

Abstract
The glomerular basement membrane is a vital component of the filtration barrier of the kidney and is primarily composed of a highly structured matrix of type IV collagen. Specific isoforms of type IV collagen, the α3(IV), α4(IV), and α5(IV) isoforms, assemble into trimers that are required for normal glomerular basement membrane function. Disruption or alteration in these isoforms leads to breakdown of the glomerular basement membrane structure and function and can lead to progressive CKD known as Alport syndrome. However, there is wide variability in phenotype among patients with mutations affecting type IV collagen that depends on a complex interplay of sex, genotype, and X-chromosome inactivation. This article reviews the genetic basis of collagen disorders of the kidney as well as potential treatments for these conditions, including direct alteration of the DNA, RNA therapies, and manipulation of collagen proteins.
Keywords: Alport syndrome, type IV collagen, gene therapy, collagen diseases, kidney genomics series
Introduction
The glomerular basement membrane (GBM) is a vital component of the filtration barrier of the kidney and is primarily composed of a highly structured matrix of type IV collagen, laminin, nidogens, agrin, and perlecan (1). Disruption or alterations in these components lead to breakdown of the GBM structure and function and can lead to progressive CKD. However, there is wide variability in phenotype among patients with mutations affecting type IV collagen that depends on a complex interplay of sex, genotype, and X-chromosome inactivation. This article reviews the genetic basis of type IV collagen disorders of the kidney as well as currently available treatments and potential future genomic treatments for these conditions.
Type IV Collagen
Six genes, COL4A1–COL4A6, encode six isoforms of type IV collagen, α1(IV) to α6(IV). The genes are arranged in three pairs, COL4A1–COL4A2, COL4A3–COL4A4, and COL4A5–COL4A6, situated in a head-to-head orientation on chromosomes 13, 2, and X, respectively. The α(IV) isoforms share structural features, including an amino-terminal sequence of approximately 25 amino acids (7S), a collagenous domain of approximately 1400 amino acids containing multiple Gly-X-Y repeats where X and Y represent nonglycine amino acids, and a carboxy-terminal (NC1) domain of approximately 230 amino acids. Type IV collagen isoforms self-assemble in the endoplasmic reticulum to form triple helices in a very specific stoichiometry. The presence of glycine at every third residue in the collagenous domain is required for assembly of the triple helix. Three heterotrimers that occur in mammalian basement membranes have been described: α1-α1-α2(IV), α3-α4-α5(IV), and α5-α5-α6(IV) (2,3). The α1-α1-α2(IV) network is predominant in the developing GBM until the capillary loop stage, when it is substantially replaced by an α3-α4-α5(IV) network (4). If any of the α3(IV), α4(IV), or α5(IV) isoforms are absent due to severe mutations (truncating mutations, for example), then the other type IV collagen isoforms are degraded, and no α3α4α5(IV) heterotrimers are deposited in the GBM, leading to Alport syndrome (2). In these patients, the α1-α1-α2(IV) network persists, increasing susceptibility to proteolytic degradation and leading to progressive deterioration of the GBM and CKD (4). Milder mutations, generally missense mutations affecting the glycine residues in the collagenous domain that are involved in triple-helix formation, may lead to abnormally folded trimers that are either degraded or lead to formation of an abnormal α3-α4-α5(IV) GBM matrix. Patients with COL4A5 variants who express the α3-α4-α5(IV) network in the GBM have a slower progression of kidney disease (median age of kidney failure >50 years) compared with those patients where the α3-α4-α5(IV) network is absent (median age of kidney failure 29 years) (5). Patients with Alport syndrome may also exhibit sensorineural hearing loss due to the dysfunction of the α3-α4-α5(IV) network in the cochlea as well as ocular findings, such as anterior lenticonus, due to presence of the α3-α4-α5(IV) network in the lens of the eye.
Variants in type IV collagen genes are distributed throughout each gene with no specific hot spots. Over 1500 unique variants have been reported in COL4A5, and over 500 each have been reported in COL4A3 and COL4A4 (6). For COL4A5, these are primarily missense substitutions in 43% (33% in regions encoding glycine within the collagenous domain and 10% other); 34% nonsense mutations (both direct and downstream); 23% splicing variants; 14% small deletions; 7% rearrangements or copy number variants; and small numbers of duplications, insertions, and indels (7). Both the phenotypic heterogeneity of Alport syndrome and the slow progression of the phenotype over decades make assigning pathogenicity using the American College of Medical Genetics and Genomics guidelines potentially problematic (8). This has been addressed for variants in COL4A5 by the development of variant databases, but phenotype genotype correlation data lag behind for variants in COL4A3 and COL4A4 (9,10). This will be addressed by the Clinical Genome Resource Variant Curation Expert Panels, an international collaboration aimed at resolving discrepancies in variant interpretation (9).
Alport Syndrome: Pathogenesis and Current Treatments
If the α3-α4-α5(IV) trimer network does not form in the GBM during the capillary loop stage of glomerular development, the α1-α1-α2(IV) network persists. Compared with the α3-α4-α5(IV) network, basement membranes containing the α1-α1-α2(IV) network are less crosslinked and more susceptible to proteolysis by matrix metalloproteases (2,11). The GBM with predominantly α1-α1-α2(IV) network is also more distensible, leading to biomechanical strain on the GBM affecting the adjacent endothelial cells and podocytes. Alport mice exposed to hypertension with increased biomechanical strain on the GBM demonstrate increased expression of matrix metalloproteinases and inflammatory cytokines compared with wild-type mice (12). In addition, biomechanical strain induces endothelin-1 expression in endothelial cells in animal models of Alport syndrome (13,14). Activation of endothelin type A receptors on mesangial cells leads to mesangial filopodial invasion of the GBM with deposition of aberrant laminins, including laminin 211, that can be blocked with endothelin receptor antagonists (13,15). Podocyte-derived invasions into the GBM in Alport mice have also been described; however, the inciting trigger is unknown (16). Finally, aberrant signaling between the abnormal GBM and the podocyte via integrins also likely plays a role in Alport pathogenesis. Alport mice that have had integrin-α1 or integrin-α2 genes additionally knocked out demonstrate reduced matrix deposition, improved life span, and reduced expression of matrix metalloproteases (17,18). Thus, the thickening of the GBM in Alport syndrome over time appears to be due to increased deposition of matrix from both podocyte and mesangial cell origin triggered by biomechanical strain and aberrant signaling between the GBM and adherent cells (Figure 1).
Figure 1.
