

PURPOSE
Rhabdomyosarcoma is the most common soft tissue sarcoma of childhood. Despite aggressive therapy, the 5-year survival rate for patients with metastatic or recurrent disease remains poor, and beyond PAX-FOXO1 fusion status, no genomic markers are available for risk stratification. We present an international consortium study designed to determine the incidence of driver mutations and their association with clinical outcome.
PATIENTS AND METHODS
Tumor samples collected from patients enrolled on Children's Oncology Group trials (1998-2017) and UK patients enrolled on malignant mesenchymal tumor and RMS2005 (1995-2016) trials were subjected to custom-capture sequencing. Mutations, indels, gene deletions, and amplifications were identified, and survival analysis was performed.
RESULTS
DNA from 641 patients was suitable for analyses. A median of one mutation was found per tumor. In FOXO1 fusion-negative cases, mutation of any RAS pathway member was found in > 50% of cases, and 21% had no putative driver mutation identified. BCOR (15%), NF1 (15%), and TP53 (13%) mutations were found at a higher incidence than previously reported and TP53 mutations were associated with worse outcomes in both fusion-negative and FOXO1 fusion-positive cases. Interestingly, mutations in RAS isoforms predominated in infants < 1 year (64% of cases). Mutation of MYOD1 was associated with histologic patterns beyond those previously described, older age, head and neck primary site, and a dismal survival. Finally, we provide a searchable companion database (ClinOmics), containing all genomic variants, and clinical annotation including survival data.
CONCLUSION
This is the largest genomic characterization of clinically annotated rhabdomyosarcoma tumors to date and provides prognostic genetic features that refine risk stratification and will be incorporated into prospective trials.
INTRODUCTION
Rhabdomyosarcoma (RMS) is the most common soft tissue sarcoma of childhood.1 With the development of multimodal chemotherapy regimens, relapse-free survival rates have improved to 70%-80% in patients with localized disease, albeit with significant toxicity.2 Unfortunately, despite aggressive therapy, the 5-year survival rate for patients with metastatic disease remains poor, but variable.3 Therapy assignment in North American and European trials is currently based on clinicopathologic features and not molecular or genetic markers, with the exception of the recent incorporation of FOXO1 fusion status by the Children's Oncology Group (COG) and European paediatric Soft tissue sarcoma Study Group (EpSSG).4–6 Although clinical features reasonably stratify patients into broad treatment cohorts, prognostic imprecision hampers efforts to successfully escalate or de-escalate therapy. Particularly problematic is the COG intermediate risk category, defined as localized FOXO1 fusion-positive (FP) RMS and localized, incompletely resected (clinical group III) FOXO1 fusion-negative (FN) RMS arising from an unfavorable anatomic site; this category comprises approximately 50% of cases and has a heterogeneous clinical outcome.4,7,8 This suggests that some of these children could be treated with less aggressive therapy or alternatively should be considered to have more aggressive disease.
CONTEXT
Key Objective
Rhabdomyosarcoma (RMS) is a sarcoma of childhood with a poor 5-year survival rate for patients with metastatic or recurrent disease. No genomic markers are currently available for risk stratification except for PAX-FOXO1 fusion gene status. This study performed sequencing to determine the mutational status of genes implicated in RMS oncogenesis and correlated these results with clinical outcomes.
Knowledge Generated
The genetic and clinical characteristics associated with primary tumors from two international cohorts are presented. Survival analysis demonstrated that TP53 and MYOD1 mutations were associated with worse event-free survival.
Relevance
This study nominates mutant genes MYOD1 and TP53 as indicators of poor prognosis in fusion-negative RMS, and TP53 alterations as a biomarker of more aggressive disease in fusion-positive RMS. Mutation of MYOD1 was not restricted to spindle histology, and the association with adverse outcome highlights the need to accurately diagnose MYOD1 mutations and develop novel treatment strategies for these patients.
Previous comprehensive genomic sequencing studies of RMS have been completed, but outcome analysis was limited by sample size or incomplete clinical annotation of the included samples.9,10 To genetically classify RMS and refine risk stratification, we formed an international collaborative group and performed standardized sequencing of a large cohort of clinically annotated cases. Herein, we report the summary findings and detail the importance of incorporation of genomic data into prospectively enrolling RMS clinical trials. Adopting molecular features to RMS risk stratification should improve clinical outcomes for patients with RMS by allowing further tailoring of therapies to match an individual patient's risk and mutational profile. To ensure that this critical data set is available to the broader research community, the generated sequencing data are available within dbGAP (accession phs000720.v4.p1) and the clinical and mutational data are publicly accessible.11
PATIENTS AND METHODS
Study Population and Clinical Annotation
Samples from the two cohorts included in this work were collected on institutional review board–approved clinical trials or tissue banking studies. COG samples included samples collected on ARST0331, ARST0431, D9602, D9803, and D9902. UK samples from patients treated on protocols through the malignant mesenchymal tumor and RMS2005 protocols12-14 were collected and approved for study through local and national ethical approvals (CCR2015 and 06/MRE04/71, respectively) (Appendix Table A1, online only). Because there are subtle differences in risk stratification between the COG and EpSSG (reviewed in Ref. 15), an overarching simplified risk stratification definition was used to enable merging data. Additional population and annotation details are provided in the Data Supplement (online only).
Gene Panel Sequencing
A custom-capture sequencing assay targeting 39 genes previously implicated in RMS (Appendix Table A2, online only) was performed, and variants were called using previously published sequencing algorithms.9,16-19 Detailed sequencing and variant calling methods are provided in the Data Supplement.
Statistical Methods
Patient characteristics were summarized using medians and ranges or frequencies and percentages. Associations between pairs of gene markers were tested using the exact conditional test of proportions (Fisher's exact test). Event-free survival (EFS) was defined as time from diagnosis (United Kingdom) or enrollment on a study (COG) until event (relapse, second malignant neoplasm, or death) or last contact. Each gene predictor variable was tested for univariate association with EFS and overall survival using the log-rank test and for association within a multivariate Cox proportional hazard regression. The survival analysis was done separately for each cohort of patients: COG and United Kingdom and within each cohort, separately for FP and FN patients. Detailed statistical methods are provided in the Data Supplement.
