

Abstract
There is an urgent need to develop antiviral drugs and alleviate the current COVID‐19 pandemic. Herein we report the design and construction of chimeric oligonucleotides comprising a 2′‐OMe‐modified antisense oligonucleotide and a 5′‐phosphorylated 2′‐5′ poly(A)4 (4A2‐5) to degrade envelope and spike RNAs of SARS‐CoV‐2. The oligonucleotide was used for searching and recognizing target viral RNA sequence, and the conjugated 4A2‐5 was used for guided RNase L activation to sequence‐specifically degrade viral RNAs. Since RNase L can potently cleave single‐stranded RNA during innate antiviral response, degradation efficiencies with these chimeras were twice as much as those with only antisense oligonucleotides for both SARS‐CoV‐2 RNA targets. In pseudovirus infection models, chimera‐S4 achieved potent and broad‐spectrum inhibition of SARS‐CoV‐2 and its N501Y and/or ΔH69/ΔV70 mutants, indicating a promising antiviral agent based on the nucleic acid‐hydrolysis targeting chimera (NATAC) strategy.
Keywords: antisense oligonucleotides, pseudoviruses, ribonuclease L, SARS-CoV-2, spike
Based on the strategy of nucleic acid hydrolysis targeting chimera, 5′‐phosphorylated 2′‐5′‐poly(A)4‐modified chimeric oligonucleotides were developed for enhanced degradation of the envelope or spike RNA of SARS‐CoV‐2 in vitro. The oligonucleotides can recognize specific viral RNA sequences and 2′‐5′ poly(A)4 can activate RNase L to cleave them specifically.

Severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) has continued to spread globally and caused the COVID‐19 pandemic since 2019. [1] This situation demands more preclinical drug candidates for further development of antiviral agents. As structural proteins of SARS‐CoV‐2, envelope (E), spike (S) and membrane (M) proteins assemble viral membranes in infected host cells, [2] thus becoming ideal drug targets to intervene virus proliferation.
RNase L participates in innate antiviral response of vertebrate cells by cleaving UN^N sites located in viral or cellular single‐stranded RNA (ssRNA). Cytoplasmic RNase L monomer only displays weak catalytic cleavage on the substrate. However, upon dimerization induced by its specific ligand 5′ phosphorylated 2′‐5′ polyA (such as 4A2‐5), RNase L is highly activated and performs intense RNA cleavage. [3] Ubiquitous activation of RNase L might cause widespread attenuation of basal mRNA and possible cell apoptosis, especially at high doses of 4A2‐5. [4] Otherwise, guided and controlled activation of RNase L could achieve more specific target RNA degradation.
RNA binding small molecules conjugated with RNase L recruiter such as 4A2‐5 have been recently designed to selectively target highly‐structured microRNA or RNA fragments of viral genomes [5] aided by the online Inforna computational server. [6] High lipophilicity of these small molecules facilitates their cellular uptake [5a] and tissue penetration, [7] while the accessibility to discover such a chemical structure to selectively bind RNA region of interest is relatively limited. As an alternative approach, sequence‐selective antisense oligonucleotides (ASOs) are more available and have broader application to bind pathogenic RNA transcripts currently not targetable by small molecules. After binding, ASO can inhibit expression of target RNA through various mechanisms such as RNase‐H1 mediated cleavage and steric interference. [8] Since the natural anionic phosphate backbone of ASO hinders its efficient tissue delivery, multiple approaches seeking to address this challenge have been developed such as bioconjugation and chemical modification. [9] As common modification approaches, phosphorothioate (PS) and 2′‐O‐methyl (2′‐OMe) can further promote ASO nuclease resistance and/or binding affinity. [9] Currently nine ASO drugs have been approved to treat rare diseases, [8] and two ASO candidates have entered clinical phase II to treat lung diseases. [8] One of them (Eluforsen) also utilized full PS backbone and full 2′‐OMe modification to alleviate lung cystic fibrosis. [10] In the early nineties the Silverman group have raised the possibility to combine ASO with 4A2‐5 for targeted viral RNA decay, such as human immunodeficiency virus. [11] Therefore, it is promising to develop ASO‐4A2‐5 chimera to target SARS‐CoV‐2 genomic RNA and inhibit viral infection based on nucleic acid‐hydrolysis targeting chimera (NATAC) strategy (Scheme 1).