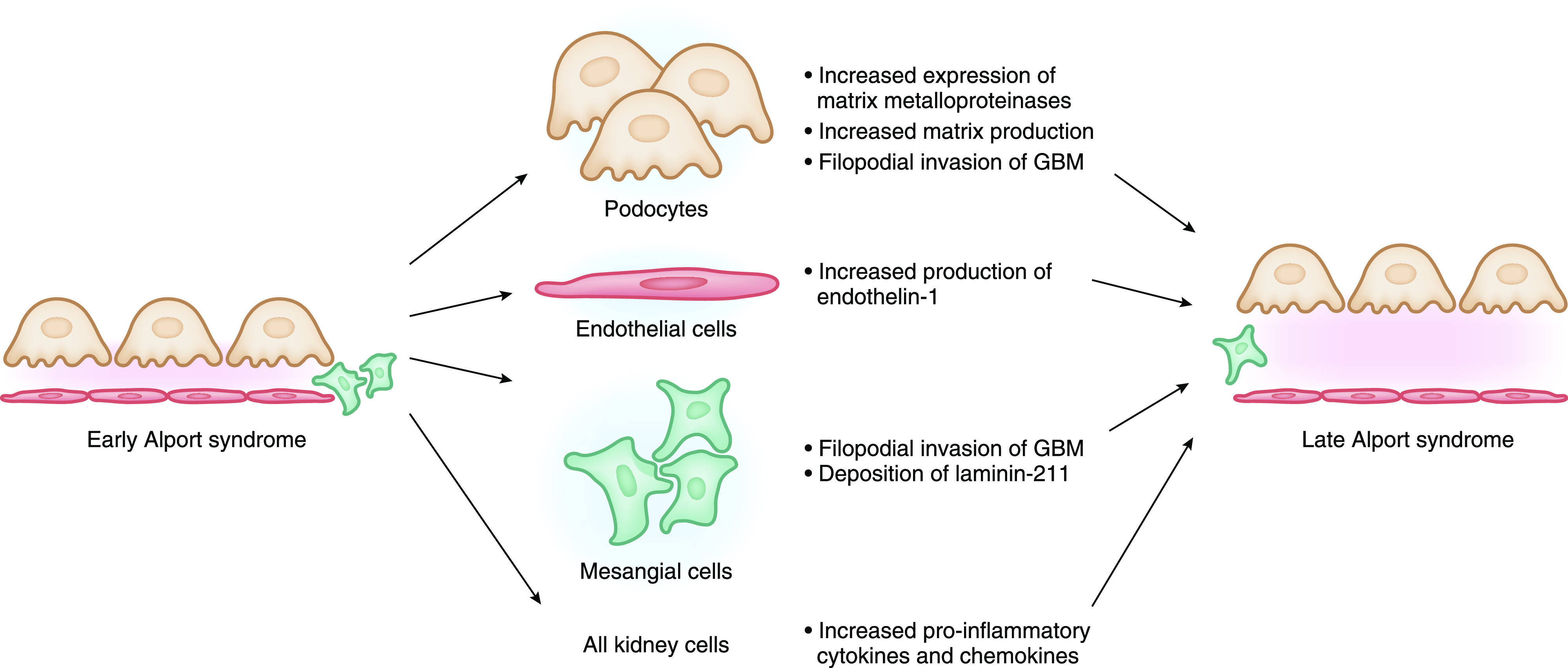
Pathogenesis of Alport syndrome kidney disease progression. GBM, glomerular basement membrane.
Current standard of care for patients with Alport syndrome includes inhibition of the renin-angiotensin-aldosterone system to reduce the biomechanical strain on the abnormal GBM. Treatment of mouse models of Alport syndrome with ACE inhibitors doubles the life span of treated animals (19). Retrospective registry studies have also shown an association between improved kidney outcomes and ACE inhibition. A study from the European Alport Registry included 283 patients with Alport syndrome and demonstrated that time to kidney failure was longer in patients who were treated with ACE inhibitors, and the benefit was greatest in those who started treatment earlier (20). These findings were confirmed in a second cohort of Japanese patients with Alport syndrome where those treated with ACE inhibitors had a median age of kidney failure of >50 years, whereas those not treated had a median age of kidney failure of 28 years (5). A prospective study of children with Alport syndrome showed a trend toward delay in progression of proteinuria in children who were treated with ramipril at very early stages of disease (urine albumin <300 mg/g creatinine or isolated hematuria) (21). Given these findings, current treatment recommendations suggest initiation of ACE inhibitors at the time of diagnosis in men or boys with X-linked Alport syndrome and all patients with autosomal recessive Alport syndrome and at initial development of albuminuria in women or girls with X-linked Alport syndrome or all patients with autosomal dominant Alport syndrome (22).
Several other novel drugs are currently in clinical trials to treat Alport kidney disease, primarily targeting later fibrosis signaling pathways. Lademirsen, an inhibitor of microRNA-21 (miRNA-21), is currently being tested in a phase 2 randomized controlled trial in patients with Alport syndrome at high risk of progression (NCT02855268). Bardoxolone is an anti-inflammatory agent that acts via activation of Nrf-2 and inhibition of NF-κB to increase eGFR. Studies in diabetic kidney disease demonstrate an increase in eGFR but were halted early due to a higher risk of hospitalization and death from heart failure (23). Bardoxolone is currently being tested in phase 2/3 randomized controlled trials in patients with Alport syndrome with careful screening to minimize risk of cardiovascular disease (NCT03019185).
Alport Syndrome: Clinical Correlation
Classic Alport syndrome is estimated to occur in 1:50,000 live births (24). Prevalence of milder forms of the disease (heterozygous mutation in COL4A3 and COL4A4) is unknown. X-linked Alport syndrome, caused by mutations in COL4A5 on the X chromosome, accounts for 70%–80% of patients with Alport syndrome. Men with X-linked Alport syndrome invariably develop kidney failure, and their rate of kidney disease progression is strongly influenced by genotype. In a European registry cohort, survival analysis demonstrated that large deletions and nonsense mutations conferred a 90% probability of kidney failure before age 30, compared with a 70% risk with splice site mutations and a 50% risk with missense mutations (25). Similar genotype-phenotype correlations were reported in a Japanese cohort with median age of kidney failure of 18 years for patients with nonsense mutations and 40 years for patients with missense mutations (5). The position of a glycine substitution may also affect the phenotype, as 5′ glycine missense mutations are associated with a more severe phenotype than 3′ glycine mutations (26). The number of side-chain carbon atoms in the substituting amino acid also influences the phenotype associated with a glycine substitution (27).
Women with heterozygous mutations in COL4A5 have a wide spectrum of disease from microscopic hematuria alone to kidney failure (28). In a large cohort of women with X-linked Alport syndrome, the risk of kidney failure was 12% by age 45, 30% by age 60, and 40% by age 80 (29). The explanation for the wide variability in outcomes for women with X-linked Alport syndrome is unclear but is determined at least in part by X inactivation (30). The α3-α4-α5(IV) heterotrimer is present in the GBM in a mosaic pattern due to random X inactivation during fetal development. If by random chance more of the mutant COL4A5 is expressed, kidney outcomes are worse.
Homozygous or compound heterozygous mutations in COL4A3 and COL4A4 cause autosomal recessive Alport syndrome, which accounts for approximately 5% of patients with Alport syndrome. Kidney outcomes in autosomal recessive Alport syndrome are similar to those in men with X-linked Alport syndrome (31). Individuals with heterozygous mutations in COL4A3 or COL4A4 also demonstrate a wide spectrum of disease from microscopic hematuria alone to progressive kidney disease and kidney failure and are categorized as autosomal dominant Alport syndrome (32). Previously, these patients may have been classified as having “thin basement membrane disease” on the basis of biopsy; however, a recent consensus report recommended including all patients with heterozygous mutations in COL4A3 or COL4A4 under the umbrella of Alport syndrome given the similarities in GBM abnormalities and risk of progression requiring ongoing monitoring (32). It has been increasingly recognized that autosomal dominant Alport syndrome accounts for a larger percentage of patients with Alport syndrome than previously recognized, up to 19%–31% of affected patients (33,34).
Digenic inheritance in Alport syndrome has also been described, including patients with dual COL4A3 and COL4A4 variants and patients with COL4A5 and COL4A4/3 variants in both cis and trans configurations (35,36). Sequencing of the coding exons of all three type IV collagen genes is important for diagnosis, even if the inheritance pattern seems clear by pedigree analysis.
Heterozygous mutations in COL4A3 and COL4A4 also may manifest as FSGS with or without classic GBM findings of thinning or thickening with lamellation. This association was first described in 2007 in a cohort of patients from Cyprus and has been reported numerous times since then (37 –39). Type IV collagen mutations are among the most common genetic mutations, identified in up to 31% of adults with familial FSGS (39) and up to 10% of a cohort of predominantly sporadic FSGS (40). It is unclear why some patients with heterozygous COL4A3 or COL4A4 mutations develop classic Alport syndrome kidney phenotype, whereas others exhibit FSGS.
