

To the Editor:
Autoinflammatory syndromes are caused by dysregulation of the innate immune system, often affecting the inflammasome and other pathogen recognition pathways that lead to overproduction of IL-1b and IL-18.1 Here we report 4 patients with severe autoinflammatory syndrome, presenting with cytopenias, rashes, and a significant increase in serum IL-18 levels secondary to novel specific mutations in cell division control protein 42 homolog (CDC42). CDC42 encodes a small Rho family GTPase that regulates multiple signaling pathways controlling cell polarity and migration, actin polarization, cytoskeletal architecture, endocytosis, and cell-cycle progression.2 All mutations affect the last 3 to 5 amino acids of the CDC42 C-terminus. Mutations predispose to the development of macrophage activation syndrome (MAS), and all patients improved significantly on therapy with IL-1 receptor inhibitors.
We report 4 patients from 3 different institutions. Clinical descriptions and biological parameters are summarized in Table 1 and detailed in the Methods section in this article’s Online Repository at www.jacionline.org. In brief, all patients presented at birth or in the neonatal period with growth restriction, hepatosplenomegaly, transaminitis, recurrent febrile episodes, urticaria-like rashes, and significant cytopenias. Patient 4 had aseptic meningitis, papilledema, and posterior retinitis and was thought to have clinical features consistent with the IL-1-mediated autoinflammatory disease neonatal-onset multisystem inflammatory disease (NOMID).1 Most patients displayed some facial dysmorphisms, such as mild frontal bossing, mild hypertelorism, and nasal bridge depression (Fig 1, B). Anemia (all patients) and thrombocytopenia (patients 3 and 4) were transfusion dependent. Skin biopsy specimens of patients 2 and 3 revealed perivascular lymphocytic and neutrophilic infiltration without vasculitis (data not shown). Further laboratory evaluation revealed for all patients a dramatic increase in C-reactive protein (CRP) levels, erythrocyte sedimentation rates (ESRs), and ferritin levels. In workup of their hyperferritinemic inflammation, all patients were found to have substantially increased peripheral blood IL-18 levels (Fig 1, D). Patients 2 and 3 met the criteria for hemophagocytic lymphohistiocytosis/MAS. None of the patients had a family history of consanguinity.
TABLE I.
Clinical phenotype and biological parameters of the reported patients before and after therapy with IL-1 inhibitors
| Patient 1 | Patient 2 | Patient 3 | Patient 4 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CDC42 variant | c.563G>A (p.C188Y) | c.563G>A (p.C188Y) | c.556C>T (p.R186C) | c.576A>C (stop lost [p.*192C*24]) | |||||||||
| Dose of anakinra (d) | Before anakinra | 1 mg/kg | 2.5 mg/kg | Before anakinra | 6 mg/kg | 5 mg/kg | Before anakinra | 3 mg/kg | 8.5 mg/kg | Canakinumab, 8 mg/kg/mo | Before anakinra | 3 mg/kg | 4.5 mg/kg |
| Clinical presentation | |||||||||||||
| Facial dysmorphism | Mild frontal bossing, mild hypertelorism, depresse nasal bridge | Frontal bossing, macrocephaly, thin sparse hair, depressed nasal bridge | None | Mild frontal bossing | |||||||||
| Weight (percentile) | <3rd | <3rd | 5th | <3rd | <3rd | 5th | 3rd | 3rd | 3rd | 5th | <3rd | <3rd | 10th |
