

Significance Statement
Hypoxia-inducible factor prolyl hydroxylase inhibitors have been shown in clinical trials to increase hemoglobin levels via the production of endogenous erythropoietin. Vadadustat, an oral agent in this drug class, is an alternative to erythropoiesis-stimulating agents for the treatment of CKD-related anemia. This appears to be the first phase 3 randomized controlled trial to demonstrate noninferiority of vadadustat to darbepoetin alfa for the treatment of anemia in Japanese patients with nondialysis-dependent CKD. In participants receiving vadadustat, mean hemoglobin increased to the target range (11.0–13.0 g/dl) and was within this range up to week 52. Vadadustat was generally well tolerated over 52 weeks of treatment and no major safety concerns were identified. These findings suggest that vadadustat may be a potential treatment for anemia in patients with nondialysis-dependent CKD.
Keywords: anemia, erythropoiesis, HIF prolyl hydroxylase inhibitor, Japan, nondialysis-dependent CKD, vadadustat
Visual Abstract

Abstract
Background
Standard care for treating anemia in patients with CKD includes use of erythropoiesis-stimulating agents, which sometimes involves increased risks of cardiovascular morbidity and mortality. Previous studies in patients with anemia and nondialysis-dependent CKD (NDD-CKD) found significantly elevated hemoglobin levels with use of vadadustat, an oral hypoxia-inducible factor prolyl hydroxylase inhibitor, compared with placebo.
Methods
In this phase 3, open-label, active-controlled noninferiority trial, we randomized 304 Japanese adults with anemia in NDD-CKD (including erythropoiesis-stimulating agent users and nonusers) to oral vadadustat or subcutaneous darbepoetin alfa for 52 weeks. The primary efficacy end point was average hemoglobin at weeks 20 and 24. Safety data included adverse events (AEs) and serious AEs.
Results
A total of 151 participants received vadadustat and 153 received darbepoetin alfa. Least squares mean of the average hemoglobin at weeks 20 and 24 was 11.66 (95% confidence interval [95% CI], 11.49 to 11.84) g/dl for vadadustat and 11.93 (95% CI, 11.76 to 12.10) g/dl for darbepoetin alfa. The 95% CIs for both treatments were within the target hemoglobin range (11.0–13.0 g/dl), and the lower 95% confidence limit for the difference between groups (−0.50 g/dl) was above the predefined noninferiority margin (−0.75 g/dl), demonstrating noninferiority of vadadustat to darbepoetin alfa. Similar proportions of patients in each group reported AEs and serious AEs. The most frequent AEs with vadadustat were nasopharyngitis, diarrhea, and constipation.
Conclusions
In Japanese patients with NDD-CKD, vadadustat was noninferior to darbepoetin alfa, was effective up to week 52 in terms of average hemoglobin, and was generally well tolerated. These results suggest that vadadustat may be a potential treatment for anemia in this patient population.
Anemia, which commonly occurs in patients with CKD, increases in prevalence as kidney function declines1–3 and is associated with kidney disease progression,4 increased mortality,4–7 cardiovascular morbidity,4–6 and reduced quality of life.8 In Japan, approximately 13% of the population are estimated to have nondialysis-dependent CKD (NDD-CKD)9 and the rates of anemia range from 10.4% to 68.4% depending on CKD stage.10 Standard care for anemia treatment in patients with CKD is erythropoiesis-stimulating agents (ESAs), with iron supplementation as needed to help achieve target hemoglobin (Hb) levels.11 Despite the benefits of ESAs in reducing blood transfusions and improving quality of life, patients with NDD-CKD who undergo subcutaneous administration are burdened by the need for outpatient visits, and there are increased risks of cardiovascular mortality and morbidity with ESA treatment to higher Hb targets.12–15 Furthermore, findings from a previous cohort study of Japanese patients with anemia in NDD-CKD showed that 67.6% of patients who required treatment for anemia did not receive ESAs.10 Thus, oral drugs could be a new option for the treatment of anemia in NDD-CKD.
Renal anemia is primarily caused by the inability of kidneys to produce sufficient levels of erythropoietin.10,16 Hypoxia-inducible factors (HIFs), which are degraded by the enzyme prolyl hydroxylase under normoxic conditions, are the main transcription factors that regulate the cellular response to hypoxia.17 Vadadustat is a once-daily, orally administered, HIF prolyl hydroxylase inhibitor (HIF-PHI) for the treatment of anemia in patients with CKD. HIF-PHIs mimic hypoxic conditions by stabilizing HIF, which induces production of endogenous erythropoietin.17,18 In addition, because HIF also induces vascular endothelial growth factor (VEGF), which may promote the onset of retinopathy, tumor growth, and metastasis,19 the safety profile of HIF-PHIs is not yet fully established.
In previous studies, vadadustat has been shown to significantly elevate and maintain Hb levels compared with placebo in patients with anemia in NDD-CKD.20,21 In the phase 2 study from Japan,21 vadadustat increased Hb at 6 weeks from baseline in a dose-dependent manner and safety was evaluated for up to 16 weeks.
The objectives of this phase 3 study were to evaluate the efficacy and safety of vadadustat compared with darbepoetin alfa in Japanese patients with anemia in NDD-CKD, with and without a recent history of ESA treatment, for up to 52 weeks.
Methods
Study Design
This was a parallel-group, randomized, phase 3, open-label, active-controlled noninferiority study in Japanese patients with anemia in NDD-CKD, conducted at 86 sites in Japan from October 2017 to August 2019 (Figure 1). The protocol was approved by each site’s institutional review board and the study was conducted in accordance with the Declaration of Helsinki, the International Conference on Harmonisation Guideline for Good Clinical Practice, and applicable laws and regulations. All patients provided written informed consent before participating in the study, and the study was registered at www.clinicaltrials.gov (NCT03329196).
Figure 1.

Study design. The study comprised a screening period, a 52-week treatment period, and a follow-up period. Patients were randomized 1:1 to oral vadadustat or subcutaneous darbepoetin alfa. Vadadustat was started at 300 mg once daily and doses were adjusted (dose range: 150–600 mg once daily) to maintain hemoglobin levels within the predefined target of 11.0–13.0 g/dl. The initial dose of darbepoetin alfa was set in accordance with previous ESAs in ESA users and was 30 μg every 2 weeks in ESA non–users. Doses of darbepoetin alfa were adjusted between 15 and 180 μg once weekly, every 2 weeks, or every 4 weeks to maintain Hb levels within the target range. The primary efficacy end point was the average Hb at weeks 20 and 24. q.d., once daily; q.w., once weekly; q.2.w, every 2 weeks; q.4.w, every 4 weeks
Study Population
Details of the inclusion and exclusion criteria are provided in Supplemental Material. In brief, patients aged ≥20 years with CKD who were not on dialysis and had an eGFR of <60 ml/min per 1.73 m2 were eligible. Both “ESA users” and “ESA non−users” were included. ESA users had average Hb levels at the last two visits during screening of 9.0–12.5 g/dl and received the same ESA for 8 weeks before screening. ESA nonusers had average Hb levels of 8.0–11.0 g/dl and had not received an ESA for 8 weeks before screening. During screening, patients were to have serum ferritin levels ≥100 ng/ml or transferrin saturation (TSAT) ≥20%. Key exclusion criteria were anemia attributable to causes other than CKD; active bleeding or blood loss ≤8 weeks before screening; received red blood cell transfusion ≤8 weeks before screening; active fundus disease or ocular fundus observations not available; uncontrolled hypertension; malignancy in the last 5 years; severe heart failure; and cerebrovascular disorder or acute coronary syndrome within 12 weeks of screening.
Treatment Protocol
The study comprised a screening period (up to 6 weeks), a 52-week treatment period, and follow-up (2 weeks) (Figure 1). Patients were enrolled by the investigator and randomized 1:1 (using a web-based system) to oral vadadustat (Akebia Therapeutics Inc., Cambridge, MA) or subcutaneous darbepoetin alfa (Kyowa Kirin Co., Ltd, Tokyo, Japan). Darbepoetin alfa was chosen as an active control for this study because it is widely used to treat renal anemia and has a well-established efficacy profile. Vadadustat was started at 300 mg once daily, regardless of previous ESA treatment, and doses were adjusted (dose range: 150–600 mg once daily) according to a dose-adjustment algorithm (Supplemental Material) to maintain Hb levels within the predefined target of 11.0–13.0 g/dl, which is recommended as a treatment target for anemia in NDD-CKD by the Japanese Society for Dialysis Therapy guidelines.11 The dose-increase interval of vadadustat was to be ≥4 weeks. The initial dose of darbepoetin alfa was set in accordance with previous ESAs in ESA users and was 30 μg once every 2 weeks in ESA non–users. Doses of darbepoetin alfa were adjusted between 15 and 180 μg once every 4 weeks, every 2 weeks, or weekly to maintain Hb levels within the target range according to an algorithm (Supplemental Material). The dose-increase interval of darbepoetin alfa was to be ≥2 weeks.
