

Abstract
Platelet-dependent mechanisms for excessive clotting and bleeding in CKD remain undefined. Moreover, platelets’ contribution to inflammation, and specifically to CKD, are equally elusive. To date, descriptions of changes in the functional properties of circulating platelets during CKD have provided confusing interpretations. Experimental approaches that can advance our understanding of platelet dysfunction in CKD are needed, and studies that provide mechanistic insights into the dynamic relationships between thrombosis, bleeding, and inflammation associated with CKD will be essential to improve clinical management and outcomes for this vulnerable population. This article summarizes existing literature characterizing platelets in CKD and identifies areas that need further investigation.
Keywords: platelets, chronic inflammation, chronic kidney disease, thrombosis, bleeding
In the early 1900s, thrombotic and bleeding complications originating from the kidney were first described.1 For patients with CKD, “uremic platelet dysfunction” was the general explanation for clinical findings of atherothrombotic cardiovascular events, hemorrhagic pleural effusions, retroperitoneal hematoma, gastrointestinal bleeding, and subdural hematoma.1 In the last five decades, studies that reported “uremic platelet dysfunction” were on the basis of in vitro assays of human platelet function, which resulted in conflicting results of platelet activation during CKD. Some studies described a prothrombotic phenotype and others a probleeding phenotype.2 With advancements in dialysis therapy and predialysis management of CKD, contemporary patients with CKD have fewer symptoms associated with kidney failure. Nevertheless, one in four patients with CKD still develops two entirely opposite hemostatic complications.3,4 The role of platelets in hemostatic complications of CKD remains poorly defined.5,6
Platelets are circulating anucleated cells that are important for hemostasis.7 A growing body of literature describes the roles of circulating platelets in modulating pathophysiologic processes outside of thrombosis, such as inflammation.8 So far, however, the pathophysiologic roles of platelets in generating inflammatory and hemostatic complications for patients with CKD remain unexplored. Why some patients present with thrombotic versus bleeding events and vice versa remains unknown. The most unifying concept to reconcile coexistence of inflammation and hemostasis in patients with CKD is a simple assumption that “the platelet is the driver of two seemingly different pathophysiologic events.”
Two vital questions remain unanswered on the role of platelets in CKD. First, what is the role of platelets in modulating inflammation associated with CKD? Second, what mechanisms lead to the paradoxical hemostatic events in CKD? There are no established platelet-dependent mechanisms to explain the hemostatic and the inflammatory imbalance in patients with CKD. This review article will focus on our understanding of platelet dysfunction in the CKD state and on identifying knowledge gaps that exist.
What is the Clinical Significance of Platelets for Nephrologists?
CKD affects 10%–16% of the adult population worldwide, with more than 14 million affected individuals in the United States alone.9 The presence of CKD increases risk for hospitalizations, cardiovascular events, and all-cause mortality because of the disturbed internal physiologic milieu from failing kidneys, resulting in increased inflammation, platelet dysfunction, CKD bone and mineral disorder, and other effects.10 Compared with the general population, patients with CKD have a 16% higher hazard of thrombotic cardiovascular events, such as acute myocardial infarction or ischemic stroke.11,12 Furthermore, compared with individuals who have normal kidney function, those with CKD have a 38% higher risk of either in-hospital or all-cause mortality after a heart attack or stroke.10,11 The rates of major cardiac adverse events (defined as a composite of nonfatal heart attack, nonfatal stroke, and cardiovascular death) increase dramatically with worsening CKD severity.13,14 This excessive risk of thrombosis and related death worsens as CKD progresses, such that many die before reaching dialysis dependence.13 This occurs despite current guideline-based treatments that include antiplatelet therapy and percutaneous coronary interventions.15–17 Recommendations of the American College of Cardiology and American Heart Association Task Force on Practice Guidelines for management of heart attacks emphasizes that, for special patient groups such as those with CKD, clinicians should be aware of treatment strategies that are alternatives to antiplatelet therapies, but such alternatives remain unclear.18,19 Of the many proposed mechanisms underlying thrombotic risks in CKD,20–23 one explanation posits that the major drivers of these clinical events are increased platelet activation21 and inflammation20 resulting from the CKD state.
CKD is also associated with bleeding risks that are approximately ten-fold higher than those of the general population.4,24 This is exacerbated by antiplatelet therapies, which are commonly given for thrombotic complications.4 Many believe that platelet defects and abnormal interactions between platelets and vessel walls are associated with the bleeding tendency.1 Not surprisingly, antiplatelet therapy is one of the top four reasons for emergency hospitalizations for patients aged ≥65 years—an age group with high prevalence of CKD.25 Thus, a dynamic imbalance in hemostasis and thrombosis is a hallmark clinical complication of CKD.
The two seemingly different pathophysiologic complications of thrombosis and bleeding likely fit with the platelet paradigm of a temporal sequence of events that involves adhesion, followed by aggregation. If adhesion is defective, then bleeding is likely; if aggregation is dysregulated, then thrombosis is likely. Because the mechanisms related to CKD complications are not understood, optimal antiplatelet therapy remains a clinical dilemma, as it requires a balance between efforts to reduce risks for thrombosis while minimizing bleeding events in this patient population.
Role of Platelets in Hemostasis
Platelets are capable of recognizing vascular damage by detecting the subendothelial matrix, which is exposed to the circulation after endothelial denudation of a blood vessel.7,26,27 On recognition of vessel damage, platelets aggregate at the site to form a hemostatic plug, which mitigates blood loss. Vascular damage results in the removal of the vessel’s endothelial lining, allowing circulating vWF to bind collagen within the subendothelium. Subsequently, vWF is denatured because of shearing. This denaturation exposes the A1 domain of vWF, which binds glycoprotein Ib-IX (GPIb-IX) on the platelet surface. The formation of GPIb-IX/vWF localizes the receptor glycoprotein VI (GPVI) to its ligand, collagen, and causes platelet activation. This elicits a conformational shift of the platelet surface integrins αIIbβ3 (GPIIb/IIIa) and α2β1 from inactive to active states and degranulation of the α and dense granules, thereby releasing additional adhesion and activation mediators. The integrin α2β1 then binds to collagen, which further anchors the platelet to the subendothelium. Likewise, αIIbβ3 binds vWF, which solidifies the platelet’s association to the damaged site; it also binds fibrinogen released from α-granules, serving as a bridge to crosslink platelets in the growing hemostatic plug. Additionally, αIIbβ3 is able to bind the C1 domain of circulating vWF. The binding of vWF to static, activated platelets causes a similar denaturation, revealing vWF’s A1 domain and prompting the recruitment of additional circulating, inactivated platelets. These newly recruited platelets are activated by soluble mediators (such as ADP) binding to P2Y receptors and by thromboxane A2 binding to its receptor, released from the activated platelet’s dense granules. This additional recruitment aids successful platelet aggregation, which is necessary to form a stable hemostatic plug capable of preventing further blood loss through the damaged vessel. In summary, platelet activation occurs via various signaling pathways and can be measured using agonists specific to each of them.
