

Abstract
Infections with filoviruses in humans are highly virulent, causing hemorrhagic fevers which result in up to 90% mortality. In addition to natural infections, the ability to use these viruses as bioterrorist weapons is of significant concern. Currently, there are no licensed vaccines or therapeutics available to combat these infections. The pathogenesis of disease involves the dysregulation of the host’s immune system, which results in impairment of the innate and adaptive immune responses, with subsequent development of lymphopenia, thrombocytopenia, hemorrhage, and death. Questions remain with regard to the few survivors of infection, who manage to mount an effective adaptive immune response. These questions concern the humoral and cellular components of this response, and whether such a response can be elicited by an appropriate prophylactic vaccine. The data reported herein describe the production and evaluation of a recombinant subunit Ebola virus vaccine candidate consisting of insect cell expressed Zaire ebolavirus (EBOV) surface glycoprotein (GP) and the matrix proteins VP24 and VP40. The recombinant subunit proteins are shown to be highly immunogenic in mice, yielding both humoral and cellular responses, as well as highly efficacious, providing up to 100% protection against a lethal challenge with live virus. These results demonstrate proof of concept for such a recombinant non-replicating vaccine candidate in the mouse model of EBOV which helps to elucidate immune correlates of protection and warrants further development.
Keywords: Zaire ebolavirus, recombinant subunit proteins, subunit vaccine, GP, VP24, VP40
1. Introduction
Although the frequency of human infections is low, the extreme virulence of filoviruses has heightened both public and scientific awareness. The most prominent members of the family are Zaire ebolavirus (EBOV) and Marburg marburgvirus (MARV) which cause fulminant hemorrhagic fevers and death in up to 90% of human infections depending on the infecting strain, route of infection and medical care provided. While state of the art medical treatment may increase the chances of survival after EBOV infection, currently no vaccine or antiviral therapy is available to prevent or cure the disease. As shown during the West African outbreak of EBOV (2013–2016), diagnostic capabilities as well as the required supportive treatment of patients is very resource demanding and therefore the development of safe and effective prophylactic vaccines is very important in preventing and combating future outbreaks. As part of the outbreak response in the affected West African countries, WHO and various industrial and government partners collaborated on expedited clinical paths for EBOV vaccines and therapeutics. The most promising reports on progress towards an efficacious EBOV vaccine have been of human clinical trials of a recombinant replication-competent Vesicular Stomatitis Virus (VSV) vectored Ebola vaccine containing the EBOV GP protein in place of the VSV G protein [1–3]. The efficacy and effectiveness of this vaccine (rVSV-ZEBOV) was assessed in a phase 3 clinical trial using the approach of ring-vaccinations in Guinea, West Africa. The interim and final reports [1, 2] showed that a single administration of the vaccine was efficacious and effective and deemed safe as well which led to recent (December 2016) public statements by the WHO declaring the vaccine trial to be successful. Indeed, the results of the ring-vaccination, cluster-randomized trial demonstrated that the vaccine efficacy was 100% based on the occurrence of new cases of Ebola Virus Disease (EVD) more than ten days after identification of an index case when comparing results from immediate- versus delayed-vaccinated trial subjects (primary and secondary contacts of EVD index cases). The occurrence of EVD cases during the first nine days after identification of the cluster was not different between the two study groups. While these developments are encouraging and seem to provide a viable path to market for the first EBOV vaccine candidate, many hurdles, particularly in regards to safety, stability, and durability of protection remain to be overcome. In contrast to many other viral infections, the pathology of filovirus hemorrhagic fevers in primate hosts is not linked to systemic viremia, but to a dysregulation of the immune system. Thus disease pathogenesis should also be viewed from an immunological perspective. An understanding of critical virus-host interactions that lead to development of a protective adaptive immune response instead of lymphocytopenia, thrombocytopenia, hemorrhage and death is essential for developing immune therapeutics or prophylactic vaccines. One possible link to EVD survival may be the kinetics of the host’s immune response. For humoral responses, faster immunoglobulin class switching in human convalescents compared to casualties in the Kikwit outbreak (1995) of EBOV has been described [4] as well as the more rapid development of cellular immunity. Whole blood transfer from human convalescents seemed to improve the outcome for treated patients [5]. These observations and the fact that non-human primate (NHP) survivors of EBOV challenge are immune to subsequent EBOV infection [6], suggest that prophylactic vaccination is possible. In a recent report from a human clinical trial of the “rVSV-ZEBOV” vaccine candidate described by Khurana et al. [7], the investigators demonstrate that the human antibody profile generated by this vaccine consists largely of IgM isotype antibody, with a lack of antibody class switching and affinity maturation. Furthermore, the antibody titers appear to decline rapidly after vaccination with only about 10–20% of peak titers remaining 84 days post vaccination and no apparent booster effect after another dose of vaccine. While the IgM antibodies demonstrated activity in a pseudovirion neutralization assay, their avidity was relatively low. This raises questions about the durability of protection afforded by this vaccine candidate and warrants further research into vaccine immunogenicity and potential prime-boost approaches.
Filoviruses are enveloped, negative strand RNA viruses. The viral RNA is packaged with viral nucleoprotein (NP) and the envelope is formed by the association of the viral matrix proteins VP40 and VP24 with the membrane containing the mature surface glycoprotein (GP) [8]. GP has been identified as the viral protein leading to cell surface binding and membrane fusion and has therefore been selected as the major candidate antigen which may also induce virus neutralizing antibodies, even though different mechanisms other than classical virus neutralization such as antibody-dependent cytotoxicity or cell-mediated immunity may also be required to clear EBOV infections [9]. Several preclinical challenge studies have demonstrated that immune responses to EBOV GP raised with various experimental approaches using viral vectors (VSV, various adenoviruses, or human parainfluenza virus (HPIV)) may be sufficient to protect NHP against death from EBOV infection [10–13]. The use of additional viral proteins (e.g., VP24, VP40, or NP) may contribute to vaccine efficacy and possibly also to the cross-protective potential of a candidate vaccine since they are more conserved amongst different filoviruses than the GPs. The cross-protective potential of additional virus proteins was shown indirectly in a comparative experiment in guinea pigs in which groups of animals were vaccinated with recombinant VSV vectors expressing only the GPs of EBOV, Sudan ebolavirus (SUDV), Tai Forest ebolavirus (TAFV) or Reston ebolavirus (RESTV) or immunized by infection with the four wild-type (non-guinea pig adapted) ebolavirus species which are non-lethal to guinea pigs. Only recipients of the recombinant VSV vaccine expressing EBOV GP were protected against challenge with guinea-pig adapted EBOV while animals immunized with the GPs of SUDV, TAFV, or RESTV succumbed to disease. In contrast, animals “immunized” by infection with each of the four non-adapted ebolaviruses were protected against lethal challenge with guinea pig-adapted EBOV virus independent of the species used for vaccination [14]. This suggests that the cross-protective potential must be found in adaptive responses raised by viral component(s) other than GP. One of these potential vaccine candidate antigens is NP which has been utilized in DNA vaccinations [15], adenovirus-vectored approaches [16] and as part of virus-like particle (VLP) vaccine development efforts [17]. NP is abundantly present in mature virions as it forms the nuclear core together with genomic RNA and has been shown to possess T-cell epitopes [18]. Studies have shown that Venezuelan Equine Encephalitis virus replicon particles (VRP) expressing NP can elicit cytotoxic T-cell responses in mice [19]. The matrix protein VP40, a major component of the virus particle, and the minor matrix protein VP24 are possible additional vaccine antigens. Both have shown protective potential in mouse challenge studies when administered in the form of VRPs [20]. Subsequent work showed that VRPs expressing VP24 or VP40 induce cytotoxic T lymphocytes (CTL) that confer protection in mice [21].
Multiple filovirus vaccine candidates employing recombinant technologies have demonstrated promise in preclinical studies; however, thus far the mechanisms by which the virus components induce protection are unknown. As expected, the GP has proven useful as a vaccine antigen in animals, including NHP, using recombinant VSV, HPIV or adenoviruses as vectors [10, 11, 13]. Recombinant protein antigens in the form of VLP’s produced in mammalian or insect cells have also been shown to induce protection in rodents and NHP [22]. In contrast to the recombinant EBOV and MARV VLP’s, inactivated MARV [23] and EBOV [24] induced only partial protection in NHPs. These results may be related to the structural damage caused by denaturation during irradiation of the viruses. The lack of efficacy may also be caused by incorrect presentation and/or processing of antigens, incorrect dosing, insufficient adjuvantation, or due to contaminating proteins.
Achieving proper conformation of complex viral proteins is often problematic and the Drosophila S2 expression system has demonstrated the ability to overcome the challenges and produce conformationally relevant envelope proteins for a number of viral vaccine targets. The native-like structure of dengue envelope proteins produced in this manner has been demonstrated through the determination of X-ray crystal structures [25, 26]. In contrast to virally vectored vaccines, DNA-vaccines or virus-like particles, formulations of recombinant subunits allow for delivery of well-defined antigen combinations to vaccinees, which are designed to achieve optimal safety and potency in diverse populations. Therefore, a detailed understanding of the mechanism by which protective responses are achieved with the individual antigens is required.
Here we present data on evaluating recombinant subunit filovirus proteins expressed in stably transformed Drosophila S2 cell lines for their potential to induce humoral and cell-mediated immune responses leading to protection against infection in the mouse model of EBOV.
2. Materials and Methods
2.1. Expression and purification
Expression vectors (pMT/BiP, Invitrogen, Carlsbad, CA) were generated by inserting the coding regions for EBOV GP (amino acids 33–647), VP40 (amino acids 1–326) or VP24 (amino acids 1–251) (all sequences are based on Zaire ebolavirus, Mayinga strain, Genbank accession number NC_002549). Drosophila S2 cells adapted to ExCell420 medium (Sigma-Aldrich, St. Louis, MO) were co-transformed with expression plasmids and selectable marker plasmid pCoHygro using the calcium phosphate coprecipitation method. Stable transformants were selected by adding hygromycin B to the medium. After selection was complete, cultures of the cell lines were induced by addition of 200 μM CuSO4 to the culture medium. Expression was verified by SDS-PAGE and western blot. For this, nitrocellulose membranes after western transfer were probed with Ebola hyperimmune mouse ascitic fluid (HMAF) obtained from the US Army Medical Research Institute of Infectious Diseases (USAMRIID), Frederick, MD. This was followed by treatment with a goat anti-mouse IgG alkaline phosphatase-conjugated secondary antibody (Southern Biotech, Birmingham, AL) and development with nitro-blue tetrazolium chloride and 5-bromo-4-chloro-3’-indolyl-phosphate (NBT/BCIP; Promega, Madison, WI) solid phase alkaline phosphatase substrate. The glycosylation status of the recombinant subunits was documented using either Peptide-N-Glycosidase F (PNGase F; NEB, Ipswich, Maine) to study N-linked glycosylation or a complete enzymatic deglycosylation kit (EDEGLY, Sigma, St. Louis, MO) following the manufacturer’s instructions.
