

Significance Statement
Whole-genome sequencing of 320 individuals with nephrotic syndrome (NS) of unclear genetic etiology and data from several independent patient cohorts provided insight into the genetic architecture of the condition. The strategy identified a disease-causing autosomal dominant mutation in regulator of calcineurin type 1 (RCAN1) that increased cellular calcineurin (CN) activity, NFAT (NF of activated T cells) activation, and susceptibility to apoptosis of podocytes in vitro. Inhibition of an RCAN regulator, GSK-3β, rescued the increased CN activation. Mutations in RCAN1 are a novel cause of NS and reveal a potential target for developing personalized therapy.
Keywords: focal segmental glomerulosclerosis, glomerular disease, genetic renal disease, nephrotic syndrome, tacrolimus, chronic kidney disease, calcineurin inhibitors, calcineurin, podocyte
Visual Abstract

Abstract
Background
Podocyte dysfunction is the main pathologic mechanism driving the development of FSGS and other morphologic types of steroid-resistant nephrotic syndrome (SRNS). Despite significant progress, the genetic causes of most cases of SRNS have yet to be identified.
Methods
Whole-genome sequencing was performed on 320 individuals from 201 families with familial and sporadic NS/FSGS with no pathogenic mutations in any known NS/FSGS genes.
Results
Two variants in the gene encoding regulator of calcineurin type 1 (RCAN1) segregate with disease in two families with autosomal dominant FSGS/SRNS. In vitro, loss of RCAN1 reduced human podocyte viability due to increased calcineurin activity. Cells expressing mutant RCAN1 displayed increased calcineurin activity and NFAT activation that resulted in increased susceptibility to apoptosis compared with wild-type RCAN1. Treatment with GSK-3 inhibitors ameliorated this elevated calcineurin activity, suggesting the mutation alters the balance of RCAN1 regulation by GSK-3β, resulting in dysregulated calcineurin activity and apoptosis.
Conclusions
These data suggest mutations in RCAN1 can cause autosomal dominant FSGS. Despite the widespread use of calcineurin inhibitors in the treatment of NS, genetic mutations in a direct regulator of calcineurin have not been implicated in the etiology of NS/FSGS before this report. The findings highlight the therapeutic potential of targeting RCAN1 regulatory molecules, such as GSK-3β, in the treatment of FSGS.
Glomerular diseases, including diabetic nephropathy, are the primary known cause of CKD in the United States and the rest of the world.1,2 Most glomerular diseases are due to primary dysfunction or secondary injury to the podocyte, the visceral epithelial cell of the trilayer glomerular filtration barrier. Primary podocyte dysfunction, referred to as podocytopathy, typically manifests as steroid-resistant nephrotic syndrome (SRNS) with morphologic changes of FSGS or minimal change disease apparent on kidney biopsy specimens.3,4
It is estimated that 5%–30% of all podocytopathies are due to mutation in single genes, especially in children and young adults.5–7 More than 60 genes have been identified as causes of monogenic SRNS; however, these genes are responsible for only 20% of all genetic SRNS, suggesting there are other, unidentified, single-gene causes of SRNS.6,8–11 Identification of these causal genes has the potential to improve our understanding of disease pathogenesis, the identification of disease biomarkers, the identification of new therapeutic agents, and the repurposing of existing agents to treat nephrotic syndrome (NS).
To identify new, single-gene causes of SRNS, we carried out whole-genome sequencing (WGS) on 320 individuals from 201 families with familial and sporadic NS, and reviewed whole-exome sequencing data from patients with NS of unclear genetic etiology. We identified two segregating, heterozygous mutations in the regulator of calcineurin (CN) type 1 (RCAN1) in two large Northern European families. There are three genes in the RCAN family RCAN1–3, all of which encode proteins capable of interacting with CN and inhibiting CN-dependent signaling pathways.12–22 Therefore, we screened families with hereditary and sporadic NS in other independent cohorts for rare variants in RCAN1–3 genes. We identified four possible disease-causing variants: three in RCAN2, and one in RCAN3.
The RCAN family of proteins form a complex with the catalytic subunit of CN, and regulate both CN phosphatase activity and its ability to bind key substrates like NF of activated T cells (NFAT).12–14,16–18,20,23–27 Unregulated CN activation is central to the pathogenesis of multiple glomerular disease processes, and CN inhibitors (CNIs) are often used to treat glomerular diseases.28–36 The rationale for treating acquired forms of NS with CNIs has historically been that the immune system was thought to play a significant role in acquired forms of NS, such as minimal change disease and FSGS.28,29 However, CN has nonimmunologic actions that are important in the pathogenesis of kidney diseases. For example, CN causes cytoskeletal instability by dephosphorylating synaptopodin and promoting its degradation.28–31,33 Moreover, podocyte loss plays a key role in pathogenesis of FSGS, and CN promotes a decrease in the number of glomerular podocytes by both genetic and nongenetic mechanisms.28–31,34,37,38
In this study, we discovered that a disease-causing RCAN1 variant in individuals with FSGS had a reduced ability to inhibit activated CN compared with wild-type (WT) RCAN1. The increase in CN activation induced by the RCAN1 variant was inhibited by treatment with antagonists of glycogen synthase kinase 3 (GSK-3). In addition, cells expressing this RCAN1 variant were more sensitive to apoptotic stimuli, which could be rescued by CNI treatment. Collectively, our findings suggest mutations in RCAN1 are a novel genetic cause of NS, and use of CNIs and GSK antagonists may represent targeted or personalized therapy for individuals with NS/FSGS due to RCAN1 mutations.
Methods
WGS
WGS was performed at GENEWIZ (South Plainfield, NJ). Briefly, genomic DNA samples were assessed for purity, quantity, and quality by using the NanoDrop 2000 Spectrophotometer (Thermo Fisher), Qubit 2.0 Fluorometer, Qubit dsDNA HS Assay Kit (Thermo Fisher), and agarose gel electrophoresis. Library construction was then performed using Illumina’s TruSeq DNA PCR-Free library preparation kit following the manufacturer’s protocol. Genomic DNA was fragmented by acoustic shearing with a Covaris S220 instrument. Sheared DNA was then end repaired and A-tailed, followed by adaptor ligation. Final libraries were analyzed on the Agilent TapeStation, for library sizing, and quantified using the Qubit dsDNA HS Assay Kit along with the KAPA Library Quantification Kit for quantitative PCR. DNA libraries were sequenced using Illumina platforms to generate ≥120 Gb of raw data per sample, with a 2×150-bp, paired-end sequencing configuration.
Variant Calling and Annotation
DNA-sequencing data were processed using fastp1 to trim low-quality bases and Illumina sequencing adapters from the 3′ end of reads.39 Reads were then aligned to the GRCh37 version of the human genome with the BWA2 algorithm.40 PCR duplicates were flagged using the PICARD Tools3 software suite.41 Alignment processing and variant calling were performed using the GATK4 toolkit following the Broad Institute’s Best Practices Workflow.42,43 Functional consequences and genotype provenances of variants were annotated using Ensembl Variant Predictor.44 After annotation, variants meeting the following criteria were selected for further analysis: having a “pass” status after GATK’s Variant Quality Score Recalibration, found to reside in a coding region, and had an allele frequency of <5% in at least one population of the Genome Aggregation Database (gnomAD).45 Second-level filtering to identify disease-causing variants is as shown in Supplemental Figure 1. Variants of interest were confirmed by Sanger sequencing.
RCAN1 Knockdown Podocytes
Multiple, conditionally immortalized, human podocyte lines (courtesy of Dr. Jeffrey Kopp) with reduced RCAN1 expression were created using lentiviral transduction of short hairpin RNA (shRNA) against RCAN1 (TRCN0000019848; Millipore Sigma). Lentiviral control lines were created using shRNA with no known target (SHC016V; Millipore Sigma). Podocyte lentiviral transduction was performed as described previously.46 RCAN1 KD was confirmed through immunoblotting (LS-C162511; LifeSpan Biosciences).
Immunoprecipitation Studies
Immunoprecipitation was performed using a protocol modified from Fuentes et al. 13 For the studies, human embryonic kidney cells (HEK293) cells were grown in DMEM supplemented with 10% FCS, penicillin (100 U/ml), and streptomycin (100 µg/ml) (all from Gibco, Gaithersburg, MD), as previously described.47 For transfection, HEK293 cells were plated in six-well Costar tissue culture plates (Corning, Corning, NY) and grown to approximately 80% confluency. Cells were then cotransfected with the FLAG-tagged CN construct (GenScript, Piscataway, NJ) and the Myc-tagged RCAN1 construct (WT or mutant as indicated; GenScript) using Lipofectamine 2000, according to the manufacturer’s recommendations (ThermoFisher Scientific, Waltham, MA). Cells were harvested 48 hours after transfection, and cell pellets were lysed in ice-cold 50 mM Tris-hydrochloride (pH 7.5), 150 mM sodium chloride, and 1% nonidet P-40 in the presence of protease inhibitors (Protease Inhibitor Cocktail; Sigma-Aldrich, St. Louis, MO). For immunoprecipitation of RCAN1 constructs, protein lysates were incubated with anti-Myc antibodies (ThermoFisher Scientific) at 4°C for 1 hour, and then Protein A Plus Protein G (Millipore, Bedford, MA) was added to the lysate and rocked for approximately 4 hours at 4°C. For immunoprecipitation of CN constructs, protein lysates were incubated with anti-FLAG antibodies linked to sepharose beads (Cell Signaling Technology, Danvers, MA) and rocked for approximately 4 hours at 4°C. After three washes with ice-cold lysis buffer, Laemmli sample buffer was added to the pellet and boiled for approximately 10 minutes, and immunoblotting was then performed. This experiment was repeated in triplicate.