Type IV Collagen Mutations in Other Glomerular Disease
Type IV collagen mutations are also frequently found in patients with CKD who were previously unknown to have Alport syndrome. In a study from Columbia University, 3% of patients with CKD who underwent whole-exome sequencing had pathogenic variants in COL4A3 (0.8%), COL4A4 (0.6%), or COL4A5 (1%), the majority of whom did not have a diagnosis of Alport syndrome (41). This finding highlights the problem of underdiagnosis of patients with type IV collagen mutations.
Type IV collagen mutations may also contribute to pathogenesis in other glomerular disorders. Thin basement membranes have been observed on kidney biopsy in patients with familial IgA nephropathy (42). Pathogenic variants in COL4A3–5 were identified in nine of 46 families with familial IgA nephropathy (43). In a genome-wide association study of over 19,000 patients with diabetic nephropathy, a common missense variant that encodes a tyrosine in place of aspartic acid at position 326 of the COL4A3 protein was identified that was protective against the development of diabetic kidney disease (44). It was hypothesized that the baseline thinning of the GBM associated with this variant prevented the GBM thickening that occurs in diabetic kidney disease and was thus protective. However, in a smaller study of nine individuals with diabetic kidney disease associated with maturity-onset diabetes in the young (MODY), variants in COL4A3 were associated with a more severe kidney phenotype (45). We are just beginning to understand the role of type IV collagen mutations in patients with not only classic Alport syndrome but other glomerular disorders as well.
Alport Syndrome Treatments: Future Genomic Strategies
Current treatments for Alport syndrome only slow the progression of kidney disease, and most patients will still require kidney transplantation; thus, there is an unmet need for novel, curative treatments. Understanding the cellular and molecular biology of the GBM opens up several avenues of genomic therapy for Alport syndrome, including gene editing, gene therapy, RNA therapy, and chaperone therapy (Figure 2).
Figure 2.
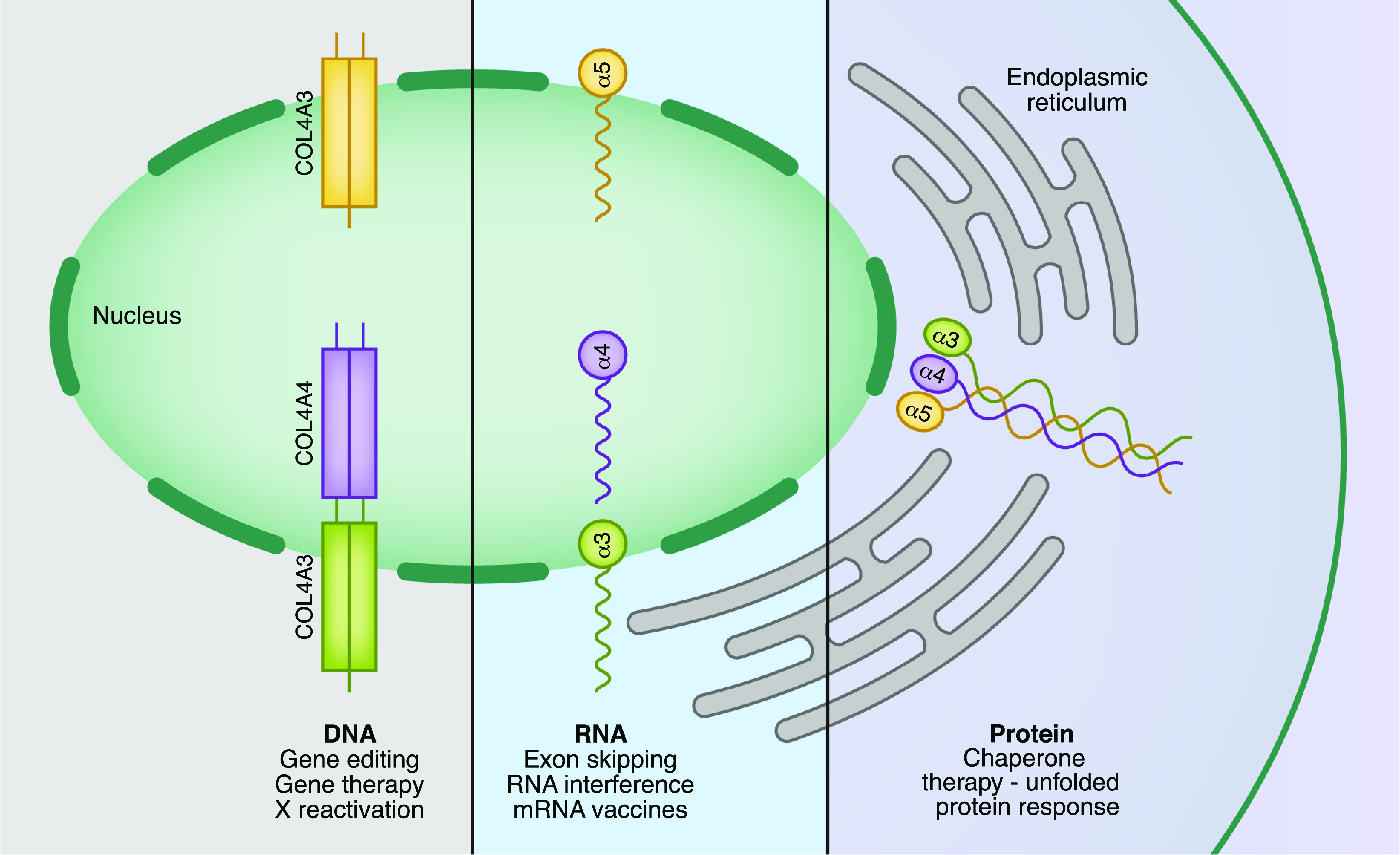
Potential genomic treatments for Alport syndrome.COL4A3, COL4A4, and COL4A5 are transcribed into RNA in the nucleus. The RNA is then translated into type IV collagen protein isoforms. These type IV collagen isomers self-assemble into trimers in the endoplasmic reticulum. After they are processed into trimers, they are secreted into the GBM. There are a number of potential targets for rectification of this process in the setting of pathogenic variants in type IV collagen.
Gene Editing
Gene editing targets disease-causing genes to permanently correct, remove, or replace a gene and, thus, cure the disease. Despite significant advances in ex vivo genome editing, it has been used in only a small number of human trials, largely due to concerns about off-target effects, which could disrupt gene function.
Stem cell transplantation is a less precise approach to gene editing and has been tried in animal models of Alport syndrome with promising results, including improvements in kidney function (46). Although mature podocytes are thought to be incapable of replication, bone marrow may act as a podocyte progenitor niche, enabling some regeneration of the GBM (47). Y chromosomes have been demonstrated in the podocytes of male (XY) recipients of female (XX) kidneys, suggesting that some limited regeneration is possible (48). Although patients with GBM disease have had improvement in proteinuria following bone marrow transplants as part of leukemia treatment through recruitment of podocytes and partial expression of α3(IV) chains, this approach is not suitable for the majority of patients (49). Care must be taken with interpretation of stem cell–based experimental results in kidney disease because injection of amniotic fluid stem cells into COL4A5 (−/−) mice before the onset of proteinuria has been shown to delay interstitial fibrosis and glomerular sclerosis, reduce the decline in kidney function, and prolong survival without podocyte differentiation, likely by reducing fibrosis (50). Bone marrow transplants carry a significant mortality risk however; thus, they cannot be considered a safe treatment for Alport syndrome at this time (51).
With experimental evidence showing it is possible to correct a clinically significant proportion of COL4A3 and COL4A5 variants in podocyte cell lines (52), and human trials for Hemophilia-B using infused CRISPR/Cas9-corrected patient-specific stem cells underway (NCT03728322), it is likely that human trials for Alport syndrome will occur in the next decade. Challenges to this approach include difficulties in manipulation of the podocyte in vivo.
