

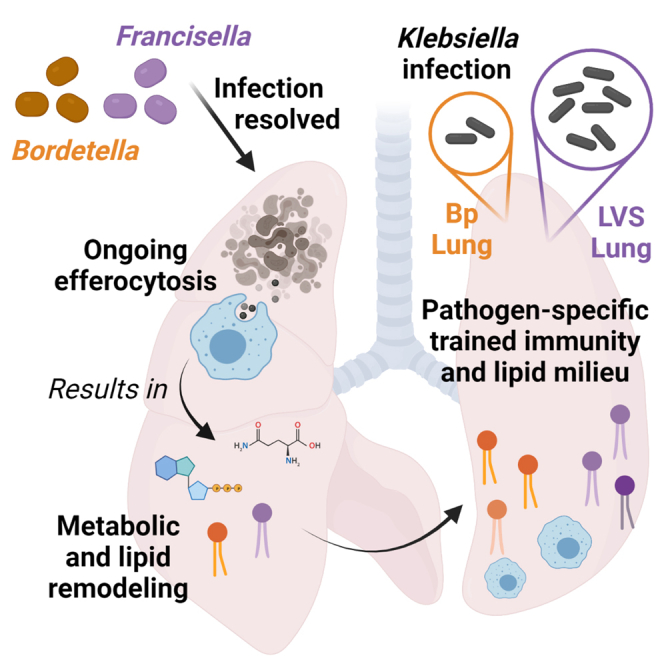
Summary
Resolution of infection results in development of trained innate immunity which is typically beneficial for defense against unrelated secondary infection. Epigenetic changes including modification of histones via binding of various polar metabolites underlie the establishment of trained innate immunity. Therefore, host metabolism and this response are intimately linked. However, little is known regarding the influence of lipids on the development and function of trained immunity. Utilizing two models of pulmonary bacterial infection combined with multi-omic approaches, we identified persistent, pathogen-specific changes to the lung lipidome that correlated with differences in the trained immune response against a third unrelated pathogen. Further, we establish the specific cellular populations in the lung that contribute to this altered lipidome. Together these results expand our understanding of the pulmonary trained innate immune response and the contributions of host lipids in informing that response.
Subject areas: Biological sciences, Immunity, Genomics, Lipidomics
Graphical abstract
Highlights
-
•
Pathogens exert differential effects on pulmonary efferocytosis post-infection
-
•
Differences in efferocytosis are mediated by macrophage subsets
-
•
Unique immune lipid mediator milieus are linked to these macrophage subsets
-
•
Changes in the lipid landscape impact trained immunity to an unrelated infection
Biological sciences; Immunity; Genomics; Lipidomics
Introduction
During pulmonary infection, innate and adaptive immune responses are carefully orchestrated to mediate pathogen clearance. Following resolution of infection, both immune compartments retain an immunological memory of the encounter. In addition to the antigen-specific adaptive immune response, a non-specific innate immune memory develops termed “trained immunity” (reviewed in Netea et al. (2020)). Trained immunity often manifests as a faster and stronger response to a secondary stimulus within myeloid cells due to epigenetic reprograming (Kleinnijenhuis et al., 2012; Saeed et al., 2014). While trained immunity triggered by infection is typically advantageous for defense against unrelated secondary infection, similar immune responses provoked by sterile inflammation can result in worsening pathology in models of transplantation, cancer, and neurodegenerative disease (Netea et al., 2020).
Alterations in cellular metabolism is an essential feature of trained immunity as the balance of metabolites, especially polar metabolites such as α-ketoglutarate, fumarate, and succinate, aid in driving the epigenetic changes observed in “trained” cells (Donohoe and Bultman, 2012; Shyer et al., 2020). However, the role for non-polar metabolites (e.g. lipids) in trained immunity is poorly understood, particularly after the host resolves an infectious insult. Lipids, with a focus on cholesterol and saturated fatty acids, have been examined in bone marrow precursors early after β-glucan stimulation (Mitroulis et al., 2018), suggesting lipid remodeling is an aspect of trained immunity. Phospholipids are key players in cellular membrane integrity and importantly also serve as precursors for potent immunoregulatory lipid mediators (LMs) such as prostaglandins (PGs), leukotrienes, lipoxins, and resolvins (Serhan, 2010). The contribution or shifts in these lipid pools and their influence on trained immunity is not well understood.
To determine if alterations in the host lipidome, including phospholipids and immune LMs, contribute to varying outcomes of trained immunity we utilized two pulmonary bacterial infection models that, upon resolution, result in strikingly different capabilities to control an unrelated bacterial infection. By combining metabolomics, single-cell RNASeq, lipidomics, and flow cytometry, we identified pathogen-specific changes in pulmonary metabolism that correlated with host’s ability to control heterologous secondary infection. Together these results provide new insight into how the pulmonary lipid environment contributes to the evolution of a successful, or not, trained immune response.
Results
Trained innate immune response is pathogen-specific
Bordetella pertussis (Bp) and Francisella tularensis live vaccine strain (LVS) are two Gram-negative bacterium that readily colonize the lung following intranasal infection. Both bacteria also trigger pulmonary inflammatory responses as the infection progresses. Further, unlike many other bacterial infections that are controlled within 7–10 days, resolution of Bp or LVS infection takes weeks before bacteria are finally cleared. Once bacteria are cleared, there is no gross pathology evident in the lung (data not shown). Given these similarities, we hypothesized that Bp and LVS would also elicit similar trained innate responses based on current understanding of these processes. As expected, both Bp and LVS experienced lungs exhibited hallmarks of trained immunity, including increased expression of MHCI and MHCII on lung CD11c+ cells compared to naive animals (Figure 1A). Notably, cells from LVS experienced lungs also expressed significantly more CD86 than naive lungs and had significantly higher expression of all three markers compared to Bp lungs, suggesting a superior trained innate immune response. To test whether these differences manifested as predicted, we intranasally inoculated naive, Bp recovered, or LVS recovered animals with the Gram-negative bacterium Klebsiella pneumoniae (Kp), and quantified lung bacterial burdens on days 1–4 after infection. Bacterial burdens were similar in naive and Bp animals on day 1 post-infection, while LVS animals had significantly lower numbers of bacteria compared with naive controls (Figure 1B). However, as the infection progressed (days 2–4 post-infection), Bp animals controlled Kp replication significantly better than either naive or LVS recovered mice (Figure 1B). Furthermore, there were no significant differences in naive and LVS bacterial burdens on days 2–4 (Figure 1B). These data indicate that there are pathogen-specific components of the trained immune response that exist beyond the apparent activated state of the macrophage populations. We proposed that this observation may be driven by differences in the pulmonary environment, specifically the shared metabolome that supports the cellular components of trained immunity. Thus, all subsequent experiments were performed on Bp or LVS experienced lungs in the absence of Kp infection to understand how prior infection alters the pulmonary environment.
Figure 1.

Trained innate immune response is pathogen-specific
(A) The geometric mean fluorescence intensity (gMFI) of CD86, MHCI, and MHCII on lung CD11c+ cells from naive, Bordetella pertussis (Bp; day 42 post-infection), or Francisella tularensis LVS (LVS; day 28 post-infection). Statistical significance for gMFIs was determined using a one-way ANOVA with a correction for multiple comparisons; ∗ indicate p < 0.05. Only statistically significant differences are indicated. Data are representative of two independent experiments n = 5 mice/group. Individual values are represented as unique points with the bars showing the mean ± SEM.
(B) Klebsiella pneumoniae (Kp) lung bacterial burdens in naive, Bp, and LVS mice. Statistical significance for bacterial burdens was determined on log-transformed data by a one-way ANOVA with a correction for multiple comparisons at each time point post-challenge. The table indicates whether bacterial burdens significantly differed between groups at each time point post-Kp infection; ∗ indicate p < 0.05. Data are combined from 3 independent experiments n = 7–14 mice/group/time point. Data are represented as mean ± SEM.
Resolved pulmonary infection activates a conserved metabolic program suggestive of cellular recycling
To examine metabolic differences in Bp and LVS experienced lungs that could explain the differential ability to control Kp infection, we first qualitatively measured central metabolic components using targeted liquid chromatography-tandem mass spectrometry (LC-MS/MS). Metabolomic analysis revealed shifts in metabolite pools between LVS and Bp recovered lungs on day 28 or 42 post-infection, respectively, compared with naive controls (Figures 2A and 2B). Significant polar metabolite changes in LVS or Bp recovered lungs occupied four general groups, namely amino acids, glycolysis, nucleic acids, and nucleic acid-derived cofactors (Figure 2C). Apart from arginine, free amino acid levels were significantly increased after recovery from Bp and LVS. Within glycolysis, terminal products such as pyruvate and lactate were also elevated while the early glycolytic intermediates were decreased in both Bp and LVS recovered lungs. Amongst nucleic acids, nucleotide triphosphates and nucleotide-derived cofactors were decreased in LVS and trended downward after Bp recovery. Interestingly, Bp recovered animals had universally elevated levels of products of purine degradation including hypoxanthine, xanthine, and urate across all experimental replicates. LVS recovered animals had much more variability in the quantities of these products suggesting less consistent flux in the dynamics of these pathways within this group. Collectively, the decrease in upstream glycolytic intermediates and energetic triphosphates suggested the lung was an energy depleted environment after resolving bacterial infection, regardless of the infecting agent. When coupled with elevated free amino acids, increased purine degradation products, and decreased levels of cofactors, these results pointed to an on-going degradative recycling process such as efferocytosis being active in the lungs of infection-recovered hosts (Yurdagul et al., 2020; Zhang et al., 2019).
Figure 2.

