

Abstract
Spinal muscular atrophy with respiratory distress type 1 (SMARD1) is an autosomal recessive disorder that develops in infancy and arises from mutation of the immunoglobulin helicase μ-binding protein 2 (IGHMBP2) gene. Whereas IGHMBP2 is ubiquitously expressed, loss or reduction of function leads to alpha motor neuron loss and skeletal muscle atrophy. We previously developed a gene therapy strategy for SMARD1 using a single-stranded AAV9-IGHMBP2 vector and compared two different delivery methods in a validated SMARD1 mouse model. An important question in the field relates to the temporal requirements for this or any potential treatment. To examine the therapeutic window, we utilized our recently developed SMARD1 model, FVB/NJ-Ighmpb2nmd-2J, to deliver AAV9-IGHMBP2 at four different time points starting at post-natal day 2 (P2) through P8. At each time point, significant improvements were observed in survival, weight gain, and motor function. Similarly, treatment improved important hallmarks of disease, including motor unit pathology. Whereas improvements were more pronounced in the early-treatment groups, even the later-treatment groups displayed significant phenotypic improvements. This work suggests that an effective gene therapy strategy could provide benefits to pre-symptomatic and early-symptomatic individuals, thereby expanding the potential therapeutic window for SMARD1.
Keywords: IGHMBP2, SMARD1, AAV9, gene therapy, neurodegeneration
Graphical abstract
For many diseases, the time at which a therapeutic is delivered impacts therapeutic effectiveness. Using a mouse model of a rare pediatric disease, the corresponding author and colleagues provide evidence that a gene therapy approach can significantly impact disease development if the treatment is given before or after symptoms develop.
Introduction
Spinal muscular atrophy with respiratory distress type 1 (SMARD1) is an infantile autosomal recessive neurodegenerative disease that is characterized by loss of alpha motor neurons and muscle atrophy.1,2 In contrast to 5q-linked spinal muscular atrophy (SMA), the first clinical symptom in SMARD1 is respiratory distress that develops between 6 weeks and 13 months of age.1,3,4 Respiratory distress is a result of diaphragm muscle atrophy,1, 2, 3,5 whereas in the related disease, SMA, intercostal muscle atrophy is typically the cause of respiratory failure. The respiratory complications in SMARD1 typically lead to complete paralysis of the diaphragm and artificial ventilation. Additional symptoms for SMARD1 include intrauterine growth delay, autonomic dysfunction, distal-to-proximal spread of muscle atrophy, and decreased diameter of myofibrils in skeletal muscles including the diaphragm.2,4,6,7
SMARD1 is caused by loss-of-function mutations in the housekeeping gene immunoglobulin μ-DNA binding protein 2 (IGHMBP2), located on chromosome 11q13.3.3,8,9 The ubiquitously expressed IGHMBP2 gene is comprised of 15 exons encoding 993 amino acids corresponding to a ~110 kDa product. IGHMBP2 consists of an ATP-binding motif, a helicase-like motif, and two nucleic-acid-binding motifs. Patient mutations are predominately located in these functional motifs; however, the helicase domain is the region most frequently mutated.2,4,8, 9, 10, 11 Based on in vitro and in vivo studies, the IGHMBP2 protein functions in immunoglobulin-class switching,9 pre-mRNA maturation,12 transcription regulation by either DNA binding activity13,14 or interaction with TATA-binding protein,15 and translation by direct interaction with tRNA and other components of translational machinery.16,17 However, the precise disease-causing role of IGHMBP2 mutation that leads to selective motor neuron loss remains unclear.
A spontaneous mutation in Ighmbp2 resulted in the initial identification and characterization of the neuromuscular degeneration (nmd2J) mouse model.18,19 The original nmd2J mouse is on the C57BLKS background and contains a mutation in Ighmbp2 intron 4, giving rise to a cryptic splice site, resulting in an aberrant splicing event in ~75%–80% of the transcripts. The altered splicing causes an addition of 23 nucleotides to the transcript that creates a premature translational termination codon.19 At 3 weeks of age, nmd2J mice become symptomatic and rapidly develop hindlimb muscle weakness to the extent that mice were paralyzed by 5 weeks of age with survival to 14 weeks.20 By postnatal day 10, nmd2J mice demonstrated over a 40% loss in motor neurons that progressed until death.20
Our lab and another group have developed a single-stranded AAV9 (ssAAV9) vector containing human IGHMBP2 cDNA that demonstrated excellent efficacy in rescuing the nmd mice.21,22 We compared the efficacy of two delivery methods (intravenous [i.v.] and intracerebroventricular [i.c.v.]) in the nmd2J model using a single low dose, which demonstrated that i.v. injections are not as effective as i.c.v. injections in rescuing motor deficits.23 We recently developed a closely related strain of nmd mice on the FVB background called FVB/NJ-Ighmpb2nmd-2J or “FVB-nmd” from this point on, resulting in a model that was consistent, severe, and well-suited for therapeutic studies, with a median survival of ~18–20 days.24 To date, AAV-mediated gene replacement has been shown to be the most effective means to rescue the SMARD1 mouse model. To advance the pre-clinical animal studies, we examined the therapeutic window to determine the relative efficacy of AAV9-IGHMBP2 at various stages of disease development by delivering vector at post-natal day 2 (P2), P4, P6, and P8. Our results suggest that the therapeutic window of IGHMBP2 gene replacement is not limited to early asymptomatic stages such as P1 or P2; rather, significant phenotypic benefit was observed in all treatment groups, including the cohort that received treatment at P8. This was surprising, since the vector is single-stranded, and maximal expression typically occurs more than 10 days after delivery. These results provide the first evidence of the effectiveness of SMARD1 gene therapy at later time points following disease onset.
