

Summary
The mechanisms by which transcriptional activation domains (tADs) initiate eukaryotic gene expression have been an enigma for decades because most tADs lack specificity in sequence, structure, and interactions with targets. Machine learning analysis of data sets of tAD sequences generated in vivo elucidated several functionality rules: the functional tAD sequences should (i) be devoid of or depleted with basic amino acid residues, (ii) be enriched with aromatic and acidic residues, (iii) be with aromatic residues localized mostly near the terminus of the sequence, and acidic residues localized more internally within a span of 20–30 amino acids, (iv) be with both aromatic and acidic residues preferably spread out in the sequence and not clustered, and (v) not be separated by occasional basic residues. These and other more subtle rules are not absolute, reflecting absence of a tAD consensus sequence, enormous variability, and consistent with surfactant-like tAD biochemical properties. The findings are compatible with the paradigm-shifting nucleosome detergent mechanism of gene expression activation, contributing to the development of the liquid-liquid phase separation model and the biochemistry of near-stochastic functional allosteric interactions.
Subject areas: Biological sciences, Molecular biology, Bioinformatics, Computational bioinformatics, gene activation, transcriptional regulation, IDR, machine learning, neural network, chromatin, nucleosome
Graphical abstract
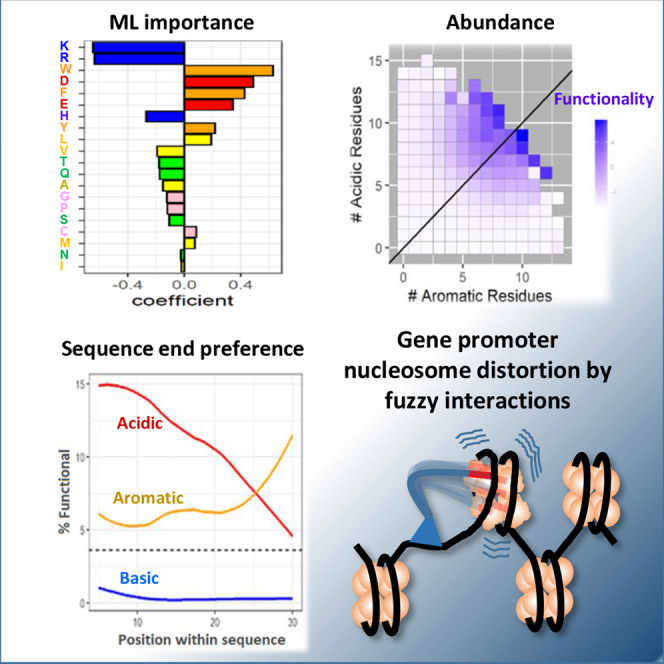
Highlights
-
•
Transcriptional activation domain features are analyzed by machine learning
-
•
Absence of basic and redundant presence of acidic and aromatic residues is important
-
•
Among hydrophobic residues, only aromatics are significantly more represented
-
•
C-terminal localization is essential for aromatic but not for acidic residues
Biological sciences; Molecular biology; Bioinformatics; Computational bioinformatics
Introduction
The function of transcriptional activation domains (tADs) of gene activators is critical for gene expression in eukaryotes. However, the mechanisms of activation have remained an enigma for decades because tADs, which are short, intrinsically disordered, highly variable protein regions, are involved in a variable number of poorly defined fuzzy interactions (Fuxreiter and Tompa, 2012; Scholes and Weinzierl, 2016) with an uncertain number of targets (Keung et al., 2014). There are two working models for the explanation of tAD function: (i) the direct recruitment model (Ferreira et al., 2005; Hahn and Young, 2011; Ptashne and Gann, 1997; Warfield et al., 2014), whereby tADs interact physically with and bring to gene promoters both coactivators and general transcription factors, and (ii) the nucleosome detergent model (Erkina and Erkine, 2016; Erkine, 2018), whereby the tADs act primarily as low-specificity nucleosome-distorting agents, which trigger the local coactivator-mediated remodeling of gene promoter chromatin that is necessary for the initiation of transcription.
The two models are fundamentally different, but both apply to frequently found tADs with an excess of acidic and hydrophobic residues, especially aromatic amino acids. In the direct recruitment model, the tADs containing an acidic/hydrophobic mini-motif scan and interact alternately with a variety of hydrophobic/basic pockets and crevices of the coactivator subunits (Ptashne and Gann, 1997; Tuttle et al., 2018; Warfield et al., 2014), thus physically bringing them to the gene promoter. In the nucleosome detergent model (Erkine, 2018), tADs act as promoter nucleosome-destabilizing agents with aromatic residues first intercalating between aromatic DNA bases, followed by acidic residues interfering with and breaking salt bridges between phosphate groups of DNA and amino groups of lysine- and arginine-rich histones. By destabilizing the local promoter nucleosome(s), tADs trigger local nucleosome modification by chromatin remodeling complexes. The direct recruitment model, accepted for decades (Ptashne and Gann, 1997), does not explain the sequence of the many possible recruitment steps, nor the low specificity of many interactions, and increasingly conflicts with experimental data indicating an uncertainty about tAD sequences, structure, and potential interacting targets. The nucleosome detergent model, which attributes the tAD function to near-stochastic detergent-like interactions, challenges fundamental biochemistry principles on the role of structure and specificity in molecular interactions.
The high variability in the tADs within even a single activator molecule is characterized by an average of 1% of the random pool of sequences screened in vivo for different activator/gene-reporter contexts (Abedi et al., 2001; Ma and Ptashne, 1987; Ravarani et al., 2018). Considering even a short 20-amino-acid stretch (Erkine, 2018) and a 20-amino-acid alphabet, the number of theoretical amino acid combinations capable of functioning as tADs is about 1% of 2020 ≈ 1024. Because of this astronomical variability which is typical for tADs (Abedi et al., 2001; Ma and Ptashne, 1987; Ravarani et al., 2018), in our work we rarely considered any specific tAD sequence or its derivatives, as it is traditionally done in conventional biochemistry. Instead, we examined large data sets of sequences and used machine learning (ML) regression and neural network (NN) approaches to analyze general tAD features to develop the ML model and elucidate the biological mechanism of tAD function.
Results
Absence of basic and presence of acidic and aromatic amino acid residues is the core of the tAD functionality
Several libraries of random sequences have been screened in vivo for functional tADs, and the resulting data sets are publicly available (Arnold et al., 2018; Erijman et al., 2020; Ravarani et al., 2018; Staller et al., 2018). The data sets have been analyzed and interpreted either strictly (Arnold et al., 2018; Erijman et al., 2020; Staller et al., 2018) or primarily (Ravarani et al., 2018) in the context of the recruitment model. We analyzed these data sets, taking into consideration both the direct recruitment and the nucleosome detergent models. In our current analysis, we used two data sets of tADs from yeast, Gcn4 (Erijman et al., 2020) and HSF1 (Ravarani et al., 2018), which were created using unbiased random pools of short peptide sequences and used for in vivo screening.