| Appetite | Poor | Improved | Normal | Poor | Improved | Improved | Poor | Improved | Improved | Normal | Poor | Decreased | Normal |
| Spleen size* (cm [patient age; SM or normal, UL for age]) | 11.4 (12 mo; SM, UL 8.0) | 8 (15 mo; normal, UL 8.0) | 8 (18 mo; normal, UL 8.0) | 11.2 (neonatal; NA SM, UL 6.0) | NA | 10.1 (12 mo; SM, UL 8.0) | 9.2 (neonatal; SM, UL 6.0) | 9.9 (5 mo; SM, UL 6.5) | 10.8 (16 mo; SM, UL 8.0) | 11.9 (6 y; SM, UL 10.0) | Hepatomegaly and SM | 6.7 (15 mo old; normal, UL 8.0) | NA |
| Painful rash | Daily | None | None | Bimonthly | Monthly | None | Daily | Resolved | Resolved | Resolved | Daily | Recurrent | None |
| Ibuprofen intake | Daily | None | None | None | None | None | None | None | None | None | None | None | None |
| Severe infections | None | None | None | None | None | None | None | Lymphadenitis | Lymphadenitis | Lymphadenitis, streptococcal sepsis | None | Yes† Lymphadenitis |
Yes‡ |
| Biological parameters | |||||||||||||
| Platelet (103/mL) | 72 | 155 | 141 | 182 | 267 | 276 | 13 | 22 | 148 | 202 | 54 | 381 | 307 |
| Hemoglobin (g/dL) | Transfusion-dependent anemia (Hb, <7 g/dL) | 13.8 | 13.3 | Transfusion-dependent anemia (Hb, <7 g/dL) | 11.2 | 11.7 | 8.1 | 7.9 | 10.1 | 12.3 | 7.4 | 13.3 | 13.5 |
| RBC transfusion | Monthly | None | None | Q2-3 mo | None | None | Weekly or biweekly | Sporadic | Rare | Rare | Multiple | None | None |
| Platelet transfusion | None | None | None | None | None | None | Multiple | Sporadic | Rare | Rare | Multiple | None | None |
| LDH (ng/mL) | 540 | NA | 547 | 584 | 269 | 271 | 817 | 998 | 1,115 | 1,633 | NA | 284 | 353 |
| ESR (mm/h) | >130 | 16 | 32 | 51 | 22 | 40 | 62 | 65 | 39 | 22 | 140 | 26 | 21 |
| CRP (mg/L) | 55 | 2 | 4 | 56 | 3.4 | 8.8 | 14.2 | 3.6 | 5.6 | 0.2 | Increased | 28.8 | 1.54 |
| Ferritin (ng/mL [<400]) | 2,364 | 801 | 680 | 614 | 316 | 126 | 5,191 | 3,682 | 1,329 | 557 | NA | 172 | 51 |
| IL-6 (pg/mL [<5]) | 18 | NA | <5 | NA | NA | NA | NA | NA | 15.9 | 11.0 | NA | NA | NA |
| IL-18§ (pg/mL [100-500]) | 35,616 | 27,803 | 22,690 | 36,479‖ | NA | 83,268 | NA | NA | 28,407 | 26,402 | NA | 19,025 | 21,535 |
| IL-18BP§ (pg/mL [1,000-6,000]) | 14,418 | 8,557 | 6,264 | NA | NA | 14,653 | NA | NA | 11,803 | NA | NA | 23,986 | 9,626 |
| CXCL9§ (pg/mL [500-3,500]) | 8,457 | 2,978 | 4,975 | NA | NA | 2,064 | NA | NA | 3,942 | NA | NA | 11,473 | 4,278 |
For patient 1, duplicate samples were run for all 3 time points of IL-18 and were 26,477, 31,134, and 25,431 pg/mL, respectively.
Hb, Hemoglobin; LDH, lactate dehydrogenase; NA, not applicable; SM, splenomegaly; UL, upper limit of normal.
Suggested upper limit splenic length in 230 infants and children (PMID: 2048509): 0 to 3 months, 6.0 cm; 3 to 6 months, 6.5 cm; 6 to 12 months, 7.0 cm; 1 to 2 years, 8.0 cm; 2 to 4 years, 9.0 cm; 4 to 6 years, 9.5 cm; 6 to 8 years, 10.0 cm; 8 to 10 years, 11.0 cm; and 10 to 12 years, 11.5 cm.
While receiving 1 mg/kg/d prednisone and anakinra, he had respiratory syncytial virus pneumonia, septic arthritis, and cervical lymphadenitis, which resolved when steroids were discontinued.
At age 9 years, when anakinra was switched to canakinumab, he had Staphylococcus aureus bacteremia and septic myositis with multiple abscesses and was switched back to anakinra with significant improvement.
