

Significance Statement
SGLT2 inhibitors represent a class of drugs that were originally developed for improving glycemic control. Cardiovascular outcome trials designed to evaluate cardiovascular safety yielded unexpected and unprecedented evidence of the cardiorenal benefits of SGLT2 inhibitors. Many hypotheses have been proposed to explain the mechanisms underlying these effects. Our study demonstrates that SGLT2 inhibition is associated with the restoration of euvolemia in nondiabetic heart failure (HF) rats by preserving GFR and renal mass and inhibiting proximal tubule NHE3-mediated sodium reabsorption. The attenuation of kidney dysfunction may constitute an essential mechanism by which SGLT2 inhibitors attenuate HF development and progression in either the presence or absence of diabetes.
Keywords: sodium-glucose cotransporter type 2, Na+/H+ exchanger isoform 3, gliflozins, renal function, cardiorenal protection
Visual Abstract

Abstract
Background
SGLT2 inhibitors reduce the risk of heart failure (HF) mortality and morbidity, regardless of the presence or absence of diabetes, but the mechanisms underlying this benefit remain unclear. Experiments with nondiabetic HF rats tested the hypothesis that the SGLT2 inhibitor empagliflozin (EMPA) inhibits proximal tubule (PT) NHE3 activity and improves renal salt and water handling.
Methods
Male Wistar rats were subjected to myocardial infarction or sham operation. After 4 weeks, rats that developed HF and sham rats were treated with EMPA or untreated for an additional 4 weeks. Immunoblotting and quantitative RT-PCR evaluated SGLT2 and NHE3 expression. Stationary in vivo microperfusion measured PT NHE3 activity.
Results
EMPA-treated HF rats displayed lower serum B-type natriuretic peptide levels and lower right ventricle and lung weight to tibia length than untreated HF rats. Upon saline challenge, the diuretic and natriuretic responses of EMPA-treated HF rats were similar to those of sham rats and were higher than those of untreated HF rats. Additionally, EMPA treatment prevented GFR decline and renal atrophy in HF rats. PT NHE3 activity was higher in HF rats than in sham rats, whereas treatment with EMPA markedly reduced NHE3 activity. Unexpectedly, SGLT2 protein and mRNA abundance were upregulated in the PT of HF rats.
Conclusions
Prevention of HF progression by EMPA is associated with reduced PT NHE3 activity, restoration of euvolemia, and preservation of renal mass. Moreover, dysregulation of PT SGLT2 may be involved in the pathophysiology of nondiabetic HF.
Sodium-glucose cotransporter type 2 (SGLT2) inhibitors, also known as gliflozins, suppress glucose reabsorption in the renal proximal tubule (PT), leading to substantial glycosuria, thereby lowering hyperglycemia in patients with type 2 diabetes (T2D).1,2 Gliflozins were initially developed as antidiabetic agents but have recently emerged as among the most impactful cardiovascular drugs. Three cardiovascular outcome trials consistently showed that SGLT2 inhibitors remarkably reduce cardiovascular mortality and hospitalization for heart failure (HF) in patients with T2D.3–5 Most recently, the phase 3 Dapagliflozin and Prevention of Adverse Outcomes in Heart Failure6 and the Empagliflozin Outcome Trial in Patients with Chronic Heart Failure with Reduced Ejection Fraction7 studies have announced that these SGLT2 inhibitors reduce cardiovascular death and HF progression when added to the standard therapy in patients with HF and reduced ejection fraction, regardless of the presence or absence of diabetes. As such, these trial findings may likely expand the clinical use of gliflozins beyond diabetes care. Nevertheless, the mechanisms underlying the unprecedented benefits of gliflozins in HF management remain elusive.
The presence of renal dysfunction portends adverse outcomes in patients with HF. HF is often associated with sodium and water retention, a reduction in renal blood flow and GFR, renal tubular damage, and proteinuria.8,9 Despite its cardioprotective actions, SGLT2 inhibitors have also been shown to confer renoprotection by preserving glomerular function, delaying progression to dialysis, reducing urinary protein excretion, and decreasing renal damage in patients with diabetes and rodent models.10–12 However, it remains to be established whether gliflozins are capable of ameliorating renal function in the setting of nondiabetic HF.
The natriuresis elicited by gliflozins, with consequent hemodynamic improvements, constitutes a plausible mechanism underpinning the positive cardiorenal outcomes of these drugs.13,14 Interestingly, although SGLT2 inhibitors cause a persistent 7% reduction in the extracellular volume of patients with T2D,15 mice lacking SGLT2 do not exhibit volume depletion, suggesting that the blockade of SGLT2-mediated sodium reabsorption per se is not sufficient to affect sodium balance and extracellular fluid homeostasis. In this regard, by coupling a mathematical model of renal function and volume homeostasis with clinical data, Hallow et al. 16 predicted that inhibition of apical PT Na+/H+ exchanger isoform 3 (NHE3) is required for the gliflozin-induced natriuretic effect. In line with the in silico predictions, we previously demonstrated that the nonselective sodium-glucose cotransporter inhibitor phlorizin remarkably reduces PT NHE3 activity.17 We also found that SGLT2, but not sodium-glucose cotransporter type 1, colocalizes with NHE3 in the apical membrane of PT cells.17 However, the influence of selective SGLT2 inhibitors on NHE3 activity under physiologic or pathophysiologic conditions has yet to be evaluated.
On the basis of these observations, this study aimed to test the hypothesis that an SGLT2 inhibitor is capable of exerting renoprotective effects in the setting of nondiabetic HF. More specifically, we investigated whether empagliflozin improves renal salt and water handling in rats with HF, and we sought to elucidate the potential mechanisms. Furthermore, we examined whether selective SGLT2 inhibition is capable of downregulating PT NHE3 in HF.
Methods
Reagents
Jardiance tablets (Boehringer Ingelheim Pharma GmbH & Co. KG) containing 25 mg of empagliflozin were purchased from a local pharmacy. These tablets were then sent to the Rhoster Company (Araçoiaba da Serra, Sao Paulo, Brazil) to be added to the rodent chow for daily empagliflozin treatment (10 mg/kg per day). Untreated animals received a control diet without empagliflozin. Chemicals were obtained from Merck (Darmstadt, Germany) unless otherwise specified.
Animal Protocols, Surgical Procedures, and Drug Treatment
All experiments were carried out following the ethical principles in animal research of the Brazilian College of Animal Experimentation and were approved by the Institutional Animal Care and Use Committee of the University of Sao Paulo Medical School (protocol no. 941/2018). Eight-week-old male Wistar rats (200–250 g; n=104) were obtained from the Institute of Biomedical Sciences, University of Sao Paulo, Sao Paulo, Brazil. Rats were randomly assigned to sham surgery (n=36) or myocardial infarction (n=68) by ligation of the left anterior descending (LAD) artery (Supplemental Figures 1 and 2) as previously described.18 Briefly, rats were anesthetized with ketamine (50 mg/kg) and xylazine (10 mg/kg) intraperitoneally and placed under positive pressure ventilation (rate: 90 breaths per minute; tidal volume: 2.5 ml on a Harvard rodent respirator [model 683; Harvard Apparatus Co., South Natick, MA]). The thoracotomy was performed at the fourth intercostal space. After that, the heart was exposed, and the LAD coronary artery was isolated and ligated approximately 3 mm from the origin of the aorta using 6–0 Prolene suture. The chest was closed, and the animals were carefully moved from ventilation support. Sham-operated rats were submitted to a similar surgical procedure, except for LAD coronary artery ligation. After recovering from surgery, the rats were maintained in a temperature- and humidity-controlled environment with a 12-hour dark/light cycle at the Heart Institute (InCor) animal facility. Food and water were supplied ad libitum.
Four weeks after surgery, HF was characterized by echocardiographic evaluation of left ventricle (LV) systolic function and serum levels of brain natriuretic peptide (BNP). HF was considered when the fractional area change (FAC; i.e., the percentage of change in the LV cross-sectional area between diastole and systole) was lower than 40% and when circulating levels of BNP were higher than 1.0 ng/ml (Supplemental Figure 3, Supplemental Table 1). Myocardial infarction rats that developed HF (n=38) and sham (n=36) rats were randomly divided into two groups and treated with empagliflozin (10 mg/kg per day supplied in the rat chow) or not treated. After 4 weeks (post-treatment), the rats were anesthetized by an intraperitoneal injection of pentobarbital (50 mg/kg) and subsequently euthanized by cervical dislocation. The study design is depicted in Supplemental Figure 2.