Polar metabolites in the post-infection lung reflect active efferocytosis and energetic depletion
(A and B) Targeted metabolomics of experienced lungs are displayed post-infection with (A), Bordetella pertussis (Bp; day 42 post-infection) or (b), Francisella tularensis LVS (LVS; day 28 post-infection) as compared to naive animals. Features above a 10% FDR cutoff line, as calculated using Benjamini–Hochberg correction, are shown in red (Bp p = 0.044, LVS p = 0.058). Vertical lines reflect a fold change of 2 and –2. Common features of interest are number as (1) PRPP, (2) NADP+, (3) NADPH, (4) Glutamine, (5) Asparagine, (6) Uracil, (7) Glutamate, (8) Aspartate.
(C) Heatmap of the autoscaled value of metabolites of interest from each biological replicate are displayed with the accompanying p value from the binary comparison of each experienced group to naive. All heatmap features listed pass the 10% FDR cutoff for either Bp vs. naive or LVS vs. naive. G6P, glucose 6-phosphate; F-1,6-BP, fructose 1,6-bisphosphate; DHAP, dihydroxyacetone phosphate; 1,3-BPG, 1,3-bisphosphogylcerate; 2/3-PG, 2- and 3-phosphoglycerate pool; PEP, phosphoenolpyruvate; PRPP, phosphoribosyl diphosphate; cAMP, cyclic-AMP; GSH, reduced glutathione. Data are representative of 3 independent experiments with n = 5/group. See also Figure S2.
Once we established that changes in the host metabolome were detectable within one week of bacterial clearance, we sought to determine how long these alterations persisted. Therefore, we assessed metabolite pools at days 70 and 56 after infection which represents an additional 28 days after elimination of either Bp or LVS, respectively. As expected, organ-wide metabolic effects were dampened over time but deviations from naive lungs were still observed. These changes included elevated levels of aspartate and glutamate along with decreases in NADP+ and NADPH for both Bp and LVS (Figure S2). Similarly, glycolytic intermediates continued to be altered in both experienced groups at these later time points including elevated glucose and decreased levels of glucose-6-phosphate (G6P), 1,3-bisphosphoglycerate (1,3-BPG), and lactate (Figure S2). Patterns across nucleic acids varied between groups, distinguishing LVS and Bp from one another. Specifically, LVS displayed higher levels of triphosphates compared with both naive and Bp, whereas Bp experience lungs had elevated levels of monophosphates compared to all other groups (Figure S2). Taken together, these results indicated that the metabolic milieu of the lung is altered in a pathogen-specific fashion and that the lung remains altered for extended periods of time following clearance of infecting microbes.
Recovered mice exhibit alterations in macrophage populations
To investigate which cell type(s) were responsible for the recycling metabolic signature observed above, we performed single-cell RNA-sequencing (scRNA-Seq) on whole lungs from Bp and LVS recovered animals at 42 or 28 days post-infection, respectively, and naive controls. Analysis of all cellular populations in the lung avoided bias for a particular cell population and provided the opportunity to capture intracellular cross talk. We first identified all major cell populations in the lung using an unbiased single-cell identification algorithm developed by Speranza et al. (2021) using the mouse cell atlas lung data as a background (Han et al., 2018) (Figure 3A). Given that our metabolomic analysis suggested on-going efferocytosis, we next analyzed each population of cells for genes associated with efferocytosis (Figure 3B). The efferocytosis GO term (GO:0043277) genes clustered into two distinct subsets across the cells (see STAR Methods and Figure S1B). Gene set one was found to be enriched in stromal and mesenchymal cells (main gene markers: Anxa1, Thbs1, C3, C2, Lrp1, Gas6, Nr1h3, Mfge8) and gene set 2 enriched in innate immune cells (main gene markers: Rac2, Tyrobp, Ccr2, Cd200lf, Rhog, Trem2, Axl, Marco), i.e. macrophages, neutrophils, pDC, and NK cells (Figures 3B and S1B). Further analysis of the cell types enriched for efferocytosis gene set 1 or gene set 2 revealed that the greatest overall transcriptional differences between naive and recovered animals were within the macrophage subset (Figure S3). Based on these findings and the role of myeloid-derived cells in trained innate immunity, we further investigated changes in pulmonary macrophage populations beyond trained immunity markers highlighted above (Figure 1A). Therefore, we next subset the single-cell data on macrophages. Macrophage data was re-analyzed to identify the principal components (PCs). Within the naive sample, alveolar macrophages (AMs) and interstitial macrophages (IMs) clearly separated along PC2 (Figure 3C). While both AMs and IMs were present in the Bp and LVS samples, we identified a population of cells that did not fall into either AM or IM clusters among recovered animals and was particularly enriched in the LVS sample (Figure 3D). Further transcriptional analysis of this cell subset suggested that these were likely recruited alveolar macrophages (recAMs). For example, they expressed markers of AMs such as high CD11c (Itgax) expression but not resident markers, e.g. marco (Aegerter et al., 2020) (Figures S1C and S1D). We further investigated the cellular origin by examining C1q1 expression, a gene associated with macrophages differentiated from monocytes (Figure S1D). The recAM and IM populations expressed C1q1, but not Marco, as expected (Gibbings et al., 2015). We then determined the percentage of the total macrophage population represented by each of these subsets. Both Bp and LVS recovered animals had fewer resAM and increased numbers of recAMs compared with naive controls (Figure 3D). While there was a trend toward an enrichment of IMs in the Bp recovered host, the LVS recovered animal had a greater proportion of these cells compared with either Bp experienced or naive (Figure 3D).
Figure 3.

Single-cell RNA-sequencing of whole lung shows changes in macrophage populations
Single-cell RNA-sequencing of whole lung from naive, Bordetella pertussis (Bp; day 42 post-infection), or Francisella tularensis LVS (LVS; day 28 post-infection) mice were collected.
(A) UMAP projection of the integrated data set with all conditions present colored based on cell type annotation; names of cell types are listed next to the main cluster.
(B) Violin plots of efferocytosis gene sets determined by clustering from the GO term for apoptosis of dead cells. The cell types are listed along the y axis and colored to match the annotations in (A). The gene module score for each cell is represented on the x axis for the two gene sets. Individual cells are not shown for clarity and only the distribution is displayed by cell type. The vertical line is where the module score is 0 for reference across the cell types.
(C) Principal component projection of the macrophage populations separated by infection and colored by cell type with interstitial macrophages (IM; green), resident alveolar macrophages (resAM; blue), and recruited alveolar macrophages (recAMs; black).
(D) Percent of the total macrophage population for each of the conditions (naive in black, Bp in orange, and LVS in purple) separated by resAM, recAMs, and IMs. Single-cell RNA-sequencing was performed on cells isolated from one mouse per group. See also Figures S1 and S3.
To validate the differences in macrophage populations identified by scRNA-Seq, we identified each macrophage population using flow cytometry. No significant differences in the overall cellularity of lungs isolated from naive, Bp, or LVS animals were detected (Figure 4A). Macrophage populations were categorized into resAMs, recAMs, and IMs by the receptor expression pattern and gating strategy indicated in Figures 4B and 4C. In agreement with scRNA-Seq data, recovered Bp and LVS animals had a significant decrease in the number of resAMs compared with naive animals and concordant increase in the number of recAMs (Figure 4D). No significant difference among the number of IMs was observed in animals that resolved Bp infection compared with naive controls (Figure 4D). In contrast, the number of IMs in LVS recovered mice was significantly increased compared with naive animals (Figure 4D).
Figure 4.

Pulmonary macrophage populations are altered after resolving bacterial infection
(A) The number of lung cells in naive, Bordetella pertussis (Bp; day 42 post-infection), and Francisella tularensis LVS (LVS; day 28 post-infection) experienced mice was determined.
(B) Table describing the surface phenotype for each macrophage population
(C) Gating scheme discriminating resident alveolar macrophages (resAMs), recruited alveolar macrophages (recAMs), and interstitial macrophages (IMs).
(D) The number of resAMs, recAMs, and IMs was determined. Statistical significance was determined by a one-way ANOVA with a correction for multiple comparisons; ∗ indicate p < 0.05. Only statistically significant differences are indicated. Data are combined from two independent experiments n = 10 mice/group total. Individual values are represented as unique points with the bars showing the mean ± SEM. See also Figure S4.
As stated above, we observed sustained differences in metabolite profiles 56 and 70 days post-bacterial clearance (Figure S2). Given that we identified markers are associated with efferocytosis within pulmonary macrophages (Figure 3B), we hypothesized that changes in the macrophage populations among recovered mice would be similarly retained at these later time points. Analysis of lungs from Bp and LVS recovered animals at days 70 and 56 after infection, respectively, revealed that resident AMs remained significantly decreased in LVS recovered mice compared with naive animals. In contrast, Bp resAMs had returned to baseline (Figure S4B). Recruited AMs remained significantly increased in Bp and LVS animals compared with naive controls (Figure S4C). Bp and LVS IM populations had returned to numbers observed in naive animals (Figure S4D). Interestingly the numbers of recruited macrophages in each experienced lung set was preserved between the early and late timepoints suggesting these cells were a stable, long-lasting phenotype, and not a transient intermediate population. Together, these data confirmed our scRNA-Seq observations of dynamic changes in pulmonary macrophage populations from mice that had resolved bacterial infection and suggest that there are elements of the response that are pathogen specific.
Macrophages from recovered mice are enriched in efferocytosis markers
The metabolomic and genomic analyses above suggested that despite elimination of LVS and Bp, pulmonary macrophages among recovered animals were still actively participating in efferocytosis and likely contributing to the altered metabolism of the experienced lungs. Thus, we next confirmed the observed genetic changes of elevated expression of efferocytosis markers within resAMs, recAMs, and IMs by flow cytometry. Trem2 and Axl are surface receptors on phagocytes that recognize apoptotic bodies as the initiating step of efferocytosis. In agreement with the scRNA-Seq results, Trem2 was significantly increased on LVS resAMs and recAMs compared with Bp and naive animals (Figures 5A and 5B). Trem2 expression on IMs was approximately 50% of resAMs in naive lungs, yet its expression was still significantly increased on IMs from LVS animals compared with naive and Bp mice (Figure 5C). Like Trem2, Axl was significantly increased on the surface of resAMs from recovered animals compared with naive controls (Figure 5D). Recruited AMs among LVS mice also had significantly more surface expression of Axl than Bp-derived recAMs recapitulating the Trem2 pattern (Figure 5E). Within the IM pool, LVS IMs expressed significantly higher levels of Axl compared with both naive and Bp IMs (Figure 5F).