Several research groups found short linear motifs (SLiMs), which are thought to be important for the physical recruitment of varying coactivators (reviewed and summarized in the study by Staby et al., 2017). We calculated the frequency of SLiMs in the functional and nonfunctional subpools for each library. Consistently for Gcn4 and HSF1 contexts, none of the SLiMs dominated the functional library pools, and all were represented more frequently among the nonfunctional sequences (Figures 1A and 1B). Although the frequency of individual SLIM was above the threshold for random functional sequences in the entire library (green bar), this is probably because all SLiMs contain one or more hydrophobic or acidic amino acids, which are important for function (see later in discussion). Additionally, the frequencies of all SLiMs were near or below the level of functional sequences that lack positively charged amino acids (red bar). This suggests that identifying an SLiM that recruits a specific coactivator, or even identifying a proposed nine-amino-acid consensus (Piskacek et al., 2007) in an individual sequence, does not accurately predict the functionality, consistent with previous conclusions (Erijman et al., 2020; Ravarani et al., 2018). When the ML algorithm used SLiMs individually or in combinations to predict the functionality of sequences in the Gcn4 or HSF1 library, the importance for prediction for individual SLiMs was less than the importance of peptide sequences that simply lacked basic amino acids (Figures 1C and 1D). Additionally, the ML prediction accuracy, measured as the area under the receiver operating characteristic (ROC) curve (AUC) using SLiMs as ML features, was much lower than the accuracy of the same algorithm, based on 20 individual amino acids as the sole feature set (Gcn4 data set: AUC, 0.73 in Figure 1C versus 0.93 in Figure 2A; HSF1 data set: AUC, 0.53 in Figure 1D versus 0.69 in Figure 2A).
Figure 1.

SLiMs of tADs are localized predominantly in nonfunctional subpools and perform poorly as an ML feature set
(A and B) Y axis: percent of tAD functional sequences in the library. X axis: (1) entire library. Sequences containing SLiMs of (2) p53; (3) RelA/p65; (4) CREBZF; (5) AR; (6) ANACO13, (7) EKLF; (8) Gcn4; (9) 9 aa tAD consensus (stringent); (10) 9 aa tAD consensus (moderate); (11) 9 aa tAD consensus (lenient); (12) all sequences devoid of K, R, and H.
(C and D) Y axis: ML feature coefficients of SLiMs, performed as features using ML ridge regression to predict function and trained on (C) Gcn4 library or (D) HSF1. X axis: SLiM source as in panel (A) and (B).
Figure 2.

The absence of basic and presence of acidic and aromatic amino acid residues are important for the prediction of tAD function by ML models
(A) Y axis: individual amino acids. X axis: the ML feature coefficients of individual amino acids as features for the ML model (ridge and lasso) performance. The accuracy of the ML algorithm (AUC) is indicated in the lower right corner of each panel.
(B) Correlation of the coefficients obtained through ridge regression for the two data sets. Y axis: the ML feature importance for the HSF1 data set. X axis: same for the Gcn4 data set.
NN ML algorithms produce high prediction accuracy (Erijman et al., 2020), while the generally less precise regression algorithms (Ravarani et al., 2018) can predict model-based feature gains. Applying two regression-type ML algorithms, ridge and lasso, using 20 natural amino acids as the only ML features (Figure 2A), we found that the accuracy of prediction (Gcn4 ridge AUC, 0.9345; lasso AUC, 0.9347) was comparable with the accuracy of the functionality prediction for tADs using NN algorithms (AUC, 0.975) (Erijman et al., 2020) or more complex sets of features for regression algorithms (Ravarani et al., 2018). The higher predictive value for the Gcn4 data set versus the HSF1 data set (Figure 2A) was due primarily to the more than 10-fold greater size of the Gcn4 data set. The top eight amino acids contributing highly to the ML functionality prediction by two different ML algorithms for both data sets were three basic (K, R, and H), two acidic (D and E), and three aromatic (W, F, and Y) amino acids, with K, R, and H contributing negatively and D, E, W, F, and Y contributing positively. Training of the ML algorithm using only these eight amino acids (Figure S2) produced an almost identical accuracy of prediction (Gcn4 ridge AUC, 0.9347 for 20 features, versus 0.9213 for 8 features). The lasso regression for the HSF1 data set also produced nonzero feature coefficients for only eight amino acids (W, F, Y, D, E, R, K, and V). The correlation of the feature importance between the two data sets (Figure 2B) indicates that for both Gcn4 and HSF1, the functionality rules are similar. These simple rules can be formulated as the absence of basic and the presence of acidic and aromatic residues, with other amino acids contributing less. Underscoring the importance of acidic and aromatic residues, D, which has the lowest pKa among acidic amino acids, and W, which has the highest number of aromatic p-electrons, which are important for pi-pi interactions, showed the highest importance for prediction as ML features and the highest representation in acidic and aromatic groups correspondingly in the subpool of functional sequences (Figure S1).
To determine whether the similarities between the Gcn4 and HSF data sets represent general rules used by two different gene activators, we trained the ridge algorithm on the HSF data set using 20 amino acids as individual features and tested it on the Gcn4 data set. The AUC for the Gcn4 data set was 0.92, which was nearly the same as the value of 0.93 observed when the algorithm was trained on the Gcn4 subset (Figure 2), suggesting that the functionality rules are indeed conserved between the two different activator contexts.
Presence of multiple aromatic and acidic residues in a tAD increases both the probability and the level of functionality
To determine whether specific combinations of amino acids positively affect tAD function, we used dipeptides as ML features and saw a slight increase in the accuracy of prediction from a Gcn4 ridge AUC value of 0.9342 for 20 individual amino acids to 0.94 for 400 possible dipeptides. The dipeptides containing W, F, D, and E contributed positively as ML features, while those containing K and R contributed negatively (Figure 3). In addition, the repetition of acidic or aromatic residues in the dipeptide had highly positive impact, while the presence of purely basic dipeptides was detrimental to function, consistent with a previous analysis of dipeptides as ML features (Erijman et al., 2020).
Figure 3.

Sequences containing aromatic and acidic dipeptides have the highest probability for function as tADs and produce the highest gain for ML model performance
(A) Percent functionality of sequences containing various dipeptides in Gcn4 data set. (B) Percent functionality of sequences containing various dipeptides in HSF1 data set. Y axis: the first amino acid of the dipeptide within an individual sequence. X axis: the second amino acid of the dipeptide within an individual sequence.