RESULTS
Patient Population
Clinically annotated cases from patient samples were assembled on COG biology study ARST14B1Q and a parallel cohort assembled from UK malignant mesenchymal tumor and RMS2005 studies, to generate two large cohorts of samples. The clinical details are summarized in Table 1. In total, 641 cases had adequate DNA to generate sequencing libraries of minimum quality to be included in the study. The median age of the combined cohort was 5.9 years (range 0.02-37.8) (Appendix Fig A1, online only). The male:female ratio was 1.9:1, slightly higher than the generally accepted ratio of 1.5:1,20 reflecting an enrichment of paratesticular tumors within this cohort. The most common anatomic locations were the parameningeal (20%) and paratesticular tumors (20%) followed by tumors of the retroperitoneum, peritoneum, or trunk (16%) and the extremity (14%). A simplified risk stratification algorithm was used to harmonize COG and UK cohorts (Data Supplement) and using this method, the population had representation of cases of the low-risk (34%), intermediate-risk (47%), and high-risk populations (18%).
TABLE 1.
Clinical Characteristics of Included Patients
Mutation Frequency Observations
Overall, the sequenced tumors had a median of one mutation call per tumor (range 0-5). The most frequently observed gene mutations are presented in Table 2. Consistent with prior reports, the genomic profiles of the FP and FN populations were distinct. The most frequently observed lesions in FP tumors were the focal amplification of CDK4 (13%) or MYCN (10%). The genes BCOR (6%), NF1 (4%), TP53 (4%), and PIK3CA (2%) were found in a small number of FP RMS cases, verifying previous observations.9 In contrast, the most frequently observed genetic alteration in FN tumors were RAS isoform mutations NRAS (17%), KRAS (9%), and HRAS (8%), with any RAS isoform mutation noted in 32% (n = 167 of 515) of FN tumors. Mutation of an RAS pathway gene (defined as NRAS, KRAS, HRAS, FGFR4, NF1, and PIK3CA) could be found in 56% (n = 288 of 515) of all FN samples. Recurrence of mutations in tumor suppressor genes in FN RMS, TP53 (13%), NF1 (15%), and BCOR (15%), was higher than reports in previous studies.9 Hotspot mutations in FGFR4 (13%), CTNNB1 (6%), PIK3CA (5%), and MYOD1 (3%) were observed at similar frequencies as previously reported and seen at similar percentages within the two independent international patient cohorts.9,10 No mutations were found in 14 genes previously associated with RMS (MTOR, PKN1, ALK, SOS1, SOS2, ROBO1, PDGFRA, GAB1, BRAF, CCND1, CCND2, ATM, AKT, and SMARCA4), although variants of unknown significance were observed in each of these genes. The median age of presentation of the patients correlated with alteration of individual genes, with a notable increase in MYOD1 mutations, CDK4 amplification, and MYCN amplification in patients older than 10 years and HRAS mutations in infants < 1 year (Fig 1A).
TABLE 2.
Mutation Frequency Observations by Cohort

FIG 1.
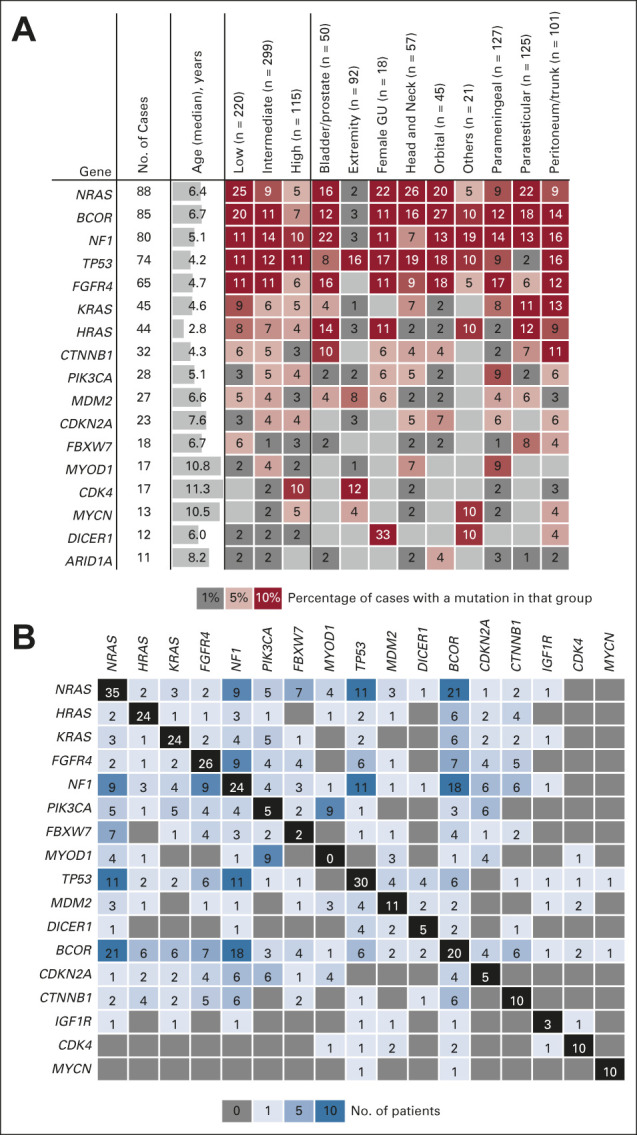
Mutations summarized by anatomic location and co-occurrence. (A) Summary of mutations by occurrence within defined risk groups (low, intermediate, or high) or by anatomic location. The reported value is a percentage of the number of cases with a mutation in that gene within each group. (B) Co-occurrence of mutated genes reported as absolute number of cases. GU, genitourinary.
Mutations Summarized by Anatomic Distribution
RMS tumors arise in diverse anatomic locations throughout the body, and the site of disease is known to correlate with clinical outcome.21 Current clinical risk stratification assigns tumors arising in the orbit, nonparameningeal head and neck, and the male or female genital tracts as favorable. The distribution of mutations by anatomic location is presented in Figure 1A. As previously described,22 DICER1 mutations had a predilection for tumors arising within the female genitourinary tract. Within the sequenced tumors, 33% (6 of 18) of all female genitourinary cases harbored a mutation in DICER1. Interestingly, FN cases of the extremity had an enrichment for TP53 mutations or MDM2 amplifications, whereby 42% (15 of 35) of FN cases with a primary tumor of the extremity had an alteration of one of these two genes (Appendix Table A3, online only). MYOD1-mutant tumors also showed a distinct anatomic enrichment with 88% (15 of 17) of MYOD1-mutant tumors observed in either the head and neck or parameningeal region.
