

Significance Statement
Abnormal extracellular matrix is a histologic feature of kidney aging and disease. However, a comprehensive molecular basis for altered matrix is not well understood. Ultrastructural and proteomic studies in mouse models of genetic kidney disease and human tissue define a molecular basis for altered matrix, which has common features across aging and disease progression. Broadly, basement membrane components are reduced, interstitial matrix proteins are increased, and this is coupled with altered cell adhesion and metabolic processes. Furthermore, a signature of altered matrix proteins appears before ultrastructural defects and could have utility as biomarkers of kidney health. Mechanistically, this altered kidney matrix may initiate abnormal kidney cell–matrix and immune cell–matrix interactions, which therapy could target.
Keywords: extracellular matrix, basement membrane, mass spectrometry, proteomics, tubulointerstitium, fibrosis, matrix signature, nephronectin, type VI collagen, type IV collagen
Visual Abstract

Abstract
Background
Accumulation of extracellular matrix in organs and tissues is a feature of both aging and disease. In the kidney, glomerulosclerosis and tubulointerstitial fibrosis accompany the decline in function, which current therapies cannot address, leading to organ failure. Although histologic and ultrastructural patterns of excess matrix form the basis of human disease classifications, a comprehensive molecular resolution of abnormal matrix is lacking.
Methods
Using mass spectrometry–based proteomics, we resolved matrix composition over age in mouse models of kidney disease. We compared the changes in mice with a global characterization of human kidneymatrix during aging and to existing kidney disease datasets to identify common molecular features.
Results
Ultrastructural changes in basement membranes are associated with altered cell adhesion and metabolic processes and with distinct matrix proteomes during aging and kidney disease progression in mice. Within the altered matrix, basement membrane components (laminins, type IV collagen, type XVIII collagen) were reduced and interstitial matrix proteins (collagens I, III, VI, and XV; fibrinogens; and nephronectin) were increased, a pattern also seen in human kidney aging. Indeed, this signature of matrix proteins was consistently modulated across all age and disease comparisons, and the increase in interstitial matrix was also observed in human kidney disease datasets.
Conclusions
This study provides deep molecular resolution of matrix accumulation in kidney aging and disease, and identifies a common signature of proteins that provides insight into mechanisms of response to kidney injury and repair.
Extracellular matrix (ECM) is organized as basement membranes (BMs), which support continuous layers of cells, or as looser interstitial matrix.1 In addition to providing a scaffold, ECM directs organ shape and sequesters growth factors and enzymes for controlled, outside-in signaling.2 This extracellular environment is complex and dynamic, with >1000 matrisome proteins annotated.3 Matrix synthesis and turnover requires tight regulation for the maintenance of organ function, and matrix accumulation is associated with a wide spectrum of human disease. In the Western world, 45% of deaths have been associated with organ fibrosis.4 Thus, understanding the drivers of matrix accumulation is key to identifying strategies to prolong organ survival.
Excess matrix is seen across a wide spectrum of pathology and is incorporated into the histologic and ultrastructural descriptions of many diseases.5 In the kidney, glomerulosclerosis is a feature of aging, and glomerular disease6 and tubulointerstitial (TI) fibrosis is seen with both primary tubular disease and as a consequence of glomerular disease7,8; however, the comprehensive molecular definition of altered matrix is limited. Using proteomics to define global protein composition has improved the resolution of matrix complexity, and this includes the human glomerular matrisome, which contains >140 structural and regulatory components.9,10 Proteomic studies in kidney disease have also revealed changes in matrix composition,11,12 but these have been limited by sample volume and have not used matrix-enrichment strategies, which improve resolution.13
Along the nephron, there is distinct matrix composition within BMs and interstitial matrices.14 BMs underlie parietal epithelial cells, separate endothelial cells from overlying podocytes, provide a scaffold for tubular epithelial cells, and form an integral component of blood vessel walls in the kidney. Interstitial matrices include the mesangial matrix and the loose TI matrix. The glomerular and tubular segments of the nephron have distinct functions, and their differential matrix composition is likely to reflect their underlying functions, including filtration and reabsorption. One example of structure supporting function is the triple helix of type IV collagen protomers, which may resist mechanical load15 and could explain the distinct localization of collagen IV isoforms along the nephron.
Here, we performed deep proteomic profiling to define global changes in kidney matrix and to identify common components and pathways. In mouse models of genetic defects in collagen IV, we found distinct structural abnormalities of the matrix, associated with altered cell adhesion and metabolic processes, and a signature of abnormal matrix composition. In young and aged human kidneys and existing kidney disease datasets, we identified a similar signature of altered matrix. Broadly, we observed a reduction in BM components and an increase in interstitial matrix proteins in both mouse models and human tissue.
Methods
Antibodies
Primary antibodies were diluted in a blocking buffer (1% donkey serum, 2% BSA, 0.1% Triton X-100 in PBS). We used primary antibodies against type VI collagen (ab6588; Abcam) at a dilution of 1:100 and 1:500 for stochastic optical reconstruction microscopy (STORM) imaging; the type IV collagen chains α1, α2, α3, α4, α5, and α6 (7070, 7071, 7076, 7073, 7077, and 7074, respectively; Chondrex, Woodinville, WA) at a dilution of 1:200; type I collagen (OARAO2579; Gentaur, Kapenhout, Belgium) at a dilution of 1:100; podocin (ab50339; Abcam) at a dilution of 1:400; fibulin-1 (sc-25281; Santa Cruz Biotechnology, Dallas, TX) at a dilution of 1:100; nidogen/entactin antibody (MAB1946, ELM1; Merck, Darmstadt, Germany) at a dilution of 1:100; laminin-β2 (ab210956; Abcam) at a dilution of 1:100; Tinag (ab67614; abcam) at a dilution of 1:100; and integrin β1 (clone 9EG7; BD Pharmigen) at a dilution of 1:500. We used secondary antibodies conjugated to Alexa Fluor 488 or 594 (Life Technologies) for immunofluorescence.
Mice
We used Col4a1+/SVC mice, which carry a point mutation causing an amino acid substitution (G1064D).16 These mice were on a BL6/C57 background. We used littermate controls, male mice for phenotypic consistency, and three mice from each group for proteomic analysis. Col4a3−/− mice were generated as described.17 In brief, the first three exons of the carboxy-terminal (NC1) domain of collagen-α3(IV) chain were deleted, causing absence of collagen-α3(IV) protein. Col4a5−/− mice were obtained from the International Mouse Phenotyping Consortium.18 The line was generated by deletion of the critical exon 36, resulting in the absence of the collagen-α5(IV) protein, using the published allele map.19 Col4a3−/− and Col4a5−/− mice were on a BL6/C57 background. We used littermate controls, male mice for phenotypic consistency, and five mice from each group for proteomic analysis. All experiments were performed in compliance with the Animals (Scientific Procedures) Act 1986 regulations of the United Kingdom Home Office.
Glomerular Isolation from Mice
Cortical tissue was dissected from frozen-thawed kidney, placed in a glass dish, and cut into small pieces (≤1 mm3) as previously described.9 Tissue pieces were washed three times with PBS. Tissue pieces were transferred onto a 100-µm cell strainer (Falcon) and mechanically disintegrated. We then added PBS and collected the flow through. The resulting tissue solution was filtered through a 70-µm cell strainer (Falcon). Glomeruli were caught on the 70-µm cell strainer. The cell strainer was inverted and caught glomeruli were washed off the sieve and collected in a container.
Human Kidney
Human kidneys (n=6) anatomically unsuitable for transplantation, and with no recorded comorbidities, were collected under ethical approval (regional ethics committee approval identifiers 05/Q0508/6 [University College London] and 06/Q1406/38 [Manchester]). Samples were grouped into young (15, 29, 37 years) and aged (61, 67, 69 years) kidneys, with n=3 per group. Five of the six kidneys were from male donors, and the sex of the kidney from the 67 year old was not recorded. We also used kidney biopsy sections from a male patient presenting at age 14 years with CKD stage 3, due to autosomal recessive (COL4A3) Alport syndrome, and control kidney tissue (nonaffected tissue of tumor nephrectomy), with the ethical approval identifier 06/Q1406/38 (Manchester).
Glomerular and TI Isolation from Human Kidney
Cortical tissue (2 g) was dissected from frozen-thawed kidney, placed in a glass dish, and cut into small pieces (≤1 mm3) as previously described.9 Graded sieves of 250, 150, and 106 µm were assembled, with the 250-µm sieve at the top, and placed on a collection container. Tissue pieces were transferred onto the largest sieve in 5 ml of ice-cold PBS (without calcium and magnesium). We used the plunger from a 10-ml syringe to press the sample through the sieves. Fragments were washed through the sieves with 60–100 ml of ice-cold PBS. Glomeruli were retained on 150- and 106-µm sieves, and were collected by inverting the sieves and washing with 20 ml of ice-cold PBS. Cortical TI fragments were retrieved from the collection container. The glomeruli and TI samples were centrifuged at 3000 × g for 3 minutes at 4°C, washed three times by centrifugation, and then resuspended in 10 ml of ice-cold PBS. After the final wash, samples were resuspended in 5 ml PBS and aliquoted into five 1-ml aliquots. One aliquot from each sample was viewed under a light microscope to confirm the separation of glomeruli and TI tissue. One aliquot was frozen at −20°C for ECM enrichment, and the remainder was stored at −80°C.
Matrix Enrichment
Samples stored at −20°C were defrosted at room temperature, homogenized using a 21G needle and 2-ml syringe, and centrifuged at 14,000 × g for 5 minutes at room temperature. We added 500 μl of ice-cold buffer solution 1 (10 mM Tris, 150 mM sodium chloride, 25 mM EDTA, 1% vol/vol Triton X-100, 25 µg/ml leupeptin, 25 µg/ml aprotinin, 0.5 mM 4-[2-aminoethyl]benzenesulfonyl fluoride hydrochloride) to the pellet and incubated the samples on ice for 1 hour. Samples were centrifuged at 14,000 × g for 10 minutes at 4°C, and the supernatant was collected and mixed with 125 μl recovery buffer (15% wt/vol SDS, 100 mM 1,4-dithiothreitol, 200 mM Tris, 30% vol/vol glycerol, 0.01% bromophenol blue). This sample was designated soluble fraction 1 (most of the cellular compartment) and stored at −80°C. The pellet was resuspended in 500 μl ice-cold buffer solution 2 (20 mM ammonium hydroxide, 0.5% vol/vol Triton X-100) and incubated on ice for 1 hour. Samples were centrifuged at 14,000 × g for 10 minutes at 4°C and the supernatant was collected and designated soluble fraction 2. The pellet was resuspended in 500 μl PBS with DNase I (25 µg/ml) and incubated for 30 minutes at room temperature. Samples were centrifuged at 14,000 × g for 10 minutes at 4°C, and the collected supernatant was designated soluble fraction 3. The final pellet was resuspended in a sample buffer and incubated for 10 minutes at 95°C to generate the ECM-enriched fraction.