(C and D) Coefficients of individual dipeptides as individual features in the ridge ML model for Gcn4 data set. (D) Coefficients of individual dipeptides as individual features in the ridge ML model for HSF1 data set.
To test if further increase of the multiplicity of aromatic and acidic amino acids contributes to the functionality of tADs, we analyzed the subpools with different count of aromatic and acidic residues within an individual sequence (Figure 4A). For both Gcn4 and HSF1 contexts, functional sequences containing multiple aromatic and acidic residues together were highly enriched in the functional part, reaching 74% for sequences containing at least 8 aromatic and at least 8 acidic residues in the case of Gcn4. A similar trend is seen for the HSF1 data set with lower numbers owing to the smaller size of this data set and the shorter span of the tAD region. These observations suggest that the abundant presence of aromatic and acidic amino acid residues in a single sequence is highly desirable for functionality. These results are consistent with the previous demonstration that the monotonous sequences such as WDWDWDWDWDWDWDWDWDWD or FDFDFDFDFDFD (Ravarani et al., 2018) are highly functional in vivo as tADs. This observation also proves that the presence of only aromatic and acidic amino acid residues without others is sufficient for the high functionality as a tAD in vivo. It also suggests that the representation of a specific aromatic moiety is not critically important. In addition, it underlines that the importance of basic residues as ML algorithm features is based on the preferred absence of them in functional tADs.
Figure 4.

Multiple aromatic and acidic residues within an individual tAD increase both the probability and the level of function
(A) Y axis: percent of functional sequences in the library. X axis: the number of acidic (D and E) and aromatic (W, F, and Y) amino acids within individual tAD sequences.
(B). Y axis: count of acidic amino acids within an individual tAD sequence. X axis: count of aromatic amino acids within an individual tAD sequence. The enrichment score for both Gcn4 and HSF1 is the average of the scores for all sequences with the specified number of acidic and aromatic amino acids. The diagonal line indicates an equal number of aromatic and acidic amino acids.
The importance of multiple representations for acidic and aromatic residues was further highlighted by the analysis of the functionality scores for individual sequences (Figure 4B). In case of the Gcn4 library, it is possible because cells expressing a higher level of the green fluorescent protein (GFP)P reporter were cell-sorted and sequenced with the subsequent calculation of the enrichment score in the GFP fluorescent subpool (Erijman et al., 2020), and in case of HSF1, the enrichment score was estimated as a read count normalized to the count of that sequence in the pool before the in vivo screen (Ravarani et al., 2018). It is evident that the functionality score for individual sequences dramatically increases with the increase of aromatic and acidic amino acid counts in the individual sequence (Figure 4B). That means that with the increasing multiplicity of aromatic and acidic amino acid residues representation, not only the probability of being functional increases (Figure 4A), but also the level of individual sequence activity rises significantly (Figure 4B).
Spreading of aromatic and acidic residues in the sequence is beneficial for function with more tolerance of an excess of acidic residues
To investigate further the relationships between aromatic and acidic residues, we tested if the balance between these two amino acid classes is important. Evidently (Figure 4B), a very high count (>10) of exclusively aromatic or acidic amino acids while the other is significantly diminished (≤3) is correlated with poor functionality. In addition, for sequences containing multiple aromatic and acidic residues, the probability of functionality is slightly higher for those with a relative excess of acidic over the aromatic residues (Figure 5A), although the functionality remained high as long as an overall excess of aromatic and acidic residues remains (Figures 4B and 5A).
Figure 5.

Spreading of aromatic and acidic residues in the tAD sequence is beneficial for function with more tolerance of the shift in imbalance toward excess of acidic residues
(A) Y axis: percent of functional sequences with specific balance/imbalance between aromatic and acidic residues. X axis: the difference between aromatic and acidic counts within the individual sequence (N = NAr – NAc). Color: The total number of aromatic and acidic amino acids within the sequence (N = NAr + NAc).
(B) Y axis: percent of functional sequences within the subpool of sequences containing five aromatic and five acidic amino acids. X axis: sequences with indicated amino acids clustered or spread within the 30-amino-acid tAD region. The number above each bar represents the total number of sequences meeting each criterion. Clustered sequences contain four acidic or four aromatic residues within a five-position window, somewhere within the tAD region. Spread sequences contain at least three spaces between each acidic or aromatic residue.
(C) Multivariate lasso regression analysis for functionality versus counts of individual amino acids and mini-motifs (dipeptides with 0–4 spaces in between them, with spacing labeled on the bar graph). Y axis: amino acid mini-motifs or individual amino acids. X axis: the value for ML feature coefficients.
We next asked whether clustering versus spreading of aromatic and acidic residues is important for function. It is evident that clustering of both acidic and aromatic residues, among sequences with the same number of these residues, negatively affects the probability of functionality, with acidic clustering being more detrimental (Figure 5B). In contrary, spreading of both acidic and aromatic residues is beneficial, leading to the above-average representation of the functional sequences for this subpool.
To determine the optimal spacing of functionally important amino acids, we used multivariate lasso regression analysis for functionality versus counts of individual amino acids and mini-motifs (dipeptides) with variable spacing (0–4) between the two important amino acids of the dipeptide. No specific spacing was either beneficial or detrimental, although aromatic and acidic residues nearby provided some benefit to function (Figure 5C). The lack of improvement of the multivariable model (AUC, 0.935) vs. simple lasso regression (Figure 2) suggested that functional sequences may contain variable spacing between amino acids. Similar to the results for the dipeptide analysis (Figure 3), for all distances acidic residues followed by aromatics were more functional than aromatic residues followed by acidic residues (Figure 5C).
Near-terminal positions of aromatic residues and internal locations of acidic residues are beneficial for tAD functionality
As has been shown previously (Erijman et al., 2020) and confirmed here in the dipeptide analysis (Figure 3C), an acidic residue followed by an aromatic residue is more preferential for function than the opposite orientation. To extend this directionality analysis, we looked at the probability of functionality for sequences with specific positioning for amino acid residues along the 30 amino acid stretch of the tAD region in the Gcn4 library (Figures 6A and S3). This analysis confirmed that acidic and aromatic residues are the two most important amino acid groups, which consistently increase the functionality of sequences containing them above the average for the library. In addition, we see that the probability of functionality decreases for sequences with C-terminal locations of acidic residues, while for aromatic residues, the trend is opposite with the C-terminal localization, increasing the probability of functionality. The opposite trends in importance for aromatic and acidic residues toward the end of the molecule is even more obvious if the subpool of sequences containing clusters of aromatic, acidic, or basic residues is separately analyzed (Figure 6C). The negative importance of basic residues increases toward the middle of the 30-amino-acid stretch and then decreases in absolute value again (Figures 6A and 6B). This observation is consistent with the negative role of basic residues neutralizing the charge of acidic ones. As the importance of acidic residues at the C-terminus decreases, the negative importance of neutralizing the charge also decreases.