Characterization of Platelet Dysfunction in CKD
The first obvious questions in understanding the role of platelets in CKD are “what is the platelet phenotype in CKD? Are platelets hyperactivated, less activated, or unchanged?” The crux of the literature over the last six decades is that the in vitro and ex vivo studies of platelet function in patients with CKD have produced conflicting reports of reduced activation, hyperactivation, or unchanged platelet activation (Supplemental Table 1).2,21,28–46 Almost all of these studies included primarily patients with CKD on chronic dialysis, with very few participants not on dialysis. However, dialysis treatment can bring about changes in platelet function.47 In addition, the studies that included patients with CKD who were not on dialysis loosely defined CKD based solely on an GFR cutoff of <60 ml/min per 1.73.2
Our studies extended findings from previous investigators and found no major differences in platelet aggregation and platelet secretion in patients with stages 4–5 CKD versus controls with normal kidney function except for minor differences in ADP-induced platelet aggregation.42 Even after study participants received 2 weeks of antiplatelet therapy with aspirin and a P2Y12 inhibitor, we found no notable differences between the two groups in AA–induced platelet aggregation (i.e., aspirin effect), and found minor differences in ADP-induced platelet aggregation (i.e., P2Y12 inhibitor effect). These results are different from those of some studies that reported residual platelet aggregation on aspirin to be higher in patients with worsening severity of CKD,48 but are similar to those of other studies measuring the effects of P2Y12 inhibitors in CKD.49,50 We also found no effect of depression or its treatment with the selective serotonin reuptake inhibitor sertraline on the platelet phenotype of this population,51 whereas others reported reduced platelet aggregation and activation markers with selective serotonin reuptake inhibitor use in patients who did not have CKD.52 More recently, we asked whether the levels of platelet surface proteins known to contribute to thrombosis—such as GPIb-IX, P2Y12, P-selectin, and GPIIb-IIIA—differ between CKD and controls. We found that levels of these proteins in samples from patients with stages 4–5 CKD versus controls did not differ before or after antiplatelet therapy. Our results are consistent with previous studies in patients on chronic hemodialysis.38,45,46
There are numerous in vitro or ex vivo laboratory assays available for platelet functional assessment, including skin bleeding time, closure time using the Platelet Function Analyzer, and traditional aggregometry, as measured by optical or impedance changes. Studies on blood samples from patients on hemodialysis demonstrated poor correlation between various assays.53 Furthermore, these assays are not readily available, are costly, and are not covered by most insurers because their use was not shown to improve clinical outcomes in the non-CKD population.18 Flow cytometry, which can measure cell-to-platelet ratios and platelet surface proteins, is a valuable tool to understand platelet pathophysiology, but remains underutilized in CKD.
Two important conclusions can be derived from the research, which spans six decades:
(1) simple changes to in vitro platelet functioning or platelet-surface receptors as a result of antiplatelet therapy do not explain the increased thrombotic risk associated with CKD, and
(2) novel approaches are needed to move the field forward.
Paradigm Shift: Interplay between Platelets and Inflammation
For decades, studies of platelet biology have focused primarily on the ability of these cells to form clots. As a result, our understanding of the roles of platelets has been limited to their participation in hemostasis and thrombosis. An emerging literature highlights platelets’ effects on the immunologic response in inflammation.8 Because mammalian platelets resemble their ancestral predecessors—thrombocytes of lower vertebrates—they are also considered an extension of the immune system. Platelets have been shown to modify the inflammatory phenotype by influencing leukocyte function directly via cell-platelet adhesion, and indirectly through the release of soluble mediators and microparticles.54 Additionally, because hemostasis and inflammation often occur in tandem, one could reason that the presence of platelets in both pathways positions this cell type at the interface of the responses.
Recent studies have shown that platelets play a critical role in modulating inflammation and thrombosis in murine models of polymicrobial sepsis.54,55 Sepsis induced via the cecal ligation and puncture technique in a mouse model lacking GPIb-IX (hIL-4R/Ibα) resulted in significantly reduced platelet associations with neutrophils/monocytes, heightened neutrophil activation, increased nonclassical monocyte production, and elevated levels of circulating cytokines (e.g., TNFα) during sepsis. This inflammatory phenotype results from loss of a regulatory function that platelets have over immune cells, and is predicated on GPIb-IX interfacing with Mac-1 (CD11b/CD18), an integrin found on many leukocytes.55–57 Interestingly, compared with controls, mice that lack the thrombotic ligand for GPIb-IX, vWF, survive longer after induction of severe sepsis because their platelets retain the ability to regulate leukocyte function through the GPIb-IX/Mac-1 axis even as they are prevented from inducing thrombosis via the GPIb-IX/vWF association.55–57 These findings suggest the platelet receptor GPIb-IX is critical for platelets to modulate inflammation.55 Consistent with these animal studies, human studies also showed that antiplatelet therapy with inhibitors of P2Y12 receptor reduces proinflammatory cytokines and platelet-leukocyte aggregates in healthy controls when sepsis is induced by injecting bacterial endotoxin.58
So, given GPIb-IX provides platelets with the ability to participate in both hemostasis and inflammation, we must be cognizant of benefits and drawbacks of targeting this receptor to modify either pathway. As previously demonstrated, GPIb-IX disruption mitigated sepsis-induced thrombosis, but additionally exacerbated the inflammatory response by increasing leukocyte activation and proinflammatory cytokine release. The fact that patients with CKD already display a prothrombotic or a probleeding state raises an important yet simple clinical question: does the participation of platelets in the CKD inflammatory response help explain its hemostatic complications?