Iron supplements were administered during the screening and treatment periods to maintain serum ferritin levels ≥100 ng/ml or TSAT ≥20%, in accordance with the Japanese Society for Dialysis Therapy guidelines.11 Patients receiving an iron-containing phosphate binder at screening continued its use at the same dose during the treatment period. Oral iron or iron-containing phosphate binders were not to be taken within 2 hours of vadadustat dosing to avoid a decline in the bioavailability of vadadustat.
Administration of ESAs, red blood cell transfusion, or phlebotomy was permitted as rescue therapy at the investigators’ discretion.
Study End Points and Outcome Measures
The primary efficacy end point was the average Hb at weeks 20 and 24. Secondary efficacy end points included mean Hb at each time point and the proportion of patients with mean Hb within the target range of 11.0–13.0 g/dl. Other end points included the doses of study drugs, the dose of iron supplementation, iron-related parameters (total iron-binding capacity [TIBC], TSAT, serum ferritin, and serum hepcidin), and red blood cell indices (mean corpuscular volume [MCV], mean corpuscular Hb [MCH], MCH concentration [MCHC], and red blood cell distribution width [RDW]).
Safety assessments included adverse events (AEs) and adverse drug reactions (ADRs), laboratory tests including plasma VEGF, vital signs, and ophthalmoscopy over 52 weeks. AEs of special interest were defined as those related to the HIF-PHI class and ESAs, including cardiovascular events/cardiac failure, thromboembolism, pulmonary hypertension, malignancy, retinal disorders, and hyperkalemia.22 The proportions of patients with a confirmed Hb ≥13.0 g/dl or ≥14.0 g/dl and with a rapid Hb rise >2.0 g/dl over 4 weeks were also evaluated.
Statistical Analyses
The plan was to randomize 300 patients (150 patients per group). This sample size was calculated to give 95% power for noninferiority with a two-sided alpha of 0.05 on the basis of a noninferiority margin ≥ −0.75 g/dl and the following assumptions: an average Hb at weeks 20 and 24 for darbepoetin alfa of 12.0 g/dl, a difference in the mean Hb between vadadustat and darbepoetin alfa of 0, and an SD of 1.78 g/dl, which was assumed from the results of the Japanese phase 2 trial of vadadustat in patients with NDD-CKD.21
The full analysis set and safety populations included all patients with efficacy and safety data after receiving study drug, respectively. For the primary end point, a mixed-model repeated measures (MMRM) method was used to calculate the least squares mean of the average Hb values and two-sided 95% confidence interval (95% CI) for the between-group difference at weeks 20 and 24. The MMRM included treatment group, visits, patient groups (ESA users, ESA non–users), treatment group-by-visit interaction, and visit-by-patient groups interaction as fixed effects; baseline values as covariate effects; and patients as a random effect. Missing data were not imputed.
For the secondary and other end points, descriptive statistics were calculated for each time point and week 52 using the last observation carried forward (LOCF). To be consistent with other efficacy end points, post hoc analyses were conducted to calculate 95% CIs for MCHC and RDW. Except for the dose of study drugs, changes from baseline or the screening period were analyzed for other end points using paired t tests at the two-sided significance level of 0.05. Data were also analyzed for ESA users and non–users. ESA users were further categorized by average Hb levels during the screening period (Hb <11.0 g/dl, ≥11.0 g/dl). AEs were coded using the Japanese Medical Dictionary for Regulatory Activities. All analyses were performed using SAS version 9.4 (SAS Institute Inc., Cary, NC).
Results
Patient Disposition
Of the 432 patients who gave informed consent, 304 were randomized to vadadustat (151 patients) or darbepoetin alfa (153 patients) (Figure 2). Of these, 271 patients (130 in the vadadustat group, 141 in the darbepoetin alfa group) completed the 24-week treatment period and 234 patients (111 in the vadadustat group, 123 in the darbepoetin alfa group) completed the 52-week treatment period. The most common reason for withdrawal from both groups was initiation of kidney replacement therapy, including chronic hemodialysis/peritoneal dialysis or kidney transplant.
Figure 2.
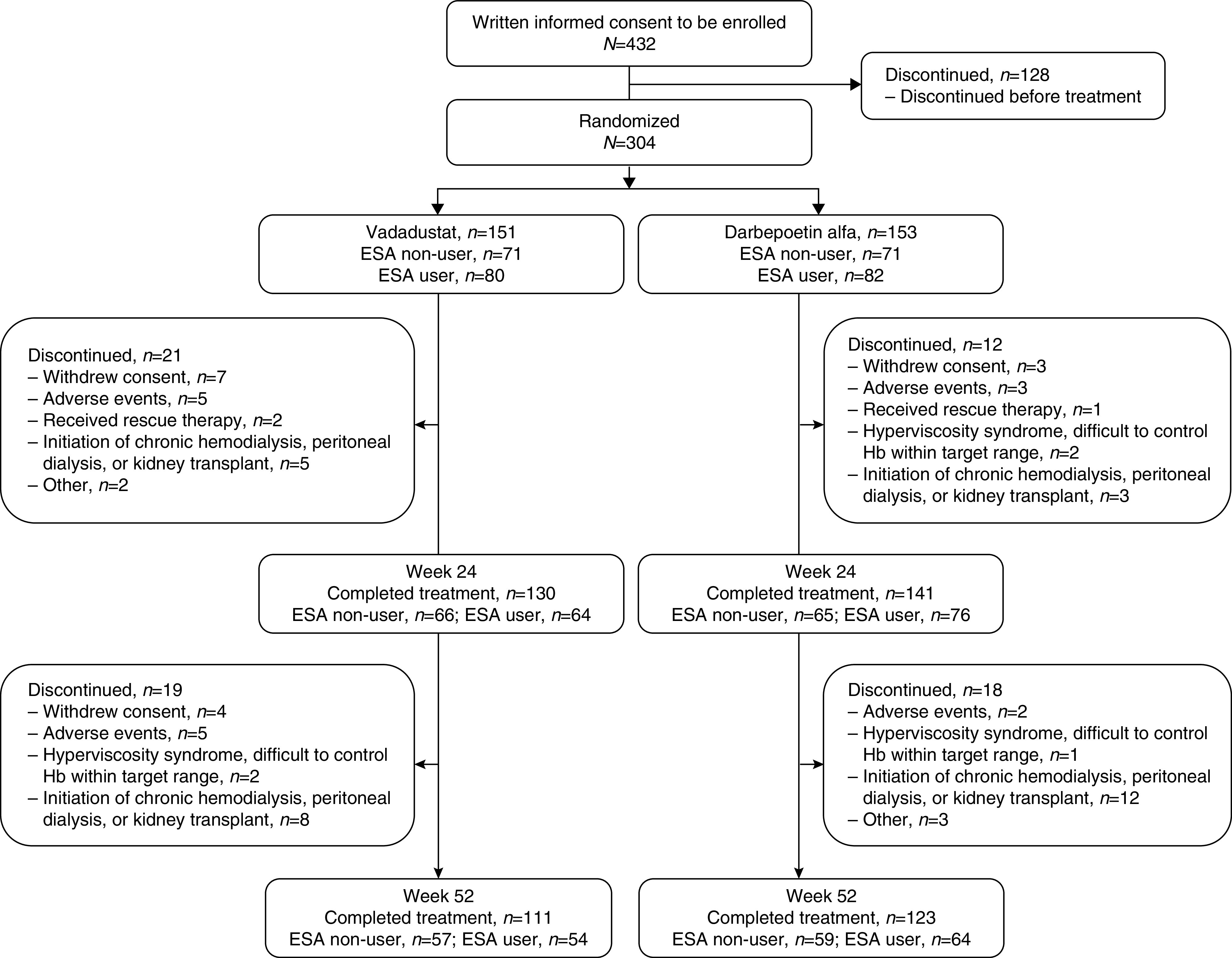
Patient flow diagram. In this study, 304 patients were randomized to vadadustat (n=151) or darbepoetin alfa (n=153). Of these, 271 patients completed the 24-week treatment period and 234 patients completed the 52-week treatment period.