Figure 6.

The near-terminal location of acidic amino acids decreases function, while for aromatic residues, the trend is opposite
(A) Average ridge regression coefficients (AUC = 0.94) for amino acid classes across every position in the TAD. Y axis: ML coefficients associating a given AA at that position with function. X axis: position within the 30 amino acid tAD region.
(B) Y axis: ML coefficients for specific amino acids. X axis: position within the 30 amino acid tAD region.
(C) Y axis: percent of functional sequences with groups of four indicated amino acids within a window of five positions; X axis: end position of the window within the 30 amino acid region of tAD. The dotted line is the average percentage of functional sequences in the entire library.
tAD function is least affected by basic residues in a basic-acidic-aromatic configuration
To better understand tADs and also increase the accuracy of ML, we used the attention mechanism (Vaswani et al., 2017) and long short-term memory (LSTM) network (Hochreiter and Schmidhuber, 1997), a type of recurrent NN useful for analysis of complex relationships among different elements in sequence information (Figure 7A and STAR methods). The attention mechanism design enables us to analyze the contribution of each amino acid to the global functionality by assigning weights to every amino acid. Compared with other methods, we showed slightly better accuracy with 98.3% AUC on the testing data set (Figure 7B). The previous NN ADpred method (Erijman et al., 2020) built and trained on this data set achieved 97.5% by using a fully connected neural network, demonstrating excellent identification for most cases, but we further improved the performance by the attention mechanism and LSTM. Compared with ADpred, we have two advantages. On the one hand, LSTM makes greater use of the order of amino acid in the sequences, which agrees better to the observation in the experiments that the order of amino acid contributes to the functionality. On the other hand, the self-attention mechanism allows some explanation of features for the NN, as the assigned weight by attention can be seen as the importance factor of the corresponding amino acids.
Figure 7.

The best function is observed with basic groups inside of the molecule, followed by acidic and then aromatic groups at the end
(A) Architecture of attention LSTM framework. The input is first encoded by the embedding layer, which is a fully connected layer. Then the LSTM network (shown in the light green rectangle) is adopted to encode the embedding of the sequence. Later, the attention matrix is calculated based on sequence embedding and element embedding . Finally, attention-weighted sums of element embeddings are processed by the FC layer to predict the functionality of the sequence.
(B) Testing AUC of regression and LSTM network methods in predicting function. Y axis: ML models; X axis: the AUC value.
(C) (Left) Numbers of sequences correctly and incorrectly predicted by LSTM network and ridge regression among functional and nonfunctional sequences from the testing set. (Right) Consensus sequences for functional and nonfunctional sequences that were correctly predicted by LSTM network and incorrectly (solid line) or correctly predicted (dotted line) by ridge regression. Y axis for subpanel: frequency of specific amino acid; X axis for subpanel: position within the 30-amino-acid tAD region.
(D) Examples of functional and nonfunctional sequences correctly predicted by LSTM network analysis and incorrectly predicted by ridge regression (RR).
(E) Bar plot of function for different orders of aromatic, acidic, and basic amino acid groups. Y axis: percentage of functional sequences in the Gcn4 data set. X axis: composition as indicated by cartoons with triangles representing the N-terminal part of the activator including the DNA-binding domain.
Although NN models provide higher precision, they generally keep tAD features hidden. The 4% difference between ridge and the aforementioned LSTM NN approach (ridge AUC, 0.94 versus LSTM AUC, 0.983; Figure 7B) potentially contains valuable information. To reveal features not realized by the regression analysis, we separated sequences that were missed by the ridge regression model as positive and falsely labeled as negative while identified correctly by LSTM NN and also sequences that were falsely labeled by the ridge model as positive while correctly identified by the LSTM NN as true negative; we then compared these sequences with those that were correctly predicted by both models (Figures 7C and S4, S5). The composition plots show that functional sequences correctly identified by the NN method had fewer aromatic residues at the N-terminus of the tAD, fewer acidic residues at the C-terminus, and more basic residues at the N-terminus. This pattern is demonstrated by three sequences in Figure 7D. Of the true nonfunctional sequences, 167,173 (83%) were correctly identified by both methods, while 22,525 (11%) were identified correctly only by NN analysis. Sequences in this group generally showed high levels of aromatic or acidic residues with few basic residues (Figure 7C). However, example sequences (Figure 7D) demonstrate that sequences containing many acidic or aromatic residues were predicted to be functional by ridge regression, while NN analysis recognized that both acidic and aromatic residues are required.
Our analysis suggested that the regression model did not recognize functional sequences with long distances between acidic and basic amino acids; therefore, using the Gcn4 data set, we calculated the probability of functionality for sequences with variable positions for aromatic, acidic, and basic amino acid regions (Figure 7E). Sequences with combinations of aromatic and acidic residues separated by basic residues showed poor functionality, but the worst combination was aromatic-basic-acidic. When acidic and aromatic residues were adjacent, the worst combination included C-terminal acidic residues, while the best combination was basic-acidic-aromatic. These results confirm the importance for function of adjacent acidic-aromatic residues with aromatic residues situated at the spatially free C-terminus, with basic residues preferably separated toward the middle of the molecule.
Within the basic microenvironment, cysteine becomes more important for tAD function
A peculiar outcome of all in vivo screens for functional tAD sequences in yeast (Abedi et al., 2001; Erijman et al., 2020; Ma and Ptashne, 1987) is an appearance of net-positively charged sequences in the functional subpool. In the case of the Gcn4 library (Erijman et al., 2020), this pool is significant, amounting to 2,292 functional sequences. These types of sequences puzzled researchers, suggesting that in this case tAD sequences might be working via an alternative mechanism. To test if the functionality rule is different in this case, we trained two different ML algorithms on the net-positively charged pool of sequences and analyzed the ML feature importance for correct prediction of sequence functionality. Similar to the entire library, the overall depletion of basic and presence of aromatic residues remain on top of the ML feature ranking, suggesting that the functionality rules are the same. The difference was observed with the diminished role of acidic residues and the increased importance of cysteine for net-positively charged tAD sequences (compare Figures 8 and 2). While lower ML coefficients for acidic residues can be explained by the bias of this subpool toward the positively charged amino acids, the role of cysteine is not as easily explained. It is likely that within the microenvironment of each particular net-positively charged K- and R-enriched sequence, the cysteine residue (with pKa = 8.1) starts to deprotonate, acquiring a negative charge, thereby contributing to the negative charge that is important for tAD function. This likely scenario again underlines the importance of a negative charge for the tAD function and confirms the same general rule.
Figure 8.