Apart from the GPIb-IX–centric approach to this paradigm, other platelet surface receptors may also be crucial, including GPVI. In mouse models of glomerulonephritis, within a few minutes after infusion of anti–glomerular basement membrane antibody, there is GPVI-mediated platelet recruitment that is not dependent on GPIb-IX.59 Exclusively expressed on platelets and megakaryocytes, GPVI binds to its ligand (collagen) and results in immunoreceptor tyrosine-based activation motif–mediated phosphorylation of tyrosine kinase Syk.60 This “outside-in” signaling is central to collagen-mediated platelet activation and the release of platelet microparticles. These membrane-bound platelet fragments, <1 µm in size, are known to augment proinflammatory and procoagulant response. Collagen-induced platelet activation and thrombus formation, mediated by the GPVI-Syk pathway, can be inhibited by a tyrosine kinase inhibitor.61 Blocking this pathway manifests as gastrointestinal bleeds in patients who are leukemic and treated with tyrosine kinase inhibitors.62 By reducing proinflammatory cytokines and macrophage infiltration of the kidney tissue, tyrosine kinase inhibitors also act as antifibrotic agents in murine models of CKD induced by unilateral ureteral obstruction.63 Thus, this lays out a novel pathway of a GPVI-mediated inflammatory cascade as a therapeutic target for future therapies in CKD. Moreover, this pathway has also been reported to determine severity of sepsis in a study of pediatric sepsis, in which a particular haplotype of GPVI, known to generate higher amounts of platelet microparticles compared with wild type, was associated with septic shock.64
Finally, platelets also play a crucial role in maintaining vascular integrity at the site of inflammation and organ development. C-type lectin receptor-2 (CLEC-2), which has no role in primary hemostasis, works as an adhesion receptor65; it inhibits bleeding at the site of inflammation and is not dependent on αIIbβ3. In certain inflammatory skin conditions, platelets control neutrophil-mediated vascular destruction by two crucial interactions: interaction between CLEC-2 and podoplanin on macrophages and stromal cells, and interaction between GPVI and collagen. In mice, deficiency of both GPVI and CLEC-2 results in bleeding in the inflamed skin due to loss of vascular integrity. Similarly, CLEC-2–deficient mice exhibit severe bleeding in the developing brain. Unlike patients with GPIb defects, such as Bernard Soulier syndrome, families living with genetic defects in GPVI do not exhibit high risk of bleeding.65 These defects could be explored as a strategy to identify novel targets for future antiplatelet therapies in patients with CKD.
On the basis of these paradigms, several parallels can be identified between severe inflammatory response syndrome (SIRS) and CKD. In both conditions, the ability to maintain a balance between inflammatory and hemostatic pathways is altered (Figure 1A). We already know that CKD begets inflammation, as shown in cross-sectional studies reporting that patients with CKD have higher levels of inflammatory markers, including high-sensitivity C-reactive protein, TNF-α, leukocyte numbers, and IL-6.66–68 Inflammation also may contribute to development of CKD and its progression in humans.69 Thus, CKD and SIRS can alter hemostasis in similar ways. These parallels lead us to posit similarities with the state of global dysregulation of inflammation and hemostasis that is observed in SIRS and the possible role of platelets in orchestrating similar complications of CKD. Therefore, elucidating the roles of platelets in modifying inflammation in patients with CKD is important to discovering why an individual with CKD develops clots or a tendency for bleeding and to unraveling the elusive mechanisms at work in these scenarios.
Figure 1.
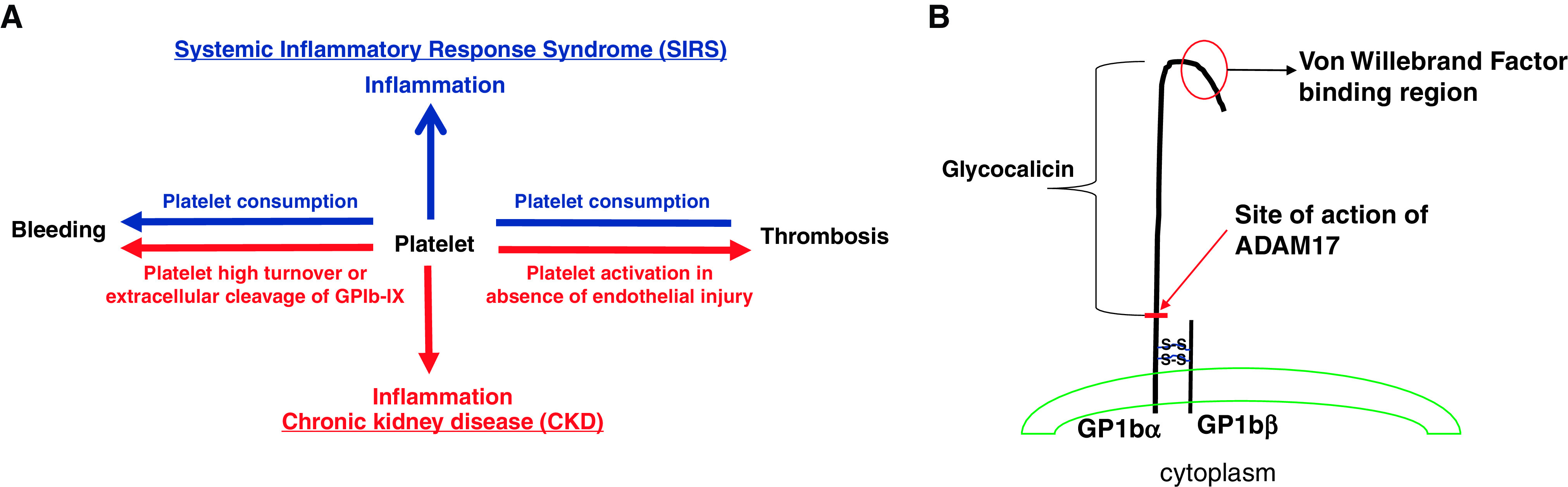
Parallels identified between systemic inflammatory response syndrome (SIRS) and CKD position platelets as possible modulators of inflammation, thrombosis and bleeding in both conditions. (A) Hypothesis on the basis of the central role of platelets in driving inflammation, thrombosis, and bleeding in CKD. Platelets are the primary contributors to the maintenance of vascular integrity by forming platelet plugs to mitigate blood loss. However, under pathologic conditions such as SIRS, one may encounter two entirely different hemostatic abnormalities: (1) dysregulated formation of thrombi in disseminated intravascular coagulation (DIC) that contributes to ischemic events and organ failure, and (2) platelet consumption, which manifests as bleeding events. These two entirely opposite hemostatic events of thrombosis and bleeding are also seen in CKD state. To date, mechanisms to explain these entirely opposite hemostatic events and inflammation remain elusive. The most unifying concept to put these events together is the concept that the platelet may be participating in modulation of systemic inflammation of the CKD state. On the basis of previous animal research, it may be possible that one of its surface receptors, GPIb-IX, may mediate a platelet-dependent inflammatory response. Depending on the inflammatory state, a patient with CKD may exhibit prothrombotic or probleeding phenotype; alternatively, high platelet turnover or GPIb-IX extracellular cleavage may result in impaired platelet-to-vessel wall interaction rendering an individual susceptible to bleeding. (B) Structure of GP-1b α and β chains on platelet surface. It is linked with two di-sulfide bonds. It also shows site of action of ADAM17 to cleave extracellular GP1bα polypeptide, termed glycocalicin.