Demographic and Baseline Clinical Characteristics
Baseline characteristics were generally balanced between treatment groups (Table 1). Overall, mean Hb was approximately 10.5 g/dl, eGFR was 21–23 ml/min per 1.73 m2, and the duration of anemia was approximately 3 years in both treatment groups. The most common comorbidities were hypertension and dyslipidemia. All ESA users in both treatment groups had received long-acting ESAs (darbepoetin alfa or epoetin beta pegol) before randomization.
Table 1.
Baseline characteristics (FAS)
| Characteristic | Overall | ESA Non–users | ESA Users | |||
|---|---|---|---|---|---|---|
| Vadadustat (n=151) | Darbepoetin Alfa (n=153) | Vadadustat (n=71) | Darbepoetin Alfa (n=71) | Vadadustat (n=80) | Darbepoetin Alfa (n=82) | |
| Sex (male), n (%) | 75 (49.7) | 73 (47.7) | 34 (47.9) | 30 (42.3) | 41 (51.3) | 43 (52.4) |
| Age, yr | 71.7 (10.3) | 72.2 (9.5) | 70.8 (10.8) | 71.7 (8.8) | 72.5 (9.9) | 72.6 (10.1) |
| Body weight, kg | 57.4 (11.9) | 57.5 (10.7) | 58.4 (13.3) | 57.9 (10.2) | 56.6 (10.5) | 57.1 (11.2) |
| Duration of anemia from CKD, yr | 2.75 (3.02) | 2.98 (6.08) | 2.54 (3.45) | 3.81 (8.62) | 2.93 (2.58) | 2.26 (1.98) |
| Hb, g/dl | 10.44 (0.91) | 10.52 (0.88) | 10.17 (0.87) | 10.10 (0.77) | 10.68 (0.89) | 10.89 (0.81) |
| eGFR, ml/min per 1.73 m2 | 21.28 (11.66) | 22.60 (11.60) | 24.33 (12.40) | 25.22 (12.39) | 18.58 (10.31) | 20.32 (10.42) |
| eGFR category, n (%) | ||||||
| <15 ml/min per 1.73 m2 | 60 (39.7) | 50 (32.7) | 22 (31.0) | 19 (26.8) | 38 (47.5) | 31 (37.8) |
| 15≤–<30 ml/min per 1.73 m2 | 54 (35.8) | 70 (45.8) | 24 (33.8) | 31 (43.7) | 30 (37.5) | 39 (47.6) |
| 30≤– <60 ml/min per 1.73 m2 | 37 (24.5) | 32 (20.9) | 25 (35.2) | 20 (28.2) | 12 (15.0) | 12 (14.6) |
| ≥60 ml/min per 1.73 m2 | 0 (0.0) | 1 (0.7) | 0 (0.0) | 1 (1.4) | 0 (0.0) | 0 (0.0) |
| Serum ferritin, ng/dl | 151.24 (119.82) | 138.12 (107.56) | 167.85 (146.65) | 142.35 (108.18) | 136.49 (87.93) | 134.45 (107.54) |
| TSAT, % | 31.1 (10.9) | 29.8 (9.5) | 29.3 (10.3) | 27.3 (8.3) | 32.7 (11.1) | 31.9 (9.9) |
| Prior ESA, n (%) | ||||||
| Epoetina | 0 (0.0) | 0 (0.0) | – | – | 0 (0.0) | 0 (0.0) |
| Darbepoetin alfa | 47 (58.8) | 45 (54.9) | – | – | 47 (58.8) | 45 (54.9) |
| Epoetin beta pegol | 33 (41.3) | 37 (45.1) | – | – | 33 (41.3) | 37 (45.1) |
| Comorbidities, n (%) | ||||||
| Hypertension | 147 (97.4) | 145 (94.8) | 69 (97.2) | 67 (94.4) | 78 (97.5) | 78 (95.1) |
| Diabetes mellitus | 57 (37.7) | 62 (40.5) | 28 (39.4) | 27 (38.0) | 29 (36.3) | 35 (42.7) |
| Dyslipidemia | 94 (62.3) | 100 (65.4) | 44 (62.0) | 47 (66.2) | 50 (62.5) | 53 (64.6) |
| Etiology of CKDb, n (%) | ||||||
| Diabetes | 43 (28.5) | 48 (31.4) | 25 (35.2) | 17 (23.9) | 18 (22.5) | 31 (37.8) |
| Hypertension | 52 (34.4) | 60 (39.2) | 23 (32.4) | 32 (45.1) | 29 (36.3) | 28 (34.1) |
| Autoimmune/glomerulonephritis/vasculitis | 20 (13.2) | 31 (20.3) | 7 (9.9) | 13 (18.3) | 13 (16.3) | 18 (22.0) |
| Interstitial nephritis/pyelonephritis | 4 (2.6) | 2 (1.3) | 3 (4.2) | 1 (1.4) | 1 (1.3) | 1 (1.2) |
| Cystic/hereditary/congenital disease | 13 (8.6) | 9 (5.9) | 6 (8.5) | 3 (4.2) | 7 (8.8) | 6 (7.3) |
| Unknown | 18 (11.9) | 10 (6.5) | 8 (11.3) | 6 (8.5) | 10 (12.5) | 4 (4.9) |
| Other | 7 (4.6) | 7 (4.6) | 3 (4.2) | 4 (5.6) | 4 (5.0) | 3 (3.7) |
Data are mean (SD) unless otherwise stated. FAS, full analysis set.
Epoetin alfa or epoetin beta.
Total percentages add to more than 100 because patients could have more than one etiology.
Efficacy Outcomes
The least squares mean of the average Hb levels at weeks 20 and 24 was 11.66 (95% CI, 11.49 to 11.84) in the vadadustat group and 11.93 (95% CI, 11.76 to 12.10) g/dl in the darbepoetin alfa group. The 95% CIs for both treatments were within the target Hb range of 11.0–13.0 g/dl and the lower 95% confidence limit for the difference between treatment groups (vadadustat − darbepoetin alfa) was −0.50 g/dl, which was above the predefined noninferiority margin of −0.75 g/dl, confirming the noninferiority of vadadustat to darbepoetin alfa (Table 2).
Table 2.
Primary end point (FAS)
| Average Hb, weeks 20 and 24 | Vadadustat (n=151) | Darbepoetin Alfa (n=153) | Difference Vadadustat − Darbepoetin Alfa |
|---|---|---|---|
| LSMeana | 11.66 | 11.93 | −0.26 |
| 95% CI | 11.49 to 11.84 | 11.76 to 12.10 | −0.50 to −0.02 |
FAS, full analysis set; LSMean, least squares mean.
MMRM included treatment group, visits, patient groups (ESA non–users, ESA users), interaction of treatment group and visits, interaction of visits and patient groups (ESA non–users, ESA users) as fixed effects, baseline values as covariate effects, and patient as a random effect (covariance matrix: unstructured).
Mean Hb increased from baseline, reached the target range at weeks 6–8 in both treatment groups, and was maintained within the target range up to week 52 (Figure 3). In ESA non–users, mean Hb increased from baseline, reached the target range at weeks 6–8 in both groups, and remained within the target range thereafter (Figure 4A). The proportions of patients with Hb levels within the target range in the vadadustat and darbepoetin alfa groups were 15.5% and 9.9%, respectively, at baseline, and were 71.4% and 84.5%, respectively, at week 52. From weeks 48 to 52, the mean dose was 335.65 mg/d (95% CI, 286.72 to 384.58) for the vadadustat group and 16.37 µg/wk (95% CI, 13.09 to 19.65) for the darbepoetin alfa group (Figure 4C).
Figure 3.
