and holds stock in Alnylam Pharmaceuticals. D. S. Milliner reports employment with the Mayo Clinic; receiving honoraria from Alnylam Pharmaceuticals and Synlogic Medical Advisory Committee personal honorarium; and serving on the advisory committee for Alnylam Pharmaceuticals, the data safety and monitoring committee for a clinical trial conducted by Dicerna, the data safety monitoring board for a clinical trial conducted by OxThera, and the editorial board for Urolithiasis. D. S. Milliner reports consultancy agreements with Allena Pharmaceutical Company, Alnylam Pharmaceuticals, Dicerna Pharmaceutical Company, and OxThera Pharmaceutical Company, with all consulting fees paid directly to the Mayo Clinic; she received no personal compensation for this work. D. S. Milliner also reports receiving research funding from Alnylam Pharmaceuticals, Dicerna, and OxThera, with all research funding provided to the Mayo Clinic and not to her personally. D. S. Milliner reports ongoing work with the Oxalosis and Hyperoxaluria Foundation, a nonprofit private foundation. This includes some research funding; there is no personal compensation. Additionally, D. S. Milliner reports ongoing work with the Kidney Health Initiative and Oxalosis & Hyperoxaluria Foundation for a KHI-sponsored project. S. Talamudupula reports employment with and ownership interest in Alnylam Pharmaceuticals. A. K. Vaishnaw is a full-time employee and stockholder of Alnylam Pharmaceuticals. W. G. van’t Hoff reports employment with the National Institute for Health Research Clinical Research Network; receiving research funding from Alnylam Pharmaceuticals with contract research income to the previous employing organization and travel/accommodation for investigator meeting; and receiving research funding from Ultragenyx with contract research income to the previous employing organization. W. G. van’t Hoff also reports travel expenses to participate in clinical research meetings and financial recompense for clinical trial participation, which was paid to his institute during the conduct of the study, and travel expenses for attending investigator meetings from Kyowa Kirin Pharmaceuticals outside of the published work.
Funding
This work was supported by Alnylam Pharmaceuticals.
Supplementary Material
Acknowledgments
The authors thank Dr. Marianne Sweetser for her valued contribution to this study. The authors acknowledge the medical writing services provided by Dr. Sofia Fountana of Adelphi Communications Ltd., Macclesfield, United Kingdom, in accordance with the Good Publication Practice guidelines, funded by Alnylam Pharmaceuticals, and the editorial assistance of Dr. Ana Camejo, of Alnylam Pharmaceuticals.
The authors who are employees of Alnylam Pharmaceuticals conducted the formal statistical analysis and validation of the data and, in conjunction with the other authors, contributed to the interpretation and presentation of the data. All authors interpreted the data, collaborated in manuscript preparation, decided to publish the manuscript, and vouch for the accuracy and completeness of the data and for the adherence of the trial to the protocol.
Data Sharing Statement
Because of the sensitive nature of the data collected for this study, the dataset will not be made available to other researchers. The redacted Study Protocol and Statistical Analysis Plan are publicly available at ClinicalTrials.gov.
Footnotes
Published online ahead of print. Publication date available at www.cjasn.org.
Supplemental Material
This article contains the following supplemental material online at http://cjasn.asnjournals.org/lookup/suppl/doi:10.2215/CJN.14730920/-/DCSupplemental.
Supplemental Material. Healthy participant eligibility criteria, healthy participant sample numbers for pharmacodynamic and pharmacokinetic analyses, additional safety outcomes, and additional details from the pharmacodynamic analysis.
Supplemental Figure 1. CONSORT diagram.
Supplemental Figure 2. Plasma pharmacokinetic profile of lumasiran (A) after a single subcutaneous injection in healthy participants and (B) after a single or multiple subcutaneous injections in patients with primary hyperoxaluria type 1.
Supplemental Figure 3. Spot urinary glycolate-creatinine ratios after a single dose of lumasiran in healthy participants (part A).
Supplemental Figure 4. Plasma oxalate after multiple doses of lumasiran in patients with primary hyperoxaluria type 1 (part B).
Supplemental Table 1. Summary of changes to the conduct of the study.
Supplemental Table 2. Clinical characteristics of the patients with primary hyperoxaluria type 1 at baseline (part B).
Supplemental Table 3. Treatment-emergent adverse events in part A healthy participants during the placebo comparison period (days 1–85).
Supplemental Table 4. Treatment-emergent adverse events in part B patients with primary hyperoxaluria type 1 during the placebo comparison period (days 1–85; additional results).