Mass Spectrometry Data Acquisition
After SDS-PAGE, gel lanes were sliced and subjected to in-gel digestion with trypsin,20 with modifications.21 Peptide samples were analyzed by liquid chromatography–tandem mass spectrometry using a nanoACQUITY UltraPerformance LC system (Waters) coupled online to an LTQ Velos mass spectrometer (Thermo Fisher Scientific), or an UltiMate 3000 Rapid Separation LC system (Thermo Fisher Scientific) coupled online to an Orbitrap Elite mass spectrometer (Thermo Fisher Scientific). Peptides were concentrated and desalted on a Symmetry C18 preparative column (20 mm × 180 μm, 5-μm particle size; Waters) and separated on a bridged ethylene hybrid C18 analytical column (250 mm × 75 μm, 1.7-μm particle size; Waters), using a 45-minute linear gradient from 1%–25% or 8%–33% (vol/vol) acetonitrile in 0.1% (vol/vol) formic acid at a flow rate of 200 nl/min. Peptides were selected for fragmentation automatically by data-dependent analysis.
Mass Spectrometry Identifications
Tandem mass spectra were extracted using extract_msn (Thermo Fisher Scientific) executed in Mascot Daemon (version 2.5.1; Matrix Science). Peak list files were searched against a modified version of the Uniprot mouse database (April 2016 database selected for Mus musculus) for mouse datasets, or Uniprot human database (November 2015 database selected for Homo sapiens) for human datasets. We set carbamidomethylation of cysteine as a fixed modification; oxidation of methionine and hydroxylation of proline and lysine were allowed as variable modifications. Only tryptic peptides were considered, with up to one missed cleavage permitted. Monoisotopic precursor mass values were used, and only doubly and triply charged precursor ions were considered. The mass tolerances for precursor and fragment ions were 5 ppm and 0.5 Da, respectively.
Mass Spectrometry Quantification
We used peptide intensity to calculate relative protein abundance. Orbitrap mass spectrometry data were entered into Progenesis LC-MS (Nonlinear Dynamics Ltd.) and automatically aligned. Spectra were extracted using extract_msn executed in Mascot Daemon (version 2.5.1) and imported back into Progenesis LC-MS to acquire intensity data. Chromatograms were aligned using the automatic alignment algorithm, followed by manual validation and adjustment of the aligned chromatograms. All features were used for peptide identifications. Progenesis LC-MS created the peak list file that was exported and searched in Mascot. Hi-N was used to infer protein abundance, using the three most abundant peptides.22 We then exported peptide and protein data from Progenesis LC-MS as .csv files to be analyzed in Excel (Microsoft). Proteins identified by one unique peptide, and a Mascot score indicating a <5% false discovery rate, were considered to be part of the definition of the human TI matrisome. The data were further filtered to include only proteins present in two out of three TI samples in a specific age group. Proteins identified by three unique peptides and a Mascot score indicating a <5% false discovery rate were considered for quantitative analysis. We inferred protein abundance from normalized peptide intensities, using the “three most abundant unique Hi-N peptides” setting in Progenesis. Conflicts were resolved using protein grouping. To compare group protein abundances across samples, we used ANOVA with post hoc Bonferroni protein lists are shown in Supplemental Tables 1 and 2. To enable Gene Ontology (GO) lists with sufficiently large numbers of proteins in all comparisons, proteins with P<0.1 and 1.4-fold increased or decreased abundance were taken forward for further analysis.
Protein Interaction Network Analysis
Cytoscape (version 3.7.2) was used for protein interaction network analysis. Matrisome proteins were selected from the Matrisome Project,3 and proteins identified in at least two biologic replicates were mapped onto a merged human, mouse, and rat interactome. This was built from the PICKLE 2.5 (released November 15, 2019; based on IntAct release 227/2019-11-04, BioGRID release 3.5.178, DIP release 20170205, and Human Protein Reference Database release 9) database merged with a matrix protein–specific interactome from the Protein Interaction Network Analysis platform; the H. sapiens network (release date, December 10, 2012); M. musculus network (release date, December 10, 2012); the Rattus norvegicus network (release date, December 10, 2012); the ECM interactions database MatrixDB (release date, April 20, 2012); and a literature-curated database of integrin-based, adhesion-associated proteins.23 For networks where enrichment/fold change is presented, Progenesis LC-MS–normalized intensity data were used. Networks were manually grouped.
Data Analysis
DAVID (version 6.7) was used for functional enrichment analysis. Keywords with fold enrichment ≥ 1.5, Bonferroni-corrected P<0.05, EASE score (modified Fisher exact test) of <0.05, and at least two proteins per keyword were considered significantly over-represented. We generated enrichment maps using all changed proteins detected, P<0.1, and fold change at least >1.4. Enrichment maps were clustered using the clusterMaker application with Markov clustering algorithm tuning, and a granularity parameter of 2.5. Principal component analysis (PCA) was performed using MATLAB (version R2019a; MathWorks). Hierarchically clustered heat maps were generated on the basis of uncentered, Euclidean complete-linkage clustering in MultiExperiment Viewer (version 4.8.1). For clustered heat maps where the z-score or fold change is presented, we used Progenesis LC-MS–normalized intensity data.
Nephroseq Analysis
We used Nephroseq version 5 for analysis. Signature matrix proteins were searched for their fold change in gene expression in disease datasets compared with associated control samples. The following filters were applied: tissue type, glomeruli and analysis type, disease versus control analyses. Microarray data were extracted with the P value selection “all” and fold change “all.” The primary data are shown in Supplemental Table 3. Data that met the following criteria: P<0.05, fold change= ±1.5 are highlighted in orange in Supplemental Table 3 and taken forward for further analysis. Data are shown as box-and-whisker plots displaying all data points. The values below every plot indicate the number of datasets in which a transcript was detected out of all analyzed datasets (21 datasets). Data from the following datasets were extracted: Berthier Lupus Glom (analysis of lupus nephritis versus healthy living donor),24 Hodgin Diabetes Mouse Glom (analyses of diabetic nephropathy versus nondiabetic kidney [mouse model db/db C57BLKS], diabetic nephropathy versus nondiabetic kidney [mouse model DBA/2], and diabetic nephropathy versus nondiabetic kidney [mouse model eNOS-deficient C57BLKS db/db]),25 Hodgin FSGS Glom (analyses of collapsing FSGS versus normal kidney, FSGS versus normal kidney, and minimal change disease versus normal kidney),26 Ju CKD Glom (analyses of arterial hypertension versus healthy living donor, diabetic nephropathy versus healthy living donor, FSGS versus healthy living donor, IgA nephropathy versus healthy living donor, lupus nephritis versus healthy living donor, minimal change disease versus healthy living donor, membranous glomerulonephropathy versus healthy living donor, tumor nephrectomy versus healthy living donor, thin BM disease versus healthy living donor, and vasculitis versus healthy living donor),27 Neusser Hypertension Glom (analysis of nephrosclerosis versus tumor nephrectomy),28 Peterson Lupus Glom (analysis of lupus nephritis versus normal kidney),29 Reich IgAN Glom (analysis of IgA nephropathy versus healthy living donor),30 and Woroniecka Diabetes Glom (analysis of diabetic nephropathy versus healthy living donor).31
Electron Microscopy
Samples were prepared as previously described.32 Briefly, mouse kidneys were harvested; immediately cut into 1 mm2 pieces; and fixed in 2.5% glutaraldehyde, 4% paraformaldehyde (PFA), and 0.1 M HEPES, pH 7.4. Samples were stained in 1% osmium tetroxide and 1.5% potassium ferrocyanide in 0.1 M cacodylate buffer, followed by 1% thiocarbohydrazide. Samples were washed and stained further in 1% osmium tetroxide and incubated in 1% uranyl acetate overnight. Samples were incubated in lead aspartate (pH 5.5) for 1 hour at 60°C, dehydrated, and finally embedded in TAAB 812 hard resin. Samples were mounted onto an aluminum cryo pin. Block surfaces were trimmed using a glass knife or diamond trimming tool. To create a conductive surface, a gold coating was applied to the specimen. The samples were analyzed using a Quanta 250 FEG (FEI Company) and Gatan 3View system. The analyzed Col4a1+/SVC mice received treatment with 4-sodium phenyl butyric acid, as described previously, which did not affect the kidney phenotype.33 Kidney samples for transmission electron microscopy were fixed similarly as for serial block-face scanning electron microscopy. Sections (70–80 nm thick) were prepared and examined using a FEI Tecnai 12G2 Biotwin transmission electron microscope. Foot process width, glomerular BM (GBM) thickness, and Bowman’s capsule BM thickness were measured using ImageJ (version 2.0.0-rc-68/1.52e). The foot process width was measured at the basal side of podocytes from ten different fields of view in the glomerulus. Measurements were taken from one glomerulus per mouse genotype. GBM thickness was measured radially from the endothelial side of the electron-dense GBM to the basal membrane of podocyte foot processes. Bowman’s capsule thickness was measured radially in different sections of the acquired serial block-face scanning electron microscopy stack from one glomerulus.
Light Microscopy
We used formaldehyde-fixed, paraffin-embedded kidney sections for immunofluorescence. Images were acquired using a Zeiss Axioimager.D2 upright microscope, with a 20×/0.50 EC Plan-Neofluar objective, and captured using a Coolsnap HQ2 camera (Photometrics) through Micromanager software version 1.4.23. Images of stained cryosections were acquired using a Zeiss AxioObserver Z1 wide-field microscope equipped with a 40×/EC Plan-Neofluar 1.3 Oil objective and an Axiocam MRm camera, controlled by Zeiss Axiovision software. Specific band pass filter sets for 4′,6-diamidino-2-phenylindole, FITC, and Texas Red were used to prevent bleed through from one channel to the next. Images were processed and analyzed using ImageJ (versions 1.51j8 and 2.0.0-rc-68/1.52e). Total positive antibody fluorescence was measured in glomeruli and the TI separately and normalized to the total background in the same area to generate corrected total fluorescence. Background was subtracted using a rolling-ball algorithm in ImageJ in the immunofluorescence images.