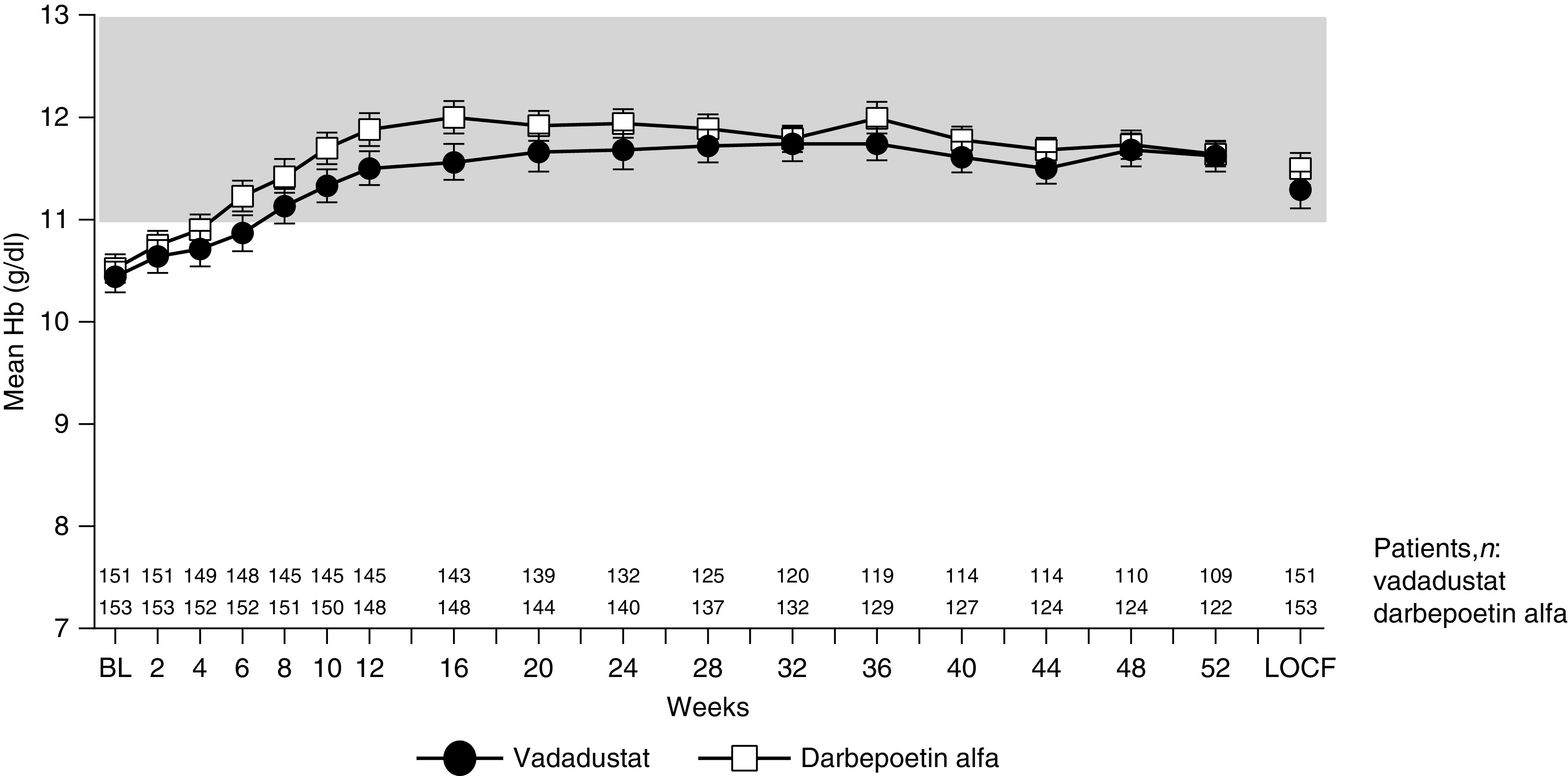
Mean Hb over time (overall population). Hb (g/dl) was measured every 2 weeks from baseline to week 12, and then every 4 weeks from week 12 to week 52. Data are displayed as mean and 95% CI. The shaded area represents the target Hb range. Mean Hb increased from baseline, reached the target range at weeks 6–8 in both treatment groups, and was maintained within the range up to week 52. BL, baseline.
Figure 4.
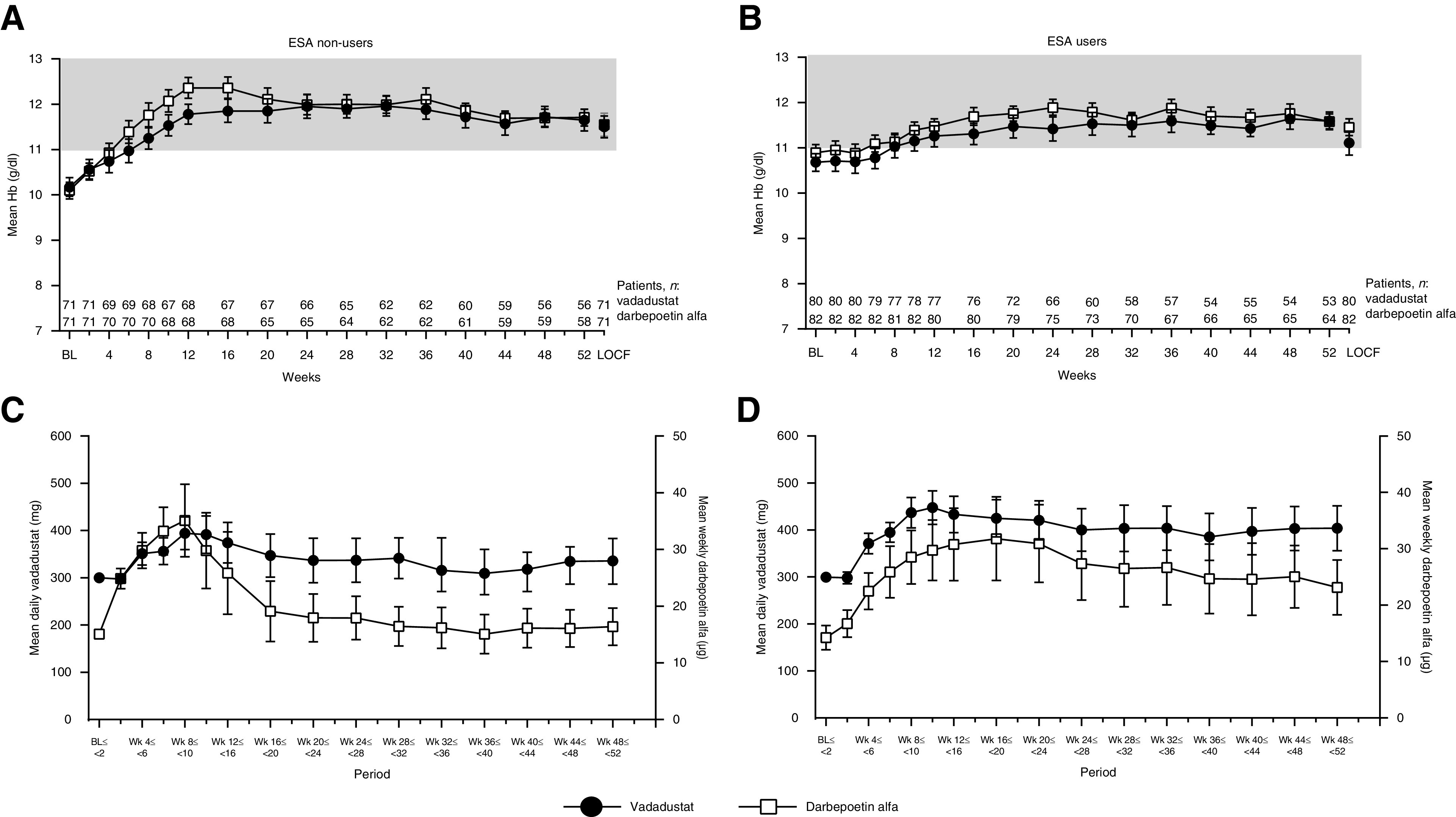
Mean Hb over time and mean dose of vadadustat (daily) or darbepoetin alfa (weekly) in the ESA non–user (A and C) and ESA user (B and D) groups. Data are displayed as mean and 95% CI. Hb (g/dl) was measured every 2 weeks from baseline to week 12, and then every 4 weeks from week 12 to week 52. The shaded area represents the target Hb range. In both ESA non–users (A) and ESA users (B), mean Hb increased from baseline, reached the target range at weeks 6–8 in both treatment groups, and remained within the range thereafter. In ESA non–users (C), the mean dose from weeks 48 to 52 was 335.65 mg/d for vadadustat and was 16.37 µg/wk for darbepoetin alfa. In ESA users (D), the mean dose from weeks 48 to 52 was 403.67 mg/d for vadadustat and was 23.15 µg/wk for darbepoetin alfa.