Antigens were produced in 400mL spinner flasks or in a WAVE Bioreactor (GE Healthcare, Piscataway, NJ) using 2 or 10 L bag sizes (and 1–5L culture volumes) and were subsequently purified by immunoaffinity chromatography (IAC). Monoclonal antibodies specific for the individual proteins (Z-AC1-BG11 (EBOV VP24), M-HD06-A10A (EBOV VP40) and EGP13C6 (EBOV GP)) were obtained from USAMRIID, purified via protein A affinity chromatography and coupled to NHS-Sepharose (GE Healthcare, Piscataway, NJ) at 10mg/ml bed volume. For antigen purification, S2 cell culture medium containing recombinant protein was clarified and sterile filtered (0.2 μm pore size). The material was then loaded onto the respective IAC column, at a linear flow rate of approximately 2 cm/min. After the medium was loaded, the matrix was washed with 10 mM phosphate buffered saline, pH 7.2, containing 0.05% (v/v) Tween® 20 (PBST, 140 mM NaCl) followed by washing with 10 mM phosphate buffer, pH 7.2 (no detergent present). Bound protein was eluted from the IAC column with 20 mM glycine buffer, pH 2.5. The eluent was neutralized with 10 mM phosphate buffer, pH 7.2, buffer exchanged into 10 mM phosphate buffered saline, pH7.2 (PBS), and concentrated using Centricon Plus-20 devices (Millipore, Billerica, MA). The purified products were analyzed by SDS-PAGE with Coomassie blue or silver staining, western blot, and quantified by UV absorption. Purified recombinant proteins were stored frozen at −80°C until used for vaccine formulation. The control “NULL” antigen was prepared by concentrating and buffer exchanging supernatants from untransformed S2 cells grown under identical conditions to the S2 cell lines expressing recombinant proteins into PBS using Centricon Plus-20 devices.
2.2. Mouse immunogenicity studies – Vaccine formulation and immunization of mice
All work with animals was conducted in compliance with the Animal Welfare Act and other Federal statutes and regulations relating to animals and experiments involving animals and adhered to the principles stated in the Guide for the Care and Use of Laboratory Animals, NRC Publication, 1996 edition. All procedures were reviewed and approved by the appropriate Institutional Animal Care and Use Committees at the University of Hawaii and USAMRIID. All work with live virus was conducted in the BSL4 animal facility at USAMRIID.
For the immunogenicity studies, mice were immunized using four different adjuvants with different modes of action. A saponin-based, TLR-4 (toll-like receptor 4) agonist, GPI-0100 (Hawaii Biotech, Inc., Honolulu, HI) [27, 28] was used at doses of 100 or 250μg. In addition to directly activating the TLR4-pathway, saponins have the ability to modulate immune responses by intercalating into the cell membranes, thus allowing soluble protein antigens to enter the endogenous antigen presentation pathway for “cross presentation” resulting in activation of cytotoxic CD8+ T cells. Three emulsion-based adjuvants were tested: 1) ISA51 (Seppic, Fairfield, NJ) used at 50% v/v; 2) CoVaccine HT™ (an emulsion of squalane with immunostimulatory sucrose fatty acid sulphate esters and an adjuvant of Protherics Medicines Development Ltd., a BTG Company, London, United Kingdom) [29] used at a dose of 1 mg; and 3) Ribi R-700 (Sigma-Aldrich, St. Louis, MO) which in each mouse dose contains 50 μg monophosphoryl lipid A and 50 μg synthetic trehalose dicorynomycolate in a squalene-Tween 80 emulsion. Emulsion-based adjuvants act by sequestering antigens thereby promoting a “depot effect” whereby antigens are slowly released from the depot and provide a longer lasting immune stimulus. In addition, adjuvants containing TLR or PRR (pattern recognition receptor) agonists such as glycans or lipid A may also activate the innate immune system resulting in cytokine release and activation of effector lymphoid cells. Groups of 10 or 15 female BALB/c mice (8 weeks old) were vaccinated subcutaneously (s.c.) three times with individual subunit proteins at the chosen dose level (between 1–10 μg as indicated in the Results section below) and formulated with one of the four selected adjuvants at 4-week intervals. Vaccine formulations were prepared fresh for each vaccination day from frozen antigen stocks, adjuvant stock solutions and sterile PBS to give the desired dose within a final volume of 0.2 mL. Serum samples were obtained 2 weeks after the second vaccination. Five mice from each group were euthanized on the fourth and/or seventh day after the third vaccination and splenectomies were performed for preparation of splenocyte cultures. The remaining five or ten mice from each group were euthanized 14 days after the third vaccination and individual serum samples collected from each animal.
2.3. Mouse Efficacy studies
Groups of ten 6 week-old female BALB/c mice were immunized s.c. 3 times at days 0, 28 and 56 with 10μg doses of VP24, VP40 and/or GP formulated with either 100 μg of GPI-0100 or 1 mg of CoVaccine HT™, or without adjuvant. Negative control groups received equivalent doses of adjuvant only. Serum samples were collected via tail bleeds 2 weeks after each immunization to determine ELISA IgG antibody titers against irradiated EBOV. Approximately one month after the last vaccination, mice were transferred into the BSL4 animal facility and challenged intraperitoneally (i.p.) with 100 pfu of mouse adapted EBOV (ma-EBOV) [30]. Mice were observed daily for signs of illness and death. Surviving animals were euthanized 28 days after challenge.
2.4. Analysis of antibodies by ELISA
Sera of individual mice were titrated for IgG specific to the recombinant VP24, VP40 and GP proteins by standard ELISA technique using plates coated with purified recombinant antigens or plates coated with irradiated whole virus [31]. The titers presented are defined as the dilution of antiserum yielding 50% maximum absorbance values (EC50) and was determined using a sigmoidal dose response curve fitting algorithm (Prism, Graphpad Software, San Diego, CA). Alternatively, endpoint titers were determined. They were defined as the highest dilution yielding an absorption (A405) of 0.2 above background.
2.5. Proliferation and cytokine analysis of immune splenocytes
Splenectomies were performed on immunized mice four and/or seven days post final vaccination and splenocyte suspensions prepared. Erythrocytes were lysed with an NH4Cl solution (0.15 M NH4Cl, 10 mM KHCO3, 0.1 mM EDTA, pH 7.3) and the splenocytes were then collected by centrifugation. The resultant cell pellet was washed and resuspended in cell culture medium. Cell counts were performed on each suspension using a cell counter (Beckman Coulter, Brea, CA), and the suspensions diluted to 4 × 106 cells/mL. For proliferation assays, 4 × 105 splenocytes (0.1 mL) were dispensed into wells of a 96-well cell culture plate. EBOV VP24, VP40 or GP antigens (1 μg /well) in a volume of 0.1 mL were then added to the cell suspensions (in quadruplicate). Unstimulated (antigen omitted) cell suspensions, phytohemagglutinin (PHA, 10 μg/mL, final concentration) stimulated cell suspensions, and “NULL” stimulated cell suspensions (buffer exchanged proteins from S2 cell cultures to document the potential effect of contaminants in antigen preparations) were included as controls. Cultures were incubated at 37°C, 5% CO2, in humidified chambers for 7 days (3 days for PHA stimulated cultures), and then one microcurie of tritiated (methyl-3H) thymidine (60 Ci/mmol; ICN Biomedicals, Inc., Irvine, CA) was added to each well (in a volume of 0.01 mL), and incubation continued for 18 hrs. Cell cultures were harvested onto glass fiber filtration plates (Filtermate Plate Harvester, PerkinElmer Instrument Co., Waltham, MA) and analyzed for radioactivity using the TopCount Microplate Scintillation and Luminescence Counter (PerkinElmer Instrument Co., Waltham, MA). The stimulation index (SI) was calculated by dividing the specific stimulation counts by the unstimulated cell counts for each suspension. An SI of 3 or greater was considered significant (positive).
For cytokine production assays, 2 ×106 splenocytes (in 0.5 mL) were dispensed into wells of a 24-well cell culture plate and stimulated with equal volumes of antigens or controls yielding final concentrations of 106 cells/mL and 5 μg/mL of antigen or pokeweed mitogen (instead of PHA) control. Unstimulated controls and “null” antigen controls were also included. The culture supernatants were harvested on day 5 post-stimulation and frozen until analyzed for secreted cytokines. The cytokines interferon-gamma (IFN-γ), tumor necrosis factor-alpha (TNF-α), and interleukins 4, 5, and 10 were assayed by standard ELISA technique or by using a flow cytometric cytokine bead array assay (BD Biosciences, San Jose, CA).
2.6. Passive protection studies in BALB/c mice
Formulations containing 10μg EBOV GP or VP24 and 1mg CoVaccine HT™ were administered s.c. three times to groups of 35 female BALB/c mice at 4-week intervals. Fourteen days after the last vaccination, 30 mice from each group were euthanized and serum samples collected by cardiac puncture. Serum samples obtained from each group were pooled and subsequently transferred i.p. to ten naïve BALB/c mice (1.0mL per mouse). Splenocytes were isolated from the spleens of immunized mice and administered i.p. to groups of ten BALB/c mice (female, 20–25 g) at 7×107 cells/mouse. T-cells were separated from other cell types contained in splenocyte populations by negative selection (using MACs separation technique; Invitrogen, Carlsbad, CA). Separated T-cells were administered (i.p.) to naïve mice at rates of 1.5×107 cells/mouse (high dose) and 1.5×106 cells/mouse (low dose). Mice were subsequently transferred into the BSL4 laboratory and challenged approximately 24 hours post serum or cell transfer by i.p. injection with 1000 pfu (30,000 LD50) of ma-EBOV. Survivors were euthanized 28 days post challenge and serum samples collected from selected groups.
2.7. Statistical analysis
Significant differences in antibody titers, stimulation indices, or cytokine production between immunized groups of mice were determined by unpaired t tests (GraphPad Prism). P < 0.05 was considered to be significant. Significant differences in survival between immunized (or non-immunized control) groups subsequently challenged were determined by the Fisher exact probability test (GraphPad Prism). P < 0.05 was considered to be significant.
3. Results
3.1. Expression of filovirus immunogens in Drosophila S2 cells
Stably transformed insect cell lines expressed proteins and showed yields between 10–15mg/L cultured in either spinner flasks or Wave bioreactor. Figure 1 illustrates the successful expression of secreted Ebola virus subunit proteins. Expression levels were estimated to be >10μg/ml for all three proteins based on SDS-PAGE gels. GP, VP24 and VP40 antigens were subsequently purified by IAC to 85–95% homogeneity (Figure 2).
Figure 1. Expression of recombinant EBOV subunits from Drosophila S2 cells.