Another approach to gene editing that may be explored in Alport syndrome is X-chromosome reactivation. X-chromosome inactivation occurs in early development and ensures that female XX cells have similar expression to male XY cells. In each cell, one X chromosome is randomly epigenetically silenced and is referred to as the inactive X chromosome (Xi), in a process initiated by a molecule called Xist (53). Because the phenotype of women with X-linked Alport syndrome can vary from hematuria to kidney impairment depending on lyonization of the X chromosome, a potential treatment of severe X-linked Alport syndrome in women could be reactivation of the healthy copy of the COL4A5 gene on the Xi. A number of factors have been identified that are involved in this pathway, and the use of small molecule inhibitors targeted at these pathways has been shown to reactivate Xi in mouse models of Rett syndrome (54). Human Genotype-Tissue Expression analysis shows COL4A5 escapes Xi in the brain, although the effect of this is not understood (55). Biallelic expression of some X-linked genes in women has been shown to contribute to a portion of their reduced cancer incidence when compared with men. Thus, Xi remains experimental at this point in time due to significant concerns about off-target effects.
Gene Therapy
In gene therapy, the effect of a mutation is offset by inserting a corrected copy of the gene into the body using a vehicle while the disease-related genes remain in the genome. If the normal gene replaces the mutant allele, then transformed cells proliferate and produce enough normal protein to restore a healthy phenotype. Effective delivery depends on a vehicle, such as a virus or nanoparticle, and an accessible tissue compartment. Thus far, it has been most successful for eye, blood, or bone marrow disease (56).
The feasibility of gene therapy has been shown in mouse models of Alport syndrome with an inducible transgene system, where secretion of α3α4α5(IV) heterotrimers by podocytes into a preformed Alport GBM was effective at restoring the missing collagen IV network, reducing proteinuria, slowing disease progression, and increasing survival (57). Adenovirus-mediated gene transfer into kidney glomeruli has also been demonstrated in mouse models of Alport syndrome (58). However, inducing expression of COL4A3 in mice using an endothelial cell–specific inducible transgenic system does not result in restoration of the α3α4α5(IV) heterotrimer or resolution of the Alport phenotype (59). Successful delivery of COL4A5 into swine kidney by an adenovirus vector has been reported with deposition of α5(IV) into the GBM (60). Unfortunately, this technique required direct infusion of vector into the renal artery, which is not feasible for translation to widespread human application.
Although CRISPR/Cas9 gene editing in the kidney has not yet moved beyond proof of concept, it may be that ocular gene therapy will be utilized at an earlier time point than kidney targeting with delivery of the viral vector directly into the eye (61), where clinical trials and licensed gene replacement therapy are further advanced (62).
Chaperone Therapy
Missense mutations can result in the production of misfolded proteins, which are retained in the endoplasmic reticulum, the quality control center of the cell. This leads to endoplasmic reticulum stress, unfolded protein response activation, and increased cellular apoptosis. Chaperone therapy uses small molecules to unfold abnormal proteins, enabling them to escape the endoplasmic reticulum and be utilized by the cell.
Examples of these agents in human use include Lumacaftor (63), which binds directly to F508del-CFTR correcting its mislocalization, ameliorating the phenotype, and transforming the outlook for people with cystic fibrosis, and Migalastat (64), which stabilizes mutant forms of α-Gal and has shown promise as an alternative to enzyme replacement for patients with Fabry disease.
Mouse models and human kidney biopsy samples of COL4A3 disease have shown unfolded protein response activation (65) as part of the pathogenesis of Alport syndrome. Treatment of cellular models of Alport syndrome with the chaperone sodium-4-phenylbutyrate has shown reduced endoplasmic reticulum stress (66) and may facilitate α5(IV) extracellular transport (67). Future clinical trials targeting Alport phenotypes that lead to unfolded protein response may result in improved outcomes when started early in disease.
RNA Therapy
There are currently three approaches to targeting RNA for treatment of disease: single-stranded antisense oligonucleotides (ASOs), short stretches of DNA that prevent mRNA from being translated into a protein; RNA interference, small interfering RNAs (siRNAs) degrade mRNA and prevent it from being translated into protein and miRNAs, small noncoding RNAs whose functions include post-transcriptional regulation of gene expression; and RNA vaccines, introducing mRNA into the body reprograming the cell to produce a specific protein.
Exon skipping therapy using an ASO has been investigated as a therapy for a small group of patients with a specific variant in COL4A5. Genotype-phenotype correlation data show that some individuals with COL4A5 variants experience a milder clinical course, raising the question of whether exon skipping could nudge the genetic code toward a milder phenotype. For example, published data have shown that specific COL4A5 gene splice site mutations with in-frame deletions showed a good kidney prognosis when compared with an out-of-frame deletion group (68). On the basis of these data, Yamamura et al. (69) have developed an exon-skipping therapy using an ASO targeting truncating variants in exon 21 of the COL4A5 gene.
In patients with truncating variants in COL4A5, the α5(IV) chain will terminate at the stop codon, and the NC1 domain is missing. In contrast, exon-skipping therapy will replace the truncating variant with an in-frame deletion variant at the transcript level, and the NC1 domain is not lost, leading to the formation of the trimer and restoration of the GBM. Mice treated with this protocol had α3α4α5(IV) triple-helix formation with clinical and pathologic improvements, including expression of the α5(IV) chain on GBM and tubular basement membrane with prolonged survival time. Exon skipping does not aim to “cure” the disease but induces a frameshift that leads to the expression of a milder phenotype. The future potential for this approach includes establishing mutant mouse models for other exons that could be targeted by an ASO and will rely on large genomic datasets of individuals with pathogenic variants in COL4A5. There may be significant risk with this approach given that benign hypomorphic variants in gnomAD, such as COL4A5 c.1871G>A, p.(Gly624Asp), have also been implicated in the development of a severe disease (70).
RNA interference can use miRNA, a small, noncoding RNA molecule whose functions include post-transcriptional regulation of gene expression. There are a number of miRNAs that are thought to play a role in the progression of CKD, with several shown to be upregulated in fibrotic kidney biopsies compared with healthy tissue or animal models of human injury. miRNA-21 is upregulated in models of mouse and human kidney disease. In mouse models of Alport syndrome, it is elevated in the kidneys prior to the development of histologic abnormalities and appears to contribute to the pathogenesis of disease by reducing TGF-β–induced fibrogenesis and inflammation (71).
Anti-RNA oligonucleotides can be delivered via subcutaneous injection and act to suppress translation of miRNA. Following injection, the oligonucleotides are found in high concentration in the kidney particularly in the proximal tubular cells, with diseased glomerular cells concentrating the oligonucleotides far more avidly than the healthy glomerulus. In mouse models of Alport syndrome, anti–miRNA-21 oligonucleotides delivered as a weekly injection significantly reduced disease progression and increased life expectancy by 50% (72,73). These agents are currently in clinical trials in humans (see above).
Dominantly inherited conditions could be treated by using specific siRNA molecules to silence the mutated gene or protein, supplying a normal copy of the gene to take over and produce a sufficient amount of the healthy protein. Animal studies have shown promise for this approach in Huntington disease using siRNA to target the degradation of abnormal huntingtin protein (74), and human clinical trials are planned.
Several new coronavirus disease 2019 vaccines involve an intramuscular injection of an mRNA molecule containing the instructions to make a severe acute respiratory syndrome coronavirus 2 spike protein, triggering an immune response and leading to immunity from coronavirus disease 2019 (75). The global drive to develop this vaccine has moved this field of research forward significantly. At the time of writing, large-scale vaccination programs are underway, and although there are not RNA-based therapies for Alport syndrome at present, this field of research is likely to yield benefits for patients with monogenic diseases over the next decade.
In the 30 years since the discovery of mutations in COL4A5 as the cause of Alport syndrome (76), huge strides have been made in understanding of the structure and function of the GBM, as well as the genomic features that affect the course of disease. A number of exciting potential treatments for Alport syndrome are in the pipeline with the aim to slow, and eventually cure, this progressive disease.
