

Graphical abstract
Keywords: Transcriptional Regulatory Networks, Gene Regulatory Networks, Transcription regulation modelling, Core transcriptional Regulatory Circuitry, Transcriptional regulatory network mapping
Abstract
Transcriptional Regulatory Networks (TRNs) are mainly responsible for the cell-type- or cell-state-specific expression of gene sets from the same DNA sequence. However, so far there are no precise maps of TRNs available for each cell-type or cell-state, and no ideal tool to map those networks clearly and in full from biological samples. In this review, major approaches and tools to map TRNs from high-throughput data are presented, depending on the type of methods or data used to infer them, and their advantages and limitations are discussed. After summarizing the main principles defining the topology and structure–function relationships in TRNs, an overview of the extensive work done to map TRNs from bulk transcriptomic data will be presented by type of methodological approach. Most recent modellings of TRNs using other types of molecular data or integrating different data types, including single-cell RNA-sequencing and chromatin information, will then be discussed, before briefly concluding with improvements expected to come in the field.
1. Introduction
Transcriptional Regulatory Networks (TRNs) can be defined as the complete set of transcriptional regulatory information [1], [2]. These networks mainly describe the interactions between transcription factor (TF) proteins and the genes they regulate, by binding to specific DNA motifs in their vicinity (Fig. 1). TRNs thus define the gene expression programs that control cell-type-specific protein expression, and condition early development, as well as terminal fate and response to environmental changes. This regulation involves formation of a chromatinian landscape, that can be inherited by daughter cells as an encoded “memory” of the fate of the cells. TRNs should be robust enough in order to maintain the identity of the cell and pass it on, but should also allow dynamics in order to respond to signaling cues. They play fundamental roles in maintaining homeostasis, and their perturbation can lead to diseases. Multiple approaches have been developed to try to decipher TRNs but none of them has yet perfectly solved the general mapping of their complexity in diverse organisms and conditions.
Fig. 1.

A) 4 representations of the same Transcriptional Regulatory Network (TRN) made of 3 genes, which TF proteins bind to each other’s DNA binding motifs present in their DNA regulatory sequences. TF1 binds to the regulatory sequences of TF1 and TF2 coding genes. TF2 binds to the regulatory sequences of TF1, TF2 and TF3 coding genes. TF3 binds to the regulatory sequences of TF1 and TF3 coding genes. B) Data sets available to infer TRNs used in the research works described in this review, placed on the scheme of the gene expression steps of a Transcription Factor.
Generation of molecular data being no longer a limitation, modelling and representation of biological networks become the next challenges of Systems Biology. Description of TRNs should provide understanding of their functions and predict their behaviors. It should also help identification of new cellular reprogramming candidates, oncogenes and drug targets. Advances in the field have been encouraged by the Dialogue for Reverse Engineering Assessments and Methods (DREAM) project [3], which provided evaluation of biomolecular network inference methods through successive challenges. In this review, advantages and limitations of different computational biology approaches and 34 tools to infer TRNs (Table 1) will be discussed. This work relies on examples of significant research work in the field, and do not intend to be exhaustive. After discussing TRN topology, the extensive work done to map TRNs from bulk transcriptomic data, which was the first layer of data available for such inference, will be presented. As gene expression can be regulated at the genetic, epigenetic, transcriptional, post-transcriptional or proteic levels, multiple molecular data sets are now available to reconstruct TRNs (Fig. 1B). Methods to infer TRNs using other types of data, such as genetic perturbation or chromatin conformation, or integrative methods using multiple data types will then be discussed.
Table 1.
Computational biology tools to map Transcriptional Regulatory Networks.
| Tools | Full names (when available) | Links to tool (when available) | Approaches and data used | References |
|---|---|---|---|---|
| ARACNe-AP | Algorithm for the Reconstruction of Accurate Cellular Networks | http://sourceforge.net/projects/aracne-ap | Information theory, bulk transcriptomics, tested on single-cell RNA-seq | [56], [60] |
| CLR | Context Likelihood of Relatedness | Information theory, bulk transcriptomics, tested on single-cell RNA-seq | [57] | |
| ANOVerence | http://www2.bio.ifi.lmu.de/~kueffner/anova.tar.gz | Correlation, bulk transcriptomics | [58] | |
| CoExpNetViz | http://bioinformatics.psb.ugent.be/webtools/coexpr/ | Information theory, bulk transcriptomics | [59] | |
| GeneXPress | Module Networks algorithm | https://pypi.org/project/GeneXpress/#files | Regression, bulk transcriptomics | [38] |
| GENIE3 | GEne Network Inference with Ensemble of trees | https://bioconductor.org/packages/release/bioc/html/GENIE3.html | Regression, bulk transcriptomics, tested on single-cell RNA-seq | [39] |
| GRNBoost | Gene Regulatory Networks Boost | http://arboreto.readthedocs.io | Regression, bulk transcriptomics, tested on single-cell RNA-seq | [40] |
| TIGRESS | Trustful Inference of Gene REgulation using Stability Selection | http://cbio.ensmp.fr/tigress | Regression, bulk transcriptomics | [42] |
| LiPLike | Linear Profile Likelihood | https://gitlab.com/Gustafsson-lab/liplike | Regression, bulk transcriptomics | [41] |
| Banjo | Bayesian Network Inference with Java Objects | Source code and simulated data are available upon request | Bayesian inference, bulk transcriptomics | [63] |
| LeMoNe | http://bioinformatics.psb.ugent.be/software | Bayesian inference, bulk transcriptomics | [64] | |
| TWNs | Transcriptome-Wide Networks | https://github.com/battle-lab/twn_tsn | Bayesian inference, splicing isoforms, bulk transcriptomics | [66] |
| NIR | Network Identification by multiple Regression | ODEs, bulk transcriptomics | [67] | |
| Inferelator | freely available upon request | ODEs, bulk transcriptomics | [68] | |
| GINsim | Gene Interaction Network simulation suite | http://ginsim.org/ | Logical modelling, bulk transcriptomics | [70], [71] |
| GNA | Genetic Network Analyzer | http://www-helix.inrialpes.fr/gna | Piecewise linear equations, bulk transcriptomics | [72] |
| Network Deconvolution | Network Deconvolution | http://compbio.mit.edu/nd/index.html | Network deconvolution, bulk transcriptomics | [73] |
| RegulonDB | http://regulondb.ccg.unam.mx | TF regulatory information, relational database, bulk transcriptomics | [34], [28] | |
| GRAM | Genetic Regulatory Modules | TF regulatory information, bulk transcriptomics | [26] | |
| DISTILLER | Data Integration System to Identify Links in Expression Regulation | TF regulatory information, bulk transcriptomics | [35] | |
| SEREND | SEmi-supervised REgulatory Network Discoverer | http://sb.cs.cmu.edu/ecoli/ | TF regulatory information, logistic regression, bulk transcriptomics | [36] |
| DeMAND | Detecting Mechanism of Action by Network Dysregulation | Bioconductor package or web based geWorkbench module | TF regulatory information, logistic regression, bulk transcriptomics | [37] |
| SIRENE | Supervised Inference of REgulatory NEtworks | http://projects.cbio.mines-paristech.fr/sirene/ | TF regulatory information, SNV classifiers, bulk transcriptomics | [33] |
| DREM | Dynamic Regulatory Events Miner | http://sb.cs.cmu.edu/drem/ | TF regulatory information, HMM based, bulk transcriptomics | [32], [29] |
| Flynet | http://compbio.mit.edu/flynet/ | Evolutionary conserved sequence motifs integrated with TF binding and chromatin modification data | [77] | |
| SCENIC | Single-CEll regulatory Network Inference and Clustering | https://aertslab.org/#scenic | Single-cell RNA-seq, based on GENIE3 and GENEBoost | [62] |
| SCINET | Single-Cell Imputation and NETwork construction | https://github.com/shmohammadi86/SCINET | Single-cell RNA-seq and a reference global interactome | [85] |
| PIDC | Partial Information Decomposition and Context | Single-cell RNA-seq, information theory, bulk transcriptomics, partial information decomposition | [86] | |
| SCNS toolkit | Single-Cell Network synthesis | http://scns.stemcells.cam.ac.uk/ | Single-cell RNA-seq, boolean logical rules | [87] |
| SCODE | scRNA-seq performed on differentiating cells by integrating the transformation of linear ODEs and linear regression | https://github.com/hmatsu1226/SCODE | Single-cell RNA-seq via regulatory dynamics based on ODEs | [88] |
| CSHMM-TF | Continuous-State Hidden Markov Models TF | https://github.com/jessica1338/CSHMM-TF-for-time-series-scRNA-Seq | Single-cell RNA-seq and TF-gene interaction, Continuous-State Hidden Markov Models | [89] |
| MARINa | Master Regulator Inference Algorithm | http://califano.c2b2.columbia.edu/marina-license | Differential expression and protein-protein interactions | [93] |
| GNAT | http://mostafavilab.stat.ubc.ca/gnat/ | Bulk transcriptomics using hierarchy of tissues, Gaussian Markov Random Fields | [95] | |
| CRCmapper | Core transcriptional Regulatory Circuitry mapper | https://github.com/ViolaineSaint-Andre/CRCmapper | Graph theory, enhancer information (H3K27ac or relevant TF ChIP-seq) and optionally expression and/or ATAC-seq data | [15] |
2. TRN topology
TRN topology is classically represented by a graph on which genes are depicted by nodes, and their regulatory interactions by edges between those nodes. In this section, I will summarize elementary structures of TRNs, and will discuss TRN conservation and hierarchical structure.