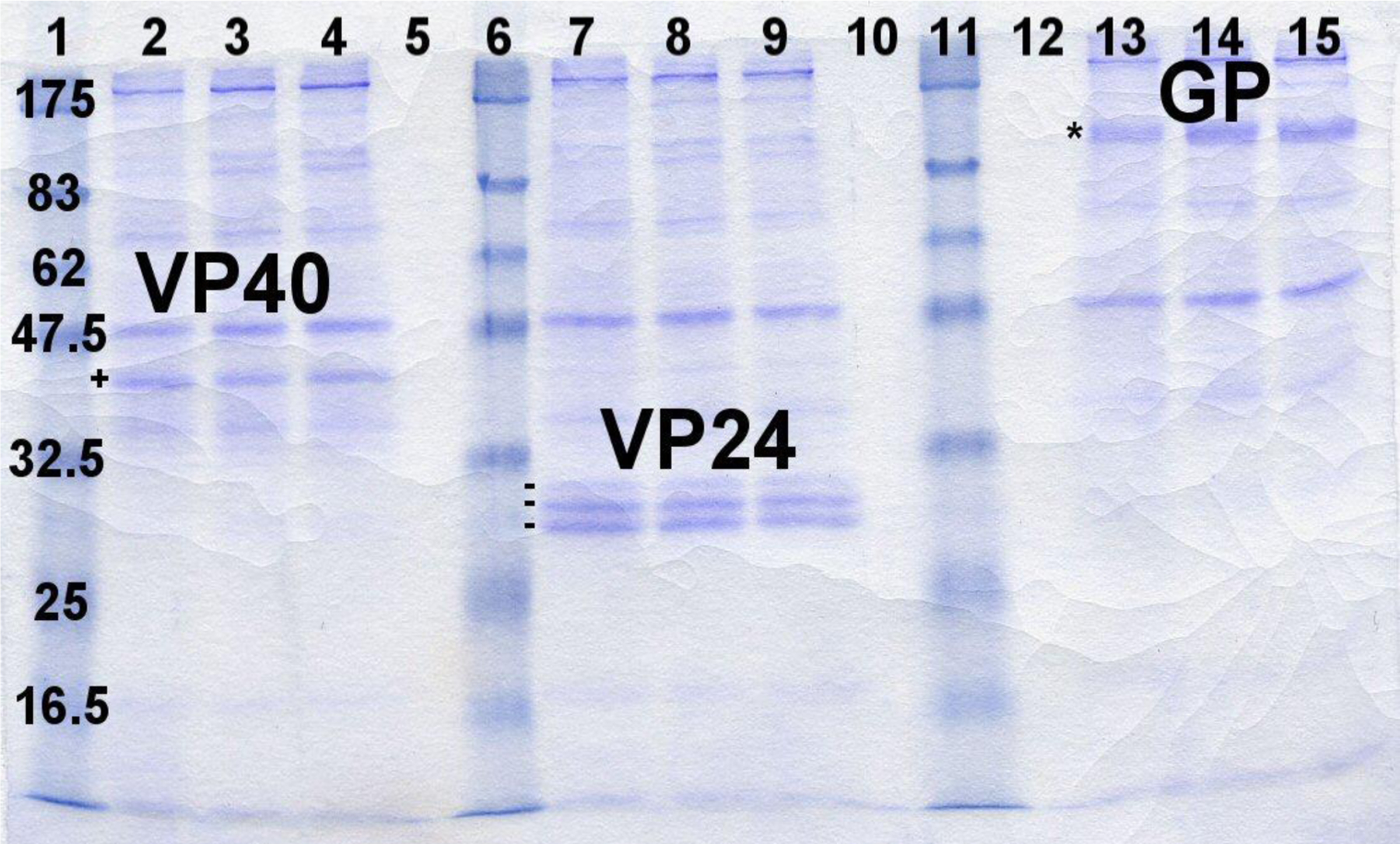
Coomassie stained SDS-PAGE gel (12%) featuring supernatants from Ebola subunit expression lines. Lanes 1, 6 and 11 – Molecular weight standard (sizes in kDa), Lanes 2–4: Ebola VP40 (one protein band marked: +), Lanes 7–9: Ebola VP24 (3 bands marked: −), Lanes 13–15: Ebola GP (one protein band marked: *)
Figure 2. Purified recombinant EBOV proteins.
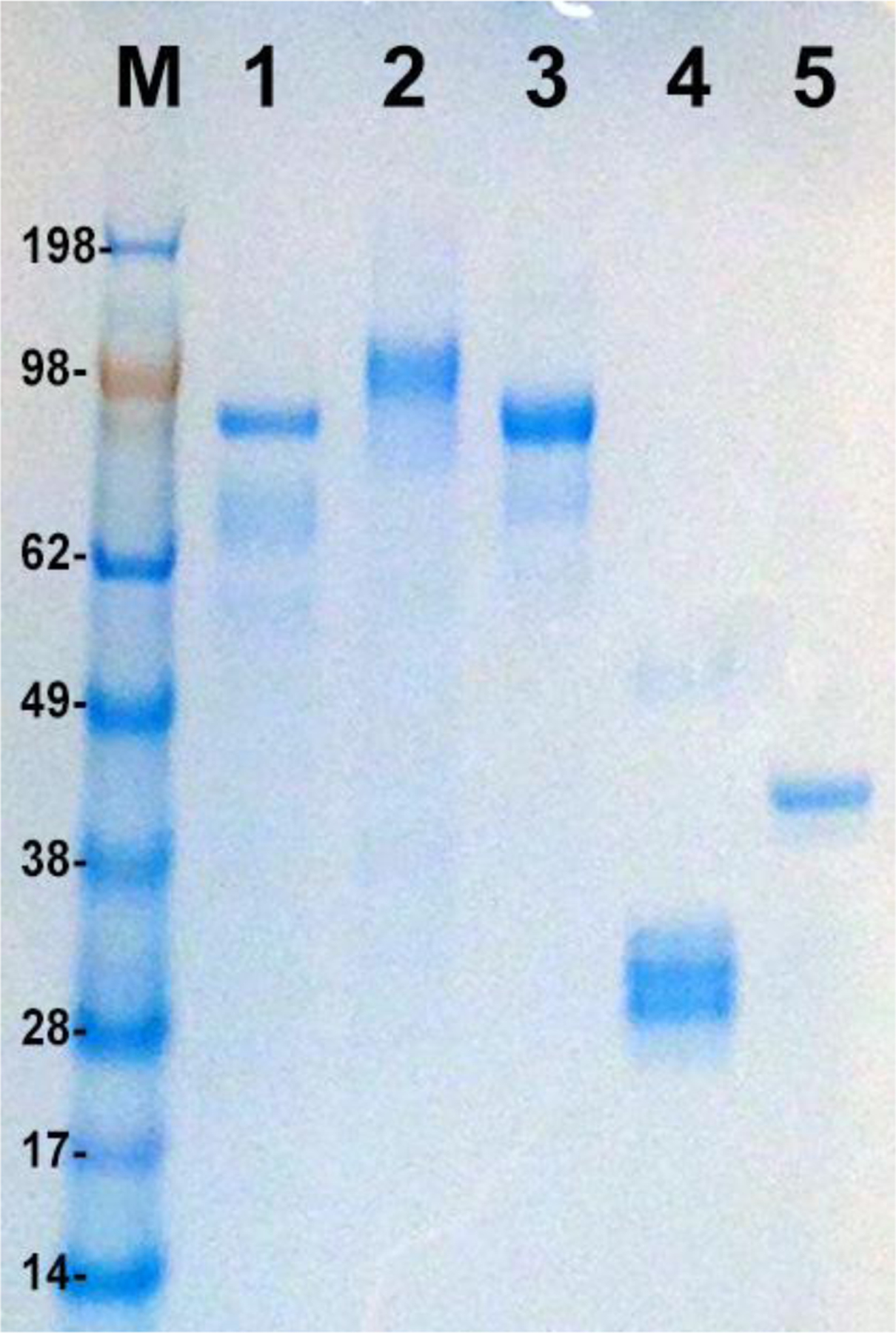
4–12% NuPAGE gel (Invitrogen, Carlsbad, CA) loaded with 1μg each of insect cell expressed, immunoaffinity purified recombinant filovirus proteins. Lane M: Prestained molecular weight marker (Seeblue Plus2, Invitrogen); Lane 1: EBOV GP, Lane 2: MARV GP, Lane 3 SUDV GP, Lane 4: EBOV VP24, lane 5: EBOV VP40. SUDV and MARV GP proteins are expressed using an analogous process to EBOV proteins and are shown here for reference.
Analysis of the glycosylation status of each of the individual antigens was conducted using enzymatic deglycosylation with analysis on protein gels. For GP, the PNGase treatment resulted in a protein which migrated faster on SDS-PAGE, consistent with the removal of the carbohydrate side chains from all N-linked glycosylation sites (supplementary Figure S1). In contrast, no evidence was found for O-linked glycosylation using the EDEGLY kit. Reduction of the GP protein results in separation of GP1 and GP2 fragments (figure S1) and confirms that the furin cleavage site is being processed completely during post-translational processing. PNGase treatment suggests that the VP40 with secretion signal is produced as a uniform product that is glycosylated at one glycosylation site (documented on protein gel, Figure S2) and by mass spectrometry (Figure S3). In contrast, VP40 expressed intracellularly is not glycosylated (data not shown). As expected based on previous work [32], recombinant VP40 in solution shows dimerization as well as higher oligomerization. VP24 contains three internal N-linked glycosylation sites which are partially processed during passage through the secretion pathway resulting in the triplet seen in Figures 1 and 2. This finding has also been confirmed by mass spectrometry (Figure S4).
3.2. Recombinant EBOV antigens raise humoral and cellular immune responses in mice
The purified candidate EBOV immunogens were first used to test their potential in generating humoral and cellular immune responses in BALB/c mice. For this, the three EBOV antigens were tested individually at 10 μg doses in formulations with two functionally different adjuvants, ISA-51 (water-in-oil emulsion) and GPI-0100 (saponin-based preparation). Antibody titers after three vaccinations observed by ELISA using homologous recombinant antigens as coating antigens are shown in Figure 3.
Figure 3. Humoral responses to recombinant EBOV antigens.