Disclosures
C. Quinlan reports employment with The Murdoch Children’s Research Institute, The Royal Children’s Hospital, and the University of Melbourne and serving as section editor for Nephrology. M.N. Rheault reports employment with the University of Minnesota; receiving research funding from Advicenne, Reata, Travere, and Sanofi; and serving on the Alport Syndrome Foundation Medical Advisory Board, NephJC (501c3) Board of Directors, and Pediatric Nephrology Research Consortium (501c3) Steering Committee.
Funding
None.
Footnotes
Published online ahead of print. Publication date available at www.cjasn.org.
References
- 1.Naylor RW, Morais MRPT, Lennon R: Complexities of the glomerular basement membrane. Nat Rev Nephrol 17: 112–127, 2021 [DOI] [PubMed] [Google Scholar]
- 2.Gunwar S, Ballester F, Noelken ME, Sado Y, Ninomiya Y, Hudson BG: Glomerular basement membrane. Identification of a novel disulfide-cross-linked network of alpha3, alpha4, and alpha5 chains of type IV collagen and its implications for the pathogenesis of Alport syndrome. J Biol Chem 273: 8767–8775, 1998 [DOI] [PubMed] [Google Scholar]
- 3.Borza DB, Bondar O, Ninomiya Y, Sado Y, Naito I, Todd P, Hudson BG: The NC1 domain of collagen IV encodes a novel network composed of the alpha 1, alpha 2, alpha 5, and alpha 6 chains in smooth muscle basement membranes. J Biol Chem 276: 28532–28540, 2001 [DOI] [PubMed] [Google Scholar]
- 4.Kalluri R, Shield CF, Todd P, Hudson BG, Neilson EG: Isoform switching of type IV collagen is developmentally arrested in X-linked Alport syndrome leading to increased susceptibility of renal basement membranes to endoproteolysis. J Clin Invest 99: 2470–2478, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yamamura T, Horinouchi T, Nagano C, Omori T, Sakakibara N, Aoto Y, Ishiko S, Nakanishi K, Shima Y, Nagase H, Takeda H, Rossanti R, Ye MJ, Nozu Y, Ishimori S, Nicnchoji T, Kaito H, Morisada N, Iijima K, Nozu K: Genotype-phenotype correlations influence the response to angiotensin-targeting drugs in Japanese patients with male X-linked Alport syndrome. Kidney Int 98: 1605–1614, 2020 [DOI] [PubMed] [Google Scholar]
- 6.Leiden Open Variation Database. Available at https://databases.lovd.nl/shared/genes/. Accessed December 7, 2020
- 7.Savige J, Storey H, Il Cheong H, Gyung Kang H, Park E, Hilbert P, Persikov A, Torres-Fernandez C, Ars E, Torra R, Hertz JM, Thomassen M, Shagam L, Wang D, Wang Y, Flinter F, Nagel M: X-linked and autosomal recessive alport syndrome: Pathogenic variant features and further genotype-phenotype correlations. PLoS One 11: e0161802, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL; ACMG Laboratory Quality Assurance Committee: Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17: 405–424, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rehm HL, Berg JS, Brooks LD, Bustamante CD, Evans JP, Landrum MJ, Ledbetter DH, Maglott DR, Martin CL, Nussbaum RL, Plon SE, Ramos EM, Sherry ST, Watson MS; ClinGen : ClinGen--The clinical genome resource. N Engl J Med 372: 2235–2242, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Savige J, Ars E, Cotton RG, Crockett D, Dagher H, Deltas C, Ding J, Flinter F, Pont-Kingdon G, Smaoui N, Torra R, Storey H; International Alport Mutation Consortium: DNA variant databases improve test accuracy and phenotype prediction in Alport syndrome. Pediatr Nephrol 29: 971–977, 2014 [DOI] [PubMed] [Google Scholar]
- 11.Zeisberg M, Khurana M, Rao VH, Cosgrove D, Rougier JP, Werner MC, Shield CF 3rd, Werb Z, Kalluri R: Stage-specific action of matrix metalloproteinases influences progressive hereditary kidney disease. PLoS Med 3: e100, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Meehan DT, Delimont D, Cheung L, Zallocchi M, Sansom SC, Holzclaw JD, Rao V, Cosgrove D: Biomechanical strain causes maladaptive gene regulation, contributing to Alport glomerular disease. Kidney Int 76: 968–976, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dufek B, Meehan DT, Delimont D, Cheung L, Gratton MA, Phillips G, Song W, Liu S, Cosgrove D: Endothelin A receptor activation on mesangial cells initiates Alport glomerular disease. Kidney Int 90: 300–310, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zallocchi M, Johnson BM, Meehan DT, Delimont D, Cosgrove D: α1β1 integrin/Rac1-dependent mesangial invasion of glomerular capillaries in Alport syndrome. Am J Pathol 183: 1269–1280, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Clark SD, Nabity MB, Cianciolo RE, Dufek B, Cosgrove D: X-linked alport dogs demonstrate mesangial filopodial invasion of the capillary tuft as an early event in glomerular damage. PLoS One 11: e0168343, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Randles MJ, Collinson S, Starborg T, Mironov A, Krendel M, Königshausen E, Sellin L, Roberts IS, Kadler KE, Miner JH, Lennon R: Three-dimensional electron microscopy reveals the evolution of glomerular barrier injury. Sci Rep 6: 35068, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cosgrove D, Rodgers K, Meehan D, Miller C, Bovard K, Gilroy A, Gardner H, Kotelianski V, Gotwals P, Amatucci A, Kalluri R: Integrin alpha1beta1 and transforming growth factor-beta1 play distinct roles in Alport glomerular pathogenesis and serve as dual targets for metabolic therapy. Am J Pathol 157: 1649–1659, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rubel D, Frese J, Martin M, Leibnitz A, Girgert R, Miosge N, Eckes B, Müller GA, Gross O: Collagen receptors integrin alpha2beta1 and discoidin domain receptor 1 regulate maturation of the glomerular basement membrane and loss of integrin alpha2beta1 delays kidney fibrosis in COL4A3 knockout mice. Matrix Biol 34: 13–21, 2014 [DOI] [PubMed] [Google Scholar]
- 19.Gross O, Beirowski B, Koepke ML, Kuck J, Reiner M, Addicks K, Smyth N, Schulze-Lohoff E, Weber M: Preemptive ramipril therapy delays renal failure and reduces renal fibrosis in COL4A3-knockout mice with Alport syndrome. Kidney Int 63: 438–446, 2003 [DOI] [PubMed] [Google Scholar]
- 20.Gross O, Licht C, Anders HJ, Hoppe B, Beck B, Tönshoff B, Höcker B, Wygoda S, Ehrich JH, Pape L, Konrad M, Rascher W, Dötsch J, Müller-Wiefel DE, Hoyer P, Knebelmann B, Pirson Y, Grunfeld JP, Niaudet P, Cochat P, Heidet L, Lebbah S, Torra R, Friede T, Lange K, Müller GA, Weber M; Study Group Members of the Gesellschaft für Pädiatrische Nephrologie: Early angiotensin-converting enzyme inhibition in Alport syndrome delays renal failure and improves life expectancy. Kidney Int 81: 494–501, 2012 [DOI] [PubMed] [Google Scholar]