2.1. Motifs
Pioneer work on gene regulatory circuits in Escherichia coli (E. coli), evidenced that these contain small sets of regulatory patterns, and the recurrence of these motifs in networks suggested putative functions associated to them [4], [5]. Network motifs have been found in many other organisms since then, from yeast [6], [7] to mouse and human [8], [9], [10], showing that TRNs can generally be decomposed in basic building blocks with putative functions. This extends the notion of structure–function relationship, best known at the level of molecules, to the level of network patterns. Even if the existence, enrichment and significance of these motifs can be arguable, in particular as these are dependent on data availability [11], some general patterns have been identified. In TRNs, most genes are regulated just one step away from their activator [12]. The main types of recurring motifs described in yeast, mouse and human [9], [13], [14], [15], can be depicted as follows. Simple regulation consists of a TF that regulates the expression of a gene, either positively or negatively, with no intermediates (Fig. 2A). It is simple auto-regulation when a gene regulates its own expression (Fig. 2B). At the cell population level, positive auto-regulation induces a bimodal distribution that can maintain a mixed phenotype for better adaptation, and negative auto-regulation speeds up the response time of gene circuits and reduce inter-cellular variation in protein levels [12]. Feedforward loops (FFL) consist of three genes: a regulator, x, which regulates y, and a gene z, which is regulated by both x and y (Fig. 2C and D). As each of the regulatory interactions in the FFL can be either positive or negative, eight structural types of FFL are possible. Coherent FFLs have two arms with the same net sign of actions and have been shown to filter out brief spurious pulses of signal. Incoherent FFLs have two arms with different net signs of action and have been shown to generate pulse-like response dynamics. Among all FFLs, the coherent type-1 FFL (Fig. 2C) and the incoherent type-1 FFL (Fig. 2D) were shown to occur more frequently in yeast TRNs than the other types [16]. The possible specific dynamical function of these loops are described in details in [13]. Feedback Loops are simple circular chains of interactions, that can be either positive (Fig. 2E) or negative (Fig. 2F), depending on the product of the signs of their constitutive interactions. Positive circuits were shown to be involved in the generation of differentiated states, whereas negative circuits may be important for homeostasis or sustained oscillatory behavior [14]. Single-Input Modules (SIMs) consist of a protein that regulates a group of target genes, positively (Fig. 2G) or negatively (Fig. 2H). This motif can allow coordinated expression of a group of genes with shared function, and can even generate a temporal expression program, depending on its target activation thresholds [13]. Dense Overlapping Regulons (DORs) consist of a set of regulators that control a set of output genes [5] (Fig. 2I). This topology has probably evolved to enable prompt response to environmental changes, considering it can take up to one cell generation time to pass a signal down each step of a cascade [13].
Fig. 2.

Transcriptional Regulatory Network motifs. A) Simple regulation B) Auto-regulation C) Coherent type-1 Feed Forward Loop D) Incoherent type-1 Feed Forward Loop E) Positive Feedback Loops F) Negative Feedback Loops G) Single-Input Module with positive regulation H) Single-Input Module with negative regulation I) Dense Overlapping Regulon J) Feedback Loop comprising two positive transcription interactions K) Feedback Loop comprising two negative transcription interactions L) Core transcriptional Regulatory Circuitry.
In addition to the motifs described above, work on the sea urchin and yeast, revealed that developmental TRNs show specific motifs of regulation that enable cell-fate decisions to last in time [13], [17]. These specific motifs may thus be the most important motifs to regulate cellular identity at steady state, and can be described as follows. Feedback Loops comprising two transcription interactions are positive-feedback loops of mutually regulated TFs. These can be double-positive loops (Fig. 2J) or double-negative loops (Fig. 2K). In both cases, a transient signal x can cause the loop to lock irreversibly into a steady state [17]. This network motif can thus provide memory of an input signal long after the input signal has stopped. Often, y and z also positively regulate themselves, strengthening the memory effects. Finally, a motif that plays a fundamental role in establishment and maintenance of cellular identity is the Core transcriptional Regulatory Circuitry (CRC) (Fig. 2L). The CRC consists of TFs that regulate each other's expression forming an interconnected auto-regulatory loop of regulation [9], [15]. The TFs that compose the CRC, or “core TFs”, bind to the regulatory sequences of their target genes and regulate their expression, generally positively. However, in some cases, these TFs can act as repressors. CRCs have been identified and characterised as major regulators of cell identity in multiple human and mouse cell-types [8], [9], [18], [19], [20]. Among the few, generally between 3 and 30, core TFs that compose the CRCs [15], are the TFs that are able to reprogram another cell-type into the cell-type in which they are identified. With this property of containing reprogramming TFs, CRC can be thought of as the most upstream motif of TRNs [15], [21]. As core TFs collectively bind to most expressed genes in each cell-type,their targets can be identified to extend the networks [15]. The CRC motif highlights the fact that when multiple TFs cooperate synergistically, it potentiates their ability to induce changes in cell fate [22]. On a broader level, cross-regulatory interactions among the core TFs could facilitate the integration of complex cellular signals, while conferring robustness to these circuits.
2.2. Modules
Studies of TRNs have shown that cellular functions are likely to be carried out in a modular manner [2], [23], [24], [25]. In general, modularity in graphs refers to clusters of nodes that participate to common biological processes [26], [27]. The network potential modularity can be reflected by the clustering coefficient, which, if modularity exists in the network, should be different from the one of randomised networks. Multiple methods have been developed to identify modules, also called communities [26], [27], [28], [29], [30], [31], [32], [33], [34], [35], [36], [37], [38], [39], [40], [41], [42]. Regulatory modules are most often identified as a set of co-expressed genes whose expression vary with the expression levels of regulators [38], [39], [40], [41], [42] or to which the same set of TFs binds [26], [29], [32], [33], [35], [36], [37], [43]. These methods will be described in more details in section 2) and 3). A DREAM challenge recently allowed to test and compare module identification methods on protein–protein interaction (PPI) data in human using Genome-Wide Association Studies (GWAS) data [44]. Kernel clustering leveraging a new diffusion state distance instead of the shortest-path metric that is usually used to predict function using PPI structure [31], was identified as the top performing method for this challenge. Modularity optimisation, with a resistance parameter that controls the granularity of modules, and random-walk based on Markov clustering, with locally adaptive granularity to balance module sizes, were also among the top performing methods, and the value of applying multiple methods to detect complementary types of modules was emphasized [44]. Module identification is still an active field of research. For example, following this DREAM challenge benchmark, Didier et al. showed that a randomization procedure, as well as the consideration of weighted edges and layers, increases the number of trait and disease community detected [30].
2.3. TRN conservation
Structural properties of TRNs seem to be conserved across species [10], [45] and tissues [10], [15]. Comparison of TRNs across 394 human samples and 662 TFs on the basis of edges overlap, showed that developmentally and functionally related lineages consistently grouped together, indicating that they share regulatory components [46]. Interestingly, between different cell-types or species, the regulatory link of TF to gene is more conserved than are the sequences [10], [47], [48], [49]. In many cases, it seems that evolution has converged independently on the same regulation circuit [50]. Network motifs may have been 'rediscovered' because they perform important functions, they are robust, and use the least number of components to carry out biological functions [13].
2.4. Hierarchy in TRNs
Reprogramming experiments in human and mouse show that as few as 3 to 5 TFs, when expressed ectopically, have the ability to rewire the gene expression program of a cell towards the one of another cell-type [21], [51]. However, TRNs are often represented as hairy balls of large number of interactions, in which it is not obvious to identify a hierarchy in the information flow. With these networks, it is difficult to read how reprogramming factors, when introduced in a cell, could easily rewire its TRN. A few studies included hierarchy in the design of TRN mapping [15], [52]. When organized into a hierarchy, genomic binding information of 119 human transcription-related factors in over 450 distinct experiments, showed that factors at different levels have different binding properties [52]. By identifying a Core transcriptional Regulatory Circuit upstream of the full TRN, CRCmapper [15] helps understand how a few TFs only are sufficient to fully reprogram a cell and durably change its phenotype. In addition, “hierarchy” is sometimes confused with “connectivity”. Indeed, numerous methods for mapping TRNs use “in-degree”, “in betweenness” or “centrality degree” as metrics to identify “hub” genes, which are often considered “master regulators” of transcription. In- and out-degree are respectively defined as the number of edges arriving to a node and the number of edges leaving a node. In a random network, the in- and out- degrees would follow a Poisson distribution, as most nodes would have roughly the same number of links. In TRNs however, the in-degree distribution, is best approximated by an exponential, showing that most genes are regulated by few TFs, and the out-degree, by a power law distribution, showing that most TFs regulate a few genes [5], [53], [54]. Thus, “in-degree”, “hub” or “centrality” metrics are not necessarily good criteria to identify master regulators of transcription and hierarchy of factors should be better considered in the design of TRN mapping.
3. Modelling TRNs from bulk transcriptomic data
Use of DNA micro-arrays enabled quantification of messenger RNA (mRNA) molecules under many experimental conditions and provided a base to develop methods to analyse TRNs. These algorithms can now be applied to RNA-sequencing (RNA-seq) data, which have become more and more affordable. Most methods to model TRNs from transcriptomic data are based on machine learning approaches. They include supervised and non-supervised methods and various types of models, as for example, artificial neural networks, decision trees, regressions or Bayesian networks [55].
3.1. Correlation and mutual information
Correlation methods can be used on time series or when multiple conditions are available. These correlate the variation in mRNA levels of a TF with sets of mRNA levels for other genes. Mutual information has been introduced to consider non-linear dependencies between TFs and their targets [56]. For correlation and mutual information methods, partial correlation coefficient and data processing inequality can be applied to distinguish direct and indirect dependencies. Examples of correlation and mutual information methods include, ARACNE (Algorithm for the Reconstruction of Accurate Cellular Networks) [56], CLR (Context Likelihood of Relatedness) [57], ANOVerence [58] and CoExpNetViz [59]. ARACNE was designed to scale up to the complexity of regulatory networks in mammalian cells. Even if it is sensitive to loops and complex topologies, it seems to work pretty well on human samples. A new version of ARACNe, ARACNe-AP [60], is less computationally demanding. The CLR algorithm, using mutual information, and controlling for false positive interactions, showed a 60% true positive rate on 3,216 known E. coli regulatory interactions. ANOVerence [58], which uses a non-parametric, non-linear correlation coefficient, is an interesting alternative to other measures of dependency, and was rated the best performer on real expression compendia in the DREAM5 challenge. Finally, a recent tool adapted to TRN mapping in plants, CoExpNetViz [59], uses mutual information and a set of query or “bait” genes to predict TRNs.