Results
Delivery of AAV9-IGHMBP2 at all time points significantly increased FVB-nmd lifespan
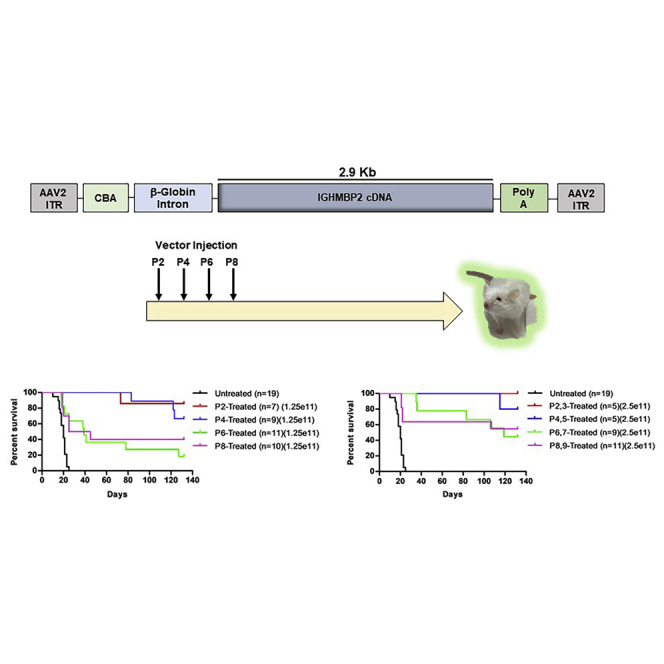
A single low dose of AAV9-IGHMBP2 at P2 via i.c.v. or i.v. injection at P2 significantly rescues the nmd phenotype.21,23 Here, we analyzed the phenotype of the severe FVB-nmd model following i.c.v. delivery of two different doses of AAV9-IGHMBP2 (Figure 1A) at 4 different time points starting at P2–P3 (asymptomatic) through P8–P9 (early symptomatic). A single low dose (1.25e11 vector genomes [vg]) was injected at P2, P4, P6, and P8, whereas the high dose (total of 2.5e11 vg) was applied on two consecutive days for each time point (P2,3; P4,5; P6,7; P8,9). Interestingly, all treated groups displayed significantly extended lifespans compared to untreated animals (Figures 1B and 1C; Figures S1A–S1D). Within the low-dose cohort, earlier P2 and P4 were similarly effective, while P6 and P8 significantly improved survival compared to the early injections. The same pattern was also observed in the cohorts that received the high-dose treatment, although the high dose reduced the number of early deaths in the P6 and P8 cohorts (Figure 1C). While one death occurred prior to P80 in the P2 and P4 low-dose treatment cohorts, all of the high-dose-treated animals in the P2 and P4 groups lived beyond 100 days (Figures 1B and 1C). Delivery of the vector at P6 and P8 time points still resulted in a significant extension in survival for treated FVB-nmd mice, albeit not as robust as the earlier treatment groups (p = 0.0002 for P6 and p = 0.001 for P8) (Figure 1B). The endpoint for these experiments was 132 days, and all remaining cohorts were euthanized.
Figure 1.

ICV injection of AAV9-IGHMBP2 in low and high dose significantly increases the survival and weight of FVB-nmd at early and late time points
(A) Map of AAV9-IGHMBP2 containing 2.9 kb of human IGHMBP2 cDNA under control of the ubiquitously expressing chicken-beta-actin (CBA) promoter. (B and C) Homozygous FVB-nmd mice were i.c.v. injected at P2, P4, P6, and P8, using a low dose (1.25 × 1011 vg) (B) and at P2,3, P4,5, P6,7, and P8,9 using a high dose (2.5 × 1011 vg) (C) of AAV9-IGHMBP2 and compared to untreated mice. Survival was determined by Kaplan-Meier curves, and p value was calculated by the log-rank (Mantel-Cox) test. Median survival of either low- or high-dose treated at P2 and P4 were undefined, since most of the animals in each group were alive at day 132, living significantly longer than untreated mice, which had a median survival of 20 days (p < 0.0001) (B and C). Low-dose P6- and P8-treated FVB-nmd mice had a median survival of 39 and 35 days, respectively (p = 0.48), which was significantly longer than untreated (P6 versus untreated, p = 0.0002; P8 versus untreated, p = 0.001) (B). The median survival of high-dose-treated mice at P6 and P8 was 119 days and undefined, respectively (P6,7 and P8,9 versus untreated, p < 0.0001) (C). (D and E) Weight gain of low- (D) and high-dose-treated (E) mice at P2, P4, P6, and P8 is assessed and compared to untreated. The average weight of both low- (D) and high-dose-treated (E) animals at every time point was significantly higher than untreated FVB-nmd mice, which weigh 3.6 ± 0.18 g (one-way ANOVA p < 0.0001). The average weight of both low- and high-dose-treated animals was significantly different between each group based on the time point of therapy. P2-treated mice weighing 17.39 ± 0.53 in low dose and 18.22 ± 0.58 g in high dose were significantly heavier than P4 mice, which weighed 14.41 ± 0.44 in low-dose and 16.06 ± 0.51 g in high-dose groups (one-way ANOVA, p < 0.001 for low dose; p < 0.01 for high dose). P4 weight was significantly higher than P6, weighing an average of 12.18 ± 0.39 in low-dose-treated mice and 13.14 ± 0.43 g in high-dose-treated mice (one-way ANOVA, p < 0.01 for low dose; p < 0.001 for high dose). P6 was drastically heavier than P8, with average weight of 10.45 ± 0.32 in low-dose-treated and 11.21 ± 0.34 g in high-dose-treated mice (one-way ANOVA, p < 0.05 for low dose; p < 0.01 for high dose). Error bars represent ± SEM.
Delivery of AAV9-IGHMBP2 at all time points significantly increased FVB-nmd weight
The weight of untreated FVB-nmd mice fluctuates between 3–4 g, whereas the nmd2J mice can weigh up to 10 g.21,24 To investigate the effect of the low- and high-dose AAV9-IGHMBP2 on the weight of FVB-nmd mice treated at different time points, daily weight was recorded starting at P2 until P132, when the survival and weight analyses were terminated. Consistent with the extension of the lifespan, both low and high doses of the viral vector led to a considerable increase in the weight of every treated group compared to untreated (p < 0.001) (Figures 1D and 1E). All treated groups gained weight until 40–50 days of age and then reached a plateau. Weight gain for each group was directly correlated to the time point of therapy rather than the dose of the vector. Treatment at earlier time points led to higher weight gain than the later time points, indicating a significant difference between each treated group regardless of the dose (P2 versus P4, p < 0.001; P4 versus P6, p < 0.001; P6 versus P8, p < 0.05; P2,3 versus P4,5, p < 0.01; P4,5 versus P6,7, p < 0.001; P6,7 versus P8,9, p < 0.01) (Figures 1D and 1E). Additionally, there was no significance in the weight gain of any individual group treated with either low or high dose (Figures S2A–S2D) except for P4-treated mice, in which the increased dose led to higher weight gain during the age of 85–115 days (p < 0.05).