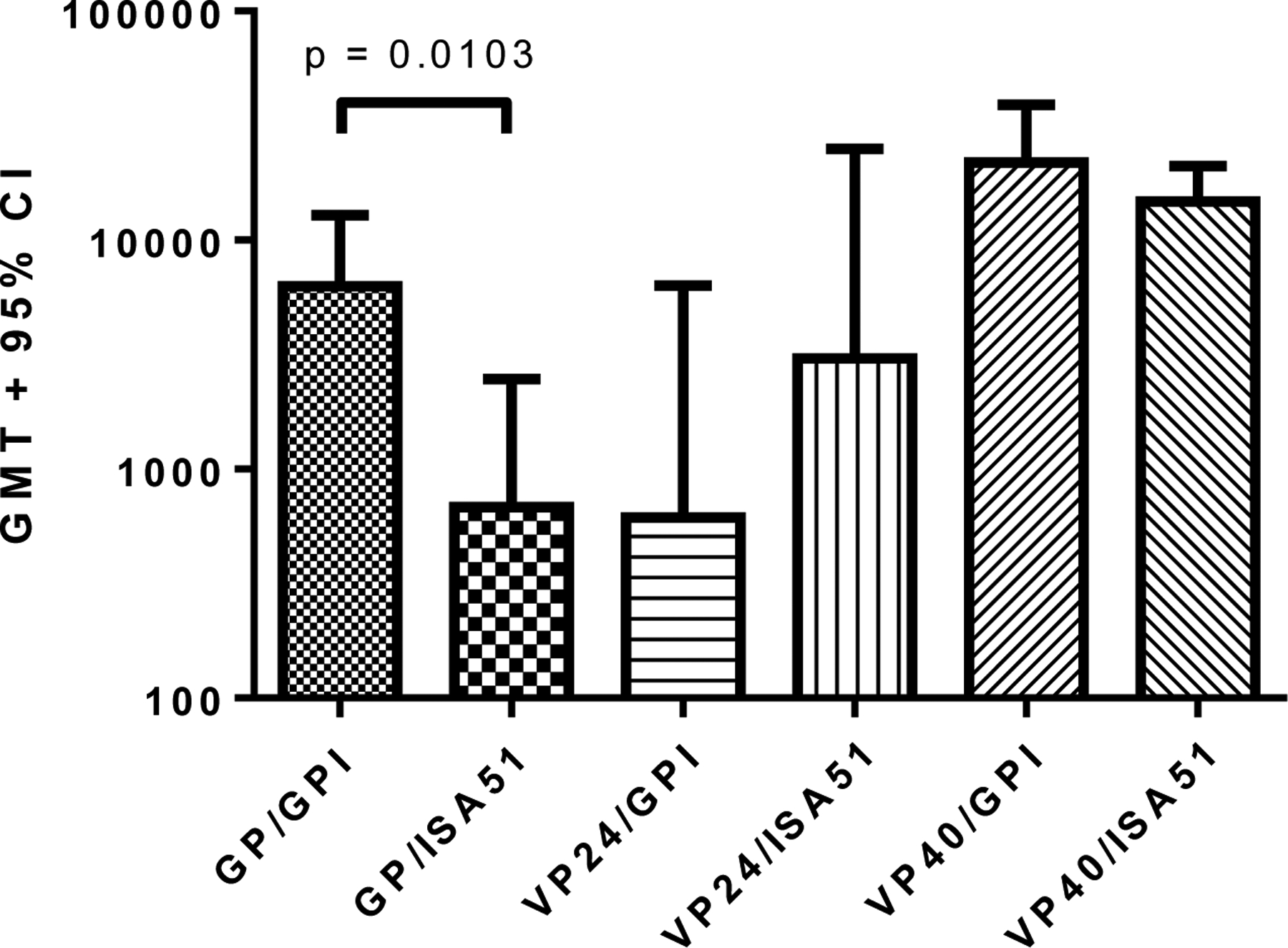
ELISA IgG antibody titers (EC50) calculated using a sigmoidal dose-response, variable slope program (Graphpad Prism). The GMT + 95% CI is plotted for each group (n=5). Plates were coated with the homologous immunizing antigen. Control groups (mice immunized with adjuvant only) were completely negative (EC50 values << lowest dilution tested, 1:250). Antibody titration curves for all groups including control groups are shown in Figure S5. Differences in antibody titers between GP/GPI-0100 and GP/ISA51 immunized groups were significant (p<0.05). Differences in antibody titers between VP24/GPI-0100 and VP24/ISA51 immunized groups, and between VP40/GPI-0100 and VP40/ISA51 immunized groups were not significant (p>0.05).
Antibody titers generated against Ebola GP and VP24 were comparatively low after the first vaccination, but increased following the second and third vaccinations (Table SI). In contrast, VP40-specific antibody titers were elicited after only one vaccination and rose above the maximum dilution tested after the third vaccination. Assays for cell-mediated immunity (after three vaccinations) demonstrated that lymphocyte proliferation and IL-4 responses from immune mouse splenocytes were higher in groups administered vaccine formulated with GPI-0100 than with ISA-51 (except for VP40 stimulated proliferation; Figure 4) as were IL-5 and IL-10 responses (data not shown). IFN-γ responses were strong in all groups and suggest the ability of the tested antigens to induce potent cell mediated immunity.
Figure 4. Cell-mediated immune responses raised by recombinant antigens.
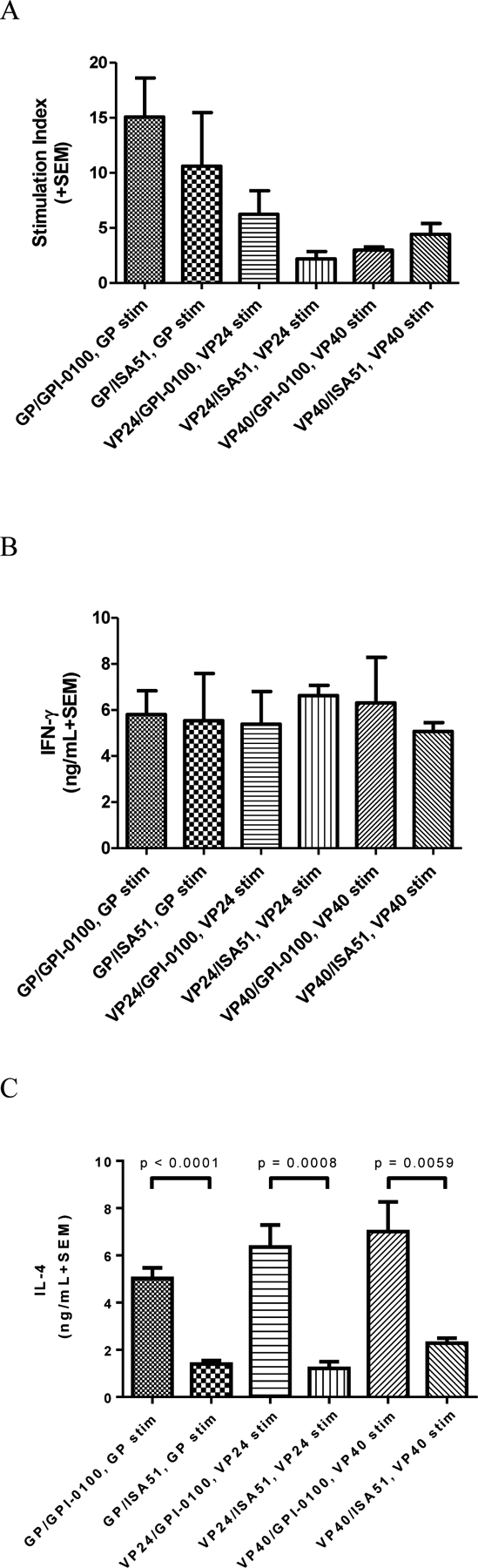
Panel A: Mean (n=5 per group) lymphocyte proliferation (indicated as stimulation index, SI) in vitro from immune splenocytes stimulated with homologous antigens. Mean SI from mitogen (PHA) stimulated cultures varied in the range of 4.2–42. Mean SI in splenocyte cultures from adjuvant only immunized mice re-stimulated with GP, VP24, or VP40 was <2.0 in all cases.
Panel B: Mean (n=5 per group) IFN-γ production in vitro from immune splenocytes re-stimulated with homologous antigens. Mean IFN-γ production from PWM stimulated cultures varied in the range of 15–58 ng/mL. Mean IFN-γ production from control (unstimulated) cultures was <0.5 ng/mL in all cases. Mean IFN-γ production in splenocyte cultures from adjuvant only immunized mice re-stimulated with GP, VP24, or VP40 was <1.0 ng/mL in all cases.
Panel C: Mean (n=5 per group) IL-4 production in vitro from immune splenocytes re-stimulated with homologous antigens. Mean IL-4 production from PWM stimulated cultures varied in the range of 5.7–11.7 ng/mL. Mean IL-4 production from control (unstimulated) cultures was <0.25 ng/mL in all cases. Mean IL-4 production in splenocyte cultures from adjuvant only immunized mice stimulated with GP, VP24, or VP40 was <0.2 ng/mL in all cases. Differences in IL-4 production between groups immunized with formulations containing GPI-0100 or ISA51 were significant (p < 0.05) between the groups immunized and re-stimulated using the same antigen. Differences in IFN-γ production or proliferation were not significant (p > 0.05) for formulations using the two different adjuvants suggesting that adjuvant has a lower effect on Th1 type responses.
3.3. Antigen dose response with selected adjuvants
BALB/c mice were immunized with varying amounts of GP antigen to determine the effect of increasing antigen doses on the immune response. The glycoprotein was formulated at three different doses (1, 3, and 9 μg) with GPI-0100, CoVaccine HT™, or Ribi R-700. Results are shown in table SII and Figure 5. Similar to the first experiment, antibody responses to GP are relatively low following the first vaccination and all groups immunized with GP showed a typical (increasing) dose-related response following the second vaccination (table SII). By the third vaccination the titers induced in the GPI-0100 adjuvanted formulation appeared to reach a plateau as dose response was no longer evident, while there was still evidence of a dose response in the groups receiving formulations containing CoVaccine HT™ or Ribi. The GPI-0100 formulation yielded the highest antibody titers, while the Ribi R-700 adjuvanted formulation yielded the lowest antibody titers (Figure 5A). In general, antigen-stimulated lymphocyte proliferation and cytokine production did not demonstrate consistent antigen dose responses (Figure 5B–D). With GPI-100 or CoVaccine HT™, there was no antigen dose effect evident at all, with the exception of IL-5 with CoVaccine HT™. With Ribi R-700, there appeared to be a large increase in lymphocyte proliferation between the 1, 3, and 9 μg doses, but these differences were not statistically significant due to the large SEM. In some cases, a decreasing tendency was observed in responses with increasing antigen dose.
Figure 5. Humoral responses are affected by adjuvant selection and antigen dose.