CN Activity Assay
CN activity was examined using the Cellular Calcineurin Activity assay kit according to the manufacturer’s protocol (Enzo Life Sciences, Farmingdale, NY). Conditionally immortalized podocytes were grown on collagen-coated, six-well dishes until confluent. HEK293 cells were grown in six-well dishes and transfected with PPP3CA and RCAN1 constructs using Lipofectamine 2000, as described above, for 48 hours. Immunoblotting was performed to ensure equal levels of PPP3CA and RCAN1 transfection between HEK293 cell samples. All experimental cells were washed with Tris-buffered saline before lysates were harvested and cleared of free nucleotides using a desalting column. Phosphate standards were loaded in duplicate on a 96-well plate. Calmodulin-supplemented buffer was added to appropriate wells for measuring the background and total phosphatase levels. EGTA-supplemented buffer was added to additional wells for each sample to measure the non-CN phosphatase activity. All sample wells received water and CN substrate (RII phosphopeptide), except for background wells in which water was substituted for substrate. A positive control (CN enzyme supplied by the kit) was used to ensure assay effectiveness. The plate was incubated at 30°C for 10 minutes, lysate was added to all sample wells, and then it was incubated at 30°C for 30 minutes. BioMol Green reagent (100 µl) was then added to all wells and incubated for 25 minutes at room temperature before reading the OD620nm using a Tecan (Männedorf, Switzerland) Infinite 200 microplate reader, with two reads per well. Background well readings were subtracted from all experimental well readings, and moles of phosphate were calculated using the phosphate standard curve. The CN-specific phosphatase activity was calculated by subtracting the phosphate present in EGTA-treated cells (non–CN-related phosphatase activity) from calmodulin-treated cells (total phosphatase activity). All experiments were repeated in triplicate.
GSK-3 Inhibition
We diluted LY2090314 (Selleck Chemicals, Houston, TX) and tideglusib (Selleck Chemicals) in water (1:2000) from DMSO stock (10 mM and 2 mM, respectively) and replaced 2 µl of water in the CN activity assay described above with an equal volume of diluted GSK inhibitors to reach a final concentrations of 200 nM LY2090314 and 1 µM tideglusib. Untreated samples wells received 2 µl of 1:2000 diluted DMSO. To accommodate the additional sample conditions for these inhibition studies, we used cell lysate(SPACE)te diluted 1:1 in lysis buffer. CN activity was calculated by subtracting the phosphate activity in EGTA-treated wells from the activity in treated or untreated wells. This experiment was repeated in triplicate.
NFAT Luciferase Assay
The NFAT luciferase assay was performed using the Dual Firefly and Renilla Luciferase Assay Kit (Biotium, Freemont, CA), according to the manufacturer’s protocol. Briefly, HEK293 cells were grown in 24-well plates and transfected with Lipofectamine 2000 according to the manufacturer’s protocol. Cells were transfected with equal parts of an NFAT-luciferase reporter construct (Promega), loading control construct (pRL-TSK; Promega), PPP3CA, and one of the RCAN1 constructs (0.6 ng DNA per construct per well). Lysates were harvested using the supplied lysis buffer after 48 hours and plated in duplicate on 96-well plates. Measurements were taken using an Infinite Pro 200 microplate reader with automated injection (Tecan), which injected 100 µl of luciferase assay reagent into each well, recorded the fluorescence, added 100 µl of Renilla assay reagent, and performed another fluorescence reading. The relative luminescence units for each well were then calculated by dividing the luciferase reagent readings by the Renilla reagent readings. The experiment was repeated in triplicate.
Automated Cell Apoptosis Imaging
To both visualize and quantify the apoptosis and total cell death, we used a Lionheart FX automated microscope from BioTek along with fluorescent apoptosis reagents. Podocytes were plated and grown to confluency before beginning the assay. HEK293 cells were grown in 96-well plates, as described above, and transfected with PPP3CA and RCAN1 constructs using Lipofectamine LTX, according to the manufacturer’s protocol, for 48 hours before beginning the assay. Cells were exposed to serum-free media containing a 1:500 dilution of NucView Caspase-3 Alexa 488 (Biotium) and a 1:2000 dilution of propidium iodide (Sigma-Aldrich). The NucView reagent consists of a substrate of caspase-3 that emits green fluorescence when cleaved, whereas propidium iodide fluoresces in late apoptotic and necrotic cells. These media also contained either 1 μM FK506 or an equal concentration of vehicle (ethanol). Bright-field images, along with green and red fluorescent images, were collected every 2 hours for 48 hours. Using automated GEN5 software from BioTek, the images were processed to remove background, and the number of fluorescent cells was quantified for each well using label-free cell counting. Wells containing full serum were used as a control to test the validity of the apoptosis readings. The experiment was repeated in quadruplicate with a total N of at least 16 for each cell type, and full videos of the representative HEK293 cell images are available in Supplemental Videos 1–3.
Three-Dimensional In Silico Protein Modeling
Molecular graphics and analyses of PDB files created in the I-TASSER software48,49 was performed with UCSF ChimeraX, which was developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco, with support from National Institutes of Health (NIH) (R01-GM129325) and the Office of Cyber Infrastructure and Computational Biology, National Institute of Allergy and Infectious Diseases.50
Illustrations
The summary graphic was created using Biorender.com.
Immunoblotting
Immunoblotting was performed using standard methods and visualized by enhanced chemiluminescence, as previously described.51 Antibodies were used at the following concentrations: 1:500 for caspase-3 (Cell Signaling Technology), 1:1000 for MYC tag (Cell Signaling Technology), 1:1000 for DYKDDDDK tag (Cell Signaling Technologies), and 1:3000 for β-actin (Sigma-Aldrich). For apoptosis experiments, HEK293 cells were grown in six-well plates and transfected with PPP3CA and RCAN1 constructs using Lipofectamine LTX, according to the manufacturer’s protocol. After 48 hours of serum starvation, the cells were washed with PBS, harvested, and the lysates were analyzed with immunoblotting. The experiments were repeated in triplicate and quantified using ImageJ software. Unmodified Western blot images are shown in the Supplemental Materials.
Quantitative Real-Time PCR
Conditionally immortalized, human podocytes (generously provided by Dr. Jeffrey Kopp) were grown until confluent in T75 and differentiated at 37° for 14 days before lysate harvesting, RNA extraction (RNAeasy kit; Qiagen), and generation of cDNA (Promega), as previously described.52 TaqMan probes (Invitrogen) were used in to analyze gene expression for CD2AP (Hs00961451_m1), RCAN1 (Hs01120954_m1), RCAN2 (Hs00195165_m1), RCAN3 (Hs00203728_m1), and PTPRO (Hs00958177_m1). The analysis was repeated in triplicate, with multiple wells per sample in each replicate.
Electron Microscopy of Renal Biopsy Specimens
The harmonic mean of the glomerular basement membrane thickness was calculated using multiple measurements, using reference ranges for men and women as reported by Das et al. 53
Statistical Analyses
The two-tailed t test was used for the comparison of RCAN1-KD podocyte immunoblotting against RCAN1 (t=9.802; degrees of freedom [df]=6) and cleaved caspase-3 (t=4.764; df=6). Two-way ANOVA, followed by a Dunnett multiple comparison analysis, was used to analyze automated live imaging of RCAN1-KD podocytes (df=2; F=7.698). One-way ANOVA, followed by a Tukey multiple comparisons test, was used to determine the differences between means for the analysis of RCAN1-variant apoptosis immunoblotting results (df=3; F=56.94). A two-tailed t test analysis was used for the variant NFAT luciferase assay (t=11.84; df=10) and the CN activity assay (t=6.579; df=22). Two-way ANOVA, followed by a Dunnett multiple comparisons analysis, was used to compare groups for RCAN1-variant automated live-cell apoptosis imaging (df=3; F=10.92) and GSK-3 inhibition experiments (df=2; F=27.03).
Results
Clinical Ascertainment and WGS
We identified 320 individuals from 201 families with familial and sporadic NS/FSGS with no pathogenic mutations in any known NS/FSGS genes. We performed WGS and filtered variants using previously published algorithms for the identification of causal variants in families with Mendelian kidney disease (Supplemental Figure 1). We identified four variants in four genes that are present in all affected individuals (Supplemental Table 1) in a large Irish family recruited as part of our collaboration with the Irish Kidney Gene Project (Figure 1).54,55 One of these variants was in RCAN1 (c. T485C, p.I162T; transcript, ENST00000313806.4; GRCh37). A search for pathogenic variants in these four genes in existing whole-exome sequencing data from 191 families with NS of unclear etiology identified a second family with another segregating variant in RCAN1 (Supplemental Figure 2), but did not find pathogenic variants in the other three candidate genes. The second variant, c.A382G, p.K128E, is rare, with a minor allele frequency of 0.0004172 in the gnomAD database, and both RCAN1 variants are conserved in evolution (Supplemental Table 2).
Figure 1.