Echocardiography
Rats were anesthetized with 1.5% isoflurane in O2 and placed in the left lateral decubitus position to obtain cardiac images. Images were captured and analyzed using Sonos 5500 ultrasound equipment (Philips Medical System, Bothell, WA) with a 12- to 14-MHz transducer (2-cm depth with fundamental and harmonic imaging). Echocardiographic images were acquired by placing the cursor of pulsed-wave Doppler in the LV outflow tract to display the end of aortic ejection and the onset of mitral inflow. Tracing the endocardial border by planimetry, excluding the papillary muscle, in end diastolic and end systolic frames provides values for left ventricle end diastolic area (LVEDA) and LV end systolic area (Supplemental Table 1). Applying this in the following equation would give an FAC of LV from diastole to systole: FAC = [LVEDA – (LV end systolic area/LVEDA)]×100%. Echocardiography was performed by an investigator who was blind to the experimental groups.
Determination of BNP Serum Levels
Before randomization, blood samples were withdrawn from the retro-orbital sinus under isoflurane anesthesia. At post-treatment, blood was collected from the abdominal aorta artery at the time of death. The samples were immediately transferred into vacuum tubes with a gel separator (BD vacutainer SST II Advance; Becton, Dickinson and Company, Franklin Lakes, NJ) and centrifugated at 4000 rpm for 10 minutes at 4°C to obtain serum. Serum levels of BNP were measured by ELISA (BNP 32 Rat ELISA kit; Abcam) according to the manufacturer’s instructions.
BP Measurements
Tail-cuff BP was measured noninvasively in conscious restrained rats by plethysmography (BP Blood Pressure Analysis System 2000; Visitech System, Apex, NC) as previously described.19
Renal Function Evaluation
The rats were individually housed and placed in metabolic cages (Tecniplast; Buguggiate, VA, Italy) as previously described.20 Food and water consumption was determined daily and subsequently normalized to body weight. Urine samples were collected for 24 hours and used to determine urinary flow, sodium, creatinine, and proteinuria. Creatinine clearance was used to estimate the GFR.
Blood and Urine Analyses
Fasting blood glucose levels were measured using the ACCU-CHECK Performa meter (Roche Diagnostics GmbH, Mannheim, Germany). The serum and urinary sodium concentrations were determined by flame photometry (DM-62; Digimed, Sao Paulo, Brazil). The serum urea, urinary glucose, proteinuria, and creatinine concentrations were measured by colorimetric methods using Labtest kits (Labtest; Lagoa Santa, Brazil). The experiments were carried out following the manufacturer’s instructions.
Saline Challenge
Animals were anesthetized with isoflurane and were injected intraperitoneally with a volume of warmed (37°C) saline (0.9% NaCl) equivalent to 10% of their body weight (vol/wt). After that, the rats were placed immediately in metabolic cages, where they quickly woke up. Urine volume, collected for 3 hours, was measured with a graduate pipette, and urinary sodium concentration was measured by flame photometry (DM-62; Digimed). The results were expressed as the percentage of the fluid and sodium load that was injected.
Biometric and Morphometric Analyses
The lungs, heart, and kidneys were excised and weighed. The organ weight was normalized by left tibia length. The lungs were stored in an oven to dry at 70°C for 48 hours. The relative water content of lung tissue was calculated using the following equation: lung water content (in percentage) = (wet lung weight − dry lung weight)/lung wet weight×100. The kidneys were immediately removed for isolation of renal cortical proteins for immunoblotting, tissue fixation for immunohistochemistry, or freezing for RNA extraction and quantitative RT-PCR.
Preparation of Renal Cortical Homogenate
The right kidney from rats was removed and cut in half on a midsagittal plane, and the cortices were isolated and homogenized in ice-cold PBS buffer (150 mM sodium chloride, 2.8 mM monobasic sodium phosphate, 7.2 mM dibasic sodium phosphate, pH 7.4) containing protease inhibitors (0.7 mg/ml pepstatin, 0.5 mg/ml leupeptin, and 40 mg/ml phenylmethanesulfonyl fluoride) and phosphatase inhibitors (50 mM sodium pyrophosphate decahydrate and 15 mM sodium fluoride) using a Potter–Elvehjem-style tissue grinder (POLIMIX PX-SR50E; Kinematica Inc., Luzern, Switzerland). The homogenate was centrifuged at 4500 rpm for 10 minutes at 4°C. The supernatant was collected and stored at −80°C. Protein concentration was determined by the Lowry method.21
SDS-PAGE and Immunoblotting
Renal cortical proteins were solubilized in Laemmli sample buffer and separated by SDS-PAGE using 7.5% polyacrylamide gels. The separated proteins were transferred from the gel to a polyvinylidene difluoride membrane (Immobilon-P; Merck Millipore, Bedford, MA) at 350 mA for 8–10 hours at 4°C with a TE 62 Transfer Cooled Unit (GE HealthCare, Piscataway, NJ) and stained with Ponceau S. Then, the polyvinylidene difluoride membranes were incubated with blocking solution (5% nonfat dry milk and 0.1% Tween 20 in PBS, pH 7.4) for 1 hour and overnight (4°C) with specific primary antibodies: a polyclonal antibody against SGLT2 (1:1000, NBP1–92384; Novus Biologicals, Centennial, CO); a mouse mAb against NHE3, clone 3H3 (1:1000; Peter Aronson, Yale University, New Haven, CT)22; a phosphospecific mAb against NHE3, PS552-NHE3, clone 14D522 (1:1000; Santa Cruz Biotech, Dallas, TX); or a polyclonal antibody against β-actin (1:5000, ab8227; Abcam). Proteins were detected using horseradish peroxidase–conjugated secondary antibodies (1:2000; Jackson ImmunoResearch, West Grove, PA). The bound antibodies were detected using an enhanced chemiluminescence system (GE HealthCare) according to the manufacturer’s protocols. The membranes were then placed in a digital imaging system (ImageQuant LAS 4000 mini; GE HealthCare). Immunoblot quantification was performed using ImageJ software (National Institutes of Health, Bethesda, MD).
Immunohistochemistry
The left kidney was cut in half on a midsagittal plane, fixed in 10% formalin for 24 hours, stored in 70% ethanol, and then embedded in paraffin. Four-micrometer kidney paraffin sections were incubated with 3% H2O2 for 3 minutes (five times at room temperature) to block endogenous peroxidase activity and then rinsed with Tris-buffered saline with 0.1% Tween 20 (TBST). Nonspecific reactions were blocked in 2% goat serum for 20 minutes and then incubated with the rabbit polyclonal anti-SGLT2 antibody (1:100) or the rabbit polyclonal anticleaved caspase-3 antibody (1:100, 9661; Cell Signaling Technology, Danvers, MA). After 18 hours of incubation at 4°C, kidney sections were washed three times for 5 minutes with TBST and incubated with a secondary antibody. After washing in TBST, tissue sections were incubated with peroxidase-conjugated universal immunoenzyme polymer, anti-rabbit solution [Histofine Simple Stain MAX PO(MULTI); Nichirei Biosciences Inc., Tokyo, Japan] for 30 minutes at room temperature. After washing in TBST, immunoreactions were detected with 3,3′-diaminobenzidine tetrahydrochloride (DAB-Zymed) for 7 minutes and counterstained with hematoxylin. The images were acquired under a ×400-magnification light microscope using the software Quantimet Leica (Leica Biosystems, Wetzlar, Germany).
Terminal Deoxynucleotidyl Transferase–Mediated Digoxigenin-Deoxyuridine Nick-End Labeling Assay
DNA fragmentation was detected using an in situ cell death detection kit (ApopTag Peroxidase In Situ Apoptosis Detection Kit S7100; Merck Millipore), according to the manufacturer’s instructions. Briefly, tissue sections were deparaffinized in xylene and ethanol, rehydrated in serial ethanol dilutions, and permeabilized with proteinase K. The reaction with terminal deoxynucleotidyl transferase and alkaline phosphatase conversion was performed, and the cross-sections were examined by light microscopy. The percentage of terminal deoxynucleotidyl transferase–mediated digoxigenin-deoxyuridine nick-end labeling–positive nuclei was quantified by an observer blinded to the four conditions using the Quantimet-Leica software (Leica Biosystems).