Bleeding Tendencies Are Also A Problem in CKD
Excessive bleeding tendencies in patients with CKD mainly are reported as gastrointestinal bleeding, subdural hematoma, retroperitoneal bleeding, and hemorrhagic pleural effusions.1 These clinical manifestations are thought to be a result of abnormal platelets and interactions between platelets and vessel walls, given that plasma coagulation and fibrinolysis factors remain unchanged.1,70 Studies in patients on chronic dialysis who experience bleeding events reported conflicting results on the platelet phenotype, including prolonged bleeding time and reduced or unchanged aggregation (Supplemental Table 2).71–75 In patients with CKD who are dialysis dependent, binding of GPIb-IX to anti-GPIb mAb may be diminished and was shown to correlate with a bleeding tendency.21,74 Possible explanations for this reduced binding of GPIb-IX could be increased platelet turnover or increased cleavage of the extracellular GPIbα polypeptide that results in release of glycocalicin into the plasma (Figure 1B). Release of glycocalicin can occur due to increased ADAM17 activity from kidney disease itself76 or from exposure of blood to the extracorporeal circuit (e.g., the dialysis machine).77 Plasma glycocalicin levels remain unexplored in patients with CKD not on dialysis. ADAM17 (also known as TNF converting enzyme) is widely implicated in several chronic diseases, including CKD progression,78 but no studies have reported a correlation between bleeding tendencies and upregulation of ADAM17 plasma activity or glycocalicin levels in patients with CKD not on dialysis.
Interaction between platelets and vessel walls also remains poorly understood in CKD.1 Binding of vWF to the platelet GPIb-IX allows platelets to anchor to the subendothelium at the site of injury. Weibel-Palade bodies store vWF multimers,79 and on stimulation, these bodies rapidly release vWF multimers and other molecules. Desmopressin binds directly to the vasopressin 2 receptor on the endothelial cell to release contents of Weibel-Palade bodies, and is thus commonly used to treat bleeding complications in von Willebrand disease, hemophilia A, several platelet disorders, uremic bleeding, and in settings of antiplatelet therapy–induced perioperative bleeding.80 However, how desmopressin reduces uremic bleeding when there is no known deficiency of vWF in the CKD state remains poorly understood.79
Effects of Uremic Toxins on Platelets
A few reports suggest gut-derived uremic toxins, molecules that accumulate in plasma as kidneys fail, block platelet activation and promote bleeding, but others suggest these toxins increase platelet activation and promote thrombosis (Supplemental Table 3).81–85 Multiple observational studies report associations between plasma levels of uremic toxins and thrombotic cardiovascular events and arteriovenous fistula thrombosis.86 Mechanisms underlying the modulation of platelet function by uremic toxins remain elusive. In platelets obtained from patients on hemodialysis, platelet fibrinogen receptors exhibit an impaired ability to undergo conformational change and to bind to the ligand due to presence of putative inhibitors, such as uremic toxins. Dialysis treatment was shown to partially correct the abnormality with possible removal of these putative inhibitors.46 However, mechanistic studies are needed to understand these complex interactions. On the one hand, murine studies have demonstrated that indoxyl sulfate increases the expression of P-selectin and αIIbβ3 in a dose-dependent manner and activation of the collagen pathway.81,83 On the other hand, platelet isolates from humans and rabbits exhibit downregulation of the thromboxane A2 pathway on exposure to p-cresol sulfate.82 All in all, our understanding of the roles of platelets in bleeding complications and the role that uremic toxins play in eliciting the platelet dysfunction seen in CKD remains unclear.
Where Do We Stand Today and Where Should We Go in Future?
In short, we do not understand what is wrong with platelets or how platelet dysfunction alters inflammatory and thrombotic pathways in patients with CKD, who experience two entirely different and paradoxical hemostatic events. The literature has not shed light on mechanisms that might be altered to explain hemostatic complications arising from the CKD state. Inherent limitations of various assays of platelet function have also hindered this field of study. Previous research and our own study findings suggest that patients with CKD and those with normal kidney function exhibit no major differences in platelet aggregation and platelet secretion that might explain the opposite hemostatic clinical events. We do not fully understand the molecular basis for these events, and as a result, this unexplained clinical paradox prevents clinicians from alleviating the burdens of their patients.