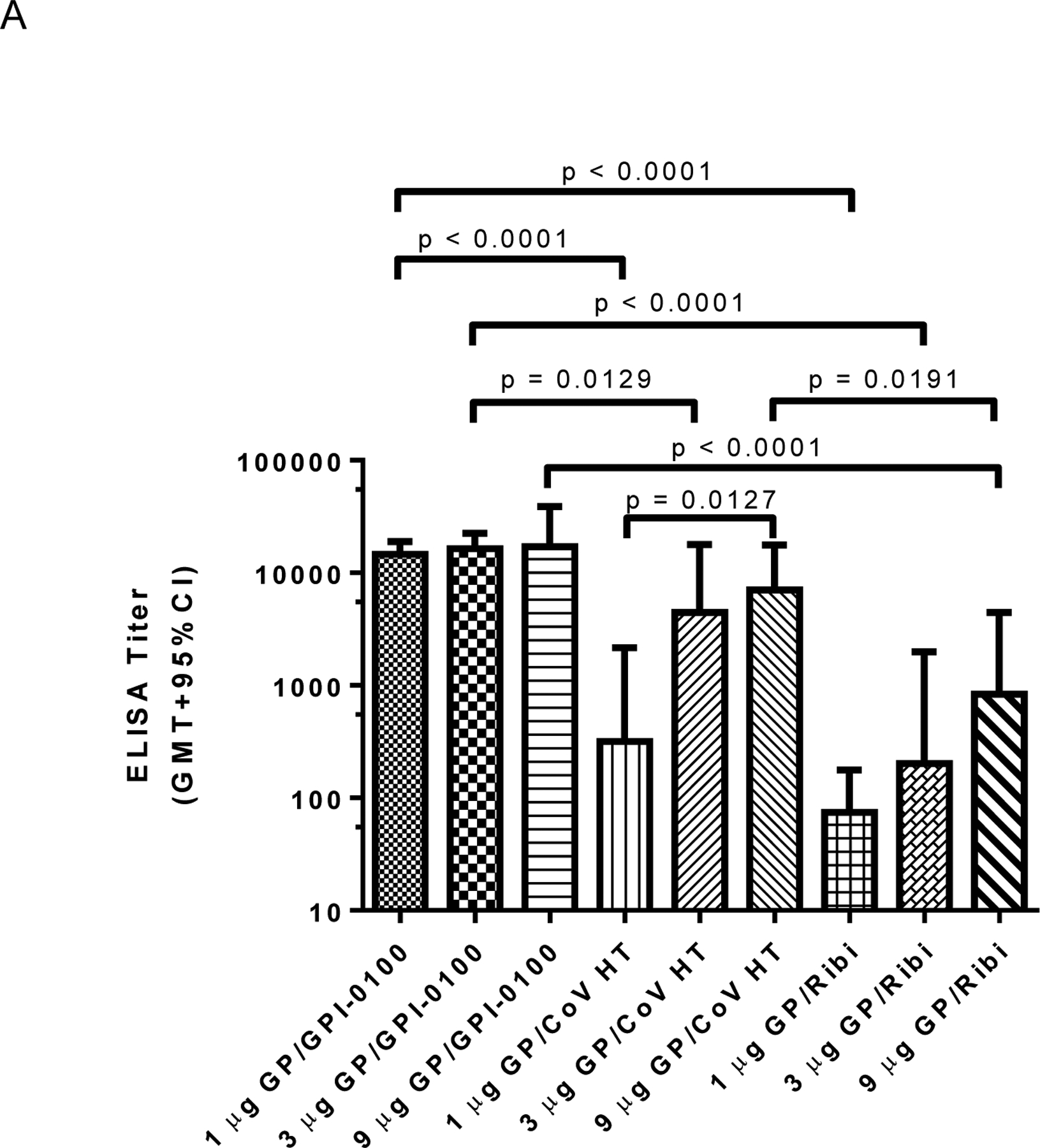
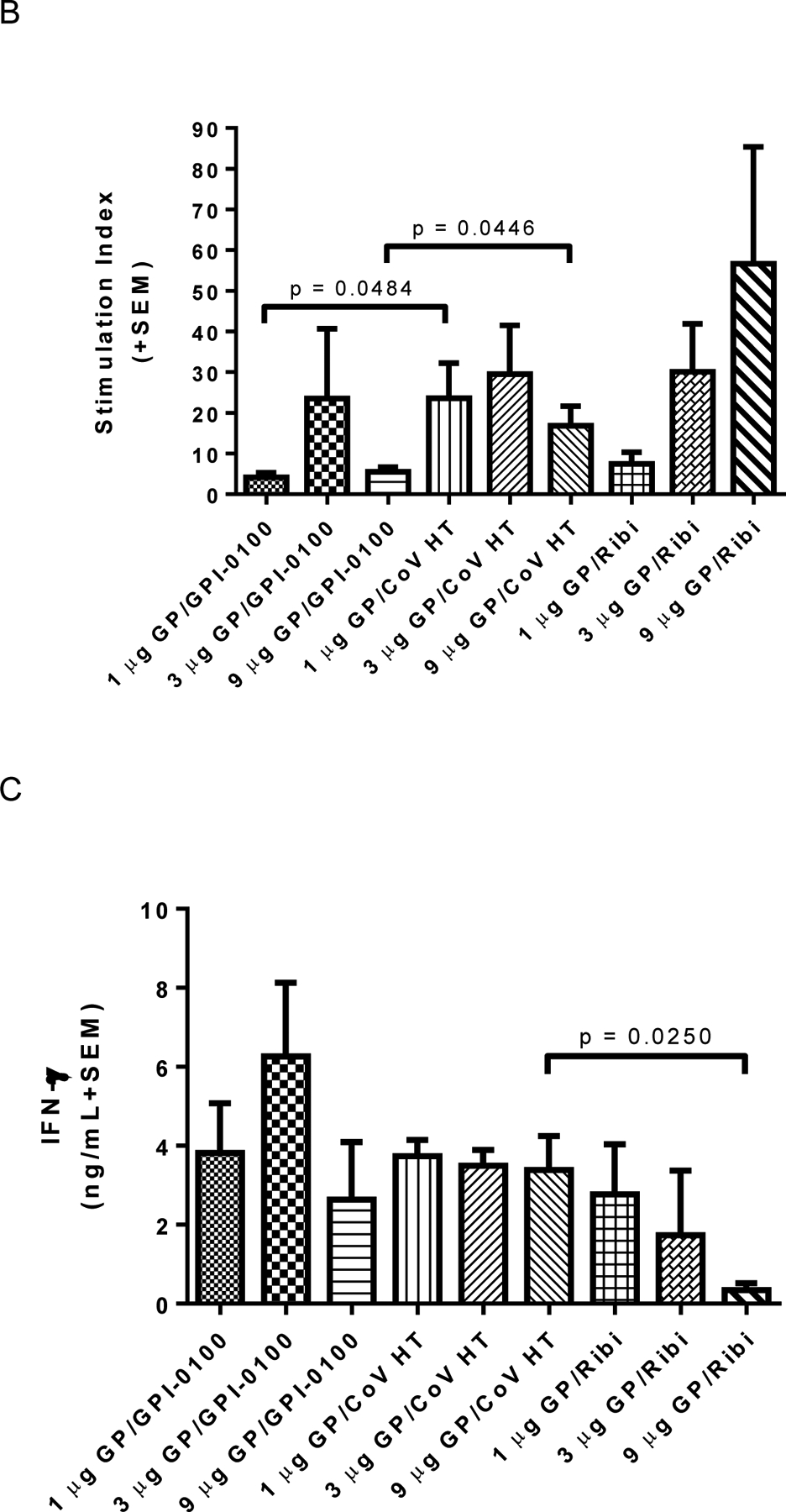
Panel A: ELISA IgG antibody titers post third vaccine dose using plates coated with homologous antigen. EC50 titers from individual animals (n=4 per group) were calculated using a sigmoidal dose-response, variable slope program (Graphpad Prism). The GMT + 95% CI is plotted for each group. Significant differences between groups are indicated by overlying horizontal bars on Figure 5A. At the same antigen dose levels of both 1 or 3 μg of EBOV GP, differences between groups immunized with formulations containing GPI-0100 showed significantly higher titers than formulations containing CoVaccine HT™ (CoV HT) or Ribi. Differences between groups immunized with 9 μg GP and GPI-0100 or Ribi and with 9 μg GP and CoVaccine HT™ or Ribi were also significant (p = 0.0191 for both comparisons). IgG titers in formulations containing CoVaccine HT™ showed the only statistically significant dose response when comparing the 1 and 9 μg doses of vaccine. Differences between all other groups were not significant (p > 0.05). Panel B: Mean (n=6 per group) lymphocyte proliferation (stimulation index, SI) from immune splenocytes stimulated with homologous antigen in vitro, harvested at day 4 (n=3) or day 7 (n=3) post booster vaccination. Mean SI from mitogen (PHA) stimulated cultures varied in the range of 2.4–50. Mean SI in splenocyte cultures from adjuvant only immunized mice stimulated with GP was <1.7 in all cases. Significant differences between groups are indicated by overlying horizontal bars and showed significant differences between CoVaccine HT™ and GPI-0100 adjuvanted formulations at the 1 and 9 μg GP dose levels. No other pairwise comparisons yielded significant differences. Panel C: Mean (n=3 per group) IFN-γ production in vitro from immune splenocytes stimulated with homologous antigen. Mean IFN-γ production from control (unstimulated) cultures was <0.35 ng/mL in all cases except the 1 μg GP/Ribi group, which had 0.97 ng/mL. Mean IFN-γ production in splenocyte cultures from adjuvant only immunized mice stimulated with GP was undetectable (<0.1 ng/mL) in all cases. Significant differences between groups are indicated by overlying horizontal bars and showed a significant difference only between GPI-0100 and Ribi adjuvanted formulations at the 9 μg GP dose level. No other pairwise comparisons yielded significant differences. Panel D: Mean (n=3 per group) IL-5 production in vitro from immune splenocytes stimulated with homologous antigen. Mean IL-5 production from control (unstimulated) cultures was undetectable (<0.1 ng/mL) in all cases. Mean IL-5 production in splenocyte cultures from adjuvant only immunized mice stimulated with GP was undetectable (<0.1 ng/mL) in all cases. Significant differences between various groups are indicated by overlying horizontal bars. No other pairwise comparisons yielded significant differences.
3.4. Recombinant EBOV antigens elicit protection against homologous challenge with ma- EBOV
Based on the results of our immunogenicity studies, lead candidate vaccines were formulated using individual recombinant EBOV proteins, or a mixture of all three, for a mouse challenge study. Figure 6 summarizes these vaccine candidates’ immunogenicity based on humoral responses and Table I provides the documentation of their protective efficacy. IgG titers verify good immunogenicity of all the proteins, especially in adjuvanted groups. While VP24 antibody titers appear lower, this is due to using irradiated (whole) virus as coating antigen instead of recombinant subunits, as the VP24 antigen is only a minor component of the virus localized inside the particle and thus would not result in as much antibody binding to coating antigen as when animals were immunized with GP or VP40. Formulations containing CoVaccine HT™ induced the highest titers with all antigens and the titers, as previously observed, reached near maximal level after two vaccinations (Table SI). Titers induced by the GPI-0100 based formulations were lower than titers generated by CoVaccine HT™ formulations, but higher than those induced with the unadjuvanted antigens (Figure 6). Three vaccinations were required to induce maximal titers in mice with either the unadjuvanted or GPI-0100 adjuvanted formulations (Table SI).
Figure 6. ELISA IgG antibody titers (endpoint) to irradiated whole virus after three immunizations and prior to virus challenge.
Mice were immunized with 10 μg of GP, VP24, VP40, or 10 μg each of GP+VP24+VP40 with and without adjuvants. Log10 antibody titers against irradiated EBOV as coating antigen are shown for all formulations containing antigens. Endpoint titers in control groups (mice immunized with either adjuvant alone) were 1.76 and 2.14 for GPI-0100 and CoVaccine HT™, respectively.
Table I.
Recombinant Ebola virus subunits protect mice against live virus challenge
| Group no. | Immunogena | Adjuvant | Survival (day 20 post challenge) | P value vs. adjuvant control groupb |
|---|---|---|---|---|
| 1 | GP | NONE | 7/10c | 0.0015 |
| 2 | GP | GPI-0100 | 9/10c | 0.0005 |
| 3 | GP | CoVaccine HT™ | 10/00 | <0.0001 |
| 4 | VP24 | NONE | 0/10 | >0.05 |
| 5 | VP24 | GPI-0100 | 0/10 | >0.05 |
| 6 | VP24 | CoVaccine HT™ | 6/10c | 0.0054 |
| 7 | VP40 | NONE | 0/10 | >0.05 |
| 8 | VP40 | GPI-0100 | 0/10 | >0.05 |
| 9 | VP40 | CoVaccine HT™ | 0/10 | >0.05 |
| 10 | GP + VP40 + VP24 | NONE | 9/10 | <0.0001 |
| 11 | GP + VP40 + VP24 | GPI-0100 | 10/10 | <0.0001 |
| 12 | GP + VP40 + VP24 | CoVaccine HT™ | 10/10 | <0.0001 |
| 13 | NONE | NONE | 0/9 | --- |
| 14 | NONE | GPI-0100 | 1/10c | --- |
| 15 | NONE | CoVaccine HT™ | 0/10% | --- |
Mice were immunized with 10 μg of each antigen by the i.m. route.
Adjuvant control groups: 13 (no adjuvant), 14 (GPI-0100), 15 (CoVaccine HT™)
Animals showed signs of illness for part of the study(e.g. ruffled fur).