In ESA users, mean Hb levels increased from baseline after conversion from a previous ESA in both treatment groups. Mean Hb reached the target range at weeks 6–8 in both groups, and remained within the target range thereafter (Figure 4B). The proportions of patients with Hb levels within the target range for the vadadustat and darbepoetin alfa groups were 45.0% and 52.4%, respectively, at baseline, and increased to 79.2% and 76.6%, respectively, at week 52. From weeks 48 to 52, the mean dose was 403.67 mg/d (95% CI, 355.84 to 451.50) for vadadustat and was 23.15 µg/wk (95% CI, 18.29 to 28.00) for darbepoetin alfa (Figure 4D).
ESA users were stratified by average Hb levels during the screening period (Hb <11.0 g/dl, ≥11.0 g/dl). For the Hb ≥11.0 g/dl subgroup, Hb levels were stable and maintained within the target range throughout the treatment period in both treatment groups. For the Hb <11.0 g/dl subgroups, mean Hb increased from baseline, reached the target range at weeks 10–12, and remained within the range thereafter (Supplemental Figure 1). For the vadadustat group, the proportions of patients with Hb levels within the target range at baseline and at week 52 were 4.7% and 78.3% for the Hb <11.0 g/dl subgroup, respectively, and 91.9% and 80.0% for the Hb ≥11.0 g/dl subgroup, respectively. For darbepoetin alfa, the proportions of patients with Hb levels within the target range at baseline and at week 52 were 22.7% and 84.4% for the Hb <11.0 g/dl subgroup, respectively, and were 86.8% and 68.8% for the Hb ≥11.0 g/dl subgroup, respectively.
Iron-Related Parameters and Red Blood Cell Indices
There was a trend toward initial decreases in serum ferritin and hepcidin in the first few weeks of treatment in both the vadadustat and darbepoetin alfa groups. However, in this study, serum ferritin and hepcidin were lower than baseline in the vadadustat group at 52 weeks LOCF (Figure 5, A and B). In the darbepoetin alfa group, there was no change in serum ferritin, and hepcidin was higher than baseline (Figure 5, A and B). There was no change in TSAT from baseline at 52 weeks LOCF in the vadadustat group, but TSAT was higher than baseline in the darbepoetin alfa group (Figure 5C). TIBC was higher than baseline at 52 weeks LOCF in the vadadustat group and was lower than baseline in the darbepoetin alfa group (Figure 5D). For both treatment groups, the mean monthly dose of oral iron was higher at weeks 48–52 compared with the screening period (Figure 5E). The proportions of patients receiving oral iron during the screening period and at weeks 48–52 were 23.8% and 33.6%, respectively, in the vadadustat group, and were 18.3% and 29.0%, respectively, in the darbepoetin alfa group.
Figure 5.
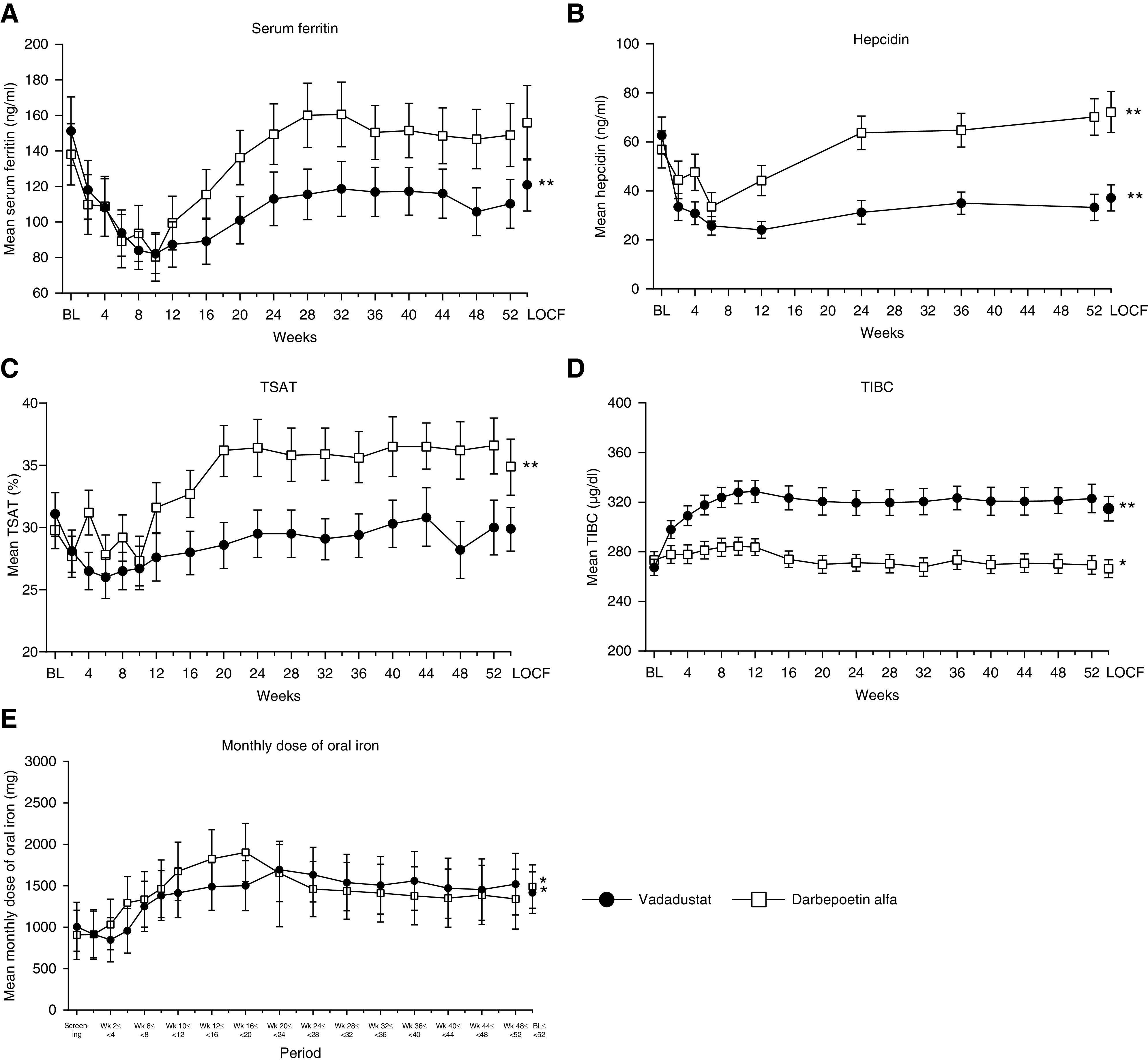
Mean iron-related parameters over time (52 weeks); (A) serum ferritin, (B) hepcidin, (C) TSAT, (D) TIBC, and (E) monthly dose of oral iron. Data are displayed as mean and 95% CI. Asterisks indicate statistically significant difference between week 52 LOCF and baseline, except for monthly dose of iron, which is between weeks 48 and 52 and screening (paired t test; *P<0.05, **P<0.01). In the vadadustat group, serum ferritin and hepcidin decreased, and TIBC increased from baseline to week 52 LOCF. In the darbepoetin alfa group, hepcidin and TSAT increased, and TIBC decreased from baseline to week 52 LOCF. The monthly iron dose was higher at weeks 48–52 than during the screening period in both treatment groups.
In the vadadustat group, MCV, MCH, and MCHC levels (Figure 6, A–C) were higher than baseline at 52 weeks LOCF and there was no change in RDW (Figure 6D). In the darbepoetin alfa group, there were no changes in these red blood cell indices at 52 weeks LOCF compared with baseline, except for RDW, which was higher than at baseline (Figure 6D).
Figure 6.

Mean red blood cell–related parameters over time. (A) MCV, (B) MCH, (C) MCHC, and (D) RDW. Data are displayed as mean and 95% CI. Asterisks indicate statistically significant difference between week 52 LOCF and baseline (paired t test; *P<0.05; **P<0.01). In the vadadustat group, MCV, MCH, and MCHC levels were higher than baseline at 52 weeks LOCF and there was no change in RDW. In the darbepoetin alfa group, there were no changes in these red blood cell indices except for RDW.
Safety
The proportions of patients reporting AEs (vadadustat group 90.1%, darbepoetin alfa group 92.2%) and serious AEs (SAEs; vadadustat group 27.8%, darbepoetin alfa group 32.0%) were similar between the treatment groups (Table 3). All SAEs were considered by investigators to be not related to the study drugs. One 56-year-old man in the darbepoetin alfa group died of acute myocardial infarction and the event was judged to be not related to darbepoetin alfa.