Scheme 1.
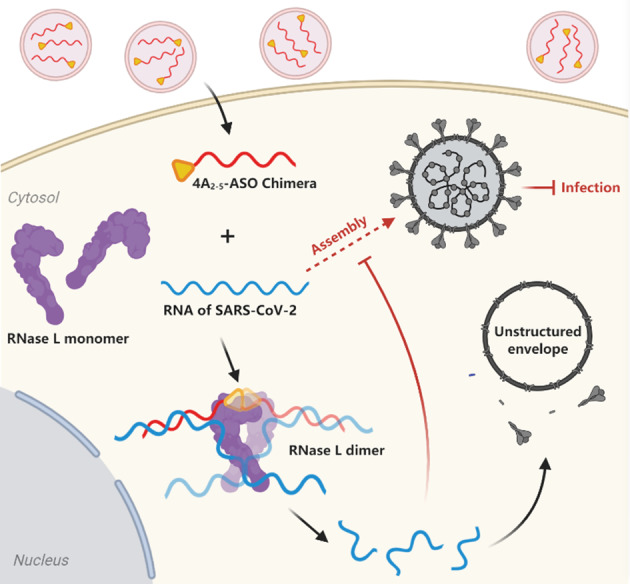
Schematic representation of 4A2‐5‐ASO chimera induced inhibition of SARS‐CoV‐2 proliferation. Black arrows: processing of viral RNA upon treatment; dashed arrow: inhibited viral assembly.
Here, we designed and selected chimeric antisense oligonucleotides targeting envelope (E‐) or spike (S‐) RNA of SARS‐CoV‐2. In vitro E‐RNA cleavage by RNase L and target RNA knockdown was first evaluated. Then in the pseudotyped SARS‐CoV‐2 infection model, ASO‐4A2‐5 chimeras targeting S‐RNA successfully inhibited pseudovirus packaging and further infection of host cells. One of these chimeras also effectively inhibited three SARS‐CoV‐2 pseudovirus mutants, including N501Y, ΔH69/ΔV70, and the recently discovered dual‐site mutations with higher transmission ability.
Our study began with the selection of complementary oligonucleotide sequences for specific RNA targets in the SARS‐CoV‐2 genome. After predicting RNA secondary structures of spike receptor binding domain (S‐RBD) and envelope (E) protein of SARS‐CoV‐2, loops composed of more than 10 nucleotides were selected as ideal target regions. In addition, considering the space required for RNase L activation and substrate cleavage, stem structure in 3′ proximity of the selected loop was limited to have less than 4 base pairs, and its 3′ pairing end should have more than 1 RNase L cleavage site (UN^N) in a bulge structure. Oligonucleotide sequences predicted with more than 70 % success probability and complementary to the selected loops were synthesized by solid phase synthesis (Table S1). Since 2′‐OMe modification of ASO is not compatible with RNase H activity, [8] PS backbone and 2′‐OMe were incorporated to avoid RNase H‐mediated target cleavage and to improve oligonucleotide nuclease resistance. A 5′ terminal poly 2′‐5′ poly(A)4 ligand (4A2‐5) was conjugated through a short PEG linker (Figure 1 A).
Figure 1.

Targeted degradation of SARS‐CoV‐2 envelope RNA (E‐RNA) with RNase L participation in vitro. A) Structures of 4A2‐5‐ASO chimera targeting E‐RNA (Chimera‐E). B) RNase L cleavage assay of a 3′‐Cy3‐labeled E‐RNA segment. C, D) Relative levels of E‐RNA and RNase L mRNA in Vero cells upon 24 h treatment, as measured by RT‐qPCR. Data represent mean ± s.e.m. (n≥3). *P<0.033, **P<0.002, ***P<0.001 as measured by a two‐tailed Student's t test.
We first tested RNase L recruitment ability of Chimera‐E‐PO that is complementary to a loop structure of Cy3‐labeled partial E‐RNA of SARS‐CoV‐2 (Table S1). In vitro cleavage assay showed RNase L alone or addition of ASO‐E did not cleave substrate RNA, while additional Chimera‐E‐PO treatment activated RNase L and produced cleavage bands in a manner different from that of 4A2‐5 treated group at 375 nM (Figure 1 B).