Mice were challenged on day 23 after the 3rd vaccination by i.p. injection with ma-EBOV. Morbidity and mortality within individual groups are shown in Table I. GP alone or formulated with GPI-0100 afforded a high level of protection against mortality but not morbidity. In contrast, GP formulated with CoVaccine HT™ showed 100% protective efficacy against both morbidity and mortality demonstrating the protective potential of the critical GP antigen. The two formulations containing the combination of three antigens co-administered with GPI-0100 or CoVaccine HT™ adjuvant showed full protection against both morbidity and mortality. Surprisingly, immunization of animals with the unadjuvanted antigen combination yielded 90% protective efficacy against morbidity and mortality. Results with unadjuvanted individual proteins generally showed either no protection or a moderate protection level, suggesting a synergistic effect of the combination.
3.5. Protective efficacy in mice is based on cellular & humoral immune responses
Since individual GP or VP24 subunits were shown to elicit protection in immunized mice, we were interested in identifying the immune mechanisms of protection for these two antigens by performing passive transfer experiments using serum or spleen cells from immunized mice. Pooled anti-GP or anti-VP24 immune sera, whole splenocyte preparations, or isolated T-cells were administered i.p. to naïve BALB/c mice which were challenged approximately 24 hours later. Pre-challenge sera analyzed for antigen specific ELISA IgG titers showed GMT (EC50 titers) >100,000 for both antigens after two or three vaccinations. Direct challenge controls verified previous findings of full protection in GP-vaccinees and partial protection in animals receiving the VP24-only formulation (Table II). Survivors were euthanized 28 days post challenge and serum samples collected from selected groups. Post-challenge antibody titers to GP and VP40 in survivors are shown in Figure 7.
Table II.
Passive transfer of immune serum or immune cells protects naïve BALB/c mice against lethal challenge.
| group | n | treatment | Survivors | P value vs. control group |
|---|---|---|---|---|
| 1 | 5 | GP + CoVaccine HT™ (direct) | 5/5 | 0.0040 |
| 2 | 5 | VP24 + CoVaccine HT™ (direct) | 2/5 | >0.05 |
| 3 | 5 | CoVaccine HT™ (direct) | 0/5 | Adjuvant control group |
| 4 | 10 | GP serum (1 ml)a | 9/10 | <0.0001 |
| 5 | 10 | VP24 serum (1 ml)a | 1/10 | >0.05 |
| 6 | 10 | Naïve | 0/10 | Challenge control group |
| 7 | 10 | GP T cells hi (1.5×10^7)b | 7/10 | p < 0.05f |
| 8 | 10 | VP24 T cells hi (1.5 ×10^7)b | 8/10 | p < 0.05f |
| 9 | 10 | GP T cells low (1.5×10^6)c | 5/10 | p < 0.05f |
| 10 | 10 | VP24 T cells low (1.5 ×10^6)c | 5/10 | p < 0.05f |
| 11 | 10 | GP+VP24 T cells hi (1.5 ×10^7 both)d | 8/10 | p < 0.05f |
| 12 | 10 | GP+VP24 T cells low (1.5×10^6 both)d | 6/10 | p < 0.05f |
| 13 | 10 | GP spleno hi (7×10^7)e | 8/10 | p < 0.05f |
| 14 | 10 | VP24 spleno hi (7 ×10^7)e | 5/10 | p < 0.05f |
| 15 | 10 | GP+VP24 spleno hi (7×10^7 both)e | 8/10 | p < 0.05f |
1 ml of immune serum per mouse administered i.p.,
1.5×107 T-cells/mouse administered i.p.,
1.5×106 T-cells/mouse administered i.p,
mixed cells from group 1 (GP immunized) + group 2 (VP24 immunized) animals; indicated amount of cells administered from both groups into each animal,
Splenocyte (unfractionated) transfers: 7×107 cells/mouse,
Normal serum, T cell or splenocyte transfers were conducted in the past and have shown that the same amount of normal serum or number of normal T cells or splenocytes administered to mice yield 100% fatalities with the identical challenge virus and dose as administered in this experiment. Thus, all groups of mice receiving anti-GP serum or immune cells in this experiment had significant protection (p < 0.05) compared to mice receiving normal serum or cells.
Figure 7. IgG ELISA antibody titers (EC50) against recombinant EBOV GP and VP40 after live virus challenge in the passive protection experiment.
Serum samples of all surviving animals in selected groups were collected at the end of the study and after irradiation analyzed for IgG titers against EBOV GP and VP40 (individually).
Panel A: Antibody titers to GP antigen. Panel B: Antibody titers to VP40 antigen.
As expected, transfer of GP-specific antiserum produced near complete protection in naïve recipients, while VP24-specific serum did not (Table II; selected Kaplan-Meier survival plots are shown in Figure S6). Protected animals receiving GP-specific serum and the directly challenged GP-vaccinees showed no weight loss (Figure S7), an indicator of morbidity in the model. Post-challenge ELISA analysis was performed as induction of GP and VP40-specific IgG responses in the naïve recipients may indicate viral replication. Anti-GP ELISA titers in serum from directly challenged mice remained steady (Figure 7A), while post challenge anti-VP40 titers observed (Figure 7B) were extremely low suggesting that no or only minimal viral replication occurred. Isolated T-cells as well as whole splenocyte preparations protected the majority of naïve recipients from death. For T-cell transfer a dose-dependency was seen for individual and mixed cell populations. The post-challenge serum samples showed equivalent IgG titers against both antigens in all groups of immune cell adoptees but one: animals receiving whole mixed splenocytes developed considerably higher anti-GP titers. This result is very likely due to activation of GP-specific memory B-cells that are part of the whole splenocyte preparation. In summary, this experiment demonstrated that recombinant GP as well as VP24 not only induce potent humoral responses, but also generate functional cellular immune responses in T-cells as well that confer protection against viral challenge.
4. Discussion
Expression of the recombinant EBOV antigens from Drosophila S2 cells yielded high quality protein secreted into the culture medium. GP appears as a single band product indicating complete processing of its (N-linked) glycosylation sites and the furin cleavage site is processed completely leading to separation of GP1 and GP2 regions upon reduction of disulfide linkages. Despite an absence of O-linked glycosylations, the purified recombinant GP demonstrates excellent immunogenic properties and also reacts with EBOV GP specific antibodies in convalescent serum or serum from immunized rodents and primates (data not shown). In contrast to the proteins present in virus infected cells, the intrinsic glycosylation sites of recombinant VP24 and VP40 are processed either partially (at three sites for VP24) or uniformly (at one site for VP40) during secretion into the culture supernatant. Nevertheless, these post-translational modifications of the proteins did not affect purification using IAC methods, their reactivity with antigen-specific antibodies from convalescent serum samples, or immunogenic potential. This eliminates the need for cell lysis and allows for use of IAC as a gentler purification method that protects native conformation of the antigens.
The use of recombinant proteins as vaccine antigens is a standard approach for contemporary vaccine development. However, in the filovirus field some earlier setbacks in experiments with inactivated viruses [24] or recombinant proteins [33] had a significant impact on application of recombinant subunits to the formulation of vaccine candidates. Expression yields of full length GP in mammalian cells are typically poor (in the range of 1 mg/L when transiently expressed from transfected cells) and purification may be problematic due to the amount of contaminants relative to target protein and the diversity of protein species achieved via processing of O-linked glycosylation sites. More recent approaches therefore use mammalian cell expressed GP fused to the Fc fragment of human IgG1 [34] or, similarly, a plant expressed Ebola Immune Complex (EIC) composed of human or murine antibodies and the GP1 region of EBOV GP [35]. Both of these chimeric antigens can be purified using standard affinity chromatography methods for immunoglobulins. GP expression from Sf9 cells infected with recombinant baculoviruses has been used as an alternative to generate fully glycosylated GP. While the MARV and EBOV GP’s derived from baculovirus expression, in conjunction with Ribi® R-700 adjuvant, have shown good immunogenicity in guinea pigs, only a moderate level of protection in the guinea pig models of Marburg and Ebola Hemorrhagic Fever was reported [33, 36, 37]. In contrast, our studies show that the IAC-purified Drosophila-expressed GP does not only result in significant humoral responses in BALB/c mice, but three vaccinations with antigen induced 70% protection, even in the absence of an adjuvant. This level of protection in mice is close to the 80% efficacy reported for another recombinant subunit approach using EIC [38]. While the EIC approach utilized a similar dose level (10μg), four immunizations and the use of an adjuvant were required to achieve this level of efficacy. With proper adjuvantation (using CoVaccine HT™) three 10 μg doses of the Drosophila expressed GP completely protected mice from ma-EBOV challenge, a result replicated in two experiments shown herein. Full protection in the mouse model has been met by all leading EBOV vaccine candidates and the immunogenicity data generated suggests that the GP antigen produces robust humoral responses over a wide dose range and that the responses can be enhanced by adjuvants with diverse modes of action. Cell-mediated responses against GP are more variable and careful adjuvant selection will be required to optimize these.
The immunogenicity of purified VP24 and VP40 subunits was strong, and while the adjuvant chosen had a significant impact on final antibody titers observed, the cell mediated responses were robust in all tested formulations. The immunogenicity of the recombinant VP40 is extraordinary, most likely linked to its propensity to assemble donut-shaped hexamers, nanoparticles which could be observed upon electron microscopic evaluation of concentrated supernatants from Drosophila cells expressing VP40 (data not shown). Therefore, given the abundance of VP40 in viral particles, it was a surprise that none of the 30 VP40 vaccinees infected with ma-EBOV survived the challenge (Table I), especially since Wilson et al. [20] reported partial protection when alphavirus replicons expressing VP40 were administered and Olinger et al identified CTL responses to VP40 [21]. This may be linked to a difference in antigen presentation and it would therefore be important to compare which cell types are primarily targeted by the two different approaches as well as by VLP’s which have been reported to directly activate dendritic cells [39, 40].
Mice immunized with VP24 in CoVaccine HT™ showed a relatively consistent percentage of survival after challenge (6/10 and 2/5, Tables I and II), although surviving animals showed clear signs of disease pathology (e.g., ruffled fur, abnormal gait, lethargy). As expected based on its localization and excellent ability to raise cell-mediated responses as indicated by cytokine release after antigen restimulation, the protective effect of VP24 is mediated by T-cell immunity as demonstrated by passive (adoptive) transfer studies here and previously using replicons [21]. This mechanism of action should be further investigated as it potentially provides insight into potential therapies to alleviate the effects of EVD.
A combination of all three recombinant antigens in the absence of adjuvant was able to protect 9/10 mice not only from mortality but also from overt EBOV-associated morbidity. The kinetics of antibody response and the ultimate titers achieved (against irradiated EBOV) were not significantly different from those found in animals immunized with GP only. These observations suggest that VP24 and VP40 induce cell-mediated responses that develop a synergy in enhancing the quality of the protective response. As expected, clinical adjuvants raised the efficacy level to 100% and therefore our vaccine candidate of GP with CoVaccine HT™ as well as the combination of three antigens with adjuvants yield equivalent or superior responses to those seen with EBOV VLPs in mice [41]. While a role of VP24 in protection has already been identified based on adoptive transfer of immunity with T cells, additional mechanistic studies will be required to determine if T-cells primed with recombinant VP40 also contribute to protection. Furthermore, assessing the compartmentalization of T cell responses (i.e., CD4+ or CD8+ T-cells) may help to elucidate if VP24 mainly induces T helper cells or also cytotoxic T cell responses aiding in viral clearance. The ability to fine-tune the immune responses against the individual vaccine components is one of the advantages of applying a deliberate mix of non-replicating virus subunits and can facilitate more mechanistic studies as required for dissection of the mechanism of protection afforded by this or similar vaccine candidates.