3.2. Regressions
Contrary to correlation and information theoretic methods, regression methods can predict directed interactions. Regression-based approaches rely on the assumption that RNA levels of a TF and their direct target genes vary linearly. This assumption is clearly not optimal, as TF are transcriptionally and post-transcriptionally highly regulated, and their binding to DNA motifs depend on the accessibility of the chromatin, as well as their interactions with co-factors. Feature selection methods are generally used to select for the TFs to be used in the regularised regression models. Multiple TRN mapping methods are based on this approach. This is the case of one of the first tool to map TRNs, GeneXPress [38]. This method identifies the small set of TFs which expression is predictive of the expression level of modules of co-expressed genes using a regression tree. Later developed, GENIE3 (GEne Network Inference with Ensemble of trees) [39] is also based on regressions. It trains random forest models to predict the expression of each gene in the data set from the expression of TFs passed in input. The models are then used to derive weights for the TF-target pairs, depending on their respective relevance. Interestingly, the use of random forest regression added value of allowing non-linear co-expression relationships between a TF and its candidate targets and GENIE3 was the top-performing method for network inference in the DREAM4 and DREAM5 challenges [61]. A faster alternative to GENIE3, GRNBoost [40], [62], is based on a regression model using gradient boosting. Another great example of regression method is TIGRESS (Trustful Inference of Gene REgulation with Stability Selection) [42], which also ranked among the top methods in the DREAM5 challenge. TIGRESS performs a collection of feature selection for each target gene, and uses randomization-based techniques to score the evidence of regulatory interactions. The main differences with the other methods are that TIGRESS aggregates the features selected by least angle regression (LARS) and not by decision tree, and introduces a novel scoring technique for stability selection, which improved the performance of LARS. Finally, in order to tackle the persistent obstacle of high correlation in expression between regulatory elements in TRNs, a recent tool, LiPLike (Linear Profile Likelihood) [41], assumes a regression model and iteratively searches for interactions that cannot be replaced by a linear combination of other predictors. The overall good results obtained by regression methods suggest that, despite their basic assumption of linear relationship between regulator and target RNA levels, regression-based formalism can be highly relevant for TRN inference.
3.3. Bayesian inferences
A Bayesian network is a probabilistic graphical model that represents a set of random variables and their dependencies. In the context of TRNs, edges represent the conditional dependencies between genes. In a first step, the structure of the network is learnt and possible model improvements, such as changes in topology, are evaluated. In a second step, TF activities are predicted from the network model, and a likelihood score is used to assign additional target genes for which the expression can be predicted by the TF activity profile. Such models are attractive for their ability to learn from observations. However, searching the space of all possible conditional dependencies is very computationally intensive. To render the task manageable, heuristic approximation methods, that use locally constraint search techniques, have been developed. An influence score for dynamic Bayesian networks that attempts to estimate both the sign and relative magnitude of interactions among variables was used in Banjo (Bayesian Network Inference with Java Objects) [63]. When faced with limited quantities of observational data, the authors found that combining this influence score with moderate data interpolation reduced a significant portion of false positive interactions in the recovered networks. Another Bayesian inference tool, LeMoNe [64] infers TRNs using a centroid clustering approach to assign genes and conditions to modules and subsequently assigns a regulatory program to the gene sets. The authors show that reliably detecting condition-specific or combinatorial regulation is particularly difficult in a single optimum, but can be achieved using ensemble averaging. Too often, TRN analyses do not consider the large variety of transcripts isoforms corresponding to splicing variants, while alternative splicing critically contributes to the transcriptome diversity of most eukaryotes. In particular, alternative splicing can be a way to promptly respond to stimulation by modifying the transcription output, using epigenetic information present in coding genes [65]. Interestingly, a recently developed framework named TWNs (Transcriptome-Wide Networks) [66], based on a Bayesian bi-clustering model, focused on the regulation of relative isoform abundance and splicing, using human samples from the Genotype-Tissue Expression (GTEx) project.
3.4. Ordinary differential equations
Methods based on Ordinary Differential Equations (ODEs) allow to consider quantitative and dynamic interactions between genes, and are thus particularly well suited to time-course data. An ODE is a differential equation containing one or more functions of an independent variable and the derivatives of those functions. The term ordinary is used in contrast with the term partial differential equation, which implies more than one independent variable. For example, NIR (Network Identification by multiple Regression) [67] and Inferelator [68] use ODEs. The NIR method is based on multiple linear regression analysis of steady-state transcription profiles. It was used to retrieve a first-order model of regulatory interactions in a nine-gene subnetwork of the SOS pathway in E. coli. Inferelator uses multiparametric logistic regression to search for co-expressed modules enriched for genes that are highly connected in metabolic and functional association networks, or that contain over-represented de novo-detected motifs. It was used to successfully predict a large portion of the regulatory network of the archaeon Halobacterium NRC-1.
3.5. Qualitative modelling
Biological regulatory interactions being usually non-linear, qualitative representation of regulatory networks are of interest to map TRNs [1], [69]. Logic modelling is based on the idea that a variable can take a discrete number of states or values, two in the case of Boolean models, and that the state of a variable is determined by a logical combination of the states of other variables. Logical modelling provides a qualitative dynamical description of the corresponding regulatory system, which can help simplify the complexity of TRNs. GINsim (Gene Interaction Network simulation suite) [70], [71], is a great example of logical modelling tool. It simulates qualitative models of genetic regulatory networks based on a discrete logical formalism. Piece-wise linear differential equations have also been used to describe gene regulatory networks [69]. Piece-wise linear models capture the regulatory effects by means of step functions that change their value in a switch-like manner at threshold concentrations of the regulatory proteins. GNA (Genetic Network Analyzer) [72] is based on these models for the simulation of genetic regulatory networks that can combine gene expression data with knowledge about regulatory interactions from multiple sources.
3.6. Network deconvolution
Recognizing direct relationships between variables connected in a network is a recurrent problem in TRN mapping. To surround this difficulty, network deconvolution can be used. Work from the Kellis lab presented a method, using a reverse approach of network convolution, by exploiting eigen-decomposition and infinite-series sums, for inferring the direct dependencies in a network [73]. The advantage of this method is that the transitive effects that result from indirect effects are subtracted in a single operation, and the effectiveness of this algorithm was demonstrated on several large-scale networks.
4. Modelling TRNs from other data types or integrating multiple data types
Approaches considering transcriptomic data only, globally lack directness and typically require large number of samples to build a network. In the following section, I will describe methods using other types of data or data integrative approaches to model TRNs, by highlighting some examples.
4.1. TF regulatory information
Early work using Chromatin Immuno-precipitation (ChIP) technology helped progresses in TRN mapping [74], [75]. Methods integrating TF-gene physical interaction knowledge are particularly interesting compared to approaches based on expression only, because, as mentioned in 2.2), TF RNA levels are not very good surrogates of their activities. One of the first tool integrating TF regulatory information is RegulonDB [34]. The latest version of this well-maintained relational database including TF-genes interactions information in E. coli, RegulonDB v9.0, integrates interactions with small RNAs [28]. Another pioneer algorithm to infer TRNs by using TF regulatory information is the GRAM algorithm [26]. It was applied to 106 TF ChIP-Chip data and 500 expression profiles to construct a genome-wide TRN in yeast Saccharomyces cerevisiae (S. cerevisiae). Other methods, such as DISTILLER (Data Integration System To Identify Links in Expression Regulation) [35], combine expression data with motifs, ChIP-chip or ChIP followed by sequencing (ChIP-seq) data, to search for co-regulated modules. Supervised (or semi-supervised) methods, such as SEREND (SEmi-supervised REgulatory-Network Discoverer) [36], DeMAND (Detecting Mechanism of Action by Network Dysregulation) [37] and SIRENE (Supervised Inference of REgulatory NEtworks) [33] have also been developed to integrate expression with TF binding information. The first two use models based on logistic regression to predict the probability that genes belong to the same regulon, and SIRENE splits network inference into multiple binary classification problems for each TF, training Support Vector Machine (SVM)-based classifiers. Computational work using GeneXPress [38] on 38 purified populations of human hematopoietic cells integrated gene expression with cis-elements in gene promoters, to identify 276 TFs differentially expressed across hematopoietic states, and modules of highly co-expressed genes [43]. This led to graphs, on which nodes represent TFs, and edges are colored according to the correlation between the expression patterns of the nodes in the specific lineage (Fig. 3A). Integration of TF binding information also led to other interesting representations of TRNs. For example, a representation based on ChIP-seq combined with expression data from the induction of the same factors in Mycobacterium tuberculosis was proposed [53] (Fig. 3B). It was made using DREM [29], [32] to model gene expression, which is a HMM-based approach to integrate static interaction data with time series gene expression. Altogether, methods coupling gene expression with TF regulatory information are efficient, but these methods generally do not consider chromatin accessibility, long-range interactions permitted by the 3D structure of the genome, or combinatorial interactions between TFs and cofactors.