Figure 5.

Macrophages from recovered mice are enriched in efferocytosis markers
(A–I) The geometric mean fluorescence intensity (gMFI) of Trem2, (D–F) Axl, and (G–I) Abca1 on resident alveolar macrophages (resAMs), recruited alveolar macrophages (recAMs), and interstitial macrophages (IMs) from naive, Bordetella pertussis (Bp; day 42 post-infection), and Francisella tularensis LVS (LVS; day 28 post-infection) experienced mice was determined using flow cytometry. Statistical significance was determined by a one-way ANOVA with a correction for multiple comparisons or t test; ∗ indicate p < 0.05. Only statistically significant differences are indicated. Data are representative of two independent experiments n = 5 mice/group. Individual values are represented as unique points with the bars showing the mean ± SEM.
After engulfing apoptotic cells, macrophages breakdown cellular components which increase the intracellular concentration of lipids. To effectively process this lipid debris, macrophages must export excess lipids to lipoprotein particles. Abca1 is an ATP-dependent efflux pump responsible for exporting cholesterol and phospholipids into lipoprotein particles in the context of efferocytosis. Abca1 was significantly increased among resAMs from Bp and LVS recovered mice compared with naive controls (Figure 5G). Both resAMs and recAMs from LVS mice expressed significantly more Abca1 compared with cells from Bp mice (Figures 5G and 5H). There was no difference in Abca1 levels between naive and Bp IMs. However, LVS-derived IMs expressed significantly more Abca1 compared with both groups (Figure 5I). We further interrogated the scRNA-Seq data set for other genes associated with lipid transport (npc1, abcg1, scarb1). These genes were more highly expressed in Bp and LVS experienced IMs compared to naive IMs (data not shown) suggesting broad changes in lipid transport and not an alteration in specific transport family. Together, these data indicated that efferocytosis and associated lipid export pathways were active within multiple pulmonary macrophage populations in mice that had recently resolved a bacterial infection. In addition, these data pointed toward pathogen-specific effects in the cellular components of this response and the phenotype of the cells involved.
Recovered lungs exhibit an altered pulmonary lipidome
As noted above, efferocytosis is regulated by, and drastically changes, the balance of lipids in the engulfing macrophages and in the surrounding organ via lipid export. The differences in efferocytosis markers between Bp and LVS lungs suggested that lipid-balances in these lungs may also vary in a pathogen-specific manner. Abca1 was significantly up regulated on multiple macrophage populations in infection-recovered animals suggesting that Bp and LVS experienced lungs would have an altered lipidome (Figure 5). Lipid products known collectively as immune LMs are known to regulate both efferocytosis and the immune response. Changes in the bulk lipidome of the lung may feedback on amount and identity substrates for the generation of LMs in the experienced lung and thus the effectiveness of the trained immune response. Therefore, we next determined if there were changes in the bulk lipidome followed by analysis of LM pools. Changes in the lung lipidome were determined using a targeted LC-MS/MS strategy to assess members of major families of phospholipids, sphingolipids, and neutral lipids with acyl-chain resolution. ANOVA analysis was utilized to select for the top one hundred features that varied between groups. These features consisted largely of neutral lipids (53% of features) and plasmalogen phosphatidylethanolamine (28% of features). Each lipid group was then sorted by their behavior across naive, Bp, or LVS experienced lungs. In general, neutral lipids were enriched in lungs from Bp and LVS recovered animals compared with naive controls (clusters 1–3). Specifically, Bp lungs exhibited enrichment in neutral lipids containing poly-unsaturated fatty acids (PUFAs) as seen in clusters 1 and 3 (Figure 6). Conversely, plasmalogen species were decreased in lungs from both LVS and Bp recovered animals. These trends were greater in magnitude in Bp experienced lungs than in lungs from LVS recovered animals (Figure 6).
Figure 6.

Targeted lipidomics indicates overlapping pathogen-specific shifts in plasmalogen and neutral lipids pools in experienced lungs
Heatmap of the autoscaled intensity of the top 100 lipids from one-way ANOVA analysis of lipid features in naive, Bordetella pertussis (Bp; day 42 post-infection), and Francisella tularensis LVS (LVS; day 28 post-infection) experienced lungs. Features are clustered broadly by their behavior between groups. The displayed percentage indicates the percentage within the cluster reflecting that identity. Poly-unsaturated fatty acids (PUFA) were defined as a length and degree of unsaturation greater than or equal to 18:3. Individual lipid species are marked with LIPID MAPS abbreviations. Neutral lipids are defined to include (TG) triacyl-, (DG) diacyl-, and (MG) monoacylglycerols. The ANOVA p value is displayed to the right. PC, phosphatidylcholine; LPC, lyso-PC; PS, phosphatidylserine; LPS, Lyso-PS; PE, phosphatidylethanolamine; LPE, lyso-PE; PE(O), plasmanyl PE plasmalogen; PE(P), plasmenyl PE plasmalogen; PG, phosphatidylglycerol; Cer, ceramide; HCer, hexosylceramide. Data are representative of 3 independent experiments with n = 5/group. See also Figure S5.
To assess the persistence of these patterns, the lipidome of Bp and LVS experienced lungs was assessed at day 70 and 56 post-infection for Bp and LVS, respectively. A similar selection and grouping of features was performed to organize lipid components by behavior between groups. Interestingly, increases in neutral lipids persisted in Bp but not among lungs from mice recovered from LVS infection (Figure S5). The pattern of decreased plasmalogen was also maintained at these later timepoints in both Bp and LVS samples (Figure S5). The conservation of patterns in both neutral lipids and plasmalogen among experienced lungs at later timepoints suggested persistent lipid rearrangement following recovery from infection that is in good agreement with observed long-lasting cellular changes in the macrophage pools (Figure S4). The representation of PUFA-containing species, which can serve as substrates for LM production, amongst the altered lipids in the experienced lung supported the idea that differences in efferocytosis may be changing the availability of LM precursors.
Infection-intrinsic LM profile after pathogen clearance
As described above, we observed persistence of differential levels of neutral lipids and PE-plasmalogen containing PUFAs, which may change the availability of these PUFAs for conversion to LMs. Numerous LMs including the D- and E-series resolvins have been previously shown to be stimulated by and subsequently regulate efferocytosis during the recovery from immune insult (Dalli and Serhan, 2012; Schif-Zuck et al., 2011). Considering the presence of efferocytotic macrophages and the rearrangement of PUFA-containing glycerolipid pools (Figures 3, 4, and 5), namely depleted PUFA-plasmalogen and increased PUFA-triacylglycerol species, we hypothesized that the LM balance of the infection-recovered lung would also be altered compared with naive tissue.
LMs in the experienced lung were measured by LC-MS/MS and a combination of spectral and standard comparison was used to distinguish between the numerous isobaric compounds in this family of lipid species. Utilizing principal component analysis (PCA), we observed that LMs in naive, Bp, and LVS lungs incompletely separated along the second and third principle component (Figure 7A). This diagonal axis of separation was defined by cyclooxygenase (COX)-derived PGs in the LVS direction and ALOX12/15 derived mono-hydroxylated products in the Bp direction with the naive lung occupying an intermediate space (Figure 7B). To further characterize groups of LMs that drove the differences between the groups in a cooperative manner, a group-biased partial least squared discriminant analysis (PLSDA) was utilized. In addition to the ALOX12/15 and COX pattern, which defined the primary axis of intergroup variance by PLSDA, a secondary axis correlated with the experienced lungs was observed as defined by the levels of free PUFAs (Figures 7C and 7D). This pattern of free PUFAs was expected as an indicator of infection and the wide distribution of recovered lungs along this axis may represent different rates of recovery from the initial infection. The LM patterns were further interrogated by univariate analysis (Figure 7E). COX-derived PGs were significantly elevated in both LVS and Bp conditioned lungs. However, the distribution of PGs among LVS lungs reached a much higher maximum. The levels of PGs varied amongst LVS lungs in line with the higher variability in this same group during aqueous metabolite analysis (Figure 2). These data suggest differential recovery kinetics from LVS infection in terms of metabolic flux. As suggested by multivariate analysis, ALOX12/15 products were significantly elevated in Bp experienced lungs while many of these same species trended downward in LVS lungs. Together, these data confirm that LM species are altered in Bp and LVS recovered lungs and that there are infection-intrinsic differences among these important immunoregulatory products.
Figure 7.

Analysis of PUFA-derived immune lipid mediators indicates pathogen-specific LM profiles in the experienced lung
Lipid mediator (LM) profile was determined in naive, Bordetella pertussis (Bp; day 42 post-infection), and Francisella tularensis LVS (LVS; day 28 post-infection) experienced lungs.
(A) PCA separation of the autoscaled LM data set of the experienced lungs.
(B) Corresponding loading plot.
(C) PLSDA analysis of the autoscaled LM data set of the experienced lungs
(D) Corresponding loading plot. For loading plots, LMs are colored by their enzyme of origin.
(E) Heatmap of the autoscaled intensities of LM features that passed a 10% FDR cutoff for one of the three possible binary comparisons. Corresponding p values from the binary comparison are displayed at right. AA, arachidonic acid; DHA, docosahexaenoic acid; DPA, docosapentaenoic acid; Rv(X), resolvin; HETE, hydroxyeicosatetraenoic acid; HEPE, hydroxyeicosapentaenoic acid; HDHA, hydroxydocosahexaenoic acid; HDPA, hydroxydocosapentaenoic acid; DiHETrE, dihydroxyeicosatrienoic acid; HHT, hydroxyheptadecatrenoic acid; PG(X), prostaglandin; TxB2, Thromboxane B2. Data are composite of 2 independent experiments with n = 6 for naive, n = 8 for Bp and LVS in total.