Early AAV9-IGHMBP2 treatment improved motor function in FVB-nmd more than later time points
Rotarod and grip strength were performed starting at P40 for 7 consecutive days following a training and acclimation period. Untreated FVB-nmd animals were not included in these studies focused upon motor function, since younger animals are not capable of performing these tasks and their lifespan is too short. Therefore, each treatment group was compared to the heterozygous unaffected littermates (HET: FVB-Ighmbp2nmd/+) and to each other in low- and high-dose-treated groups. Rotarod performance, as determined by the time an animal remained on the rotating cylinder, of early-treated animals (P2 and P4 treated) were indistinguishable from unaffected HET counterparts in both low- and high-dose groups (Figures 2A and 2B; Figures S3A and S3B). The rotarod performance of P6- and P8-treated mice, though improved, was significantly less compared to the HET and early-treated littermates in the low- and high-dose groups (p < 0.001) (Figures 2A and 2B). High-dose delivery slightly improved the rotarod performance of the P6- and P8-treated compared to low dose, but it did not differ statistically (Figures S3C and S3D).
Figure 2.

I.c.v. delivery of AAV9-IGHMBP2 in low and high dose significantly increases the motor function of FVB-nmd mice at early and late time points
(A and B) Hindlimb strength measured by rotarod performance, recording the riding time parameter in seconds, of low- (A) and high-dose-treated (B) mice at P2, P4, P6, and P8 compared to the age-matched HET cohort. (A) Rotarod performance of low-dose P2-treated mice was improved significantly compared to every treated group and HET (one-way ANOVA, P2 versus P4 and HET, p < 0.01; P2 versus P6 and P8, p < 0.001). Rotarod performance of low-dose P4-treated mice was identical to HET and significantly more improved than P6- and P8-treated mice (one-way ANOVA, P4 versus P6 and P8, p < 0.001), while P6- and P8-treated mice were not statistically different in the low-dose group. (B) High-dose P2- and P4-treated mice were statistically similar to the HET cohort and drastically improved over P6- and P8-treated mice (p < 0.001), whereas P6-treated mice performed notably better than P8 (p < 0.05). P6- and P8-treated animals were drastically weaker than HET in either low- or high-dose group (p < 0.001) (B and C). (C and D) Grip strength measured in grams for low-dose-treated (C) and high-dose-treated (D) mice at P2, P4, P6, and P8 and compared to age-matched HET cohort. (C and D) The grip strength of all the treated animals, in both low- and high-dose groups, was significantly weaker than HET (one-way ANOVA, p < 0.001). Early-treated groups were significantly stronger than late-treated littermates (one-way ANOVA, P2 and P4 versus P6 and P8, p < 0.001; P2 versus P4, p < 0.001 in low dose). High-dose P2- and P4-treated mice did not differ statistically (p > 0.05); P6- and P8-treated mice had similar grip strength in low- and high-dose groups (p > 0.05). Measurements were taken for 7 consecutive days starting from P40 through P46. Error bars represent ± SEM.
In contrast to the rotarod performance, forelimb strength in all treated groups was significantly weaker than unaffected animals (p < 0.001) (Figures 2C and 2D). This reduced level of rescue in forelimb strength is consistent with the original nmd mice, demonstrating that the i.c.v. delivery of the vector is capable of repairing hindlimb activity more efficiently than forelimb,21,23 although the grip strength in each treated group improved significantly using the high dose of the viral vector when compared to low-dose group (Figures 2C and 2D). Single comparison of each time point treatment in low- and high-dose groups revealed that improvements in forelimb strength, resulting from high-dose administration, are more obvious in early-treated (P2 versus p2,3 and P4 versus P4,5, p < 0.001) (Figures S4A and S4B) but still significant in the P6-treated (P6,7 versus P6, p < 0.01) (Figure S4C) and P8-treated mice (P8,9 versus P8, p < 0.05) (Figure S4D). Nevertheless, the high dose could not sufficiently increase the grip strength to reach the level of the HET littermates in any treated group (p < 0.001) (Figures 2D; Figures S4A–S4D). Western blot analysis of early- and late-treated mice (P2, P8) showed no statistical difference among the various cohorts (Figure S5).
Muscle pathology improved in all treatment groups
FVB-nmd mice have a severe hindlimb phenotype characterized by contracture in the hindlimbs and an inability to splay their legs by P17.24 In our previous study, we showed that FVB-nmd mice have significantly decreased gastrocnemius muscle fiber size compared to FVB mice.24 Additionally, we have previously reported that P2-treated nmd2J/2J have a high level of rescue in their gastrocnemius muscle.21,23 To investigate whether the early (P2) and late (P8) time point treatment led to muscle fiber rescue in FVB-nmd, we compared laminin-immunostained cross-sectioned gastrocnemius muscles of high-dose-treated animals with those of the wild-type (WT) FVB at 42 days of age. The muscles of phenotypically unaffected age-matched HET mice were used as a control for untreated FVB-nmd harvested at 14 days of age, a late symptomatic stage. Consistent with our previous results, muscle fiber area and perimeter of the gastrocnemius muscle are significantly decreased in untreated FVB-nmd mice compared to HET (p < 0.0001) (Figures 3A–3C). A blinded quantitative assessment of muscles revealed that muscle fiber area and perimeter of the gastrocnemius in P2-treated mice were significantly larger than P8-treated mice (p < 0.0001) but still notably smaller than WT (p < 0.0001) (Figures 3A–3C). Diaphragm muscle fiber has been shown to be significantly smaller in SMARD1 patients. We wanted to determine if the FVB-nmd mice have reduced diaphragm muscle fiber area and evaluate the extent AAV-IGHMBP2 treatment rescues. A blinded quantitative assessment of muscles showed that diaphragm muscle area and perimeter are significantly reduced in P14 FVB-nmd mice compared to age-matched HET mice (p < 0.0001) (Figures 3D and 3E). AAV-IGHMBP2 treatment at both P2 and P8 improved the diaphragm muscle fiber; however, were both significantly smaller than WT (p < 0.0001) (Figures 3D and 3E). Additionally, P2-treated mice had a significantly larger diaphragm size than P8-treated mice (p < 0.0001) (Figures 3D and 3E). These results demonstrate that AAV-IGHMBP2 treatment at P2 and P8 significantly improves the skeletal muscle phenotype in SMARD1 model mice; however, consistent with the other parameters, earlier delivery provides an even greater degree of protection.