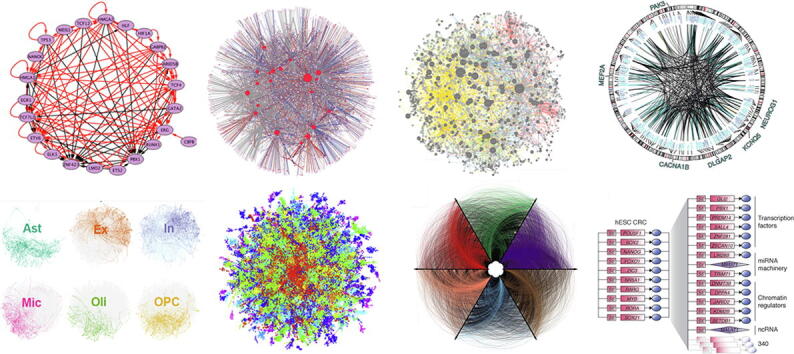
Fig. 3.

Diverse representations of Transcriptional Regulatory Networks. Images adapted from : A) Novershtern et al., 2011 B) Galagan et al., 2013 C) Kemmeren et al., 2014 D) Wang et al., 2018 E) Mohammadi et al., 2019 F) Sandhu et al., 2012 G) Neph et al., 2012 H) Saint-André et al., 2016.
4.2. Evolutionary conserved DNA binding motifs for TFs
Transcriptional enhancers, which are DNA regulatory elements able to recruit TFs and the transcriptional apparatus to activate expression of their target gene, are more likely to be functional when they are under purifying evolutionary selection [76]. This conservation feature of regulatory sequence, in addition to gene expression, TF binding and chromatin marks, was used as input to map TRNs from 12 Drosophila melanogaster species with flynet [77]. In contrast to methods which require training data for each considered TF, flynet trains a global classifier to predict regulatory interactions by integrating motif, binding, chromatin, and expression data as input features. Interestingly, this analysis showed that chromatin profiles were more informative than the commonly used expression profiles.
4.3. Genetic perturbation
Genetic knockout and perturbation were also used to reconstruct TRNs and won the DREAM3 challenge [78]. More recently, Kemmeren et al. monitored mRNA expression using micro-arrays after individual deletions of one-quarter of S. cerevisiae genes [79]. From this data, positive and negative regulations could be identified and the resulting network was displayed with Cytoscape [80] (Fig. 3C). Genetic perturbations can be interesting for mapping or validating TRNs, but it is important to note that these approaches can only capture expression changes compatible with viability and may induce deregulation of the TRNs of the cells.
4.4. eQTL data
Some studies used inter-individual variation in gene expression, defining eQTLs (expression Quantitative Trait Loci), to infer TRNs [81], [82]. For example, Fairfax et al. created TRNs based on eQTLs identified upon monocytes stimulation, which revealed multiple master regulators of innate immune responses [81]. Another example, is recent work performed on adult brain cell samples across 1866 individuals, for which regulatory connections were identified by relating the activity of TFs to target genes through eQTL mapping [82]. For each TF, a regulatory link was created if the TF had a DNA binding motif within 1 kb of the promoter or associated enhancers of the target and if it had a high coefficient in a regularised elastic net regression relating TF activity to target expression. The final TF-enhancer-target gene list constitutes the gene regulatory network. Linkages from the full network targeting cell-type-specific biomarker genes, were displayed with Circos [83], as exemplified for excitatory neurons (Fig. 3D).
4.5. Single-cell RNA-seq data
Recent advances in microfluidics have opened opportunities for generating single-cell RNA-seq (scRNA-seq) data sets. Computational biology approaches to map TRNs from these data have to adjust to their inherent stochastic variation and sparsity. These approaches are designed to map TRNs at the level of unique cells [62], [84], [85], [86] or to help reconstruct dynamic cell trajectories or putative TF order of action [87], [88], [89]. SCENIC (Single-Cell regulatory Network Inference and Clustering) [62] identifies sets of genes that are co-expressed with TFs, using GENIE3 [39] and the faster variant of it, GRNBoost [40]. Also, to reduce false positive and indirect targets found with GENIE3, SCENIC performs cis-regulatory motif analysis on each co-expression module, and only modules with significant motif enrichment of the correct upstream regulator are retained. Another computational framework, SCINET (Single-I mputation and NETwork construction) [85], enables inference of single-cell interactomes by integrating a reference protein interactome with single-cell gene expression data (Fig. 3E). PIDC (Partial Information Decomposition and Context) [86] uses partial information decomposition to identify potential regulatory relationships between genes, and outputs a weighted network from an expression matrix. Finally, SCODE (scRNA-seq performed on differentiating cells by integrating the transformation of linear ODEs and linear regression) [88], SCNS (Single-Cell Network Synthesis) [87] and CSHMM-TF (Continuous-State Hidden Markov Models TF) [89] interpret scRNA-seq as time-course expression data, where the pseudo-time corresponds to the time information, and are relevant for biological systems undergoing dynamic transcriptional changes. A recent benchmark was performed on six scRNA-seq network inference methods, primarily developed for bulk RNA-seq, based on their ability to infer similar networks when applied to two independent data sets for the same biological condition [90]. For networks with up to 100,000 links, GENIE3 resulted to be the most reproducible algorithm and, together with GRNBoost2, showed higher intersection with ground-truth biological interactions. However, even the best performing methods showed reproducibility scores that were below 54%, indicating that further improvements are still needed in the design of network inference methods for scRNA-seq data. Other benchmarks including both methods targeting bulk RNA-seq data and methods specifically designed for scRNA-seq data [91], [92], also concluded a rather lack of accuracy of these methods, even for approaches specifically developed for single-cell data, and that methods that do not require pseudo-time-ordered cells are generally more accurate.
4.6. Protein interactomes
Protein interactome have also been used for TRN mapping. For example, MARINa (Master Regulator Inference Algorithm) [93] generates a TRN and infers master regulators, from a regulatory model of protein–protein interactions and a list of genes ranked by their differential expression in two phenotypes. Others have used whole-exome sequencing and a global proteome network aggregated from different network resources [94], or integrated a reference protein interactome with scRNA-seq data [85], for TRN inference.
4.7. Hierarchy of tissues
The GTEx Project generated RNA-seq expression data for more than 30 distinct human tissues. Within the frame of this project, GNAT [95] addresses the problem of having a small number of samples for a majority of the tissues, by inferring tissue-specific gene co-expression networks using a hierarchy of tissues. The networks are modelled for each tissue in the hierarchy, using a Gaussian Markov Random Field (GMRF) transfer learning approach, which increases the accuracy with which networks are learnt. The resulting networks from this original approach show that tissue-specific TFs are hubs that preferentially connect to genes with tissue-specific functions.
4.8. Chromatin conformation
Recent evidences suggest a reciprocal interplay between fine-scale 3D genome structure and transcription [96]. 3D structure high-throughput methods such as chromosome conformation capture (Hi-C) or Chromatin Interaction Analysis by Paired-End Tag sequencing (ChIA-PET), have revealed Topologically Associating Domains (TADs) [97], [98] and insulated neighborhoods [99] mediated by specific proteins, such as CTCF, for insulation and regulation of transcription. ChIA-PET is a method enriching for chromatin interactions involving a specific protein. It was used to examine RNA polymerase-II-associated chromatin interactions in human cells [100]. Interacting genomic sites were defined as nodes of a TRN and connected as per their connectivity in the ChIA-PET data set. The resulting graph consists of a giant network, organized into chromatin communities (Fig. 3F). The color gradient represents the structural organization of the chromatin communities, that were found to be enriched in specific functions and syntenic through evolution. Chromatin conformation data may be useful to consider for TRN inference, especially for accurate identification of enhancer-promoter interactions, but do not per se permit a full reconstruction of transcriptional regulatory interactions.
4.9. DNase hyper sensitivity
Study of the chromatin state, which modulates DNA accessibility, has provided essential information to understand gene expression regulation in space and time, and is highly relevant for inferring TRNs. Chromatin openness can be measured by DNase I hypersensitivity followed by sequencing (DNase-seq). DNase I treatment preferentially cleaves the genome within highly accessible DNA regions, called DNase I-hypersensitive sites (DHSs), leaving “footprints” that can be used to infer TF occupancy. Genome-wide maps of DNase I footprints were used to assemble a human regulatory network, and to analyse the dynamics of these connections across 41 cell samples [101]. DNA elements in all DHSs within a 10 kb interval centered on the transcriptional start site were screened for TF binding motifs. Repeating this process for every sample, disclosed a total of 38,393 edges between 475 TFs, with an average of 11,193 edges per cell sample, that were rendered with Circos [83] for 6 cell-types (Fig. 3G). This study identified many widely expressed factors that impact TRNs in a cell-selective manner. These results also show the ability of DNase I footprinting to generate TRNs without the use of gene expression data. However, the predicted regulatory links may be overestimated, as all motifs, even located in open chromatin regions are not bound by their cognate TFs. Also, only a subset of TFs is included on these networks, and yet they can be difficult to read.
4.10. ChIP-seq for histone marks
Work from consortium, such as ENCODE [102], have compiled wide arrays of ChIP-seq samples and detailed genomic annotations. Most recent compendium, Epimap [48] assembled epigenomic data with the aim to identify trait-relevant tissues and putative causal nucleotide variants. For Epimap, active enhancers were defined as the intersection of DHS consensus elements, determined from 733 DNase-seq experiments, which collectively span more than 20% of the genome, with imputed enhancer annotations, and importantly, high signal of H3K27ac. Pearson correlations between gene expression and six histone mark activity of nearby enhancers, for the samples with paired expression data, enabled prediction of 3.3 million tissue specific enhancer-gene links. For this integrative work, the large majority of samples were imputed. Imputation may result in increased homogeneity, especially for marks that are highly cell-type specific, and is less accurate for broad chromatin marks, such as H3K27ac, than for ChIP-seq data with sharp peaks. Rather than providing clear transcriptional regulatory circuitry maps for a large number of cell-types, the major interest of this study is to predict causal variants and trait-relevant tissues. Resolution of such resource should soon be improved with increasing availability of single-cell datasets and more tissue types or environmental perturbations related samples.