Identification of ALOX12/15 expressing pulmonary cells
Since ALOX12/15 LM products were the key distinguishing feature between LVS and Bp lungs, we interrogated expression patterns of the ALOX12/15 associated pathways to determine which cell type(s) were responsible for synthesizing this specific family of LMs. We limited the C2CP gene set from MSig DB (Subramanian et al., 2005) to only those containing Alox5, Alox12, Alox12e, Alox15, or Ptgs2 and were completely contained within the Lipid Metabolism reactome pathway. We then looked at the expression of these pathways across each cell type. In addition to macrophages, we identified a small cluster of cells containing neutrophils and pDCs with high expression of several pathways, related to ALOX12/15 and ALOX5 activity (Figure 8). Alveolar type 1 and 2 cells and stromal/mesenchymal cells also displayed expression of ALOX12/15 pathways but not ALOX5. Using a bootstrapping method to estimate p values, we found that Bp recovered lungs had an elevation of ALOX12/15-associated pathways, but not the ALOX5 pathways, specifically within the macrophage populations but not in neutrophils or pDCs. These data suggest that the macrophage populations in Bp recovered animals are partially responsible for the high production of LMs that require ALOX12/15 for their synthesis and may contribute to favorable trained immunity against Kp infection among Bp experienced mice.
Figure 8.

Analysis of Alox-associated pathways in single-cell sequencing
cRNA-Seq data from naive, Bordetella pertussis (Bp; day 42 post-infection), and Francisella tularensis LVS (LVS; day 28 post-infection) experienced lungs was analyzed for pathways involved in lipid mediator synthesis.
(A) Heatmap of the mean module gene score across the individual cell types (columns) in each of the Alox associated pathways contained in lipid metabolism (rows). The scale bar shows the mean expression with darker blue being low expression and orange being high expression. The yellow box highlights the cluster of cells and pathways of interest.
(B) Violin plots for the three pathways from the yellow box across the cell types from the yellow box in (A) Each cell type is divided between LVS (left, darker color) and Bp (right, lighter color). The gene module score is shown on the y axis. Individual points are not shown for clarity. ∗ represents when the estimated p value from bootstrapping showed an elevation in the Bp recovered animals (<5% chance it was random) and # represents the same for when the LVS immune animals were elevated.
Discussion
Trained innate immunity is an important element of host responsiveness to secondary infection. The elements of trained immunity manifesting within the myeloid compartment are largely due to epigenetic re-programming allowing cells to respond more effectively to secondary stimuli (reviewed in Netea et al. (2020)). However, the same myeloid-derived cells responsible for trained immunity also play an important role in repairing structural damage caused by the inflammatory response. This process, called efferocytosis, consists of the engulfment and processing of apoptotic bodies and debris followed by the recycling and export of small molecule components such as amino acids, nucleotides, and lipids. Herein, we establish a previously unrecognized link between the cellular and lipid regulators of efferocytosis and the trained immune response. Further, we define patterns in the host lipidome and generation of immune LMs in the lung that correlate with the ability of the host to either respond favorably to secondary infection or fail to promote enhanced clearance of the pathogen.
Prior to defining pathogen specific patterns in the lipidome, we first observed some general shifts in lipid profiles in both Bp and LVS recovered lungs. Regardless of the primary infection, the lipidomic pattern in experienced lungs was dominated by decreases in phosphatidylethanolamine plasmalogen and increases in neutral lipids, many of which contained PUFAs. The dynamics in both of these PUFA-containing lipid classes suggests sequestration of PUFAs by decreasing membrane localized plasmalogen pools and increasing intracellular or lipoprotein-associated triacylglycerol pools (Astudillo et al., 2019; Olzmann and Carvalho, 2019; Schlager et al., 2015). Despite the similarities in PUFA-containing pools, analysis of LMs derived from PUFAs revealed that these products were defining features of either a Bp or LVS experienced lung.
The differential generation of LMs in response to unique bacterial stimuli has been examined in isolated cell culture but has not been examined in depth in animal infection models (Werz et al., 2018). Our in vivo data supports and extends these previous findings. First, animals which had cleared LVS infection were enriched in PGs and other COX products. These results are consistent with the induction of PGs during acute LVS infection, e.g. within the first 7 days of infection (Woolard et al., 2008). Together this suggests that the balance of acute phase LMs influence the resultant LM profile after bacterial clearance. Second, we established that pulmonary bacterial infections generate distinct LM pools. Specifically, in contrast to LVS, animals that had cleared Bp had higher levels of ALOX12/15 derived products. Finally, we provide evidence that the LM milieu present at the time of bacterial challenge correlated with the ability of the host to control replication of an unrelated bacterium with greater resistance to Kp infection in Bp recovered mice. The increase in monohydroxlated products of ALOX12/15 indicates this pathway is more active in Bp experienced lungs. Certain observed molecules such as 17-HDHA and 14-HDHA have been shown to participate in the resolution phase as the molecular precursors of resolvins (Serhan and Levy, 2018). In the acute phase of inflammation, ALOX12/15 is known to regulate the pro-inflammatory response (Wen et al., 2007). While it is unclear if it is the pro-resolving, pro-inflammatory, or balance of both of these products driven by ALOX12/15 that may be responsible for superior resistance to Kp in Bp experienced lungs, the increased activity of this pathway will allow for more rapid generation of ALOX12/15 products that may drive or regulate inflammation in a manner that is beneficial to the host. Regardless of the outcome of Kp infection, it is important to consider that the challenge pathogen will factor into this phenomenon in the same way as the initial pathogen. For instance, when a pathogen requires a strong cyclooxygenase response, an LVS experienced lung would be predicted to be more resistant than a Bp experienced lung.
In addition to identifying LMs in recovered animals that are associated with resolution of secondary infection, we also used a multi-omics approach to ask which cell type(s) were responsible for the generation of these lipids. Cellular debris and apoptotic bodies taken up via efferocytosis can serve as precursors for LMs (Dalli and Serhan, 2012). Therefore, we determined which cellular families were undergoing active efferocytosis in Bp and LVS recovered lungs compared with naive controls. This analysis brought out some interesting features that suggested cross talk among specific cellular populations in the recovered lung. For example, both the stromal/mesenchymal and myeloid populations showed transcriptomic signatures that suggested regulation of the lipidome and efferocytosis through interaction with each other. Specifically, Gas6 expression on stromal/mesenchymal cells acts as the ligand for Axl and Trem2 found on macrophages (Nagata et al., 1996). Further, lipid-driven communication between the two efferocytotic cellular families is evident by the presence of the lipoprotein-receptor Lrp1 on stromal/mesenchymal cells and the observed upregulation of the lipoprotein loading enzyme Abca1 on macrophages. Together, these proteins work in concert to regulate the generation and content of lipoprotein particles. In addition, both of these cell types are likely contributing to LM production either by direct generation or through controlling substrate availability. While myeloid cells have been identified as a major producer of LMs, stromal/mesenchymal cells have recently been identified as a potentially important source of LM synthesis (Holopainen et al., 2019; Hyvarinen et al., 2018). We found both myeloid and stromal/mesenchymal cells express lipoxygenases though stromal/mesenchymal cells express much less ALOX5 relative to myeloid cells. We also observe localization of the glucocorticoid-stimulated protein Annexin A1 (Anxa1) to the stromal cell group. Anxa1 directly regulates phospholipase A2 (PLA2), which liberates PUFAs for LM generation, and changes in PLA2 activity will control how much and from what cell type PUFAs can be sourced (Kwon et al., 2012; Perucci et al., 2017; Schaloske and Dennis, 2006; Solito et al., 1998). Thus, the pathogen-specific changes in the lipidome and LM pools observed herein are likely tied to coordination between macrophages and stromal/mesenchymal cells. These data highlight the complementary role that distinct cellular components play in the recovered lung and the utility of thorough, whole tissue analysis.
Our analysis demonstrated resAMs and recAMs are removing and recycling cellular debris via efferocytosis. While being active participants in this process, it is also likely resAMs are being turned over after undergoing apoptosis because of the infections modeled herein. We found the number of resAMs was significantly lower in infection-experienced lungs compared with naive mice. This finding wasn’t surprising given AMs are the first cells infected with F. tularensis and would undergo cell death as a consequence of intracellular bacteria replication (Roberts et al., 2014). Further, although B. pertussis doesn’t directly target AMs for infection, this cell type is also subject to apoptosis after bacterial exposure, presumably via the action of encoded toxins (Khelef et al., 1993). The elevated numbers of recAMs in our study suggests these monocyte-derived cells are primarily responsible for re-populating the resident alveolar macrophage pool as also demonstrated after Influenza A infection (Aegerter et al., 2020). The biological consequences of replacing yolk-sac-derived alveolar macrophages with monocyte-derived alveolar macrophages will depend on whether these two cell types are functionally distinct once fully conditioned by the pulmonary milieu. Understanding these processes is an important further line of study.
Polar metabolite measurements further supported efferocytotic activity in both LVS and Bp experienced lungs, at day 28 and 42 post-infection, respectively, through increased levels of free amino acids and base units of nucleotide metabolism. As expected, given the clearance of infecting agent and lack of gross pathology, changes in polar metabolites did not exceed a fold change of 3 between groups. Despite the subtlety of these patterns, certain differences were still present at the later measured time point for LVS (day 56 post-infection) and Bp (day 70 post-infection). These persistent metabolite changes including NADP(H), aspartate, and glutamate may indicate infection responsive processes that are still active well after infection clearance. NADPH oxidase activity has been previously connected to LM-associated processes in the lung including COX2 and phospholipases (Hsu et al., 2019; Lin et al., 2010). Glutamate and aspartate are the upstream intermediate points for de novo synthesis of numerous amino acids and their elevation may indicate a persistent change in amino acid synthesis in response to active efferocytosis and molecular recycling. In addition, glutamate is a well-established signaling molecule in lung injury and may be playing a role as a soluble mediator of inflammation and resolution (Bai et al., 2017a, 2017b). Further understanding the behavior of these polar metabolites during the transition between the acute and resolving phases of infection will be essential in defining their causal role in the cellular-molecular network established here.