Figure 3.

I.c.v. delivery of AAV9-IGHMBP2 in high dose repaired the gastrocnemius muscle fibers of early-treated mice more efficiently than late-treated mice
(A) Laminin immuno-stained cross sections of gastrocnemius muscles of high-dose-treated mice at P2 and P8 (n = 4) were examined under 40× magnification and compared to age-matched WT littermates (P42) (n = 3), while untreated FVB-nmd sections (n = 5) were evaluated in comparison to the age-matched HET mice (P14) (n = 3). (B and C) The area and perimeter of gastrocnemius muscle fibers in untreated FVB-nmd mice are drastically smaller than their HET counterparts (one-way ANOVA, p < 0.0001). The area and perimeter of P2-treated gastrocnemius muscle fibers are significantly larger than P8-treated mice (p < 0.0001) but markedly smaller than the age-matched WT cohort (p < 0.0001). (D and E) The area and perimeter of diaphragm muscle fibers in untreated FVB-nmd mice are significantly smaller than their HET counterparts (one-way ANOVA, p < 0.0001). The area and perimeter of P2-treated diaphragm muscle fibers are significantly larger than P8-treated (p < 0.0001) but are not rescued to age-matched WT size (p < 0.0001). Scale bar, 30 μm. Error bars represent mean ± SEM. ∗∗∗∗p < 0.0001.
Neuromuscular junction (NMJ) pathology of the gastrocnemius improved in early- and late-treated FVB-nmd mice
NMJs from the gastrocnemius muscle exhibit severe denervation defects in FVB-nmd mice, but these can be partially rescued by delivery of AAV9-IGHBMP2 on P1.23,24 To examine how early and late delivery of AAV9-IGHMBP2 impacts the FVB-nmd NMJ phenotype, we conducted a blinded quantitative assessment of NMJ pathology from the gastrocnemius of P2- and P8-treated FVB-nmd mice and age-matched, untreated WT mice at P42. The untreated FVB-nmd and their age-matched HET counterparts harvested at P14 were compared to each other at a relatively early time point. As expected, NMJs from untreated FVB-nmd mice displayed severe pathology, with less than 40% fully innervated endplates and more than 50% fully denervated endplates compared to age-matched HET mice (p < 0.0001) (Figures 4A and 4B). The P2-treated mice had similar level of innervation and denervation to age-matched WT counterparts, with almost 85% fully innervated endplates (p > 0.05) (Figures 4A and 4B). Interestingly, the P8-treated NMJs, albeit not as improved as P2-treated, were 65% fully innervated (P8-treated versus WT and P2-treated, p < 0.0001) and approximately 22% fully denervated endplates (P8-treated versus WT, p < 0.01; P8-treated versus P2-treated, p < 0.05) (Figures 4A and 4B). These results are consistent with the other phenotypic parameters and further demonstrate that early delivery is the most efficacious, but even the symptomatic cohort (P8) significantly benefits from this effective treatment.
Figure 4.

I.c.v. delivery of high-dose AAV9-IGHMBP2 improved the NMJ pathology of FVB-nmd mice at early and late time points
(A) Representative images of NMJs from gastrocnemius muscles of high-dose P2- and P8-treated mice (n = 4) at P42 compared to those of age-matched WT mice (n = 3), while the NMJ of untreated FVB-nmd mice (n = 4) at P14 was analyzed in comparison to the age-matched unaffected HET (n = 3). Muscles were labeled with α-bungarotoxin (α-BTX) for acetylcholine receptors (AChRs), anti-neurofilament, and anti-synaptic vesicle protein 2 (SV2) for nerve terminals. Fluorescent images were taken at 40× magnification. (B) Percentage of fully innervated, partially denervated, and fully denervated muscles. Percentage of fully innervated in FVB-nmd mice was less than 40%, compared to 90% in HET counterparts (two-way ANOVA, p < 0.0001). P2-treated NMJs were ~85% fully innervated and ~10% fully denervated versus 95% and 5% in WT cohort (two-way ANOVA, p > 0.5). P8-treated mice exhibited ~65% full innervation (two-way ANOVA, P8-treated versus P2-treated and WT, p < 0.0001) and ~22% full denervation (two-way ANOVA, P8- versus P2-treated, p < 0.05; P8 versus WT, p < 0.01). Scale bar, 50 μm. Error bars represent mean ± SEM. ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗∗p < 0.0001.
AAV9-IGHMBP2 increases the motor neuron numbers in the spinal cord (SP) of the early- and late-treated FVB-nmd mice
Based on our previous analysis, the number of motor neurons in the lumbar region of the original nmd2J mice was reduced nearly 65% at 8 weeks of age.21 We investigated whether the delivery of AAV9-IGHMBP2 prevents motor neuron loss and pathology of motor neurons in the FVB-nmd model. Lumbar spinal cord sections representing L3–L5 regions from P14 cohorts, FVB-nmd and HET, and P42 cohorts, WT, P2 treated, and P8 treated, were analyzed through a blinded quantification of immunofluorescent images. As predicted, FVB-nmd contained a significantly reduced number of motor neurons, with an average of ~5 motor neurons per section, compared to ~6.6 or 8 motor neurons per section in the age-matched HET or WT mice (p < 0.001) (Figures 5A and 5B). However, the area and perimeter of the FVB-nmd motor neurons were only slightly smaller than those in HET (p > 0.05), suggesting that even though motor neurons at P14 are reduced in numbers, the remaining motor neurons do not yet exhibit dramatic cellular pathology (Figures 5A–5D). Interestingly, the average motor neuron numbers in P2-treated, P8-treated, and WT mice were similar to each other, with an average of 8 motor neurons per section (p > 0.05) (Figures 5A and 5B). Although the P2-treated motor neuron area and perimeter were smaller than WT (p < 0.05), they were significantly larger than the P8-treated in area (p < 0.01) and similar to the P8-treated in perimeter (p > 0.05) (Figures 5A–5D). P8-treated motor neurons were significantly smaller than WT in area and perimeter (p < 0.001) (Figures 5A–5D). These results confirm the effectiveness of early treatment in rescuing the numbers and, to some extent, the size of motor neurons, whereas late treatment was only able to rescue the number of motor neurons but not the size.
Figure 5.

