

Abstract
In this information era, there is an urgent need for tighter integration of bioinformatics and experimental biology. The enormous amount of data generated by biological experiments calls for extensive computational analysis. Many bioinformatics textbooks at present mainly focus on theories, which hinders the vigorous development of scientific research. As a result, most students are simply familiar with the bioinformatics theories but lack the opportunity to put them into practice. Here, we present our bioinformatics docking project conducted during the self‐isolation period of the COVID‐19 pandemic. Five students used the RBD–ACE2 complex as a benchmark to conduct a systematic comparison of several open‐source online molecular docking programs. The virus surface spike protein mediates the entry of the SARS‐CoV‐2 virus into human cells by binding to its receptor, angiotensin‐converting enzyme 2 (ACE2), through its receptor‐binding domain (RBD). Through docking and comparing predicted structures to the crystal structure, students gained the opportunity to practice different bioinformatics tools independently and conduct research collaboratively. It opens a window for students to reach out to the state‐of‐the‐art bioinformatics techniques and to keep up with the research trends. The online workshop has also proven to be an innovative method for bioinformatics teaching. We hope our work can inspire other educators to develop strategies to expose undergraduate students to modern bioinformatics and turn every temporary difficulty into a possible learning opportunity.
Keywords: COVID‐19, molecular docking, protein complex, SARS‐CoV‐2
1. INTRODUCTION
Novel coronavirus disease 2019 (COVID‐19) first arose in December 2019 and quickly turned into the most damaging pandemic in the 21st century by far. 1 , 2 This highly transmittable viral pandemic was caused by a new coronavirus named severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2), which belongs to the Betacoronavirus genus. 3 Among this genus, SARA‐CoV, and MERS‐CoV caused severe acute respiratory syndrome (SARS) in 2003 and middle east respiratory syndrome (MERS) in 2012, respectively. 4 , 5 Similar to other coronaviruses, SARS‐CoV‐2 has many crown‐like spike proteins that protrude from the viral surface. The spike protein is made of two subunits, S1 and S2. The S1 subunit contains a receptor‐binding domain (RBD), which can recognize and bind to the receptor angiotensin‐converting enzyme 2 (ACE2) on the cell surface, while the S2 subunit mediates viral cell membrane fusion. 6 After successful binding of the spike protein to ACE2, a serine protease from the host cell membrane, named transmembrane protease serine 2, promotes fusion of the viral and host membrane by activating the spike protein. 7
Since the interaction between RBD and ACE2 is critical for the viral life cycle, the inhibition of this interaction can be an efficient treatment. 8 Computational designs allow not only virtual screening to discover potential small molecule inhibitors, but also the design of peptide inhibitors of SARS‐CoV‐2 based on ACE2 sequence. For example, Benítez‐Cardoza et al. used the structure of RDB–ACE2 complex to screen out 20 possible compounds for developing a drug 9 and Han et al. designed possible inhibitor peptides originated from the two sequential α‐helices from the RBD–ACE2 binding interface of ACE2 protein. 10 However, all these works can hardly proceed without the critical RBD–ACE2 complex structure. Therefore, structural biologists succeeded in solving the RBD–ACE2 complex structure only 2 months after the COVID‐19 outbreak. 11
Molecular docking is a widely used method in structural biology and computational biology. This method can be used to model interactions between two molecules, such as protein to protein, protein to peptide, or protein to small molecule interaction at the atomic level. 12 , 13 Predicted complex conformations allow the characterization of behaviors of the two binding partners, thereby elucidating the fundamental biochemical principles underlying their interaction. Although the structure of the RBD–ACE2 complex can be obtained through laborious experiments, for example, Cryo‐EM and X‐ray crystallography, docking can still provide critical information about the formation of this key complex, such as conformations and orientations of two proteins and the energy of the interaction. In other scenarios when prior experimental evidence is not available, docking may be the only way to provide a quick solution to an urgent task. For these reasons, molecular docking constitutes a critical component of bioinformatics.