Filoviruses induce a disease in the immune system of primates in which the symptomatic (hemorrhagic) phase is primarily a secondary reaction to a dysregulated immune response [42]. The current knowledge of EBOV pathogenesis has been reviewed in detail by Falasca et al. [43]. However, the mechanisms of how filoviruses evade the immune system or, most importantly, why the few survivors develop an immune response protecting them from death are still poorly understood. In human cases a correlation was made which indicated that patients with an IgM response maintained for a long period of time had a lower chance of survival than patients who showed a faster maturation towards IgG responses [4]. A potential explanation could be a lack or delay of IL-12 responses from virus-infected monocyte-derived dendritic cells [44] which would have an impact on development of helper T cells and subsequently delay the maturation of the antibody response. EBOV infection of monocytes and macrophages has in contrast been shown to actually increase activation of pro-inflammatory cytokine responses [45] and may therefore delay development of adaptive responses. The answer to the question of why innate mechanisms of protection cannot clear the virus may lie within the components of EBOV that seem to mislead or suppress the immune system, for example due to the presence of soluble glycoprotein (sGP) and truncation variants of the mature GP [46]. EBOV infection also induces apoptosis in primary antigen-presenting cells which unquestionably slows down the host’s ability to mount an adaptive response. By contact with macrophages and monocytes, filoviruses appear to trigger inflammatory responses independent of virus replication [45] that ultimately can cause hemorrhage and death of the primate host. One possible explanation for this may be the presence of an immunosuppressive region (mucin-like domain) identified in the GP [47]. In addition to possible effects linked to GP, VP35 [48, 49] and VP24 [50, 51] have both been shown to act as potent inhibitors of IFN type 1 signaling. Mice infected by wild type EBOV show normal IFN-signaling, enabling a protective immune response to develop [52]. In contrast, ma-EBOV inhibits type I interferon stimulated antiviral responses causing increased virulence in mice. This increased virulence may possibly be related to mutations observed in VP24 and NP of ma-EBOV [53]. Similarly, the lower virulence of RESTV compared to EBOV (or MARV) could also be linked to the level of inhibition of type I interferon responses [54], based on a genomic analysis of the host responses in EBOV infected primates.
While the efficacy data of the rVSV-ZEBOV vaccine candidate are impressive, safety of this vaccine is one of the main concerns reported by Huttner et al. [3], who examined the effects of vaccine dose on safety and immunogenicity in a phase 1/2 clinical trial. Three dose levels of vaccine were evaluated: 3 × 105, 1 × 107, and 5 × 107 pfu and safety was assessed by reactogenicity using multiple parameters. After administering the two higher doses of vaccine to 51 subjects, viral oligoarthritis was observed in 11 of them. At that point the studies with the two higher doses were stopped and only the lowest dose level continued. While there was less reactogenicity observed at the lowest dose, the immunogenicity was also decreased in that there was a significant drop in antibody titers at the lowest dose compared to the higher doses. It should be pointed out that the dose demonstrating efficacy by Henao-Restrepo et al. [1, 2] in the Guinea ring vaccination trial was 2 × 107 pfu. While the identification of a protective antibody titer has not been determined, it is likely that higher antibody titers would yield better efficacy. This is of high relevance in this context, as a recombinant subunit vaccine could further be used to design a successful prime-boost approach, enhancing the fast onset of immunity of a virally vectored vaccine candidate with a consistent boost of IgG titers and increased durability of protection.
In summary, the data presented here suggests that a carefully designed vaccine candidate based on recombinant virus subunits can be used to effectively elicit protective responses which allows the host to battle the arsenal of “molecular weapons” which the Ebola virus deploys to stifle the immune system while maintaining a desirable safety profile.
Supplementary Material
Acknowledgements
Funding for the described work was provided through contract DAMD 17-00-C-0032 between Hawaii Biotech, Inc. and the United States Army Medical Research Institute for Infectious Diseases (USAMRIID), Ft. Detrick, MD, through NIH phase I SBIR grant 1R43AI066616, through intramural funds at USAMRIID, and through corporate funds at Hawaii Biotech, Inc. and PanThera Biopharma, LLC. We also greatly acknowledge the gift of adjuvant (CoVaccine HT™) from CoVaccine NV and Protherics, a BTG company. We furthermore would like to acknowledge Dr. Beth-Ann Coller for her contributions to scientific management of this project at Hawaii Biotech, Inc. and would like to thank Charmaine Aniya, Beverly Orillo, and David Waller (Hawaii Biotech, Inc.) for excellent technical support. Opinions, conclusions, interpretations, and recommendations are those of the authors and are not necessarily endorsed by the U.S. Department of the Army and the U.S. Department of Defense.
References
- [1].Henao-Restrepo AM, Camacho A, Longini IM, Watson CH, Edmunds WJ, Egger M, Carroll MW, Dean NE, Diatta I, Doumbia M, Draguez B, Duraffour S, Enwere G, Grais R, Gunther S, Gsell PS, Hossmann S, Watle SV, Konde MK, Keita S, Kone S, Kuisma E, Levine MM, Mandal S, Mauget T, Norheim G, Riveros X, Soumah A, Trelle S, Vicari AS, Rottingen JA, and Kieny MP, Efficacy and effectiveness of an rVSV-vectored vaccine in preventing Ebola virus disease: final results from the Guinea ring vaccination, open-label, cluster-randomised trial (Ebola Ca Suffit!). Lancet, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Henao-Restrepo AM, Longini IM, Egger M, Dean NE, Edmunds WJ, Camacho A, Carroll MW, Doumbia M, Draguez B, Duraffour S, Enwere G, Grais R, Gunther S, Hossmann S, Konde MK, Kone S, Kuisma E, Levine MM, Mandal S, Norheim G, Riveros X, Soumah A, Trelle S, Vicari AS, Watson CH, Keita S, Kieny MP, and Rottingen JA, Efficacy and effectiveness of an rVSV-vectored vaccine expressing Ebola surface glycoprotein: interim results from the Guinea ring vaccination cluster-randomised trial. Lancet, 2015. 386(9996): p. 857–66. [DOI] [PubMed] [Google Scholar]
- [3].Huttner A, Dayer JA, Yerly S, Combescure C, Auderset F, Desmeules J, Eickmann M, Finckh A, Goncalves AR, Hooper JW, Kaya G, Krahling V, Kwilas S, Lemaitre B, Matthey A, Silvera P, Becker S, Fast PE, Moorthy V, Kieny MP, Kaiser L, Siegrist CA, and Consortium VS-E, The effect of dose on the safety and immunogenicity of the VSV Ebola candidate vaccine: a randomised double-blind, placebo-controlled phase 1/2 trial. Lancet Infect Dis, 2015. 15(10): p. 1156–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Ksiazek TG, Rollin PE, Williams AJ, Bressler DS, Martin ML, Swanepoel R, Burt FJ, Leman PA, Khan AS, Rowe AK, Mukunu R, Sanchez A, and Peters CJ, Clinical virology of Ebola hemorrhagic fever (EHF): virus, virus antigen, and IgG and IgM antibody findings among EHF patients in Kikwit, Democratic Republic of the Congo, 1995. J Infect Dis, 1999. 179Suppl 1: p. S177–87. [DOI] [PubMed] [Google Scholar]
- [5].Mupapa K, Massamba M, Kibadi K, Kuvula K, Bwaka A, Kipasa M, Colebunders R, and Muyembe-Tamfum JJ, Treatment of Ebola hemorrhagic fever with blood transfusions from convalescent patients. International Scientific and Technical Committee. J Infect Dis, 1999. 179Suppl 1: p. S18–23. [DOI] [PubMed] [Google Scholar]
- [6].Qiu X, Audet J, Wong G, Fernando L, Bello A, Pillet S, Alimonti JB, and Kobinger GP, Sustained protection against Ebola virus infection following treatment of infected nonhuman primates with ZMAb. Sci Rep, 2013. 3: p. 3365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Khurana S, Fuentes S, Coyle EM, Ravichandran S, Davey RT Jr., and Beigel JH, Human antibody repertoire after VSV-Ebola vaccination identifies novel targets and virus-neutralizing IgM antibodies. Nat Med, 2016. 22(12): p. 1439–1447. [DOI] [PubMed] [Google Scholar]
- [8].Noda T, Sagara H, Suzuki E, Takada A, Kida H, and Kawaoka Y, Ebola virus VP40 drives the formation of virus-like filamentous particles along with GP. J Virol, 2002. 76(10): p. 4855–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Takada A, Ebihara H, Jones S, Feldmann H, and Kawaoka Y, Protective efficacy of neutralizing antibodies against Ebola virus infection. Vaccine, 2007. 25(6): p. 993–9. [DOI] [PubMed] [Google Scholar]
- [10].Jones SM, Feldmann H, Stroher U, Geisbert JB, Fernando L, Grolla A, Klenk HD, Sullivan NJ, Volchkov VE, Fritz EA, Daddario KM, Hensley LE, Jahrling PB, and Geisbert TW, Live attenuated recombinant vaccine protects nonhuman primates against Ebola and Marburg viruses. Nat Med, 2005. 11(7): p. 786–90. [DOI] [PubMed] [Google Scholar]
- [11].Sullivan NJ, Geisbert TW, Geisbert JB, Xu L, Yang ZY, Roederer M, Koup RA, Jahrling PB, and Nabel GJ, Accelerated vaccination for Ebola virus haemorrhagic fever in non-human primates. Nature, 2003. 424(6949): p. 681–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Herbert AS, Kuehne AI, Barth JF, Ortiz RA, Nichols DK, Zak SE, Stonier SW, Muhammad MA, Bakken RR, Prugar LI, Olinger GG, Groebner JL, Lee JS, Pratt WD, Custer M, Kamrud KI, Smith JF, Hart MK, and Dye JM, Venezuelan equine encephalitis virus replicon particle vaccine protects nonhuman primates from intramuscular and aerosol challenge with ebolavirus. J Virol, 2013. 87(9): p. 4952–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Bukreyev A, Rollin PE, Tate MK, Yang L, Zaki SR, Shieh WJ, Murphy BR, Collins PL, and Sanchez A, Successful topical respiratory tract immunization of primates against Ebola virus. J Virol, 2007. 81(12): p. 6379–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Marzi A, Ebihara H, Callison J, Groseth A, Williams KJ, Geisbert TW, and Feldmann H, Vesicular stomatitis virus-based Ebola vaccines with improved cross-protective efficacy. J Infect Dis, 2011. 204Suppl 3: p. S1066–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Xu L, Sanchez A, Yang Z, Zaki SR, Nabel EG, Nichol ST, and Nabel GJ, Immunization for Ebola virus infection. Nat Med, 1998. 4(1): p. 37–42. [DOI] [PubMed] [Google Scholar]