- 21.Gross O, Tonshoff B, Weber LT, Pape L, Latta K, Fehrenbach H, Lange-Sperandio B, Zappel H, Hoyer P, Staude H, Konig S, John U, Gellermann J, Hoppe B, Galiano M, Hoecker B, Ehren R, Lerch C, Kashtan CE, Harden M, Boeckhaus J, Friede T; German Pediatric Nephrology (GPN) Study Group and EARLY PRO-TECT Alport Investigators:A multicenter, randomized, placebo-controlled, double-blind phase 3 trial with open-arm comparison indicates safety and efficacy of nephroprotective therapy with ramipril in children with Alport’s syndrome. Kidney Int 97: 1275–1286, 2020 [DOI] [PubMed] [Google Scholar]
- 22.Kashtan CE, Gross O: Clinical practice recommendations for the diagnosis and management of Alport syndrome in children, adolescents, and young adults-an update for 2020 [published correction appears in Pediatr Nephrol 36: 731, 2021 10.1007/s00467-020-04892-x]. Pediatr Nephrol 36: 711–719, 2021 [DOI] [PubMed] [Google Scholar]
- 23.de Zeeuw D, Akizawa T, Audhya P, Bakris GL, Chin M, Christ-Schmidt H, Goldsberry A, Houser M, Krauth M, Lambers Heerspink HJ, McMurray JJ, Meyer CJ, Parving HH, Remuzzi G, Toto RD, Vaziri ND, Wanner C, Wittes J, Wrolstad D, Chertow GM; BEACON Trial Investigators: Bardoxolone methyl in type 2 diabetes and stage 4 chronic kidney disease. N Engl J Med 369: 2492–2503, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Levy M, Feingold J: Estimating prevalence in single-gene kidney diseases progressing to renal failure. Kidney Int 58: 925–943, 2000 [DOI] [PubMed] [Google Scholar]
- 25.Jais JP, Knebelmann B, Giatras I, De Marchi M, Rizzoni G, Renieri A, Weber M, Gross O, Netzer KO, Flinter F, Pirson Y, Verellen C, Wieslander J, Persson U, Tryggvason K, Martin P, Hertz JM, Schröder C, Sanak M, Krejcova S, Carvalho MF, Saus J, Antignac C, Smeets H, Gubler MC: X-linked Alport syndrome: Natural history in 195 families and genotype-phenotype correlations in males. J Am Soc Nephrol 11: 649–657, 2000 [DOI] [PubMed] [Google Scholar]
- 26.Gross O, Netzer KO, Lambrecht R, Seibold S, Weber M: Meta-analysis of genotype-phenotype correlation in X-linked Alport syndrome: Impact on clinical counselling. Nephrol Dial Transplant 17: 1218–1227, 2002 [DOI] [PubMed] [Google Scholar]
- 27.Tsiakkis D, Pieri M, Koupepidou P, Demosthenous P, Panayidou K, Deltas C: Genotype-phenotype correlation in X-linked Alport syndrome patients carrying missense mutations in the collagenous domain of COL4A5. Clin Genet 82: 297–299, 2012 [DOI] [PubMed] [Google Scholar]
- 28.Rheault MN: Women and Alport syndrome. Pediatr Nephrol 27: 41–46, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jais JP, Knebelmann B, Giatras I, De Marchi M, Rizzoni G, Renieri A, Weber M, Gross O, Netzer KO, Flinter F, Pirson Y, Dahan K, Wieslander J, Persson U, Tryggvason K, Martin P, Hertz JM, Schröder C, Sanak M, Carvalho MF, Saus J, Antignac C, Smeets H, Gubler MC: X-linked Alport syndrome: Natural history and genotype-phenotype correlations in girls and women belonging to 195 families: A “European Community Alport Syndrome Concerted Action” study. J Am Soc Nephrol 14: 2603–2610, 2003 [DOI] [PubMed] [Google Scholar]
- 30.Rheault MN, Kren SM, Hartich LA, Wall M, Thomas W, Mesa HA, Avner P, Lees GE, Kashtan CE, Segal Y: X-inactivation modifies disease severity in female carriers of murine X-linked Alport syndrome. Nephrol Dial Transplant 25: 764–769, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Storey H, Savige J, Sivakumar V, Abbs S, Flinter FA: COL4A3/COL4A4 mutations and features in individuals with autosomal recessive Alport syndrome. J Am Soc Nephrol 24: 1945–1954, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kashtan CE, Ding J, Garosi G, Heidet L, Massella L, Nakanishi K, Nozu K, Renieri A, Rheault M, Wang F, Gross O: Alport syndrome: A unified classification of genetic disorders of collagen IV α345: A position paper of the Alport Syndrome Classification Working Group. Kidney Int 93: 1045–1051, 2018 [DOI] [PubMed] [Google Scholar]
- 33.Fallerini C, Dosa L, Tita R, Del Prete D, Feriozzi S, Gai G, Clementi M, La Manna A, Miglietti N, Mancini R, Mandrile G, Ghiggeri G, Piaggio G, Brancati F, Diano L, Frate E, Pinciaroli A, Giani M, Castorina P, Bresin E, Giachino D, De Marchi M, Mari F, Bruttini M, Renieri A, Ariani F: Unbiased next generation sequencing analysis confirms the existence of autosomal dominant Alport syndrome in a relevant fraction of cases. Clin Genet 86: 252–257, 2014 [DOI] [PubMed] [Google Scholar]
- 34.Morinière V, Dahan K, Hilbert P, Lison M, Lebbah S, Topa A, Bole-Feysot C, Pruvost S, Nitschke P, Plaisier E, Knebelmann B, Macher MA, Noel LH, Gubler MC, Antignac C, Heidet L: Improving mutation screening in familial hematuric nephropathies through next generation sequencing. J Am Soc Nephrol 25: 2740–2751, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mencarelli MA, Heidet L, Storey H, van Geel M, Knebelmann B, Fallerini C, Miglietti N, Antonucci MF, Cetta F, Sayer JA, van den Wijngaard A, Yau S, Mari F, Bruttini M, Ariani F, Dahan K, Smeets B, Antignac C, Flinter F, Renieri A: Evidence of digenic inheritance in Alport syndrome. J Med Genet 52: 163–174, 2015 [DOI] [PubMed] [Google Scholar]
- 36.Fallerini C, Baldassarri M, Trevisson E, Morbidoni V, La Manna A, Lazzarin R, Pasini A, Barbano G, Pinciaroli AR, Garosi G, Frullanti E, Pinto AM, Mencarelli MA, Mari F, Renieri A, Ariani F: Alport syndrome: Impact of digenic inheritance in patients management. Clin Genet 92: 34–44, 2017 [DOI] [PubMed] [Google Scholar]
- 37.Voskarides K, Damianou L, Neocleous V, Zouvani I, Christodoulidou S, Hadjiconstantinou V, Ioannou K, Athanasiou Y, Patsias C, Alexopoulos E, Pierides A, Kyriacou K, Deltas C: COL4A3/COL4A4 mutations producing focal segmental glomerulosclerosis and renal failure in thin basement membrane nephropathy. J Am Soc Nephrol 18: 3004–3016, 2007 [DOI] [PubMed] [Google Scholar]
- 38.Malone AF, Phelan PJ, Hall G, Cetincelik U, Homstad A, Alonso AS, Jiang R, Lindsey TB, Wu G, Sparks MA, Smith SR, Webb NJ, Kalra PA, Adeyemo AA, Shaw AS, Conlon PJ, Jennette JC, Howell DN, Winn MP, Gbadegesin RA: Rare hereditary COL4A3/COL4A4 variants may be mistaken for familial focal segmental glomerulosclerosis. Kidney Int 86: 1253–1259, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gast C, Pengelly RJ, Lyon M, Bunyan DJ, Seaby EG, Graham N, Venkat-Raman G, Ennis S: Collagen (COL4A) mutations are the most frequent mutations underlying adult focal segmental glomerulosclerosis. Nephrol Dial Transplant 31: 961–970, 2016 [DOI] [PubMed] [Google Scholar]