FN Tumors Frequently Harbor More Than One Genetic Driver Alteration
Mutational heterogeneity has previously been described in FN RMS.23 In this study, the presence of multiple driver mutations within individual tumors was evident within FN samples with 41% (213 of 515) of tumors having one mutation, 37% (193 of 515) of tumors having two or more mutations, and 21% (109 of 515) containing no alteration of a candidate gene (Appendix Fig A2A, online only). Greater than two mutations within a tumor was a significant marker in terms of worse EFS (P = .01, hazard ratio [HR] 2.014 [1.010-4.015]) within the COG cohort; however, this observation was not replicated in the UK cohort for EFS (P = .39, HR 1.098 [0.633-1.904]) (Appendix Fig A2B). To establish the most common pairings of gene interactions that drive RMS, we analyzed mutational data across the cohort and observed that mutations of tumor suppressor genes such as NF1, TP53, and BCOR frequently co-occurred with other mutations. Significant interactions included BCOR with NRAS (P = .01) or NF1 (P = .03), and MYOD1 with PIK3CA (P < .0001) or CDKN2A (P = .0049) (Fig 2B, Appendix Fig A2C). MYOD1 mutations were exceptional in that they always occurred with an additional gene being mutated. Although NRAS, KRAS, HRAS, and FGFR4 were mutually exclusive in most cases, surprisingly, there were individual tumors with co-occurrence of multiple hotspot mutations in these genes.
FIG 2.
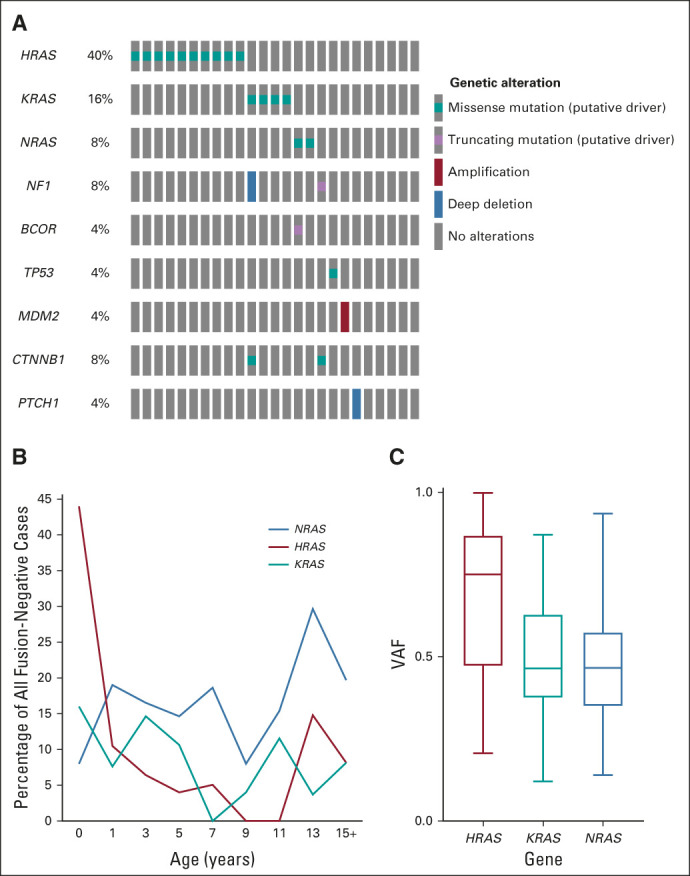
RAS isoform mutations. (A) Oncoprint of mutations observed in infants < 1 year old (n = 25) showed an enrichment for RAS mutations. (B) Distribution of RAS isoform mutations by age with a distinct peak of HRAS mutant cases discovered in the infant population. (C) HRAS mutations were frequently found to have a higher VAF indicating that the mutation occurred before a loss-of-heterozygosity event on chromosome 11p. VAF, variant allele frequency.
CDK4, MYCN, and TP53 in FP Tumors
Although PAX3-FOXO1 fusion is itself of prognostic value, no molecular markers are currently available for risk substratification of FP tumors. In total, 126 FP tumors were evaluated (69 COG and 57 UK) with a dedicated fusion assay performed on 80 of 126 (64%) and centrally reviewed alveolar histology used as a proxy in the remaining cases. Small numbers of CDK4- and MYCN-mutant cases (COG: n = 14 and n = 10; UK: n = 2 and n = 3, respectively) were observed between the two cohorts, limiting conclusions about the prognostic significance of these genes (Appendix Fig A3A, online only). Interestingly, a small number of FP cases (COG: n = 3; UK: n = 2) had mutations in TP53 and were universally fatal (Appendix Fig A3B).
Survival Analysis of RAS Isoforms and Enrichment of RAS Mutations in Infants
A driving hypothesis of this study was that the presence of a mutation in a RAS isoform or RAS pathway gene would correlate with poor outcomes in FN RMS, because of the observation in previous smaller cohorts of enrichment of RAS isoform mutations in high-risk cases.10 Therefore, we examined each RAS isoform and individual RAS pathway members for correlation with survival. Neither mutation of any RAS isoform nor a RAS pathway gene was associated with a worse EFS or overall survival across the two cohorts (Appendix Fig A4, online only). One striking observation was the correlation of RAS isoform mutations longitudinally with age. Most notable was the enrichment of RAS isoform mutations in FN cases that occurred under the age of 1 year (64% of cases, P = .0068) with a clear peak incidence of HRAS mutations (40% of all FN infants, P < .0001) within this age group and no enrichment of secondary mutations (Fig 2A). Although not significant, KRAS-mutant tumors were frequent within the toddler period (15% of cases at 3 years, P = .2368) and a peak of NRAS-mutant tumors was observed in adolescence (30% of all cases at 13 years, P = .4407) (Fig 2B). NRAS, HRAS, and KRAS isoforms had distinct codon and amino acid profiles, consistent with previous studies (Appendix Fig A5, online only). Interrogation of the observed allele frequency of RAS isoform mutations showed that the majority of HRAS mutations occur at variant allele frequency > 0.5, likely reflecting the occurrence of this mutation within the frequently observed uniparental disomy event that occurs on chromosome 11p (Fig 2C).24
Association of TP53 Alterations With Survival in FN Tumors
TP53 was found to be altered in 13% (n = 69 of 515) of the FN cohort, and the observed lesions included deep deletions, truncating mutations, and point mutations (Appendix Fig A6A, online only). The mutations occurred throughout the gene body with some enrichment seen within the DNA-binding domain of the protein (Appendix Fig A6B). The most recurrent mutations were found at the codons G245S (six cases), R248Q or W (six cases), R175H (four cases), and P72A (four cases). Given the lack of a matched normal sample, no determination was made if these lesions represent somatic or germline events. Univariate (EFS P = .0083; HR 2.067 [1.192-3.585]) and risk-stratified analysis (EFS P = .0146; HR 1.973 [1.132-3.438]) of survival data within the COG cohort demonstrated that the presence of a TP53 mutation imparted a worse EFS (Figs 3A and 3B). Evaluation of the UK cohort verified the significance of this observation in both non–risk-stratified (EFS P = .0079; HR 2.006 [1.187-3.390]) and risk-stratified (EFS P = .0055; HR 2.105 [1.230-3.604]) analysis (Figs 3C and 3D).