4.11. Enhancer information
Enhancer information was recently used to anchor maps of Core transcriptional Regulatory Circuitry (CRC) [15], the small network of TFs that was shown to be most upstream of multiple cell-type’s gene expression programs. In the cell-types in which they have been identified, the TFs forming the CRC bind to each-other’s DNA regulatory sequences (Fig. 2L) [8], [9], [15], [18], [19], [21]. These core TFs also co-bind the regulatory sequences of most cell identity genes in a given cell-type [15] and contribute to the formation of super-enhancers (SEs) in the vicinity of their targets, including their own genes [15], [103]. Since core TFs are not known for most cell-types, ChIP-seq targeting H3K27ac, the best chromatin mark to identify active enhancers [104], can be used as a surrogate to identify SEs with the ROSE program [103], [105]. In Saint-André et al. [15], we used SE information and motif binding data for 1207 TFs, to predict CRC models for 85 human samples, corresponding to 75 cell and tissue types. A CRC example of human embryonic stem cells and first layer of target genes is shown in Fig. 3H. These models recapitulate and expand on previously described CRCs for well-studied cell-types and provide novel and readable TRN models for a broad range of human cell-types. Over hundreds of mouse and human samples, when computing TF interactions, a CRC (fully interconnected regulatory loop of at least 3 TFs) is always found from any ChIP-seq sample of sufficient sequencing depth and quality. Interestingly, there are not multiple independent loops identified, but rather one major CRC is emerging from each sample, with sometimes some regulatory edges missing, but always made of about the same TFs, supporting the idea that one CRC per cell-type or cell-state may be a general rule. Predicted CRCs contain already characterized master TFs, proto-oncogenes, and terminal TFs of signaling pathways, and are largely supported by ChIP-seq data and functional tests in corresponding cell-types [15]. The CRCmapper program [15] has been designed so that users just need to input a ChIP-seq track to get a CRC map of a sample of interest. The program performs DNA motif analysis of the sum of the promoter and the enhancer sequences within SEs (not the full SE) to predict regulatory interactions, uses a graph algorithm to compute all maximal cliques from a graph, and identifies the most representative fully interconnected loop of TFs as the CRC. For a refined analysis, gene expression data, such as RNA-seq, and DNA accessibility data, such as Assay for Transposase Accessible Chromatin followed by sequencing (ATAC-seq), can be passed to the program, to respectively, improve enhancer to gene associations, and reduce the search space for motifs within the individual enhancers that compose a SE. To best use ATAC-seq information, the corresponding peaks should be passed to the program instead of the peaks output from MACS. CRCmapper was successfully used to map core TRNs in normal [20], [106] or cancer samples [107], [108]. Importantly, as SEs cluster in the cell to form phase-separated condensates, these can be targeted with drugs to rewire transcriptional addictions in cancer [109], [110], [111].
5. Summary and outlook
Over the last 20 years, multiple approaches have been developed to map TRNs. They mostly followed technological developments allowing production of different types of molecular data. First based on bulk transcriptomic data, different mathematical modelling methods have been tested and compared, in particular during the DREAM challenges. Following ChIP-seq development, TF regulatory information has then been integrated to improve accuracy of the models. Other types of data, such as genetic perturbation, eQTL data, chromatin conformation or chromatin accessibility data have also been used on their own, more or less successfully, to model TRNs. Most recent developments adapted to integrate multiple data types to gain in prediction quality. In particular, approaches integrating chromatin derived information have proven to be extremely useful in mapping TRNs, transcriptional networks derived from enhancer information accurately predicting master regulators of cell identity, or proto-oncogenes when applied to cancer samples.
TRN mapping has made great progresses with more data being available, but the field still faces a number of limitations. One of them concerns the use of DNA binding motifs for TFs, which are defined for a subset of the TFs only, and are not great predictors of TF binding, unless these are present in open chromatin regions. Indeed, for example in the human genome, about 2% only of the motifs are bound by their cognate TF when picked randomly, while around 30 % of the motifs are bound by the corresponding TF when these occur in active enhancer regions [15]. Another limitation is that, even when TF binding is observed though experimental assays, such as ChIP-seq, this does not necessarily mean actual binding of the factor at the identified genomic location, nor automatically regulation of the closest gene [11], [99]. It is also important to keep in mind when making predictions from specific data sets that chromatin accessibility is cell-type specific and condition dependent [15], [101]. Finally, reproducibility still often remains a challenge for these approaches [90] and integration of multi-layers of data asks the question of convergence of the results [11] and of some data types being much more informative than others.
Multiple technological and analytical developments should help improve current mapping of TRNs. New developments in identification of TF and transcription co-regulator binding motifs [112], [113], in indirect TF binding [114], [115], and availability of high-quality motif databases [116], [117], [118], should allow better resolution of TRN mapping. Improvement in chromatin conformation identification techniques [119] may also allow better association of enhancers to the genes they regulate. In addition, integration of diverse regulatory elements, such as non-coding RNAs, and of signaling pathways, post-transcriptional regulation, or metabolic data, should add additional layers of complexity to already available TRN models. Altogether, these expected improvements should lead to help better interpret transcriptional signatures [120], [121], [122], and identify actionable targets [109]. Detailed TRN maps of cell and tissue types and disease- or condition-specific networks, may also help better understand GWAS variant functions [46], [48]. Future approaches to map TRNs will necessarily be integrative, to make best use of the amount of available data. Although individual networks, such as TRNs, protein–protein, metabolic or signaling networks, are far from being extensively mapped yet, comprehensive whole-cell models are developed to integrate multiple layers of regulation together [123]. In particular, data integration using multiple networks are promising to bring insight in systemic understanding of cancer [90]. Up-coming deep learning approaches should also develop to help stratify patients based on gene regulation [124]. The ability to make accurate predictions from TRNs should thus continue to further our understanding of disease circuitries and favor future advances in precision medicine.
6. Author statement
This work was funded by Institut Pasteur, Paris, France.
Declaration of Competing Interest
The author declares to have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- 1.Thieffry D., Thomas R. Qualitative analysis of gene networks. Pac Symp Biocomput. 1998:77–88. [PubMed] [Google Scholar]
- 2.Levine M., Davidson E.H. Gene regulatory networks for development. Proc Natl Acad Sci U S A. 2005;102(14):4936–4942. doi: 10.1073/pnas.0408031102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stolovitzky G, Monroe D, Califano A. Dialogue on reverse-engineering assessment and methods: The DREAM of high-throughput pathway inference. Ann. N. Y. Acad. Sci., vol. 1115, Blackwell Publishing Inc.; 2007, p. 1–22. https://doi.org/10.1196/annals.1407.021. [DOI] [PubMed]
- 4.Thieffry D., Huerta A.M., Pérez-Rueda E., Collado-Vides J. From specific gene regulation to genomic networks: A global analysis of transcriptional regulation in Escherichia coli. BioEssays. 1998;20:433–440. doi: 10.1002/(SICI)1521-1878(199805)20:5<433::AID-BIES10>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 5.Shen-Orr S.S., Milo R., Mangan S., Alon U. Network motifs in the transcriptional regulation network of Escherichia coli. Nat Genet. 2002;31(1):64–68. doi: 10.1038/ng881. [DOI] [PubMed] [Google Scholar]
- 6.Milo R., Shen-Orr S., Itzkovitz S., Kashtan N., Chklovskii D., Alon U. Network motifs: simple building blocks of complex networks. Science (80-) 2002;298:824–827. doi: 10.1126/science.298.5594.824. [DOI] [PubMed] [Google Scholar]
- 7.Lee T.I., Rinaldi N.J., Robert F., Odom D.T., Bar-Joseph Z., Gerber G.K., et al. Transcriptional regulatory networks in Saccharomyces cerevisiae. Science. 2002;298:799–804. doi: 10.1126/science.1075090. [DOI] [PubMed] [Google Scholar]