I.c.v. delivery of AAV9-IGHMBP2 in high dose improved the motor neuron number and area size in the lumbar region of FVB-nmd mice at early and late time points
(A) Nissl body (NeuroTrace) and anti-choline acetyl transferase (ChAT) were used to immunostain motor neurons from cross-sections of lumbar spinal cord (L3–L5). High-dose P2- and P8-treated mice (n = 4) were compared to age-matched WT littermates (P42) (n = 3), while untreated FVB-nmd mice (n = 4) were evaluated in comparison to the age-matched HET mice (P14) (n = 3). Fluorescent representative images were taken at 40× magnification. (B) The number of motor neurons in FVB-nmd mice was drastically less than HET (one-way ANOVA, p < 0.001), while the motor neuron numbers in P2 and P8 were similar to the WT cohort (p > 0.5). (C and D) The area and perimeter of the motor neurons in FVB-nmd mice was not statistically different from HET (p > 0.5). P2- and P8-treated motor neurons were smaller than WT in area and perimeter with varying degrees (one-way ANOVA, P2 versus WT, p < 0.05; P8 versus WT, p < 0.001). P2-treated and P8-treated mice differed only in the area of the motor neuron soma (p < 0.05) (C and D). Scale bar, 50 μm. Error bars represent mean ± SEM.
Discussion
While there is no approved treatment for SMARD1, AAV9-IGHMBP2 gene therapy has made significant progress in pre-clinical mouse models. However, understanding the temporal requirements of this, or any, potential therapeutic is critical as it advances through pre-clinical evaluation. To begin to understand the temporal requirements of IGHMBP2 from a therapeutic perspective, we devised a series of experiments that delivered a previously characterized gene therapy vector, AAV9-IGHMBP2. I.c.v. injections of the vector were administered to FVB-nmd cohorts on P2, P4, P6, and P8. This time frame spanned pre-symptomatic through early symptomatic days. Additionally, two different doses were administered: a low dose and a high dose, corresponding to 1.25e11 and 2.5e11 vector genomes, with mice receiving either a single injection or double injection to accommodate the small volume of the neonatal ventricles, respectively. Previous studies in nmd2J mice have shown a significant extension of lifespan, improvements in hindlimb splay, and significant reduction of cellular pathology following a single treatment with AAV9-IGHMBP2 immediately after birth.21, 22, 23 While we have previously observed that P2 pre-symptomatic delivery of this vector protects from disease development in the B6.BKS-nmd2J mouse model, it was surprising to observe a robust response in the later treatment time points of P6 and P8, with symptomatic cohorts significantly responding to treatment. The FVB-nmd mice present with a severe phenotype and a relatively short lifespan, with an average lifespan of ~18–20 days. This dramatic extension in survival was unexpected due to the severity of the disease and because single-stranded AAV vectors such as the AAV-IGHMBP2 vector typically do not achieve maximal levels of expression until 10 days post-delivery. It is important to note that in this animal model, there is a baseline (~20%) of Ighmbp2 protein that is full-length and WT. This baseline level may be very important as it relates to therapies that further increase IGHMBP2 levels, since achieving a therapeutic threshold should theoretically be easier than if the genetic context was purely mutant in nature, such as any number of the SMARD1-causing IGHMBP2 point mutations. The development of additional models that reflect these patient-based contexts would be another important step toward understanding the requirements of a potential AAV9-IGHMBP2 therapeutic.
We examined Ighmbp2/IGHMBP2 protein levels in the spinal cord from P2 and P8 at 10 days after treatment, respectively. In this mouse model, we did not see a significant change in protein between the unaffected, the untreated SMARD1 model mice, and the two treated cohorts. Clearly, there is a difference in the phenotype of these animals; however, the western blots do not appear to be capturing these differences. A potential explanation for this is that the relative amount of protein is tightly regulated by the cell, but in specific cell types (perhaps motor neuron populations), a critical increase in full-length IGHMBP2 is achieved that significantly improves the phenotype.
In the 5q SMA context, several studies have examined the temporal requirements of SMN-dependent therapeutics in severe SMA mice (SMNΔ7 mice), including a gene therapy strategy.25, 26, 27 Consistent results were observed in different labs, each demonstrating that SMNΔ7 mice do not respond well to SMN induction past ~P6. While improvements were observed in the cohorts that received treatment at P7–P8, the magnitude of rescue was significantly reduced. The SMA phenotype was largely recalcitrant to SMN increases when delivered post-symptomatically, suggesting that SMNΔ7 mice with lifespans of 14–15 days may have progressed to a point in their disease state that has permanent damage despite rapid expression of SMN1. However, in the instance of the P7–P8-treated FVB-nmd cohorts, AAV9-IGHMBP2 results in ~40% survival past 50 days of age. This suggests that in the FVB-nmd2J mice, P7–P8 delivery of full-length IGHMBP2 is sufficient to have a protective effect.
Through our time point study, we were able to demonstrate that the early injection time points, P2 and P4, resulted in a more robust phenotypic rescue compared to P6- and P8-treated mice. Our cellular pathology analyses show that P8 AAV9-IGHMBP2-treated mice do not differ in the number of lumbar motor neurons when compared to WT and P2-treated mice, which indicates that the specific motor neurons targeted by AAV9-IGHMBP2 are present at the time of delivery. Additionally, we show that NMJ innervation is significantly protected in P8 AAV9-IGHMBP2-treated mice, with ~20% increase in innervated endplates compared to FVB-nmd. The gastrocnemius and diaphragm muscle fiber size additionally showed improvement in P2-treated and P8-treated mice; however, the muscle fiber size did not reach WT levels, indicating that AAV9-IGHMBP2 does not result in a full rescue but does have a protective effect.
Like many other related neurodegenerative diseases, the disease-specific role for IGHMBP2 is poorly understood. The best-described function for IGHMBP2 is its role in the translational machinery and ribosome biogenesis. IGHMBP2 has been shown to interact with tRNATyr and TFIIIC220, which are essential factors for tRNA transcription.16 Additionally, IGHMBP2 also interacts with Reptin/Pontin and activator of basal transcription 1 (ABT1), which has an important role for ribosome biogenesis.17 Although it still remains unknown, we speculate that AAV9-IGHMBP2 allows for sufficient restoration of translation-related pathways that are dysregulated in Ighmbp2-deficient tissues, such as sub-populations of motor neurons. It remains unclear why motor neurons are the most susceptible cell in SMARD1, though dysregulation of pre-mRNA, rRNA, and tRNA appears to be a common feature among motor neuron diseases.