Because of the opposite events that occur on the spectrum of hemostasis among CKD patients, it is difficult to formulate testable hypotheses for future research. As part of the kidney research community, we need to support bold, “out-of-the-box,” clinically relevant research ideas to propel this understudied field forward. We need to study a new paradigm for platelet functions in CKD, focusing on platelets as modulators of inflammation that may explain the prothrombotic state. We also need a multidisciplinary approach for investigating molecules in CKD that may explain the probleeding state.
To date, neither changes in platelet phenotype nor aggregability can explain the excessive thrombotic risk found in patients with CKD. Therefore, there is an urgent need to discover the mechanisms by which patients with CKD have a proinflammatory and a prothrombotic state. Discovering the biology underlying platelet dysfunction and altered interaction with the immune system in CKD remains our goal.
Disclosures
All authors have nothing to disclose.
Funding
This study was supported by a American Society of Nephrology (ASN) Foundation for Kidney Research Joseph V. Bonventre Career Development grant (to N. Jain). This study was also supported by the Translational Research Institute and the National Center for Advancing Translational Sciences of the National Institutes of Health through grant KL2 TR003108.
Acknowledgments
A.L. Corken, N. Jain, and J. Ware conceptualized the study; C.L. Davis, N. Jain, and A. Kumar were responsible for the literature review; A.L. Corken, C.L. Davis, N. Jain, and A. Kumar developed tables and figures; J.M. Arthur, A.L. Corken, N. Jain, and J. Ware critically appraised the literature; and J.M. Arthur, N. Jain, and J. Ware wrote the manuscript. The views expressed here are those of the authors and do not necessarily represent the views of the American Society of Nephrology.
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
Supplemental Material
This article contains the following supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2020121806/-/DCSupplemental.
Supplemental Table 1. Summary of the studies reporting unchanged and changed platelet phenotypes due to chronic kidney disease.
Supplemental Table 2. Summary of studies reporting platelet phenotype for bleeding complications in patients with chronic kidney disease.
Supplemental Table 3. Summary of studies reporting effects of uremic toxins on platelet function.
References
- 1.Boccardo P, Remuzzi G, Galbusera M: Platelet dysfunction in renal failure. Semin Thromb Hemost 30: 579–589, 2004 [DOI] [PubMed] [Google Scholar]
- 2.Ballard HS, Marcus AJ: Primary and secondary platelet aggregation in uraemia. Scand J Haematol 9: 198–203, 1972 [DOI] [PubMed] [Google Scholar]
- 3.United States Renal Data System: U.S. renal data system chapter 7 for patients with chronic kidney disease and chapter 10 for patients with end stage renal disease. In: United States Renal Data System 2017 Annual Data Report, Bethesda, MD, National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases, 2014. Available at https://www.usrds.org/annual-data-report/previous-adrs/. Accessed December 11, 2017 [Google Scholar]
- 4.Hiremath S, Holden RM, Fergusson D, Zimmerman DL: Antiplatelet medications in hemodialysis patients: A systematic review of bleeding rates. Clin J Am Soc Nephrol 4: 1347–1355, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jain N, Reilly RF: Oral P2Y12 receptor inhibitors in hemodialysis patients undergoing percutaneous coronary interventions: Current knowledge and future directions. Semin Dial 29: 374–381, 2016 [DOI] [PubMed] [Google Scholar]
- 6.Jain N, Hedayati SS, Sarode R, Banerjee S, Reilly RF: Antiplatelet therapy in the management of cardiovascular disease in patients with CKD: What is the evidence? Clin J Am Soc Nephrol 8: 665–674, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rivera J, Lozano ML, Navarro-Núñez L, Vicente V: Platelet receptors and signaling in the dynamics of thrombus formation. Haematologica 94: 700–711, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thomas MR, Storey RF: The role of platelets in inflammation. Thromb Haemost 114: 449–458, 2015 [DOI] [PubMed] [Google Scholar]
- 9.Coresh J, Selvin E, Stevens LA, Manzi J, Kusek JW, Eggers P, et al. : Prevalence of chronic kidney disease in the United States. JAMA 298: 2038–2047, 2007 [DOI] [PubMed] [Google Scholar]
- 10.Levey AS, de Jong PE, Coresh J, El Nahas M, Astor BC, Matsushita K, et al. : The definition, classification, and prognosis of chronic kidney disease: A KDIGO Controversies Conference report. Kidney Int 80: 17–28, 2011 [DOI] [PubMed] [Google Scholar]
- 11.Foley RN, Murray AM, Li S, Herzog CA, McBean AM, Eggers PW, et al. : Chronic kidney disease and the risk for cardiovascular disease, renal replacement, and death in the United States Medicare population, 1998 to 1999. J Am Soc Nephrol 16: 489–495, 2005 [DOI] [PubMed] [Google Scholar]
- 12.Parfrey PS, Foley RN: The clinical epidemiology of cardiac disease in chronic renal failure. J Am Soc Nephrol 10: 1606–1615, 1999 [DOI] [PubMed] [Google Scholar]
- 13.Foley RN, Parfrey PS, Sarnak MJ: Clinical epidemiology of cardiovascular disease in chronic renal disease. Am J Kidney Dis 32[Suppl 3]: S112–S119, 1998 [DOI] [PubMed] [Google Scholar]
- 14.Levey AS, Beto JA, Coronado BE, Eknoyan G, Foley RN, Kasiske BL, et al. : Controlling the epidemic of cardiovascular disease in chronic renal disease: What do we know? What do we need to learn? Where do we go from here? National Kidney Foundation Task Force on Cardiovascular Disease. Am J Kidney Dis 32: 853–906, 1998 [DOI] [PubMed] [Google Scholar]