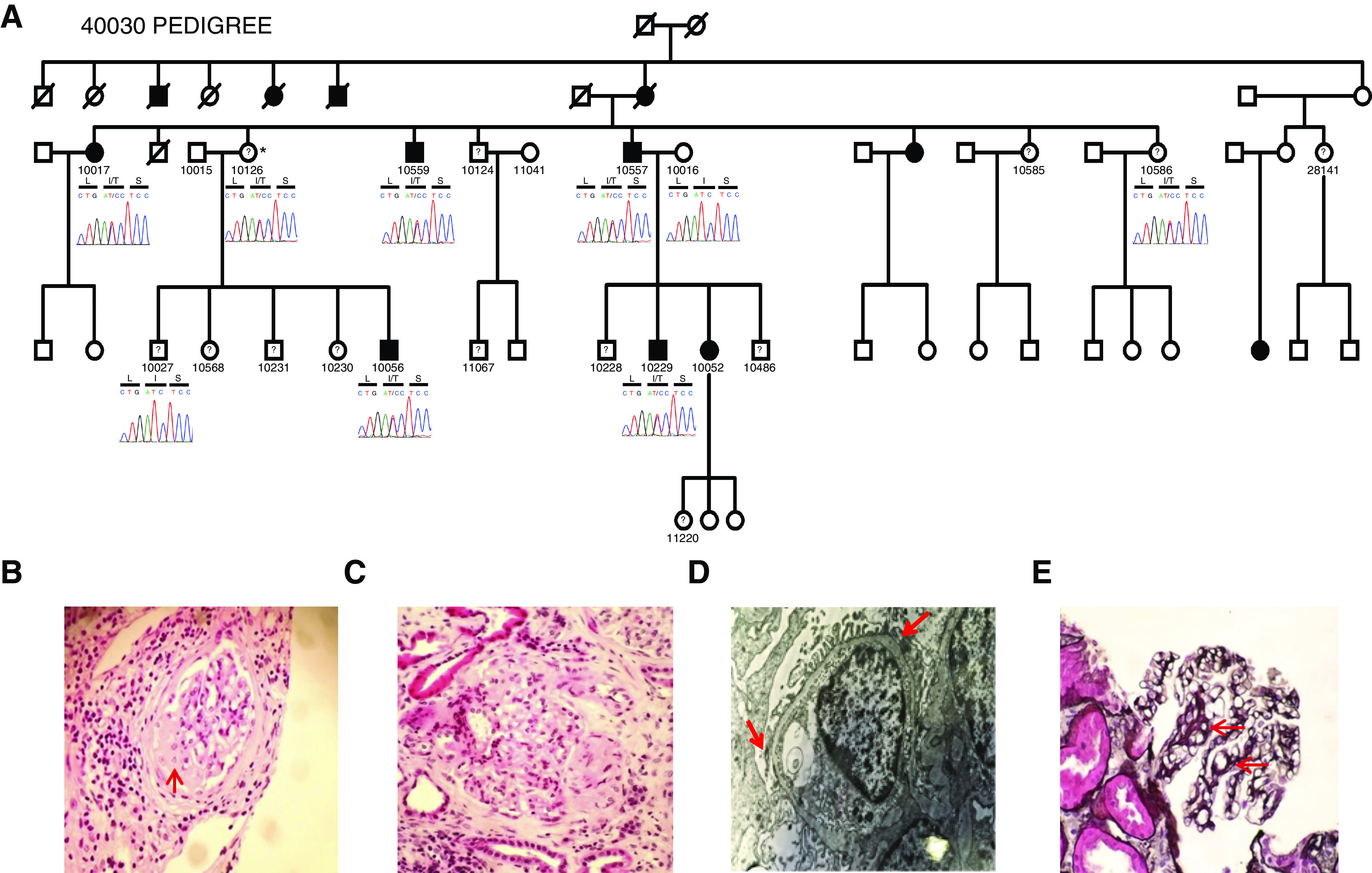
The RCAN1 p.I162T mutation segegrates with disesase in a family with FSGS. (A) Pedigree of European family 40030 with FSGS and the RCAN1 I162T variant segregating with the disease in the family. Family members that are currently unnaffected but may develop disease later in life are depicted with a question mark. Sequenced individuals are shown with a chromatogram and associated amino acid sequence (L=Leucine, I= Isoleucine, T= Threonine, S=Serine). Asterisk indicates obligate carrier. (B–E) Kidney histology from individual 10557 in family 40030. (B and C) FSGS on hematoxylin and eosin staining at (red arrow). (D) Mild foot process effacement (red arrows) and thinned glomerular basement membrane. (E) Capillary loop double contour formation (red arrows) on silver staining. Original magnification, ×20 in (B), ×40 in (C) and (D).
In Silico Modeling
In silico modeling revealed the p.I162T RCAN1 variant is predicted to be damaging by at least three prediction tools, with a Combined Annotation Dependent Deletion score >20 (Table 1). Modeling the mutation in the three-dimensional structure of RCAN1, using I-TASSER and ChimeraX software, revealed that this mutation is predicted to disrupt both the amino and carboxy termini of the protein involved in CN binding, and the highly conserved serine-proline (SP) motif, which is required for regulation of CN activity (Figure 2, Supplemental Videos 1 and 2).48,49,56 The SP motif also contains GSK-3 and other kinase phosphorylation sites that can alter the activity of RCAN1.19,26,57,58
Table 1.
In silico prediction of disease-causing RCAN1 heterozygous variant
| Variant | gnomAD (EUR) | MAF (EUR) | MAF (all population) | CADD | PolyPhen | SIFT | MutationTaster |
|---|---|---|---|---|---|---|---|
| RCAN1 c. T485C, p. I162T | 2 of 128,738 | 0.00001 | 0.000007 | 26.7 | Probably damaging | Damaging | Disease causing |
GRCH37: RCAN1-001, transcript ENST00000313806.4. EUR, European; MAF, minor allele frequency; CADD, combined annotation dependent depletion; SIFT, sorting intolerant from tolerant.
Figure 2.

Potentially pathogenic variants disrupt conserved RCAN protein domains. (A) Depictions of RCAN1, RCAN2, and RCAN3 peptides showing the conserved protein domains within the RCAN1 family of proteins and locations (arrows) of the newly identified variants. The variants identified in these patients are all located in or near conserved domains, including the RNA recognition motif (RRM; RNA binding–like) domain (blue-green); the carboxy-terminal CN binding motifs (orange and red); and the SP motif (green-yellow), which contains the LxxP, FLISPPxSPP, and ExxxP sequences. (B) Three-dimensional modeling of WT RCAN1 and the p.I162T variant revealed disruption of the amino- and carboxy-terminal regions (blue and red, respectively), and the region around the SP motif (green), when compared with WT RCAN1.
Phenotype of Individuals with RCAN1 Mutations
The phenotypes of the eight affected individuals from the two families with RCAN1 variants are displayed in Table 2. Affected individuals presented between the ages of 5 and 65 years. Six individuals had proteinuria at presentation, four of which presented with nephrotic-range proteinuria. However, one of the two individuals with unknown proteinuria at presentation ultimately progressed to ESKD. Biopsy specimen reports were available for three individuals, and all had FSGS on light-microscopy results and podocyte foot process effacement on electron-microscopy results (Figure 1). Three patients had undergone a kidney transplant, follow-up data are available in two of these individuals, and none of these patients developed recurrence of primary disease after the kidney transplant. Two individuals from the index family carry an RCAN1 variant but did not present with kidney disease at the time of ascertainment and follow-up, whereas one individual in the second family also carries the second RCAN1 variant without any evidence of kidney disease (Table 2). Our data suggest an autosomal dominant pattern of inheritance with incomplete penetrance.
Table 2.
Phenotype of individuals with RCAN1 mutations
| Family Number | Individual Number | Age at Onset (yr) | Proteinuria (quantity) | Biopsy (histology) | CKD Stage | Transplant | Recurrence |
|---|---|---|---|---|---|---|---|
| 40030 | 10056 | 29 | UNK | N | 5 | Y | N |
| 40030a | 10126 | NA | N | N | 0 | N | NA |
| 40030 | 10559 | 48 | Y | Y (UNK) | 5 | Y | UNK |
| 40030 | 10552 | 35 | Y (825 mg) | Y (FSGS) | 5 | N | NA |
| 40030 | 10557 | 65 | Y (5000 mg) | Y (FSGS) | 5 | N | NA |
| 40030a | 10586 | NA | N | N | 0 | N | NA |
| 40030 | 10017 | UNK | UNK | UNK | UNK | UNK | UNK |
| 40030 | 10229 | 45 | Y (>3000 mg) | N | 1 | N | NA |
| 6559 | 1 | 5 | Y (1860 mg) | Y (FSGS) | 5 | Y | N |
| 6559 | 101 | 40 | Y (2410 mg) | UNK | 1 | N | NA |
| 6559a | 0106 | NA | N | N | 0 | N | NA |
UNK, unknown; N, no; Y, yes; NA, not available.
Asymptomatic.
Sequencing RCAN1, RCAN2, and RCAN3 in Independent Cohorts
There are three genes in the RCAN family of genes, RCAN1 (chromosome 21q 22.11), RCAN2 (chromosome 6p12.3), and RCAN3 (chromosome 1p 36.11).13,21,22 Because all of the proteins encoded by RCAN genes interact with CN and modulate CN-dependent signaling pathways, and to provide supportive evidence for the pathogenicity of RCAN1 variants, we screened for variants in RCAN1–3 in additional cohorts of patients with NS/FSGS. We screened patients for rare variants in the three genes in five other independent cohorts (cohorts from the Boston Children’s Hospital and Beth Israel Hospital Boston, the Nephrotic Syndrome Study Network Consortium [NEPTUNE] cohort, UK NephroS cohort, and a cohort from the Toronto General Hospital) (Supplemental Table 3). We identified four potentially pathogenic variants in RCAN2 and RCAN3 (Table 3). The RCAN3 variant and one of the three RCAN2 variants are novel and they are not found in the gnomAD (approximately 250,000 chromosomes analyzed). The other two variants in RCAN2 have minor allele frequency of ≤0.00001 in gnomAD. All of the variants are predicted to be damaging by three in silico prediction tools, and they are all conserved in evolution. Other missense variants found in the three genes are listed in Supplemental Table 4.
Table 3.