Supplemental Table 5. Treatment-emergent adverse events in part B patients with primary hyperoxaluria type 1 during the lumasiran treatment experience (days 1–197).
Supplemental Table 6. Pharmacokinetic parameters (part A healthy participants).
Supplemental Table 7. Pharmacokinetic parameters (part B patients with primary hyperoxaluria type 1).
Supplemental Table 8. Oxalate and glycolate parameters in multiple ascending dose cohorts of lumasiran in patients with primary hyperoxaluria type 1 from days 1–197 (part B).
Supplemental Table 9. Study collaborators.
References
- 1.Williams HE, Smith LH Jr: L-glyceric aciduria: A new genetic variant of primary hyperoxaluria. N Engl J Med 278: 233–238, 1968 [DOI] [PubMed] [Google Scholar]
- 2.Danpure CJ, Jennings PR: Peroxisomal alanine:glyoxylate aminotransferase deficiency in primary hyperoxaluria type I. FEBS Lett 201: 20–24, 1986 [DOI] [PubMed] [Google Scholar]
- 3.Hoppe B: An update on primary hyperoxaluria. Nat Rev Nephrol 8: 467–475, 2012 [DOI] [PubMed] [Google Scholar]
- 4.Zhao F, Bergstralh EJ, Mehta RA, Vaughan LE, Olson JB, Seide BM, Meek AM, Cogal AG, Lieske JC, Milliner DS; Investigators of Rare Kidney Stone Consortium: Predictors of incident ESRD among patients with primary hyperoxaluria presenting prior to kidney failure. Clin J Am Soc Nephrol 11: 119–126, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cochat P, Deloraine A, Rotily M, Olive F, Liponski I, Deries N: Epidemiology of primary hyperoxaluria type 1: Société de Néphrologie and the Société de Néphrologie Pédiatrique. Nephrol Dial Transplant 10[Suppl 8]: 3–7, 1995 [DOI] [PubMed] [Google Scholar]
- 6.van Woerden CS, Groothoff JW, Wanders RJ, Davin JC, Wijburg FA: Primary hyperoxaluria type 1 in The Netherlands: Prevalence and outcome. Nephrol Dial Transplant 18: 273–279, 2003 [DOI] [PubMed] [Google Scholar]
- 7.Hopp K, Cogal AG, Bergstralh EJ, Seide BM, Olson JB, Meek AM, Lieske JC, Milliner DS, Harris PC; Rare Kidney Stone Consortium: Phenotype-genotype correlations and estimated carrier frequencies of primary hyperoxaluria. J Am Soc Nephrol 26: 2559–2570, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cochat P, Rumsby G: Primary hyperoxaluria. N Engl J Med 369: 649–658, 2013 [DOI] [PubMed] [Google Scholar]
- 9.Kamoun A, Lakhoua R: End-stage renal disease of the Tunisian child: Epidemiology, etiologies, and outcome. Pediatr Nephrol 10: 479–482, 1996 [DOI] [PubMed] [Google Scholar]
- 10.Harambat J, Fargue S, Acquaviva C, Gagnadoux MF, Janssen F, Liutkus A, Mourani C, Macher MA, Abramowicz D, Legendre C, Durrbach A, Tsimaratos M, Nivet H, Girardin E, Schott AM, Rolland MO, Cochat P: Genotype-phenotype correlation in primary hyperoxaluria type 1: The p.Gly170Arg AGXT mutation is associated with a better outcome. Kidney Int 77: 443–449, 2010 [DOI] [PubMed] [Google Scholar]
- 11.Perinpam M, Enders FT, Mara KC, Vaughan LE, Mehta RA, Voskoboev N, Milliner DS, Lieske JC: Plasma oxalate in relation to eGFR in patients with primary hyperoxaluria, enteric hyperoxaluria and urinary stone disease. Clin Biochem 50: 1014–1019, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Milliner DS, Harris PC, Cogal AG, Lieske JC: Primary hyperoxaluria type 1. In: GeneReviews, edited by Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A, Seattle, WA, University of Washington, Seattle, 2017 [Google Scholar]
- 13.Cochat P, Hulton SA, Acquaviva C, Danpure CJ, Daudon M, De Marchi M, Fargue S, Groothoff J, Harambat J, Hoppe B, Jamieson NV, Kemper MJ, Mandrile G, Marangella M, Picca S, Rumsby G, Salido E, Straub M, van Woerden CS; OxalEurope: Primary hyperoxaluria Type 1: Indications for screening and guidance for diagnosis and treatment. Nephrol Dial Transplant 27: 1729–1736, 2012 [DOI] [PubMed] [Google Scholar]