- 15.Best PJ, Lennon R, Ting HH, Bell MR, Rihal CS, Holmes DR, et al. : The impact of renal insufficiency on clinical outcomes in patients undergoing percutaneous coronary interventions. J Am Coll Cardiol 39: 1113–1119, 2002 [DOI] [PubMed] [Google Scholar]
- 16.Machecourt J, Danchin N, Lablanche JM, Fauvel JM, Bonnet JL, Marliere S, et al. ; EVASTENT Investigators: Risk factors for stent thrombosis after implantation of sirolimus-eluting stents in diabetic and nondiabetic patients: The EVASTENT Matched-Cohort Registry. J Am Coll Cardiol 50: 501–508, 2007 [DOI] [PubMed] [Google Scholar]
- 17.Zhu ZB, Zhang RY, Zhang Q, Zhang JS, Hu J, Yang ZK, et al. : Moderate-severe renal insufficiency is a risk factor for sirolimus-eluting stent thrombosis. The RIFT study. Cardiology 112: 191–199, 2009 [DOI] [PubMed] [Google Scholar]
- 18.Wright RS, Anderson JL, Adams CD, Bridges CR, Casey DE Jr., Ettinger SM, et al. : 2011 ACCF/AHA focused update of the guidelines for the management of patients with unstable angina/ non-ST-elevation myocardial infarction (updating the 2007 guideline): A report of the American College of Cardiology Foundation/American Heart Association task force on practice guidelines [published correction appears in Circulation 123: e625–e626, 2011]. Circulation 123: 2022–2060, 2011 [DOI] [PubMed] [Google Scholar]
- 19.Wolff T, Miller T, Ko S: Aspirin for the primary prevention of cardiovascular events: An update of the evidence for the U.S. Preventive Services Task Force. Ann Intern Med 150: 405–410, 2009 [DOI] [PubMed] [Google Scholar]
- 20.Oberg BP, McMenamin E, Lucas FL, McMonagle E, Morrow J, Ikizler TA, et al. : Increased prevalence of oxidant stress and inflammation in patients with moderate to severe chronic kidney disease. Kidney Int 65: 1009–1016, 2004 [DOI] [PubMed] [Google Scholar]
- 21.Sloand JA, Sloand EM: Studies on platelet membrane glycoproteins and platelet function during hemodialysis. J Am Soc Nephrol 8: 799–803, 1997 [DOI] [PubMed] [Google Scholar]
- 22.Kozek-Langenecker SA, Masaki T, Mohammad H, Green W, Mohammad SF, Cheung AK: Fibrinogen fragments and platelet dysfunction in uremia. Kidney Int 56: 299–305, 1999 [DOI] [PubMed] [Google Scholar]
- 23.Muntner P, He J, Astor BC, Folsom AR, Coresh J: Traditional and nontraditional risk factors predict coronary heart disease in chronic kidney disease: Results from the atherosclerosis risk in communities study. J Am Soc Nephrol 16: 529–538, 2005 [DOI] [PubMed] [Google Scholar]
- 24.Holden RM, Harman GJ, Wang M, Holland D, Day AG: Major bleeding in hemodialysis patients. Clin J Am Soc Nephrol 3: 105–110, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Budnitz DS, Lovegrove MC, Shehab N, Richards CL: Emergency hospitalizations for adverse drug events in older Americans. N Engl J Med 365: 2002–2012, 2011 [DOI] [PubMed] [Google Scholar]
- 26.Estevez B, Du X: New concepts and mechanisms of platelet activation signaling. Physiology (Bethesda) 32: 162–177, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brass LF, Diamond SL, Stalker TJ: Platelets and hemostasis: A new perspective on an old subject. Blood Adv 1: 5–9, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zeck J, Schallheim J, Lew SQ, DePalma L: Whole blood platelet aggregation and release reaction testing in uremic patients. BioMed Res Int 2013: 486290, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cetin O, Bekpinar S, Unlucerci Y, Turkmen A, Bayram C, Ulutin T: Hyperhomocysteinemia in chronic renal failure patients: Relation to tissue factor and platelet aggregation. Clin Nephrol 65: 97–102, 2006 [DOI] [PubMed] [Google Scholar]
- 30.Ho SJ, Gemmell R, Brighton TA: Platelet function testing in uraemic patients. Hematology 13: 49–58, 2008 [DOI] [PubMed] [Google Scholar]
- 31.Rabiner SF, Molinas F: The role of phenol and phenolic acids on the thrombocytopathy and defective platelet aggregation of patients with renal failure. Am J Med 49: 346–351, 1970 [DOI] [PubMed] [Google Scholar]
- 32.Zwaginga JJ, IJsseldijk MJ, de Groot PG, Vos J, de Bos Kuil RL, Sixma JJ: Defects in platelet adhesion and aggregate formation in uremic bleeding disorder can be attributed to factors in plasma. Arterioscler Thromb 11: 733–744, 1991 [DOI] [PubMed] [Google Scholar]
- 33.Pluta J, Nicińska B, Grzeszczyk M, Kołacz M, Jureczko L, Kwiatkowski A, et al. : Assessment of the hemostatic parameters and platelet function on thromboelastometry and impedance aggregometry in hemodialysis patients qualified for kidney transplantation: Preliminary report. Transplant Proc 48: 1431–1434, 2016 [DOI] [PubMed] [Google Scholar]
- 34.Zhu P, Tang XF, Xu JJ, Song Y, Liu R, Zhang Y, et al. : Platelet reactivity in patients with chronic kidney disease undergoing percutaneous coronary intervention. Platelets 30: 901–907, 2019 [DOI] [PubMed] [Google Scholar]
- 35.Mavrakanas TA, Alam A, Reny JL, Fontana P: Platelet reactivity in stable cardiovascular patients with chronic kidney disease. Platelets 29: 455–462, 2018 [DOI] [PubMed] [Google Scholar]
- 36.Kim HY, Oak CY, Kim MJ, Kim CS, Choi JS, Bae EH, et al. : Prevalence and associations for abnormal bleeding times in patients with renal insufficiency. Platelets 24: 213–218, 2013 [DOI] [PubMed] [Google Scholar]
- 37.Dudley A, Byron JK, Burkhard MJ, Warry E, Guillaumin J: Comparison of platelet function and viscoelastic test results between healthy dogs and dogs with naturally occurring chronic kidney disease. Am J Vet Res 78: 589–600, 2017 [DOI] [PubMed] [Google Scholar]