Currently, bioinformatics courses are available to students majoring in life sciences in most Chinese universities. However, most courses mainly focus on theory but ignore case practices. Therefore, during the COVID‐19 self‐isolation period, we conducted an online workshop focusing on molecular docking. In this workshop, four sophomores and one graduate student collaborated to systematically compare different open‐source online docking programs using the RBD–ACE2 complex as a benchmark. The students not only mastered most of the basic usage of the molecular visualization program, PyMOL but also learned how to perform docking operations using different online programs. They also learned to use Python scripts to calculate the root mean square deviations (RMSDs), which is a standard quantitative measure of similarity between two protein structures. 14 , 15 Throughout the online workshop, students not only deepened their knowledge of bioinformatics but also gained teamwork skills through collaboration and discussion with each other. We envision that in the context of the COVID‐19 pandemic, this teaching approach will provide a great example of distance education and inspire other educators.
2. MATERIALS AND METHODS
2.1. Docking procedure
In each docking program, the preprocessed RBD and ACE2 structure files were uploaded to a specific server, and results were retrieved through emails. For ZDOCK, we conducted blind docking and constraint docking with ZDOCK server 3.0.2 (http://zdock.umassmed.edu/). For SwarmDock, we conducted blind docking, using Particle Swarm Optimization mode (PSO mode) and Normal mode in SwarmDock server 15.04.01 (https://bmm.crick.ac.uk/~svc-bmm-swarmdock/submit.cgi). For HDock, we conducted template‐free docking and template‐based docking in HDock server (http://hdock.phys.hust.edu.cn/). For ClusPro, we used four restraint modes, namely Balanced, Hydrophobic‐favored, Electrostatic‐favored, and Van der Waals & electrostatics, to conduct docking in ClusPro 2.0 server (https://cluspro.org). For pyDockWEB, we used user‐defined constraint docking in pyDockWEB server (https://life.bsc.es/pid/pydockweb/). For HADDOCK, we defined the constraint residues as active residues and all solvent accessible neighbor residues as passive residues. After defining the active and passive residues, we performed single‐constraint and double‐constraint docking in HADDOCK 2.4 server (https://www.bonvinlab.org/software/haddock2.4/). For PatchDock, the surface of the two molecules was first divided into patches according to their geometric shapes, which were then matched to give candidate complex structures using the built‐in shape matching algorithms in PatchDock server (http://bioinfo3d.cs.tau.ac.il/PatchDock/). All the results were categorized and further manually inspected in PyMOL.
2.2. Automated RMSD calculations
RMSD is used to quantify the quality of reproduction of a known structure by a computational method, such as homology modeling and docking. RMSD is defined by the standard deviation of the difference in atom positions between the predicted structure and the crystal structure (Equation 1), which can be obtained in PyMOL using the align command.
| (1) |
In Equation (1), N is the total number of atoms, i is the index of each atom, and d i is the distance between atom i and its equivalent position in the crystal structure.
Calculating RMSDs of a considerable number of predicted structures is repetitive and monotonous, so we wrote a Python script to run in PyMOL for automated RMSD calculation. The script imports the built‐in os python module to list PDB files in the target folder. The script uses the cmd.load function from the pymol module to load the predicted structures into PyMOL. Then, the program uses cmd.align to align the predicted structure to the crystal structure (PDB: 6LZG) and calculates the RMSD value. Finally, it repeats the aforementioned steps until all the PDB files are analyzed. Between each cycle, the cmd.reinitialize function from pymol module is applied to refresh PyMOL in order to prevent interference from the last structure.
2.3. Programming
Anaconda distribution of Python 3.7.3 and Visual Studio Code editor were used to program all the python scripts for automated RMSD calculation. Figures were generated by ggplot2 3.3.2 package with R language 3.5.3.
3. RESULTS AND DISCUSSION
3.1. Workflow of docking
In this project, we evaluated the performance of several docking programs by comparing the predicted structure of the RBD–ACE2 complex with its crystal structure (PDB: 6LZG). The workflow encompasses four steps (Figure 1). In step 1, students retrieved the crystal structure of RBD–ACE2 from the RCSB Protein Data Bank. Then, they used PyMOL to process the complex structure and separated the RBD and ACE2 structure into individual files. These individual files were then used as input for docking. In step 2, students attempted to identify important interacting residues at the RBD–ACE2 complex interface by analyzing the forces at the complex interface and by mining the literature. These interacting residues were used as constraints in the following docking procedures. In step 3, students learned to use ZDOCK, a typical docking program, to predict possible structures of the RBD–ACE2 complex with or without constraints. In step 4, each student was assigned to evaluate several different docking programs. In this project, seven popular docking programs (ZDOCK, 16 ClusPro, 17 , 18 , 19 HDock, 20 HADDOCK, 21 SwarmDock, 22 pyDockWeb, 23 and PatchDock 24 ) were selected according to their citation ranking in Google Scholar. The complex structures predicted by the different programs were compared. At the end of each step, each student prepared a report and shared it weekly via online meetings.