Quantitative Real-Time RT-PCR
Total RNA was isolated from the left kidney using Trizol (Thermo Fisher Scientific, Carlsbad, CA) according to the manufacturer’s instructions, quantified (ND-1000 spectrophotometer; NanoDrop Technologies, Inc.), and treated with DNase-I. First-strand cDNA synthesis was performed using Super-Script III Reverse Transcription (Thermo Fisher Scientific) following the manufacturer’s guidelines. Details about the oligonucleotide primers used in this study are listed in Supplemental Table 2. Reactions were carried using SYBR Green PCR Master Mix-PE (Thermo Fisher Scientific) on an ABI Prism 7500 Fast Sequence Detection System (Applied Biosystem, Foster City, CA). The comparative threshold cycle method was used for data analyses. All samples were assayed in triplicate. Transcripts for three reference genes (Gapdh, Actb, and Ppia) were determined (Supplemental Table 2). The BestKeeper software23 was used to identify the best suit reference gene (Gapdh) for data normalization under our experimental conditions. Relative expression was analyzed by the 2−ΔΔCT method.
In Vivo Stationary Microperfusion
In vivo stationary microperfusion was used to determine NHE3 activity in the PT to test the hypothesis that selective SGLT2 inhibition is capable of downregulating PT NHE3 in HF. Rats (four to five per group) were anesthetized by intramuscular administration of tiletamine/zolazepam (50 mg/kg) and xylazine (5 mg/kg). After tracheostomy, the left jugular vein was cannulated for infusion of 3% mannitol in isotonic saline solution at a rate of 0.05 ml/min. The kidney was isolated using a lumbar approach, immobilized in situ using Ringer agar in a Lucite cup under a microscope, and adequately illuminated. PTs were punctured using a double-barreled micropipette, one barrel being used to inject FDC green-colored Ringer perfusion solution (100 mM NaCl, 5 mM KCl, 25 mM NaHCO3, 1 mM CaCl2, 1.2 mM MgSO4, FDC green, and raffinose to reach isotonicity) and the other being used to inject Sudan black-colored castor oil, the latter used to block the injected fluid column in the lumen. The tubules were perfused with the Ringer solution in the presence or absence of the selective NHE3 inhibitor, S3226 (10 µM).24 The voltage between the microelectrode barrels, representing luminal H+ activity, was continuously recorded by a computer equipped with an analog to digital conversion board (Lynx, Sao Paulo, Brazil) for data acquisition and processing.
To measure luminal pH, PTs were impaled by a double-barreled asymmetric microelectrode, the larger barrel containing H+ ion-sensitive ion exchange resin silanized with hexamethyldisilazane (Sigma Fluka, Buchs, Switzerland).
Tubular acidification rate was assessed by injecting a droplet of the perfusion solution between the oil columns and following the luminal pH changes toward the steady-state level (stationary perfusion). The luminal acidification was also assessed in the presence or absence of the selective NHE3 inhibitor, S3226 (10 µM), in the control perfusion solution. For evaluation of acidification kinetics, luminal [H+] values from each pH recovery curve were fitted with Origin 2020 (Origin Lab, Northampton, MA) by using the following equation:
| (1) |
where [H+]stat represents stationary luminal [H+], k stands for the intrinsic kinetic constant of each fitted curve, and b is a constant that determines the initial pH values for each fitted curve and can be mathematically defined as .
We then calculated the rate of luminal acidification until the half-maximum point of each curve by
| (2) |
where t1/2 is the predicted time at that point [] and .
Statistical Analyses
The results are reported as the mean ± SEM, with n indicating the number of rats. Comparisons among the means were assessed using a two-way ANOVA followed by the Tukey post hoc test. A P value <0.05 was considered significant, considering the main effect of empagliflozin treatment, the main effect of HF induction, the interaction between empagliflozin treatment and HF induction, and the differences among groups.
Results
Empagliflozin Lowers Serum BNP Levels and Reduces the Right Ventricle and Lung Weight-Tibia Length Ratio in Nondiabetic HF Rats
As shown in Figure 1, untreated HF rats displayed higher serum BNP levels and lower FAC than sham animals. Importantly, treatment with empagliflozin restored serum BNP to levels similar to those of sham groups (Figure 1A). Also, empagliflozin treatment induced a modest but significant improvement in FAC compared with untreated HF rats (23%±2% versus 17%±1%, P=0.02) (Figure 1B). As expected, serum BNP levels and FAC were similar between untreated and empagliflozin-treated sham rats.
Figure 1.

Treatment with empagliflozin (EMPA) normalizes serum BNP concentration and slightly improves LV systolic function in nondiabetic rats with induced HF. Doppler echocardiography and quantitative determination of serum BNP were performed in sham and HF rats after 4 weeks of treatment with EMPA or no treatment. (A) The circulating levels of BNP and (B) the FAC of sham and HF rats treated with EMPA or untreated. The values represent individual measurements and the means ± SEM. ***P<0.001 versus sham; ††† P<0.001 versus sham + EMPA; # P=0.02 versus HF; ### P<0.001 versus HF.
The biometric characteristics of the animals are shown in Table 1. The average body weights were similar among the untreated HF, empagliflozin-treated HF, and empagliflozin-treated sham groups (but not the untreated sham group). Therefore, organ weight was normalized by tibial length, which remained unchanged among the four groups of rats. The biometric analysis showed that untreated HF rats exhibited a higher right ventricle (RV) weight-tibia length ratio, a higher lung-tibia length ratio, and more pulmonary congestion than the sham groups. In contrast, empagliflozin prevented pulmonary congestion and RV hypertrophy.
Table 1.
Biometric parameters of sham and nondiabetic rats with myocardial-induced HF treated with the SGLT2 inhibitor empagliflozin or untreated
| Biometric Parameters | Sham, n=16 | Sham + EMPA, n=20 | HF, n=14 | HF + EMPA, n=17 |
|---|---|---|---|---|
| Body weight, g | 450±10 | 403±10a | 398±5a | 406±9b |
| Tibia length, mm | 41.2±0.3 | 40.4±0.3 | 40.5±0.2 | 40.7±0.4 |
| LV/tibia, mg/mm | 20.4±0.8 | 17.6±0.4a | 20.1±0.5c | 20.6±0.4d |
| RV/tibia, mg/mm | 6.26±0.24 | 5.22±0.16 | 8.90±0.96e, f | 6.39±0.30g |
| LV + RV/tibia, mg/mm | 26.7±1.1 | 22.8±0.5a | 29.0±1.1f | 27.0±0.6d |
| Lung/tibia, mg/mm | 32.2±1.5 | 31.8±1.1 | 44.7±3.3e, f | 31.7±1.7g |
| Lung water content, % | 78.3±0.4 | 77.9±0.4 | 80.6±0.2e, f | 79.1±0.3g |
| LK + RK/tibia, mg/mm | 64±1 | 67±1 | 58±1a, f | 67±1g |
The values represent individual measurements and the means ± SEM. EMPA, empagliflozin; LK, left kidney; RK, right kidney.
P=0.005 versus sham.
P=0.01 versus sham.
P=0.01 versus sham + EMPA.
P=0.002 versus sham + EMPA.
P<0.001 versus sham.
P<0.001 versus sham + EMPA.
P<0.001 versus HF.
Glycosuria, Diuresis, and Natriuresis Induced by Empagliflozin Were Higher in HF Rats than in Sham Rats
Glycosuria markedly increased in both the sham and HF groups treated with empagliflozin compared with untreated animals (Figure 2A). Interestingly, the effect of empagliflozin on glycosuria was more pronounced in HF rats than in sham rats (Figure 2A). Changes in urinary glucose excretion among the four groups of rats were not associated with changes in blood glucose concentration (Figure 2B). Similar to the findings of urinary glucose excretion, urinary sodium (Figure 2C) and urinary flow (Figure 2D) were higher in the empagliflozin-treated sham group and the HF group than in the untreated groups. Moreover, urinary sodium was greater in empagliflozin-treated HF rats than in empagliflozin-treated sham animals (Figure 2C). The increase in urinary flow and urinary sodium in empagliflozin-treated rats was accompanied by increased water and sodium intake (Supplemental Figure 4).