Performed by Dr Scott Canna.
IL-18 levels were measured after 1 dose of anakinra.
FIG 1.
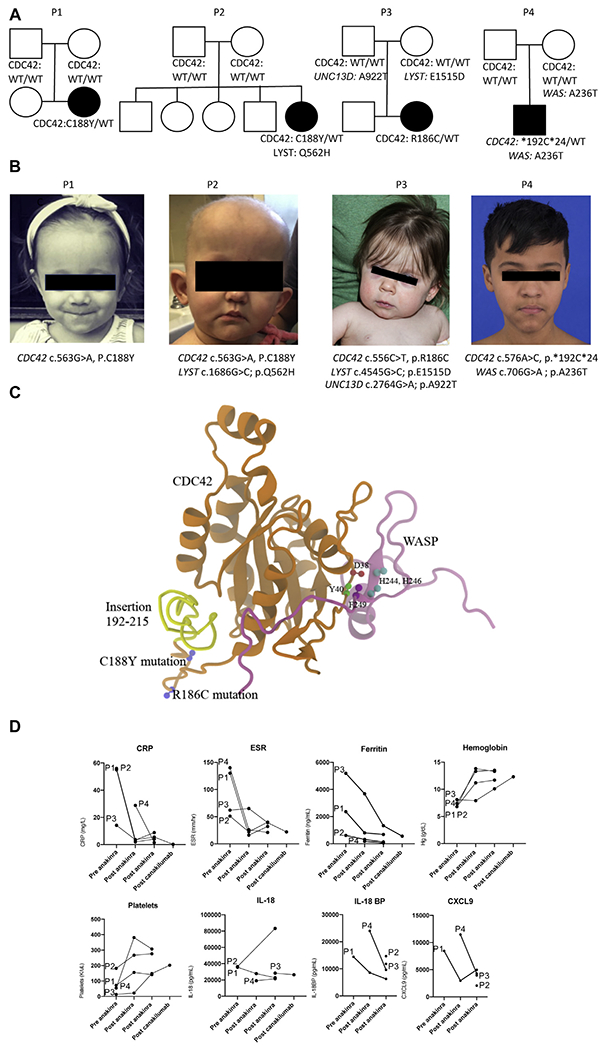
A and B, Picture (Fig 1, A) and pedigree (Fig 1, B) of 4 patients showing a de novo monoallelic mutation in CDC42. The affected subjects are indicated as follows: circles, female subjects; squares, male subjects. WT, Wild-type. C and D, Crystal structure of CDC42 (orange). Highlighted are the Wiskott-Aldrich syndrome protein (WASP) binding domain (Fig 1, C) and the 3 reported C-terminal variants (Fig 1, D). Laboratory parameters measured were CRP, ESR, ferritin, hemoglobin, platelet, and cytokines (IL-18, IL-18BP, and CXCL9).
Because of the complex nature of their disease, whole-exome sequencing (WES) was performed in all patients and identified 3 distinct monoallelic, heterozygous, possibly pathogenic de novo variants in CDC42. Patients 1 and 2 carried CDC42 c.563G>A (p.C188Y), patient 3 carried CDC42 c.556C>T (p.R186C), and patient 4 carried CDC42 c.576A>C (p.*192C*24), which causes read-through of the stop codon and adds 24 amino acids to the C-terminus of the protein (Fig 1, C).