FIG 3.

TP53 mutations and association with survival. (A) KM analysis of EFS within the COG FN cohort (n = 275) by the presence of a TP53 Mut or absence of a TP53 lesion (TP53 WT). (B) KM analysis of EFS within the COG FN cohort by TP53 status and RMS risk category. Total case numbers: low, n = 124; intermediate, n = 126; high, n = 35. (C) KM analysis of EFS within the UK FN cohort (n = 240) by the presence of a TP53 Mut or absence of a TP53 lesion (TP53 WT). (D) KM analysis of EFS within the UK FN cohort by TP53 status and RMS risk category. Total case numbers: low, n = 96; intermediate, n = 113; high, n = 25. Presented P values are log-rank and BH adj. HR with 95% CI. BH adj, Benjamini-Hochberg–adjusted; COG, Children's Oncology Group; EFS, event-free survival; FN, fusion-negative; HR, hazard ratio; KM, Kaplan-Meier; RMS, rhabdomyosarcoma; TP53 Mut, TP53 mutation; TP53 WT, TP53 wild type.
Association of MYOD1 Mutations With Survival in FN Tumors
Mutations in the transcription factor MYOD1 were found in 3% (n = 17 of 515) of all FN cases and no FP cases (Fig 4A). The observed mutations were confined to the previously reported hotspot codon change L122R.25 As noted, MYOD1-mutant tumors within this pediatric cohort occurred at an older mean age of 10.8 years (2.1-21.1 years) when compared with the rest of the cohort. Centrally reviewed pathology from COG and review of UK samples frequently noted spindle or sclerosing features of the tumor (Figs 4B1 and 4B2); however, interestingly, cases with densely packed cells that mimicked the dense pattern of embryonal rhabdomyosarcoma (ERMS) or RMS not otherwise specified were also found (Figs 4B3 and 4B4). EFS of those patients with MYOD1 mutations within the COG cohort was dismal and associated with rapid progression in non–risk-stratified (EFS P < .0001; HR 6.839 [3.463-13.507]) and risk-stratified (EFS P < .0001; HR 5.579 [2.791-11.151]) analysis (Figs 4C and 4D). Parallel survival analysis of the UK cohort verified this observation in non–risk-stratified (EFS P = .0133; HR 3.320 [1.212-9.099]) and risk-stratified (EFS P = .0111; HR 3.455 [1.247-9.571]) analysis (Figs 4E and 4F). Importantly, within the MYOD1-mutant group, 23% of patients were identified before treatment as low-risk and 65% were identified as intermediate-risk. Consistent with previous reports,26 53% of cases had a corresponding alteration of PIK3CA and MYOD1 mutations were not found to be mutually exclusive with RAS mutations, with coexisting lesions seen in NRAS (n = 4), HRAS (n = 1), and NF1 (n = 1). Interestingly, MYOD1-mutant tumors also frequently harbored deep deletions in CDKN2A (n = 4 of 17, 24%). Deep deletions or deleterious mutations of CDKN2A were present in 4% of all FN tumors and associated with a worse EFS (COG: P = .0031, HR 2.737 [1.363 to 5.494]; UK: P = .0031, HR 4.7 [1.896 to 11.648]) (Appendix Fig A7A, online only). This observation was independent of the co-occurrence of MYOD1 mutations (Appendix Fig A7B), however was specific for cases within the intermediate-risk group (Appendix Fig A7C).
FIG 4.

MYOD1 mutations and survival. (A) All tumors with a mutation in MYOD1 within the entire sequenced cohort (N = 641) are shown to visualize co-occurrence of MYDO1 mutations with other genes. All MYOD1 cases were observed within FN cases. (B) Histology of MYOD1 mutant cases demonstrated the frequent presence of sclerosing or spindle morphology (case 1 and case 2). Cases 3 and 4 show that some cases demonstrated areas more typical of dense embryonal histology. (C) KM analysis of EFS within the COG FN cohort (n = 275) by the presence of a MYOD1 Mut or absence of a MYDO1 lesion (MYOD1 WT). (D) KM analysis of EFS within the COG FN cohort by MYOD1 status and RMS risk category. Total case numbers: low, n = 124; intermediate, n = 126; high, n = 35. (E) KM analysis of EFS within the UK FN cohort (n = 240) by the presence of a MYOD1 Mut or absence of a MYDO1 lesion (MYOD1 WT). (F) KM analysis of EFS within the UK FN cohort by MYOD1 status and RMS risk category. Total case numbers: low, n = 96; intermediate, n = 113; high, n = 25. Presented P values are log-rank and BH adj. HR with 95% CI. BH adj, Benjamini-Hochberg–adjusted; COG, Children's Oncology Group; EFS, event-free survival; FN, fusion-negative; HR, hazard ratio; KM, Kaplan-Meier; MYOD1 Mut, MYOD1 mutation; MYOD1 WT, MYOD1 wild type; RMS, rhabdomyosarcoma.
DISCUSSION
Integration of molecular features into risk stratification and therapeutic decision making remains a major challenge to improving the care of any patient with a rare tumor. Decades of clinical trials led to the development of a complicated system for risk stratification of patients with RMS, on the basis of information from both the pretreatment tumor staging and surgical grouping.4 The imprecision of these assignments is known, which has important implications for how current therapy is delivered and how clinical trials incorporating novel agents are designed. Our international collaboration generated the largest cohort of clinically annotated and genomically characterized RMS tumors analyzed to date. The effort discovered critical genetic insights into the underpinnings of the disease and significant molecular markers that provide refinement to the current risk stratification of patients with RMS. On the basis of our results, we propose a new framework for the classification and treatment of RMS, using TP53 and MYOD1 mutations in addition to the FOXO1 fusion status,5-7 which could be tested in prospective clinical trials (Appendix Table A4, online only).