- 40.Yao T, Udwan K, John R, Rana A, Haghighi A, Xu L, Hack S, Reich HN, Hladunewich MA, Cattran DC, Paterson AD, Pei Y, Barua M: Integration of genetic testing and pathology for the diagnosis of adults with FSGS. Clin J Am Soc Nephrol 14: 213–223, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Groopman EE, Marasa M, Cameron-Christie S, Petrovski S, Aggarwal VS, Milo-Rasouly H, Li Y, Zhang J, Nestor J, Krithivasan P, Lam WY, Mitrotti A, Piva S, Kil BH, Chatterjee D, Reingold R, Bradbury D, DiVecchia M, Snyder H, Mu X, Mehl K, Balderes O, Fasel DA, Weng C, Radhakrishnan J, Canetta P, Appel GB, Bomback AS, Ahn W, Uy NS, Alam S, Cohen DJ, Crew RJ, Dube GK, Rao MK, Kamalakaran S, Copeland B, Ren Z, Bridgers J, Malone CD, Mebane CM, Dagaonkar N, Fellström BC, Haefliger C, Mohan S, Sanna-Cherchi S, Kiryluk K, Fleckner J, March R, Platt A, Goldstein DB, Gharavi AG: Diagnostic utility of exome sequencing for kidney disease. N Engl J Med 380: 142–151, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Frascá GM, Soverini L, Gharavi AG, Lifton RP, Canova C, Preda P, Vangelista A, Stefoni S: Thin basement membrane disease in patients with familial IgA nephropathy. J Nephrol 17: 778–785, 2004 [PubMed] [Google Scholar]
- 43.Li Y, Groopman EE, D’Agati V, Prakash S, Zhang J, Mizerska-Wasiak M, Caliskan Y, Fasel D, Karnib HH, Bono L, Omran SA, Sabban EA, Kiryluk K, Caridi G, Ghiggeri GM, Sanna-Cherchi S, Scolari F, Gharavi AG: Type IV collagen mutations in familial IgA nephropathy. Kidney Int Rep 5: 1075–1078, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Salem RM, Todd JN, Sandholm N, Cole JB, Chen WM, Andrews D, Pezzolesi MG, McKeigue PM, Hiraki LT, Qiu C, Nair V, Di Liao C, Cao JJ, Valo E, Onengut-Gumuscu S, Smiles AM, McGurnaghan SJ, Haukka JK, Harjutsalo V, Brennan EP, van Zuydam N, Ahlqvist E, Doyle R, Ahluwalia TS, Lajer M, Hughes MF, Park J, Skupien J, Spiliopoulou A, Liu A, Menon R, Boustany-Kari CM, Kang HM, Nelson RG, Klein R, Klein BE, Lee KE, Gao X, Mauer M, Maestroni S, Caramori ML, de Boer IH, Miller RG, Guo J, Boright AP, Tregouet D, Gyorgy B, Snell-Bergeon JK, Maahs DM, Bull SB, Canty AJ, Palmer CNA, Stechemesser L, Paulweber B, Weitgasser R, Sokolovska J, Rovīte V, Pīrāgs V, Prakapiene E, Radzeviciene L, Verkauskiene R, Panduru NM, Groop LC, McCarthy MI, Gu HF, Möllsten A, Falhammar H, Brismar K, Martin F, Rossing P, Costacou T, Zerbini G, Marre M, Hadjadj S, McKnight AJ, Forsblom C, McKay G, Godson C, Maxwell AP, Kretzler M, Susztak K, Colhoun HM, Krolewski A, Paterson AD, Groop PH, Rich SS, Hirschhorn JN, Florez JC; SUMMIT Consortium, DCCT/EDIC Research Group, GENIE Consortium: Genome-wide association study of diabetic kidney disease highlights biology involved in glomerular basement membrane collagen. J Am Soc Nephrol 30: 2000–2016, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang Y, Zhang J, Zhao Y, Wang S, Zhang J, Han Q, Zhang R, Guo R, Li H, Li L, Wang T, Tang X, He C, Teng G, Gu W, Liu F: COL4A3 gene variants and diabetic kidney disease in MODY. Clin J Am Soc Nephrol 13: 1162–1171, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Prodromidi EI, Poulsom R, Jeffery R, Roufosse CA, Pollard PJ, Pusey CD, Cook HT: Bone marrow-derived cells contribute to podocyte regeneration and amelioration of renal disease in a mouse model of Alport syndrome. Stem Cells 24: 2448–2455, 2006 [DOI] [PubMed] [Google Scholar]
- 47.Wanner N, Hartleben B, Herbach N, Goedel M, Stickel N, Zeiser R, Walz G, Moeller MJ, Grahammer F, Huber TB: Unraveling the role of podocyte turnover in glomerular aging and injury. J Am Soc Nephrol 25: 707–716, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Becker JU, Hoerning A, Schmid KW, Hoyer PF: Immigrating progenitor cells contribute to human podocyte turnover. Kidney Int 72: 1468–1473, 2007 [DOI] [PubMed] [Google Scholar]
- 49.Sugimoto K, Sakata N, Fujita S, Miyazawa T, Nishi H, Takemura T, Okada M: Cure of relapsing nephrosis by an allogeneic marrow graft for chronic myelogenous leukemia. Pediatr Nephrol 28: 975–978, 2013 [DOI] [PubMed] [Google Scholar]
- 50.Sedrakyan S, Da Sacco S, Milanesi A, Shiri L, Petrosyan A, Varimezova R, Warburton D, Lemley KV, De Filippo RE, Perin L: Injection of amniotic fluid stem cells delays progression of renal fibrosis. J Am Soc Nephrol 23: 661–673, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McDonald GB, Sandmaier BM, Mielcarek M, Sorror M, Pergam SA, Cheng GS, Hingorani S, Boeckh M, Flowers MD, Lee SJ, Appelbaum FR, Storb R, Martin PJ, Deeg HJ, Schoch G, Gooley TA: Survival, nonrelapse mortality, and relapse-related mortality after Allogeneic hematopoietic cell transplantation: Comparing 2003-2007 versus 2013-2017 cohorts. Ann Intern Med 172: 229–239, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Daga S, Donati F, Capitani K, Croci S, Tita R, Giliberti A, Valentino F, Benetti E, Fallerini C, Niccheri F, Baldassarri M, Mencarelli MA, Frullanti E, Furini S, Conticello SG, Renieri A, Pinto AM: New frontiers to cure Alport syndrome: COL4A3 and COL4A5 gene editing in podocyte-lineage cells. Eur J Hum Genet 28: 480–490, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Petropoulos S, Edsgärd D, Reinius B, Deng Q, Panula SP, Codeluppi S, Reyes AP, Linnarsson S, Sandberg R, Lanner F: Single-cell RNA-Seq reveals lineage and X chromosome dynamics in human preimplantation embryos. Cell 167: 285, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Przanowski P, Wasko U, Zheng Z, Yu J, Sherman R, Zhu LJ, McConnell MJ, Tushir-Singh J, Green MR, Bhatnagar S: Pharmacological reactivation of inactive X-linked Mecp2 in cerebral cortical neurons of living mice. Proc Natl Acad Sci U S A 115: 7991–7996, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yu B, Qi Y, Li R, Shi Q, Satpathy AT, Chang HY: B cell-specific XIST complex enforces X-inactivation and restrains atypical B cells. Cell 184: 1790–1803.E17, 2021 10.1016/j.cell.2021.02.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Russell S, Bennett J, Wellman JA, Chung DC, Yu ZF, Tillman A, Wittes J, Pappas J, Elci O, McCague S, Cross D, Marshall KA, Walshire J, Kehoe TL, Reichert H, Davis M, Raffini L, George LA, Hudson FP, Dingfield L, Zhu X, Haller JA, Sohn EH, Mahajan VB, Pfeifer W, Weckmann M, Johnson C, Gewaily D, Drack A, Stone E, Wachtel K, Simonelli F, Leroy BP, Wright JF, High KA, Maguire AM: Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: A randomised, controlled, open-label, phase 3 trial. Lancet 390: 849–860, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lin X, Suh JH, Go G, Miner JH: Feasibility of repairing glomerular basement membrane defects in Alport syndrome. J Am Soc Nephrol 25: 687–692, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Heikkila P, Parpala T, Lukkarinen O, Weber M, Tryggvason K: Adenovirus-mediated gene transfer into kidney glomeruli using an ex vivo and in vivo kidney perfusion system - First steps towards gene therapy of Alport syndrome. Gene Ther 3: 21–27, 1996 [PubMed] [Google Scholar]