Tissue Preparation and Staining for STORM Imaging
Tissue was fixed and stained as describedpreviously.34,35 In brief, kidneys were fixed in 4% PFA in PBS. Samples were washed in PBS incubated in a cryoprotectant solution of 2.3 M sucrose plus 10% polyvinylpyrrolidone in 0.1 M piperazine-N,N′-bis(2-ethanesulfonic acid) at pH 7.2 at 4°C overnight. Tissue pieces were mounted on a metal sectioning pin and frozen in liquid nitrogen. Frozen tissue was sectioned using an EM-FC6 ultracryomicrotome (Leica) and collected on carbon-coated coverslips. Sections were fixed in 4% PFA for 20 minutes, washed with PBS, and PFA was quenched using 50 mM glycine in PBS. For immunofluorescence, sections were blocked in 2% BSA in PBS overnight, followed by another overnight incubation with primary antibodies diluted in 2% BSA in PBS. After washing, sections were incubated with secondary antibodies diluted in 2% BSA in PBS for 2 hours at room temperature. Secondary antibodies for direct STORM were purchased from Jackson ImmunoResearch Laboratories and custom conjugated to Andy Fluor 488, Cy3, or Cy5 reporter dyes, asdescribed previously.35 Sections were washed again in PBS and postfixed in 3% PFA plus 0.05% glutaraldehyde in PBS. After a final washing series, samples were imaged by Nikon N-STORM according to the manufacturer’s instructions, as described previously. 35
Histology
Paraffin-embedded mouse tissue sections were stained using a Shandon Linistain GLX stainer (Thermo Fisher) for hematoxylin and eosin. Samples were mounted using Histokitt solution (Roth). For Picrosirius red staining, paraffin-embedded tissue sections were dewaxed using a Leica ST5010 autostainer. Tissue sections were stained by incubation of slides in Picrosirius red solution (0.5 g sirius red and 500 ml saturated aqueous solution of picric acid; Sigma) for 1 hour. Slides were washed in acidified water. Slides were dehydrated and mounted using Histokitt solution (Roth). Images were collected on an Olympus BX63 upright microscope, using a 60×/1.42 PlanApo N objective, and captured and white-balanced using a DP80 camera (Olympus) in color/polarized light mode through CellSens Dimension version 1.16 (Olympus). For TriPAS staining, paraffin sections were dewaxed and placed into 1% Periodic acid for 10 minutes, and washed with tap and distilled water. Nuclei were stained with Gills No.2 hematoxylin for 1.5 minutes and washed in tepid tap water. Sections were stained in 2% orange G dissolved in 100 ml of 5% phosphotungstic acid for 1 minute. Sections were washed, dehydrated, and coverslips were applied. For human and mouse sections, a specialist kidney pathologist performed percentage glomerulosclerosis analysis and percentage interstitial fibrosis and tubular atrophy scoring on the TriPAS-stained tissue sections.
Urinary Albumin and Creatinine Analysis
Spot urine was collected at the same time of day for each group of mice. Urinary albumin was measured using a mouse albumin ELISA kit (E99-134; Bethyl Laboratories). Creatinine was measured using a creatinine ELISA assay according to the manufacturer’s instructions (KGE005; R&D Systems).
Statistical Analyses
Data are presented as bar charts (mean±SEM) or box-and-whisker plots, and were analyzed using GraphPad Prism (version 7; GraphPad Software, La Jolla, CA). Data were analyzed using the Shapiro–Wilk test for normality and analyzed using the Kruskal–Wallis test or ANOVA with post hoc Bonferroni correction or t test, as appropriate. Statistical significance was accepted at P<0.05. PCA was performed in MATLAB. Unsupervised hierarchic clustering was performed in T4MeV using a Euclidean, distance-based, complete-linkage matrix.
Results
Structural and Compositional Change in Matrix
Type IV collagen is an essential component of BM in the kidney and exists as three networks comprised of α1.α1.α2(IV), α3.α4.α5(IV), and α5.α5.α6(IV) heterotrimers. We confirmed the glomerular localization of these three networks with α1.α1.α2(IV) in the mesangium, GBM, and Bowman’s capsule; α3.α4.α5(IV) in the GBM; and α5.α5.α6(IV) in Bowman’s capsule (Supplemental Figure 1). To understand the effect of glomerular disease on matrix, we investigated three mouse models with defects in the different collagen IV heterotrimers. First, Col4a1+/SVC mice that harbor a glycine to aspartic acid variant (G1064D) and have phenotypic overlap with human COL4A1 variants, including defects in Bowman’s capsule, kidney cysts, and tubular dysfunction.33,36 Second, Col4a3−/− mice (autosomal recessive Alport syndrome) that have progressive glomerular disease associated with GBM defects.17 Third, a new Col4a5−/− model of X-linked Alport syndrome. We characterized the Col4a5−/− mice and demonstrated abnormal GBM thickness, podocyte effacement, and progressive albuminuria (Supplemental Figure 2). To investigate the effect of Col4a defects on matrix ultrastructure, we performed serial block-face scanning electron microscopy of young (6–8 weeks), adult (16–18 weeks), and aged (28 weeks) mice (Figure 1, A–D, and Supplemental Videos 1–4). GBM was similar to wild type in adult Col4a1+/svc mice, but thickened in adult Col4a3−/− and Col4a5−/−mice. Podocyte foot process width was preserved in Col4a1+/svc mice, but increased with age in Col4a3−/− and Col4a5−/− mice. Bowman’s capsule BM thickness was increased in adult Col4a3−/− mice compared with adult wild type and Col4a5−/− mice, and markedly thickened in aged Col4a3−/− mice (Supplemental Video 3). The Bowman’s capsule BM of adult Col4a1+/svc mice was of a normal thickness, but there was infiltration of parietal epithelial cells. Given these structural changes in the matrix, we analyzed matrix composition using mass spectrometry–based proteomics. To interrogate changes that precede—and also coincide with—ultrastructural changes, we analyzed glomerular proteomes in young and adult mice. We isolated glomeruli and separated cellular and matrix fractions from three Col4a1+/svc, five Col4a3−/−, and five Col4a5−/− mice, and their respective littermate controls, as previously described.9 Proteins were taken forward if identified by three unique peptides, with a Mascot score indicating a <5% false discovery rate. GO enrichment analysis highlighted increases in cell matrix and cell adhesion ontologies and a reduction in mitochondrial and metabolic components in all three Col4a models, compared with controls (Supplemental Figure 1E). Equivalent analyses of individual Col4a datasets highlighted similar cellular pathways (Supplemental Figure 3A), and PCA demonstrated model and age segregation (Figure 2A). Taken together, these findings highlight both structural and compositional changes in matrix in models of glomerular disease.
Figure 1.

Identification of glomerular basement membrane and Bowman's capsule abnormalities in Col4a mutant mice. (A) Segmentation and modeling of serial block-face scanning electron microscopy data. Glomerular structure is visualized in Alport (Col4a3−/− and Col4a5−/−) and Col4a1+/SVC mice. Col4a1+/SVC mice demonstrated splitting of Bowman’s capsule with enclosed nuclei. Col4a3−/− mice presented irregular GBM and progressive thickening of Bowman’s capsule, whereas Bowman’s capsule was conserved in the Col4a5−/− Alport mouse model. GBM (yellow), podocyte cell body with foot processes (light blue), and Bowman’s capsule BM (green) were reconstructed. Dark blue lines highlight the thickness of the Bowman’s capsule BM. Nuclei of cells intercalating the BM in Col4a1+/SVC are highlighted in pink. The analyzed Col4a1+/SVC mice received treatment with 4-sodium phenyl butyric acid, as described previously, which did not affect the kidney phenotype.33 Quantification of (B) GBM thickness, (C) podocyte foot process width, and (D) Bowman’s capsule BM (BCBM) thickness. Young (Y) mice were aged 6–8 weeks, adult (A) mice were 16–18 weeks old, and aged (A+) mice were 28 weeks old. Box-and-whisker plots represent the median, where the box represents interquartile range and whiskers represent the tenth to 90th percentile. n=1 biologic sample per model. (E) GO enrichment map analysis was performed on 719 proteins detected by mass spectrometry with altered abundance (>1.4-fold; P<0.1) in Col4a1+/svc, Col4a3−/−, and Col4a5−/− mice at all analyzed ages compared with aged-matched control mice. Over-represented biological processes are presented as nodes, and color represents Bonferroni-corrected P value (the lower the P value, the more intense the node color). Edge weight represents the overlap between the proteins in the connected nodes. n=5 biologic samples per mouse model studied. WT, wild type.
Figure 2.

Changes in glomerular matrix composition are exacerbated with aging in Col4a mutant mice. Mouse glomerular extracts were fractionated to cellular and matrix fractions. All fractions were analyzed by mass spectrometry; proteins were identified with Mascot and quantified using Progenesis. (A) MATLAB was used for PCA of all detected proteins. Components 1 and 2 are presented; the percentage variance explained by component 1 is indicated on the x axis, and that of component 2 on the y axis. Samples were fractionated and the matrix fraction was further analyzed. (B–E) Volcano plots of proteins detected in matrix fractions demonstrate differential expression shown as log2(fold change) compared with age-matched control on the x axis and −log10(P value) onthe y axis. (B) Col4a3−/− mice at 6–8 weeks old, (C) Col4a3−/− mice at 16–18 weeks, (D) Col4a5−/− mice at 6–8 weeks old, and (E) Col4a5−/− mice at 16–18 weeks old. (F) Detected matrix proteins at 6–8 weeks of age were clustered in MeV using Euclidean distance complete-linkage clustering. The clustered dendrogram is presented as a heat map, with orange representing increased abundance in Col4a mutant relative to control. FC, fold change; WT, wild type.
Evolution of Altered Matrix in Aging and Disease
To explore the altered glomerular matrix, we focused on the matrix-enriched fractions in which 707 (Col4a1+/svc), 334 (Col4a3−/−), and 600 (Col4a5−/−) proteins were detected. Volcano plots revealed age- and genotype-specific changes in protein identifications (Figure 2, B–E). In keeping with predictions based on genotype, collagen-α1(IV) and collagen-α2(IV) were reduced in Col4a1+/svc glomeruli, but moderately increased in both Col4a3−/− and Col4a5−/− glomeruli (Figure 2F and Supplemental Figure 4A). In addition, both collagen-α3.α4.α5(IV) and laminin 521 were reduced in Col4a3−/− and Col4a5−/− in contrast to Col4a1+/svc glomeruli (Figure 2F and Supplemental Figure 4A), and immunofluorescence confirmed reduced levels of collagen-α3(IV) and collagen-α4(IV) in Col4a5−/− mice (Supplemental Figure 4B). Although ultrastructure was preserved in young Alport mice (Figure 1), we observed altered matrix composition (Figure 2, B and D, and Supplemental Figures 5A and 6A), with a broad reduction in BM components and an increase in interstitial matrix. This pattern was exaggerated in the older mice (Figure 2, C and E, and Supplemental Figures 5B and 6B). Although less pronounced, there were also changes during aging in wild type mice, which showed reduced collagen IV and increased collagen VI (Supplemental Figure 3B). We performed immunofluorescence on Col4a5−/− kidney sections and also observed increased collagen VI and collagen I, and reduced laminin-β2 and Tinag (Figure 3, A and B). For greater spatial resolution, we used STORM to determine the localization of increased collagen VI. Control, heterozygous Col4a3+/− mice have normal kidney function,37 and we detected regular integrin β1 fluorescence and no GBM collagen VI. However, in Col4a3−/− mice, we detected collagen VI in expanded regions of GBM (Figure 3C). Overall, we found altered matrix composition before overt ultrastructural changes, and these changes increased in aging and disease progression.