We have demonstrated that a higher dose of AAV9-IGHMBP2 in FVB-nmd mice resulted in higher peak weight gain, lowered the number of early deaths following treatment, and improved their overall forelimb grip strength. The next step would be to understand the relationship between dosage and the extent of rescue in cellular pathology defects. Collectively, this pre-clinical study addresses the temporal requirements for AAV9-IGHMBP2 in FVB-nmd mice by illustrating that treatment pre-symptomatically results in the most therapeutic benefit but that a significant phenotypic improvement was also observed in the cohorts that received the vector even after symptoms developed. While it is important to stress that these are experiments performed in mice, it is a step forward for this potential therapeutic. This is particularly important as, unlike SMA, the majority of SMARD1 patients are diagnosed after symptomatic onset due to lack of a newborn genetic test and the infrequency of SMARD1 cases.
Materials and methods
Animal procedures, viral vector, and injections
All experimental procedures were approved by the University of Missouri’s Institutional Animal Care and Use Committee and were performed following the regulations established by the National Institute of Health’s Guide for the Care and Use of Laboratory Animals. The FVB-nmd mouse model was created in the Animal Modeling Core of the University of Missouri, Columbia as previously described in detail, and the colony was established.24 FVB-nmd animals were genotyped at P1. The viral vector (single-stranded AAV9-IGHMBP2) has been previously described in detail.21,23 Vector was delivered via an i.c.v. injection with 1.25 × 1011 (low dose) viral genomes at four time points (P2, P4, P6, and P8) and 2.5 × 1011 (high dose) on two consecutive days for each time point (P2,3; P4,5; P6,7; P8,9).28,29 At 14 days of age, two groups of FVB-nmd mice (unaffected heterozygous HET [n = 3] and untreated [n = 4]) and at 6 weeks of age, three groups (WT [n = 3], P2,3-treated, and P8,9-treated [n of 4 per group]) were anesthetized with 2.5% isoflurane and then perfused transcardially with cold 0.1 M phosphate-buffered saline (PBS) (pH 7.4), followed by 4% paraformaldehyde (PFA) in phosphate buffer (0.1 M, pH 7.4). The legs were incised following the perfusion and post-fixed for additional 2 h in 4% PFA. The remaining bodies of the perfused mice were post-fixed in 4% PFA for 48 h and the spinal cords were harvested.
Motor function tests
Motor activity and coordination were quantified using rotarod treadmill for mice (IITC Rotarod Series 8, IITC Life Science, Woodland Hills, CA, USA) as previously described.21,23 In brief, the animals were placed on textured rotating drums. When the tested animal fell onto the individual sensing platform, the test results were recorded in seconds. For forelimb grip strength measurement, a grasping response test was utilized. Each pup’s front paws were placed on a wire mesh (1-cm2 grids) and gently dragged horizontally along the mesh (BioSeb Model BP32025, Vitrolles, France). Any resistance felt was scored as a positive response. The strength of the animal holding onto the mesh before release was recorded in grams.21,23 Grip strength values (grams) were normalized against animal body weight. Both measurements were performed every day for 7 consecutive days starting on P40.
Muscle immunostaining
Two groups of FVB-nmd (HET [n = 3] and homozygous untreated [n = 5]) at 14 days of age and three groups of FVB-nmd (WT [n = 3], P2,3-treated [n = 4], and P8,9-treated [n = 4]) at 42 days of age were transcardially perfused and gastrocnemius muscles harvested after 24 h. 16-μm cross-sections were prepared from gastrocnemius and diaphragm muscles followed by immunohistochemistry with anti-laminin. Images were collected using a Leica DM5500 B fluorescent microscope (Leica Microsystems). A blinded quantification assessments of muscle fibers were performed using ImageJ as previously described.30
NMJ immunostaining
This procedure was described in detail previously.31 Two groups (HET [n = 3] and homozygous untreated [FVB-nmd] [n = 4]) at 14 days of age and three groups (WT [n = 3], P2,3-treated [n = 4], and P8,9-treated [n = 4]) at 42 days of age were used for NMJ analysis. Following transcardial perfusion with ice-cold 4% PFA and post-fixing for 24 h at 4°C, the gastrocnemius muscle of each mouse was dissected. Anti-neurofilament heavy chain (NF-H) (1:2,000; catalog AB5539 Chemicon, EMD Millipore) and antisynaptic vesicle 2 (SV2) (1:200; catalog YE269, Life Technologies) primary antibodies followed by donkey anti-chicken Alexa Fluor 488 (1:400; Jackson ImmunoResearch) and goat anti-rabbit Alexa Fluor 488 (1:200; Jackson ImmunoResearch) secondary antibodies were used to label the axon and synaptic terminal. Acetylcholine receptors were labeled with Alexa Fluor 594-conjugated alpha-bungarotoxin (1:200; Life Technologies). Representative images for the NMJs of gastrocnemius muscles were obtained using laser scanning confocal microscope at 40× magnification with Leica TCS SP8 (Leica Microsystems). NMJ analyses were performed in a blinded manner on three randomly selected fields of view per muscle at 20× magnification with Leica DM5500 B (Leica Microsystems). Images were analyzed based on endplate overlap with the synaptic terminals and were placed into three categories: fully innervated, partially denervated, and fully denervated, using Fiji Software (NIH).
Motor neuron count
Two groups of FVB-nmd (HET [n = 3] and homozygous untreated [n = 4]) at 14 days of age and three groups of FVB-nmd (WT [n = 3], P2,3-treated [n = 4], and P8,9-treated [n = 3]) at 42 days of age were transcardially perfused with ice-cold 4% PFA followed by subsequent post-fixing at 4% PFA for 24 h at 4°C. Lumbar (L3–L5) regions from spinal cord tissue were dissected and cryoprotected in 30% sucrose solution overnight before being embedded in optimal cutting temperature (OCT) media. Embedded tissues were cryosectioned at 16 μm thickness with every 10th section from the spinal cord tissue being collected for immunohistochemistry. Sections were stained with choline acetyltransferase (ChAT) primary antibody (1:100; catalog AB144P; Millipore Sigma), donkey anti-goat Alexa Fluor-594 secondary antibody (1:250; Jackson ImmunoResearch), and NeuroTrace Green Fluorescent Nissl (1:100; catalog N21480; Thermo Fisher Scientific) for motor neuron identification. Images were collected using a Leica DM5500 B fluorescent microscope (Leica Microsystems) under 20× magnification. Motor neuron counts, cell body perimeter, and area measurements were performed manually with Fiji Software (NIH) in a blinded manner from 14 sections per mouse.