Cleavage preferences of Chimera‐E‐PO for these specific cleavage sites indicated its specific binding to RNA substrate. At 750 nM, 4A2‐5 could induce so intense E‐RNA cleavage that short RNA fragments as cleavage products couldn't be retained and analyzed in this 12.5 % denaturing PAGE gel. We then evaluated E‐RNA degradation in Vero cells using Chimera‐E with a full‐length PS backbone to further improve the nuclease resistance and chemical stability in vitro. Treatment of 20 nM ASO‐E alone only downregulated E‐RNA level to 83 %, while the treatment of 20 nM Chimera‐E downregulated the E‐RNA level to 35 % (Figure 1 C). This improved degradation efficiency induced by Chimera‐E was attributed to intracellular RNase L activation. Transcription level of RNase L was also elevated when further increasing Chimera‐E concentration (Figure 1 D), which might result from some regulatory mechanisms amplifying RNase L activity to enhance cellular immune response. However, at 320 nM, Chimera‐E and ASO‐E showed similarly potent effects on E‐RNA degradation. Since ASO‐E alone could induce intense target RNA silencing through post‐RNA‐binding mechanisms at such a high concentration, [8] Chimera‐E showed no improvement of E‐RNA degradation via RNase L compared with that of ASO‐E. Therefore, we chose concentrations below 80 nM in the following evaluation. These promising results inspired us to choose spike protein, a more promising target to inhibit SARS‐CoV‐2 infection.
We then evaluated the on‐target effects of three previously designed chimeric oligonucleotides (Table S1) against S‐RNA. All chimeras (Chimera‐S) and antisense oligonucleotides without 4A2‐5 conjugation (ASO‐S) decreased S‐RNA down to less than 50 % level at 80 nM (Figure 2 A). More than 2‐fold enhancement of efficiency was also observed for Chimera‐S compared with ASO‐S. Furthermore, RNase L transcription levels were elevated with Chimera‐S treatment, while ASO‐S didn't change it obviously (Figure 2 B). In a pseudotyped SARS‐CoV‐2 infection model (Figure 2 C), each Chimera‐S was applied to HEK293T packaging cells. Pseudoviral titration was performed by measuring two reporter genes, GFP and firefly luciferase in HEK293T‐hACE2 host cells. At 40 nM and 80 nM, Chimera‐S4 treatment reduced the firefly luminescence to 24 % and 6 % respectively, more potent than Chimera‐S5 (45 % and 14 %) and Chimera‐S6 (50 % and 28 %) (Figure 2 D). Results of GFP measurement were consistent with luciferase assay in HEK293T‐hACE2 cells (Figure 2 E). Meanwhile, the scrambled oligonucleotide showed no inhibitory effect on pseudovirus, thus confirming the on‐target effect of Chimera‐S. Similar GFP fluorescence in HEK293T cells indicated similar transfection efficiencies of plasmids in the first step between groups (Figure S2).
Figure 2.

Screening the most effective 4A2‐5‐ASO chimeric oligonucleotides to target spike RNA (S‐RNA) of SARS‐CoV‐2. A, B) Relative transcription level of S‐RNA and RNase L mRNA upon 24 h treatment, as measured by RT‐qPCR. C) Experimental procedure to evaluate the inhibitory effect of oligonucleotides on virus infection in a pseudotyped SARS‐CoV‐2 model. D, E) Relative expression levels of firefly luciferase and GFP in infected HEK293T‐hACE2 cells. Negative control (NC): group transfected with only virus‐constructing plasmids. Scramble: group treated with the plasmids and a nonsense oligonucleotide. Blank: group without exogenous transfection. Scale bar=100 μm. Data represent mean ± s.e.m. (n≥3). *P<0.033, **P<0.002, ***P<0.001 as measured by a two‐tailed Student's t test.