- 38.Mourikis P, Helten C, Dannenberg L, Hohlfeld T, Stegbauer J, Petzold T, et al. : Platelet reactivity in patients with chronic kidney disease and hemodialysis. J Thromb Thrombolysis 49: 168–172, 2020 [DOI] [PubMed] [Google Scholar]
- 39.Gäckler A, Rohn H, Lisman T, Benkö T, Witzke O, Kribben A, et al. : Evaluation of hemostasis in patients with end-stage renal disease. PLoS One 14: e0212237, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mekawy MA, Habashy DM, Abd El-Mohsen WA: Effect of hemodialysis on platelet function in end-stage renal disease Egyptian patients using in vitro closure time test (PFA-100 analyzer). Platelets 26: 443–447, 2015 [DOI] [PubMed] [Google Scholar]
- 41.Ando M, Iwata A, Ozeki Y, Tsuchiya K, Akiba T, Nihei H: Circulating platelet-derived microparticles with procoagulant activity may be a potential cause of thrombosis in uremic patients. Kidney Int 62: 1757–1763, 2002 [DOI] [PubMed] [Google Scholar]
- 42.Jain N, Li X, Adams-Huet B, Sarode R, Toto RD, Banerjee S, et al. : Differences in whole blood platelet aggregation at baseline and in response to aspirin and aspirin plus clopidogrel in patients with versus without chronic kidney disease. Am J Cardiol 117: 656–663, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huang MJ, Wei RB, Wang Y, Su TY, Di P, Li QP, et al. : Blood coagulation system in patients with chronic kidney disease: A prospective observational study. BMJ Open 7: e014294, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Forsythe LT, Jackson ML, Meric SM: Whole blood platelet aggregation in uremic dogs. Am J Vet Res 50: 1754–1757, 1989 [PubMed] [Google Scholar]
- 45.Moal V, Brunet P, Dou L, Morange S, Sampol J, Berland Y: Impaired expression of glycoproteins on resting and stimulated platelets in uraemic patients. Nephrol Dial Transplant 18: 1834–1841, 2003 [DOI] [PubMed] [Google Scholar]
- 46.Gawaz MP, Dobos G, Späth M, Schollmeyer P, Gurland HJ, Mujais SK: Impaired function of platelet membrane glycoprotein IIb-IIIa in end-stage renal disease. J Am Soc Nephrol 5: 36–46, 1994 [DOI] [PubMed] [Google Scholar]
- 47.Tangvoraphonkchai K, Riddell A, Davenport A: Platelet activation and clotting cascade activation by dialyzers designed for high volume online hemodiafiltration. Hemodial Int 22: 192–200, 2018 [DOI] [PubMed] [Google Scholar]
- 48.Polzin A, Dannenberg L, Sansone R, Levkau B, Kelm M, Hohlfeld T, et al. : Antiplatelet effects of aspirin in chronic kidney disease patients. J Thromb Haemost 14: 375–380, 2016 [DOI] [PubMed] [Google Scholar]
- 49.Htun P, Fateh-Moghadam S, Bischofs C, Banya W, Müller K, Bigalke B, et al. : Low responsiveness to clopidogrel increases risk among CKD patients undergoing coronary intervention. J Am Soc Nephrol 22: 627–633, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Angiolillo DJ, Bernardo E, Capodanno D, Vivas D, Sabaté M, Ferreiro JL, et al. : Impact of chronic kidney disease on platelet function profiles in diabetes mellitus patients with coronary artery disease taking dual antiplatelet therapy. J Am Coll Cardiol 55: 1139–1146, 2010 [DOI] [PubMed] [Google Scholar]
- 51.Jain N, Wan F, Kothari M, Adelodun A, Ware J, Sarode R, et al. : Association of platelet function with depression and its treatment with sertraline in patients with chronic kidney disease: Analysis of a randomized trial. BMC Nephrol 20: 395, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shirazian S, Grant CD, Aina O, Mattana J, Khorassani F, Ricardo AC: Depression in chronic kidney disease and end-stage renal disease: Similarities and differences in diagnosis, epidemiology, and Management. Kidney Int Rep 2: 94–107, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zupan IP, Sabovic M, Salobir B, Ponikvar JB, Cernelc P: Utility of in vitro closure time test for evaluating platelet-related primary hemostasis in dialysis patients. Am J Kidney Dis 42: 746–751, 2003 [DOI] [PubMed] [Google Scholar]
- 54.Franco AT, Corken A, Ware J: Platelets at the interface of thrombosis, inflammation, and cancer. Blood 126: 582–588, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Corken A, Russell S, Dent J, Post SR, Ware J: Platelet glycoprotein Ib-IX as a regulator of systemic inflammation. Arterioscler Thromb Vasc Biol 34: 996–1001, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang Y, Gao H, Shi C, Erhardt PW, Pavlovsky A, A Soloviev D, et al. : Leukocyte integrin Mac-1 regulates thrombosis via interaction with platelet GPIbα. Nat Commun 8: 15559, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lisman T: Platelet-neutrophil interactions as drivers of inflammatory and thrombotic disease. Cell Tissue Res 371: 567–576, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Thomas MR, Storey RF: Effect of P2Y12 inhibitors on inflammation and immunity. Thromb Haemost 114: 490–497, 2015 [DOI] [PubMed] [Google Scholar]
- 59.Devi S, Kuligowski MP, Kwan RY, Westein E, Jackson SP, Kitching AR, et al. : Platelet recruitment to the inflamed glomerulus occurs via an alphaIIbbeta3/GPVI-dependent pathway. Am J Pathol 177: 1131–1142, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li Z, Delaney MK, O’Brien KA, Du X: Signaling during platelet adhesion and activation. Arterioscler Thromb Vasc Biol 30: 2341–2349, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jooss NJ, De Simone I, Provenzale I, Fernández DI, Brouns SLN, Farndale RW, et al. : Role of platelet glycoprotein VI and tyrosine kinase Syk in thrombus formation on collagen-like surfaces. Int J Mol Sci 20: 2788, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Quintás-Cardama A, Han X, Kantarjian H, Cortes J: Tyrosine kinase inhibitor-induced platelet dysfunction in patients with chronic myeloid leukemia. Blood 114: 261–263, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Liu F, Wang L, Qi H, Wang J, Wang Y, Jiang W, et al. : Nintedanib, a triple tyrosine kinase inhibitor, attenuates renal fibrosis in chronic kidney disease. Clin Sci (Lond) 131: 2125–2143, 2017 [DOI] [PubMed] [Google Scholar]