Statistical analysis
The statistical significance in comparing all the experimental groups (unaffected [HET, WT], untreated, and treated FVB-nmd at different time points) in all the experiments was calculated using one-way ANOVA (two-way ANOVA in NMJ analysis) with Newman-Keuls multiple comparison post hoc test. Analyses were performed with GraphPad Prism software. Error bars represent means ± standard error of the mean (SEM). Significance in survival was determined with log-ranked (Mantel-Cox) test. Percentages were calculated as values from unaffected and were standardized as 100%.
Acknowledgments
We would like to thank members of the Lorson lab who contributed to the early stages of this project and to animal colony maintenance and Dr. Monique Lorson for editing. We are grateful for technical assistance from the MU Molecular Cytology Core. This work was supported by the Sims’ Fund to Cure SMARD (M.S. and C.L.L.), NIH/NINDS grant R21NS109762 (C.L.L.), NIH Training Grant T32 GM008396 (C.E.S. and S.M.R.H.), and NIH PREP R25GM064120 (J.M.).
Author contributions
M.S., C.E.S., and C.L.L. designed the experiments and wrote and edited the manuscript. M.S., C.E.S., E.V., J.M., and M.O.G.-K. conducted the experiments; Z.A. and S.M.R.H quantified the immuno-stained images.
Declaration of interests
C.L.L. is co-founder and Chief Scientific Officer of Shift Pharmaceuticals. All other authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtm.2021.07.008.
Supplemental information
References
- 1.Grohmann K., Schuelke M., Diers A., Hoffmann K., Lucke B., Adams C., Bertini E., Leonhardt-Horti H., Muntoni F., Ouvrier R. Mutations in the gene encoding immunoglobulin mu-binding protein 2 cause spinal muscular atrophy with respiratory distress type 1. Nat. Genet. 2001;29:75–77. doi: 10.1038/ng703. [DOI] [PubMed] [Google Scholar]
- 2.Grohmann K., Varon R., Stolz P., Schuelke M., Janetzki C., Bertini E., Bushby K., Muntoni F., Ouvrier R., Van Maldergem L. Infantile spinal muscular atrophy with respiratory distress type 1 (SMARD1) Ann. Neurol. 2003;54:719–724. doi: 10.1002/ana.10755. [DOI] [PubMed] [Google Scholar]
- 3.Grohmann K., Wienker T.F., Saar K., Rudnik-Schöneborn S., Stoltenburg-Didinger G., Rossi R., Novelli G., Nürnberg G., Pfeufer A., Wirth B. Diaphragmatic spinal muscular atrophy with respiratory distress is heterogeneous, and one form Is linked to chromosome 11q13-q21. Am. J. Hum. Genet. 1999;65:1459–1462. doi: 10.1086/302636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pitt M., Houlden H., Jacobs J., Mok Q., Harding B., Reilly M., Surtees R. Severe infantile neuropathy with diaphragmatic weakness and its relationship to SMARD1. Brain. 2003;126:2682–2692. doi: 10.1093/brain/awg278. [DOI] [PubMed] [Google Scholar]
- 5.Pitt M.A., Kim W., Myung I.J. Flexibility versus generalizability in model selection. Psychon. Bull. Rev. 2003;10:29–44. doi: 10.3758/bf03196467. [DOI] [PubMed] [Google Scholar]
- 6.Kaindl A.M., Guenther U.P., Rudnik-Schoneborn S., Varon R., Zerres K., Schuelke M., Hubner C., von Au K. Spinal muscular atrophy with respiratory distress type 1 (SMARD1) J. Child. Neurol. 2008;23:199–204. doi: 10.1177/0883073807310989. [DOI] [PubMed] [Google Scholar]
- 7.Rudnik-Schöneborn S., Stolz P., Varon R., Grohmann K., Schächtele M., Ketelsen U.P., Stavrou D., Kurz H., Hübner C., Zerres K. Long-term observations of patients with infantile spinal muscular atrophy with respiratory distress type 1 (SMARD1) Neuropediatrics. 2004;35:174–182. doi: 10.1055/s-2004-820994. [DOI] [PubMed] [Google Scholar]
- 8.Viollet L., Barois A., Rebeiz J.G., Rifai Z., Burlet P., Zarhrate M., Vial E., Dessainte M., Estournet B., Kleinknecht B. Mapping of autosomal recessive chronic distal spinal muscular atrophy to chromosome 11q13. Ann. Neurol. 2002;51:585–592. doi: 10.1002/ana.10182. [DOI] [PubMed] [Google Scholar]
- 9.Fukita Y., Mizuta T.R., Shirozu M., Ozawa K., Shimizu A., Honjo T. The human S mu bp-2, a DNA-binding protein specific to the single-stranded guanine-rich sequence related to the immunoglobulin mu chain switch region. J. Biol. Chem. 1993;268:17463–17470. [PubMed] [Google Scholar]
- 10.Maystadt I., Zarhrate M., Landrieu P., Boespflug-Tanguy O., Sukno S., Collignon P., Melki J., Verellen-Dumoulin C., Munnich A., Viollet L. Allelic heterogeneity of SMARD1 at the IGHMBP2 locus. Hum. Mutat. 2004;23:525–526. doi: 10.1002/humu.9241. [DOI] [PubMed] [Google Scholar]
- 11.Lim S.C., Bowler M.W., Lai T.F., Song H. The Ighmbp2 helicase structure reveals the molecular basis for disease-causing mutations in DMSA1. Nucleic Acids Res. 2012;40:11009–11022. doi: 10.1093/nar/gks792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Molnar G.M., Crozat A., Kraeft S.K., Dou Q.P., Chen L.B., Pardee A.B. Association of the mammalian helicase MAH with the pre-mRNA splicing complex. Proc. Natl. Acad. Sci. USA. 1997;94:7831–7836. doi: 10.1073/pnas.94.15.7831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen N.N., Kerr D., Chang C.F., Honjo T., Khalili K. Evidence for regulation of transcription and replication of the human neurotropic virus JCV genome by the human S(mu)bp-2 protein in glial cells. Gene. 1997;185:55–62. doi: 10.1016/s0378-1119(96)00630-0. [DOI] [PubMed] [Google Scholar]