Since Chimera‐S4 showed more efficient inhibition of spike RNA transcription and virus packaging/infection than Chimera‐S5 and Chimera‐S6, we further investigated its concentration dependence of S‐RNA degradation and pseudovirus inhibition. Chimera‐S4 induced a reduction of S‐RNA up to 80 % at 20 nM. Increasing its concentration to 80 nM only led to slight enhancement of S‐RNA reduction, but would cause an approximately 2‐fold up‐regulation of RNase L expression (Figure 3 A, 3B). Surprisingly, the titers of SARS‐CoV‐2 pseudovirus dropped sharply from 60 % to 6 % when the concentration of Chimera‐S4 increased from 20 to 80 nM (Figure 3 C). In comparison to the individual ASO‐S4 and 4A2‐5, Chimera‐S4 degraded S‐RNA in Vero cells with up to 4.5‐ and 2.1‐fold higher efficiency at 40 nM concentration (Figure 3 A). Compared with physically mixed 4A2‐5 and ASO‐S4 (4A2‐5 + ASO‐S4), Chimera‐S4 led to a similar reduction profile of S‐RNA from 20 nM to 80 nM. However, Chimera‐S4 displayed 2.2‐ and 6.1‐fold higher inhibitory effects on viral titers at 40 nM and 80 nM than those of 4A2‐5 + ASO‐S4 group (Figure 3 C). In the infection model, the positive rate of GFP fluorescent cells treated with 40 nM Chimera‐S4 was 29.07 %, indicating the lowest viral titer at 40 nM (Figure 3 D), and could be further enhanced at higher concentrations (Figure S2). Fluorescent images of the infected HEK293T‐hACE2 cells were consistent with the results of flow cytometry (Figure 3 E, Figure S2). Similarly, GFP fluorescence in HEK293T packaging cells confirmed the consistency of transfection efficiency across different groups (Figure S2).
Figure 3.

Chimeric‐S4 can effectively degrade SARS‐CoV‐2 S‐RNA and inhibit pseudoviral infection in vitro. A, B) Concentration‐dependent degradation of S‐RNA and increase of RNase L mRNA in Vero cells upon 24 h treatment. Firefly luminescence (C), cytometry analysis of GFP signals (D, 40 nM), GFP fluorescence images (E, 40 nM) in HEK293T‐hACE2 cells after 48 h infection of the collected SARS‐CoV‐2 pseudovirus. Negative control (NC): transfection of virus‐constructing plasmids. Blank: without exogenous transfection. 4A2‐5 + ASO‐S4: co‐transfection of virus‐constructing plasmids and physically mixed 4A2‐5 and ASO‐S4 with each final concentration of 20 nM, 40 nM and 80 nM. Scale bar=100 μm. Data represent mean ± s.e.m. (n≥3). *P<0.033, **P<0.002, ***P<0.001 as measured by a two‐tailed Student's t test.
Therefore, Chimera‐S4 could efficiently reduce S‐RNA level and inhibit SARS‐CoV‐2 pseudovirus at moderate doses, without severely damage to cell status or viability (Figure S2, Figure S3). Low concentrations of 4A2‐5 component of Chimera S4 could also lower the risk of ubiquitous basal RNA decay caused by 4A2‐5 induced RNase L activation.[ 4 , 12 ] In addition, Chimera‐S4 increased the relative mRNA levels of IFN‐β and IL‐6 in A549 cells up to 5.9‐ and 2.0‐ fold, respectively at 40 nM, both higher than those of individual 4A2‐5, ASO‐S4, and 4A2‐5 + ASO‐S4 mixture (Figure S3). The result was consistent with literature and demonstrated that RNase L activation could stimulate interferon (IFN) production, [13] which in turn induces positive feedback on expression of interferon stimulated genes (ISGs) including RNase L, to enhance antiviral response.[ 13b , 14 ]
Mutations ΔH69/ΔV70 and N501Y on the spike protein of SARS‐CoV‐2 have been reported to cause S‐gene target failure (SGTF) and greatly increase viral transmissibility. [15] To assess the broad‐spectrum inhibitory effect on SARS‐CoV‐2 mutants, Chimera‐S4 was then co‐transfected with pseudovirus packaging plasmids carrying ΔH69/ΔV70, N501Y or dual‐site mutations into HEK293T packaging cells. Transfection efficiencies in HEK293T cells across different groups were consistent (Figure S5). The luciferase assay showed that titers of all three mutants were reduced to less than 20 % after 48 hours treatment of 40 nM Chimera‐S4 (Figure 4). GFP fluorescence analysis in infected HEK293T‐hACE2 cells also displayed the same inhibiting mode (Figure S4). Since mutation sites on S‐RNA corresponding to N501Y and ΔH69/ΔV70 are not overlaid with targeted region of Chimera‐S4, unaffected high potent inhibition of these mutants was still successfully achieved.