The overall gene mutation frequency that was observed is consistent with previous sequencing studies9,27 with the notable exception of an increased frequency of tumor suppressor genes TP53, NF1, and BCOR. The observed increase in frequency of mutation of these genes likely results from improved depth of sequencing using a targeted assay approach. Of interest are the approximately 20% of FN tumors that have no driver mutation of a candidate gene. Beyond genome-wide aneuploidy and focal loss of heterozygosity of 11p15, the previous comprehensive analyses,9,10,23,28 and this focused analysis, have failed to discover recurrent genetic driver genes in this group of tumors. This suggests the need for continued comprehensive genomic and epigenetic evaluations that would allow identification of genes mutated at a low recurrence frequency, as well as alternative mechanisms of oncogenesis. The discovery that FN tumors are frequently driven by alteration of multiple coexisting mutations mirrors previous work that showed FN tumors are composed of multiple subclones that follow an evolutionary selection.23 This finding suggests that clonal evolution of these tumors may be significant, and elegant genomic work has highlighted how these processes might drive relapsed or refractory disease.10 Although the trend toward a worse outcome in patients with multiple mutations in the COG cohort is intriguing, this observation was not replicated in the UK cohort and may be confounded by the different therapeutic regimens that the patients received. Comprehensive, prospective assessment is required to address the validity of the survival correlations. In addition, studies designed to assay sequential tumor biopsies will be required to fully interrogate the mechanisms of metastasis and relapse in RMS.
Mutations in RAS isoforms have long been described as a driver of FN RMS.29 Our study clearly determines that the presence at diagnosis of a mutation in an RAS isoform or RAS pathway gene does not portend a poor prognosis, in contrast to previous smaller cohorts finding enrichment for RAS isoform mutations in high-risk cases.10 Although RAS was not found to be a prognostic predictor, we highlight an interesting observation that RAS isoform mutations appear to have some age-specific correlations, with HRAS occurring in the infants, KRAS occurring in the toddlers, and NRAS mutations with a peak in adolescence. Infants have previously been shown to have an inferior 5-year failure-free survival as compared with older patients (67% v 81%).30 This difference is attributed to the general reluctance to use more aggressive local control, including radiation, in these patients.31 Our results indicate that incorporation of targeted therapeutic agents such as tipifarnib (NCT04284774) or AMG510 (NCT03600883) may be particularly beneficial to this vulnerable and high-risk population. In addition, further mechanistic surveys of the developmental biology underlying these observations might have important implications for the generation of accurate preclinical models of RMS.
No molecular markers are currently used for risk stratification of FP RMS tumors. Amplification of the chromosomal regions 2p24 and 12q13-q15 and the implicated genes MYCN and CDK4, respectively, are the most recurrent lesions associated with a FOXO1 fusion. Previous work dedicated to assigning the prognostic value of these lesions identified amplification of 12q13-q14, but not 2p24, as a marker of an aggressive subset of FP tumors.32 Other efforts discovered that in ARMS, overexpression or gain of genomic copies of MYCN was significantly associated with adverse outcome.33 The current study found inconsistent results for MYCN and CDK4 amplification, with nonreproducible correlations noted between the two cohorts. There is evidence of a small subset of FP tumors that harbor a mutation of TP53 at diagnosis and appear to be particularly aggressive. Ultimately, prospective consortium-level trials should include profiling of each of these genes to define their prognostic value and the biologic role they play in FP RMS.
From the seminal report of Li-Fraumeni syndrome,34 the role of TP53 in ERMS oncogenesis has long been established; however, the association of TP53 mutations with clinical outcome has previously been unknown. Given the lack of a corresponding germline sample, our study could not determine whether the discovered TP53 mutation was germline or somatic. Despite this, we demonstrated that the presence of a TP53 mutation was predictive of a worse outcome. This finding is consistent with reports from several cancer types that found mutation of TP53 is associated with poor response and survival.35 This is also consistent with higher levels of TP53 protein in metastatic versus localized ERMS36 and also observations in zebra fish models of ERMS, where tp53 mutations are linked to more aggressive and metastatic disease.37 Determination of TP53 status in all cases of RMS therefore is critical, both for prognostic value and the implications that germline mutations have for genetic counseling.
MYOD1 mutation of the L122R codon was reported by independent groups in 2014.25,38 MYOD1-mutant tumors make up only 3% of FN RMS, and this study highlights the importance of MYOD1 mutations within the RMS population. These tumors have unique demographic, anatomic, and histologic characteristics, but none of these appear to definitively capture all MYOD1-mutant tumors. This suggests the need to incorporate sequencing of this gene into the diagnostic workup of FN RMS. Our observation that MYOD1 mutations invariably co-occur with mutation in a second gene, most notably PIK3CA and CDKN2A, is consistent with a recent report.26 The co-occurrence with CDKN2A is of interest given that a recent large survey of soft tissue sarcomas of multiple histologies, including a small number of RMS tumors, implicated CDKN2A as a biomarker of poor prognosis.39 Although our study indicates that CDKN2A alterations may have prognostic value independent of MYOD1, this conclusion is based on low overall numbers. Therefore, we recommend that CDKN2A alterations be evaluated in prospective studies. Regardless, MYOD1-mutant tumors have an aggressive nature and have limited responses to current therapeutic regimens, which highlights the impetus to identify these cases and develop novel therapeutic trials for this rare subset of patients with RMS.
ACKNOWLEDGMENT
The authors thank the core sequencing facility of the NCI Division of Cancer Epidemiology and Genetics. Processing of the data was made possible by the NIH Helix computer cluster. The authors also thank the Children's Cancer and Leukaemia Group for facilitating sample collection, the National Cancer Research Institute Young Onset Sarcoma Subgroup and EpSSG for endorsing this study, and the Clinical Trials Unit in Birmingham, UK, for providing clinical trial data.
Appendix
FIG A1.
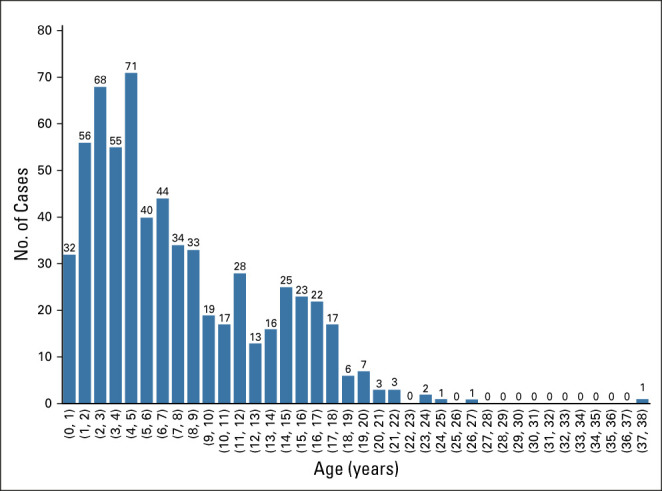
Histogram presentation of the age distribution of cases within the cohort (N = 641).
FIG A2.