Within the subpool of net-positively charged sequences, Cysteine gains importance for prediction of tAD functionality
Y axis: individual amino acids as ML features. X axis: the ML feature coefficients of individual amino acids as features for the ML ridge model performance.
Discussion
General rules of tAD function
The bioinformatics analysis of the data sets obtained by in vivo screening of random sequence pools for functional tADs suggests that, at least in yeast, while being extremely variable, the functional sequences should be (i) devoid of or depleted with basic amino acid residues, (ii) enriched with aromatic and acidic residues, (iii) with aromatic residues localized mostly near the terminus of the sequence, and acidic residues localized more internally within a span of 20–30 amino acids, and (iv) with both aromatic and acidic residues preferably spread out in the sequence and not clustered. (v) Importantly, the separation of acidic and aromatic residues by several basic ones not always but generally is detrimental for functionality. The specific and very accurate prediction of functionality based on these, and additional less obvious subtle rules can be done using algorithms described by us and others (Erijman et al., 2020; Ravarani et al., 2018). None of the rules formulated above are absolute, which is not surprising given that the theoretical amount of functional sequences based on extrapolation of the in vivo screening results (Erijman et al., 2020; Ravarani et al., 2018) for the stretch of 20 amino acids is ∼1% of 2020 ≈ 1024. The additional subtle rules can contribute to the range of activity within the pool of functional sequences and may be elucidated in future investigations.
Comparison of models describing gene activation
The conventional model of gene activation (Ferreira et al., 2005; Hahn and Young, 2011; Ptashne and Gann, 1997; Warfield et al., 2014) suggests the direct physical interaction and recruitment by tADs of transcriptional coactivators such as chromatin-remodeling and histone-modifying complexes, as well as components of general transcriptional machinery including Mediator complex. This model, however, is increasingly inconsistent with accumulating experimental data (Arnold et al., 2018; Erijman et al., 2020; Ravarani et al., 2018; Staller et al., 2018) demonstrating enormous variability of tADs, absence of a consensus sequence among different swapping sequence variants even within the context of a single activator, intrinsically disordered nature of tAD structures, and absence of a definitive target and/or sequence of interactions with potential targets. The tAD interactions are increasingly described as fuzzy and uncertain (Fuxreiter and Tompa, 2012; Scholes and Weinzierl, 2016).
Out of the two mechanisms of tAD function described in the introduction, the nucleosome detergent model is the most consistent one with the results of our analysis. The direct recruitment model presumes specificity, if not for the tAD sequence or structure, then at least for its interactions with targets. This specificity led to the proposal that the WxxLF motif is critical for Gcn4 tAD function by its interaction with Mediator complex subunits (Tuttle et al., 2018; Warfield et al., 2014). However, we have shown here that neither this nor any other previously proposed SLiMs or consensus sequences (Staby et al., 2017) are critical for tAD function (Figure 1) and none rank high in importance as a feature for ML. Instead, the high functionality can be achieved by the combination of simple rules formulated earlier, with the monotonous sequences such as (WD)n or (FD)n exemplifying the dominant acidic-aromatic rule without any other amino acids.
The direct recruitment model assumes that acidic and hydrophobic residues scan and probe coactivators, with the acidic residues forming an initial strong salt bridge bond during the scanning phase of coactivator recruitment, while hydrophobic interactions as a secondary step strengthen the bond (Ferreira et al., 2005). This model requires high functionality for acidic residues situated at the end of the molecule where there is the greatest spatial freedom. However, our data demonstrated that acidic residues at the C-terminus functioned poorly and were somewhat detrimental (Figures 6 and 7), while near-terminal aromatic residues improved function. These results are consistent with the nucleosome detergent model, whereby terminal aromatic moieties first anchor the tAD by intercalation between aromatic bases in the nucleosomal DNA; then the acidic residues adjacent to salt bridges between DNA and histones trigger chromatin remodeling performed by the chromatin-remodeling and histone-modifying complexes (Erkine, 2018). Terminal acidic residues would significantly decrease the initial interactions with DNA owing to the repulsion between acidic amino acid residues and similarly charged phosphate groups of the DNA, which is consistent with our data (Figure 6).
For the direct recruitment model, hydrophobicity plays an important role in tAD interactions, which alternate between crevices and surface pockets of targets. There are several experimental hydrophobicity scales (Hessa et al., 2005; Kyte and Doolittle, 1982; Moon and Fleming, 2011; Zhao and London, 2006) that place amino acids I, V, and L as higher or comparable with aromatics (W, F, and Y) in their hydrophobicity values. Contrary to expectations based on the direct recruitment model, we found that aliphatic amino acids were not important for tAD function (Figure 2 and (WD)10 sequence). While the direct recruitment model does not distinguish between the hydrophobicity of aromatic and aliphatic residues, as is evident from the composition of SLiMs of tADs (Staby et al., 2017), the nucleosome detergent model requires aromatic residues, which, via pi-pi interactions, mediate the aromatic ring intercalation in the DNA. This tAD anchoring by aromatic residues is a critical initial step, which is highly probable due to the overabundance of available aromatic DNA bases on the nucleosome surface. In contrary, for the direct recruitment model, these interactions are unlikely, as aromatic residues are relatively rare and seldom localized at the protein (coactivator) surface, but instead are buried inside the hydrophobic protein core.
Given the high variability and absence of a definitive sequence and structure for tAD sequences, the direct recruitment model cannot readily explain the management of the multiple low-affinity low-specificity recruitment steps or how choices are made among the many possible targets, while also ignoring equally possible irrelevant or detrimental interactions (Hahn and Young, 2011; Staby et al., 2017). In contrast, the nucleosome detergent model is compatible with and requires the low specificity of tAD sequence interactions with multiple sites on the surface of promoter nucleosomes (Erkine, 2018). A major difference between the two models is in the proposed function of the DNA-binding domain (DBD) of the gene activator. In the direct recruitment model, the DBD simply targets the activator with its tAD to the cognate gene promoter. In the nucleosome detergent model, the DBD in addition to targeting has a catalytic role by greatly increasing (inversely proportional to the Kd of the DBD) the local concentration of the tAD (Erkine, 2018). This increase of local concentration makes local and only local interactions orders of magnitude more likely than general, true-noise interactions with karyoplasm components. If, in contrary, the tAD interactions were strong and site-specific, the nucleosome would be anchored rather than destabilized, leading to the repression of transcription initiation. This explains why tADs lack specificity, are variable in sequence, are structurally intrinsically disordered, and are fuzzy in their target specificity. The local promoter nucleosome distortions achieved by tADs are similar and achievable by interactions with a variable spectrum of acidic-aromatic sequences. The high representation of acidic-aromatic, detergent-like mini-motifs (Ravarani et al., 2018) likely makes this nucleosome distortion more effective without significantly changing the specificity and affinity (Figure 4).