- 8.Odom D.T., Zizlsperger N., Gordon D.B., Bell G.W., Rinaldi N.J., Murray H.L., et al. Control of pancreas and liver gene expression by HNF transcription factors. Science. 2004;303:1378–1381. doi: 10.1126/science.1089769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boyer L.A., Lee T.I., Cole M.F., Johnstone S.E., Levine S.S., Zucker J.P., et al. Core transcriptional regulatory circuitry in human embryonic stem cells. Cell. 2005;122(6):947–956. doi: 10.1016/j.cell.2005.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stergachis A.B., Neph S., Sandstrom R., Haugen E., Reynolds A.P., Zhang M., et al. Conservation of trans-acting circuitry during mammalian regulatory evolution. Nature. 2014;515(7527):365–370. doi: 10.1038/nature13972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Monteiro P.T., Pedreira T., Galocha M., Teixeira M.C., Chaouiya C. Assessing regulatory features of the current transcriptional network of Saccharomyces cerevisiae. 2020;10(1) doi: 10.1038/s41598-020-74043-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rosenfeld N., Elowitz M.B., Alon U. Negative autoregulation speeds the response times of transcription networks. J Mol Biol. 2002;323(5):785–793. doi: 10.1016/S0022-2836(02)00994-4. [DOI] [PubMed] [Google Scholar]
- 13.Alon U. Network motifs: theory and experimental approaches. Nat Rev Genet. 2007;8(6):450–461. doi: 10.1038/nrg2102. [DOI] [PubMed] [Google Scholar]
- 14.Thieffry D. Dynamical roles of biological regulatory circuits. Brief Bioinform. 2007;8:220–225. doi: 10.1093/bib/bbm028. [DOI] [PubMed] [Google Scholar]
- 15.Saint-André V., Federation A.J., Lin C.Y., Abraham B.J., Reddy J., Lee T.I., et al. Models of human core transcriptional regulatory circuitries. Genome Res. 2016;26(3):385–396. doi: 10.1101/gr.197590.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mangan S., Itzkovitz S., Zaslaver A., Alon U. The incoherent feed-forward loop accelerates the response-time of the gal system of Escherichia coli. J Mol Biol. 2006;356(5):1073–1081. doi: 10.1016/j.jmb.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 17.Davidson E.H., Rast J.P., Oliveri P., Ransick A., Calestani C., Yuh C.H., et al. A genomic regulatory network for development. Science (80-) 2002;295:1669–1678. doi: 10.1126/science.1069883. [DOI] [PubMed] [Google Scholar]
- 18.Odom D.T., Dowell R.D., Jacobsen E.S., Nekludova L., Rolfe P.A., Danford T.W., et al. Core transcriptional regulatory circuitry in human hepatocytes. Mol Syst Biol. 2006;2(1) doi: 10.1038/msb4100059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sanda T., Lawton L., Barrasa M.I., Fan Z., Kohlhammer H., Gutierrez A., et al. Core transcriptional regulatory circuit controlled by the TAL1 complex in human T cell acute lymphoblastic leukemia. Cancer Cell. 2012;22(2):209–221. doi: 10.1016/j.ccr.2012.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aldiri I., Xu B., Wang L., Chen X., Hiler D., Griffiths L., et al. The dynamic epigenetic landscape of the retina during development, reprogramming, and tumorigenesis. Neuron. 2017;94(3):550–568.e10. doi: 10.1016/j.neuron.2017.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Young R. Control of the embryonic stem cell state. Cell. 2011;144(6):940–954. doi: 10.1016/j.cell.2011.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Niwa H. The principles that govern transcription factor network functions in stem cells 2018. https://doi.org/10.1242/dev.157420. [DOI] [PubMed]
- 23.Ihmels J., Friedlander G., Bergmann S., Sarig O., Ziv Y., Barkai N. Revealing modular organization in the yeast transcriptional network. Nat Genet. 2002;31(4):370–377. doi: 10.1038/ng941. [DOI] [PubMed] [Google Scholar]
- 24.Berman B.P., Nibu Y., Pfeiffer B.D., Tomancak P., Celniker S.E., Levine M., et al. Exploiting transcription factor binding site clustering to identify cis-regulatory modules involved in pattern formation in the Drosophila genome. Proc Natl Acad Sci U S A. 2002;99(2):757–762. doi: 10.1073/pnas.231608898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thieffry D., Romero D. The modularity of biological regulatory networks. BioSystems. 1999;50(1):49–59. doi: 10.1016/S0303-2647(98)00087-2. [DOI] [PubMed] [Google Scholar]
- 26.Bar-Joseph Z., Gerber G.K., Lee T.I., Rinaldi N.J., Yoo J.Y., Robert F., et al. Computational discovery of gene modules and regulatory networks. Nat Biotechnol. 2003;21(11):1337–1342. doi: 10.1038/nbt890. [DOI] [PubMed] [Google Scholar]
- 27.Dutkowski J., Kramer M., Surma M.A., Balakrishnan R., Cherry J.M., Krogan N.J., et al. A gene ontology inferred from molecular networks. Nat Biotechnol. 2013;31(1):38–45. doi: 10.1038/nbt.2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gama-Castro S., Salgado H., Santos-Zavaleta A., Ledezma-Tejeida D., Muñiz-Rascado L., García-Sotelo J.S., et al. RegulonDB version 9.0: High-level integration of gene regulation, coexpression, motif clustering and beyond. Nucleic Acids Res. 2016;44(D1):D133–D143. doi: 10.1093/nar/gkv1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schulz M.H., Devanny W.E., Gitter A., Zhong S., Ernst J., Bar-Joseph Z. DREM 2.0: Improved reconstruction of dynamic regulatory networks from time-series expression data. BMC Syst Biol. 2012;6(1):104. doi: 10.1186/1752-0509-6-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Didier G, Brun C, Baudot A. Identifying communities from multiplex biological networks. PeerJ 2015;2015. https://doi.org/10.7717/peerj.1525. [DOI] [PMC free article] [PubMed]
- 31.Cao M., Zhang H., Park J., Daniels N.M., Crovella M.E., Cowen L.J., et al. Going the distance for protein function prediction: a new distance metric for protein interaction networks) going the distance for protein function prediction: a new distance metric for protein interaction networks. PLoS ONE. 2013;8(10):e76339. doi: 10.1371/journal.pone.0076339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ernst J., Vainas O., Harbison C.T., Simon I., Bar‐Joseph Z. Reconstructing dynamic regulatory maps. Mol Syst Biol. 2007;3(1):74. doi: 10.1038/msb4100115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mordelet F, Vert JP. SIRENE: Supervised inference of regulatory networks. Bioinformatics, vol. 24, Bioinformatics; 2008. https://doi.org/10.1093/bioinformatics/btn273. [DOI] [PubMed]
- 34.Huerta AM, Salgado H, Thieffry D, Collado-Vides J. RegulonDB: a database on transcriptional regulation in Escherichia coli. vol. 26. 1998. [DOI] [PMC free article] [PubMed]
- 35.Lemmens K., De Bie T., Dhollander T., De Keersmaecker S.C., Thijs I.M., Schoofs G., et al. DISTILLER: A data integration framework to reveal condition dependency of complex regulons in Escherichia coli. Genome Biol. 2009;10(3):R27. doi: 10.1186/gb-2009-10-3-r27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ernst J., Beg Q.K., Kay K.A., Balázsi G., Oltvai Z.N., Bar-Joseph Z., et al. A semi-supervised method for predicting transcription factor-gene interactions in Escherichia coli. PLoS Comput Biol. 2008;4(3):e1000044. doi: 10.1371/journal.pcbi.1000044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Woo J.H., Shimoni Y., Yang W.S., Subramaniam P., Iyer A., Nicoletti P., et al. Elucidating compound mechanism of action by network perturbation analysis. Cell. 2015;162(2):441–451. doi: 10.1016/j.cell.2015.05.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Segal E., Shapira M., Regev A., Pe'er D., Botstein D., Koller D., et al. Module networks: Identifying regulatory modules and their condition-specific regulators from gene expression data. Nat Genet. 2003;34(2):166–176. doi: 10.1038/ng1165. [DOI] [PubMed] [Google Scholar]
- 39.Huynh-Thu V.A., Irrthum A., Wehenkel L., Geurts P., Isalan M. Inferring regulatory networks from expression data using tree-based methods. PLoS ONE. 2010;5(9):e12776. doi: 10.1371/journal.pone.0012776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Moerman T, Aibar Santos S, Bravo González-Blas C, Simm J, Moreau Y, Aerts J, et al. GRNBoost2 and Arboreto: efficient and scalable inference of gene regulatory networks. Bioinformatics 2019;35:2159–61. https://doi.org/10.1093/bioinformatics/bty916. [DOI] [PubMed]
- 41.Magnusson R, Gustafsson M. LiPLike: towards gene regulatory network predictions of high certainty. Bioinformatics 2020;36:2522–9. https://doi.org/10.1093/bioinformatics/btz950. [DOI] [PMC free article] [PubMed]
- 42.Haury A.-C., Mordelet F., Vera-Licona P., Vert J.-P. Trustful Inference of Gene REgulation using Stability Selection. BMC Syst Biol. 2012;6(1):145. doi: 10.1186/1752-0509-6-145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Novershtern N., Subramanian A., Lawton L.N., Mak R.H., Haining W.N., McConkey M.E., et al. Densely interconnected transcriptional circuits control cell states in human hematopoiesis. Cell. 2011;144(2):296–309. doi: 10.1016/j.cell.2011.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Choobdar S., Ahsen M.E., Crawford J., Tomasoni M., Fang T., Lamparter D., et al. Assessment of network module identification across complex diseases. Nat Methods. 2019;16(9):843–852. doi: 10.1038/s41592-019-0509-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Boyle A.P., Araya C.L., Brdlik C., Cayting P., Cheng C., Cheng Y., et al. Comparative analysis of regulatory information and circuits across distant species. Nature. 2014;512(7515):453–456. doi: 10.1038/nature13668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Marbach D., Lamparter D., Quon G., Kellis M., Kutalik Z., Bergmann S. Tissue-specific regulatory circuits reveal variable modular perturbations across complex diseases. Nat Methods. 2016;13(4):366–370. doi: 10.1038/nmeth.3799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dunham I., Kundaje A., Aldred S.F., Collins P.J., Davis C.A., Doyle F., et al. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Boix C.A., James B.T., Park Y.P., Meuleman W., Kellis M. Regulatory genomic circuitry of human disease loci by integrative epigenomics. Nature. 2021;590(7845):300–307. doi: 10.1038/s41586-020-03145-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pérez-Rico Y.A., Boeva V., Mallory A.C., Bitetti A., Majello S., Barillot E., et al. Comparative analyses of super-enhancers reveal conserved elements in vertebrate genomes. Genome Res. 2017;27(2):259–268. doi: 10.1101/gr.203679.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Babu M.M., Luscombe N.M., Aravind L., Gerstein M., Teichmann S.A. Structure and evolution of transcriptional regulatory networks. Curr Opin Struct Biol. 2004;14(3):283–291. doi: 10.1016/j.sbi.2004.05.004. [DOI] [PubMed] [Google Scholar]