Rare heterozygous RCAN variants in NS cohorts
| Study Number | Phenotype | Variant | Allele Count | gnomAD MAF (All) | PolyPhen | SIFT | MutationTaster | Conservation |
|---|---|---|---|---|---|---|---|---|
| 159 | SRNS, MCD | RCAN2 | 0 | 0.000000 | Damaging | Probably damaging | Disease causing | Zebrafish |
| c.C445A | ||||||||
| p.P149T | ||||||||
| 260 | SRNS, FSGS | RCAN2 | 2 of 248,944 | 0.000008 | Damaging | Probably damaging | Disease causing | Frog |
| c.A728C | ||||||||
| p.N243H | ||||||||
| 3 | NS | RCAN2 | 3 of 249,306 | 0.00001 | Damaging | Probably damaging | Disease causing | Zebrafish |
| c.C700T | ||||||||
| p.R234H | ||||||||
| 459 | SSNS, FR/SD | RCAN3 | 0 | 0.000000 | Tolerated | Probably damaging | Disease causing | Zebrafish |
| c.T517G | ||||||||
| p.C173G |
GRCH37: RCAN1-001, transcript ENST00000313806.4; RCAN2-002, transcript ENST00000371374.1; RCAN3-001, transcript ENST00000374395.4. MAF, minor allele frequency; MCD, minimal change disease; SSNS, steroid-sensitive NS; FR/SD, frequent relapsing/steroid-dependent; SIFT, sorting intolerant from tolerant.
Loss of RCAN1 Disrupts Podocyte CN Regulation and Decreases Podocyte Viability
To determine the relevance of RCAN1 in the maintenance of podocyte functional integrity, we first confirmed expression in podocytes through quantitative real-time PCR of conditionally immortalized, human podocyte cell lines. RCAN1, RCAN2, and RCAN3 are all expressed in podocytes at comparable levels with key podocyte genes, such as CD2AP and PTPRO (GLEPP1) (Supplemental Figure 3). With the known role of RCAN proteins in CN regulation, we examined the effects of loss-of-function RCAN1 mutations on podocyte CN activity using shRNA-mediated RCAN1 KD in conditionally immortalized podocytes. As expected, podocytes with reduced functional RCAN1 displayed increased CN activity compared with WT controls (Figure 3).
Figure 3.
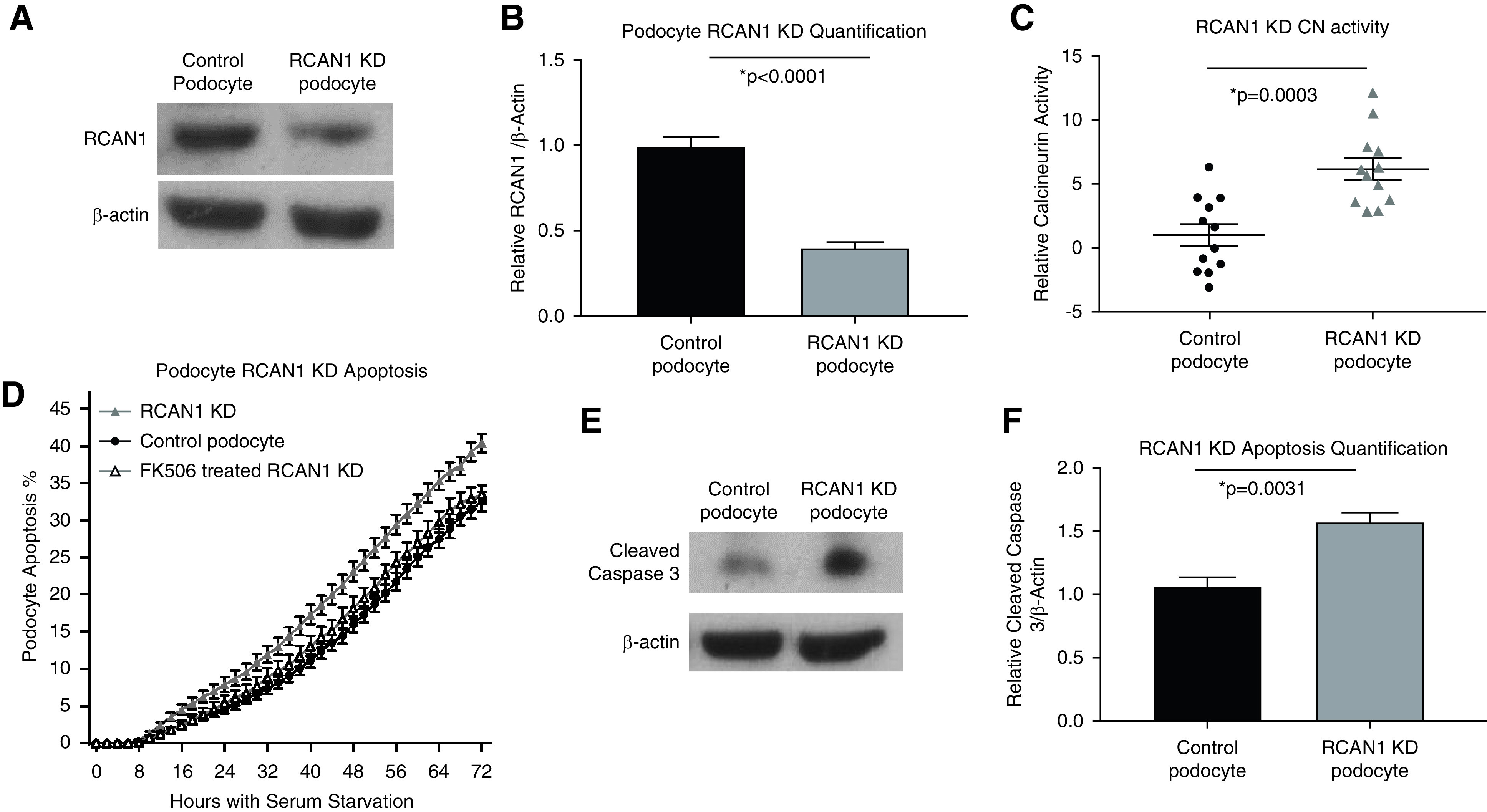
Loss of RCAN1 increases CN activity and reduces viability in human podocytes. (A) Immunoblotting and (B) subsequent quantification revealed a significant reduction in RCAN1 expression in conditionally immortalized podocyte cell lines transduced with shRNA against RCAN1 (RCAN1 KD) when compared with lentiviral control podocytes (P<0.001, n=4, t test). (C) RCAN1 KD podocytes had increased levels of CN activity compared with controls (P=0.0003, n=12, t test). (D) RCAN1 KD podocytes exposed to serum starvation displayed an increased susceptibility to apoptosis, as measured by automated live-cell imaging of a fluorescent reporter of cleaved caspase-3 activity (P<0.01 for all time points after 30 hours, n>16 for each group, two-way ANOVA) and (E and F) immunoblotting (P=0.003, n=4, t test). This apoptosis could be rescued by CNI treatment (1 µM FK506), demonstrating the contributions of altered CN regulation to podocyte viability (P>0.35 for all time points).
Increased CN activity is known to induce podocyte apoptosis both in vitro and in vivo, a key phenotype associated with FSGS.38 To examine the effects of decreased RCAN1-mediated CN regulation on podocyte viability, we examined the susceptibility of podocytes to serum starvation using automated live-cell imaging and quantification of cleaved caspase-3 activity. RCAN1-KD podocytes displayed increased apoptosis compared with WT controls that could be rescued by treatment with CN inhibition (FK506) (Figure 3). These data suggest loss-of-function variants in RCAN1 have the potential to alter podocyte CN regulation and reduce cell viability.
RCAN1 Variants and CN Binding
Regulation of CN by RCAN1 is mediated by direct protein-protein interactions between RCAN1 and the catalytic subunit of the phosphatase.12,13,16–18 To determine if the RCAN1 mutations identified in the affected families altered docking of RCAN1 and CN, we performed immunoprecipitation studies in HEK293 cells expressing PPP3CA (CN catalytic subunit) and WT RCAN1 or the p.I162T variant, as described in the Methods . As shown in Supplemental Figure 4, this RCAN1 mutation did not disrupt the protein-protein interaction between RCAN1 and CN.
RCAN1 p.I162T Enhances CN Activation
To investigate the effects of the p.I162T variant on RCAN1’s function as a regulator of CN activity, we examined the CN activity of HEK293 cells that were transfected with PPP3CA and RCAN1 constructs (WT or p.I162T). Evaluation of these cell lysates using a cellular CN activity assay revealed the p.I162T variants disrupted the regulatory function of RCAN1, resulting in increased CN activity compared with WT RCAN1–expressing cells (P<0.001). An NFAT luciferase assay confirmed this increased CN activity resulted in elevated NFAT activation compared with WT RCAN1–expressing cells (P<0.001) (Figure 4). To verify the effectiveness of our constructs, we also examined CN activity in cells transfected with either PPP3CA or RCAN1 WT alone to ensure CN activation increased and decreased, respectively, in these assays, as compared with untransfected controls (Supplemental Figure 5).
Figure 4.

CN and NFAT activity is increased in RCAN1 mutant cell lines. (A) HEK293 cells were transfected with constructs containing PPP3CA (CN) and either WT RCAN1 or the p.I162T RCAN1 variant. At 48 hours after transfection, the RCAN1 p.I162T variant–expressing cells displayed increased CN activity (P<0.001, n=12, t test) and (B) NFAT expression (P<0.001, n=6, t test) compared with WT RCAN1–expressing cells. For the NFAT luciferase assay, cells were also transfected with constructs to allow for both Firefly and Renilla dual-luminescence detection.