Functional near-noise interactions as a new biochemistry branch
While the function of tAD sequences is often for simplicity considered by us and others research groups as binary (functional/nonfunctional), within the functional pool, the activities of individual sequences may vary dramatically (Erijman et al., 2020; Ravarani et al., 2018). This fact is important for the understanding of the plasticity in the evolutionary development of gene expression in complex biological systems and seems to be an important consideration for further development of ML algorithms predicting the level of functional activity for any specific sequence as a tAD.
An additional consideration that makes the nucleosome detergent model attractive is that it fits well with the liquid-liquid phase separation (LLPS) model of gene regulation (Boija et al., 2018). The surfactant-like aspect of tAD interactions during transcription seems to be important for the formation or dissociation of phases. Additionally, the charge of the tAD and its activity may be regulated by posttranslational modifications, such as phosphorylation or acetylation, which also play an important role in the LLPS (Hofweber and Dormann, 2019).
The long-standing enigma of tADs roots in the clash, on the one hand, of conventional biochemical mentality and methodology based on the specificity principle and general consideration of near-stochastic interactions as a detrimental noise, and, on the other hand, growing amount of experimental data demonstrating near absence in tADs of specificity in sequence, structure, and interaction targets. Our study provides a solution showing how near-noise interactions can be central for the fundamental function of gene expression initiation. While the astronomical number of near-stochastic interactions is impossible to study using conventional biochemistry, the development of the new AI technologies in combination with breakthroughs of synthetic biology provides a methodological answer to this challenge. Recognition of the importance of near-noise interactions in the context of new AI methodology helps to define a new branch of biochemistry (Erkine, 2018), studying the near-noise interaction not only in context of gene regulation but also in application to, for example, such areas as the allosteric regulation of enzyme activity, function of molecular chaperones, and generally intrinsically disordered protein regions.
Limitations of the study
Both libraries of tADs analyzed, the Gcn4 (1,048,575 sequences) and the HSF (52,710 sequences), represent random samples from the entire combinatorial space, which is actually 2030 for the Gcn4 library and 2020 for the HSF library (based on the length of the tAD sequence in each specific library and the 20-amino-acid alphabet). As such both libraries represent a miniscule fraction of possible combinations. This fact limits the precision of ML features deduction. Thus, the features we elucidated have general characteristics, while future investigations of additional data sets might uncover more nuanced features.
From the biochemical perspective, while analyzing relatively large sets of in vivo tested and verified tAD sequences, this study is performed using bioinformatics and ML methods. The outcome of the analysis is consistent with the formulated paradigm-shifting mechanism of gene expression activation. However, backing up the findings by in vitro experiments demonstrating nucleosome distortion by tADs would be important and are planned for the future.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Deposited data | ||
| Gcn4 transcription activation domain functionality data | Erijman et al., 2020 | 10.1016/j.molcel.2020.04.020 |
| HSF1 transcription activation domain functionality data | Ravarani et al., 2018 | 10.15252/msb.20188190 |
| Software and algorithms | ||
| Machine Learning analysis of transcriptional activation domains | (Broyles et al., 2021) | 10.5281/zenodo.5148655 |
| Attention LSTM neural network software | (Wang et al., 2021) | 10.5281/zenodo.5146895 |
Resource availability
Lead contact
Further information and requests should be directed to and will be fulfilled by the Lead Contact, Alexandre Erkine (aerkine@butler.edu).
Materials availability
This study did not generate new unique reagents.
Method details
Exploratory and regression analyses were conducted in R version 4.0.2 (R Core Team, 2017). Transcriptional activation domain (tAD) functionality data sets in the Gcn4 and HSF1 contexts were downloaded from the supplemental information files of their respective publications (Erijman et al., 2020; Ravarani et al., 2018). In both publications, a quantitative estimate of the extent of function was provided for each sequence, and a call of functional versus non-functional was made based on comparison with a defined cutoff value. The Gcn4 data set included 1,048,575 sequences, 37,923 of which were identified as functional. From the HSF1 set, only sequences of at least five amino acids in length were included. This resulted in a library of 52,710 sequences, 649 of which were identified as functional. These calls were used to calculate the percentage of tAD functional sequences meeting certain characteristics, such as the percentage of functional sequences containing known mini-motifs or dipeptides.
Mini-motif analysis
To identify the percentage of functional sequences among sequences containing known published mini-motifs (Staby et al., 2017), a search of regular expressions was used to identify sequences containing the patterns of interest, as defined in the table below. The same general method was used to identify sequences containing all possible dipeptides, tripeptides, or patterns of spread and clustered amino acids for a given class.
| Motif | Regular Expression |
| p53 | [ALVIMWYF]..[ALVIMWYF][ALVIMWYF] |
| RelA_2 | [ALVIMWYF][ALVIMWYF]..[ALVIMWYF]..[ALVIMWYF] |
| CREBZF | D[VILM][VILM][RKDEQNHSTYC][RKDEQNHSTYC][VILM][VILFWYM] |
| AR | F..LF |
| ANAC013 | [DE].[YF].[DE]L |
| EKLF | [RKDEQNHSTYC][ALVIMWYF][ALVIMWYF]..[ALVIMWYF]..[RKDEQNHSTYC][RKDEQNHSTYC] |
| WxxLF | W..LF |
| stringent_9aa | [MDENQSTYG][ˆKRHCGP][ILVFWM][ˆKRHCGP][ˆCGP][ˆKRHCGP][ILVFWM][ILVFWMAY][ˆKRHC] |
| moderate_9aa | [MDENQSTYG][ˆKRHCGP][ILVFWM][ˆKRHCGP][ˆCGP][ˆCGP][ILVFWM][ˆCGP][ˆCGP] |
| lenient_9aa | [MDENQSTYCPGA].[ILVFWMAY][ˆKRHCGP][ˆCGP][ˆCGP][ILVFWMAY].. |
Regression models
Generalized linear models (GLM’s) were optimized with the ‘caret’ package in R (Kuhn et al., 2020) using lasso and ridge regression to explore the validity of proposed features in tAD function. For Figure 1, we used 10 known motifs (as well as a “no_basics” feature, which was one if a sequence had no R, H, or K residues; or zero otherwise), as features to determine whether these motifs predicted function. For Figure 2, function was regressed against the counts of each of the 20 amino acids present in each sequence. For Figure 3, regression was conducted for each of the 400 possible dipeptides. For Figure 6, function was regressed against 600 features corresponding to the presence (one, versus zero if not present) of a certain amino acid at a certain position (30 positions × 20 amino acid alphabet). A simple model also grouped the amino acids into categories as features (categories: basic, acidic, aromatic, aliphatic, polar, glycine, and proline).