FIGURE 1.
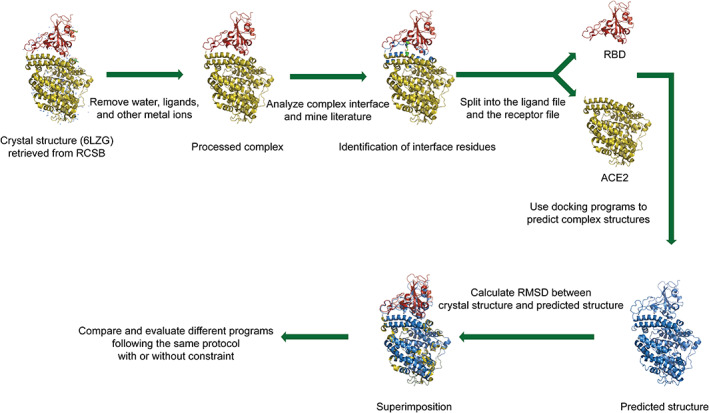
Workflow of the docking‐based training program. In the cartoon representation of the RBD–ACE2 crystal structure, the ACE2 is colored in olive and the RBD is colored in red, and the interface residues are colored in green. The predicted complex structure is colored in blue. Through aligning the predicted structure and the crystal structure, we calculated the RMSDs and then evaluated the performance of different docking programs
3.2. Acquisition and visualization of the RBD–ACE2 complex structure
The Protein Data Bank (http://www.rcsb.org/) is an open‐source archive for structures of proteins and nucleic acids. The structures are mostly acquired from X‐ray crystallography, NMR spectroscopy, and Cryo‐EM. 25 Students were informed that a considerable amount of protein structures are publicly available, and they are supposed to search for open‐source data to support their study in the future. As a test case, the structure of the RBD–ACE2 complex is retrieved from the Protein Data Bank (PDB: 6LZG). Typically, protein structures are stored in a specific file format with the .pdb suffix. This file format is a readable text file that contains thousands of lines. Students can directly open it with a text editor, such as Notepad and Microsoft Word, to check its anisotropic temperature factors, atom coordinates, connectivity records, conformation information, etc. Furthermore, students were informed that dedicated python scripts could process PDB files to fulfill specific needs, such as changing the chain index.
PDB files can also be processed by software with a graphical user interface, such as PyMOL, VMD, UCSF Chimera, and Mastero, of which PyMOL is known for its user‐friendly graphical interface and outstanding rendering ability. Mastering PyMOL is thus critical for researchers. Students learned to perform basic operations on protein structures such as moving, zooming, rotating, and aligning two structures with PyMOL. To prepare for docking, students first removed the water, ligands, and metal ions from the protein structure in PyMOL to avoid interference. Then, students split the RBD–ACE2 complex into two separated files with the Selection and Save functionality in PyMOL. These files were used as input for the following docking procedure.
3.3. Analysis of RBD–ACE2 interaction interface to obtain possible docking constraints
To prepare constraints for docking, we further analyzed the RBD–ACE2 complex interface. After loading the PDB file in PyMOL, students learned to measure the distance between two atoms in a protein structure (Figure 2). Students used Action‐Find functionality to identify critical interacting residues at the complex interface by setting a cutoff value. The interacting residues within the cutoff distance were further categorized by the type of interactions (Table 1). Students used the Show functionality to visualize and color bonds, atoms, and chains. Students also learned to deploy open‐source Python scripts to highlight the interface residues. By mapping the identified interactions to the structure, we picked 13 interacting residue pairs as constraints for docking from three regions of the RBD–ACE2 interface. Notably, three identified residues in region 3 including K353 on ACE2, Q493, and N401 on RBD (marked with * in Table 1) were reported to abolish the interaction between RBD and ACE2 upon mutations. 26
FIGURE 2.