- 14.Miao M., Chan S.L., Fletcher G.L., Hew C.L. The rat ortholog of the presumptive flounder antifreeze enhancer-binding protein is a helicase domain-containing protein. Eur. J. Biochem. 2000;267:7237–7246. doi: 10.1046/j.1432-1327.2000.01836.x. [DOI] [PubMed] [Google Scholar]
- 15.Zhang Q., Wang Y.C., Montalvo E.A. Smubp-2 represses the Epstein-Barr virus lytic switch promoter. Virology. 1999;255:160–170. doi: 10.1006/viro.1998.9588. [DOI] [PubMed] [Google Scholar]
- 16.de Planell-Saguer M., Schroeder D.G., Rodicio M.C., Cox G.A., Mourelatos Z. Biochemical and genetic evidence for a role of IGHMBP2 in the translational machinery. Hum. Mol. Genet. 2009;18:2115–2126. doi: 10.1093/hmg/ddp134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guenther U.P., Handoko L., Laggerbauer B., Jablonka S., Chari A., Alzheimer M., Ohmer J., Plöttner O., Gehring N., Sickmann A. IGHMBP2 is a ribosome-associated helicase inactive in the neuromuscular disorder distal SMA type 1 (DSMA1) Hum. Mol. Genet. 2009;18:1288–1300. doi: 10.1093/hmg/ddp028. [DOI] [PubMed] [Google Scholar]
- 18.Cook S.A., Johnson K.R., Bronson R.T., Davisson M.T. Neuromuscular degeneration (nmd): a mutation on mouse chromosome 19 that causes motor neuron degeneration. Mamm. Genome. 1995;6:187–191. doi: 10.1007/BF00293010. [DOI] [PubMed] [Google Scholar]
- 19.Cox G.A., Mahaffey C.L., Frankel W.N. Identification of the mouse neuromuscular degeneration gene and mapping of a second site suppressor allele. Neuron. 1998;21:1327–1337. doi: 10.1016/s0896-6273(00)80652-2. [DOI] [PubMed] [Google Scholar]
- 20.Grohmann K., Rossoll W., Kobsar I., Holtmann B., Jablonka S., Wessig C., Stoltenburg-Didinger G., Fischer U., Hübner C., Martini R., Sendtner M. Characterization of Ighmbp2 in motor neurons and implications for the pathomechanism in a mouse model of human spinal muscular atrophy with respiratory distress type 1 (SMARD1) Hum. Mol. Genet. 2004;13:2031–2042. doi: 10.1093/hmg/ddh222. [DOI] [PubMed] [Google Scholar]
- 21.Shababi M., Feng Z., Villalon E., Sibigtroth C.M., Osman E.Y., Miller M.R., Williams-Simon P.A., Lombardi A., Sass T.H., Atkinson A.K. Rescue of a Mouse Model of Spinal Muscular Atrophy With Respiratory Distress Type 1 by AAV9-IGHMBP2 Is Dose Dependent. Mol. Ther. 2016;24:855–866. doi: 10.1038/mt.2016.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nizzardo M., Simone C., Rizzo F., Salani S., Dametti S., Rinchetti P., Del Bo R., Foust K., Kaspar B.K., Bresolin N. Gene therapy rescues disease phenotype in a spinal muscular atrophy with respiratory distress type 1 (SMARD1) mouse model. Sci. Adv. 2015;1:e1500078. doi: 10.1126/sciadv.1500078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shababi M., Villalón E., Kaifer K.A., DeMarco V., Lorson C.L. A Direct Comparison of IV and ICV Delivery Methods for Gene Replacement Therapy in a Mouse Model of SMARD1. Mol. Ther. Methods Clin. Dev. 2018;10:348–360. doi: 10.1016/j.omtm.2018.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shababi M., Smith C.E., Kacher M., Alrawi Z., Villalón E., Davis D., Bryda E.C., Lorson C.L. Development of a novel severe mouse model of spinal muscular atrophy with respiratory distress type 1: FVB-nmd. Biochem. Biophys. Res. Commun. 2019;520:341–346. doi: 10.1016/j.bbrc.2019.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Foust K.D., Wang X., McGovern V.L., Braun L., Bevan A.K., Haidet A.M., Le T.T., Morales P.R., Rich M.M., Burghes A.H., Kaspar B.K. Rescue of the spinal muscular atrophy phenotype in a mouse model by early postnatal delivery of SMN. Nat. Biotechnol. 2010;28:271–274. doi: 10.1038/nbt.1610. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 26.Robbins K.L., Glascock J.J., Osman E.Y., Miller M.R., Lorson C.L. Defining the therapeutic window in a severe animal model of spinal muscular atrophy. Hum. Mol. Genet. 2014;23:4559–4568. doi: 10.1093/hmg/ddu169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lutz C.M., Kariya S., Patruni S., Osborne M.A., Liu D., Henderson C.E., Li D.K., Pellizzoni L., Rojas J., Valenzuela D.M. Postsymptomatic restoration of SMN rescues the disease phenotype in a mouse model of severe spinal muscular atrophy. J. Clin. Invest. 2011;121:3029–3041. doi: 10.1172/JCI57291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Coady T.H., Baughan T.D., Shababi M., Passini M.A., Lorson C.L. Development of a single vector system that enhances trans-splicing of SMN2 transcripts. PLoS ONE. 2008;3:e3468. doi: 10.1371/journal.pone.0003468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shababi M., Lorson C.L. Optimization of SMN Trans-Splicing Through the Analysis of SMN Introns. J. Mol. Neurosci. 2012;46:459–469. doi: 10.1007/s12031-011-9614-3. [DOI] [PubMed] [Google Scholar]
- 30.Kaifer K.A., Villalón E., Osman E.Y., Glascock J.J., Arnold L.L., Cornelison D.D.W., Lorson C.L. Plastin-3 extends survival and reduces severity in mouse models of spinal muscular atrophy. JCI Insight. 2017;2:e89970. doi: 10.1172/jci.insight.89970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cobb M.S., Rose F.F., Rindt H., Glascock J.J., Shababi M., Miller M.R., Osman E.Y., Yen P.F., Garcia M.L., Martin B.R. Development and characterization of an SMN2-based intermediate mouse model of Spinal Muscular Atrophy. Hum. Mol. Genet. 2013;22:1843–1855. doi: 10.1093/hmg/ddt037. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.