- [16].Sullivan NJ, Sanchez A, Rollin PE, Yang ZY, and Nabel GJ, Development of a preventive vaccine for Ebola virus infection in primates. Nature, 2000. 408(6812): p. 605–9. [DOI] [PubMed] [Google Scholar]
- [17].Warfield KL, Posten NA, Swenson DL, Olinger GG, Esposito D, Gillette WK, Hopkins RF, Costantino J, Panchal RG, Hartley JL, Aman MJ, and Bavari S, Filovirus-like particles produced in insect cells: immunogenicity and protection in rodents. J Infect Dis, 2007. 196Suppl 2: p. S421–9. [DOI] [PubMed] [Google Scholar]
- [18].Simmons G, Lee A, Rennekamp AJ, Fan X, Bates P, and Shen H, Identification of murine T-cell epitopes in Ebola virus nucleoprotein. Virology, 2004. 318(1): p. 224–30. [DOI] [PubMed] [Google Scholar]
- [19].Wilson JA and Hart MK, Protection from Ebola virus mediated by cytotoxic T lymphocytes specific for the viral nucleoprotein. J Virol, 2001. 75(6): p. 2660–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Wilson JA, Bray M, Bakken R, and Hart MK, Vaccine potential of Ebola virus VP24, VP30, VP35, and VP40 proteins. Virology, 2001. 286(2): p. 384–90. [DOI] [PubMed] [Google Scholar]
- [21].Olinger GG, Bailey MA, Dye JM, Bakken R, Kuehne A, Kondig J, Wilson J, Hogan RJ, and Hart MK, Protective cytotoxic T-cell responses induced by venezuelan equine encephalitis virus replicons expressing Ebola virus proteins. J Virol, 2005. 79(22): p. 14189–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Warfield KL, Swenson DL, Olinger GG, Kalina WV, Aman MJ, and Bavari S, Ebola virus-like particle-based vaccine protects nonhuman primates against lethal Ebola virus challenge. J Infect Dis, 2007. 196Suppl 2: p. S430–7. [DOI] [PubMed] [Google Scholar]
- [23].Ignatyev GM, Agafonov AP, Streltsova MA, and Kashentseva EA, Inactivated Marburg virus elicits a nonprotective immune response in Rhesus monkeys. J Biotechnol, 1996. 44(1–3): p. 111–8. [DOI] [PubMed] [Google Scholar]
- [24].Geisbert TW, Pushko P, Anderson K, Smith J, Davis KJ, and Jahrling PB, Evaluation in nonhuman primates of vaccines against Ebola virus. Emerg Infect Dis, 2002. 8(5): p. 503–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Modis Y, Ogata S, Clements D, and Harrison SC, A ligand-binding pocket in the dengue virus envelope glycoprotein. Proc Natl Acad Sci U S A, 2003. 100(12): p. 6986–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Modis Y, Ogata S, Clements D, and Harrison SC, Structure of the dengue virus envelope protein after membrane fusion. Nature, 2004. 427(6972): p. 313–9. [DOI] [PubMed] [Google Scholar]
- [27].Marciani DJ, Press JB, Reynolds RC, Pathak AK, Pathak V, Gundy LE, Farmer JT, Koratich MS, and May RD, Development of semisynthetic triterpenoid saponin derivatives with immune stimulating activity. Vaccine, 2000. 18(27): p. 3141–51. [DOI] [PubMed] [Google Scholar]
- [28].Marciani DJ, Reynolds RC, Pathak AK, Finley-Woodman K, and May RD, Fractionation, structural studies, and immunological characterization of the semi-synthetic Quillaja saponins derivative GPI-0100. Vaccine, 2003. 21(25–26): p. 3961–71. [DOI] [PubMed] [Google Scholar]
- [29].Hilgers LA and Blom AG, Sucrose fatty acid sulphate esters as novel vaccine adjuvant. Vaccine, 2006. 24 Suppl 2: p. S2-81-2. [DOI] [PubMed] [Google Scholar]
- [30].Bray M, Davis K, Geisbert T, Schmaljohn C, and Huggins J, A mouse model for evaluation of prophylaxis and therapy of Ebola hemorrhagic fever. J Infect Dis, 1998. 178(3): p. 651–61. [DOI] [PubMed] [Google Scholar]
- [31].Ksiazek TG, West CP, Rollin PE, Jahrling PB, and Peters CJ, ELISA for the detection of antibodies to Ebola viruses. J Infect Dis, 1999. 179Suppl 1: p. S192–8. [DOI] [PubMed] [Google Scholar]
- [32].Timmins J, Schoehn G, Ricard-Blum S, Scianimanico S, Vernet T, Ruigrok RW, and Weissenhorn W, Ebola virus matrix protein VP40 interaction with human cellular factors Tsg101 and Nedd4. J Mol Biol, 2003. 326(2): p. 493–502. [DOI] [PubMed] [Google Scholar]
- [33].Mellquist-Riemenschneider JL, Garrison AR, Geisbert JB, Saikh KU, Heidebrink KD, Jahrling PB, Ulrich RG, and Schmaljohn CS, Comparison of the protective efficacy of DNA and baculovirus-derived protein vaccines for EBOLA virus in guinea pigs. Virus Res, 2003. 92(2): p. 187–93. [DOI] [PubMed] [Google Scholar]
- [34].Konduru K, Bradfute SB, Jacques J, Manangeeswaran M, Nakamura S, Morshed S, Wood SC, Bavari S, and Kaplan GG, Ebola virus glycoprotein Fc fusion protein confers protection against lethal challenge in vaccinated mice. Vaccine, 2011. 29(16): p. 2968–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Phoolcharoen W, Bhoo SH, Lai H, Ma J, Arntzen CJ, Chen Q, and Mason HS, Expression of an immunogenic Ebola immune complex in Nicotiana benthamiana. Plant Biotechnol J, 2011. 9(7): p. 807–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Hevey M, Negley D, Geisbert J, Jahrling P, and Schmaljohn A, Antigenicity and vaccine potential of Marburg virus glycoprotein expressed by baculovirus recombinants. Virology, 1997. 239(1): p. 206–16. [DOI] [PubMed] [Google Scholar]
- [37].Hevey M, Negley D, VanderZanden L, Tammariello RF, Geisbert J, Schmaljohn C, Smith JF, Jahrling PB, and Schmaljohn AL, Marburg virus vaccines: comparing classical and new approaches. Vaccine, 2001. 20(3–4): p. 586–93. [DOI] [PubMed] [Google Scholar]
- [38].Phoolcharoen W, Dye JM, Kilbourne J, Piensook K, Pratt WD, Arntzen CJ, Chen Q, Mason HS, and Herbst-Kralovetz MM, A nonreplicating subunit vaccine protects mice against lethal Ebola virus challenge. Proc Natl Acad Sci U S A, 2011. 108(51): p. 20695–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Bosio CM, Moore BD, Warfield KL, Ruthel G, Mohamadzadeh M, Aman MJ, and Bavari S, Ebola and Marburg virus-like particles activate human myeloid dendritic cells. Virology, 2004. 326(2): p. 280–7. [DOI] [PubMed] [Google Scholar]
- [40].Ye L, Lin J, Sun Y, Bennouna S, Lo M, Wu Q, Bu Z, Pulendran B, Compans RW, and Yang C, Ebola virus-like particles produced in insect cells exhibit dendritic cell stimulating activity and induce neutralizing antibodies. Virology, 2006. 351(2): p. 260–70. [DOI] [PubMed] [Google Scholar]
- [41].Warfield KL, Bosio CM, Welcher BC, Deal EM, Mohamadzadeh M, Schmaljohn A, Aman MJ, and Bavari S, Ebola virus-like particles protect from lethal Ebola virus infection. Proc Natl Acad Sci U S A, 2003. 100(26): p. 15889–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Geisbert TW and Jahrling PB, Exotic emerging viral diseases: progress and challenges. Nat Med, 2004. 10(12 Suppl): p. S110–21. [DOI] [PubMed] [Google Scholar]
- [43].Falasca L, Agrati C, Petrosillo N, Di Caro A, Capobianchi MR, Ippolito G, and Piacentini M, Molecular mechanisms of Ebola virus pathogenesis: focus on cell death. Cell Death Differ, 2015. 22(8): p. 1250–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Bosio CM, Aman MJ, Grogan C, Hogan R, Ruthel G, Negley D, Mohamadzadeh M, Bavari S, and Schmaljohn A, Ebola and Marburg viruses replicate in monocyte-derived dendritic cells without inducing the production of cytokines and full maturation. J Infect Dis, 2003. 188(11): p. 1630–8. [DOI] [PubMed] [Google Scholar]
- [45].Stroher U, West E, Bugany H, Klenk HD, Schnittler HJ, and Feldmann H, Infection and activation of monocytes by Marburg and Ebola viruses. J Virol, 2001. 75(22): p. 11025–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Escudero-Perez B, Volchkova VA, Dolnik O, Lawrence P, and Volchkov VE, Shed GP of Ebola virus triggers immune activation and increased vascular permeability. PLoS Pathog, 2014. 10(11): p. e1004509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Yaddanapudi K, Palacios G, Towner JS, Chen I, Sariol CA, Nichol ST, and Lipkin WI, Implication of a retrovirus-like glycoprotein peptide in the immunopathogenesis of Ebola and Marburg viruses. FASEB J, 2006. 20(14): p. 2519–30. [DOI] [PubMed] [Google Scholar]
- [48].Basler CF, Mikulasova A, Martinez-Sobrido L, Paragas J, Muhlberger E, Bray M, Klenk HD, Palese P, and Garcia-Sastre A, The Ebola virus VP35 protein inhibits activation of interferon regulatory factor 3. J Virol, 2003. 77(14): p. 7945–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Basler CF, Wang X, Muhlberger E, Volchkov V, Paragas J, Klenk HD, Garcia-Sastre A, and Palese P, The Ebola virus VP35 protein functions as a type I IFN antagonist. Proc Natl Acad Sci U S A, 2000. 97(22): p. 12289–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Reid SP, Valmas C, Martinez O, Sanchez FM, and Basler CF, Ebola virus VP24 proteins inhibit the interaction of NPI-1 subfamily karyopherin alpha proteins with activated STAT1. J Virol, 2007. 81(24): p. 13469–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Reid SP, Leung LW, Hartman AL, Martinez O, Shaw ML, Carbonnelle C, Volchkov VE, Nichol ST, and Basler CF, Ebola virus VP24 binds karyopherin alpha1 and blocks STAT1 nuclear accumulation. J Virol, 2006. 80(11): p. 5156–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Bray M, The role of the Type I interferon response in the resistance of mice to filovirus infection. J Gen Virol, 2001. 82(Pt 6): p. 1365–73. [DOI] [PubMed] [Google Scholar]
- [53].Ebihara H, Takada A, Kobasa D, Jones S, Neumann G, Theriault S, Bray M, Feldmann H, and Kawaoka Y, Molecular determinants of Ebola virus virulence in mice. PLoS Pathog, 2006. 2(7): p. e73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Kash JC, Muhlberger E, Carter V, Grosch M, Perwitasari O, Proll SC, Thomas MJ, Weber F, Klenk HD, and Katze MG, Global suppression of the host antiviral response by Ebola- and Marburgviruses: increased antagonism of the type I interferon response is associated with enhanced virulence. J Virol, 2006. 80(6): p. 3009–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.