Figure 4.

Determination of the viral titer of SARS‐CoV‐2 mutants upon Chimera‐S4 treatment. Efficiently inhibited infection of three mutated SARS‐CoV‐2 pseudoviruses, N501Y, ΔH69/ΔV70 and their combined mutants (Dual) in HEK293T‐hACE2 cells after Chimera‐S4 treatment (40 nM, 48 h), as measured by luciferase assay.
In summary, we developed a group of 4A2‐5 chimeric oligonucleotides based on nucleic acid‐hydrolysis targeting chimera (NATAC) strategy, which successfully down‐regulated target SARS‐CoV‐2 RNAs. Among them, Chimera‐S4 showed the most potent ability to degrade S‐RBD RNA and to inhibit SARS‐CoV‐2 pseudovirus. Compared with classic ASO silencing strategy, Chimera‐S4 also activated RNase L, which significantly improved RNA degradation efficiency and induced additional antiviral immune responses after being recruited by 4A2‐5 ligand. This chimeric sequence still showed robust inhibiting capability toward three highly transmissible SARS‐CoV‐2 mutants involving N501Y and ΔH69/ΔV70 mutations. Since viruses can also evolve multiple mechanisms to evade RNase L activity and block IFN production, [16] such as a 2′,5′ phosphodiesterase (PDE) that can degrade native 4A2‐5 in murine coronavirus, [17] it is necessary to evaluate the antiviral efficiency of Chimera‐S4 modified with phosphorothioated 4A2‐5 ligand in a real SARS‐CoV‐2 infection model when biosafety level 4 was available in future. In addition, inhalation formulations have been reported to achieve deep lung deposition of naked nucleic acids in forms of inhaled powder or nanoparticles, [18] which helps to further deliver these exogenous oligonucleotides in vivo. Antisense oligonucleotides have been characterized by sequence‐specific targeting, convenient design and synthesis, which make this 4A2‐5‐ASO chimera suitable for further development of nucleic acid drugs combating foreseeable evolving COVID‐19 pandemics.
Conflict of interest
A patent application was filed (application No. 202110225438.X). The authors declare no other interests.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
This work was supported by National Natural Science Foundation of China (Grants No. 81821004, 21877001, 22077005), and National Major Scientific and Technological Special Project for “Significant New Drugs Development” (Grant No. 2017ZX09303013).
X. Su, W. Ma, D. Feng, B. Cheng, Q. Wang, Z. Guo, D. Zhou, X. Tang, Angew. Chem. Int. Ed. 2021, 60, 21662.
A previous version of this manuscript has been deposited on a preprint server (https://doi.org/10.1101/2021.03.04.433849).
References
- 1. Wu F., Zhao S., Yu B., Chen Y. M., Wang W., Song Z. G., Hu Y., Tao Z. W., Tian J. H., Pei Y. Y., Yuan M. L., Zhang Y. L., Dai F. H., Liu Y., Wang Q. M., Zheng J. J., Xu L., Holmes E. C., Zhang Y. Z., Nature 2020, 579, 265–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Keam S., Megawati D., Patel S. K., Tiwari R., Dhama K., Harapan H., Rev. Med. Virol. 2020, 30, e2123–e2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Han Y., Whitney G., Donovan J., Korennykh A., Cell Rep. 2012, 2, 902–913. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Burke J. M., Moon S. L., Matheny T., Parker R., Mol. Cell 2019, 75, 1203–1217; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4b. Rath S., Prangley E., Donovan J., Demarest K., Wingreen N. S., Meir Y., Korennykh A., Mol. Cell 2019, 75, 1218–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.