RCAN1 p.I162T Induces Increased Apoptosis that Can Be Rescued by CN Inhibition
Transfected HEK293 cell lines expressing PPP3CA and WT or mutant RCAN1 were exposed to serum starvation–induced apoptosis and evaluated using both automated live-cell imaging and Western blot quantification of caspase-3 activity. Overexpression of RCAN1 p.I162T induced a significant increase in apoptosis and total cell death relative to the WT RCAN1–expressing cells (Figure 5, Supplemental Figures 6 and 7, and Supplemental Videos 3 and 4). Pretreatment with FK506 rescued the increased apoptosis phenotype in the mutant cell lines, confirming the increased apoptosis in the RCAN1 p.I162T–expressing cell lines is due to increased CN activity (Figure 5, Supplemental Video 5).
Figure 5.
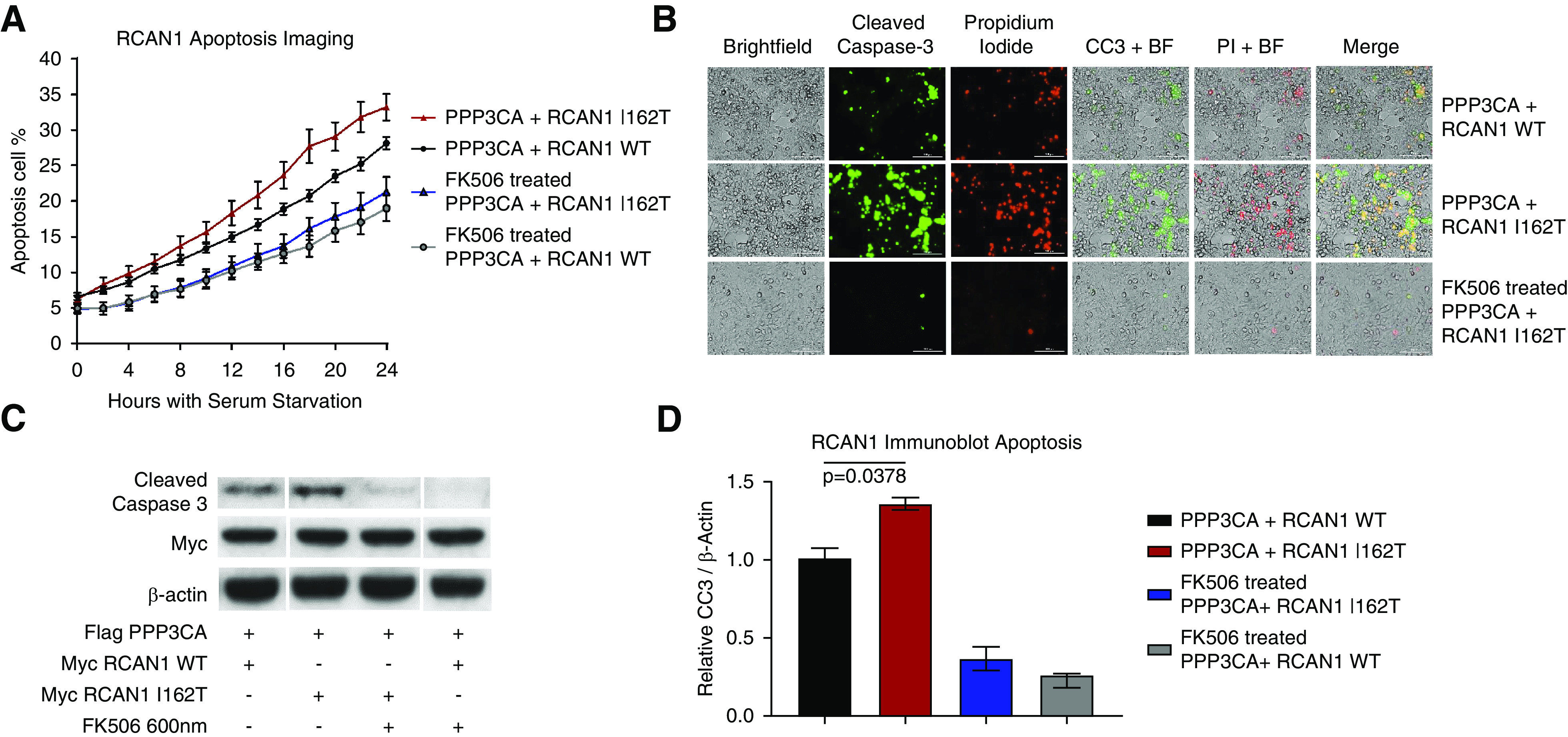
Mutant RCAN1 causes increased apoptosis that can be rescue by CNI FK506. (A) HEK293 cells were transfected with constructs containing PPP3CA (CN) and either WT RCAN1 or the p.I162T variant, and the cells were exposed to serum deprivation. We analyzed the susceptibility to apoptosis using a fluorescent reporter of caspase-3 activity over 24 hours. RCAN1 I162T–expressing cells (red) displayed increased apoptosis compared with WT RCAN1 cells (black) (P<0.02 for all time points between 18 and 24 hours, two-way ANOVA). This increased apoptosis in the RCAN1 mutants was rescued by treatment with 1 µM FK506 (P>0.3 for all time points), demonstrating this aberrant apoptosis in mutant cells is due to the increased CN activity (n>16 for all samples). (B) This increased apoptosis could be seen in still images taken 24 hours after serum starvation, which showed increased apoptosis (green, cleaved caspase-3 [CC3]) and necrosis (red, propidium iodide [PI]) in RCAN1 I162T–expressing cells compared with WT. (C and D) The increased apoptosis and rescue was confirmed through Western blot analysis of cleaved caspase-3 expression after 48 hours of serum starvation (P=0.02, n=3, one-way ANOVA). BF, bright-field imaging.
RCAN1 Mutations Can Cause Disease through Altered GSK-3 Signaling
Having ruled out deficiencies in CN binding as a driving force of increased CN activity in mutant RCAN1–expressing cells, we examined the regulation of RCAN1 activity. A key feature of proteins in the RCAN family is the presence of an SP motif with its “signature” amino acid sequence FLISPPxSPP that begins at amino acid 160 of RCAN1 isoform 1. This highly conserved region of the protein is known to be phosphorylated by GSK-3 kinases, although the consequences of these modifications on RCAN1 protein function and CN activity may be context dependent and have yet to be fully elucidated. The p.I162T variant disrupts this motif directly and is predicted to alter the structure of this region to make the GSK-3β site at serine 163 more accessible for modification (Figure 6, A and B). To determine if GSK-3 activity is a component of the pathogenic CN activation, we examined CN activity of HEK293 cells overexpressing CN and RCAN1 constructs in the presence of a dual GSK-3α/β inhibitor, LY2090314, and GSK-3β–specific inhibitor, tideglusib (Figure 6C). Both of these potent GSK-3 inhibitors were able to correct the aberrant CN activity of RCAN1 p.I162T, suggesting GSK-3β activity likely plays a pivotal role in the pathogenesis of RCAN1-mediated kidney disease.
Figure 6.

Inhibition of GSK-3β can rescue the aberrant CN activation caused by mutant RCAN1. (A) Three-dimensional modeling of the RCAN1 p.I162T variant revealed alterations to the positioning of the serine 163 (S163) residue (gold sphere marker) that is a target of GSK-3 kinase. (B) Analysis of the surface structure of the protein shows that the S163 residue appears to be more accessible for modification in RCAN1 p.I162T compared with WT. (C) HEK293 cells were transfected with constructs containing PPP3CA and either WT RCAN1 or the p.I162T variant, and the lysates were analyzed for CN activity after 48 hours using a CN cellular activity assay. The CN activity is increased in untreated I162T samples (red) compared with WT RCAN1 (P=0.005). The CN activity was restored to WT levels when the lysates were treated with 0.2 µM of the dual GSK-3α/β inhibitor LY2090314 (P=0.67) and 1 µM of the GSK-3β–specific inhibitor tideglusib (P=0.09, n=6 for all samples, one-way ANOVA).
On the basis of the combined data, we propose that genetic variants in RCAN1 can induce glomerular disease due to dysregulated CN activity, which promotes apoptosis in podocytes. Furthermore, the deficiencies in CN regulation caused by RCAN1 p.I162T are likely due to structural changes that affect critical interactions with GSK-3β. These particular RCAN1 mutations disrupt the balance of the feedback cycle of CN regulation by promoting phosphorylation of RCAN1 proteins by GSK-3, ultimately leading to increased CN activation and apoptosis (Figure 7).26
Figure 7.

RCAN1 mutants decrease cell viability by altering the GSK-3β–mediated regulation of CN activity. On the basis of the data acquired in this study, we posit that in WT cells (top), RCAN1 regulates CN activity through a regulatory feedback loop that requires GSK-3β kinase activity. When RCAN1 is phosphorylated (P) by GSK-3β, it either activates CN or dampens RCAN1’s ability to fully inhibit CN, both of which promote cellular apoptosis. The increased phosphatase activity of CN also decreases the levels of phosphorylated RCAN1, which then allows RCAN1 to resume inhibition of CN activity. In cells with mutant RCAN1 (bottom), the structural changes in RCAN1 promote increased phosphorylation by GSK-3β, which shifts the balance of the feedback loop in favor of increased CN activity and, ultimately, apoptosis.
Discussion
In this study, we carried out WGS in a cohort of families with hereditary FSGS/NS that is not due to pathogenic variants in >60 known FSGS/NS genes, and identified a loss-of-function variant in RCAN1 in a family with autosomal dominant FSGS. Despite the widely known deleterious effects of uncontrolled activation of CN in the glomerulus and other compartments of the kidney, this is the first time that mutations in genes encoding a direct regulator of CN will be implicated in the etiology of NS/FSGS.34,35,38 Although RCAN1 is the most plausible candidate based on the genetic data and the function of RCAN1, the role of the other variants that segregated with the FSGS phenotype observed in the leading family still remains unknown.