To date, trained immunity has been primarily examined as an epigenetic phenomenon within innate immune cells. Epigenetic maturation relies on metabolic processes that provide chemical moieties such as succinate, fumarate, and α-ketoglutarate for the modification of genetically associated elements like histones (reviewed in Etchegaray and Mostoslavsky (2016)). While we detected these molecules, we did not observe significant elevation in pathogen-experienced lungs compared to naive. Since metabolic changes have been shown to track with the intensity of the inflammatory response during acute pulmonary infection, epigenetic changes would be expected to operate at the peak of the inflammatory response (Tisoncik-Go et al., 2016). It is possible that changes in these metabolites could persist past the acute inflammatory phase. However, the data here suggests that their levels have returned to homeostatic baseline upon resolution of infection.
The associated differences in trained immunity may also be extended to alterations the lipidome and LMs that are not driven by previous infections. For example, obese individuals have significant alterations in lipid pools present in both adipose tissue and in circulation (as reviewed in Dahik et al. (2020)). Obesity is recognized as a comorbidity for worsening outcomes for a variety of infections and there are some examples of dysregulation of LMs correlating with severity of disease (Honce and Schultz-Cherry, 2019; Schwarz et al., 2021). Conversely, obesity has also been shown to be clinically associated with decreased severity in sepsis highlighting the potential of the pathogen-specific effects (reviewed in Ng and Eikermann (2017)). It is unknown if LMs contribute causally to these differential effects and if LMs potentially involved are present at the outset of infection, are produced as a consequence of infection, or a combination thereof. Data presented herein suggests that, at a minimum, the LM pool present prior to infection in a healthy host plays a role in defense against disease. Examination of LMs in scenarios that are not specifically linked to previous infections and how these LM pools might predispose individuals to severity of disease presents an important target for new treatment strategies.
As presented herein, our study revealed persistent, pathogen-specific changes in macrophage populations and the pulmonary lipidome, including LMs after resolution of infection. Further, these alterations correlated with the ability of the host to respond successfully to an unrelated infection suggesting that they play a role in the trained immune response. Together, these data suggest a previously unrecognized connection between efferocytosis, lipid metabolism, and trained innate immunity and highlight the importance of an individual’s infection history and their ability to successfully resolve subsequent immune insults. Finally, our data suggest that continued analysis of LMs that persist following clearance of microbial insults will provide important new insight into the generation of trained immunity.
Limitations of the study
While the use of multiple pathogens within this study is a strength, in vivo infectious disease models are dynamic and complex. Consequently, our study identified correlations relating to improved trained immunity using several approaches (flow cytometry, metabolomics/lipidomics, and scRNA-Seq) because causative elements were difficult to define in the models examined here. It would be useful to follow-up in sterile injury models that recapitulate these persistent lipidomic changes as these models would not be subject to the same limitations as when an organism is infected with a pathogen. Identifying causative molecular candidates is further limited by the inability to knockout individual LMs due to the complexity of the PUFA, enzyme, and receptor network that generates these mediators.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-Abca1 PE-Cy7; polyclonal | Novus | Cat. # NB400-105PECY7; RRID: AB_1643846 |
| anti-Axl AF405; clone 175128 | R&D | Cat. # FAB8541V; RRID: AB_2062573 |
| anti-CD11b BV510; clone M1/70 | Biolegend | Cat. # 101263; RRID: AB_2629529 |
| anti-CD11c PerCP-Cy5.5; clone N418 | Biolegend | Cat. # 117328: RRID: AB_2129641 |
| anti-CD45 BUV395; clone 30-F11 | BD | Cat. # 564279; RRID: AB_2651134 |
| anti-CD86 APC; clone GL-1 | Biolegend | Cat. # 105012; RRID: AB_493342 |
| anti-CX3CR1 BV785; clone SAO11F11 | Biolegend | Cat. # 149029; RRID: AB_2565938 |
| anti-MHCI PE-Cy7; clone 28-8-6 | Biolegend | Cat. # 114616 |
| anti-MHCII BV421; clone M5/114.15.2 | Biolegend | Cat. # 107632 |
| anti-Siglec F PE; clone S17007L | Biolegend | Cat. # 155506; RRID: AB_2750234 |
| anti-Trem2 APC; clone 237920 | R&D | Cat. #FAB17291A; RRID: AB_884527 |
| Bacterial and virus strains | ||
| Bordetella pertussis strain D420 | Dr. Todd Merkel (Center for Biologics Evaluation and Research, Food and Drug Administration, Silver Springs, MD) | NA |
| Francisella tularensis subsp holarctica Live Vaccine Strain (LVS) | Dr. Jean Celli (Washington State University, Pullman, WA) | NA |
| Klebsiella pneumoniae subspecies pneumoniae strain Schroeter Trevisan | ATCC (Manassas, VA) | NA |
| Liberase TM | Millipore Sigma | Cat #:5401127001 |
| HPLC grade Water | Fisher Scientific | Cat #: W6 4 |
| HPLC grade Methanol | Fisher Scientific | Cat #: A456 4 |
| HPLC grade Isopropanol | Fisher Scientific | Cat #: A461 4 |
| HPLC grade Hexanes | Fisher Scientific | Cat #: 270504-2L |
| HPLC grade Methyl-Formate | Fisher Scientific | Cat #: 259705-2L |
| HPLC grade Chloroform | Fisher Scientific | Cat #: C606SK-4 |
| HPLC grade Acetic Acid | Fisher Scientific | Cat #: A11350 |
| D8-5-HETE | Cayman Chemical | Cat #: 334230 |
| D5-RvD2 | Cayman Chemical | Cat #: 11184 |
| D5-LXA4 | Cayman Chemical | Cat #: 24936 |
| D4-LTB4 | Cayman Chemical | Cat #: 29629 |
| D4-PGE2 | Cayman Chemical | Cat #: 314010 |
| RvE1 | Cayman Chemical | Cat #: 10007848 |
| LXA4 | Cayman Chemical | Cat #: 90410 |
| LXA5 | Cayman Chemical | Cat #: 10011453 |
| LXB4 | Cayman Chemical | Cat #: 90420 |
| PGE2 | Cayman Chemical | Cat #: 14010 |
| PGF2 | Cayman Chemical | Cat #: 16010 |
| PGD2 | Cayman Chemical | Cat #: 12010 |
| TxB2 | Cayman Chemical | Cat #: 19030 |
| PD1 | Cayman Chemical | Cat #: 10010390 |
| RvD5 | Cayman Chemical | Cat #: 10007280 |
| Maresin | Cayman Chemical | Cat #: 10878 |
| LTB4 | Cayman Chemical | Cat #: 20110 |
| 5,15-DiHETE | Cayman Chemical | Cat #: 35280 |
| 14-HDHA | Cayman Chemical | Cat #: 33550 |
| 18-HEPE | Cayman Chemical | Cat #: 32840 |
| Arachidonic Acid | Cayman Chemical | Cat #: 90010 |
| EPA | Cayman Chemical | Cat #: 90110 |
| DHA | Cayman Chemical | Cat #: 90310 |
| Ammonium Acetate | Fisher Scientific | Cat #: A639-500 |
| Deposited data | ||
| Raw and analyzed scRNA data | This paper | GEO: GSE171510 |
| Prefiltered Metabolite and lipid data | This paper | Mendeley |
| Experimental models: Organisms/strains | ||
| Mouse: C57Bl/6J | Jackson Laboratories | Stock #: 000664 |
| Software and algorithms | ||
| Prism 8 | GraphPad Software | NA |
| BD Diva | BD | NA |
| FlowJo v10 | BD | NA |
| Sciex Analyst v1.7 | Sciex | NA |
| Sciex MultiQuant v3.0.2 | Sciex | NA |
| MarkerView v1.3 | Sciex | NA |
| R v3.6.2 | https://www.r-project.org/index.html | https://www.r-project.org/ |
| Seurat | Stuart et al., 2019 | https://satijalab.org/seurat/ |
| Other | ||
| Sep-Pak C18 cartridges | Waters Corporation | Cat #: WAT054945 |
| Waters Atlantis T3 Column | Waters Corporation | Cat #: 186003722 |
| Waters XBridge Amide Column | Waters Corporation | Cat #: 186004864 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dr. Catharine M. Bosio, PhD (bosioc@niaid.nih.gov).
Materials availability
This study did not generate any unique reagents.
Experimental model and subject details
In vivo animal studies
Five to six-week old female C57Bl/6J mice were purchased from The Jackson Laboratories (stock #000664) and housed at BSL-2 with a 12-hour light/dark cycle and water/food ad libitum. Animal studies were approved by and conducted in accordance with Rocky Mountain Laboratories’ Animal Care and Use Committee (ASP#2018-012E). All animals within a cage were randomly assigned to a single experimental group. Investigators were not blinded to the experimental group assigned to a cage of mice due to housing uninfected and infected mice on different cage racks within the same room. Further, the time points utilized within this study differed between the two infected groups making blinding not feasible.
Methods details
Mice and infections
Five- to six-week-old specific-pathogen free, female C57Bl/6J (B6) mice were purchased from Jackson Laboratories and housed at ABSL-2 at Rocky Mountain Laboratories. Mice were intranasally (i.n.) inoculated with 1x105 CFU Bordetella pertussis D420 (Tod Merkel, Center for Biologics Evaluation and Research, Food and Drug Administration, Silver Springs, MD), 150 CFU of F. tularensis subspecies holarctica Live Vaccine Strain (LVS; Jean Celli, Washington State University, Pullman, WA), or 3500 CFU of Klebsiella pneumoniae subspecies pneumoniae (Schroeter Trevisan, ATCC, Manassas, VA) in 25 μl of PBS delivered to a single nare after anesthesia with ketamine/xylazine. All bacterial inocula were confirmed by serial dilution and plating on Bordet-Gengou blood agar (B. pertussis), Modified Mueller Hinton (LVS), or Luria broth agar (K. pneumoniae). Animal studies were approved by and conducted in accordance with Rocky Mountain Laboratories’ Animal Care and Use Committee (ASP#2018-012E).