For Figure 5C, a lasso regression model combining individual amino acid counts with the counts of mini-motifs containing two of an aromatic (W, Y, F), acidic (D, E), or basic (R, H, K) amino acid nearby (with zero to four spaces in between, identified using regular expressions). This was done to estimate the marginal benefit (or cost) of having various types of residues close to each other. For example, a D followed by a W with one space in between would have individual positive contributions from the D and W residues, plus an additional positive contribution from the acidic-aromatic (one space) feature. For all GLM analyses, 20% of the sequences were held out as a testing set, and models were trained on the remaining sequences using 10-fold cross-validation, tuning lambda with a grid of 100 values.
Attention-LSTM network
Inspired by previous work on sequence processing with the attention mechanism (Shen and Lee, 2016), we proposed our attention-based long short-term memory (LSTM) architecture, whose architecture is shown in Figure 7A, for identifying the functionality of sequences. The input of each element (amino acid) is one-hot encoding to denote its amino acid types. It will be further processed by a fully connected layer to obtain element embedding . Then we selected the LSTM network, a type of recurrent NN known for its greater capacity for the complex relationships of different elements, to process inputs sequentially (Hochreiter and Schmidhuber, 1997). The sequence representation is a vector output from LSTM after processing all the element embeddings.
We then calculated the cosine similarity between the sequence embedding and element embedding :
where is the cosine operator, is the attention output for element , and is the normalized attention weight for element .
Then the normalized attention weight applied to the embedding of every element and the weighted sum embedding, based on attention weight, was the final sequence embedding. This embedding was further processed by fully connected layer and activation function sigmoid to predict the functionality.
To allow comparison between this method and regression methods, the same training and testing set sequences from the Gcn4 data set were used for this analysis. To optimize the network, we used cross-entropy loss with SGD optimizer (Polyak and Juditsky, 1991). The learning rate was set as 0.01 while the weight decay was set as 1e-4 and the momentum was 0.9 for the optimizer. In the training process, the batch size was set as 256 with training of 100 epochs. The final model was selected based on the accuracy of the validation set.
Comparison of ridge regression and LSTM network
The regression ML models ridge and lasso used in this study allow analyses of gains for individual features in each ML algorithm, while in the case of the NN approach, which produces higher accuracy, the features are formulated by the algorithm itself and remain largely unavailable for the analysis, although some advances are being made in this area (Olden and Jackson, 2002). We compared the common testing sets from the LSTM network against the ridge regression analysis for individual amino acid counts in the Gcn4 data set to see which sequences were correctly or incorrectly predicted by each model and whether any patterns of functionality were identified by the LSTM network approach (Figures 7C and 7D). For each method, a cutoff of the predicted functionality probability was selected to maximize TPR + TNR (true positive plus true negative rate). For the LSTM network, the cutoff identified was 0.52, very close to the expected p = 0.5 cutoff (TPR = 0.93, TNR = 0.94). For ridge regression, the cutoff identified was 0.045 (TPR = 0.90, TNR = 0.84). Confusion matrices were generated separately for true functional and true non-functional sequences to show the number of sequences correctly or incorrectly predicted by each method. Then consensus matrices were generated and plotted for the sequences that were correctly predicted by the LSTM network and correctly or incorrectly predicted by ridge regression to allow us to compare the prevalence, positions, and combinations of different amino acids in each group (consensus matrices for sequences incorrectly predicted by the LSTM network are provided in the supplemental information).
Acknowledgments
We thank Marcos Oliveira for discussions and suggestions. The work was supported by NSF grants MCB-1925646 (to A.M.E.) and MCB-1925643 (to D.K.) and by Butler University Innovation and HAC grants.
Author contributions
A.M.E. conceived the project. B.K.B., A.T.G., T.P.M., D.A.C., T.M.W., X.W., D.K., and C.A.C. performed data analysis. C.A.C. oversaw methods and visualizations with contributions from X.W. and D.K. A.M.E. wrote the manuscript. All authors edited and approved the manuscript.
Declaration of interests
The authors declare no competing interests.
Published: September 24, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.103017.
Supplemental information
Data and code availability
-
•
All data analyzed in this work are publicly available (Erijman et al., 2020; Ravarani et al., 2018). DOIs are listed in the Key Resources Table.
-
•
All original code used to conduct analyses and generate figures have been deposited at Zenodo and are publicly available (Broyles et al., 2021; Wang et al., 2021). DOIs are listed in the Key Resources Table.Any additional information required to reanalyze the data reported in this work paper is available from the Lead Contact upon request.
References
- Abedi M., Caponigro G., Shen J., Hansen S., Sandrock T., Kamb A. Transcriptional transactivation by selected short random peptides attached to lexA-GFP fusion proteins. BMC Mol. Biol. 2001;2:10. doi: 10.1186/1471-2199-2-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold C.D., Nemcko F., Woodfin A.R., Wienerroither S., Vlasova A., Schleiffer A., Pagani M., Rath M., Stark A. A high-throughput method to identify trans-activation domains within transcription factor sequences. EMBO J. 2018;37 doi: 10.15252/embj.201798896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boija A., Klein I.A., Sabari B.R., Dall'Agnese A., Coffey E.L., Zamudio A.V., Li C.H., Shrinivas K., Manteiga J.C., Hannett N.M. Transcription factors activate genes through the phase-separation capacity of their activation domains. Cell. 2018;175:1842–1855.e16. doi: 10.1016/j.cell.2018.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broyles B.K., Gutierrez A.T., Maris T.P., Coil D.A., Wagner T.M., Wang X., Kihara D., Class C.A., Erkine A.M. Zenodo; 2021. calebclass/tAD_analysis_2021: Initial Release (v1.0.0) [Google Scholar]