The gene encoding RCAN1 is located in chromosome 21q22.12, the region classically referred to as the minimal candidate region for the Down syndrome phenotype. RCAN1 has been reported to be overexpressed in the brain of babies with Down syndrome during development and it has been associated with neurofibrillary tangles in patients with Alzheimer disease. RCAN2 and RCAN3, located on chromosome 6p12.3 and 1p33.11, respectively, are also highly expressed in the developing brain and the heart.13,21,22,61,62 Previous studies have reported that RCAN1, RCAN2, and RCAN3 are expressed in the podocyte and other kidney cellular components; however, their role in human kidney disease is unknown.63–65 In mice with doxorubicin-induced nephropathy, a murine model of human FSGS, knockout of RCAN1 increased susceptibility to podocyte injury and albuminuria.63
The regulation of RCAN activity by molecules such as GSK-3β and its subsequent regulation of CN activation are complex, with most studies to date limited to RCAN1. Numerous potential regulators of RCAN1 activity have been identified, including a potential priming phosphorylation by Big MAP kinase 1 at serine 167 in RCAN1-1 (serine 112 in isoform RCAN1-4) that precedes phosphorylation of serine 163 by GSK-3β (serine 108 in isoform RCAN1-4).19,57,58 RCAN1 has also been shown to be a potential facilitator of CN activity when phosphorylated by TGF-β–activated kinase 1 and phosphorylation by NF-κB–inducing kinase can increase RCAN1 stability.66,67 Whereas RCAN1 phosphorylation may activate or repress CN activity, depending on the context, phosphorylated RCAN1 is also a target of CN (Figure 7).68 Furthermore, RCAN1 expression can be altered by the NFAT transcriptional network, providing an additional feedback regulatory mechanism.69,70
Regulation of CN by RCAN1 can either inhibit or activate CN, depending on the context.20,24–26 The highly conserved SP motif with its signature amino acid sequence FLISPPxSPP is able to inhibit CN activation in vitro, although RCAN1 mutants truncated after the SP motif display a lower affinity than the full-length RCAN1 protein.12,16,17,19,26 In vivo, however, the SP motif is not sufficient for inhibition of CN.18,19 In this regard, inhibition of CN by RCAN1 requires: (1) the LxxP motif within the SP domain; and (2) the PxIxIT-like domain, which is also used for docking of many CN substrates.18 Moreover, stimulation of CN signaling requires both the LxxP and Exx(x)P domains within the SP motif, and the highly conserved GSK-3 phosphorylation site within the FLISPPxSPP sequence.18,26 In addition to multiple RCAN family members, each RCAN gene produces multiple splicing variants. Isoform RCAN1-4, in particular, has been highly studied in cardiovascular disease and may also contribute to CN regulation in the kidney.24,71–73
We have shown that rare variants in RCAN1 can cause FSGS through uncontrolled activation of CN. Due to the similar protein functions between RCAN family members, it is reasonable to expect that variants in RCAN2 and RCAN3 are also capable of disrupting CN regulation. With the known roles of CN activation and NFAT signaling in the regulation of immune responses and podocyte cytoskeletal dynamics, CN regulatory molecules make attractive therapeutic targets for kidney disease. CNIs are widely used in the treatment of FSGS and other morphologic forms of NS. However, CNIs are not uniformly effective, for example, only about 30%–50% of patients with SRNS will achieve partial or full remission after CNI treatment.74 There are currently no biomarkers to predict response to CNI therapy, despite major renal and nonrenal toxicities. Our in vitro studies suggest that certain functional variants in RCAN genes may be able to identify patients with SRNS who are likely to respond to CNIs. Unfortunately, none of the patients in the two index RCAN1 families were treated with CNIs; therefore, we do not have human data to corroborate our cell culture findings.
With limited treatment options available for patients with glomerular disease, the identification of new and repurposed pharmaceutical therapies is critical to increasing therapeutic options for these conditions. In this study, we found that GSK-3 inhibitors could reverse the increased CN activity induced by RCAN1 mutations. The GSK-3 inhibitors used in this study, tideglusib and LY2093014, are potent and highly selective small-molecule inhibitors that have both been examined in human phase 2 clinical trials for a variety of diseases. Although GSK-3 activity is an attractive therapeutic target, any potential inhibition in patients would need to be carefully titrated due to the importance of GSK-3 activity for maintaining podocyte viability and kidney function.75 With numerous molecules implicated in the regulation of RCAN1 activity, additional therapeutic targets will likely emerge as the RCAN protein interactome becomes more defined.
In summary, we identified, for the first time, contributions to causality from mutations in RCAN1 in families with autosomal dominant FSGS. We showed that the RCAN1 variant, p.I162T, disrupts the ability of RCAN1 to regulate CN activation, resulting in reduced cell survival that can be rescued by CNIs. In addition, GSK-3 inhibitors can rescue the increased CN activation caused by the RCAN1 mutation. Therefore, the use of CNI and GSK antagonists may represent targeted or personalized therapy for individuals with NS due to RCAN1 mutations.
Disclosures
M. Barua reports having ownership interest in AstraZeneca; serving on the editorial board of Glomerular Diseases; and receiving research funding from Otsuka, Regulus, and Sanofi. K. Benson reports serving as chair of the ClinGen Kidney Cystic and Ciliopathy Disorders Variant Curation Expert Panel. R. Gbadegesin reports receiving research funding from AstraZeneca, Bristol Myers Squibb, and Goldfinch Biotech; and having consultancy agreements with Keryx Pharmaceutical. F. Hildebrandt reports having consultancy agreements with, ownership interest in, and serving as a scientific advisor for or member of Goldfinch Bio as cofounder; and receiving honoraria from Sanofi. S. Murray reports receiving research funding from Amgen. M. Pollak reports having ownership interest in Apolo1Bio; having patents and inventions with Athena Diagnostics; serving on the NephCure Foundation scientific advisory board; having consultancy agreements with, and receiving research funding from, Vertex; and receiving honoraria from various academic talks. Because M. Pollak is an editor of the JASN, he was not involved in the peer review process for this manuscript. A guest editor oversaw the peer review and decision-making process for this manuscript. M. Saleem reports receiving research funding from Evotec, Retrophin, and UCB; having consultancy agreements with Mission Therapeutics, Pfizer, and Retrophin; having ownership interest in Purespring Therapeutics; and receiving honoraria from Purespring Therapeutics as director and chief scientific officer. M. G. Sampson reports having consultancy agreements with Janssen Pharmaceutical, Kohlberg Kravis Roberts & Co.; and serving as a scientific advisor for, or member of, Natera. R. Spurney reports serving as scientific advisor for, or member of, the American Journal of Physiology; and having consultancy agreements with Amgen and Tectonic. Additional funding and/or programmatic support for NEPTUNE has also been provided by the University of Michigan, NephCure Kidney International, and the Halpin Foundation.Additional funding and/or programmatic support for NEPTUNE has also been provided by the University of Michigan, NephCure Kidney International, and the Halpin Foundation. All remaining authors have nothing to disclose.
Funding
R. Gbadegesin is supported by National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) grants 5R01DK098135 and 5R01DK094987, Doris Duke Charitable Foundation Clinical Scientist Development Award 2009033, Borden Scholars Award, and the Duke Health Scholars Award. B. M. Lane is supported by NIDDK Duke Nephrology Award, grant T32-DK007731. A. Bierzynska is funded by Kidney Research UK (personal nonclinical fellowship). M. Barua has received Canadian Institutes of Health Research grant 432980, McLaughlin Accelerator Award (2019), NephCure Kidney International–NEPTUNE Ancillary Studies Grant (2016), and Physicians Services Incorporated Health Research Grant 14-04 (2015); and support from the Can-SOLVE CKD Network (https://www.cansolveckd.ca/) and Toronto General Hospital Foundation. M.G. Sampson is supported by National Institutes of Health (NIH) grants R01-DK108805 and R01-DK119380. NEPTUNE is a part of the NIH Rare Disease Clinical Research Network, supported through a collaboration between the Office of Rare Diseases Research, National Center for Advancing Translational Sciences, and NIDDK, under grant U54‐DK‐083912.
Supplementary Material
Acknowledgments
The authors acknowledge all of the participants in the study. We appreciate the technical support provided by the Duke Molecular Physiology Institute Genomics Core and the personnel of Duke Center for Genomic and Computational Biology. The authors acknowledge Goldfinch Bio for their support with WGS of patient samples. The authors acknowledge the Irish Kidney Gene Project and the Genome England consortium for their collaboration.
The full list of members of NEPTUNE are listed in Supplemental Summary 1.
B.M. Lane, R. Spurney, and R. Gbadegesin designed the experiments and wrote the manuscript; B.M. Lane, R. Gbadegesin, G. Wu, L. Wang, M. Chryst-Stangl, S. Murray, S. Conlon, K. Benson, and R. Spurney performed the experiments. Subject enrollment, sequencing, and analysis of sequencing data were carried out by M. Chryst-Stangl, S. Murray, K. Benson, A. Bierzynska, G. Cavalleri, B. Doyle, N. Fennelly, S. Conlon, V. Vega-Warner, D. Fermin, P. Vijayan, M. A. Qureshi, S. Shril, M. Barua, F. Hildebrandt, M. Pollak, M.G. Sampson, M. Saleem, P.J. Conlon, and R. Gbadegesin. D. Howell and A. Dorman evaluated kidney biopsy samples. All of the authors read, edited, and approved the manuscript for submission.