Figure 3.

Imaging reveals altered matrix localization in Alport syndrome. Immunofluorescence demonstrated reduced BM proteins and increased interstitial matrix proteins in Alport mouse glomeruli. (A) Kidney sections of 8-week-old Col4a5−/− and wild type (WT) mice were immunolabeled for collagen I (Col I), collagen VI, laminin-β2 (Lamb2), Tinag, and nidogen (Nid). (B) Quantification of (A). Mean fluorescence intensity was normalized to background and measured using ImageJ software. Images were acquired from n=4 mice per genotype and between ten and 15 glomeruli were analyzed per mouse, pooled data are shown. Background was subtracted from immunofluorescence images using a rolling-ball algorithm in ImageJ. (C) STORM was performed for integrin β1 (green) and collagen VI (purple) on Col4a3+/− and Col4a3−/− mouse sections. Region of interest (ROI) highlights collagen VI localization to the GBM in Col4a3−/− glomeruli, specifically to areas of tickened GBM (arrowhead). Scale bars, 20 μm. ***P<0.01***P<0.001, ****P<0.0001
Increased Cell-Matrix Adhesion in Disease
To connect the altered matrix with cell-matrix adhesion, we used the consensus adhesome38 to select integrin adhesion complex components from all three disease model datasets (Figure 4). The consensus adhesome is centered around four dominant signaling hubs: ILK-PINCH-kindlin, FAK-paxillin, talin-vinculin, and α-actinin-zyxin-VASP. When examining proteins detected in cellular and matrix fractions across these hubs, we found a global increase in abundance of integrin adhesion components and their respective ligands. In particular, we found parallel increases in the collagen binding integrins (α1β1, α2β1) with collagen VI, collagen I, and collagen III, in addition to the parallel increase in α8β1 and its ligand, nephronectin. The observed global increase in adhesion components may reflect a greater receptor-ligand interaction associated with podocyte effacement.
Figure 4.
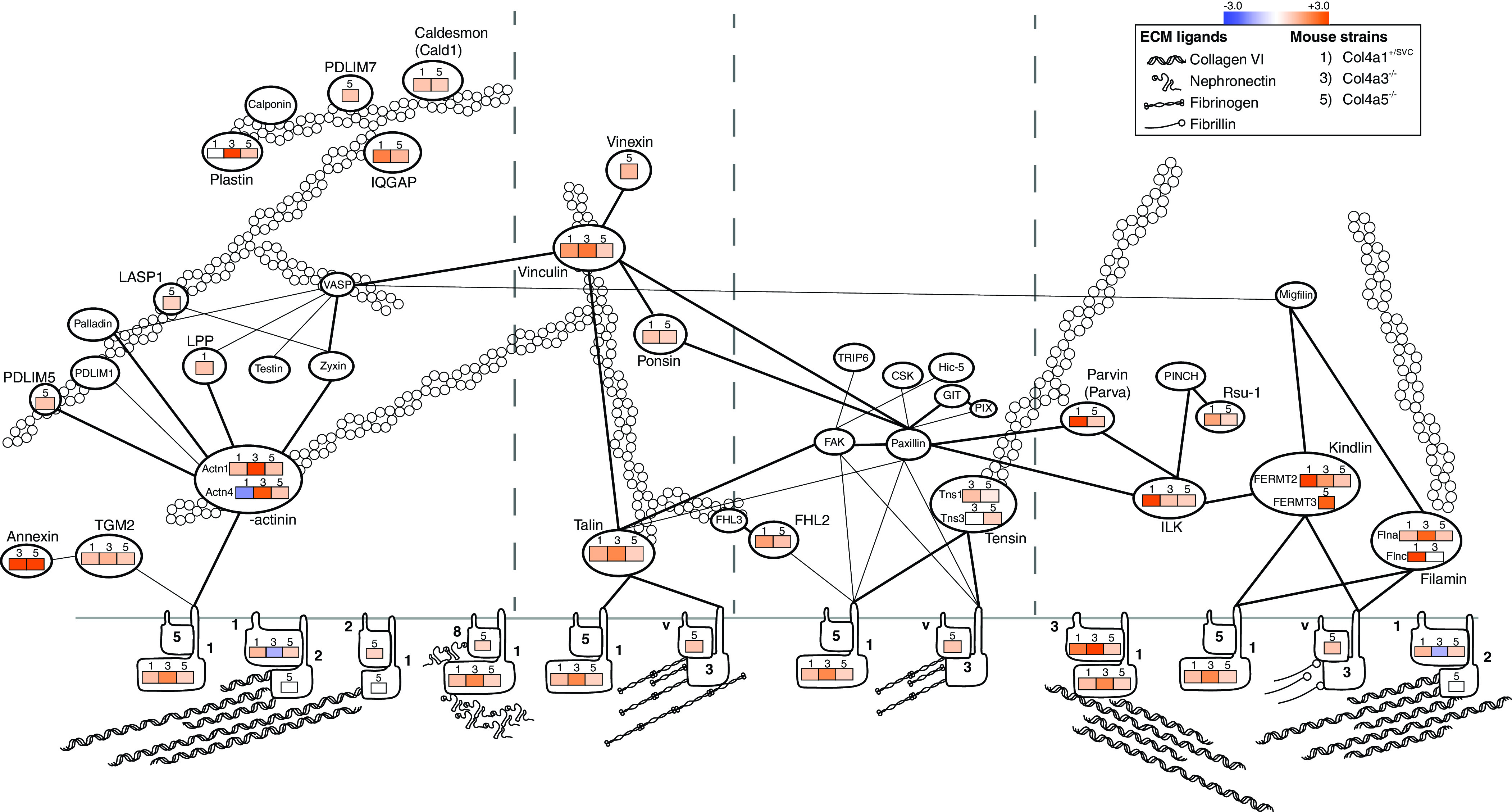
Protein abundance of cell-matrix adhesion components increases in Col4a mutants. Unsupervised hierarchical clustered heat map displaying the fold enrichment of adhesion proteins detected in 6- to 8-week-old Col4a1+/svc, Col4a3−/−, and Col4a5−/− mass spectrometry datasets compared with their respective control. Orange represents increased (+) abundance in disease relative to control; blue represents decreased (−) abundance in disease relative to control. Adhesion proteins are displayed along the four different signaling axes to the actin cytoskeleton, as described previously.38
Altered Glomerular Matrix in Human Kidney Aging
Having observed altered matrix in aging and disease in mice, we examined human tissue. Glomerulosclerosis, tubular atrophy, and interstitial fibrosis are observed in human aging.39,40 Therefore, we investigated changes in matrix over age in unused human donor kidneys. Sections of young (15, 29, 37 years) and older (61, 67, 69 years) human kidneys were assessed blindly by a kidney pathologist, who reported occasional focal glomerular and interstitial damage, consistent with age. From these kidneys, glomeruli were isolated, subjected to matrix fractionation, and analyzed by mass spectrometry. PCA of matrisome proteins showed good separation by age (Figure 5A), and GO enrichment analysis of all detected proteins highlighted matrix and cell-matrix ontologies (Supplemental Figure 7A). Volcano plots and hierarchical clustering highlighted glomerular matrix proteins changing with age (Figure 5, B and C). There was a skewed distribution with a greater number of increased abundance proteins in aged samples, including collagen VI and TIMP3 (Figure 5B). Fewer proteins were enriched in younger glomeruli, including filaggrin-2, ECM1, and agrin. Protein-protein network mapping highlighted a reduction in BM components, including laminins and collagen IV, and an increase in interstitial matrix proteins (Figure 5D). Overall, these findings highlight altered composition of matrix during human glomerular aging, with similarity to aging and disease in mice.
Figure 5.
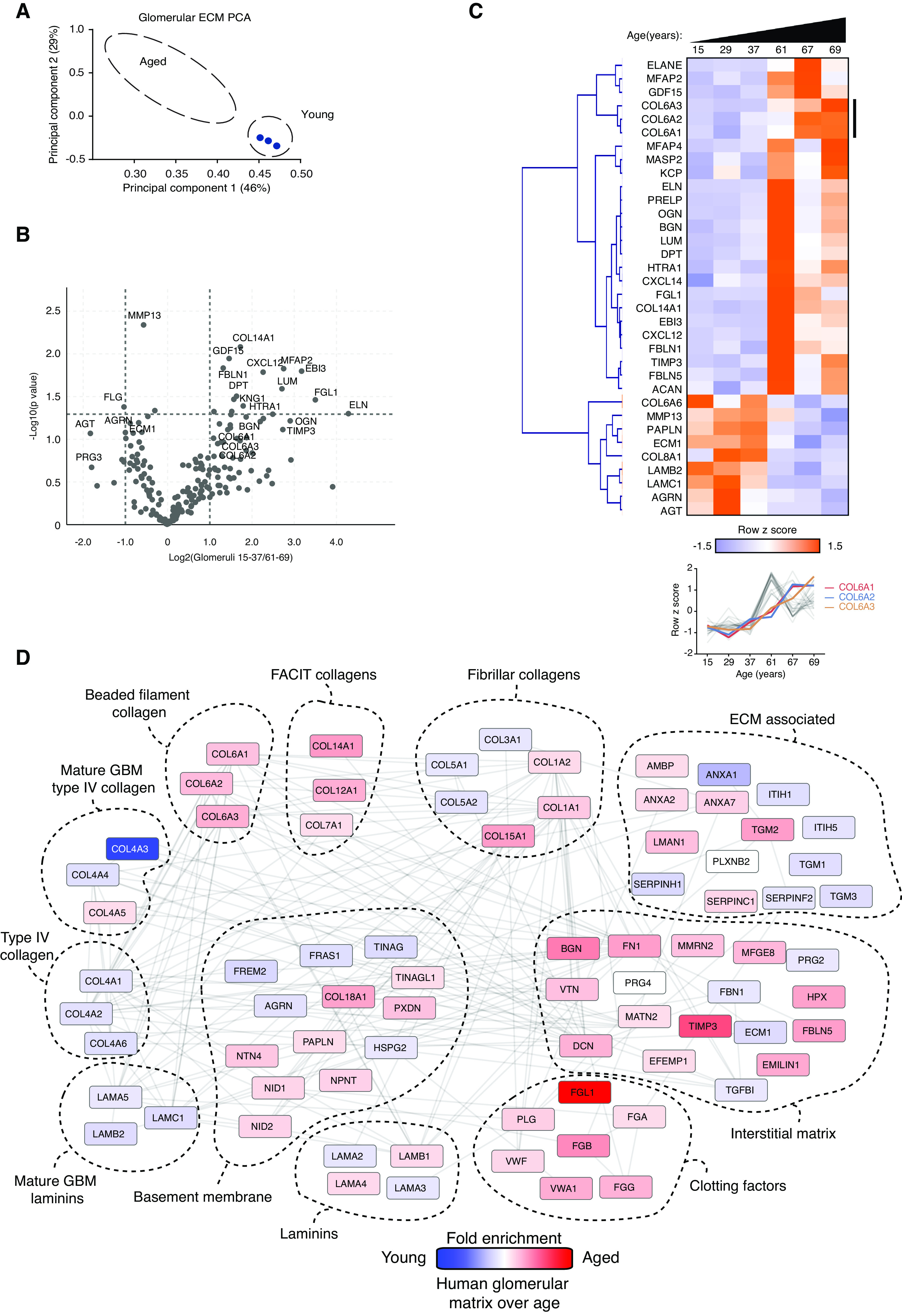
Human aging is associated with marked alterations in glomerular matrix composition. Glomeruli were isolated from young (15, 29, 37 years) and aged (61, 67, 69 years) human kidneys, subjected to matrix fractionation, and analyzed by mass spectrometry. (A) MATLAB was used for PCA of matrisome proteins. Components 1 and 2 are presented and the percentage variance explained by each component is indicated on the relevant axis. (B) Volcano plots of proteins detected in matrix fractions demonstrate differential protein abundance shown as the log2(fold change) of aged over young glomeruli on the x axis and −log10(P value) on the y axis. (C) Unsupervised hierarchical clustered heat map displaying ECM proteins detected in young and aged glomerular datasets. Data were standardized by row z-score and grouped in MeV using Pearson correlation complete-linkage clustering. Inset line graph shows increased type VI collagen protein abundance with age. (D) Protein-protein interaction network of altered human glomerular matrisome proteins. Mass spectrometry data from matrix-enriched protein fractions were filtered for known matrisome proteins. These proteins were mapped onto a merged human, mouse, and rat interactome. Nodes represent proteins and the edges represent reported protein-protein interactions. Color represents fold enrichment to datasets, with increased abundance in aged human glomerular samples illustrated in red and increased abundance in young human glomerular samples illustrated in blue. Proteins are grouped by function.