Table 3.
AEs during 52 wk of treatment (safety population)
| Overview | Vadadustat (n=151) | Darbepoetin Alfa (n=153) |
|---|---|---|
| Patients, n (%) | ||
| ≥1 AE | 136 (90.1) | 141 (92.2) |
| ≥1 adverse drug reaction | 20 (13.2) | 7 (4.6) |
| ≥1 serious AE | 42 (27.8) | 49 (32.0) |
| ≥1 serious adverse drug reaction | 0 (0.0) | 0 (0.0) |
| ≥1 AE leading to discontinuation | 10 (6.6) | 6 (3.9) |
| AE leading to dose reduction or interruption | 11 (7.3) | 4 (2.6) |
| AE leading to rescue therapy | 2 (1.3) | 1 (0.7) |
| Deaths | 0 (0.0) | 1 (0.7) |
| AEs reported in ≥5% of patients in either group, n (%) | ||
| Nasopharyngitis | 37 (24.5) | 43 (28.1) |
| Diarrhea | 18 (11.9) | 8 (5.2) |
| Constipation | 14 (9.3) | 11 (7.2) |
| Contusion | 11 (7.3) | 7 (4.6) |
| Peripheral edema | 11 (7.3) | 5 (3.3) |
| Vomiting | 10 (6.6) | 3 (2.0) |
| CKD | 9 (6.0) | 14 (9.2) |
| Renal impairment | 8 (5.3) | 8 (5.2) |
| Pyrexia | 8 (5.3) | 1 (0.7) |
| Pruritus | 7 (4.6) | 8 (5.2) |
| Cystitis | 6 (4.0) | 9 (5.9) |
| Eczema | 5 (3.3) | 8 (5.2) |
| Hypertension | 2 (1.3) | 11 (7.2) |
In the vadadustat group, the five most frequently occurring AEs were nasopharyngitis, diarrhea, constipation, contusion, and peripheral edema (Table 3). Two patients in the vadadustat group reported hypertension. ADRs occurred more frequently in the vadadustat group (13.2%) than in the darbepoetin alfa group (4.6%) (Table 3) and few patients reported AEs of special interest in either treatment group (Table 4). In the vadadustat group, nine patients reported a cardiovascular event or cardiac failure, four reported at least one retinal disorder event, two reported a malignancy, one reported an event of hyperkalemia, and one reported a thromboembolism. There were no patients with pulmonary hypertension. All AEs of special interest, except one retinal hemorrhage, were considered to be not related to vadadustat. The patient with the retinal hemorrhage event was withdrawn from the study at week 12, and had plasma VEGF levels of 16.1 pg/ml at baseline and 18.9 pg/ml at week 12.
Table 4.
AEs of special interest during 52 wk of treatment (safety population)
| Category, n (%) | Vadadustat (n=151) | Darbepoetin Alfa (n=153) |
|---|---|---|
| Cardiovascular event, cardiac failurea | 9 (6.0) | 5 (3.3) |
| Cardiac failure chronic | 3 (2.0) | 0 (0.0) |
| Cardiac failure congestive | 2 (1.3) | 0 (0.0) |
| Intracranial aneurysm | 1 (0.7) | 1 (0.7) |
| Cardiac failure | 1 (0.7) | 0 (0.0) |
| Myocardial ischemia | 1 (0.7) | 0 (0.0) |
| Subarachnoid hemorrhage | 1 (0.7) | 0 (0.0) |
| Cardiac failure acute | 0 (0.0) | 2 (1.3) |
| Lacunar infarctionb | 0 (0.0) | 2 (1.3) |
| Cerebral infarctionb | 0 (0.0) | 1 (0.7) |
| Retinal disorders | 4 (2.6) | 12 (7.8) |
| Retinal hemorrhage | 2 (1.3) | 5 (3.3) |
| Macular edema | 1 (0.7) | 0 (0.0) |
| Retinal exudates | 1 (0.7) | 0 (0.0) |
| Retinal vein occlusionb | 1 (0.7) | 0 (0.0) |
| Age-related macular degeneration | 1 (0.7) | 0 (0.0) |
| Diabetic retinopathy | 0 (0.0) | 3 (2.0) |
| Scintillating scotoma | 0 (0.0) | 1 (0.7) |
| Vitreous floaters | 0 (0.0) | 1 (0.7) |
| Retinal aneurysm | 0 (0.0) | 1 (0.7) |
| Macular fibrosis | 0 (0.0) | 1 (0.7) |
| Malignancy | 2 (1.3) | 6 (3.9) |
| Colon adenoma | 1 (0.7) | 0 (0.0) |
| Oral papilloma | 1 (0.7) | 0 (0.0) |
| Basal cell carcinoma | 0 (0.0) | 1 (0.7) |
| Gastric cancer | 0 (0.0) | 1 (0.7) |
| Keratoacanthoma | 0 (0.0) | 1 (0.7) |
| Renal cancer | 0 (0.0) | 1 (0.7) |
| Seborrheic keratosis | 0 (0.0) | 1 (0.7) |
| Skin papilloma | 0 (0.0) | 1 (0.7) |
| Renal cancer metastatic | 0 (0.0) | 1 (0.7) |
| Kidney angiomyolipoma | 0 (0.0) | 1 (0.7) |
| Hyperkalemia | 1 (0.7) | 5 (3.3) |
| Thromboembolism | 1 (0.7) | 6 (3.9) |
| Retinal vein occlusion | 1 (0.7) | 0 (0.0) |
| Lacunar infarction | 0 (0.0) | 2 (1.3) |
| Cerebral infarction | 0 (0.0) | 1 (0.7) |
| Acute myocardial infarction | 0 (0.0) | 1 (0.7) |
| Pulmonary embolism | 0 (0.0) | 1 (0.7) |
| Shunt occlusion | 0 (0.0) | 1 (0.7) |
| Pulmonary hypertension | 0 (0.0) | 0 (0.0) |
Combined data for cardiovascular events and cardiac failure.
Also reported in thromboembolism.
During treatment, a rapid rise in Hb was seen only in ESA non–users. The proportion of patients who experienced a rapid increase in Hb >2.0 g/dl over 4 weeks was 2.7% (four patients) in the vadadustat group and 1.3% (two patients) in the darbepoetin alfa group. The proportions of patients with Hb ≥13.0 g/dl and Hb ≥14.0 g/dl were 31.1% (47 patients) and 2.6% (four patients), respectively, in the vadadustat group, and were 49.0% (75 patients) and 8.5% (13 patients), respectively, in the darbepoetin alfa group.
Discussion
This is the first phase 3, randomized active-controlled trial that demonstrates noninferiority of oral vadadustat to subcutaneous darbepoetin alfa for the treatment of anemia in Japanese patients with NDD-CKD. In the vadadustat group, mean Hb increased and reached the predefined target of 11.0–13.0 g/dl in patients who were and were not previously treated with an ESA, and was maintained within the target range for up to 52 weeks. Furthermore, vadadustat was generally well tolerated and no major safety concerns were observed during the 52-week treatment period. Because vadadustat is to be administered orally, it is likely to overcome some of the barriers to ESA injection for patients with anemia in NDD-CKD.
Because this study was conducted in patients with anemia in NDD-CKD who require treatment, an active-controlled, noninferiority study design was adopted to avoid the potential disadvantage of administering placebo to patients. However, there are several perspectives inherent to noninferiority study designs that should be considered when interpreting results from these studies. These perspectives include the following: the active control that is selected should have demonstrated efficacy in the target disease and must be clinically effective in the study; the noninferiority margin must be statistically and clinically appropriate; and missing data, including early discontinuation of patients, should be minimized during the study. In this study, the 95% CI of the average Hb at weeks 20 and 24 for the darbepoetin alfa group was within the target range (11.0–13.0 g/dl), confirming the validity of darbepoetin alfa as an active control and the noninferiority margin for this study (−0.75 g/dl) was set in accordance with a study on the variance in Hb levels in patients with ESKD.23 Also, vadadustat demonstrated noninferiority to darbepoetin alfa as measured by average Hb at weeks 20 and 24. Although the percentage of patients who discontinued from the trial was slightly higher in the vadadustat group than in the darbepoetin alfa group (14%, 21 of 151 versus 8%, 12 of 153) at week 24, this imbalance is unlikely to have influenced the study outcomes because the primary analysis was conducted using the MMRM method. The MMRM method minimizes the effects of missing data by assuming all missing data are missing at random and all dropouts would behave in a similar way to other patients who had not discontinued in the same treatment group.24,25 In addition, we conducted a sensitivity analysis under the missing not at random assumption, which showed that vadadustat remained noninferior to darbepoetin alfa, confirming the primary analysis (data not shown).