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
Data Sharing Statement
Material requests and all correspondence should be sent to rasheed.gbadegesin@duke.edu.
Supplemental Material
This article contains the following supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2020101525/-/DCSupplemental.
Supplemental Figure 1. Filtering of rare variants in WGS data from family 40030.
Supplemental Figure 2. Pedigree of second FSGS family with segregating RCAN1 variant.
Supplemental Figure 3. RCAN gene expression in cultured human podocytes.
Supplemental Figure 4. RCAN1 and CN binding.
Supplemental Figure 5. RCAN1 single transfection calcineurin activity.
Supplemental Figure 6. Late apoptosis/necrosis quantification.
Supplemental Figure 7. Unmodified Western blots.
Supplemental Table 1. Segregating heterozygous variants found in family 40030.
Supplemental Table 2. Evolutionary conservation of RCAN1 variant residues.
Supplemental Table 3. Description of patient cohorts.
Supplemental Table 4. Heterozygous missense variants in RCAN1-3 genes.
Supplemental Summary 1. Members of the Nephrotic Syndrome Study (NEPTUNE).
Supplemental Video 1. RCAN1 WT protein modeling.
Supplemental Video 2. RCAN1 p.I162T protein modeling.
Supplemental Video 3. RCAN1 WT apoptosis.
Supplemental Video 4. RCAN1 p.I162T apoptosis.
Supplemental Video 5. RCAN1 p.I162T apoptosis rescue with FK506.
References
- 1.Bikbov B, Purcell CA, Levey AS, Smith M, Abdoli A, Abebe M, et al.; GBD Chronic Kidney Disease Collaboration: Global, regional, and national burden of chronic kidney disease, 1990-2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 395: 709–733, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.USRDS annual data report: Epidemiology of kidney disease in the United States. Available at: https://www.usrds.org/annual-data-report. Accessed May 20, 2020
- 3.Barisoni L, Kriz W, Mundel P, D’Agati V: The dysregulated podocyte phenotype: A novel concept in the pathogenesis of collapsing idiopathic focal segmental glomerulosclerosis and HIV-associated nephropathy. J Am Soc Nephrol 10: 51–61, 1999 [DOI] [PubMed] [Google Scholar]
- 4.Wiggins R-C: The spectrum of podocytopathies: A unifying view of glomerular diseases. Kidney Int 71: 1205–1214, 2007 [DOI] [PubMed] [Google Scholar]
- 5.Lovric S, Fang H, Vega-Warner V, Sadowski CE, Gee HY, Halbritter J, et al.; Nephrotic Syndrome Study Group: Rapid detection of monogenic causes of childhood-onset steroid-resistant nephrotic syndrome. Clin J Am Soc Nephrol 9: 1109–1116, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sadowski CE, Lovric S, Ashraf S, Pabst WL, Gee HY, Kohl S, et al.; SRNS Study Group: A single-gene cause in 29.5% of cases of steroid-resistant nephrotic syndrome. J Am Soc Nephrol 26: 1279–1289, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yao T, Udwan K, John R, Rana A, Haghighi A, Xu L, et al.: Integration of genetic testing and pathology for the diagnosis of adults with FSGS. Clin J Am Soc Nephrol 14: 213–223, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vivante A, Hildebrandt F: Exploring the genetic basis of early-onset chronic kidney disease. Nat Rev Nephrol 12: 133–146, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Preston R, Stuart HM, Lennon R: Genetic testing in steroid-resistant nephrotic syndrome: Why, who, when and how? Pediatr Nephrol 34: 195–210, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Trautmann A, Bodria M, Ozaltin F, Gheisari A, Melk A, Azocar M, et al.; PodoNet Consortium: Spectrum of steroid-resistant and congenital nephrotic syndrome in children: The PodoNet registry cohort. Clin J Am Soc Nephrol 10: 592–600, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Warejko JK, Tan W, Daga A, Schapiro D, Lawson JA, Shril S, et al.: Whole exome sequencing of patients with steroid-resistant nephrotic syndrome. Clin J Am Soc Nephrol 13: 53–62, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rothermel B, Vega RB, Yang J, Wu H, Bassel-Duby R, Williams RS: A protein encoded within the Down syndrome critical region is enriched in striated muscles and inhibits calcineurin signaling. J Biol Chem 275: 8719–8725, 2000 [DOI] [PubMed] [Google Scholar]
- 13.Fuentes JJ, Genescà L, Kingsbury TJ, Cunningham KW, Pérez-Riba M, Estivill X, et al.: DSCR1, overexpressed in Down syndrome, is an inhibitor of calcineurin-mediated signaling pathways. Hum Mol Genet 9: 1681–1690, 2000 [DOI] [PubMed] [Google Scholar]
- 14.Mulero MC, Aubareda A, Schlüter A, Pérez-Riba M: RCAN3, a novel calcineurin inhibitor that down-regulates NFAT-dependent cytokine gene expression. Biochim Biophys Acta 1773: 330–341, 2007 [DOI] [PubMed] [Google Scholar]
- 15.Cao X, Kambe F, Miyazaki T, Sarkar D, Ohmori S, Seo H: Novel human ZAKI-4 isoforms: Hormonal and tissue-specific regulation and function as calcineurin inhibitors. Biochem J 367: 459–466, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Görlach J, Fox DS, Cutler NS, Cox GM, Perfect JR, Heitman J: Identification and characterization of a highly conserved calcineurin binding protein, CBP1/calcipressin, in Cryptococcus neoformans. EMBO J 19: 3618–3629, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kingsbury TJ, Cunningham KW: A conserved family of calcineurin regulators. Genes Dev 14: 1595–1604, 2000 [PMC free article] [PubMed] [Google Scholar]
- 18.Mehta S, Li H, Hogan PG, Cunningham KW: Domain architecture of the regulators of calcineurin (RCANs) and identification of a divergent RCAN in yeast. Mol Cell Biol 29: 2777–2793, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vega RB, Yang J, Rothermel BA, Bassel-Duby R, Williams RS: Multiple domains of MCIP1 contribute to inhibition of calcineurin activity. J Biol Chem 277: 30401–30407, 2002 [DOI] [PubMed] [Google Scholar]
- 20.Sanna B, Brandt EB, Kaiser RA, Pfluger P, Witt SA, Kimball TR, et al.: Modulatory calcineurin-interacting proteins 1 and 2 function as calcineurin facilitators in vivo . Proc Natl Acad Sci U S A 103: 7327–7332, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Strippoli P, Lenzi L, Petrini M, Carinci P, Zannotti M: A new gene family including DSCR1 (Down Syndrome Candidate Region 1) and ZAKI-4: Characterization from yeast to human and identification of DSCR1-like 2, a novel human member (DSCR1L2). Genomics 64: 252–263, 2000 [DOI] [PubMed] [Google Scholar]
- 22.Miyazaki T, Kanou Y, Murata Y, Ohmori S, Niwa T, Maeda K, et al.: Molecular cloning of a novel thyroid hormone-responsive gene, ZAKI-4, in human skin fibroblasts. J Biol Chem 271: 14567–14571, 1996 [DOI] [PubMed] [Google Scholar]
- 23.Chang KT, Shi Y-J, Min K-T: The Drosophila homolog of Down’s syndrome critical region 1 gene regulates learning: Implications for mental retardation. Proc Natl Acad Sci U S A 100: 15794–15799, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vega RB, Rothermel BA, Weinheimer CJ, Kovacs A, Naseem RH, Bassel-Duby R, et al.: Dual roles of modulatory calcineurin-interacting protein 1 in cardiac hypertrophy. Proc Natl Acad Sci U S A 100: 669–674, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ryeom S, Greenwald RJ, Sharpe AH, McKeon F: The threshold pattern of calcineurin-dependent gene expression is altered by loss of the endogenous inhibitor calcipressin. Nat Immunol 4: 874–881, 2003 [DOI] [PubMed] [Google Scholar]
- 26.Hilioti Z, Gallagher DA, Low-Nam ST, Ramaswamy P, Gajer P, Kingsbury TJ, et al.: GSK-3 kinases enhance calcineurin signaling by phosphorylation of RCNs. Genes Dev 18: 35–47, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Martínez-Martínez S, Genescà L, Rodríguez A, Raya A, Salichs E, Were F, et al.: The RCAN carboxyl end mediates calcineurin docking-dependent inhibition via a site that dictates binding to substrates and regulators. Proc Natl Acad Sci U S A 106: 6117–6122, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Spurney RF: Non-immunologic actions of calcineurin inhibitors in proteinuric kidney diseases. Front Endocrinol (Lausanne) 5: 181, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schönenberger E, Ehrich JH, Haller H, Schiffer M: The podocyte as a direct target of immunosuppressive agents. Nephrol Dial Transplant 26: 18–24, 2011 [DOI] [PubMed] [Google Scholar]
- 30.Hall G, Wang L, Spurney RF: TRPC channels in proteinuric kidney diseases. Cells 9: 44, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bensman A, Niaudet P: Non-immunologic mechanisms of calcineurin inhibitors explain its antiproteinuric effects in genetic glomerulopathies. Pediatr Nephrol 25: 1197–1199, 2010 [DOI] [PubMed] [Google Scholar]
- 32.Yoo T-H, Fornoni A: Nonimmunologic targets of immunosuppressive agents in podocytes. Kidney Res Clin Pract 34: 69–75, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Faul C, Donnelly M, Merscher-Gomez S, Chang YH, Franz S, Delfgaauw J, et al.: The actin cytoskeleton of kidney podocytes is a direct target of the antiproteinuric effect of cyclosporine A. Nat Med 14: 931–938, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang Y, Jarad G, Tripathi P, Pan M, Cunningham J, Martin DR, et al.: Activation of NFAT signaling in podocytes causes glomerulosclerosis. J Am Soc Nephrol 21: 1657–1666, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gooch JL, Barnes JL, Garcia S, Abboud HE: Calcineurin is activated in diabetes and is required for glomerular hypertrophy and ECM accumulation. Am J Physiol Renal Physiol 284: F144–F154, 2003 [DOI] [PubMed] [Google Scholar]