Defining the Human TI Matrisome
Together with altered glomerular matrix in kidney aging and disease, there are changes in the tubulointerstitium.8 The matrix in this kidney compartment has received far less research attention; therefore, we first defined the human TI matrisome. We isolated TI matrix from six human kidneys (as above) for analysis by mass spectrometry. We included proteins identified by one unique peptide, a Mascot score indicating a <5% false discovery rate, and being present in two out of three samples in each age group. We robustly identified 141 proteins (Supplemental Figure 8, A and B, and Supplemental Table 2), and 90% of all of the components were also found in the glomerular matrix from this study and our previous investigation.9 Comparison of human glomerular and TI matrix revealed distinct proteomes (Supplemental Figure 8C). Collagen-α3.α4.α5(IV) was abundant in the glomerular matrix, but also detected in the tubulointerstitium, which we validated by immunofluorescence (Supplemental Figure 9A). We screened the Human Protein Atlas41 to confirm localization of 67 components in the tubulointerstitium and 80 in both glomerular and TI compartments (Supplemental Figure 9, B and C). By combining the glomerular and TI datasets, we defined the combined human kidney matrisome, dominated by proteoglycans, collagens, and a wide variety of matrix regulators and secreted factors (Supplemental Figure 10). Together, these data highlight the complexity of the TI matrix and key compositional differences with the adjacent glomerular compartment.
Altered TI Matrix in Human Kidney Aging
After defining the human TI matrisome, we compared TI matrices from young (15, 29, 37 years) and aged (61, 67, 69 years) human kidneys. GO enrichment map analysis of all proteins highlighted increased matrix and cell-matrix adhesion ontologies, and a reduction in mitochondrial components (Figure 6A). We found age-associated increases in the structural components collagen VI, fibulin-1, and fibronectin, and the matrix regulators TIMP3 and ADAMTS5 (Figure 6B). Indeed, several fibulin isoforms were increased in our mouse and human datasets, and immunofluorescence confirmed increased fibulin-1 in the tubulointerstitium (Figure 6, C and D). Furthermore, a number of age-related changes were found in both glomerular and TI matrix, including fibulin-1, TIMP3, and collagen VI (Figure 7, A and B). There were also distinct compartment changes, with reduced BM components observed in glomerular aging but not seen in the aged TI matrix, suggesting tubular BMs are less exposed to damage during aging. When relating these molecular changes to histology, we observed that aging was only accompanied by a small increase in glomerulosclerosis and interstitial fibrosis and tubular atrophy scores (Figure 7, C and D), in keeping with our finding in the mouse disease models that altered matrix composition precedes overt or widespread structural changes.
Figure 6.
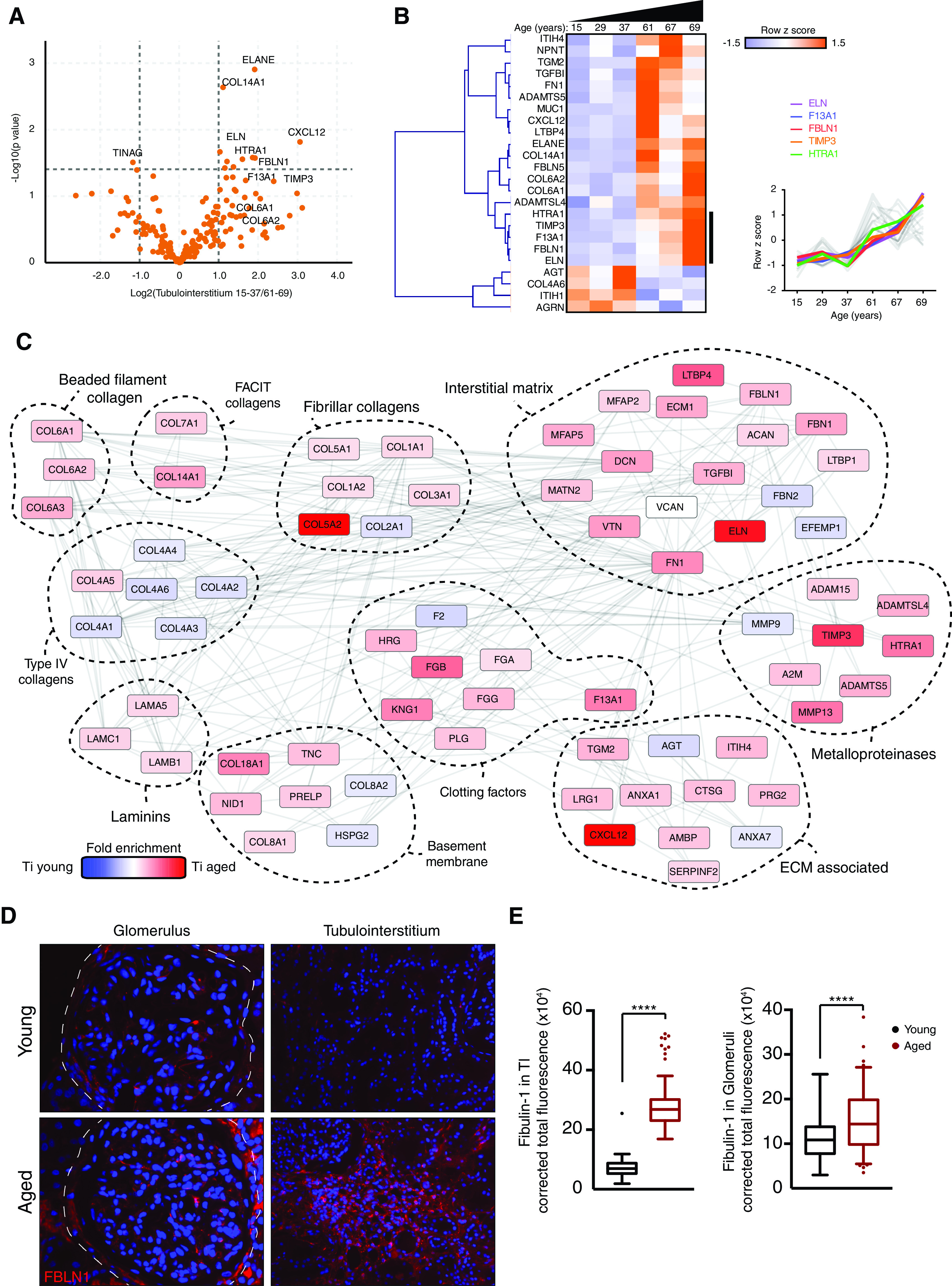
Matrix composition of the human tubulointerstitium changes with age. TI matrix was isolated from young (15, 29, 37 years) and aged (61, 67, 69 years) human kidney samples. (A) Volcano plots of proteins detected in ECM fractions demonstrate differential protein abundance shown as log2(fold change) of aged over young TI on the x axis and −log10(P value) on the y axis. (B) Unsupervised hierarchical clustered heat map displaying ECM proteins detected in young and aged glomerular datasets. Data were standardized by row z-score and grouped in MeV using Pearson correlation complete-linkage clustering. Inset line graph shows increased collagen VI protein abundance with age. (C) Protein-protein interaction network of altered human TI matrisome proteins. Mass spectrometry data from matrix-enriched protein fractions were filtered for known matrisome proteins. These proteins were mapped onto a merged human, mouse, and rat interactome. Nodes represent proteins and edges represent reported protein-protein interactions. Color represents fold enrichment to datasets, with increased abundance in aged human TI samples illustrated in red, and increased abundance in young human TI samples illustrated in blue. Proteins are grouped functionally. (D) Sections of normal human kidneys were stained with an antibody to fibulin-1 (FBLN1; red) and nuclei were stained with Hoechst 33342 (blue). Representative images are shown. Original magnification, ×40 in left panel; ×20 in right panel. (E) The fluorescent signal in 20 fields per kidney was quantified using ImageJ. ***P<0.001, ****P<0.0001.
Figure 7.