Figure 2.

Treatment with empagliflozin (EMPA) induces higher glycosuria, urinary flow, and sodium excretion in HF rats than in sham rats. Rats were individually placed into metabolic cages for 24-hour urine collection to measure glycosuria, urinary flow, and urinary sodium. Blood was collected from 12-hour fasting rats for the determination of blood glucose. (A) Glycosuria. (B) Blood glucose concentration. (C) Urinary sodium. (D) Urinary flow. Experiments were performed 4 weeks after treatment with EMPA or no treatment. The values represent individual measurements and the means ± SEM. **P=0.005 versus sham; ***P<0.001 versus sham; † P=0.02 versus sham + EMPA; ††† P<0.001 versus sham + EMPA; ### P<0.001 versus HF.
PT SGLT2 is Overexpressed in Nondiabetic HF Rats
SGLT2 expression and activity are upregulated in the diabetic kidney25,26; however, how this transporter is regulated in nondiabetic HF remains unknown. Therefore, we tested the hypothesis that HF rats display increased protein and mRNA-SGLT2 expression compared with controls. SGLT2 protein expression in the PT of the four groups of rats was first evaluated by immunohistochemistry. Representative photomicrographs of SGLT2-stained sections of the renal PTs of HF and sham rats are presented in Figure 3A. The results of this qualitative analysis strongly suggested that SGLT2 was upregulated in nondiabetic HF rats compared with sham rats and HF rats treated with empagliflozin (Figure 3A).
Figure 3.
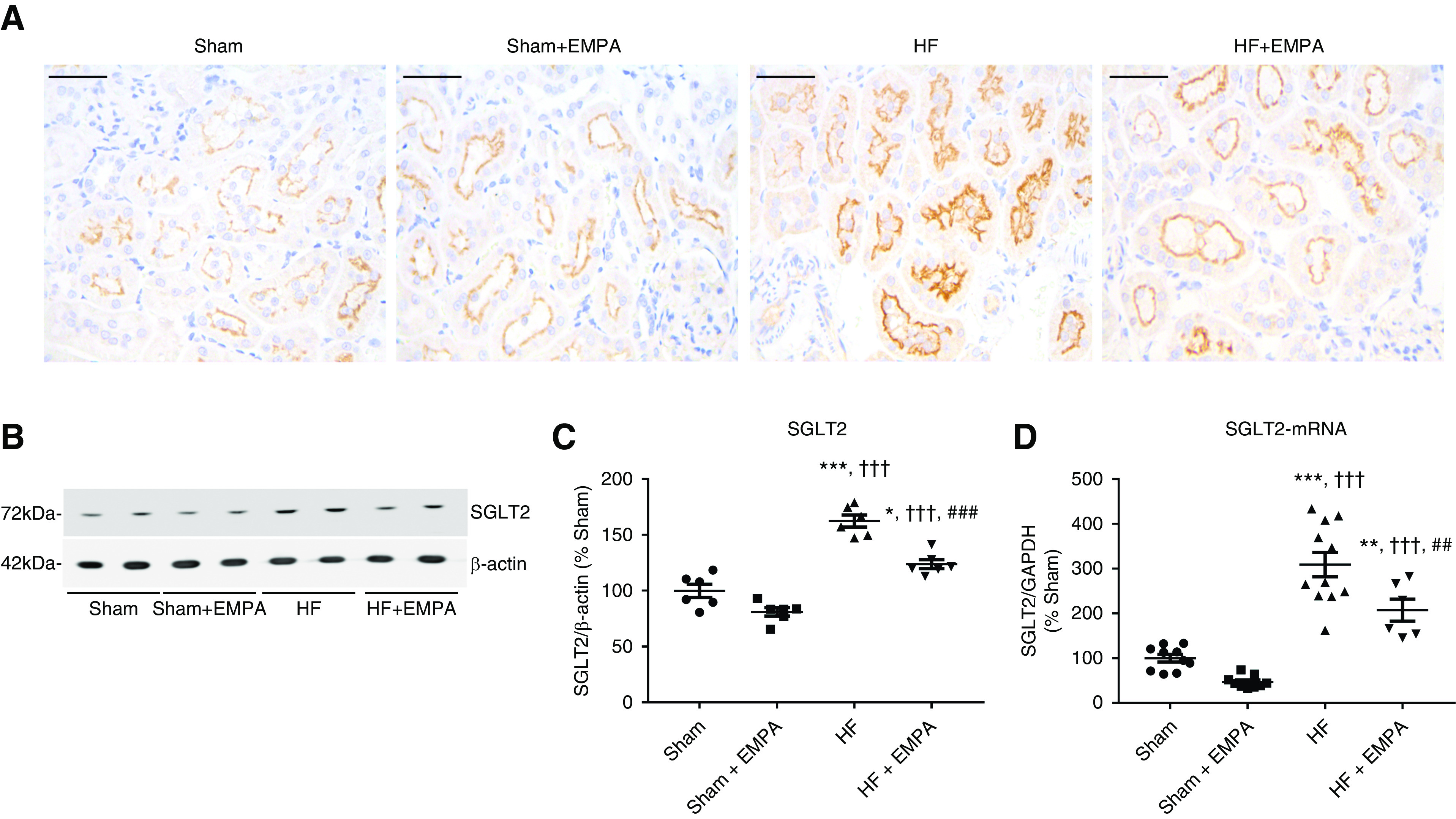
SGLT2 is overexpressed in the PT of nondiabetic HF rats. (A) Representative immunohistochemical staining of SGLT2 in the PT of sham and HF rats treated with empagliflozin (EMPA) or untreated. Scale bar: 50 µm. (B) Representative immunoblots from SDS-PAGE of renal cortical proteins isolated from sham and HF rats treated with EMPA or untreated and probed with antibodies against SGLT2 and β-actin. (C) Graphic representation of the relative levels of SGLT2 protein abundance in the renal cortex of the four groups of rats. (D) Graphic representation of the relative mRNA expression of SGLT2 in the renal cortex of sham and HF rats treated with EMPA or untreated. The levels of SGLT2 mRNA were measured using quantitative PCR, and GAPDH (glyceraldehyde 3-phosphate dehydrogenase) was used as an internal control. The values represent individual measurements and the means ± SEM (Supplemental Material). *P=0.01 versus sham; **P=0.006 versus sham; ***P<0.001 versus sham; ††† P<0.001 versus sham + EMPA; ## P=0.008 versus HF; ### P<0.001 versus HF.
Second, SGLT2 protein abundance was evaluated in the renal cortex by immunoblotting. As seen in Figure 3, B and C, SGLT2 protein abundance was enhanced in the renal cortex of HF rats compared with both untreated and empagliflozin-treated sham rats.
Finally, we found that higher SGLT2 protein abundance was accompanied by higher SGLT2 mRNA expression levels in the renal cortices of HF rats compared with sham animals (Figure 3D). Interestingly, empagliflozin treatment lowered SGLT2 expression at both the protein and mRNA levels compared with untreated HF rats (Figure 3, B–D). However, SGLT2 expression remained higher in empagliflozin HF rats than in sham rats (Figure 3, B–D).
Empagliflozin Restores Euvolemia in Nondiabetic HF Rats
First, we tested the acute renal capability of handling salt and water of HF and sham rats that were either treated with empagliflozin or untreated (Figure 4).
Figure 4.

Treatment with empagliflozin (EMPA) improves the volume status in nondiabetic HF rats. (A and B) Rats were challenged with an intraperitoneal bolus of warm saline equivalent to 10% of their body weight and were then placed in metabolic cages for 3-hour urine collection. (A) The percentage of the fluid load that was excreted within 3 hours of the saline challenge. (B) The percentage of the sodium load that was excreted within 3 hours of the saline challenge. (C) Blood samples were collected to measure hematocrit. (D) BP was measured using tail-cuff plethysmography. Experiments were conducted 4 weeks after treatment with EMPA or no treatment. The values represent individual measurements and the means ± SEM. *P=0.03 versus sham; **P=0.005 versus sham; ***P<0.001 versus sham; † P=0.02 versus sham + EMPA; †† P=0.005 versus sham + EMPA; ††† P<0.001 versus sham + EMPA; # P=0.02 versus HF; ## P=0.003 versus HF; ### P<0.001 versus HF.