- 59.Funk SD, Bayer RH, Miner JH: Endothelial cell-specific collagen type IV-α3 expression does not rescue Alport syndrome in Col4a3-/- mice. Am J Physiol Renal Physiol 316: F830–F837, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Heikkilä P, Tibell A, Morita T, Chen Y, Wu G, Sado Y, Ninomiya Y, Pettersson E, Tryggvason K: Adenovirus-mediated transfer of type IV collagen alpha5 chain cDNA into swine kidney in vivo: Deposition of the protein into the glomerular basement membrane. Gene Ther 8: 882–890, 2001 [DOI] [PubMed] [Google Scholar]
- 61.Colella P, Trapani I, Cesi G, Sommella A, Manfredi A, Puppo A, Iodice C, Rossi S, Simonelli F, Giunti M, Bacci ML, Auricchio A: Efficient gene delivery to the cone-enriched pig retina by dual AAV vectors. Gene Ther 21: 450–456, 2014 [DOI] [PubMed] [Google Scholar]
- 62.De Silva SR, Arno G, Robson AG, Fakin A, Pontikos N, Mohamed MD, Bird AC, Moore AT, Michaelides M, Webster AR, Mahroo OA: The X-linked retinopathies: Physiological insights, pathogenic mechanisms, phenotypic features and novel therapies [published online ahead of print August 26, 2020]. Prog Retin Eye Res 10.1016/j.preteyeres.2020.100898 [DOI] [PubMed] [Google Scholar]
- 63.Elborn JS, Ramsey BW, Boyle MP, Konstan MW, Huang X, Marigowda G, Waltz D, Wainwright CE; VX-809 TRAFFIC and TRANSPORT study groups: Efficacy and safety of lumacaftor/ivacaftor combination therapy in patients with cystic fibrosis homozygous for Phe508del CFTR by pulmonary function subgroup: A pooled analysis. Lancet Respir Med 4: 617–626, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Germain DP, Hughes DA, Nicholls K, Bichet DG, Giugliani R, Wilcox WR, Feliciani C, Shankar SP, Ezgu F, Amartino H, Bratkovic D, Feldt-Rasmussen U, Nedd K, Sharaf El Din U, Lourenco CM, Banikazemi M, Charrow J, Dasouki M, Finegold D, Giraldo P, Goker-Alpan O, Longo N, Scott CR, Torra R, Tuffaha A, Jovanovic A, Waldek S, Packman S, Ludington E, Viereck C, Kirk J, Yu J, Benjamin ER, Johnson F, Lockhart DJ, Skuban N, Castelli J, Barth J, Barlow C, Schiffmann R: Treatment of Fabry’s disease with the pharmacologic chaperone migalastat. N Engl J Med 375: 545–555, 2016 [DOI] [PubMed] [Google Scholar]
- 65.Pieri M, Stefanou C, Zaravinos A, Erguler K, Stylianou K, Lapathitis G, Karaiskos C, Savva I, Paraskeva R, Dweep H, Sticht C, Anastasiadou N, Zouvani I, Goumenos D, Felekkis K, Saleem M, Voskarides K, Gretz N, Deltas C: Evidence for activation of the unfolded protein response in collagen IV nephropathies. J Am Soc Nephrol 25: 260–275, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Murray LS, Lu Y, Taggart A, Van Regemorter N, Vilain C, Abramowicz M, Kadler KE, Van Agtmael T: Chemical chaperone treatment reduces intracellular accumulation of mutant collagen IV and ameliorates the cellular phenotype of a COL4A2 mutation that causes haemorrhagic stroke. Hum Mol Genet 23: 283–292, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wang D, Mohammad M, Wang Y, Tan R, Murray LS, Ricardo S, Dagher H, van Agtmael T, Savige J: The chemical chaperone, PBA, reduces ER stress and autophagy and increases collagen IV α5 expression in cultured fibroblasts from men with X-linked alport syndrome and missense mutations. Kidney Int Rep 2: 739–748, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nozu K, Vorechovsky I, Kaito H, Fu XJ, Nakanishi K, Hashimura Y, Hashimoto F, Kamei K, Ito S, Kaku Y, Imasawa T, Ushijima K, Shimizu J, Makita Y, Konomoto T, Yoshikawa N, Iijima K: X-linked Alport syndrome caused by splicing mutations in COL4A5. Clin J Am Soc Nephrol 9: 1958–1964, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yamamura T, Horinouchi T, Adachi T, Terakawa M, Takaoka Y, Omachi K, Takasato M, Takaishi K, Shoji T, Onishi Y, Kanazawa Y, Koizumi M, Tomono Y, Sugano A, Shono A, Minamikawa S, Nagano C, Sakakibara N, Ishiko S, Aoto Y, Kamura M, Harita Y, Miura K, Kanda S, Morisada N, Rossanti R, Ye MJ, Nozu Y, Matsuo M, Kai H, Iijima K, Nozu K: Development of an exon skipping therapy for X-linked Alport syndrome with truncating variants in COL4A5. Nat Commun 11: 2777, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Macheroux EP, Braunisch MC, Pucci Pegler S, Satanovskij R, Riedhammer KM, Günthner R, Gross O, Nagel M, Renders L, Hoefele J: The hypomorphic variant p.(Gly624Asp) in COL4A5 as a possible cause for an unexpected severe phenotype in a family with X-linked alport syndrome. Front Pediatr 7: 485, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Buscaglia LE, Li Y: Apoptosis and the target genes of microRNA-21. Chin J Cancer 30: 371–380, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gomez IG, MacKenna DA, Johnson BG, Kaimal V, Roach AM, Ren S, Nakagawa N, Xin C, Newitt R, Pandya S, Xia TH, Liu X, Borza DB, Grafals M, Shankland SJ, Himmelfarb J, Portilla D, Liu S, Chau BN, Duffield JS: Anti-microRNA-21 oligonucleotides prevent Alport nephropathy progression by stimulating metabolic pathways. J Clin Invest 125: 141–156, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gomez IG, Nakagawa N, Duffield JS: MicroRNAs as novel therapeutic targets to treat kidney injury and fibrosis. Am J Physiol Renal Physiol 310: F931–F944, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Stanek LM, Sardi SP, Mastis B, Richards AR, Treleaven CM, Taksir T, Misra K, Cheng SH, Shihabuddin LS: Silencing mutant huntingtin by adeno-associated virus-mediated RNA interference ameliorates disease manifestations in the YAC128 mouse model of Huntington’s disease. Hum Gene Ther 25: 461–474, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mulligan MJ, Lyke KE, Kitchin N, Absalon J, Gurtman A, Lockhart S, Neuzil K, Raabe V, Bailey R, Swanson KA, Li P, Koury K, Kalina W, Cooper D, Fontes-Garfias C, Shi PY, Türeci Ö, Tompkins KR, Walsh EE, Frenck R, Falsey AR, Dormitzer PR, Gruber WC, Şahin U, Jansen KU: Phase I/II study of COVID-19 RNA vaccine BNT162b1 in adults. Nature 586: 589–593, 2020 [DOI] [PubMed] [Google Scholar]
- 76.Barker DF, Hostikka SL, Zhou J, Chow LT, Oliphant AR, Gerken SC, Gregory MC, Skolnick MH, Atkin CL, Tryggvason K: Identification of mutations in the COL4A5 collagen gene in Alport syndrome. Science 248: 1224–1227, 1990 [DOI] [PubMed] [Google Scholar]