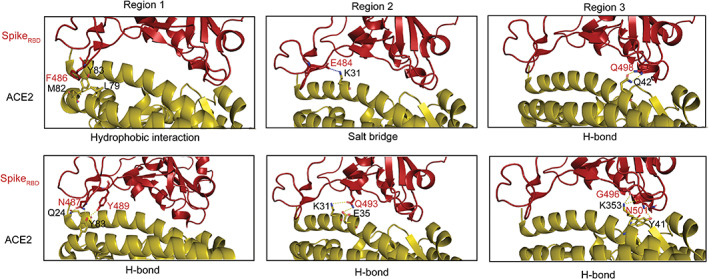
Identification of RBD–ACE2 complex interface residues. Residues from RBD are labeled in red and those from ACE2 in black. The interface residues distribute in three regions. Region 1 encompasses hydrophobic interactions between residue pairs F486‐M82 and Y83‐L79, and H‐bonds between N487‐Q24 and Y489‐Y83. Region 2 shows a salt bridge between E484 and K31, as well as two H‐bonds formed between Q493 and residues K31/E35. Region 3 shows three H‐bonds between Q498‐Q42, G496‐K353, and N501‐Y41, respectively
TABLE 1.
Docking constraints chosen by identifying the interactions at the RBD–ACE2 interface
| Constraint ID | RBD | ACE2 | Type of interaction |
|---|---|---|---|
| 1 | F486 | M82 | Hydrophobic |
| 2 | F486 | Y83 | Hydrophobic |
| 3 | F486 | L79 | Hydrophobic |
| 4 | Y489 | Y83 | H‐bond |
| 5 | N487 | Y83 | H‐bond |
| 6 | N487 | Q24 | H‐bond |
| 7 | E484 | K31 | Salt bridge |
| 8 | Q493* | K31 | H‐bond |
| 9 | Q493* | E35 | H‐bond |
| 10 | Q498 | Q42 | H‐bond |
| 11 | N501* | Y41 | H‐bond |
| 12 | N501* | K353* | H‐bond |
| 13 | G496 | K353* | H‐bond |
3.4. Predicting the complex structure using ZDOCK
ZDOCK is one of the popular docking programs in academia for its usability, robustness, and cherished history. We first conducted blind docking using ZDOCK. Except for one predicted structure with RMSD = 1.8 Å, RMSDs of the complex structures generated by blind docking ranges from 31.4 to 36.2 Å, which is generally not acceptable. We therefore continued to perform one‐constraint docking operations using the 13 identified constraints. Then, we aligned each of the predicted complex structures to the crystal structure of RBD–ACE2 and calculated the RMSD. Here, according to a previously reported standard, we consider the crystal structure is accurately reproduced in a specific prediction if the RMSD value is less than 3 Å. 27
Docking with one constraint improved the best RMSD to 1.01 Å. ZDOCK reports the top 10 complex structures in a ranked order that is based on a built‐in empirical force field. 14 However, because the RMSD values are not consistent with the rank order reported by ZDOCK, we set up a standard to evaluate which constraint performs better to obtain a more accurate structure in ZDOCK. First, we calculated the RMSDs of the 10 predicted structures obtained by using each constraint. Then, for those with RMSD values less than the 3 Å cutoff, we summed up their rank number reported by ZDOCK to obtain the accumulated rank sum. The smaller the accumulated rank‐sum, the higher the ranking of these structures in ZDOCK, which indicated that this constraint could better guide the prediction by ZDOCK. We found that except for constraint ID 11 and 12, other constraint ID docking results all yielded three results with RMSD less than 3 Å to mostly reproduce the crystal structure. In addition, constraints 1–6 performed better than others (Figure 3a).
FIGURE 3.