- Erijman A., Kozlowski L., Sohrabi-Jahromi S., Fishburn J., Warfield L., Schreiber J., Noble W.S., Soding J., Hahn S. A high-throughput screen for transcription activation domains reveals their sequence features and permits prediction by deep learning. Mol. Cell. 2020;78:890–902.e6. doi: 10.1016/j.molcel.2020.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erkina T.Y., Erkine A.M. Nucleosome distortion as a possible mechanism of transcription activation domain function. Epigenetics Chromatin. 2016;9:40. doi: 10.1186/s13072-016-0092-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erkine A.M. Nonlinear' biochemistry of nucleosome detergents. Trends Biochem. Sci. 2018;43:951–959. doi: 10.1016/j.tibs.2018.09.006. [DOI] [PubMed] [Google Scholar]
- Ferreira M.E., Hermann S., Prochasson P., Workman J.L., Berndt K.D., Wright A.P. Mechanism of transcription factor recruitment by acidic activators. J. Biol. Chem. 2005;280:21779–21784. doi: 10.1074/jbc.M502627200. [DOI] [PubMed] [Google Scholar]
- Fuxreiter M., Tompa P. Fuzzy complexes: a more stochastic view of protein function. Adv. Exp. Med. Biol. 2012;725:1–14. doi: 10.1007/978-1-4614-0659-4_1. [DOI] [PubMed] [Google Scholar]
- Hahn S., Young E.T. Transcriptional regulation in Saccharomyces cerevisiae: transcription factor regulation and function, mechanisms of initiation, and roles of activators and coactivators. Genetics. 2011;189:705–736. doi: 10.1534/genetics.111.127019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hessa T., Kim H., Bihlmaier K., Lundin C., Boekel J., Andersson H., Nilsson I., White S.H., von Heijne G. Recognition of transmembrane helices by the endoplasmic reticulum translocon. Nature. 2005;433:377–381. doi: 10.1038/nature03216. [DOI] [PubMed] [Google Scholar]
- Hochreiter S., Schmidhuber J. Long short-term memory. Neural Comput. 1997;9:1735–1780. doi: 10.1162/neco.1997.9.8.1735. [DOI] [PubMed] [Google Scholar]
- Hofweber M., Dormann D. Friend or foe-Post-translational modifications as regulators of phase separation and RNP granule dynamics. J. Biol. Chem. 2019;294:7137–7150. doi: 10.1074/jbc.TM118.001189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keung A.J., Bashor C.J., Kiriakov S., Collins J.J., Khalil A.S. Using targeted chromatin regulators to engineer combinatorial and spatial transcriptional regulation. Cell. 2014;158:110–120. doi: 10.1016/j.cell.2014.04.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn M., Wing J., Weston S., Williams A., Keefer C., Engelhardt A., Cooper T., Mayer Z., Kenkel B., Benesty M., R Core Team . R Foundation for statistical computing; 2020. Caret: Classification and Regression Training. R Package Version 6.0-86. [Google Scholar]
- Kyte J., Doolittle R.F. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 1982;157:105–132. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- Ma J., Ptashne M. A new class of yeast transcriptional activators. Cell. 1987;51:113–119. doi: 10.1016/0092-8674(87)90015-8. [DOI] [PubMed] [Google Scholar]
- Moon C.P., Fleming K.G. Side-chain hydrophobicity scale derived from transmembrane protein folding into lipid bilayers. Proc. Natl. Acad. Sci. U S A. 2011;108:10174–10177. doi: 10.1073/pnas.1103979108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olden J.D., Jackson D.A. Illuminating the “Black Box”: a randomization approach for understanding variable contributions in artificial neural networks. Ecol. Modell. 2002;154:135–150. [Google Scholar]
- Piskacek S., Gregor M., Nemethova M., Grabner M., Kovarik P., Piskacek M. Nine-amino-acid transactivation domain: establishment and prediction utilities. Genomics. 2007;89:756–768. doi: 10.1016/j.ygeno.2007.02.003. [DOI] [PubMed] [Google Scholar]
- Polyak B.T., Juditsky A.B. Acceleration of stochastic approximation by averaging. SIAM J. Control Optimization. 1991;30:838–855. [Google Scholar]
- Ptashne M., Gann A. Transcriptional activation by recruitment. Nature. 1997;386:569–577. doi: 10.1038/386569a0. [DOI] [PubMed] [Google Scholar]
- R Core Team . R Foundation for statistical computing; 2017. R: A Language and Environment for Statistical Computing.https://www.R-project.org/ [Google Scholar]
- Ravarani C.N., Erkina T.Y., De Baets G., Dudman D.C., Erkine A.M., Babu M.M. High-throughput discovery of functional disordered regions: investigation of transactivation domains. Mol. Syst. Biol. 2018;14:e8190. doi: 10.15252/msb.20188190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholes N.S., Weinzierl R.O. Molecular dynamics of "Fuzzy" transcriptional activator-coactivator interactions. PLoS Comput. Biol. 2016;12:e1004935. doi: 10.1371/journal.pcbi.1004935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen S.-S., Lee H.-Y. 2016. Neural Attention Models for Sequence Classification: Analysis and Application to Key Term Extraction and Dialogue Act Detection. [Google Scholar]
- Staby L., O'Shea C., Willemoes M., Theisen F., Kragelund B.B., Skriver K. Eukaryotic transcription factors: paradigms of protein intrinsic disorder. Biochem. J. 2017;474:2509–2532. doi: 10.1042/BCJ20160631. [DOI] [PubMed] [Google Scholar]
- Staller M.V., Holehouse A.S., Swain-Lenz D., Das R.K., Pappu R.V., Cohen B.A. A high-throughput mutational scan of an intrinsically disordered acidic transcriptional activation domain. Cell Syst. 2018;6:444–455 e446. doi: 10.1016/j.cels.2018.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuttle L.M., Pacheco D., Warfield L., Luo J., Ranish J., Hahn S., Klevit R.E. Gcn4-Mediator specificity is mediated by a large and dynamic fuzzy protein-protein complex. Cell Rep. 2018;22:3251–3264. doi: 10.1016/j.celrep.2018.02.097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaswani A., Shazeer N., Parmar N., Uszkoreit J., Jones L., Gomez A.N., Kaiser L., Polosukhin I. Vol. 30. Advances in Neural Information Processing Systems; 2017. Attention Is All You Need; pp. 6000–6010. [Google Scholar]
- Wang X., Broyles B.K., Gutierrez A.T., Maris T.P., Coil D.A., Wagner T.M., Kihara D., Class C.A., Erkine A.M. zenodo; 2021. kiharalab/Attention_AD: Attention_AD Official v1.0 (v1.0) [Google Scholar]
- Warfield L., Tuttle L.M., Pacheco D., Klevit R.E., Hahn S. A sequence-specific transcription activator motif and powerful synthetic variants that bind Mediator using a fuzzy protein interface. Proc. Natl. Acad. Sci. U S A. 2014;111:E3506–E3513. doi: 10.1073/pnas.1412088111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao G., London E. An amino acid "transmembrane tendency" scale that approaches the theoretical limit to accuracy for prediction of transmembrane helices: relationship to biological hydrophobicity. Protein Sci. 2006;15:1987–2001. doi: 10.1110/ps.062286306. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All data analyzed in this work are publicly available (Erijman et al., 2020; Ravarani et al., 2018). DOIs are listed in the Key Resources Table.
-
•
All original code used to conduct analyses and generate figures have been deposited at Zenodo and are publicly available (Broyles et al., 2021; Wang et al., 2021). DOIs are listed in the Key Resources Table.Any additional information required to reanalyze the data reported in this work paper is available from the Lead Contact upon request.