Given their inflammation, MAS-like features, and concern for NOMID (in patient 4), therapy with the IL-1 receptor antagonist anakinra was initiated in all 4 patients, with the rationale that local release of IL-1β from monocytes and in tissues promotes systemic inflammation and is associated with chronically increased CRP levels and ESR (Fig 1, D). On anakinra, fever and rashes entirely resolved, and the spleen and liver size of all patients significantly decreased. In all patients failure to thrive significantly improved with respect to both longitudinal growth and weight gain. Three of the 4 patients started to meet developmental milestones. Anemia and thrombocytopenia resolved in 3 of the 4 patients, and CRP and ferritin levels decreased significantly in all 4 patients. Patient 3 continued with intermittent flares. Facial dysmorphisms persisted. In the 1 patient analyzed, pretherapy abnormalities in monocytes and natural killer cells improved after anakinra (see Fig E1 in this article’s Online Repository at www.jacionline.org). Remarkably, peripheral blood IL-18 levels showed little or no diminution, even after months of treatment (Fig 1, D, and Table 1). Although patient 1 has never experienced significant infections over 1 year of therapy, patients 2, 3 and 4 had recurrent gastrointestinal and respiratory tract infections (patient 2, over 1 year of therapy), staphylococcal lymphadenitis (patients 3 and 4, over 6 and 11 years of therapy, respectively), group A streptococcal sepsis with purpura fulminans (patient 3), and Staphylococcus bacteremia and abscesses (patient 4). Infections in patient 3 occurred during canakinumab treatment and in patient 4 during anakinra and prednisone treatment at approximately 1 mg/kg/d and during a short treatment period when he was switched from anakinra to canakinumab (see the Methods section in this article’s Online Repository).
Heterozygous mutations in the CDC42 gene were recently reported to cause Takenouchi-Kosaki syndrome (TKS), which manifests with growth retardation, developmental delay, facial dysmorphism (diverse features depending on the type of CDC42 mutation, see Table E1 and the Methods section in this article’s Online Repository at www.jacionline.org), and macrothrombocytopenia.3 Like TKS, some of our patients have mild facial dysmorphic features (see Table E1 and the Methods section in this article’s Online Repository). However, inflammatory disease manifestations, including rashes, laboratory evidence of systemic inflammation, and development of MAS/hemophagocytic lymphohistiocytosis have not been reported in patients with TKS.
The high serum IL-18 level increase and the predisposition to development of MAS in our autoinflammatory cohort are laboratory and clinical features that overlap with patients with NLRC4-MAS and X-linked inhibitor of apoptosis deficiency.4,5 The significant clinical improvement with IL-1–blocking treatment supports a critical role for IL-1 driving the disease process and together with chronically increased IL-18 levels suggests inflammasome activation as a pathomechanism.4,6
This points to the need for further investigations to mechanistically connect the C-terminal CDC42 mutations with overproduction of IL-18 and IL-1 activation. The novel C-terminal variants in CDC42, which are associated with severe autoinflammation, all occur within or in close spatial proximity to a C-terminal diarginine motif implicated in membrane binding and lipidation of CDC42. Indeed, it has been reported that this diarginine motif (Arg186 and Arg187) plays an essential role in binding of CDC42 to liposomes containing phosphatidylinositol 4,5-bisphosphonate (PIP2).7 The interaction of CDC42 and PIP2 is critical in mediating the PIP2-induced actin assembly.8
Emerging evidence highlights the importance of the actin cytoskeleton in modulating inflammatory responses. A growing number of pathogenic mutations cause aberrant actin depolymerization, activate the inflammasome, and modulate other innate immune functions.6,9 Illustrating the functional effect of actin polymerization defects, loss-of-function mutations in WDR1, encoding actin-interacting protein 1, cause defective neutrophil mobilization, opportunistic infections, and activation of monocyte/macrophages, leading to autoinflammation.6 How exactly defects in actin depolarization predispose to exaggerated inflammasome activation or propagation remains the subject of ongoing studies.
Interestingly, 3 of the 4 patients with novel C-terminal CDC42 variants have mutations in additional genes that might modify the clinical phenotypes. These mutations include heterozygous lysosomal trafficking regulator (LYST) variants (patients 2 and 3), a rare UNC13D variant (patient 3), and a Wiskott-Aldrich syndrome (WAS) mutation that has previously been associated with thrombocytopenia and a mild Wiskott-Aldrich syndrome phenotype (patient 4, see Table E2 in this article’s Online Repository at www.jacionline.org). However, one of the 4 patients, patient 1, did not have any other mutations on WES (minor allele frequency, <1%) that are expected to confound the disease phenotype. Patient 1 also had the most complete response to anakinra, with reversal of clinical symptoms and rapid improvement of laboratory markers. Taken together, these data suggest that the C-terminal CDC42 variants might be sufficient to trigger an autoinflammatory syndrome that is responsive to IL-1 inhibition, although a modifying role for the other rare variants cannot fully be ruled out without more patients and further mechanistic studies.