As the baseline characteristics of patients enrolled in this study were similar to those of large postmarketing studies of darbepoetin alfa or epoetin beta pegol in Japan,26,27 these findings may be generalized to treatment of anemia in Japanese patients with NDD-CKD in clinical practice. Furthermore, evaluation of patients who did and did not receive ESAs is clinically meaningful, because many Japanese patients who are in the predialysis phase have not yet started treatment for anemia and, of those who receive ESAs, many have Hb levels that are below the target recommended by Japanese guidelines.10 In this study, vadadustat was effective in ESA non–users with Hb <11, which is the level recommended for initiation of treatment for anemia by Japanese guidelines.11 For ESA users, vadadustat improved Hb levels in patients with Hb <11.0 g/dl at screening and maintained Hb levels in those who had Hb ≥11.0 g/dl. This indicates treatment with vadadustat can improve Hb, even in patients who have not achieved the recommended Hb levels despite using an ESA, and can maintain Hb levels in patients with well-controlled Hb when treatment is switched from an ESA. Taken together, these findings suggest vadadustat is expected to be effective in a wide range of patients with NDD-CKD in clinical practice settings.
In addition to insufficient production of erythropoietin to compensate for a decline in Hb levels,16 disordered iron homeostasis might be involved in the onset and progression of anemia in patients with CKD.16 Consistent with previous studies,20,21,28,29 an increase in TIBC and decreases in serum ferritin and hepcidin were observed with vadadustat in this study. Vadadustat treatment also resulted in slight increases in erythrocyte indices including MCV, MCH, and MCHC. It is uncertain, however, whether vadadustat improves iron homeostasis, and further studies are needed.
In general, the safety of vadadustat in this 52-week study was consistent with shorter-term phase 2 studies conducted in White20,28,29 and Japanese patients.21 The rates of AEs and SAEs in this study were almost similar between treatment groups, and no SAEs were attributable to vadadustat or darbepoetin alfa. ADRs were more frequent in the vadadustat group than in the darbepoetin alfa group and, similar to those of previous studies in White patients,20,28,29 the most frequently reported AEs and ADRs with vadadustat were gastrointestinal events. Although more patients reported diarrhea in the vadadustat group than in the darbepoetin alfa group, diarrhea was not considered to be a major safety concern because most events were mild, and almost all patients recovered without study discontinuation. In addition, although hypertension is a known adverse effect associated with ESA treatment,30 only two patients reported hypertension in the vadadustat group, which was not numerically higher than in the darbepoetin alfa group.
The AEs of special interest assessed in this study were known to be related to the mechanism of action of HIF-PHIs or were AEs previously observed with the HIF-PHI class or ESAs.22 However, few AEs of special interest were reported in either treatment group and there were no clinically meaningful differences between the vadadustat and darbepoetin alfa groups. One case of retinal hemorrhage that was considered by the investigator to be related to vadadustat was reported, but the symptoms were mild and resolved after discontinuation of the study drug. There were no apparent changes in plasma VEGF in this patient. Additional studies will be needed to fully assess the effects of vadadustat and other HIF-stabilizing drugs on the retina. An increased incidence of hyperkalemia has been reported in clinical trials of another HIF-PHI for anemia treatment.31,32 However, in this study, there was only one case of hyperkalemia in the vadadustat group, which was not considered to be related to vadadustat.
The following limitations should be considered when interpreting the results of this study. First, it was conducted using an active-controlled, noninferiority study design because it was not ethically appropriate to include a placebo arm. Therefore, although vadadustat was shown to be noninferior to darbepoetin alfa in terms of efficacy in patients with anemia in NDD-CKD, the long-term superiority of vadadustat over placebo has not been demonstrated. Second, because this study had a relatively small sample size and short treatment period, larger and longer-term studies are required to fully investigate the safety of HIF-PHIs with regard to events that may occur at relatively low incidence rates. Finally, because this study was open label, it is possible that subjective end points may have been affected by reporting bias. Despite this, many of the end points, including the primary end point, Hb, are laboratory test values that are not easily affected by subjectivity.
In conclusion, this study is the first to demonstrate the noninferiority of vadadustat to darbepoetin alfa and to confirm the durability of efficacy of vadadustat for up to 52 weeks of treatment for anemia in Japanese patients with NDD-CKD. Despite the limitations discussed above, no new safety concerns were identified with the use of vadadustat in this study. Overall, these findings support the potential of vadadustat for the treatment of anemia in Japanese patients in the predialysis phase with and without receiving ESA therapy.
Disclosures
M. Nangaku reports receiving grants or honoraria from Akebia, Alexion, Astellas, AstraZeneca, Bayer, Boehringer Ingelheim, Chugai, Daiichi Sankyo, GlaxoSmithKline (GSK), Japan Tobacco (JT), Kyowa Kirin, Mitsubishi Tanabe, Ono, Taisho, Takeda, and Torii; reports being a scientific advisor or member of Akebia, Astellas, Bayer, Boehringer-Ingelheim, Daiichi-Sankyo, GSK, JT, Kyowa-Kirin, and Mitsubishi-Tanabe. Y. Komatsu reports receiving honoraria from AstraZeneca, Baxter, Chugai, and Kyowa Kirin; and reports consultancy agreements with Mitsubishi Tanabe. G. Kaneko, K. Kondo, K. Ueta, T. Tandai, Y. Kawaguchi, and Y. Kokado are employees of Mitsubishi Tanabe Pharma Corporation.
Funding
This study was funded by Mitsubishi Tanabe Pharma Corporation.
Data Sharing Statement
The deidentified datasets generated and/or analyzed during this study, protocols, annotated case report form, dataset specifications, and clinical study report may be available from Mitsubishi Tanabe Pharma Corporation on reasonable request to Mitsubishi Tanabe or the corresponding author, and after the product and indication have obtained marketing approval in the United States, European Union (including European Economic Area), and Japan, and after an agreement to disclose clinical data is gained from the codevelopment partner for this product. The request must not extend beyond the limitation of the participants’ informed consent. External research is limited to the conduct of approved research. Access to data may be declined by Mitsubishi Tanabe, for example, where there is a potential conflict of interest, or an actual or potential competitive risk, and other conditions.
Supplementary Material
Acknowledgments
The authors thank the study team and staff for the conduct of the study, their colleagues at Akebia Therapeutics Inc. for editorial input and support with the development of this manuscript, and the patient volunteers for their participation in the study. Mitsubishi Tanabe Pharma Corporation was involved in the study design, data collection, data analysis, and preparation of the manuscript. All authors participated in the interpretation of study results, and in the drafting, critical revision, and approval of the final version of the manuscript. G. Kaneko, Y. Kawaguchi, Y. Komatsu, K. Kondo, M. Nangaku, and T. Tandai were involved in the study design; K. Kondo and M. Nangaku were investigators in the study; and Y. Kawaguchi conducted the statistical analysis. Medical writing assistance was provided by Dr. Serina Stretton, Certified Medical Publication Professional, and Dr. Tania Dickson, Certified Medical Publication Professional, of ProScribe–Envision Pharma Group, and was funded by Mitsubishi Tanabe Pharma Corporation. ProScribe’s services complied with international guidelines for Good Publication Practice.
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
Supplemental Material
This article contains the following supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2020091311/-/DCSupplemental.
Supplemental Material. Protocol summary.
Supplemental Figure 1. Mean Hb over time (52 weeks) and mean dose of vadadustat (daily) or darbepoetin alfa (weekly) in the ESA user subgroups categorized by Hb during screening.
CONSORT Checklist.