Evidence for distinct age-associated alterations in glomerular compared with TI matrix. (A) Comparison of human glomerular and human TI matrix proteins with age. Unsupervised hierarchical clustered heat map displaying ECM proteins detected in human glomerular and TI samples analyzed by mass spectrometry. Data were standardized by row z-score, clustered using Pearson uncentered distance matrix, and visualized in MeV. Line graphs show laminin proteins, with increased abundance in the human glomerular matrix relative to human TI samples. (B) Volcano plots of proteins detected in ECM fractions demonstrate differential protein abundance shown as log2(fold change) of glomerular over tubulointerstitiumfractions on the x axis and −log10(P value) on the y axis. (c) TriPAS staining of young and older kidney sections highlighting increased matrix in the older kidney section. Scale bar, 100 μm. (D) Glomerulosclerosis and interstitial fibrosis and tubular atrophy (IFTA) scoring in n=3 young and n=3 aged kidneys.
A Signature of Altered Kidney Matrix in Aging and Disease
Across our aging and disease datasets, we observed consistent increases in collagen VI, nephronectin, and fibrinogens, and decreased BM components (Figure 8, A and B). To investigate the relevance in human disease, we examined kidney biopsy sections from a young male patient with autosomal recessive Alport syndrome. Compared with control kidney sections, we observed increased levels of collagen VI and decreased levels of laminin-β2 in the glomerulus (Figure 8C). To determine whether this signature applied to a wider spectrum of human kidney disease, we searched for the signature proteins in published proteomic datasets. In a study of IgA nephropathy,11 we found concordant increases in collagen VI and fibrinogen chains and, in diabetic kidney disease (DKD),12 there was increased collagen VI and nephronectin (Figure 8D). Data from a biopsy specimen study of human FSGS identified increased interstitial collagen XV and Tgm2.42 We also observed reduced BM components in IgA nephropathy and FSGS, but not in DKD (Figure 8D), which may reflect the overall matrix expansion seen in DKD. To provide insight into the pathways associated with the matrix signature proteins, we created a one-hop interaction network by connecting upregulated signature proteins with their nearest neighbors identified in protein interaction databases (Supplemental Figure 11). This highlighted biological processes associated with matrix and adhesion but also with immune system processes. We also compared the matrix signature proteins to RNA levels using the Nephroseq database, and across a variety of phenotypes we observed similar increases in the signature transcripts (Figure 8E and Supplemental Tables 3 and 4). Finally, we combined all primary, processed data from this study to allow direct evaluation (Supplemental Table 5).
Figure 8.
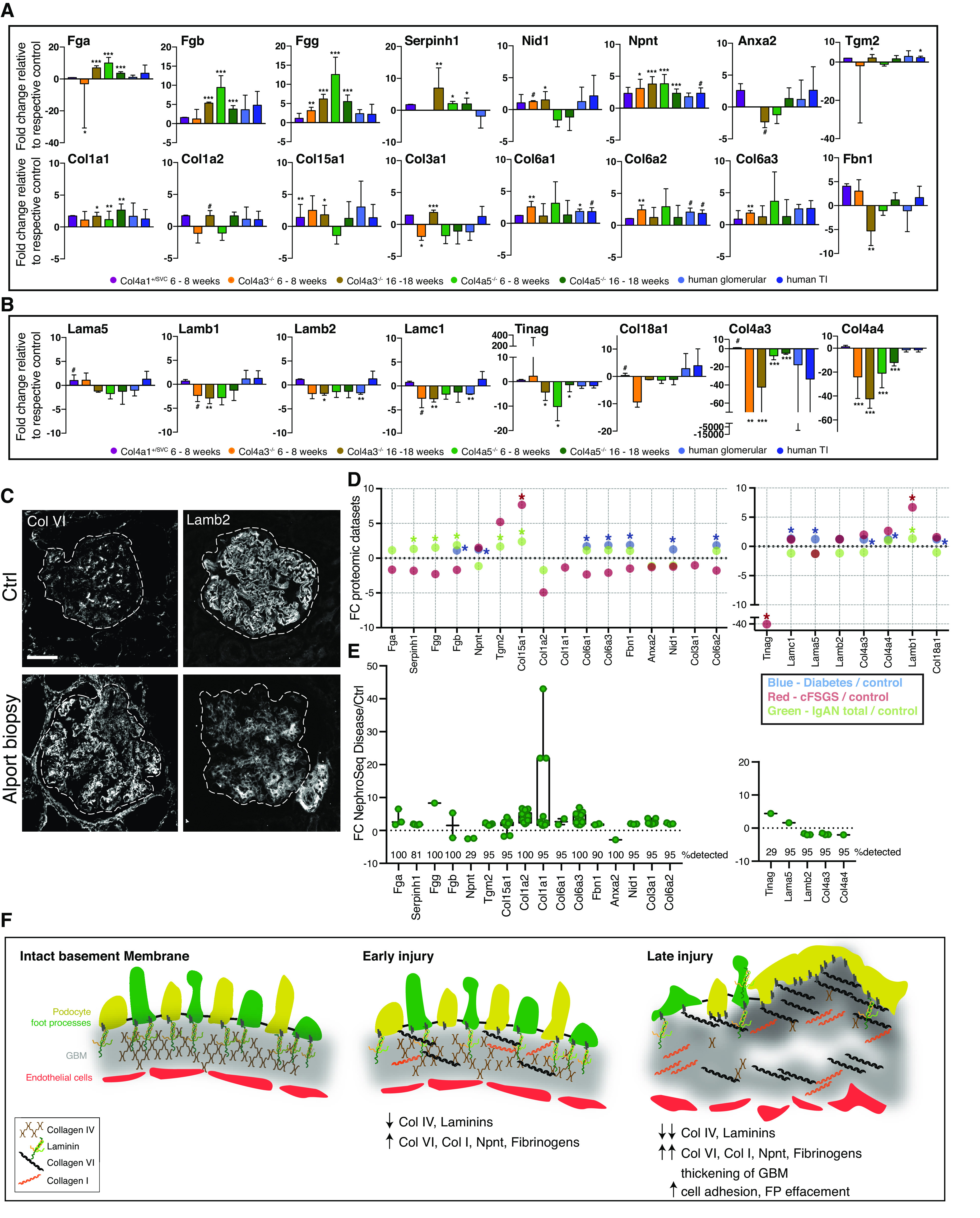
Holistic analysis reveals a signature of altered kidney matrix in aging and disease. (A–B) Bar graphs displaying protein fold change of consistently altered matrix signature proteins relative to their respective controls. Error bars represent SD. #P<0.1, *P<0.05, **P<0.01, ***P<0.001. (A) Upregulated signature proteins. (B) Downregulated signature proteins. (C) Biopsy sections from a patient with autosomal recessive Alport syndrome and control (Ctrl) kidney tissue (nonaffected tissue of a tumor nephrectomy sample) Background was subtracted from immunofluorescence images using a rolling-ball algorithm in ImageJ.C were stained for type VI collagen (Col VI), laminin-β2 (Lamb2). Scale bar, 50 μm. (D) Dot plot comparison of signature matrix proteins to their protein levels in microdissected human IgA nephropathy (IgAN) samples (IgAN total and progressive IgAN),11 human diabetic samples,12 and human FSGS samples (collapsing FSGS [cFSGS] and FSGS not otherwise specified).42 Fold change (FC) is presented on the y axis and dataset is highlighted by color. *P<0.05. (E) Analysis of signature protein transcript levels in different disease datasets using the Nephroseq version 5 database. Glomerular datasets were extracted and altered transcript levels with fold change ±1.5 and P<0.05 are shown. The percentage detection of searched gene transcript is shown (number of datasets=21). Full details for Nephroseq datasets and transcript fold changes used for each signature gene can be found in Supplemental Table 3. (F) Schematic of hypothesized BM injury process. FP, foot process.
Discussion
In this study, we aimed to define altered matrix at the molecular level in kidney aging and disease. We (1) found ultrastructural change in matrix coupled with altered composition in models of glomerular disease; (2) found similar patterns of altered glomerular matrix in human aging, with a reduction in BM proteins and an increase in interstitial matrix; (3) defined the human TI matrix as a network of 141 components, with a distinct pattern of altered matrix in aging; and (4) observed a signature of altered matrix in kidney aging and disease, and provide evidence for shared pathobiology.
The processes that drive abnormal matrix in tissues remain unclear, although this understanding could lead to targeted therapies. In this study, we describe altered matrix structure with evolution over age and disease progression. The GBM of Col4a3−/− and Col4a5-/- mice at 6–8 weeks appeared normal; however, proteomic analysis identified compositional change at this early stage. With age, these disease models progressed with thickened GBM, and the separation between control and disease matrix composition was more apparent. One aspect of the altered matrix was a reduction in BM components, including collagen IV and laminins. In dynamic studies of endogenously tagged proteins in Caenorhabditis elegans, laminin and collagen IV orthologs have slower recovery after photobleaching compared with nidogens, agrin, and perlecan.43 With this in mind, the reduction in BM components we observed could represent damage to these structures and an inability to replenish. We also observed increases of interstitial matrix components nephronectin and collagen VI within the glomerular matrix. Moreover, we identified integration of collagen VI into the GBM in Col4a3−/− mice. Genetic variants in COL6A1/2/3 cause Bethlem and Ulrich myopathies,44 but no reported kidney phenotype. However, collagen VI degradation has been associated with CKD,45 and its cleavage product, endotrophin, triggers fibrosis and metabolic dysfunction.46
Glomerular capillaries are exposed to mechanical load during filtration. Loss of a key BM component may affect the mechanical properties of the GBM. Equally, loss of collagen VI has been shown to alter stiffness in the pericellular matrix of cartilage.40 Therefore, collagen VI deposition in aging and disease may occur to restore the mechanical properties of the GBM. Furthermore, collagen VI is a prominent downstream regulator of myofibroblast activity, with knockdown reducing fibrosis.47 Our data show collagen VI increasing early, where it may act to strengthen BM defects while, paradoxically, promoting fibrosis (Figure 8E). However, the role of increased collagen VI requires further investigation, as does the potential for targeted inhibition.
In parallel with altered matrix, we observed changes in cell adhesion components. The complex of proteins that assemble upon adhesion receptor and ligand engagement has been described for interactions with fibronectin,21 in addition to the BM ligands collagen IV and laminin.48 The consensus adhesome identified a series of four signaling hubs,38 and we found a global increase in components across all four signaling hubs. In addition, we found increases in receptors and their paired matrix ligands, including integrin α1β1, integrin α2β1 with increased fibrillar collagens (I and III), and collagen VI and integrin α8β1 with nephronectin. Nephronectin is predominantly localized to the mesangium in the glomerulus and regulates mesangial cell adhesion49; however, increased GBM localization of nephronectin is reported in DKD.12 Our analysis of matrix receptors focused on the integrin adhesion complexes because we did not detect discoid domain receptors or syndecans. This could be due to low abundance or due to factors limiting their detection by mass spectrometry. Overall, the changes we observed in adhesion components could be explained by increased cell-matrix adhesion, associated with podocyte effacement, and increased interaction with the GBM.