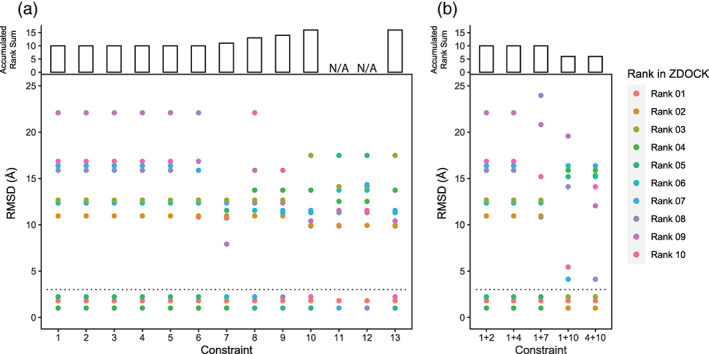
ZDOCK performance in predicting RBD–ACE2 complex with single constraint (a) and double constraints (b). (a) single‐constraint docking in ZDOCK. A dashed line on RMSD = 3.0 represents the cutoff. The predicted complex structures are considered valid and used for comparison if RMSD is smaller than 3.0. (b) Double‐constraint docking in ZDOCK. Constraint pairs were generated by combining two constraints of ID 1–13
We further tested whether docking with double constraints can further improve the predictions. Five constraints were carefully chosen based on their positions, types of the bond, and their one‐constraint‐docking performance. However, we found that docking with double constraints has a marginal advantage over one‐constraint docking (Figure 3b). Nevertheless, constraint pairs, 1 + 10 and 4 + 10, are better compared to others, which might imply that distant H‐bonds constraints are more helpful to obtaining better ZDOCK docking results.
3.5. Predicting the complex structure using other docking programs
After we successfully performed a docking trial in ZDOCK as a group, we further trained students to explore more docking programs individually. Each student was assigned to one or two docking programs. Students were informed that each program has its own online manual, which they should refer to and study. Students should note that docking programs typically have different preferences for protein files, and they should read the manuals carefully and follow the instructions to modify PDB files to meet the program's requirements if necessary. After obtaining the predicted structures from different online docking programs, students wrote a Python script to automate the calculation of the RMSDs between the crystal structure and predicted structures. The overall procedure is summarized in Figure 4, using HADDOCK as an example.
FIGURE 4.
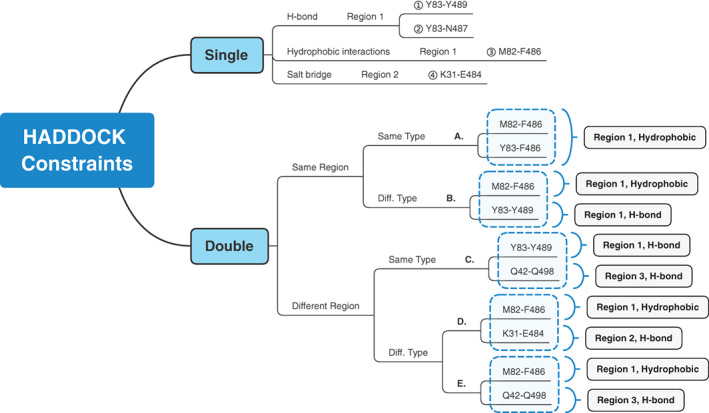
Detailed HADDOCK procedure exemplified docking in other programs. In HADDOCK, we used single‐constraint docking and double‐constraint docking. By distinguishing the interaction types and interaction regions, we obtained different results. Students also practiced other docking programs following customized procedures similar to the HADDOCK procedure
SwarmDock requires the input of all the residues constituting the binding site because the server only produces solutions in that region defined by users. The performance under the PSO mode was better than the Normal mode, while docking with constraints performed better than blind docking. In general, it performed better than ZDOCK.
HDock was performed under both template‐dependent and template‐free procedures. We found that all the minimum RMSDs of template‐free docking are smaller than the minimum RMSDs of the template‐dependent procedure.
ClusPro provides users with four types of energy minimization: Balanced, Hydrophobic‐favored, Electrostatic‐favored, and Van der Waals & electrostatics for docking. Results showed that H‐bond and salt bridge outperformed other types.
pyDockWEB gave better predictions using the double‐constraint strategy than the single‐constraint strategy. In general, we found that pyDockWEB performed better than ZDOCK.
HADDOCK results showed that there is no accurate predicted complex from single‐constraint docking. Double‐constraint docking results are generally better than single‐constraints docking. The best RMSD value reached 0.56 Å.