References
- 1.Astor BC, Muntner P, Levin A, Eustace JA, Coresh J: Association of kidney function with anemia: The Third National Health and Nutrition Examination Survey (1988-1994). Arch Intern Med 162: 1401–1408, 2002 [DOI] [PubMed] [Google Scholar]
- 2.McClellan W, Aronoff SL, Bolton WK, Hood S, Lorber DL, Tang KL, et al.: The prevalence of anemia in patients with chronic kidney disease. Curr Med Res Opin 20: 1501–1510, 2004 [DOI] [PubMed] [Google Scholar]
- 3.Imai E, Matsuo S, Makino H, Watanabe T, Akizawa T, Nitta K, et al.: Chronic Kidney Disease Japan Cohort study: Baseline characteristics and factors associated with causative diseases and renal function. Clin Exp Nephrol 14: 558–570, 2010 [DOI] [PubMed] [Google Scholar]
- 4.Portolés J, Gorriz JL, Rubio E, de Alvaro F, García F, Alvarez-Chivas V, et al.; NADIR-3 Study Group: The development of anemia is associated to poor prognosis in NKF/KDOQI stage 3 chronic kidney disease. BMC Nephrol 14: 2, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Iseki K, Kohagura K: Anemia as a risk factor for chronic kidney disease. Kidney Int Suppl 72: S4–S9, 2007 [DOI] [PubMed] [Google Scholar]
- 6.Iimori S, Naito S, Noda Y, Nishida H, Kihira H, Yui N, et al.: Anaemia management and mortality risk in newly visiting patients with chronic kidney disease in Japan: The CKD-ROUTE study. Nephrology (Carlton) 20: 601–608, 2015 [DOI] [PubMed] [Google Scholar]
- 7.Sato Y, Fujimoto S, Konta T, Iseki K, Moriyama T, Yamagata K, et al.: Anemia as a risk factor for all-cause mortality: Obscure synergic effect of chronic kidney disease. Clin Exp Nephrol 22: 388–394, 2018 [DOI] [PubMed] [Google Scholar]
- 8.Finkelstein FO, Story K, Firanek C, Mendelssohn D, Barre P, Takano T, et al.: Health-related quality of life and hemoglobin levels in chronic kidney disease patients. Clin J Am Soc Nephrol 4: 33–38, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Imai E, Horio M, Watanabe T, Iseki K, Yamagata K, Hara S, et al.: Prevalence of chronic kidney disease in the Japanese general population. Clin Exp Nephrol 13: 621–630, 2009 [DOI] [PubMed] [Google Scholar]
- 10.Akizawa T, Makino H, Matsuo S, Watanabe T, Imai E, Nitta K, et al.; Chronic Kidney Disease Japan Cohort Study Group: Management of anemia in chronic kidney disease patients: Baseline findings from Chronic Kidney Disease Japan Cohort Study. Clin Exp Nephrol 15: 248–257, 2011 [DOI] [PubMed] [Google Scholar]
- 11.Yamamoto H, Nishi S, Tomo T, Masakane I, Saito K, Nangaku M, et al.: 2015 Japanese society for dialysis therapy: Guidelines for renal anemia in chronic kidney disease. Ren Replace Ther 3: 36, 2017 [Google Scholar]
- 12.Pfeffer MA, Burdmann EA, Chen CY, Cooper ME, de Zeeuw D, Eckardt KU, et al.; TREAT Investigators: A trial of darbepoetin alfa in type 2 diabetes and chronic kidney disease. N Engl J Med 361: 2019–2032, 2009 [DOI] [PubMed] [Google Scholar]
- 13.Singh AK, Szczech L, Tang KL, Barnhart H, Sapp S, Wolfson M, et al.; CHOIR Investigators: Correction of anemia with epoetin alfa in chronic kidney disease. N Engl J Med 355: 2085–2098, 2006 [DOI] [PubMed] [Google Scholar]
- 14.Thamer M, Zhang Y, Kshirsagar O, Cotter DJ, Kaufman JS: Erythropoiesis-stimulating agent use among non-dialysis-dependent CKD patients before and after the trial to Reduce Cardiovascular Events With Aranesp Therapy (TREAT) using a large US health plan database. Am J Kidney Dis 64: 706–713, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Besarab A, Bolton WK, Browne JK, Egrie JC, Nissenson AR, Okamoto DM, et al.: The effects of normal as compared with low hematocrit values in patients with cardiac disease who are receiving hemodialysis and epoetin. N Engl J Med 339: 584–590, 1998 [DOI] [PubMed] [Google Scholar]
- 16.Babitt JL, Lin HY: Mechanisms of anemia in CKD. J Am Soc Nephrol 23: 1631–1634, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Locatelli F, Fishbane S, Block GA, Macdougall IC: Targeting hypoxia-inducible factors for the treatment of anemia in chronic kidney disease patients. Am J Nephrol 45: 187–199, 2017 [DOI] [PubMed] [Google Scholar]
- 18.Kurata Y, Tanaka T, Nangaku M: Prolyl hydroxylase domain inhibitors: A new era in the management of renal anemia. Ann Transl Med 7[Suppl 8]: S334, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Apte RS, Chen DS, Ferrara N: VEGF in signaling and disease: Beyond discovery and development. Cell 176: 1248–1264, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pergola PE, Spinowitz BS, Hartman CS, Maroni BJ, Haase VH: Vadadustat, a novel oral HIF stabilizer, provides effective anemia treatment in nondialysis-dependent chronic kidney disease. Kidney Int 90: 1115–1122, 2016 [DOI] [PubMed] [Google Scholar]
- 21.Nangaku M, Farag YMK, deGoma E, Luo W, Vargo D, Khawaja Z: Vadadustat, an oral hypoxia-inducible factor prolyl hydroxylase inhibitor, for treatment of anemia of treating chronic kidney disease: Two randomized phase 2 trials in Japanese patients [published online ahead of print July 28, 2020]. Nephrol Dial Transplant 10.1093/ndt/gfaa060 [DOI] [PubMed] [Google Scholar]
- 22.Sanghani NS, Haase VH: Hypoxia-inducible factor activators in renal anemia: Current clinical experience. Adv Chronic Kidney Dis 26: 253–266, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lacson E Jr, Ofsthun N, Lazarus JM: Effect of variability in anemia management on hemoglobin outcomes in ESRD. Am J Kidney Dis 41: 111–124, 2003 [DOI] [PubMed] [Google Scholar]
- 24.Siddiqui O, Hung HM, O’Neill R: MMRM vs. LOCF: A comprehensive comparison based on simulation study and 25 NDA datasets. J Biopharm Stat 19: 227–246, 2009 [DOI] [PubMed] [Google Scholar]
- 25.Prakash A, Risser RC, Mallinckrodt CH: The impact of analytic method on interpretation of outcomes in longitudinal clinical trials. Int J Clin Pract 62: 1147–1158, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tanaka T, Nangaku M, Imai E, Tsubakihara Y, Kamai M, Wada M, et al.: Safety and effectiveness of long-term use of darbepoetin alfa in non-dialysis patients with chronic kidney disease: A post-marketing surveillance study in Japan. Clin Exp Nephrol 23: 231–243, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hayashi T, Uemura Y, Kumagai M, Kimpara M, Kanno H, Ohashi Y; MIRACLE-CKD Study Group: Effect of achieved hemoglobin level on renal outcome in non-dialysis chronic kidney disease (CKD) patients receiving epoetin beta pegol: MIRcerA CLinical Evidence on Renal Survival in CKD patients with renal anemia (MIRACLE-CKD Study). Clin Exp Nephrol 23: 349–361, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Haase VH, Chertow GM, Block GA, Pergola PE, deGoma EM, Khawaja Z, et al.: Effects of vadadustat on hemoglobin concentrations in patients receiving hemodialysis previously treated with erythropoiesis-stimulating agents. Nephrol Dial Transplant 34: 90–99, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Martin ER, Smith MT, Maroni BJ, Zuraw QC, deGoma EM: Clinical trial of vadadustat in patients with anemia secondary to stage 3 or 4 chronic kidney disease. Am J Nephrol 45: 380–388, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Palmer SC, Navaneethan SD, Craig JC, Johnson DW, Tonelli M, Garg AX, et al.: Meta-analysis: Erythropoiesis-stimulating agents in patients with chronic kidney disease. Ann Intern Med 153: 23–33, 2010 [DOI] [PubMed] [Google Scholar]
- 31.Chen N, Hao C, Liu BC, Lin H, Wang C, Xing C, et al.: Roxadustat treatment for anemia in patients undergoing long-term dialysis. N Engl J Med 381: 1011–1022, 2019 [DOI] [PubMed] [Google Scholar]
- 32.Chen N, Hao C, Peng X, Lin H, Yin A, Hao L, et al.: Roxadustat for anemia in patients with kidney disease not receiving dialysis. N Engl J Med 381: 1001–1010, 2019 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.