To relate our findings in mice to human tissue, we investigated matrix isolated from young and older human kidneys. We also found a reduction in BM components and increased collagen VI and nephronectin in the aged glomerular matrix. Indeed, these components were also increased in proteomic studies of IgA nephropathy11 and DKD,12 and collagen VI was also upregulated in aged human dentin.50(preprint) A Nephroseq analysis provided further correlation between matrix signature proteins and transcriptional change in a variety of glomerular phenotypes. Kidney disease and aging is associated with altered TI matrix and, therefore, we defined the human TI matrisome. We confirmed localization of 70% of these proteins in different kidney matrix compartments, using the Human Protein Atlas, and thereby demonstrate the value of proteomics for increasing the known composition of matrix compartments as an adjunct to immunolocalization. We further examined the change in TI matrix in aging and defined a profile of altered matrix. Interestingly, we did not observe the same reduction in stable BM components seen in the glomerular matrix and hypothesize this is due to differential mechanical load on glomerular and tubular BMs.51 However, this could also depend on the pathological process, and a reduction of BM components was recently described in a proteomic analysis of transplant kidney rejection.52
In addition to resident kidney cells secreting abnormal matrix, transiting or infiltrating immune cells may also contribute. Macrophage to fibroblast crosstalk has been shown to direct new matrix synthesis, and the direct contribution of macrophages to collagen scar formation in the heart is described in zebrafish.53 Starting with the matrix signature proteins, we identified experimentally confirmed protein interactors and demonstrated three clusters of enriched biological processes. Unsurprisingly, these included cell matrix and cell adhesion, but also processes related to complement, TNF, NF-κB, and immune response to infection. These findings suggest the signature of an altered matrix has greater interaction with immune system components than previously recognized.
In conclusion, we describe a molecular basis for altered matrix in the kidney, with common features across aging and disease. Further investigations will focus on the cellular origins of the altered matrix and on the role of mechanical load on BM stability.
Disclosures
M.J. Humphries reports having consultancy agreements with Alpha-5; receiving honoraria from the Biotechnology and Biological Sciences Research Council; and serving as a scientific advisor for, or member of, the Biotechnology and Biological Sciences Research Council, Journal of Cell Biology, Journal of Cell Science, and Matrix Biology. R. Lennon reports serving as a trustee for Alport UK and Kidneys for Life; serving on the Scientific Advisor Research Network for the Alport Syndrome Foundation and on the Kidney Research UK grants panel; receiving studentship funding from GlaxoSmithKline; having other interests in/relationships with Kidney Research UK; serving as a consultant for, and having consultancy agreements with, Travere Therapeutics; and receiving funding from the Wellcome Trust. D.A. Long reports serving as an editorial board member of JASN and as an academic editor for PLOS One; and having a patent held with UCL Business related to vascular endothelial growth factor C therapy in polycystic kidney disease (JP6261617). J.H. Miner reports having consultancy agreements with Alpha Insights, Bridge Bio, Deerfield Management, Janssen Biotech Inc., Mantra Bio, and The Planning Shop; serving as the president-elect of the American Society for Matrix Biology; having patents and inventions with Angion, Kerafast, and Maze Therapeutics; receiving honoraria from the Japanese Society of Pharmacology and University of Kansas Medical Center; and serving as a consulting editor for the Journal of Clinical Investigation and on the editorial board of Kidney International, Matrix Biology, and Matrix Biology Plus. J.T. Norman reports receiving studentship funding from AstraZeneca and UCB; and serving as associate editor for BMC Nephrology, associate editor for the European Journal of Clinical Investigation, as a committee member of the Kidney Research UK Research Strategy Committee, as a trustee of Kidney Research UK, as a committee member of Medical Research Council Genetically Engineered Mice for Medicine, on the editorial board for Nature Scientific Reports, and as cochair of the UK Renal Fibrosis Network. T. Van Agtmael reports serving on the editorial board of the journal Bioscience Reports and the editorial board for the cardiovascular therapeutics section of the journal Frontiers in Cardiovascular Medicine. All remaining authors have nothing to disclose.
Funding
This work was supported by Wellcome Trust Senior Fellowship Award 202860/Z/16/Z (to R. Lennon) and grant 092015 (to M.J. Humphries), Kidney Research UK https://doi.org/10.13039/501100000291 grant RP52/2014 (awarded to R. Lennon and J.H. Miner to support a postdoctoral research assistant position for M.J. Randles), and Cancer Research UK grant C13329. D.A. Long’s laboratory is supported by Medical Research Council grant MR/P018629/1; Diabetes UK grants 13/0004763, 15/0005283, and 17/0005733; Kidney Research UK grant RP36/2015; and by the NIHR Imperial Biomedical Research Centre at Great Ormond Street Hospital for Children University College London Hospitals NHS Foundation Trust.T. Van Agtmael’s laboratory is supported by Kidney Research UK grant RP 19/2012, British Heart Foundation grant PG/15/92/31813, and Medical Research Council grant MR/R005567-1. The authors also acknowledge Wellcome Trust core funding under grant 203128/Z/16/Z to the Wellcome Centre for Cell-Matrix Research at the University of Manchester.
Supplementary Material
Acknowledgments
Mass spectrometry was performed in the Biomolecular Analysis Core Facility, Faculty of Life Sciences, University of Manchester, and we thank Dr. David Knight and Dr. Stacey Warwood for advice and technical support and Mr. Julian Selley for bioinformatic support. Special thanks goes to Dr. Peter March and Mr. Roger Meadows for their help with the microscopy. The authors thank the staff in the Electron Microscopy Core Facility in the Faculty of Biology, Medicine and Health for their assistance. Special thanks goes to Dr. Aleksandr Mironov and Dr. Tobias Starborgand the Wellcome Trust for equipment grant support to the Electron Microscopy Core Facility.
The mass spectrometer and microscopes used in this study were purchased with grants from the Biotechnology and Biological Sciences Research Council, the Wellcome Trust, and the University of Manchester Strategic Fund.
R. Lennon, M.J. Randles, J.H. Miner, T. Von Agtmael, J.T. Norman, D.A. Long, P. Potter, and M.J. Humphries conceived and designed the studies; M.J. Randles, F. Lausecker, Q. Kong, M. Kolatsi-Joannou, P. Tian, and S. Falcone acquired the data; G. Reid performed histologic analysis on kidney sections.; and all authors played an important role in interpreting the results, and in drafting and revising the manuscript. The authors have full access to the data in the study.
Data Sharing Statement
The proteomics data were deposited in the ProteomeXchange Consortium54 via the Proteomics Identifications Database partner repository with the dataset identifier PXD022363 (Col4a1 mouse glomerular datasets), PXD022392 (Col4a3 mouse glomerular datasets), PXD022227 (Col4a5 mouse glomerular ECM and cellular fraction), and PXD022219 (human kidney proteomics, glomerular, tubular, cellular, and matrix fractions).
Footnotes
M.J.R. and F.L. contributed equally to this work.
See related editorial “Too Little or Too Much? Extracellular Matrix Remodeling in Kidney Health and Disease,” on pages 1541–1543.
Published online ahead of print. Publication date available at www.jasn.org.
Present address: Dr. Michael J. Randles, Faculty of Medicine, and Life Sciences, Chester Medical School, University of Chester, Chester CH14AR, United Kingdom.
Supplemental Material
This article contains the following supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2020101442/-/DCSupplemental.
Supplemental Figure 1. Glomerular localization of type IV collagen isoforms.
Supplemental Figure 2. Characterization of Col4a5−/− mice.
Supplemental Figure 3. Overview of glomerular proteomic data.
Supplemental Figure 4. Altered matrisome proteins in Col4a mice
Supplemental Figure 5. Altered matrisome proteins in Col4a3−/− mice.
Supplemental Figure 6. Altered matrisome proteins in Col4a5−/− mice.
Supplemental Figure 7. Human kidney data enrichment analysis.
Supplemental Figure 8. Defining the tubulointerstitial matrisome
Supplemental Figure 9. Identification of new tubulointerstitial matrix proteins.
Supplemental Figure 10. Defining the human kidney extracellular matrix.
Supplemental Figure 11. One-hop interactors of the upregulated matrix signature proteins.
Supplemental Video 1. SV1-Col4a1-16weeks.
Supplemental Video 2. SV2-Col4a3-16weeks.
Supplemental Video 3. SV3-Col4a3-28weeks.
Supplemental Video 4. SV4-Col4a5-16weeks.
Supplemental Table 1. Abundance of matrix proteins in mouse models.
Supplemental Table 2. This is in the methods section Mass Spectrometry Quantification.
Supplemental Table 3. Nephroseq v5 analysis.
Supplemental Table 4. Human disease comparison.
Supplemental Table 5. Normalized abundance for all primary datasets.