Harvest of lung tissue and sample processing
Lungs were aseptically harvested, minced, and digested for 45 minutes at 37°C into a single-cell suspension using Liberase TM purchased from Sigma-Aldrich (St. Louis, MO) at a final concentration of 0.175 mg/ml in PBS (Anderson et al., 2010). The total number of viable cells was determined by trypan blue exclusion using a hemocytometer or TC20 Automated Cell Counter (Bio-Rad).
scRNA-Seq libraries and sequencing
Single-cell suspensions of lungs cells and gel beads were loaded into the chip and Chromium Controller according to the manufacturer’s instructions (10X Genomics). The target number of captured cells per sample was 10,000. Sequencing libraries were generated using the 10X Single-Cell 3’ v2 kit according to the manufacturer’s instructions (10X Genomics). Libraries were analyzed using High Sensitivity DNA Kits and the Agilent 2100 Bioanalyzer (Agilent). Samples were sequenced on an Illumina NextSeq550 High Throughput instrument.
scRNA-Seq data analysis
Samples were aligned to the mm10 genome from Ensembl using the cellRanger pipeline. Data was then read into R V3.6.2 using Seurat V3.1.5 (Stuart et al., 2019). To account for any batch effects from sample collection or infections, we performed data integration on the global data using the IntegrateData function in Seurat. Cells were first filtered to remove those that contained abnormally high or low RNA counts. Using the calculated quantiles of the unique molecular identifier (UMI) per cell cells in the bottom 5% and top 5% were removed. Next, cells with less than 10% of reads belonging to mitochondrial genes were retained. Once filtering was completed, the top 2000 variable features were calculated using the FindVariableFeatures function, and the data was scaled using the ScaleData function. The top 30 principal components were calculated using the top variable features and the umap projections were calculated based off the principal components. The nearest neighbor (knn) graph was then generated and used to identify clusters. Similar analysis was performed on any subset of cell analyzed but only using the top 500 variable genes within the normalized RNA data.
To identify specific cell types within the single-cell RNA-Seq dataset, an unbiased algorithm was developed in-house to define either cluster or individual cell level identity using the known transcriptional profiles of cells. First, data from the mouse cell atlas (Han et al., 2018) was pulled and subset to the lung. Marker genes were then selected for each of the annotated cell types based on a high differential expression value (greater than 1 log fold change) and a high difference in the percentage of cells expressing the gene in the cell type compared to all other cell types in the lung dataset (> 0.6). The average expression for the marker gene set was calculated for the annotated dataset. Either on a per-cell or per-cluster level, gene expression of marker genes in the unknown cell was compared to the annotated dataset average expression to generate a correlation value. The cell type with the highest correlation match to the annotated cell type was used as the initial annotation. Initial cell type assignment was further confirmed using an iterative method to correct falsely identified cells. Each cell was analyzed for its closest neighbors on the knn graph. Cell identify was assigned when greater than 70% of the neighbors shared a single-cell type identity. This analysis was run until a stable annotation was reached, i.e. no cell changed its annotation in a given run. Finally, all annotations were validated by examining known marker genes (Figure S1A). Only cell annotations that contained at least 50 cells were kept for analysis.
To determine which cell types showed the greatest changes between experimental conditions, we performed an unbiased analysis of the various cell types. After sub-setting on a given cell type, we determined the principal components that made up 95% of the explained variance within that cell population of interest. Then for each of the principal components, the mean location of the cells along the principal component of each experimental group was calculated and the difference between the two groups of interest was measured. This was done across all cell types comparing two experimental conditions to each other. To find the principal components and cell types that showed the strongest difference between groups, we identified the PCs that were greater than 3 standard deviations away from the mean of the whole population.
Efferocytosis gene marker development
Gene marker sets associated with efferocytosis were developed by identifying all genes contained in apoptotic cells (GO:0043277) in the MGI database and differential gene expression analysis was performed within the single-cell sequencing dataset. The efferocytosis gene set was filtered for genes that showed differential expression (log fold change > 1) in at least one cell type. Clusters of genes were identified by clustering the genes across the cells independent of the cell label (Figure S1B). Gene expression scoring for efferocytosis gene sets was done using the AddModuleScore function in Seurat.
Bootstrapping for comparing gene modules
A bootstrapping method was used to estimate the p-values when comparing gene module scores. The difference in the means between two groups being tested was calculated. This was compared to taking the same data and randomly shuffling the labels 1000 times and calculating the difference in the means. The p-value was estimated as less than or greater than 1% of the randomly shuffled differences.
Lipid and metabolite analysis
LCMS or HPLC grade water, methanol, isopropanol, hexanes, methyl-formate, chloroform, and acetic acid were purchased through Fisher Scientific (Waltham, MA). All lipid mediator standards were purchased from Cayman Chemical (Ann Arbor, MI). Organ sections were collected in 500 μL of ice-cold methanol and stored at –80oC until further processing. Polar metabolites and bulk lipids were extracted consistent with established protocols(Schwarz et al., 2021). Prior to extraction of polar metabolites and bulk lipids, each tissue section was homogenized through a screen while on ice, washed with 500 μL of ice-cold water and both the resultant suspension and debris were collected. Five hundred microliters of ice-cold chloroform was added to each sample. Samples were agitated for 20 minutes by shaking at 4oC and then spun at > 10,000 x g for 10 min at 4oC to induce layering. The top (aqueous) layer and bottom (organic layer) were collected and the aqueous layer was directly diluted 1:10 in 50% methanol in water for analysis. The organic layer was taken to dryness in a Savant™ DNA120 SpeedVac™ concentrator (Thermo Fisher). Organic fractions were resuspended in 500 μL of 5 μg/mL butylated hydroxytoluene in 6:1 isopropanol:methanol and further diluted 1:3 for analysis.
Immune lipid mediators were extracted from a separate section of tissue collected in 500 μL of ice-cold methanol containing 1 ng each of d8-5-HETE, d5-RvD2, d5-LXA4, d4-LTB4, d4-PGE2. Samples were gently homogenized using a micro-pestle until and then centrifuged at > 10,000 x g for 10 min at 4oC. The supernatant was transferred to a fresh tube. For solid-phase extraction, Sep-Pak® 3 mL, 200 mg, C18 cartridges (Waters Corporation, Milford, MA) were conditioned with 10 mL of methanol and 10 mL of water. Samples were primed for loading one at a time by addition of 9 mL of acidified water (pH 3.5 with hydrochloric acid) and then quickly loaded onto the column. Each sample was washed with 3 mL of water prior to proceeding to the next sample. After all sample were loaded, they were washed with an additional 5 mL of water followed by 5 mL of hexanes. Samples were eluted with 10 mL of methyl-formate and dried under nitrogen at 55oC. Each sample was resuspended in 200 μL of 1:1 water:methanol and 30 μL of each sample was injected for analysis.
LC/MS analysis
All analyses utilized previously established multi-reaction monitoring (MRM) strategies (Schwarz et al., 2021). All chromatography utilized a Sciex ExionLC™ AC system and data was acquired using a Sciex 5500 QTRAP® mass spectrometer. Polar metabolites were analyzed from the aqueous sample fraction using an ion pairing method. Quality control samples and blanks were injected to assess signal stability. Samples were injected onto a Waters Atlantis T3 column (100Å, 3 μm, 3 mm X 100 mm) and elution consisted of a gradient from 5 mM tributylamine, 5 mM acetic acid in 2% isopropanol, 5% methanol, 93% water (v/v) to 100% isopropanol over 15 minutes. Two distinct MRMs in negative mode were utilized to measure each analyte and provide confidence of identity but only one signal was carried forward for statistical analysis. Enhanced product ion scans were collected on QC injections and collected spectra were compared available references. Only relative quantification was performed.
Bulk lipids were analyzed from the organic sample fraction using a previously established HILIC method (Schwarz et al., 2021). Samples were injected onto a Water XBridge® Amide column (3.5 μm, 3 mm X 100 mm) and separated over 12 minutes with a binary gradient from (A) 100% 5 mM ammonium acetate, 5% water in acetonitrile apparent pH 8.4 to (B) 95% 5 mM ammonium acetate, 50% water in acetonitrile apparent pH 8.0. Targets were detected using scheduled MRM. Negative mode and positive mode targets were analyzed using subsequent injections and holding polarity constant for each run. The positive and negative lipid datasets were separately filtered and preprocessed before combining for statistical analysis. Lipid classes were distinguished by retention time. MRMs in negative mode consisted of molecular ion peaks for PE, PI, PS, PG, PE-plasmalogen and the product ion corresponding to an intact fatty acid. PC signals utilized an acetate adduct primary ion coupled with a fatty acid daughter ion. Fatty acids were detected via the molecular anion and the neutral loss 44 daughter ion corresponding the loss of a carboxyl group. In positive mode, TG, DG, and MG species were detected by the ammonium adduct combined with the neutral loss of the ammonium and a specified fatty acid. Cer, HCer, LCer, DCer, SM, and CE were detected with the molecular cation and the invariant chain daughter ion of 264.4 for Cer, HCer, and LCer, 266.4 for DCer, 184 for SM, and 369.4 for CE.