Untreated HF rats excreted less fluid (Figure 4A) and salt (Figure 4B) than sham rats and empagliflozin-treated HF rats. Similar fluid and salt load percentages were excreted by untreated sham- and empagliflozin-treated HF rats (Figure 4, A and B).
Second, changes in hematocrit were evaluated. The hematocrit levels of untreated HF rats were lower than those of sham rats (Figure 4C). Also, treatment with empagliflozin restored the hematocrit of HF rats to levels similar to those of sham rats (Figure 4C).
Third, as seen in Figure 4D, the effects of empagliflozin on reducing extracellular volume in HF rats were not accompanied by reduced BP. Untreated and empagliflozin-treated HF rats exhibited lower tail-cuff BP than sham rats (Figure 4D). In addition, empagliflozin treatment did not affect BP in either sham or HF rats (Figure 4D).
Empagliflozin Prevents the Reduction in GFR, Attenuates Proteinuria, and Preserves Kidney Mass in Nondiabetic HF Rats
As depicted in Figure 5, untreated HF rats exhibited lower GFR (Figure 5A), higher levels of serum urea (Figure 5B), and greater proteinuria (Figure 5C) than the other three groups of rats. In contrast, empagliflozin-treated HF rats exhibited GFR, serum urea, and proteinuria similar to sham groups (Figure 5). Interestingly, HF rats exhibited a lower kidney weight-tibia length ratio than sham rats, whereas empagliflozin treatment prevented HF-induced kidney atrophy (Table 1).
Figure 5.

Treatment with empagliflozin (EMPA) prevents the reduction in GFR and attenuates proteinuria in HF rats. Rats were individually placed into metabolic cages for 24-hour urine collection to measure GFR and proteinuria. Blood was collected for the determination of serum urea and creatinine. (A) The GFR was estimated by creatinine clearance and normalized per gram of kidney weight (KW). (B) Serum urea was measured by colorimetry. (C) Graphic representation of the urine protein-creatinine ratio. Proteinuria was measured by colorimetry. Experiments were conducted 4 weeks after treatment with EMPA or no treatment. The values represent individual measurements and the means ± SEM. **P=0.001 versus sham; ***P<0.001 versus sham; ††† P<0.001 versus sham + EMPA; ## P=0.001 versus HF; ### P<0.001 versus HF.
The lower kidney weight-tibia length ratio in HF rats was associated with a higher percentage of terminal deoxynucleotidyl transferase–mediated digoxigenin-deoxyuridine nick-end labeling–positive apoptotic renal cells and a higher abundance of cleaved caspase-3 (Figure 6). The extents of renal apoptosis and cleaved caspase-3 protein expression in HF rats were normalized by empagliflozin treatment.
Figure 6.

Treatment with empagliflozin (EMPA) normalizes renal apoptosis and cleaved caspase-3 abundance in HF rats. (A and B) Representative photomicrographs of kidney sections (×400 magnification) from HF and sham rats treated with EMPA or untreated. Scale bar: 50 µm. (A) Terminal deoxynucleotidyl transferase–mediated digoxigenin-deoxyuridine nick-end labeling assay was used to detect apoptotic cells that undergo extensive DNA degradation. (B) Immunohistochemical staining of cleaved caspase-3. (C) Quantification of apoptotic nuclei and (D) cleaved caspase-3–positive areas were determined in five random fields per slide using the ImageJ software. These analyses were performed by two investigators blinded to the experimental groups. The values represent individual measurements and the means ± SEM. *P=0.04 versus sham; **P=0.005 versus sham; ***P<0.001 versus sham; † P=0.02 versus sham + EMPA; ††† P<0.001 versus sham + EMPA; # P=0.02 versus HF; ### P<0.001 versus HF.
Empagliflozin Inhibits PT NHE3
PT luminal pH was recorded during stationary in vivo microperfusion in the presence or absence of the specific NHE3 inhibitor S3226 to assess modulation of NHE3 by empagliflozin in both sham and HF rats. Representative pH recovery recordings for each experimental group are presented in Figure 7, A and B. As illustrated in Figure 7C, the PT acidification rate (Δ[H+]/Δt) was higher in untreated HF rats than in untreated sham rats. Treatment with empagliflozin prominently reduced PT luminal acidification of both HF and sham rats (Figure 7C). Nevertheless, the inhibitory effect of empagliflozin on tubular acidification was more pronounced in HF rats (approximately 70% inhibition versus untreated HF rats) than in sham rats (approximately 45% inhibition versus untreated sham rats). In addition, no differences were observed in tubular acidification among the four groups of rats when their PTs were perfused with the selective NHE3 inhibitor S3226, confirming that differences in H+ secretion induced by empagliflozin were due to modulation of NHE3 activity (Figure 7D). The increase in PT NHE3 activity in HF rats was accompanied by enhanced NHE3 mRNA levels (Figure 7E) and NHE3 protein abundance (Figure 7, F and G). Conversely, even though empagliflozin reduced PT NHE3 activity, this SGLT2 inhibitor did not affect either cortical NHE3 protein abundance or the mRNA levels of NHE3 in HF or sham rats (Figure 7, E–G). NHE3 phosphorylation at serine 552 (PS552-NHE3) is considered a surrogate for NHE3 inhibition.22,27,28 Consistent with higher PT NHE3 activity, HF rats displayed lower levels of PS552-NHE3 to total NHE3 than the other three groups of rats (Figure 7, F and H). The levels of PS552-NHE3 to total NHE3 were higher in empagliflozin-treated HF rats than in untreated HF rats but lower than in the rats in the sham groups. Importantly, although empagliflozin inhibited NHE3 activity in the PT of sham rats, no differences were found in the levels of PS552-NHE3 to total NHE3 between untreated and empagliflozin-treated sham rats (Figure 7, F and H).
Figure 7.