In summary, these 4 patients’ mutations, biomarker assessment, and response to IL-1 inhibition all support that C-terminal variants in CDC42 cause a novel IL-1 inhibition–responsive autoinflammatory syndrome. All patients had chronic and extreme increases in IL-18 levels, likely predisposing to development of MAS. The phenotypic overlap with patients with X-linked inhibitor of apoptosis deficiency and activating NLRC4 mutations points to a shared pathogenesis.4,5 Given the remarkable treatment response to IL-1-blocking therapies, the recognition of this novel disease will hopefully facilitate rapid diagnosis and early targeted therapies. Further studies are necessary to elucidate the mechanistic basis and test the efficacy of other potential therapeutic agents, including human recombinant IL-18BP.
Supplementary Material
Acknowledgments
We thank the donors of the Center for Genetic Immune Diseases for their generous philanthropic funding. We thank Eyal Grunebaum, MD; Elizabeth Hoyte, NP; Kenneth Weinberg, MD; Maria Grazia Roncarolo, MD; Matthew Kirbey; Jay Balagas, MD; and Mansi Narula for constructive discussions and direct support of patient care. We thank Andrew Oler, PhD, and Zuoming Deng PhD, for help with the genetic analysis. We thank the patients and their families for participating in the research studies.
This research was supported in part by the Intramural Research Program of the National Institutes of Health, National Institute of Allergy and Infectious Diseases.
Footnotes
Disclosure of potential conflict of interest: The authors declare that they have no relevant conflicts of interest.
REFERENCES
- 1.de Jesus AA, Canna SW, Liu Y, Goldbach-Mansky R. Molecular mechanisms in genetically defined autoinflammatory diseases: disorders of amplified danger signaling. Annu Rev Immunol 2015;33:823–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Qadir MI, Parveen A, Ali M. Cdc42: role in cancer management. Chem Biol Drug Des 2015;86:432–9. [DOI] [PubMed] [Google Scholar]
- 3.Martinelli S, Krumbach OHF, Pantaleoni F, Coppola S, Amin E, Pannone L, et al. Functional dysregulation of CDC42 causes diverse developmental phenotypes. Am J Hum Genet 2018;102:309–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wada T, Kanegane H, Ohta K, Katoh F, Imamura T, Nakazawa Y, et al. Sustained elevation of serum interleukin-18 and its association with hemophagocytic lymphohistiocytosis in XIAP deficiency. Cytokine 2014;65:74–8. [DOI] [PubMed] [Google Scholar]
- 5.Weiss ES, Girard-Guyonvarc’h C, Holzinger D, de Jesus AA, Tariq Z, Picarsic J, et al. Interleukin-18 diagnostically distinguishes and pathogenically promotes human and murine macrophage activation syndrome. Blood 2018;131:1442–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Standing AS, Malinova D, Hong Y, Record J, Moulding D, Blundell MP, et al. Autoinflammatory periodic fever, immunodeficiency, and thrombocytopenia (PFIT) caused by mutation in actin-regulatory gene WDR1. J Exp Med 2017;214:59–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Johnson JL, Erickson JW, Cerione RA. C-terminal di-arginine motif of Cdc42 protein is essential for binding to phosphatidylinositol 4,5-bisphosphate-containing membranes and inducing cellular transformation. J Biol Chem 2012;287:5764–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen F, Ma L, Parrini MC, Mao X, Lopez M, Wu C, et al. Cdc42 is required for PIP(2)-induced actin polymerization and early development but not for cell viability. Curr Biol 2000;10:758–65. [DOI] [PubMed] [Google Scholar]
- 9.Kuhns DB, Fink DL, Choi U, Sweeney C, Lau K, Priel DL, et al. Cytoskeletal abnormalities and neutrophil dysfunction in WDR1 deficiency. Blood 2016;128:2135–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.