- 14.Jamieson NV; European PHI Transplantation Study Group: A 20-year experience of combined liver/kidney transplantation for primary hyperoxaluria (PH1): The European PH1 transplant registry experience 1984-2004. Am J Nephrol 25: 282–289, 2005 [DOI] [PubMed] [Google Scholar]
- 15.Khorsandi SE, Samyn M, Hassan A, Vilca-Melendez H, Waller S, Shroff R, Koffman G, Van’t Hoff W, Baker A, Dhawan A, Heaton N: An institutional experience of pre-emptive liver transplantation for pediatric primary hyperoxaluria type 1. Pediatr Transplant 20: 523–529, 2016 [DOI] [PubMed] [Google Scholar]
- 16.Shapiro R, Weismann I, Mandel H, Eisenstein B, Ben-Ari Z, Bar-Nathan N, Zehavi I, Dinari G, Mor E: Primary hyperoxaluria type 1: Improved outcome with timely liver transplantation: A single-center report of 36 children. Transplantation 72: 428–432, 2001 [DOI] [PubMed] [Google Scholar]
- 17.Liebow A, Li X, Racie T, Hettinger J, Bettencourt BR, Najafian N, Haslett P, Fitzgerald K, Holmes RP, Erbe D, Querbes W, Knight J: An investigational RNAi therapeutic targeting glycolate oxidase reduces oxalate production in models of primary hyperoxaluria. J Am Soc Nephrol 28: 494–503, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dutta C, Avitahl-Curtis N, Pursell N, Larsson Cohen M, Holmes B, Diwanji R, Zhou W, Apponi L, Koser M, Ying B, Chen D, Shui X, Saxena U, Cyr WA, Shah A, Nazef N, Wang W, Abrams M, Dudek H, Salido E, Brown BD, Lai C: Inhibition of glycolate oxidase with dicer-substrate siRNA reduces calcium oxalate deposition in a mouse model of primary hyperoxaluria type 1. Mol Ther 24: 770–778, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Clifford-Mobley O, Rumsby G, Kanodia S, Didi M, Holt R, Senniappan S: Glycolate oxidase deficiency in a patient with congenital hyperinsulinism and unexplained hyperoxaluria. Pediatr Nephrol 32: 2159–2163, 2017 [DOI] [PubMed] [Google Scholar]
- 20.Frishberg Y, Zeharia A, Lyakhovetsky R, Bargal R, Belostotsky R: Mutations in HAO1 encoding glycolate oxidase cause isolated glycolic aciduria. J Med Genet 51: 526–529, 2014 [DOI] [PubMed] [Google Scholar]
- 21.McGregor TL, Hunt KA, Nioi P, Mason D, Ticau S, Pelosi M, Loken PR, Finer S, Griffiths CJ, MacArthur DG, Trembath RC, Oglesbee D, Lieske JC, Wright J, Erbe DV, van Heel DA: Deep phenotyping of a healthy human HAO1 knockout informs therapeutic development for primary hyperoxaluria type 1. eLife 9: e54363, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Holmes RP: Pharmacological approaches in the treatment of primary hyperoxaluria. J Nephrol 11[Suppl 1]: 32–35, 1998 [PubMed] [Google Scholar]
- 23.Wilson DM, Liedtke RR: Modified enzyme-based colorimetric assay of urinary and plasma oxalate with improved sensitivity and no ascorbate interference: Reference values and sample handling procedures. Clin Chem 37: 1229–1235, 1991 [PubMed] [Google Scholar]
- 24.Springer AD, Dowdy SF: GalNAc-siRNA conjugates: Leading the way for delivery of RNAi therapeutics. Nucleic Acid Ther 28: 109–118, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Milliner DS, McGregor TL, Thompson A, Dehmel B, Knight J, Rosskamp R, Blank M, Yang S, Fargue S, Rumsby G, Groothoff J, Allain M, West M, Hollander K, Lowther WT, Lieske JC: End points for clinical trials in primary hyperoxaluria. Clin J Am Soc Nephrol 15: 1056–1065, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fargue S, Harambat J, Gagnadoux MF, Tsimaratos M, Janssen F, Llanas B, Berthélémé JP, Boudailliez B, Champion G, Guyot C, Macher MA, Nivet H, Ranchin B, Salomon R, Taque S, Rolland MO, Cochat P: Effect of conservative treatment on the renal outcome of children with primary hyperoxaluria type 1. Kidney Int 76: 767–773, 2009 [DOI] [PubMed] [Google Scholar]
- 27.Cochat P: Primary hyperoxaluria type 1. Kidney Int 55: 2533–2547, 1999 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.