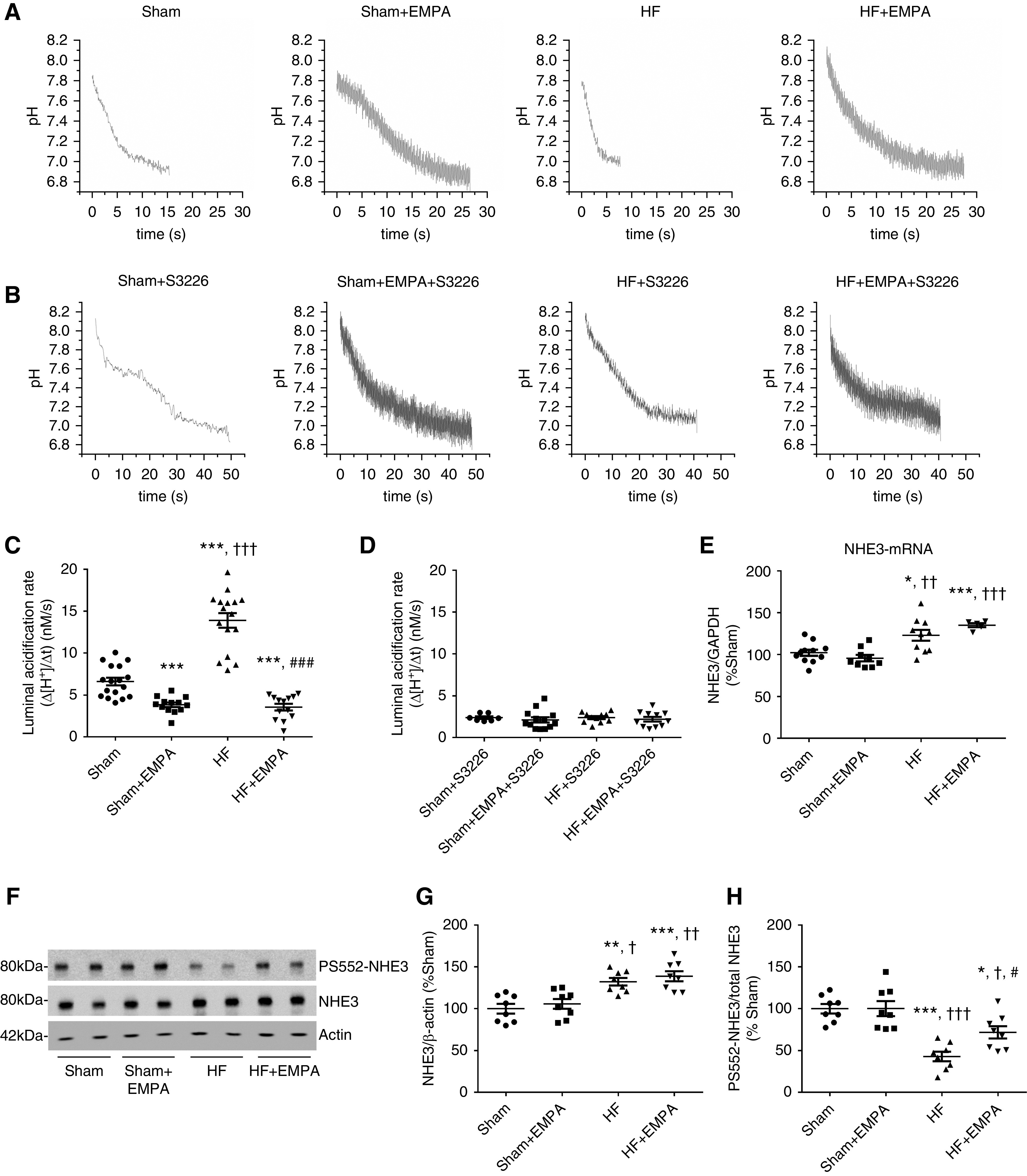
Treatment with empagliflozin (EMPA) inhibits PT NHE3 activity. (A–D) Modulation of NHE3 activity by EMPA was determined by assessing the PT luminal acidification in the presence or absence of 10 µM S3226, a selective inhibitor of NHE3. (A and C) Representative luminal pH recordings and the rate of luminal acidification in sham and HF rats treated with EMPA or untreated in the absence of S3226. (B and D) Representative luminal pH recordings and the rate of luminal acidification in sham and HF rats treated with EMPA or untreated in the presence of S3226. (E) Graphic representation of the relative mRNA expression of NHE3 in the renal cortex of sham and HF rats treated with EMPA or untreated. The levels of NHE3 mRNA were measured using quantitative PCR, and GAPDH (glyceraldehyde 3-phosphate dehydrogenase) was used as an internal control. (F) Renal cortical proteins isolated from the four groups of rats were resolved by SDS-PAGE and transferred to polyvinylidene difluoride membranes. The membranes were probed with a primary antibody against total NHE3, a phosphospecific antibody that recognizes NHE3 only when it is phosphorylated at serine 552 (PS552-NHE3), and an antibody against β-actin as an internal control. (G) Graphic representation of the relative expression of total NHE3 and (H) the ratio of phosphorylated NHE3 at serine 552 to total NHE3 (PS552-NHE3/total NHE3). The values represent individual measurements and the means ± SEM (Supplemental Material). *P=-0.02 versus sham; **P=0.002 versus sham; ***P<0.001 versus sham; † P=0.02 versus sham + EMPA; †† P=0.001 versus sham + EMPA; ††† P<0.001 versus sham + EMPA; # P=0.03 versus HF; ### P<0.001 versus HF.
Discussion
This study provides novel experimental evidence that SGLT2 inhibition by empagliflozin exerts renal benefits that may contribute to attenuate HF progression in a nondiabetic setting. Our results demonstrate that empagliflozin preserves glomerular function, prevents HF-induced renal apoptosis and loss of renal mass, and exerts a profound reduction of PT NHE3-mediated sodium reabsorption in an experimental model of myocardial infarction–induced HF. These effects were accompanied by restoration of euvolemia, a decrease in circulating levels of BNP, prevention of pulmonary congestion, and mitigation of RV hypertrophy. Moreover, to the best of our knowledge, this is the first report to reveal that SGLT2 is overexpressed in the renal PT of nondiabetic HF rats. Interestingly, empagliflozin not only suppressed SGLT2-mediated glucose reabsorption but also, reduced SGLT2 expression in the renal cortex of nondiabetic HF rats at both the protein and mRNA levels.
Excessive renal sodium avidity and extracellular volume overload are hallmark features of HF that are associated with disease progression and poor prognosis.29 The sodium and water retention that occurs with advanced LV failure can lead to pulmonary edema, increased mean pulmonary arterial pressure, and consequently, RV hypertrophy that is ultimately followed by dysfunction.30 Accordingly, we found that untreated HF rats exhibited a lower natriuretic and diuretic response to a saline challenge, a lower hematocrit, and much higher circulating BNP levels than sham rats. As expected, volume overload in untreated HF rats was accompanied by pulmonary congestion and RV hypertrophy. Conversely, treatment with empagliflozin restored euvolemia in HF rats, as evidenced by the improved sodium and water handling by the kidneys and normalization of hematocrit and serum BNP levels. The reduction in volume overload by empagliflozin prevented pulmonary congestion and RV hypertrophy in nondiabetic HF rats. In line with these findings, Chowdhury et al. 31 found that empagliflozin treatment in rats with severe experimental pulmonary hypertension reduced mean pulmonary artery pressure, RV systolic pressure, and RV hypertrophy. Importantly, these hemodynamic benefits were associated with prolonged survival in this model of pulmonary arterial hypertension. These data support the hypothesis that reduced volume overload is one of the mechanisms underlying the observed decrease in HF events in cardiovascular trials with SGLT2 inhibitors.
The potential causes of salt and water retention by the kidneys in HF may include a decreased GFR, increased tubular reabsorption of sodium, or both. This study demonstrates that empagliflozin prevents a decrease in GFR in nondiabetic HF rats. Such a result may appear contradictory at first glance because one would expect that the increased delivery of sodium and chloride to the macula densa, produced by SGLT2 inhibitors, would activate the tubuloglomerular feedback, leading to a decline in GFR. However, GFR preservation in HF rats treated with empagliflozin may be explained, at least in part, by the finding that this SGLT2 inhibitor can also prevent HF-induced renal atrophy, renal apoptosis, and consequently, kidney dysfunction. Additionally, the preservation of GFR in empagliflozin-treated HF rats might also be due to an improvement in cardiac performance. In this regard, we have found that empagliflozin modestly but significantly improves systolic function in HF rats. It is important to emphasize that, in this study, we performed echocardiography in anesthetized rats as opposed to pharmacologic stress echocardiography, which may constitute a limitation that precluded us from detecting a more pronounced cardiac function benefit in response to empagliflozin treatment.
Reduced renal blood flow due to decreased cardiac output and increased venous congestion is probably the primary mediator of GFR decline and loss of renal mass in HF. However, it remains to be determined whether SGLT2 inhibitors are capable of improving renal perfusion in the setting of nondiabetic cardiac dysfunction. Interestingly, treatment with the SGLT2 inhibitor luseogliflozin prevented endothelial rarefaction, renal hypoxia, and the development of renal fibrosis after renal ischemia-reperfusion in mice through a vascular endothelial growth factor–dependent pathway.32 Furthermore, Pirklbauer et al. 33 provided evidence that SGLT2 inhibition blocks the expression of key mediators of renal fibrosis and kidney disease progression in two distinct lines of human PT cells. Therefore, it could be speculated that the renoprotective effects of SGLT2 in nondiabetic HF are mediated not only by improvements in renal hemodynamics but also, by local actions at the level of PT cells.