PatchDock with blind docking methods yielded no accurate predicted complex (the best RMSD = 11.87 Å). With the binding site‐constraint algorithm, none of the binding site constraint strategies achieved reliable docking results. However, with the distance‐constraint algorithm using carefully designed atom distance, PatchDock can produce better results (RMSD = 0.95 Å).
4. STUDENTS' PARTICIPATION AND DISCUSSION
Four undergraduate students participated in this project with the assistant of a graduate student. They learned how to retrieve open‐source resources, basic principles of molecular docking, the usage of PyMOL, and some Python programming skills. In the context of COVID‐19, rather than panic about the number of infections, students learned to stay calm and tried to join the cutting‐edge research on SARS‐CoV‐2. Unlike on‐scene experiments, molecular docking and literature review were not affected by quarantine or isolation. Through online meetings, students and their supervisors kept sharing their results and checking the status of each other. The online workshop was proven to be a feasible way of teaching under unconventional conditions.
5. CONCLUDING REMARKS AND PERSPECTIVES
Bioinformatics is still in its infancy. Through this docking benchmark, we found that no docking program can achieve the same level of accuracy compared to the experimental results in blind docking. Nevertheless, there may be some silver lining that some programs did predict decent complex structures under the use of defined constraints. Although this swift procedure still cannot outperform experimental methods to provide emergent support against the pandemic, from another standpoint, docking expands our understanding of how RBD can possibly interact with ACE2 and reveals many energetically possible conformations that the complex may adopt. Blocking certain interactions may be the key to drug design and vaccine development. With a tighter combination of experimental biology and computational biology, we believe that progressive algorithms and rapidly growing compute power will completely change our understanding of disease and cure. Also, docking provides perfect introductory materials for bioinformatics teaching. It spans structural biology, bioinformatics, and biochemistry, which can broaden students' insight and help nurture future researchers.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
AUTHOR CONTRIBUTIONS
Shu Quan conceived the study. Wenxuan Fan and Jun Mencius designed and guided the whole docking workflow. Wenxuan Fan, Wenjing Du, Hongjin Zhu, and Huangyunxian Fan conducted the docking experiments. Wenxuan Fan, Jun Mencius, Wenjing Du, and Shu Quan prepared the figures. All authors participated in the writing the manuscript. All authors read and approved the final manuscript.
ACKNOWLEDGMENTS
This work was supported by the “Shanghai First‐class Undergraduate Courses” program, the “Shanghai Exemplary all‐English Teaching Undergraduate Courses” program, the “Prominent Educator” project in East China University of Science and Technology, and the National Natural Science Foundation of China (NSFC) (Grant Nos. 31661143021 and 31670802 to S. Quan). The authors also appreciate the assistance from Alibaba Cloud during the isolation of COVID‐19.
Fan W, Mencius J, Du W, Fan H, Zhu H, Wei D, et al. Online bioinformatics teaching practice: Comparison of popular docking programs using SARS‐CoV‐2 spike RBD–ACE2 complex as a benchmark. Biochem Mol Biol Educ. 2021;49:833–840. 10.1002/bmb.21566
Wenxuan Fan and Jun Mencius contributed equally to this work.