Specialized pro-resolving lipid mediator samples were separated on a Waters Atlantis T3 column (100Å, 3 μm, 3 mm X 100 mm) and using a binary gradient of A: 0.01 % acetic acid in water and B: 0.01 % acetic acid in methanol (Schwarz et al., 2021). A 20 min gradient from 40-100 % B was utilized to separate species. Samples were detected in negative MRM mode with triggered enhanced-product ion scans and rolling collision energy embedded in the method. Spectral information was compared to standards and a spectral library for identification. A blank and a standard mix were serially injected every 10 injections. Standard mix consisted of each of the following compounds at 10 ng/mL: RvE3, LXA4, LXA5, LXB4, PGE2, PGD2, PGF2a, TxB2, PD1, RvD5, Maresin 1, LTB4, 5,15-DiHETE, 14-HDHA, 18-HEPE, AA, EPA, DHA. Spectral confirmation was not possible for RvD3, RvD4, 20-OH-LTB4, 11,12-DiHETrE, 14,15-DiHETrE, 14,15-EET, 12-HHT, 11-HETE, 14-HDPA, 13-HDHA, 17-HDHA and 21-HDHA but identity was assessed by comparison to related standards based on spectra and expected retention time.
All data was processed using MultiQuant® Software 3.0.3. For polar metabolites and bulk lipids, data was filtered with 50 % missing value and 30 % QC coefficient of variance cutoffs. Remaining missing values were replaced with the minimum group value for that feature. Following filtering, the polar metabolite and bulk lipids were separately total sum normalized and the two bulk lipid datasets (positive and negative) were combined for further analysis. For lipid mediator datasets, signal quality was judged visually, and signal stability was assessed by repeat injection of a standard mix. Lipid mediator data was normalized to internal heavy isotope standards (Schwarz et al., 2021). All multivariate and multivariate analysis was performed in MarkerView® Software 1.3.1 or Metaboanalyst.
Flow cytometry analysis
A single-cell suspension of approximately 2x106 lung cells was stained with Zombie Near Infrared dye (Biolegend) according to the manufacturer’s instructions to discriminate live and dead cells. The following directly conjugated antibodies were used for analysis: Abca1 PE-Cy7, Axl AF405, CD11b BV510, CD11c PerCP-Cy5.5, CD45 BUV395, CD86 APC, CX3CR1 BV785, Siglec F PE, and TREM2 APC. Antibodies were purchased from Biolegend, BD Biosciences, or R&D Systems. All antibody staining was performed in the presence of 2.4G2 Fc block (2.4G2 B cell hybridoma a gift from Jeffrey Frelinger, University of Arizona). Samples were stained for 20 minutes at 4°C, washed 3 times with FACS Wash (2% FBS in PBS) to remove unbound antibody, and then fixed with 2% paraformaldehyde for 10 minutes. Samples were run on a BD Symphony flow cytometer and analyzed in FlowJo v10 (BD Biosciences).
Statistical analysis
Statistical significance when comparing three groups was determined by a one-way ANOVA with a correction for multiple comparisons. An unpaired student’s t-test was used to determine statistical significance when two groups were compared. p < 0.05 was considered statistically significant. For univariate analysis of polar metabolites and bulk lipids a Benjamini-Hochberg correction was applied with a false discovery rate cut-off of 10 %. Datasets were autoscaled (z-scaled) for display where indicated by mean-centering and normalization to the standard deviation across all displayed samples for the metabolite or lipid in question.
Quantification and statistical analysis
The statistical test, exact value of n, definition of a statistically significant p value, and dispersion/precision measures are indicated within each figure legend. Statistical significance when comparing three groups was determined by a one-way ANOVA with a correction for multiple comparisons. An unpaired student’s t-test was used to determine statistical significance when two groups were compared. p < 0.05 was considered statistically significant. For univariate analysis of polar metabolites and bulk lipids a Benjamini-Hochberg correction was applied with a false discovery rate cut-off of 10 %. Datasets were autoscaled (z-scaled) for display where indicated by mean-centering and normalization to the standard deviation across all displayed samples for the metabolite or lipid in question. A Grubb's test was performed to remove one outlier value per group within metabolomics data as indicated in the associated figure legend. No data was excluded from flow cytometry or bacterial burden analysis. A group size of 10 (2 experiments of 5 animals each) is appropriate to determine statistically significant differences in cellular populations, using unpaired, two tailed student’s t-test with significance set at p<0.05 per a power calculation where the difference in the means is 20%. (Power calculation website https://www.stat.ubc.ca/∼rollin/stats/ssize/n2.html).
Acknowledgments
We would like the thank Ronald N. Germain of NIH/NIAID for helpful discussions. Prof. Charles N. Serhan and his group including, K. Boyle, A. Shay, C. Jouvene, X. de la Rosa, S. Libreros, and N. Chiang generously provided methodology, consultation, and extensive training for the assessment of lipid mediators by LC-MS/MS. AB Sciex, in particular M. Pearson, P. Norris, B. Ubhi, and P. Baker (currently Avanti Polar Lipids), provided LC-MS/MS consultation and methods. The graphical abstract was created using Biorender.com. This work was supported by the Intramural Research Program of the National Institutes of Health, National Institute of Allergy and Infectious Diseases.
Author contributions
These authors contributed equally, L.M.R., B.S., and E.S.; conceived and designed experiments, L.R., B.S., E.S., and C.M.B.; performed experiments, L.R., B.S., I.L., and T.W.; sequencing analysis, E.S.; metabolomics analysis, B.S.; wrote manuscript, L.R., B.S., E.S., and C.M.B.; supervision and funding, S.B. and C.M.B.
Declaration of interests
The authors declare no competing interests.
Inclusion and diversity
One or more of the authors of this paper self-identifies as a member of the LGBTQ+ community.
Published: September 24, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.103025.
Supplemental information
Data and code availability
Single-cell RNA-sequencing data has been deposited in the National Center for Biotechnology Information’s Gene Expression Omnibus under GEO series accession number GSE171510. Metabolomics and lipidomics data are available via Mendeley Data https://doi.org/10.17632/m9sfjxn8c5.1. Any additional data that support the study’s findings are available from the lead contact upon request.
This paper does not report original code. All code utilized was publicly available.
References
- Aegerter H., Kulikauskaite J., Crotta S., Patel H., Kelly G., Hessel E.M., Mack M., Beinke S., Wack A. Influenza-induced monocyte-derived alveolar macrophages confer prolonged antibacterial protection. Nat. Immunol. 2020;21:145–157. doi: 10.1038/s41590-019-0568-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson R.V., Crane D.D., Bosio C.M. Long lived protection against pneumonic tularemia is correlated with cellular immunity in peripheral, not pulmonary, organs. Vaccine. 2010;28:6562–6572. doi: 10.1016/j.vaccine.2010.07.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Astudillo A.M., Balboa M.A., Balsinde J. Selectivity of phospholipid hydrolysis by phospholipase A2 enzymes in activated cells leading to polyunsaturated fatty acid mobilization. Biochim. Biophys. Acta Mol. Cell Biol. Lipids. 2019;1864:772–783. doi: 10.1016/j.bbalip.2018.07.002. [DOI] [PubMed] [Google Scholar]
- Bai W., Li W., Ning Y.L., Li P., Zhao Y., Yang N., Jiang Y.L., Liang Z.P., Jiang D.P., Wang Y. Blood glutamate levels are closely related to acute lung injury and prognosis after stroke. Front. Neurol. 2017;8:755. doi: 10.3389/fneur.2017.00755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai W., Zhu W.L., Ning Y.L., Li P., Zhao Y., Yang N., Chen X., Jiang Y.L., Yang W.Q., Jiang D.P. Dramatic increases in blood glutamate concentrations are closely related to traumatic brain injury-induced acute lung injury. Sci. Rep. 2017;7:5380. doi: 10.1038/s41598-017-05574-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahik V.D., Frisdal E., Le Goff W. Rewiring of lipid metabolism in adipose tissue macrophages in obesity: impact on insulin resistance and type 2 diabetes. Int. J. Mol. Sci. 2020;21:5505. doi: 10.3390/ijms21155505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalli J., Serhan C.N. Specific lipid mediator signatures of human phagocytes: microparticles stimulate macrophage efferocytosis and pro-resolving mediators. Blood. 2012;120:e60–e72. doi: 10.1182/blood-2012-04-423525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donohoe D.R., Bultman S.J. Metaboloepigenetics: interrelationships between energy metabolism and epigenetic control of gene expression. J. Cell. Physiol. 2012;227:3169–3177. doi: 10.1002/jcp.24054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etchegaray J.P., Mostoslavsky R. Interplay between metabolism and epigenetics: a nuclear adaptation to environmental changes. Mol. Cell. 2016;62:695–711. doi: 10.1016/j.molcel.2016.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbings S.L., Goyal R., Desch A.N., Leach S.M., Prabagar M., Atif S.M., Bratton D.L., Janssen W., Jakubzick C.V. Transcriptome analysis highlights the conserved difference between embryonic and postnatal-derived alveolar macrophages. Blood. 2015;126:1357–1366. doi: 10.1182/blood-2015-01-624809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han X., Wang R., Zhou Y., Fei L., Sun H., Lai S., Saadatpour A., Zhou Z., Chen H., Ye F. Mapping the mouse cell atlas by microwell-seq. Cell. 2018;173:1307. doi: 10.1016/j.cell.2018.05.012. [DOI] [PubMed] [Google Scholar]
- Holopainen M., Colas R.A., Valkonen S., Tigistu-Sahle F., Hyvarinen K., Mazzacuva F., Lehenkari P., Kakela R., Dalli J., Kerkela E. Polyunsaturated fatty acids modify the extracellular vesicle membranes and increase the production of proresolving lipid mediators of human mesenchymal stromal cells. Biochim. Biophys. Acta Mol. Cell Biol. Lipids. 2019;1864:1350–1362. doi: 10.1016/j.bbalip.2019.06.010. [DOI] [PubMed] [Google Scholar]