- 51.Graf T., Enver T. Forcing cells to change lineages. Nature. 2009;462(7273):587–594. doi: 10.1038/nature08533. [DOI] [PubMed] [Google Scholar]
- 52.Gerstein M.B., Kundaje A., Hariharan M., Landt S.G., Yan K.-K., Cheng C., et al. Architecture of the human regulatory network derived from ENCODE data. Nature. 2012;489(7414):91–100. doi: 10.1038/nature11245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Galagan J.E., Minch K., Peterson M., Lyubetskaya A., Azizi E., Sweet L., et al. The Mycobacterium tuberculosis regulatory network and hypoxia. Nature. 2013;499(7457):178–183. doi: 10.1038/nature12337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Guelzim N., Bottani S., Bourgine P., Képès F. Topological and causal structure of the yeast transcriptional regulatory network. Nat Genet. 2002;31(1):60–63. doi: 10.1038/ng873. [DOI] [PubMed] [Google Scholar]
- 55.Forghani R., Savadjiev P., Chatterjee A., Muthukrishnan N., Reinhold C., Forghani B. Radiomics and artificial intelligence for biomarker and prediction model development in oncology. Comput Struct Biotechnol J. 2019;17:995–1008. doi: 10.1016/j.csbj.2019.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Margolin A.A., Wang K., Lim W.K., Kustagi M., Nemenman I., Califano A. Reverse engineering cellular networks. Nat Protoc. 2006;1(2):662–671. doi: 10.1038/nprot.2006.106. [DOI] [PubMed] [Google Scholar]
- 57.Faith J.J., Hayete B., Thaden J.T., Mogno I., Wierzbowski J., Cottarel G., et al. Large-scale mapping and validation of Escherichia coli transcriptional regulation from a compendium of expression profiles. PLoS Biol. 2007;5(1):e8. doi: 10.1371/journal.pbio.0050008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Küffner R., Petri T., Tavakkolkhah P., Windhager L., Zimmer R. Inferring gene regulatory networks by ANOVA. Bioinformatics. 2012;28:1376–1382. doi: 10.1093/bioinformatics/bts143. [DOI] [PubMed] [Google Scholar]
- 59.Tzfadia O., Diels T., De Meyer S., Vandepoele K., Aharoni A., Van De Peer Y. CoExpNetViz: Comparative co-expression networks construction and visualization tool. Front Plant Sci. 2016;6:1–7. doi: 10.3389/fpls.2015.01194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lachmann A., Giorgi F.M., Lopez G., Califano A. ARACNe-AP: Gene network reverse engineering through adaptive partitioning inference of mutual information. Bioinformatics. 2016;32(14):2233–2235. doi: 10.1093/bioinformatics/btw216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Marbach D., Costello J.C., Küffner R., Vega N.M., Prill R.J., Camacho D.M., et al. Wisdom of crowds for robust gene network inference. Nat Methods. 2012;9(8):796–804. doi: 10.1038/nmeth.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Aibar S., González-Blas C.B., Moerman T., Huynh-Thu V.A., Imrichova H., Hulselmans G., et al. SCENIC: Single-cell regulatory network inference and clustering. Nat Methods. 2017;14(11):1083–1086. doi: 10.1038/nmeth.4463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yu J., Smith V.A., Wang P.P., Hartemink A.J., Jarvis E.D. Advances to Bayesian network inference for generating causal networks from observational biological data. Bioinformatics. 2004;20(18):3594–3603. doi: 10.1093/bioinformatics/bth448. [DOI] [PubMed] [Google Scholar]
- 64.Joshi A., De Smet R., Marchal K., Van de Peer Y., Michoel T. Module networks revisited: computational assessment and prioritization of model predictions. Bioinformatics. 2009;25(4):490–496. doi: 10.1093/bioinformatics/btn658. [DOI] [PubMed] [Google Scholar]
- 65.Saint-André V., Batsché E., Rachez C., Muchardt C. Histone H3 lysine 9 trimethylation and HP1γ favor inclusion of alternative exons. Nat Struct Mol Biol. 2011;18(3):337–344. doi: 10.1038/nsmb.1995. [DOI] [PubMed] [Google Scholar]
- 66.Saha A., Kim Y., Gewirtz A., Jo B., Gao C., McDowell I., et al. Co-expression networks reveal the tissue-specific regulation of transcription and splicing. Co-expression networks reveal tissue-specific. Regul Transcr Splicing. 2016;078741 doi: 10.1101/078741. [DOI] [Google Scholar]
- 67.Gardner T.S., Di Bernardo D., Lorenz D., Collins J.J. Inferring genetic networks and identifying compound mode of action via expression profiling. Science (80-) 2003;301:102–105. doi: 10.1126/science.1081900. [DOI] [PubMed] [Google Scholar]
- 68.Bonneau R., Reiss D.J., Shannon P., Facciotti M., Hood L., Baliga N.S., et al. The inferelator: An algorithn for learning parsimonious regulatory networks from systems-biology data sets de novo. Genome Biol. 2006;7 doi: 10.1186/gb-2006-7-5-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kauffman S.A. Control circuits for determination and transdetermination. Science (80-) 1973;181(4097):310–318. doi: 10.1126/science:181.4097.310. [DOI] [PubMed] [Google Scholar]
- 70.Naldi A., Berenguier D., Fauré A., Lopez F., Thieffry D., Chaouiya C. Logical modelling of regulatory networks with GINsim 2.3. BioSystems. 2009;97(2):134–139. doi: 10.1016/j.biosystems.2009.04.008. [DOI] [PubMed] [Google Scholar]
- 71.Naldi A., Hernandez C., Levy N., Stoll G., Monteiro P.T., Chaouiya C., et al. The CoLoMoTo interactive notebook: Accessible and reproducible computational analyses for qualitative biological networks. Front Physiol. 2018;9 doi: 10.3389/fphys.2018.00680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Batt G., Besson B., Ciron P.E., De Jong H., Dumas E., Geiselmann J., et al. Genetic network analyzer: A tool for the qualitative modeling and simulation of bacterial regulatory networks. Methods Mol Biol. 2012;804:439–462. doi: 10.1007/978-1-61779-361-5_22. [DOI] [PubMed] [Google Scholar]
- 73.Feizi S., Marbach D., Médard M., Kellis M. Network deconvolution as a general method to distinguish direct dependencies in networks. Nat Biotechnol. 2013;31(8):726–733. doi: 10.1038/nbt.2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ren B., Robert F., Wyrick J.J., Aparicio O., Jennings E.G., Simon I., et al. Genome-wide location and function of DNA binding proteins. Science (80-) 2000;290:2306–2309. doi: 10.1126/science.290.5500.2306. [DOI] [PubMed] [Google Scholar]
- 75.Zeitlinger J., Zinzen R.P., Stark A., Kellis M., Zhang H., Young R.A., et al. Whole-genome ChIP-chip analysis of Dorsal, Twist, and Snail suggests integration of diverse patterning processes in the Drosophila embryo. Genes Dev. 2007;21(4):385–390. doi: 10.1101/gad.1509607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kheradpour P., Stark A., Roy S., Kellis M. Reliable prediction of regulator targets using 12 Drosophila genomes. Genome Res. 2007;17(12):1919–1931. doi: 10.1101/gr.7090407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Marbach D., Roy S., Ay F., Meyer P.E., Candeias R., Kahveci T., et al. Predictive regulatory models in Drosophila melanogaster by integrative inference of transcriptional networks. Genome Res. 2012;22(7):1334–1349. doi: 10.1101/gr.127191.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yip K.Y., Alexander R.P., Yan K.-K., Gerstein M., Rattray M. Improved reconstruction of in silico gene regulatory networks by integrating knockout and perturbation data. PLoS ONE. 2010;5(1):e8121. doi: 10.1371/journal.pone.0008121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kemmeren P., Sameith K., van de Pasch L.L., Benschop J., Lenstra T., Margaritis T., et al. Large-scale genetic perturbations reveal regulatory networks and an abundance of gene-specific repressors. Cell. 2014;157(3):740–752. doi: 10.1016/j.cell.2014.02.054. [DOI] [PubMed] [Google Scholar]
- 80.Shannon P., Markiel A., Ozier O., Baliga N.S., Wang J.T., Ramage D., et al. Cytoscape: A software Environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Fairfax BP, Humburg P, Makino S, Naranbhai V, Wong D, Lau E, et al. Innate immune activity conditions the effect of regulatory variants upon monocyte gene expression. Science 2014;343:1246949. https://doi.org/10.1126/science.1246949. [DOI] [PMC free article] [PubMed]
- 82.Wang D, Liu S, Warrell J, Won H, Shi X, Navarro FCP, et al. Comprehensive functional genomic resource and integrative model for the human brain. Science (80-) 2018;362. https://doi.org/10.1126/science.aat8464. [DOI] [PMC free article] [PubMed]
- 83.Krzywinski M., Schein J., Birol I., Connors J., Gascoyne R., Horsman D., et al. Circos: An information aesthetic for comparative genomics. Genome Res. 2009;19(9):1639–1645. doi: 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Mohammadi S., Ravindra V., Gleich D.F., Grama A. A geometric approach to characterize the functional identity of single cells. Nat Commun. 2018;9(1) doi: 10.1038/s41467-018-03933-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Mohammadi S., Davila-Velderrain J., Kellis M. Reconstruction of cell-type-specific interactomes at single-cell resolution. Cell Syst. 2019;9(6):559–568.e4. doi: 10.1016/j.cels.2019.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Chan T.E., Stumpf M.P.H., Babtie A.C. Gene regulatory network inference from single-cell data using multivariate information measures. Cell Syst. 2017;5(3):251–267.e3. doi: 10.1016/j.cels.2017.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Moignard V., Woodhouse S., Haghverdi L., Lilly A.J., Tanaka Y., Wilkinson A.C., et al. Decoding the regulatory network of early blood development from single-cell gene expression measurements. Nat Biotechnol. 2015;33(3):269–276. doi: 10.1038/nbt.3154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Matsumoto H, Kiryu H, Furusawa C, Ko MSH, Ko SBH, Gouda N, et al. SCODE: An efficient regulatory network inference algorithm from single-cell RNA-Seq during differentiation. Bioinformatics 2017;33:2314–21. https://doi.org/10.1093/bioinformatics/btx194. [DOI] [PMC free article] [PubMed]