Funding information National Natural Science Foundation of China, Grant/Award Numbers: 31661143021, 31670802
REFERENCES
- 1. Li Q, Guan XH, Wu P, Wang XY, Zhou L, Tong YQ, et al. Early transmission dynamics in Wuhan, China, of novel coronavirus–infected pneumonia. N Engl J Med. 2020;382:1199–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Huang CL, Wang YM, Li XW, Ren LL, Cao B. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395:497–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zhu N, Zhang DY, Wang WL, Li XW, Yang B, Song JD, et al. A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med. 2020;382:727–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zhong NS, Zheng BJ, Li YM, Poon LLM, Xie ZH, Chan KH, et al. Epidemiology and cause of severe acute respiratory syndrome (SARS) in Guangdong, People's Republic of China, in February, 2003. Lancet. 2003;362:1353–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zaki AM, Sander VB, Bestebroer TM, Oserhaus ADME, Fouchier RAM. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N Engl J Med. 2012;367:1814–20. [DOI] [PubMed] [Google Scholar]
- 6. Walls AC, Tortorici MA, Snijder J, Xiong XL, Veesler D. Tectonic conformational changes of a coronavirus spike glycoprotein promote membrane fusion. PNAS. 2017;114:11157–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hoffmann M, Kleine‐Weber H, Schroeder S, Krüger N, Phlmann S. SARS‐CoV‐2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181:271–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yang JS, Petitjean SJL, Koehler M, Zhang QR, Dumitru AC, Chen WZ, et al. Molecular interaction and inhibition of SARS‐CoV‐2 binding to the ACE2 receptor. Nat Commun. 2020;11:4541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Benítez‐Cardoza CG, Vique‐Sánchez JL. Potential inhibitors of the interaction between ACE2 and SARS‐CoV‐2 (RBD), to develop a drug. Life Sci. 2020;256:117970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Han YX, Král P. Computational design of ACE2‐based peptide inhibitors of SARS‐CoV‐2. ACS Nano. 2020;14:5143–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yan RH, Zhang YY, Li YN, Xia L, Zhou Q. Structural basis for the recognition of SARS‐CoV‐2 by full‐length human ACE2. Science. 2020;367:1444–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rosell M, Fernández‐Recio J. Docking approaches for modeling multi‐molecular assemblies. Curr Opin Struct Biol. 2020;64:59–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Led P, Caflisch A. Protein structure‐based drug design: from docking to molecular dynamics. Curr Opin Struct Biol. 2017;48:93–102. [DOI] [PubMed] [Google Scholar]
- 14. Kabsh W. A discussion of the solution for the best rotation to relate two sets of vectors. Acta Crystallogr A. 1978;34:827–8. [Google Scholar]
- 15. Zhang Y, Skolnick J. TM‐align: a protein structure alignment algorithm based on the TM‐score. Nucleic Acids Res. 2005;33:2302–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pierce BG, Wiehe K, Hwang H, Kim BH, Vreven T, Weng Z. ZDOCK server: interactive docking prediction of protein–protein complexes and symmetric multimers. Bioinformatics. 2104;30:1771–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Xia B, Vajda S, Kozakov D. Accounting for pairwise distance restraints in FFT‐based protein–protein docking. Bioinformatics. 2016;32:3342–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kozakov D, Hall DR, Xia B, Porter KA, Dzmitry P, Christine Y, et al. The ClusPro web server for protein–protein docking. Nat Protoc. 2017;12:255–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kozakov D, Beglov D, Bohnood T, Mottarella SE, Vajda S. How good is automated protein docking? Proteins. 2013;81:2159–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yan Y, Tao HY, He JH, Huang SY. The HDOCK server for integrated protein–protein docking. Nat Prot. 2020;15:1829–52. [DOI] [PubMed] [Google Scholar]
- 21. Zundert VGCP, Rodrigues JPGLM, Trellet M, Schmitz C, Vries SJD. The HADDOCK2.2 web server: user‐friendly integrative modeling of biomolecular complexes. J Mol Biol. 2016;428:720–5. [DOI] [PubMed] [Google Scholar]
- 22. Torchala M, Moal IH, Chaleil RAG, Fernandez‐Recio J, Bates PA. SwarmDock: a server for flexible protein–protein docking. Bioinformatics. 2013;29:807–9. [DOI] [PubMed] [Google Scholar]
- 23. Jiménez‐García B, Pons C, Fernández‐Recio J. pyDockWEB: a web server for rigid‐body protein–protein docking using electrostatics and desolvation scoring. Bioinformatics. 2013;29:1698–9. [DOI] [PubMed] [Google Scholar]
- 24. Schneidman‐Duhovny D, Inbar Y, Nussinov R, Wolfson HJ. PatchDock and SymmDock: servers for rigid and symmetric docking. Nucleic Acids Res. 2005;33:W363–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. wwwPDB consortium . Protein Data Bank: the single global archive for 3D macromolecular structure data. Nucleic Acids Res. 2019;47:D520–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Shang J, Ye G, Shi K, Wan YS, Luo CM, Aihara H, et al. Structural basis of receptor recognition by SARS‐CoV‐2. Nature. 2020;581:221–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hevener KE, Zhao W, Ball DM, Babaoglu K, Qi JJ, White SW, et al. Validation of molecular docking programs for virtual screening against dihydropteroate synthase. J Chem Inf Model. 2009;49:444–60. [DOI] [PMC free article] [PubMed] [Google Scholar]