Survival analysis on the basis of number of called mutations. (A) All FN samples (n = 515) were distributed into groups having no observed mutations (black), one mutation (red), or two or more genes mutated (blue). (B) Kaplan-Meier analysis of EFS from the COG and UK FN cohorts on the basis of the number of observed mutations within a tumor. The COG cohort (n = 275) had 49 patients with no observed mutation, 113 patients with one observed mutation, and 113 patients with two or more observed mutations. The UK cohort (n = 240) had 60 patients with no observed mutation, 100 patients with one observed mutation, and 80 patients with two or more observed mutations. (C) Pairwise testing of the significance of gene-gene interactions was performed to identify interactions that occurred at higher (red and purple) or lower (green) frequencies than expected. COG, Children's Oncology Group; EFS, event-free survival; FN, fusion-negative; HR, hazard ratio.
FIG A3.

CDK4, MYCN, and TP53 mutations in FP RMS. (A) Survival analysis for all three genes within the COG and UK cohorts with and without risk stratification. The log-rank and BH adj P value are presented. (B) Kaplan-Meier analysis of EFS and OS in FP cases within the COG cohort (n = 69) and UK cohort (n = 57) for TP53 Mut or WT. COG cohort has three TP53 mutant cases and UK cohort has two TP53 mutant cases. BH adj, Benjamini-Hochberg–adjusted; COG, Children's Oncology Group; EFS, event-free survival; FP, fusion-positive; HR, hazard ratio; Mut, mutated; OS, overall survival; RMS, rhabdomyosarcoma; WT, wild type.
FIG A4.

RAS isoform and RAS pathway analysis. (A) RAS isoforms and pathway member P values for both EFS and OS. The log-rank P value and the BH adj value are presented. BH adj, Benjamini-Hochberg–adjusted; COG, Children's Oncology Group; EFS, event-free survival; OS, overall survival.
FIG A5.
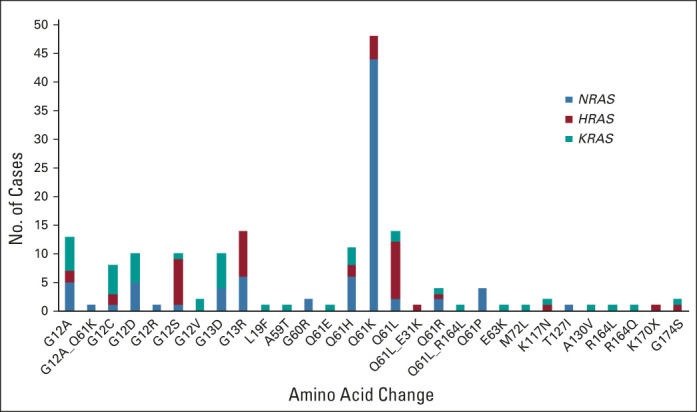
RAS codon and amino acid usage summary. Absolute number of cases with a mutation in the specified codon by amino acid change and isoform (NRAS, blue; HRAS, red; KRAS, green).
FIG A6.

TP53 mutations. (A) Association of TP53 mutations with other gene mutations across the entire rhabdomyosarcoms cohort. (B) Location and number of mutations across TP53 protein.
FIG A7.

CDKN2A and association with survival. (A) EFS and OS by CDKN2A deletion in the COG cohort (blue) and the UK cohort (orange). (B) EFS and OS by CDKN2A status after removal of any cases with a co-occurring mutation of MYOD1: COG cohort (blue) and UK cohort (orange). (C) EFS within the COG cohort demonstrated that the observed effect of CDKN2A alteration was largely within the intermediate-risk group. BH adj, Benjamini-Hochberg–adjusted; COG, Children's Oncology Group; EFS, event-free survival; HR, hazard ratio; Mut, mutated; OS, overall survival; WT, wild type.
TABLE A1.
Number of Patients Profiled From Each Trial
TABLE A2.
Included Genes in the Capture Assay

TABLE A3.
Fusion-Negative Cases Summarized by Mutation and Anatomic Location

TABLE A4.
Proposed Risk Stratification With the Incorporation of Genetic Markers

Donald A. Barkauskas
Employment: Genentech
Stock and Other Ownership Interests: Genentech
Patents, Royalties, Other Intellectual Property: US patent on the basis of PhD research in glioblastoma
Corinne M. Linardic
Leadership: Grid Therapeutics
Stock and Other Ownership Interests: Pfizer, LabCorp, Thermo Fisher Scientific
Honoraria: AstraZeneca
Patents, Royalties, Other Intellectual Property: My spouse has a patent for his therapeutic antibody developed in his laboratory at Duke and licensed by Grid
Julia Chisholm
Consulting or Advisory Role: Roche, Bayer, Roche/Genentech
Belynda Hicks
Employment: United Therapeutics
Stock and Other Ownership Interests: United Therapeutics
Travel, Accommodations, Expenses: United Therapeutics
Michael Hubank
Honoraria: Boehringer Ingelheim, Incyte, Qiagen
Consulting or Advisory Role: Guardant Health, Illumina, AstraZeneca, Bristol Myers Squibb Foundation, Bayer, Roche, Lilly, Amgen
Anna Kelsey
Honoraria: Bayer
Travel, Accommodations, Expenses: Bayer
Susanne A. Gatz
Consulting or Advisory Role: Tesaro, Bayer
Travel, Accommodations, Expenses: AstraZeneca
Douglas S. Hawkins
Research Funding: Loxo, Bristol Myers Squibb, Merck Sharp & Dohme, Bayer, Lilly, Eisai, Amgen, Seattle Genetics, Incyte, Jazz Pharmaceuticals
Travel, Accommodations, Expenses: Loxo, Bayer, AstraZeneca
Javed Khan
Research Funding: Lentigen, Taiho Oncology
Patents, Royalties, Other Intellectual Property: Monoclonal antibody-based therapeutics targeting fibroblast growth factor receptor 4 (FGFR4) for potential treatment of human cancers expressing FGFR4
No other potential conflicts of interest were reported.
See accompanying editorial on page 2851
DISCLAIMER
The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement of the US government.
PRIOR PRESENTATION
Presented at the Pediatric Oncology Session of the ASCO annual meeting, June 3, 2018; the plenary session of the Children's Oncology Group annual meetings on October 3, 2018 and March 19, 2020; and the plenary session of the EpSSG Winter meeting, December 6, 2018.
SUPPORT
Supported by the St Baldrick's Hero Fund in memory of Peyton Arens and by Grants No. U10CA098543, U10CA098413, U10CA180899, and U10CA180886 from the National Cancer Institute, Bethesda, MD, to the Children's Oncology Group. United Kingdom funding was provided by the Chris Lucas Trust, the Talan's Trust, Alice's Arc charity, and Cancer Research UK (Grant No. C5066/A10399).