The upregulation of NHE3 expression and renal PT activity has been implicated in the pathogenesis of HF, which includes sodium retention, volume overload, and peripheral edema.34 Consistent with previous findings from our group,35 we observed increased NHE3 activity, protein abundance, and mRNA levels and decreased PS552-NHE3 in the PT of HF rats. Importantly, in this report, we show for the first time that a selective SGLT2 inhibitor suppressed the activity of PT NHE3. Empagliflozin treatment prominently reduced PT NHE3 function in both HF and sham rats; however, NHE3 inhibition was more pronounced in HF rats. Empagliflozin-induced NHE3 inhibition did not affect NHE3 protein abundance or NHE3 mRNA levels in either sham or HF rats. In contrast, a small but significant increase in PS552-NHE3 levels was observed in empagliflozin-treated HF rats compared with untreated HF rats. As no difference was found in the levels of PS552-NHE3 between untreated and empagliflozin-treated sham rats, one may speculate that the effect of empagliflozin on PS552-NHE3 in HF rats might be secondary to the attenuation of the maladaptive neurohormonal response in HF. Indeed, both activation of the renal sympathetic nervous system and angiotensin II are known to activate NHE3, at least partly due to a decrease in PS552-NHE3.36,37 Nevertheless, the principal mechanism by which SGLT2 inhibition downregulates PT NHE3 activity remains unknown.
SGLT2 expression and activity appear to be upregulated in PT cells of patients with T2D as a result of a maladaptive regulatory mechanism that contributes to the maintenance of hyperglycemia.25,26 Here, we report the unprecedented finding that the expression of SGLT2 mRNA and protein is increased in nondiabetic HF. The upregulation of SGLT2 expression in nondiabetic HF may involve hyperactivation of the sympathetic nervous system and/or the renal angiotensin system (RAS), as reviewed elsewhere.13 Notably, in vitro studies have suggested that the relationship between SGLT2 and RAS may be bidirectional because increased glucose uptake by PT leads to RAS activation.38,39 The vicious cycle of SGLT2 upregulation and intrarenal RAS hyperactivation may aggravate extracellular fluid volume disorders and therefore, contribute to HF progression. Likewise, gliflozins may break this vicious cycle by lowering PT glucose uptake and decreasing RAS activation, thereby mitigating the potential stimuli for SGLT2 overexpression.
In summary, our findings demonstrated that empagliflozin confers therapeutic benefits in nondiabetic HF rats by restoring the euvolemic status, most likely due to the preservation of GFR and the inhibition of NHE3-mediated sodium reabsorption. Furthermore, our study provides novel evidence that SGLT2 overexpression may constitute a mechanism for PT dysfunction implicated in HF syndrome development and progression. Aligned with recent clinical data showing that dapagliflozin and empagliflozin confer cardioprotection in nondiabetic patients with HF,6,7 our report supports the recommendation of SGLT2 inhibitors in future treatment paradigms for HF management, independent of diabetes status.
Disclosures
A.C.C. Girardi reports scientific advisor or membership with American Journal of Physiology Cell Physiology, Current Research in Physiology, and Frontiers in Physiology. All remaining authors have nothing to disclose.
Funding
This work was supported by Fundação de Amparo à Pesquisa do Estado de São Paulo Grant 2016/22140-7, by Conselho Nacional de Desenvolvimento Científico e Tecnológico Grant 307156/2018-4, and by Coordenação de Aperfeiçoamento de Pessoal de Nível Superior Finance Code 001.
Supplementary Material
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
Supplemental Material
This article contains the following supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2020071029/-/DCSupplemental.
Supplemental Table 1. Values for LV end diastolic area and LV end systolic area.
Supplemental Table 2. Sequences of oligonucleotides used in this study.
Supplemental Figure 1. CONSORT-style diagram.
Supplemental Figure 2. Schematic timeline of the study.
Supplemental Figure 3. Evaluation of BNP and FAC in sham and HF rats before randomization.
Supplemental Figure 4. Evaluation of food and water intake.
Supplemental Material. Full unedited gels for Figures 3 and 7.
References
- 1.Ghezzi C, Yu AS, Hirayama BA, Kepe V, Liu J, Scafoglio C, et al.: Dapagliflozin binds specifically to sodium-glucose cotransporter 2 in the proximal renal tubule. J Am Soc Nephrol 28: 802–810, 2017 10.1681/ASN.2016050510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vallon V, Platt KA, Cunard R, Schroth J, Whaley J, Thomson SC, et al.: SGLT2 mediates glucose reabsorption in the early proximal tubule. J Am Soc Nephrol 22: 104–112, 2011 10.1681/ASN.2010030246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zinman B, Wanner C, Lachin JM, Fitchett D, Bluhmki E, Hantel S, et al.; EMPA-REG OUTCOME Investigators: Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N Engl J Med 373: 2117–2128, 2015 10.1056/NEJMoa1504720 [DOI] [PubMed] [Google Scholar]
- 4.Neal B, Perkovic V, Mahaffey KW, de Zeeuw D, Fulcher G, Erondu N, et al.; CANVAS Program Collaborative Group: Canagliflozin and cardiovascular and renal events in type 2 diabetes. N Engl J Med 377: 644–657, 2017 10.1056/NEJMoa1611925 [DOI] [PubMed] [Google Scholar]
- 5.Wiviott SD, Raz I, Bonaca MP, Mosenzon O, Kato ET, Cahn A, et al.; DECLARE–TIMI 58 Investigators: Dapagliflozin and cardiovascular outcomes in type 2 diabetes. N Engl J Med 380: 347–357, 2019 10.1056/NEJMoa1812389 [DOI] [PubMed] [Google Scholar]
- 6.McMurray JJV, Solomon SD, Inzucchi SE, Køber L, Kosiborod MN, Martinez FA, et al.; DAPA-HF Trial Committees and Investigators: Dapagliflozin in patients with heart failure and reduced ejection fraction. N Engl J Med 381: 1995–2008, 2019 10.1056/NEJMoa1911303 [DOI] [PubMed] [Google Scholar]
- 7.Packer M, Anker SD, Butler J, Filippatos G, Pocock SJ, Carson P, et al.; EMPEROR-Reduced Trial Investigators: Cardiovascular and renal outcomes with empagliflozin in heart failure. N Engl J Med 383: 1413–1424, 2020 10.1056/NEJMoa2022190 [DOI] [PubMed] [Google Scholar]
- 8.Damman K, Testani JM: The kidney in heart failure: An update. Eur Heart J 36: 1437–1444, 2015 10.1093/eurheartj/ehv010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brisco MA, Testani JM: Novel renal biomarkers to assess cardiorenal syndrome. Curr Heart Fail Rep 11: 485–499, 2014 10.1007/s11897-014-0226-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Neuen BL, Young T, Heerspink HJL, Neal B, Perkovic V, Billot L, et al.: SGLT2 inhibitors for the prevention of kidney failure in patients with type 2 diabetes: A systematic review and meta-analysis. Lancet Diabetes Endocrinol 7: 845–854, 2019 10.1016/S2213-8587(19)30256-6 [DOI] [PubMed] [Google Scholar]
- 11.Nespoux J, Vallon V: SGLT2 inhibition and kidney protection. Clin Sci (Lond) 132: 1329–1339, 2018 10.1042/CS20171298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kashihara N, Kidokoro K, Kanda E: Renoprotective effects of sodium-glucose cotransporter-2 inhibitors and underlying mechanisms. Curr Opin Nephrol Hypertens 29: 112–118, 2020 10.1097/MNH.0000000000000561 [DOI] [PubMed] [Google Scholar]
- 13.Silva Dos Santos D, Polidoro JZ, Borges-Júnior FA, Girardi ACC: Cardioprotection conferred by sodium-glucose cotransporter 2 inhibitors: A renal proximal tubule perspective. Am J Physiol Cell Physiol 318: C328–C336, 2020 10.1152/ajpcell.00275.2019 [DOI] [PubMed] [Google Scholar]