- 64.Asfari A, Dent JA, Corken A, Herington D, Kaliki V, Sra N, et al. : Platelet glycoprotein VI haplotypes and the presentation of paediatric sepsis. Thromb Haemost 119: 431–438, 2019 [DOI] [PubMed] [Google Scholar]
- 65.Rayes J, Watson SP, Nieswandt B: Functional significance of the platelet immune receptors GPVI and CLEC-2. J Clin Invest 129: 12–23, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Festa A, D’Agostino R, Howard G, Mykkänen L, Tracy RP, Haffner SM: Inflammation and microalbuminuria in nondiabetic and type 2 diabetic subjects: The Insulin Resistance Atherosclerosis Study. Kidney Int 58: 1703–1710, 2000 [DOI] [PubMed] [Google Scholar]
- 67.Stuveling EM, Hillege HL, Bakker SJ, Gans RO, De Jong PE, De Zeeuw D: C-reactive protein is associated with renal function abnormalities in a non-diabetic population. Kidney Int 63: 654–661, 2003 [DOI] [PubMed] [Google Scholar]
- 68.Keller CR, Odden MC, Fried LF, Newman AB, Angleman S, Green CA, et al. : Kidney function and markers of inflammation in elderly persons without chronic kidney disease: The health, aging, and body composition study. Kidney Int 71: 239–244, 2007 [DOI] [PubMed] [Google Scholar]
- 69.Sarnak MJ, Poindexter A, Wang SR, Beck GJ, Kusek JW, Marcovina SM, et al. : Serum C-reactive protein and leptin as predictors of kidney disease progression in the Modification of Diet in Renal Disease Study. Kidney Int 62: 2208–2215, 2002 [DOI] [PubMed] [Google Scholar]
- 70.Remuzzi G: Bleeding in renal failure. Lancet 1: 1205–1208, 1988 [DOI] [PubMed] [Google Scholar]
- 71.Castaldi PA, Rozenberg MC, Stewart JH: The bleeding disorder of uraemia. A qualitative platelet defect. Lancet 2: 66–69, 1966 [DOI] [PubMed] [Google Scholar]
- 72.Horowitz HI, Stein IM, Cohen BD, White JG: Further studies on the platelet-inhibitory effect of guanidinosuccinic acid and its role in uremic bleeding. Am J Med 49: 336–345, 1970 [DOI] [PubMed] [Google Scholar]
- 73.Di Minno G, Martinez J, McKean ML, De La Rosa J, Burke JF, Murphy S: Platelet dysfunction in uremia. Multifaceted defect partially corrected by dialysis. Am J Med 79: 552–559, 1985 [DOI] [PubMed] [Google Scholar]
- 74.Eknoyan G, Wacksman SJ, Glueck HI, Will JJ: Platelet function in renal failure. N Engl J Med 280: 677–681, 1969 [DOI] [PubMed] [Google Scholar]
- 75.Remuzzi G, Livio M, Marchiaro G, Mecca G, de Gaetano G: Bleeding in renal failure: Altered platelet function in chronic uraemia only partially corrected by haemodialysis. Nephron 22: 347–353, 1978 [DOI] [PubMed] [Google Scholar]
- 76.Bergmeier W, Piffath CL, Cheng G, Dole VS, Zhang Y, von Andrian UH, et al. : Tumor necrosis factor-alpha-converting enzyme (ADAM17) mediates GPIbalpha shedding from platelets in vitro and in vivo. Circ Res 95: 677–683, 2004 [DOI] [PubMed] [Google Scholar]
- 77.Mellgren K, Friberg LG, Hedner T, Mellgren G, Wadenvik H: Blood platelet activation and membrane glycoprotein changes during extracorporeal life support (ECLS). In vitro studies. Int J Artif Organs 18: 315–321, 1995 [PubMed] [Google Scholar]
- 78.Gooz M: ADAM-17: The enzyme that does it all. Crit Rev Biochem Mol Biol 45: 146–169, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kaufmann JE, Vischer UM: Cellular mechanisms of the hemostatic effects of desmopressin (DDAVP). J Thromb Haemost 1: 682–689, 2003 [DOI] [PubMed] [Google Scholar]
- 80.Desborough MJ, Oakland KA, Landoni G, Crivellari M, Doree C, Estcourt LJ, et al. : Desmopressin for treatment of platelet dysfunction and reversal of antiplatelet agents: A systematic review and meta-analysis of randomized controlled trials. J Thromb Haemost 15: 263–272, 2017 [DOI] [PubMed] [Google Scholar]
- 81.Yang K, Du C, Wang X, Li F, Xu Y, Wang S, et al. : Indoxyl sulfate induces platelet hyperactivity and contributes to chronic kidney disease-associated thrombosis in mice. Blood 129: 2667–2679, 2017 [DOI] [PubMed] [Google Scholar]
- 82.Chang MC, Wang TM, Yeung SY, Jeng PY, Liao CH, Lin TY, et al. : Antiplatelet effect by p-cresol, a uremic and environmental toxicant, is related to inhibition of reactive oxygen species, ERK/p38 signaling and thromboxane A2 production. Atherosclerosis 219: 559–565, 2011 [DOI] [PubMed] [Google Scholar]
- 83.Karbowska M, Kaminski TW, Marcinczyk N, Misztal T, Rusak T, Smyk L, et al. : The uremic toxin indoxyl sulfate accelerates thrombotic response after vascular injury in animal models. Toxins (Basel) 9: 229, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bazilinski N, Shaykh M, Dunea G, Mamdani B, Patel A, Czapek E, et al. : Inhibition of platelet function by uremic middle molecules. Nephron 40: 423–428, 1985 [DOI] [PubMed] [Google Scholar]
- 85.Davis JW, McField JR, Phillips PE, Graham BA: Guanidinosuccinic acid on human platelet. Effects of exogenous urea, creatinine, and aggregation in vitro. Blood 39: 388–397, 1972 [PubMed] [Google Scholar]
- 86.Addi T, Dou L, Burtey S: Tryptophan-derived uremic toxins and thrombosis in chronic kidney disease. Toxins (Basel) 10: 412, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]