J.F.S. and J.S. contributed equally to this work. J.M.S. and J.K. are equal senior authors.
STUDY GROUPS
International Consortium: Children's Oncology Group; Children's Cancer and Leukaemia Group, Young Onset Sarcoma Subgroup; and the National Cancer Institute.
AUTHOR CONTRIBUTIONS
Conception and design: Jack F. Shern, Susanne A. Gatz, Douglas S. Hawkins, Janet M. Shipley, Javed Khan
Financial support: Jack F. Shern, Janet M. Shipley, Javed Khan
Administrative support: Jack F. Shern, Javed Khan
Provision of study materials or patients: Xinyu Wen, Paola Angelini, Louis Chesler, Anna Kelsey
Collection and assembly of data: Jack F. Shern, Joanna Selfe, Young K. Song, Sivasish Sindiri, Jun Wei, Xinyu Wen, Erin R. Rudzinski, Tammy Lo, David Hall, Corinne M. Linardic, Debbie Hughes, Julia Chisholm, Rebecca Brown, Kristine Jones, Belynda Hicks, Paola Angelini, Sally George, Louis Chesler, Michael Hubank, Anna Kelsey, Douglas S. Hawkins, Janet M. Shipley, Javed Khan
Data analysis and interpretation: Jack F. Shern, Joanna Selfe, Elisa Izquierdo, Rajesh Patidar, Hsien-Chao Chou, Marielle E. Yohe, Sivasish Sindiri, Jun Wei, Xinyu Wen, Erin R. Rudzinski, Donald A. Barkauskas, David Hall, Corinne M. Linardic, Sabri Jamal, Meriel Jenney, Stephen X. Skapek, Douglas S. Hawkins, Janet M. Shipley, Javed Khan
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Genomic Classification and Clinical Outcome in Rhabdomyosarcoma: A Report From an International Consortium
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/jco/authors/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
Donald A. Barkauskas
Employment: Genentech
Stock and Other Ownership Interests: Genentech
Patents, Royalties, Other Intellectual Property: US patent on the basis of PhD research in glioblastoma
Corinne M. Linardic
Leadership: Grid Therapeutics
Stock and Other Ownership Interests: Pfizer, LabCorp, Thermo Fisher Scientific
Honoraria: AstraZeneca
Patents, Royalties, Other Intellectual Property: My spouse has a patent for his therapeutic antibody developed in his laboratory at Duke and licensed by Grid
Julia Chisholm
Consulting or Advisory Role: Roche, Bayer, Roche/Genentech
Belynda Hicks
Employment: United Therapeutics
Stock and Other Ownership Interests: United Therapeutics
Travel, Accommodations, Expenses: United Therapeutics
Michael Hubank
Honoraria: Boehringer Ingelheim, Incyte, Qiagen
Consulting or Advisory Role: Guardant Health, Illumina, AstraZeneca, Bristol Myers Squibb Foundation, Bayer, Roche, Lilly, Amgen
Anna Kelsey
Honoraria: Bayer
Travel, Accommodations, Expenses: Bayer
Susanne A. Gatz
Consulting or Advisory Role: Tesaro, Bayer
Travel, Accommodations, Expenses: AstraZeneca
Douglas S. Hawkins
Research Funding: Loxo, Bristol Myers Squibb, Merck Sharp & Dohme, Bayer, Lilly, Eisai, Amgen, Seattle Genetics, Incyte, Jazz Pharmaceuticals
Travel, Accommodations, Expenses: Loxo, Bayer, AstraZeneca
Javed Khan
Research Funding: Lentigen, Taiho Oncology
Patents, Royalties, Other Intellectual Property: Monoclonal antibody-based therapeutics targeting fibroblast growth factor receptor 4 (FGFR4) for potential treatment of human cancers expressing FGFR4
No other potential conflicts of interest were reported.
REFERENCES
- 1.Skapek SX Ferrari A Gupta AA, et al. : Rhabdomyosarcoma. Nat Rev Dis Primers 5:1, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Malempati S, Hawkins DS: Rhabdomyosarcoma: Review of the Children's Oncology Group (COG) Soft-Tissue Sarcoma Committee experience and rationale for current COG studies. Pediatr Blood Cancer 59:5-10, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Oberlin O Rey A Lyden E, et al. : Prognostic factors in metastatic rhabdomyosarcomas: Results of a pooled analysis from United States and European cooperative groups. J Clin Oncol 26:2384-2389, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hibbitts E Chi YY Hawkins DS, et al. : Refinement of risk stratification for childhood rhabdomyosarcoma using FOXO1 fusion status in addition to established clinical outcome predictors: A report from the Children's Oncology Group. Cancer Med 8:6437-6448, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Missiaglia E Williamson D Chisholm J, et al. : PAX3/FOXO1 fusion gene status is the key prognostic molecular marker in rhabdomyosarcoma and significantly improves current risk stratification. J Clin Oncol 30:1670-1677, 2012 [DOI] [PubMed] [Google Scholar]
- 6.Selfe J Olmos D Al-Saadi R, et al. : Impact of fusion gene status versus histology on risk-stratification for rhabdomyosarcoma: Retrospective analyses of patients on UK trials. Pediatr Blood Cancer 64, 2017 [DOI] [PubMed] [Google Scholar]
- 7.Skapek SX Anderson J Barr FG, et al. : PAX-FOXO1 fusion status drives unfavorable outcome for children with rhabdomyosarcoma: A Children's Oncology Group report. Pediatr Blood Cancer 60:1411-1417, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Meza JL Anderson J Pappo AS, et al. : Analysis of prognostic factors in patients with nonmetastatic rhabdomyosarcoma treated on intergroup rhabdomyosarcoma studies III and IV: The Children's Oncology Group. J Clin Oncol 24:3844-3851, 2006 [DOI] [PubMed] [Google Scholar]
- 9.Shern JF Chen L Chmielecki J, et al. : Comprehensive genomic analysis of rhabdomyosarcoma reveals a landscape of alterations affecting a common genetic axis in fusion-positive and fusion-negative tumors. Cancer Discov 4:216-231, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen X Stewart E Shelat AA, et al. : Targeting oxidative stress in embryonal rhabdomyosarcoma. Cancer Cell 24:710-724, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.ClinOmics Genomics Portal. https://clinomics.ccr.cancer.gov/clinomics/public/
- 12.Stevens MC Rey A Bouvet N, et al. : Treatment of nonmetastatic rhabdomyosarcoma in childhood and adolescence: Third study of the International Society of Paediatric Oncology—SIOP Malignant Mesenchymal Tumor 89. J Clin Oncol 23:2618-2628, 2005 [DOI] [PubMed] [Google Scholar]
- 13.Oberlin O Rey A Sanchez de Toledo J, et al. : Randomized comparison of intensified six-drug versus standard three-drug chemotherapy for high-risk nonmetastatic rhabdomyosarcoma and other chemotherapy-sensitive childhood soft tissue sarcomas: Long-term results from the International Society of Pediatric Oncology MMT95 study. J Clin Oncol 30:2457-2465, 2012 [DOI] [PubMed] [Google Scholar]