- 36.D’Agati VD, Kaskel FJ, Falk RJ: Focal segmental glomerulosclerosis. N Engl J Med 365: 2398–2411, 2011 [DOI] [PubMed] [Google Scholar]
- 37.Pedigo CE, Ducasa GM, Leclercq F, Sloan A, Mitrofanova A, Hashmi T, et al.: Local TNF causes NFATc1-dependent cholesterol-mediated podocyte injury. J Clin Invest 126: 3336–3350, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang L, Chang J-H, Paik S-Y, Tang Y, Eisner W, Spurney RF: Calcineurin (CN) activation promotes apoptosis of glomerular podocytes both in vitro and in vivo . Mol Endocrinol 25: 1376–1386, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen S, Zhou Y, Chen Y, Gu J: fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34: i884–i890, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li H, Durbin R: Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25: 1754–1760, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Toolkit Picard2019. Broad Institute, GitHub Repository. Available at: http://broadinstitute.github.io/picard. Accessed February 17, 2020
- 42.DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, et al.: A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 43: 491–498, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A, et al.: From FastQ data to high confidence variant calls: The genome analysis toolkit best practices pipeline. Curr Protoc Bioinformatics 43: 11.10.1–11.10.33, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GRS, Thormann A, et al.: The Ensembl variant effect predictor. Genome Biol 17: 122, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, et al.: The mutational constraint spectrum quantified from variation in 141,456 humans [published correction appears in Nature 590: E53, 2021 10.1038/s41586-020-03174-8]. Nature 581: 434–443, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hall G, Lane BM, Khan K, Pediaditakis I, Xiao J, Wu G, et al.: The human FSGS-causing ANLN R431C mutation induces dysregulated PI3K/AKT/mTOR/Rac1 signaling in podocytes. J Am Soc Nephrol 29: 2110–2122, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Spurney RF: Role of C-terminal serines in desensitization and phosphorylation of the mouse thromboxane receptor. J Biol Chem 273: 28496–28503, 1998 [DOI] [PubMed] [Google Scholar]
- 48.Zhang Y: I-TASSER: Fully automated protein structure prediction in CASP8. Proteins 77[Suppl 9]: 100–113, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang J, Zhang Y: Protein structure and function prediction using I-TASSER. Curr Protoc Bioinformatics 52: 5.8.1–5.8.15, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Goddard TD, Huang CC, Meng EC, Pettersen EF, Couch GS, Morris JH, et al.: UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci 27: 14–25, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang L, Chang J-H, Buckley AF, Spurney RF: Knockout of TRPC6 promotes insulin resistance and exacerbates glomerular injury in Akita mice. Kidney Int 95: 321–332, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hall G, Lane B, Chryst-Ladd M, Wu G, Lin J-J, Qin X, et al.: Dysregulation of WTI (-KTS) is associated with the kidney-specific effects of the LMX1B R246Q mutation. Sci Rep 7: 39933, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Das AK, Pickett TM, Tungekar MF: Glomerular basement membrane thickness—a comparison of two methods of measurement in patients with unexplained haematuria. Nephrol Dial Transplant 11: 1256–1260, 1996 [PubMed] [Google Scholar]
- 54.Connaughton DM, Bukhari S, Conlon P, Cassidy E, O’Toole M, Mohamad M, et al.: The Irish kidney gene project--prevalence of family history in patients with kidney disease in Ireland. Nephron 130: 293–301, 2015 [DOI] [PubMed] [Google Scholar]
- 55.Connaughton DM, Kennedy C, Shril S, Mann N, Murray SL, Williams PA, et al.: Monogenic causes of chronic kidney disease in adults. Kidney Int 95: 914–928, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Roy A, Yang J, Zhang Y: COFACTOR: An accurate comparative algorithm for structure-based protein function annotation. Nucleic Acids Res 40: W471–W477, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Abbasi S, Su B, Kellems RE, Yang J, Xia Y: The essential role of MEKK3 signaling in angiotensin II-induced calcineurin/nuclear factor of activated T-cells activation. J Biol Chem 280: 36737–36746, 2005 [DOI] [PubMed] [Google Scholar]
- 58.Abbasi S, Lee J-D, Su B, Chen X, Alcon JL, Yang J, et al.: Protein kinase-mediated regulation of calcineurin through the phosphorylation of modulatory calcineurin-interacting protein 1. J Biol Chem 281: 7717–7726, 2006 [DOI] [PubMed] [Google Scholar]
- 59.Turro E, Astle WJ, Megy K, Gräf S, Greene D, Shamardina O, et al.; NIHR BioResource for the 100,000 Genomes Project: Whole-genome sequencing of patients with rare diseases in a national health system. Nature 583: 96–102, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bierzynska A, McCarthy HJ, Soderquest K, Sen ES, Colby E, Ding WY, et al.: Genomic and clinical profiling of a national nephrotic syndrome cohort advocates a precision medicine approach to disease management. Kidney Int 91: 937–947, 2017 [DOI] [PubMed] [Google Scholar]
- 61.Porta S, Martí E, de la Luna S, Arbonés ML: Differential expression of members of the RCAN family of calcineurin regulators suggests selective functions for these proteins in the brain. Eur J Neurosci 26: 1213–1226, 2007 [DOI] [PubMed] [Google Scholar]
- 62.Canaider S, Facchin F, Griffoni C, Casadei R, Vitale L, Lenzi L, et al.: Proteins encoded by human Down syndrome critical region gene 1-like 2 (DSCR1L2) mRNA and by a novel DSCR1L2 mRNA isoform interact with cardiac troponin I (TNNI3). Gene 372: 128–136, 2006 [DOI] [PubMed] [Google Scholar]
- 63.Li H, Zhang W, Zhong F, Das GC, Xie Y, Li Z, et al.: Epigenetic regulation of RCAN1 expression in kidney disease and its role in podocyte injury. Kidney Int 94: 1160–1176, 2018 [DOI] [PubMed] [Google Scholar]
- 64.Jang C, Lim JH, Park CW, Cho Y-J: Regulator of calcineurin 1 isoform 4 (RCAN1.4) is overexpressed in the glomeruli of diabetic mice. Korean J Physiol Pharmacol 15: 299–305, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Grgic I, Hofmeister AF, Genovese G, Bernhardy AJ, Sun H, Maarouf OH, et al.: Discovery of new glomerular disease-relevant genes by translational profiling of podocytes in vivo . Kidney Int 86: 1116–1129, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Liu Q, Busby JC, Molkentin JD: Interaction between TAK1-TAB1-TAB2 and RCAN1-calcineurin defines a signalling nodal control point. Nat Cell Biol 11: 154–161, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lee EJ, Seo SR, Um JW, Park J, Oh Y, Chung KC: NF-kappaB-inducing kinase phosphorylates and blocks the degradation of Down syndrome candidate region 1. J Biol Chem 283: 3392–3400, 2008 [DOI] [PubMed] [Google Scholar]
- 68.Porta S, Serra SA, Huch M, Valverde MA, Llorens F, Estivill X, et al.: RCAN1 (DSCR1) increases neuronal susceptibility to oxidative stress: A potential pathogenic process in neurodegeneration. Hum Mol Genet 16: 1039–1050, 2007 [DOI] [PubMed] [Google Scholar]
- 69.Minami T, Horiuchi K, Miura M, Abid MR, Takabe W, Noguchi N, et al.: Vascular endothelial growth factor- and thrombin-induced termination factor, Down syndrome critical region-1, attenuates endothelial cell proliferation and angiogenesis. J Biol Chem 279: 50537–50554, 2004 [DOI] [PubMed] [Google Scholar]
- 70.Stathatos N, Bourdeau I, Espinosa AV, Saji M, Vasko VV, Burman KD, et al.: KiSS-1/G protein-coupled receptor 54 metastasis suppressor pathway increases myocyte-enriched calcineurin interacting protein 1 expression and chronically inhibits calcineurin activity. J Clin Endocrinol Metab 90: 5432–5440, 2005 [DOI] [PubMed] [Google Scholar]
- 71.Cheng L, Li P, Wang H, Yang X, Zhou H, Tao W, et al.: Decreased activity of RCAN1.4 is a potential risk factor for congenital heart disease in a Han Chinese population. Protein Cell 9: 1039–1044, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Qin L, Zhao D, Liu X, Nagy JA, Hoang MV, Brown LF, et al.: Down syndrome candidate region 1 isoform 1 mediates angiogenesis through the calcineurin-NFAT pathway. Mol Cancer Res 4: 811–820, 2006 [DOI] [PubMed] [Google Scholar]
- 73.Lange AW, Yutzey KE: NFATc1 expression in the developing heart valves is responsive to the RANKL pathway and is required for endocardial expression of cathepsin K. Dev Biol 292: 407–417, 2006 [DOI] [PubMed] [Google Scholar]
- 74.Gipson DS, Trachtman H, Kaskel FJ, Greene TH, Radeva MK, Gassman JJ, et al.: Clinical trial of focal segmental glomerulosclerosis in children and young adults. Kidney Int 80: 868–878, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hurcombe JA, Hartley P, Lay AC, Ni L, Bedford JJ, Leader JP, et al.: Podocyte GSK3 is an evolutionarily conserved critical regulator of kidney function. Nat Commun 10: 403, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.