- 5a. Costales M. G., Matsumoto Y., Velagapudi S. P., Disney M. D., J. Am. Chem. Soc. 2018, 140, 6741–6744; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5b. Haniff H. S., Tong Y., Liu X., Chen J. L., Suresh B. M., Andrews R. J., Peterson J. M., O'Leary C. A., Benhamou R. I., Moss W. N., Disney M. D., ACS Cent. Sci. 2020, 6, 1713–1721; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5c. Costales M. G., Suresh B., Vishnu K., Disney M. D., Cell Chem. Biol. 2019, 26, 1180–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Disney M. D., Winkelsas A. M., Velagapudi S. P., Southern M., Fallahi M., Childs-Disney J. L., ACS Chem. Biol. 2016, 11, 1720–1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Costales M. G., Aikawa H., Li Y., Childs-Disney J. L., Abegg D., Hoch D. G., Pradeep Velagapudi S., Nakai Y., Khan T., Wang K. W., Yildirim I., Adibekian A., Wang E. T., Disney M. D., Proc. Natl. Acad. Sci. USA 2020, 117, 2406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Crooke S. T., Baker B. F., Crooke R. M., Liang X. H., Nat. Rev. Drug Discovery 2021, 20, 427—453. [DOI] [PubMed] [Google Scholar]
- 9. Roberts T. C., Langer R., Wood M. J. A., Nat. Rev. Drug Discovery 2020, 19, 673–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sermet-Gaudelus I., Clancy J. P., Nichols D. P., Nick J. A., De Boeck K., Solomon G. M., Mall M. A., Bolognese J., Bouisset F., den Hollander W., Paquette-Lamontagne N., Tomkinson N., Henig N., Elborn J. S., Rowe S. M., J. Cystic Fibrosis 2019, 18, 536–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.
- 11a. Cirino N. M., Li G., Xiao W., Torrence P. F., Silverman R. H., Proc. Natl. Acad. Sci. USA 1997, 94, 1937–1942; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11b. Torrence P. F., Maitra R. K., Lesiak K., Khamnei S., Zhou A., Silverman R. H., Proc. Natl. Acad. Sci. USA 1993, 90, 1300–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Donovan J., Rath S., Kolet-Mandrikov D., Korennykh A., RNA 2017, 23, 1660–1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.
- 13a. Luthra P., Sun D., Silverman R. H., He B., Proc. Natl. Acad. Sci. USA 2011, 108, 2118–2123; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13b. Manivannan P., Siddiqui M. A., Malathi K., J. Virol. 2020, 94, e00205–e00220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Keam S., Megawati D., Patel S. K., Tiwari R., Dhama K., Harapan H., Rev. Med. Virol. 2020, 30, e2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.
- 15a. Galloway S. E., Paul P., MacCannell D. R., Johansson M. A., Brooks J. T., MacNeil A., Slayton R. B., Tong S., Silk B. J., Armstrong G. L., Biggerstaff M., Dugan V. G., Morb. Mortal. Wkly. Rep. 2021, 70, 95–99; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15b. Zhao S., Lou J., Cao L., Zheng H., Chong M. K. C., Chen Z., Chan R. W. Y., Zee B. C. Y., Chan P. K. S., Wang M. H., J. Travel Med. 2021, 10.1093/jtm/taab011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.
- 16a. Deng X., Hackbart M., Mettelman R. C., O'Brien A., Mielech A. M., Yi G., Kao C. C., Baker S. C., Proc. Natl. Acad. Sci. USA 2017, 114, e4251–e4260; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16b. Ezelle H. J., Malathi K., Hassel B. A., Int. J. Mol. Sci. 2016, 17, 74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhao L., Jha B. K., Wu A., Elliott R., Ziebuhr J., Gorbalenya A. E., Silverman R. H., Weiss S. R., Cell Host Microbe 2012, 11, 607–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.
- 18a. Liang W., Chan A. Y. L., Chow M. Y. T., Lo F. F. K., Qiu Y., Kwok P. C. L., Lam J. K. W., Asian J. Pharm. Sci. 2018, 13, 163–172; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18b. Osman G., Rodriguez J., Chan S. Y., Chisholm J., Duncan G., Kim N., Tatler A. L., Shakesheff K. M., Hanes J., Suk J. S., Dixon J. E., J. Controlled Release 2018, 285, 35–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information