- 14.Bisogno G De Salvo GL Bergeron C, et al. : Vinorelbine and continuous low-dose cyclophosphamide as maintenance chemotherapy in patients with high-risk rhabdomyosarcoma (RMS 2005): A multicentre, open-label, randomised, phase 3 trial. Lancet Oncol 20:1566-1575, 2019 [DOI] [PubMed] [Google Scholar]
- 15.Chen C Dorado Garcia H Scheer M, et al. : Current and future treatment strategies for rhabdomyosarcoma. Front Oncol 9:1458, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Izquierdo E Yuan L George S, et al. : Development of a targeted sequencing approach to identify prognostic, predictive and diagnostic markers in paediatric solid tumours. Oncotarget 8:112036-112050, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.George SL Izquierdo E Campbell J, et al. : A tailored molecular profiling programme for children with cancer to identify clinically actionable genetic alterations. Eur J Cancer 121:224-235, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chang W Brohl AS Patidar R, et al. : MultiDimensional ClinOmics for precision therapy of children and adolescent young adults with relapsed and refractory cancer: A report from the center for cancer research. Clin Cancer Res 22:3810-3820, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Talevich E Shain AH Botton T, et al. : CNVkit: Genome-wide copy number detection and visualization from targeted DNA sequencing. PLoS Comput Biol 12:e1004873, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ognjanovic S Linabery AM Charbonneau B, et al. : Trends in childhood rhabdomyosarcoma incidence and survival in the United States, 1975-2005. Cancer 115:4218-4226, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Crist W Gehan EA Ragab AH, et al. : The third intergroup rhabdomyosarcoma study. J Clin Oncol 13:610-630, 1995 [DOI] [PubMed] [Google Scholar]
- 22.Dehner LP, Jarzembowski JA, Hill DA: Embryonal rhabdomyosarcoma of the uterine cervix: A report of 14 cases and a discussion of its unusual clinicopathological associations. Mod Pathol 25:602-614, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen L Shern JF Wei JS, et al. : Clonality and evolutionary history of rhabdomyosarcoma. PLoS Genet 11:e1005075, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Robbins KM Stabley DL Holbrook J, et al. : Paternal uniparental disomy with segmental loss of heterozygosity of chromosome 11 are hallmark characteristics of syndromic and sporadic embryonal rhabdomyosarcoma. Am J Med Genet A 170:3197-3206, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kohsaka S Shukla N Ameur N, et al. : A recurrent neomorphic mutation in MYOD1 defines a clinically aggressive subset of embryonal rhabdomyosarcoma associated with PI3K-AKT pathway mutations. Nat Genet 46:595-600, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Agaram NP LaQuaglia MP Alaggio R, et al. : MYOD1-mutant spindle cell and sclerosing rhabdomyosarcoma: An aggressive subtype irrespective of age. A reappraisal for molecular classification and risk stratification. Mod Pathol 32:27-36, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Seki M Nishimura R Yoshida K, et al. : Integrated genetic and epigenetic analysis defines novel molecular subgroups in rhabdomyosarcoma. Nat Commun 6:7557, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Casey DL Wexler LH Pitter KL, et al. : Genomic determinants of clinical outcomes in rhabdomyosarcoma. Clin Cancer Res 26:1135-1140, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stratton MR Fisher C Gusterson BA, et al. : Detection of point mutations in N-ras and K-ras genes of human embryonal rhabdomyosarcomas using oligonucleotide probes and the polymerase chain reaction. Cancer Res 49:6324-6327, 1989 [PubMed] [Google Scholar]
- 30.Malempati S Rodeberg DA Donaldson SS, et al. : Rhabdomyosarcoma in infants younger than 1 year: A report from the Children's Oncology Group. Cancer 117:3493-3501, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Joshi D Anderson JR Paidas C, et al. : Age is an independent prognostic factor in rhabdomyosarcoma: A report from the soft tissue sarcoma Committee of the Children's Oncology Group. Pediatr Blood Cancer 42:64-73, 2004 [DOI] [PubMed] [Google Scholar]
- 32.Barr FG Duan F Smith LM, et al. : Genomic and clinical analyses of 2p24 and 12q13-q14 amplification in alveolar rhabdomyosarcoma: A report from the Children's Oncology Group. Genes Chromosomes Cancer 48:661-672, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Williamson D Lu YJ Gordon T, et al. : Relationship between MYCN copy number and expression in rhabdomyosarcomas and correlation with adverse prognosis in the alveolar subtype. J Clin Oncol 23:880-888, 2005 [DOI] [PubMed] [Google Scholar]
- 34.Li FP, Fraumeni JF, Jr: Rhabdomyosarcoma in children: Epidemiologic study and identification of a familial cancer syndrome. J Natl Cancer Inst 43:1365-1373, 1969 [PubMed] [Google Scholar]
- 35.Petitjean A Achatz MI Borresen-Dale AL, et al. : TP53 mutations in human cancers: Functional selection and impact on cancer prognosis and outcomes. Oncogene 26:2157-2165, 2007 [DOI] [PubMed] [Google Scholar]
- 36.Leuschner I Langhans I Schmitz R, et al. : p53 and mdm-2 expression in rhabdomyosarcoma of childhood and adolescence: Clinicopathologic study by the Kiel Pediatric Tumor Registry and the German Cooperative Soft Tissue Sarcoma Study. Pediatr Dev Pathol 6:128-136, 2003 [DOI] [PubMed] [Google Scholar]
- 37.Ignatius MS Hayes MN Moore FE, et al. : tp53 deficiency causes a wide tumor spectrum and increases embryonal rhabdomyosarcoma metastasis in zebrafish. Elife 7:e37202, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Szuhai K de Jong D Leung WY, et al. : Transactivating mutation of the MYOD1 gene is a frequent event in adult spindle cell rhabdomyosarcoma. J Pathol 232:300-307, 2014 [DOI] [PubMed] [Google Scholar]
- 39.Bui NQ Przybyl J Trabucco SE, et al. : A clinico-genomic analysis of soft tissue sarcoma patients reveals CDKN2A deletion as a biomarker for poor prognosis. Clin Sarcoma Res 9:12, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]