- Honce R., Schultz-Cherry S. Impact of obesity on influenza A virus pathogenesis, immune response, and evolution. Front. Immunol. 2019;10:1071. doi: 10.3389/fimmu.2019.01071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu P.S., Lin C.M., Chang J.F., Wu C.S., Sia K.C., Lee I.T., Huang K.Y., Lin W.N. Participation of NADPH oxidase-related reactive oxygen species in leptin-promoted pulmonary inflammation: regulation of cPLA2alpha and COX-2 expression. Int. J. Mol. Sci. 2019;20:1078. doi: 10.3390/ijms20051078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyvarinen K., Holopainen M., Skirdenko V., Ruhanen H., Lehenkari P., Korhonen M., Kakela R., Laitinen S., Kerkela E. Mesenchymal stromal cells and their extracellular vesicles enhance the anti-inflammatory phenotype of regulatory macrophages by downregulating the production of interleukin (IL)-23 and IL-22. Front. Immunol. 2018;9:771. doi: 10.3389/fimmu.2018.00771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khelef N., Zychlinsky A., Guiso N. Bordetella pertussis induces apoptosis in macrophages: role of adenylate cyclase-hemolysin. Infect Immun. 1993;61:4064–4071. doi: 10.1128/iai.61.10.4064-4071.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinnijenhuis J., Quintin J., Preijers F., Joosten L.A., Ifrim D.C., Saeed S., Jacobs C., van Loenhout J., de Jong D., Stunnenberg H.G. Bacille Calmette–Guerin induces NOD2-dependent nonspecific protection from reinfection via epigenetic reprogramming of monocytes. Proc. Natl. Acad. Sci. U. S. A. 2012;109:17537–17542. doi: 10.1073/pnas.1202870109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon J.H., Lee J.H., Kim K.S., Chung Y.W., Kim I.Y. Regulation of cytosolic phospholipase A2 phosphorylation by proteolytic cleavage of annexin A1 in activated mast cells. J. Immunol. 2012;188:5665–5673. doi: 10.4049/jimmunol.1102306. [DOI] [PubMed] [Google Scholar]
- Lin C.C., Lee I.T., Yang Y.L., Lee C.W., Kou Y.R., Yang C.M. Induction of COX-2/PGE(2)/IL-6 is crucial for cigarette smoke extract-induced airway inflammation: role of TLR4-dependent NADPH oxidase activation. Free Radic. Biol. Med. 2010;48:240–254. doi: 10.1016/j.freeradbiomed.2009.10.047. [DOI] [PubMed] [Google Scholar]
- Mitroulis I., Ruppova K., Wang B., Chen L.S., Grzybek M., Grinenko T., Eugster A., Troullinaki M., Palladini A., Kourtzelis I. Modulation of myelopoiesis progenitors is an integral component of trained immunity. Cell. 2018;172:147–161 e112. doi: 10.1016/j.cell.2017.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagata K., Ohashi K., Nakano T., Arita H., Zong C., Hanafusa H., Mizuno K. Identification of the product of growth arrest-specific gene 6 as a common ligand for Axl, Sky, and Mer receptor tyrosine kinases. J. Biol. Chem. 1996;271:30022–30027. doi: 10.1074/jbc.271.47.30022. [DOI] [PubMed] [Google Scholar]
- Netea M.G., Dominguez-Andres J., Barreiro L.B., Chavakis T., Divangahi M., Fuchs E., Joosten L.A.B., van der Meer J.W.M., Mhlanga M.M., Mulder W.J.M. Defining trained immunity and its role in health and disease. Nat. Rev. Immunol. 2020;20:375–388. doi: 10.1038/s41577-020-0285-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng P.Y., Eikermann M. The obesity conundrum in sepsis. BMC Anesthesiol. 2017;17:147. doi: 10.1186/s12871-017-0434-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olzmann J.A., Carvalho P. Dynamics and functions of lipid droplets. Nat. Rev. Mol. Cell Biol. 2019;20:137–155. doi: 10.1038/s41580-018-0085-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perucci L.O., Sugimoto M.A., Gomes K.B., Dusse L.M., Teixeira M.M., Sousa L.P. Annexin A1 and specialized proresolving lipid mediators: promoting resolution as a therapeutic strategy in human inflammatory diseases. Expert Opin. Ther. Targets. 2017;21:879–896. doi: 10.1080/14728222.2017.1364363. [DOI] [PubMed] [Google Scholar]
- Roberts L.M., Tuladhar S., Steele S.P., Riebe K.J., Chen C.J., Cumming R.I., Seay S., Frothingham R., Sempowski G.D., Kawula T.H. Identification of early interactions between Francisella and the host. Infect. Immun. 2014;82:2504–2510. doi: 10.1128/IAI.01654-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saeed S., Quintin J., Kerstens H.H., Rao N.A., Aghajanirefah A., Matarese F., Cheng S.C., Ratter J., Berentsen K., van der Ent M.A. Epigenetic programming of monocyte-to-macrophage differentiation and trained innate immunity. Science. 2014;345:1251086. doi: 10.1126/science.1251086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaloske R.H., Dennis E.A. The phospholipase A2 superfamily and its group numbering system. Biochim. Biophys. Acta. 2006;1761:1246–1259. doi: 10.1016/j.bbalip.2006.07.011. [DOI] [PubMed] [Google Scholar]
- Schif-Zuck S., Gross N., Assi S., Rostoker R., Serhan C.N., Ariel A. Saturated-efferocytosis generates pro-resolving CD11b low macrophages: modulation by resolvins and glucocorticoids. Eur. J. Immunol. 2011;41:366–379. doi: 10.1002/eji.201040801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlager S., Goeritzer M., Jandl K., Frei R., Vujic N., Kolb D., Strohmaier H., Dorow J., Eichmann T.O., Rosenberger A. Adipose triglyceride lipase acts on neutrophil lipid droplets to regulate substrate availability for lipid mediator synthesis. J. Leukoc. Biol. 2015;98:837–850. doi: 10.1189/jlb.3A0515-206R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz B., Sharma L., Roberts L., Peng X., Bermejo S., Leighton I., Casanovas-Massana A., Minasyan M., Farhadian S., Ko A.I. Cutting edge: severe SARS-CoV-2 infection in humans is defined by a shift in the serum lipidome, resulting in dysregulation of eicosanoid immune mediators. J. Immunol. 2021;206:329–334. doi: 10.4049/jimmunol.2001025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serhan C.N. Novel lipid mediators and resolution mechanisms in acute inflammation: to resolve or not? Am. J. Pathol. 2010;177:1576–1591. doi: 10.2353/ajpath.2010.100322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serhan C.N., Levy B.D. Resolvins in inflammation: emergence of the pro-resolving superfamily of mediators. J. Clin. Invest. 2018;128:2657–2669. doi: 10.1172/JCI97943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shyer J.A., Flavell R.A., Bailis W. Metabolic signaling in T cells. Cell Res. 2020;30:649–659. doi: 10.1038/s41422-020-0379-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solito E., Raguenes-Nicol C., de Coupade C., Bisagni-Faure A., Russo-Marie F. U937 cells deprived of endogenous annexin 1 demonstrate an increased PLA2 activity. Br. J. Pharmacol. 1998;124:1675–1683. doi: 10.1038/sj.bjp.0701991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speranza E., Williamson B.N., Feldmann F., Sturdevant G.L., Perez-Perez L., Meade-White K., Smith B.J., Lovaglio J., Martens C., Munster V.J. Single-cell RNA sequencing reveals SARS-CoV-2 infection dynamics in lungs of African green monkeys. Sci. Transl. Med. 2021;13:eabe8146. doi: 10.1126/scitranslmed.abe8146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart T., Butler A., Hoffman P., Hafemeister C., Papalexi E., Mauck W.M., 3rd, Hao Y., Stoeckius M., Smibert P., Satija R. Comprehensive integration of single-cell data. Cell. 2019;177:1888–1902 e1821. doi: 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U. S. A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tisoncik-Go J., Gasper D.J., Kyle J.E., Eisfeld A.J., Selinger C., Hatta M., Morrison J., Korth M.J., Zink E.M., Kim Y.M. Integrated omics analysis of pathogenic host responses during pandemic H1N1 influenza virus infection: the crucial role of lipid metabolism. Cell Host Microbe. 2016;19:254–266. doi: 10.1016/j.chom.2016.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen Y., Gu J., Chakrabarti S.K., Aylor K., Marshall J., Takahashi Y., Yoshimoto T., Nadler J.L. The role of 12/15-lipoxygenase in the expression of interleukin-6 and tumor necrosis factor-alpha in macrophages. Endocrinology. 2007;148:1313–1322. doi: 10.1210/en.2006-0665. [DOI] [PubMed] [Google Scholar]
- Werz O., Gerstmeier J., Libreros S., De la Rosa X., Werner M., Norris P.C., Chiang N., Serhan C.N. Human macrophages differentially produce specific resolvin or leukotriene signals that depend on bacterial pathogenicity. Nat. Commun. 2018;9:59. doi: 10.1038/s41467-017-02538-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolard M.D., Hensley L.L., Kawula T.H., Frelinger J.A. Respiratory Francisella tularensis live vaccine strain infection induces Th17 cells and prostaglandin E2, which inhibits generation of gamma interferon-positive T cells. Infect. Immun. 2008;76:2651–2659. doi: 10.1128/IAI.01412-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yurdagul A., Jr., Subramanian M., Wang X., Crown S.B., Ilkayeva O.R., Darville L., Kolluru G.K., Rymond C.C., Gerlach B.D., Zheng Z. Macrophage metabolism of apoptotic cell-derived arginine promotes continual efferocytosis and resolution of injury. Cell Metab. 2020;31:518–533 e510. doi: 10.1016/j.cmet.2020.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S., Weinberg S., DeBerge M., Gainullina A., Schipma M., Kinchen J.M., Ben-Sahra I., Gius D.R., Yvan-Charvet L., Chandel N.S. Efferocytosis fuels requirements of fatty acid oxidation and the electron transport chain to polarize macrophages for tissue repair. Cell Metab. 2019;29:443–456 e445. doi: 10.1016/j.cmet.2018.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Single-cell RNA-sequencing data has been deposited in the National Center for Biotechnology Information’s Gene Expression Omnibus under GEO series accession number GSE171510. Metabolomics and lipidomics data are available via Mendeley Data https://doi.org/10.17632/m9sfjxn8c5.1. Any additional data that support the study’s findings are available from the lead contact upon request.
This paper does not report original code. All code utilized was publicly available.