- 89.Lin C., Ding J., Bar-Joseph Z., Aerts S. Inferring TF activation order in time series scRNA-Seq studies. PLOS Comput Biol. 2020;16(2):e1007644. doi: 10.1371/journal.pcbi.1007644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Cantini L., Zakeri P., Hernandez C., Naldi A., Thieffry D., Remy E., et al. Benchmarking joint multi-omics dimensionality reduction approaches for the study of cancer. Nat Commun. 2021;12:1–12. doi: 10.1038/s41467-020-20430-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Chen S., Mar J.C. Evaluating methods of inferring gene regulatory networks highlights their lack of performance for single cell gene expression data. BMC Bioinf. 2018;19:232. doi: 10.1186/s12859-018-2217-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Pratapa A., Jalihal A.P., Law J.N., Bharadwaj A., Murali T.M. Benchmarking algorithms for gene regulatory network inference from single-cell transcriptomic data. Nat Methods. 2020;17(2):147–154. doi: 10.1038/s41592-019-0690-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lefebvre C., Rajbhandari P., Alvarez M.J., Bandaru P., Lim W.K., Sato M., et al. A human B-cell interactome identifies MYB and FOXM1 as master regulators of proliferation in germinal centers. Mol Syst Biol. 2010;6(1):377. doi: 10.1038/msb:2010.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Novarino G., Fenstermaker A.G., Zaki M.S., Hofree M., Silhavy J.L., Heiberg A.D., et al. Exome sequencing links corticospinal motor neuron disease to common neurodegenerative disorders. Science (80-) 2014;343(6170):506–511. doi: 10.1126/science:1247363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Pierson E., Koller D., Battle A., Mostafavi S., Rigoutsos I. Sharing and specificity of co-expression networks across 35 human tissues. PLoS Comput Biol. 2015;11(5):1–19. doi: 10.1371/journal.pcbi.1004220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.van Steensel B., Furlong E.E.M. The role of transcription in shaping the spatial organization of the genome. Nat Rev Mol Cell Biol. 2019;20:327–337. doi: 10.1038/s41580-019-0114-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Dixon J.R., Selvaraj S., Yue F., Kim A., Li Y., Shen Y., et al. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 2012;485(7398):376–380. doi: 10.1038/nature11082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Nora E.P., Lajoie B.R., Schulz E.G., Giorgetti L., Okamoto I., Servant N., et al. Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature. 2012;485(7398):381–385. doi: 10.1038/nature11049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Dowen J., Fan Z., Hnisz D., Ren G., Abraham B., Zhang L., et al. Contro155osomes. Cell. 2014;159(2):374–387. doi: 10.1016/j.cell.2014.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sandhu K., Li G., Poh H., Quek Y., Sia Y., Peh S., et al. Large-scale functional organization of long-range chromatin interaction networks. Cell Rep. 2012;2(5):1207–1219. doi: 10.1016/j.celrep.2012.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Neph S., Vierstra J., Stergachis A.B., Reynolds A.P., Haugen E., Vernot B., et al. An expansive human regulatory lexicon encoded in transcription factor footprints. Nature. 2012;489(7414):83–90. doi: 10.1038/nature11212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Moore J.E., Purcaro M.J., Pratt H.E., Epstein C.B., Shoresh N., Adrian J., et al. Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature. 2020;583(7818):699–710. doi: 10.1038/s41586-020-2493-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hnisz D., Abraham B., Lee T., Lau A., Saint-André V., Sigova A., et al. Super-enhancers in the control of cell identity and disease. Cell. 2013;155(4):934–947. doi: 10.1016/j.cell.2013.09.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Creyghton M.P., Cheng A.W., Welstead G.G., Kooistra T., Carey B.W., Steine E.J., et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci U S A. 2010;107(50):21931–21936. doi: 10.1073/pnas.1016071107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Lin C., Lovén J., Rahl P., Paranal R., Burge C., Bradner J., et al. Transcriptional amplification in tumor cells with elevated c-Myc. Cell. 2012;151(1):56–67. doi: 10.1016/j.cell.2012.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Wang L., Hiler D., Xu B., AlDiri I., Chen X., Zhou X., et al. Retinal cell type DNA methylation and histone modifications predict reprogramming efficiency and retinogenesis in 3D organoid cultures. Cell Rep. 2018;22(10):2601–2614. doi: 10.1016/j.celrep.2018.01.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Chen Y.e., Xu L., Mayakonda A., Huang M.-L., Kanojia D., Tan T.Z., et al. Bromodomain and extraterminal proteins foster the core transcriptional regulatory programs and confer vulnerability in liposarcoma. Nat Commun. 2019;10(1) doi: 10.1038/s41467-019-09257-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Sin-Chan P., Mumal I., Suwal T., Ho B., Fan X., Singh I., et al. A C19MC-LIN28A-MYCN oncogenic circuit driven by hijacked super-enhancers is a distinct therapeutic vulnerability in ETMRs: a lethal brain tumor. Cancer Cell. 2019;36(1):51–67.e7. doi: 10.1016/j.ccell.2019.06.002. [DOI] [PubMed] [Google Scholar]
- 109.Bradner J.E., Hnisz D., Young R.A. Transcriptional addiction in cancer. Cell. 2017;168(4):629–643. doi: 10.1016/j.cell.2016.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Boija A., Klein I.A., Young R.A. Biomolecular condensates and cancer. Cancer Cell. 2021;39(2):174–192. doi: 10.1016/j.ccell.2020.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Sanda T., Leong W.Z. TAL1 as a master oncogenic transcription factor in T-cell acute lymphoblastic leukemia. Exp Hematol. 2017;53:7–15. doi: 10.1016/j.exphem.2017.06.001. [DOI] [PubMed] [Google Scholar]
- 112.Nguyen NTT, Contreras-Moreira B, Castro-Mondragon JA, Santana-Garcia W, Ossio R, Robles-Espinoza CD, et al. RSAT 2018: Regulatory sequence analysis tools 20th anniversary. Nucleic Acids Res 2018;46:W209–14. https://doi.org/10.1093/nar/gky317. [DOI] [PMC free article] [PubMed]
- 113.Mariani L., Weinand K., Gisselbrecht S.S., Bulyk M.L. MedeA: Analysis of transcription factor binding motifs in accessible chromatin. Genome Res. 2020;30(5):736–748. doi: 10.1101/gr.260877.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Mariani L., Weinand K., Vedenko A., Barrera L.A., Bulyk M.L. Identification of human lineage-specific transcriptional coregulators enabled by a glossary of binding modules and tunable genomic backgrounds. Cell Syst. 2017;5(3):187–201.e7. doi: 10.1016/j.cels.2017.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Gheorghe M, Sandve GK, Khan A, Chèneby J, Ballester B, Mathelier A. A map of direct TF-DNA interactions in the human genome. Nucleic Acids Res 2019;47. https://doi.org/10.1093/nar/gky1210. [DOI] [PMC free article] [PubMed]
- 116.Liu T., Ortiz J.A., Taing L., Meyer C.A., Lee B., Zhang Y., et al. Cistrome: An integrative platform for transcriptional regulation studies. Genome Biol. 2011;12(8):R83. doi: 10.1186/gb-2011-12-8-r83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Subramanian A., Narayan R., Corsello S.M., Peck D.D., Natoli T.E., Lu X., et al. A next generation connectivity map: L1000 platform and the first 1,000,000 profiles. Cell. 2017;171(6):1437–1452.e17. doi: 10.1016/j.cell.2017.10.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Qin Q., Fan J., Zheng R., Wan C., Mei S., Wu Q., et al. Lisa: inferring transcriptional regulators through integrative modeling of public chromatin accessibility and ChIP-seq data. Genome Biol. 2020;21(1) doi: 10.1186/s13059-020-1934-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Krijger P.H.L., Geeven G., Bianchi V., Hilvering C.R.E., de Laat W. 4C-seq from beginning to end: A detailed protocol for sample preparation and data analysis. Methods. 2020;170:17–32. doi: 10.1016/j.ymeth.2019.07.014. [DOI] [PubMed] [Google Scholar]
- 120.Yosef N., Shalek A.K., Gaublomme J.T., Jin H., Lee Y., Awasthi A., et al. Dynamic regulatory network controlling TH 17 cell differentiation. Nature. 2013;496(7446):461–468. doi: 10.1038/nature11981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Alculumbre S.G., Saint-André V., Di Domizio J., Vargas P., Sirven P., Bost P., et al. Diversification of human plasmacytoid predendritic cells in response to a single stimulus article. Nat Immunol. 2018;19(1):63–75. doi: 10.1038/s41590-017-0012-z. [DOI] [PubMed] [Google Scholar]
- 122.Piasecka B., Duffy D., Urrutia A., Quach H., Patin E., Posseme C., et al. Distinctive roles of age, sex, and genetics in shaping transcriptional variation of human immune responses to microbial challenges. Proc Natl Acad Sci. 2018;115(3):E488–E497. doi: 10.1073/pnas.1714765115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Jiang P., Lee W., Li X., Johnson C., Liu J.S., Brown M., et al. Genome-scale signatures of gene interaction from compound screens predict clinical efficacy of targeted cancer therapies. Cell Syst. 2018;6(3):343–354.e5. doi: 10.1016/j.cels.2018.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Kalinin A.A., Higgins G.A., Reamaroon N., Soroushmehr S., Allyn-Feuer A., Dinov I.D., et al. Deep learning in pharmacogenomics: From gene regulation to patient stratification. Pharmacogenomics. 2018;19(7):629–650. doi: 10.2217/pgs-2018-0008. [DOI] [PMC free article] [PubMed] [Google Scholar]