References
- 1.Yurchenco PD: Basement membranes: Cell scaffoldings and signaling platforms. Cold Spring Harb Perspect Biol 3: a004911, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li X, Chen Y, Schéele S, Arman E, Haffner-Krausz R, Ekblom P, et al. : Fibroblast growth factor signaling and basement membrane assembly are connected during epithelial morphogenesis of the embryoid body. J Cell Biol 153: 811–822, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Naba A, Clauser KR, Hoersch S, Liu H, Carr SA, Hynes RO: The matrisome: In silico definition and in vivo characterization by proteomics of normal and tumor extracellular matrices. Mol Cell Proteomics 11: M111.014647, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wynn TA: Cellular and molecular mechanisms of fibrosis. J Pathol 214: 199–210, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Haas M, Rastaldi MP, Fervenza FC: Histologic classification of glomerular diseases: Clinicopathologic correlations, limitations exposed by validation studies, and suggestions for modification [published correction appears in Kidney Int 86: 648, 2014 10.1038/ki.2014.230]. Kidney Int 85: 779–793, 2014. [DOI] [PubMed] [Google Scholar]
- 6.Remuzzi G, Bertani T: Is glomerulosclerosis a consequence of altered glomerular permeability to macromolecules? Kidney Int 38: 384–394, 1990 [DOI] [PubMed] [Google Scholar]
- 7.Zeisberg M, Neilson EG: Mechanisms of tubulointerstitial fibrosis. J Am Soc Nephrol 21: 1819–1834, 2010 [DOI] [PubMed] [Google Scholar]
- 8.O’Sullivan ED, Hughes J, Ferenbach DA: Renal aging: Causes and consequences. J Am Soc Nephrol 28: 407–420, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lennon R, Byron A, Humphries JD, Randles MJ, Carisey A, Murphy S, et al. : Global analysis reveals the complexity of the human glomerular extracellular matrix. J Am Soc Nephrol 25: 939–951, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hobeika L, Barati MT, Caster DJ, McLeish KR, Merchant ML: Characterization of glomerular extracellular matrix by proteomic analysis of laser-captured microdissected glomeruli. Kidney Int 91: 501–511, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Paunas FTI, Finne K, Leh S, Osman TA, Marti HP, Berven F, et al. : Characterization of glomerular extracellular matrix in IgA nephropathy by proteomic analysis of laser-captured microdissected glomeruli. BMC Nephrol 20: 410, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nakatani S, Wei M, Ishimura E, Kakehashi A, Mori K, Nishizawa Y, et al. : Proteome analysis of laser microdissected glomeruli from formalin-fixed paraffin-embedded kidneys of autopsies of diabetic patients: Nephronectin is associated with the development of diabetic glomerulosclerosis. Nephrol Dial Transplant 27: 1889–1897, 2012 [DOI] [PubMed] [Google Scholar]
- 13.Randles MJ, Humphries MJ, Lennon R: Proteomic definitions of basement membrane composition in health and disease. Matrix Biol 57-58: 12–28, 2017 [DOI] [PubMed] [Google Scholar]
- 14.Miner JH: Renal basement membrane components. Kidney Int 56: 2016–2024, 199910.1046/j.1523-1755.1999.00785.x [DOI] [PubMed] [Google Scholar]
- 15.Khoshnoodi J, Pedchenko V, Hudson BG: Mammalian collagen IV. Microsc Res Tech 71: 357–370, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Van Agtmael T, Schlötzer-Schrehardt U, McKie L, Brownstein DG, Lee AW, Cross SH, et al. : Dominant mutations of Col4a1 result in basement membrane defects which lead to anterior segment dysgenesis and glomerulopathy. Hum Mol Genet 14: 3161–3168, 2005 [DOI] [PubMed] [Google Scholar]
- 17.Miner JH, Sanes JR: Molecular and functional defects in kidneys of mice lacking collagen alpha 3(IV): Implications for Alport syndrome. J Cell Biol 135: 1403–1413, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dickinson ME, Flenniken AM, Ji X, Teboul L, Wong MD, White JK, et al. ; International Mouse Phenotyping Consortium; Jackson Laboratory; Infrastructure Nationale PHENOMIN, Institut Clinique de la Souris (ICS); Charles River Laboratories; MRC Harwell; Toronto Centre for Phenogenomics; Wellcome Trust Sanger Institute; RIKEN BioResource Center: High-throughput discovery of novel developmental phenotypes. Nature 537: 508–514, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.IMP C: Allele Col4a5 tm1b (EUCOMM)Wtsi, Available at: Meehan, T. F. et al. Disease model discovery from 3,328 gene knockouts by the International Mouse Phenotyping Consortium. Nat. Genet. 49, (2017). [DOI] [PMC free article] [PubMed]
- 20.Shevchenko A, Wilm M, Vorm O, Mann M: Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal Chem 68: 850–858, 1996 [DOI] [PubMed] [Google Scholar]
- 21.Humphries JD, Byron A, Bass MD, Craig SE, Pinney JW, Knight D, et al. : Proteomic analysis of integrin-associated complexes identifies RCC2 as a dual regulator of Rac1 and Arf6. Sci Signal 2: ra51, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nesvizhskii AI, Keller A, Kolker E, Aebersold R: A statistical model for identifying proteins by tandem mass spectrometry. Anal Chem 75: 4646–4658, 2003 [DOI] [PubMed] [Google Scholar]
- 23.Zaidel-Bar R, Geiger B: The switchable integrin adhesome. J Cell Sci 123: 1385–1388, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Berthier CC, Bethunaickan R, Gonzalez-Rivera T, Nair V, Ramanujam M, Zhang W, et al. : Cross-species transcriptional network analysis defines shared inflammatory responses in murine and human lupus nephritis. J Immunol 189: 988–1001, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hodgin JB, Nair V, Zhang H, Randolph A, Harris RC, Nelson RG, et al. : Identification of cross-species shared transcriptional networks of diabetic nephropathy in human and mouse glomeruli. Diabetes 62: 299–308, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hodgin JB, Borczuk AC, Nasr SH, Markowitz GS, Nair V, Martini S, et al. : A molecular profile of focal segmental glomerulosclerosis from formalin-fixed, paraffin-embedded tissue. Am J Pathol 177: 1674–1686, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ju W, Greene CS, Eichinger F, Nair V, Hodgin JB, Bitzer M, et al. . Defining cell-type specificity at the transcriptional level in human disease. Genome Res 23: 1862–1873, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Neusser MA, Lindenmeyer MT, Moll AG, Segerer S, Edenhofer I, Sen K, et al. : Human nephrosclerosis triggers a hypoxia-related glomerulopathy. Am J Pathol 176: 594–607, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Peterson KS, Huang JF, Zhu J, D’Agati V, Liu X, Miller N, et al. : Characterization of heterogeneity in the molecular pathogenesis of lupus nephritis from transcriptional profiles of laser-captured glomeruli. J Clin Invest 113: 1722–1733, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reich HN, Tritchler D, Cattran DC, Herzenberg AM, Eichinger F, Boucherot A, et al. : A molecular signature of proteinuria in glomerulonephritis. PLoS One 5: e13451, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Woroniecka KI, Park AS, Mohtat D, Thomas DB, Pullman JM, Susztak K: Transcriptome analysis of human diabetic kidney disease. Diabetes 60: 2354–2369, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Starborg T, Kalson NS, Lu Y, Mironov A, Cootes TF, Holmes DF, et al. : Using transmission electron microscopy and 3View to determine collagen fibril size and three-dimensional organization. Nat Protoc 8: 1433–1448, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jones FE, Murray LS, McNeilly S, Dean A, Aman A, Lu Y, et al. : 4-Sodium phenyl butyric acid has both efficacy and counter-indicative effects in the treatment of Col4a1 disease. Hum Mol Genet 28: 628–638, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Suleiman H, Zhang L, Roth R, Heuser JE, Miner JH, Shaw AS, et al. : Nanoscale protein architecture of the kidney glomerular basement membrane. eLife 2: e01149, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Suleiman HY, Roth R, Jain S, Heuser JE, Shaw AS, Miner JH: Injury-induced actin cytoskeleton reorganization in podocytes revealed by super-resolution microscopy. JCI Insight 2: e94137, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jones FE, Bailey MA, Murray LS, Lu Y, McNeilly S, Schlötzer-Schrehardt U, et al. : ER stress and basement membrane defects combine to cause glomerular and tubular renal disease resulting from Col4a1 mutations in mice. Dis Model Mech 9: 165–176, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tsuji K, Suleiman H, Miner JH, Daley JM, Capen DE, Păunescu TG, et al. : Ultrastructural characterization of the glomerulopathy in Alport mice by helium ion scanning microscopy (HIM). Sci Rep 7: 11696, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Horton ER, Byron A, Askari JA, Ng DHJ, Millon-Frémillon A, Robertson J, et al. : Definition of a consensus integrin adhesome and its dynamics during adhesion complex assembly and disassembly. Nat Cell Biol 17: 1577–1587, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhou XJ, Saxena R, Liu Z, Vaziri ND, Silva FG: Renal senescence in 2008: progress and challenges. Int Urol Nephrol 40: 823–839, 2008 [DOI] [PubMed] [Google Scholar]
- 40.Alexopoulos LG, Youn I, Bonaldo P, Guilak F: Developmental and osteoarthritic changes in Col6a1-knockout mice: biomechanics of type VI collagen in the cartilage pericellular matrix. Arthritis Rheum 60: 771–779, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Uhlen M, Oksvold P, Fagerberg L, Lundberg E, Jonasson K, Forsberg M, et al. : Towards a knowledge-based Human Protein Atlas. Nat Biotechnol 28: 1248–1250, 2010 [DOI] [PubMed] [Google Scholar]
- 42.Merchant ML, Barati MT, Caster DJ, Hata JL, Hobeika L, Coventry S, et al. : Proteomic analysis identifies distinct glomerular extracellular matrix in collapsing focal segmental glomerulosclerosis. J Am Soc Nephrol 31: 1883–1904, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Keeley DP, Hastie E, Jayadev R, Kelley LC, Chi Q, Payne SG, et al. : Comprehensive endogenous tagging of basement membrane components reveals dynamic movement within the matrix scaffolding. Dev Cell 54: 60–74.e7, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bönnemann CG: The collagen VI-related myopathies Ullrich congenital muscular dystrophy and Bethlem myopathy. Handb Clin Neurol 101: 81–96, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rasmussen DGK, Boesby L, Nielsen SH, Tepel M, Birot S, Karsdal MA, et al. : Collagen turnover profiles in chronic kidney disease. Sci Rep 9: 16062, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sun K, Park J, Gupta OT, Holland WL, Auerbach P, Zhang N, et al. : Endotrophin triggers adipose tissue fibrosis and metabolic dysfunction. Nat Commun 5: 3485, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Williams LM, McCann FE, Cabrita MA, Layton T, Cribbs A, Knezevic B, et al. : Identifying collagen VI as a target of fibrotic diseases regulated by CREBBP/EP300. Proc Natl Acad Sci U S A 117: 20753–20763, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Randles MJ, Lausecker F, Humphries JD, Byron A, Clark SJ, Miner JH, et al. : Basement membrane ligands initiate distinct signalling networks to direct cell shape. Matrix Biol 90: 61–78, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zimmerman SE, Hiremath C, Tsunezumi J, Yang Z, Finney B, Marciano DK: Nephronectin regulates mesangial cell adhesion and behavior in glomeruli. J Am Soc Nephrol 29: 1128–1140, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Reis M, Lee F, Bedran-Russo AK, Naba A: Proteomic profiling of the human dentin identifies age-related differences in the composition and solubility of the matrisome. BioRxiv 10.1101/2020.05.27.116624 (Preprint posted December 4, 2020) [Google Scholar]
- 51.Kriz W, Lemley KV: A potential role for mechanical forces in the detachment of podocytes and the progression of CKD. J Am Soc Nephrol 26: 258–269, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Clotet-Freixas S, McEvoy CM, Batruch I, Pastrello C, Kotlyar M, Van JAD, et al. : Extracellular matrix injury of kidney allografts in antibody-mediated rejection: A proteomics study. J Am Soc Nephrol 31: 2705–2724, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Simões FC, Cahill TJ, Kenyon A, Gavriouchkina D, Vieira JM, Sun X, et al. : Macrophages directly contribute collagen to scar formation during zebrafish heart regeneration and mouse heart repair. Nat Commun 11: 600, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vizcaíno JA, Csordas A, Del-Toro N, Dianes JA, Griss J, Lavidas I, et al. : 2016 update of the PRIDE database and its related tools [published correction appears in Nucleic Acids Res 44: 11033, 2016 10.1093/nar/gkw880]. Nucleic Acids Res 44: D447–D456, 2016. 26527722 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.