- 14.Griffin M, Rao VS, Ivey-Miranda J, Fleming J, Mahoney D, Maulion C, et al.: Empagliflozin in heart failure: Diuretic and cardiorenal effects. Circulation 142: 1028–1039, 2020 10.1161/CIRCULATIONAHA.120.045691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lambers Heerspink HJ, de Zeeuw D, Wie L, Leslie B, List J: Dapagliflozin a glucose-regulating drug with diuretic properties in subjects with type 2 diabetes. Diabetes Obes Metab 15: 853–862, 2013 10.1111/dom.12127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hallow KM, Greasley PJ, Helmlinger G, Chu L, Heerspink HJ, Boulton DW: Evaluation of renal and cardiovascular protection mechanisms of SGLT2 inhibitors: Model-based analysis of clinical data. Am J Physiol Renal Physiol 315: F1295–F1306, 2018 10.1152/ajprenal.00202.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pessoa TD, Campos LC, Carraro-Lacroix L, Girardi AC, Malnic G: Functional role of glucose metabolism, osmotic stress, and sodium-glucose cotransporter isoform-mediated transport on Na+/H+ exchanger isoform 3 activity in the renal proximal tubule. J Am Soc Nephrol 25: 2028–2039, 2014 10.1681/ASN.2013060588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Johns TN, Olson BJ, et al.: Experimental myocardial infarction. I. A method of coronary occlusion in small animals. Ann. Surg 140: 675–682, 1954 10.1097/00000658-195411000-00006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Martins FL, Bailey MA, Girardi ACC: Endogenous activation of glucagon-like peptide-1 receptor contributes to blood pressure control: Role of proximal tubule Na+/H+ exchanger isoform 3, renal angiotensin II, and insulin sensitivity. Hypertension 76: 839–848, 2020 10.1161/HYPERTENSIONAHA.120.14868 [DOI] [PubMed] [Google Scholar]
- 20.Inoue BH, Arruda-Junior DF, Campos LC, Barreto AL, Rodrigues MV, Krieger JE, et al.: Progression of microalbuminuria in SHR is associated with lower expression of critical components of the apical endocytic machinery in the renal proximal tubule. Am J Physiol Renal Physiol 305: F216–F226, 2013 10.1152/ajprenal.00255.2012 [DOI] [PubMed] [Google Scholar]
- 21.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ: Protein measurement with the Folin phenol reagent. J Biol Chem 193: 265–275, 1951 [PubMed] [Google Scholar]
- 22.Kocinsky HS, Girardi AC, Biemesderfer D, Nguyen T, Mentone S, Orlowski J, et al.: Use of phospho-specific antibodies to determine the phosphorylation of endogenous Na+/H+ exchanger NHE3 at PKA consensus sites. Am J Physiol Renal Physiol 289: F249–F258, 2005 10.1152/ajprenal.00082.2004 [DOI] [PubMed] [Google Scholar]
- 23.Pfaffl MW, Tichopad A, Prgomet C, Neuvians TP: Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: BestKeeper--Excel-based tool using pair-wise correlations. Biotechnol Lett 26: 509–515, 2004 10.1023/b:bile.0000019559.84305.47 [DOI] [PubMed] [Google Scholar]
- 24.Vallon V, Schwark JR, Richter K, Hropot M: Role of Na(+)/H(+) exchanger NHE3 in nephron function: Micropuncture studies with S3226, an inhibitor of NHE3. Am J Physiol Renal Physiol 278: F375–F379, 2000 10.1152/ajprenal.2000.278.3.F375 [DOI] [PubMed] [Google Scholar]
- 25.Wang XX, Levi J, Luo Y, Myakala K, Herman-Edelstein M, Qiu L, et al.: SGLT2 protein expression is increased in human diabetic nephropathy: SGLT2 protein inhibition decreases renal lipid accumulation, inflammation, and the development of nephropathy in diabetic mice. J Biol Chem 292: 5335–5348, 2017 10.1074/jbc.M117.779520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rahmoune H, Thompson PW, Ward JM, Smith CD, Hong G, Brown J: Glucose transporters in human renal proximal tubular cells isolated from the urine of patients with non-insulin-dependent diabetes. Diabetes 54: 3427–3434, 2005 10.2337/diabetes.54.12.3427 [DOI] [PubMed] [Google Scholar]
- 27.Veiras LC, Girardi ACC, Curry J, Pei L, Ralph DL, Tran A, et al.: Sexual dimorphic pattern of renal transporters and electrolyte homeostasis. J Am Soc Nephrol 28: 3504–3517, 2017 10.1681/ASN.2017030295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Crajoinas RO, Lessa LMA, Carraro-Lacroix LR, Davel APC, Pacheco BPM, Rossoni LV, et al.: Posttranslational mechanisms associated with reduced NHE3 activity in adult vs. young prehypertensive SHR. Am J Physiol Renal Physiol 299: F872–F881, 2010 10.1152/ajprenal.00654.2009 [DOI] [PubMed] [Google Scholar]
- 29.Mullens W, Verbrugge FH, Nijst P, Tang WHW: Renal sodium avidity in heart failure: From pathophysiology to treatment strategies. Eur Heart J 38: 1872–1882, 2017 10.1093/eurheartj/ehx035 [DOI] [PubMed] [Google Scholar]
- 30.Guazzi M, Naeije R: Pulmonary hypertension in heart failure: Pathophysiology, pathobiology, and emerging clinical perspectives. J Am Coll Cardiol 69: 1718–1734, 2017 10.1016/j.jacc.2017.01.051 [DOI] [PubMed] [Google Scholar]
- 31.Chowdhury B, Luu AZ, Luu VZ, Kabir MG, Pan Y, Teoh H, et al.: The SGLT2 inhibitor empagliflozin reduces mortality and prevents progression in experimental pulmonary hypertension. Biochem Biophys Res Commun 524: 50–56, 2020 10.1016/j.bbrc.2020.01.015 [DOI] [PubMed] [Google Scholar]
- 32.Zhang Y, Nakano D, Guan Y, Hitomi H, Uemura A, Masaki T, et al.: A sodium-glucose cotransporter 2 inhibitor attenuates renal capillary injury and fibrosis by a vascular endothelial growth factor-dependent pathway after renal injury in mice. Kidney Int 94: 524–535, 2018 10.1016/j.kint.2018.05.002 [DOI] [PubMed] [Google Scholar]
- 33.Pirklbauer M, Schupart R, Fuchs L, Staudinger P, Corazza U, Sallaberger S, et al.: Unraveling reno-protective effects of SGLT2 inhibition in human proximal tubular cells. Am J Physiol Renal Physiol 316: F449–F462, 2019 10.1152/ajprenal.00431.2018 [DOI] [PubMed] [Google Scholar]
- 34.Packer M: Activation and inhibition of sodium-hydrogen exchanger is a mechanism that links the pathophysiology and treatment of diabetes mellitus with that of heart failure. Circulation 136: 1548–1559, 2017 10.1161/CIRCULATIONAHA.117.030418 [DOI] [PubMed] [Google Scholar]
- 35.Inoue BH, dos Santos L, Pessoa TD, Antonio EL, Pacheco BPM, Savignano FA, et al.: Increased NHE3 abundance and transport activity in renal proximal tubule of rats with heart failure. Am J Physiol Regul Integr Comp Physiol 302: R166–R174, 2012 10.1152/ajpregu.00127.2011 [DOI] [PubMed] [Google Scholar]
- 36.Pontes RB, Crajoinas RO, Nishi EE, Oliveira-Sales EB, Girardi AC, Campos RR, et al.: Renal nerve stimulation leads to the activation of the Na+/H+ exchanger isoform 3 via angiotensin II type I receptor. Am J Physiol Renal Physiol 308: F848–F856, 2015 10.1152/ajprenal.00515.2014 [DOI] [PubMed] [Google Scholar]
- 37.Crajoinas RO, Polidoro JZ, Carneiro de Morais CP, Castelo-Branco RC, Girardi AC: Angiotensin II counteracts the effects of cAMP/PKA on NHE3 activity and phosphorylation in proximal tubule cells. Am J Physiol Cell Physiol 311: C768–C776, 2016 10.1152/ajpcell.00191.2016 [DOI] [PubMed] [Google Scholar]
- 38.Zhang SL, Filep JG, Hohman TC, Tang SS, Ingelfinger JR, Chan JS: Molecular mechanisms of glucose action on angiotensinogen gene expression in rat proximal tubular cells. Kidney Int 55: 454–464, 1999 10.1046/j.1523-1755.1999.00271.x [DOI] [PubMed] [Google Scholar]
- 39.Wang J, Shibayama Y, Kobori H, Liu Y, Kobara H, Masaki T, et al.: High glucose augments angiotensinogen in human renal proximal tubular cells through hepatocyte nuclear factor-5. PLoS One 12: e0185600, 2017 10.1371/journal.pone.0185600 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.