

Abstract
The pandemic generated by SARS‐Cov‐2 has caused a large number of cases and deaths in the world, but South America has been one of the continents that were most hard hit. The appearance of new variants causes concern because of the possibility that they may evade the protection generated by vaccination campaigns, their greater capacity to be transmitted, or their higher virulence. We analyzed the circulating variants in Peru after improving our Genomic Surveillance program. The results indicate a steep increase of the lambda lineage (C.37) until becoming predominant between January and April 2021, despite the cocirculation of other variants of concern or interest. Lambda lineage deserves close monitoring and could probably become a variant of concern in the near future.
Keywords: genome sequencing, molecular epidemiology, SARS‐Cov‐2
1. INTRODUCTION
The SARS‐CoV‐2 has generated epidemics of different social and sanitary impacts and characteristics throughout the world. From December 2020, the emergence of viral variants of SARS‐CoV‐2 has been associated with higher transmissibility, immune escape, or increased pathogenicity. The variants of concern (VOC) and variants of interest (VOI), having different epidemiological and clinical characteristics, represent an important threat to the global containment of the pandemic. 1 , 2
Peru has been one of the countries hardest hit by the pandemic. From January to June, a devastating second wave caused more than 980 485 reported cases and 98 837 deaths. 3 However, due to its limited capacities of genomic sequencing, tracing the circulating variants was very hard. The alpha (B.1.1.7) variant was first reported in our country in December 2020, 4 whereas in January 2021, the gamma (P.1) variant was identified in three regions of our country, most of all in the Amazonian regions. 5
In April 2021, the lambda (C.37) lineage was reported in patients from the capital city of Lima. 6 The first patient having this lineage can be traced in the Global Initiative on Sharing Avian Influenza Data (GISAID) back to November 2020 (Lima). Currently, this lineage has also been identified in the United States, Chile, Brazil, Argentina, Ecuador, Mexico, Spain, and Germany, among other countries. 5
Since May, Peru has scaled up its capacity of genomic surveillance through the implementation of a genomic platform using the Illumina COVIDSEQ test. The aim of this report is to describe the main SARS‐CoV‐2 lineages circulating in our country during the second wave, from January to April 2021, and to model their effective population variations to contribute to the epidemiologic characterization of the lambda variant.
2. MATERIALS AND METHODS
2.1. Design of study and data collection
In May 2021, the National Institute of Health of Peru strengthened the National Program for Genomic Surveillance of SARS‐CoV‐2 to scale up genomic sequencing as a means of better understating the circulation and behavior of the different variants in our country. As part of this initiative, samples from patients having symptoms compatible with COVID‐19 and positive nasal/pharyngeal swabs were retrieved from all regions of the country. For this analysis, 953 samples from patients diagnosed through real‐time reverse‐transmission polymerase chain reaction from February to April 2021 in the 24 regions of Peru were included. These samples were collected from outpatients and inpatients from all our 24 regions, trying to keep proportionality with the number of cases in them (the capital city of Lima had around 50% of the cases) and tried to include all age groups. Only samples having a C t less than 30, corresponding to a high presence of the virus in the samples, were included for analysis.
In addition, relevant genomes were mined from the GISAID database 5 : 1246 genomes registered from Peru and other relevant genomes reported in the database were selected randomly.
2.2. Sample processing and data analysis
All samples were processed using Illumina's COVIDSeq platform according to a previously described protocol. 7 Processing of the fastq files generated by the NextSeq 550 sequencer was performed in Illumina's BaseSpace environment using the DRAGEN v05.021 algorithm.
To assess effective population variations in alpha, lambda, and gamma SARS‐CoV‐2 lineages in Peru, we conducted an evolutionary phylodynamic approach under the coalescent Bayesian skyline model implemented in BEASTv.1.10.4. 8 Sequences were first aligned using the MAFFT software. 9 Alignment was manually checked and edited. An uncorrelated lognormal relaxed molecular clock with a mean rate of 5.0483 × 10−4 substitutions/site/year was considered with the GTR+I+G nucleotide substitution model. Two independent Markov Chain Monte Carlo were run for 100 million generations sampled every 2000 generations. Convergence was inspected using an effective sampling size greater than 200 in TRACER software. Log and Tree files were independently merged with 10% of burning in Log‐Combiner software.
3. RESULTS
One thousand sixty‐six genomes were obtained from patients diagnosed in 2020 and 1133 genomes were analyzed from 2021. In 2021, the predominant overall variant in April is lambda with 78%, followed by gamma at 15%, and B.1.1.7 at 1.5%. A progressive increase in the percentage of lambda was observed starting in January (20.7%), February (35.1%), March (66.5%) up to April (78%) (Figure 1A,B). On the contrary, gamma shows a stable overall frequency between February and April 2021 and the alpha lineage shows low prevalence in January (1.2%), February (0%), March (1.4%), and April (1.5%).
Figure 1.
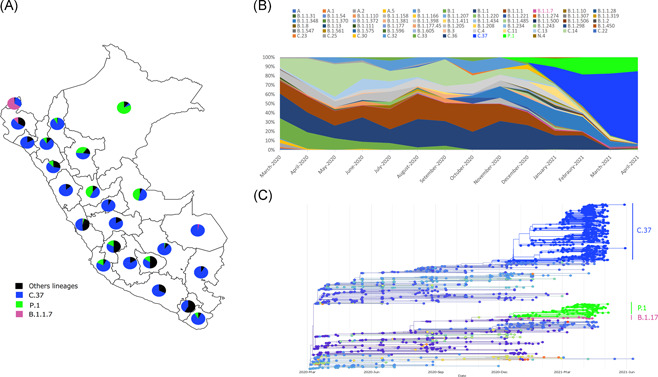
(A) Distribution of lineages present in Peru during 2021 (January–April). (B) Relative frequency of the main variants detected in Peru until April 2021. (C) Phylogenetic analysis of the main circulating variants in Peru implemented in NextStrain platform considering the genomes previously reported in the Global Initiative on Sharing Avian Influenza Data and those sequenced in this article
The lambda variant was present in the 24 analyzed regions, whereas the gamma was detected in 19 of the 24 analyzed regions. The alpha variant was detected only in five regions (Figure 1A and Table S1).
In April 2021, the gamma variant showed high prevalence in the Amazon regions of Loreto (100%), Ucayali (66.7%), San Martín (46.7%), Huancavelica (34.5%), and Apurímac (30%), whereas the alpha variant circulated in Tumbes (30%), Ayacucho (3.3%), Junín (3.3%), and Lima (1.4%).
The phylogenetic analysis shows that the studied genomes are grouped into clusters with good support and an association that was previously reported for Peru and other countries (Figure 1C).
The main mutations that characterize the lambda lineage are located in the N gene (P13L, R203K, G204R, and G214C), in ORF1a (T1246I, P2287S, F2387V, L3201P, T3255I, and G3278S), in the ORF1b gene (P314L and deletion of three amino acids SGF in positions 3675–3677), in the ORF9b gene (P10S), in the S gene (G75V, T76I, R246N, a deletion of seven amino acids SYLTPGD in positions 247–253, L452Q, F490S, D614G, and T859N).
Bayesian calculation of the mean divergence time of the lambda lineage indicates that it could have appeared around October 21, 2020 (95% highest posterior density (HPD): September 2−November 30), the gamma lineage could have been introduced around November 16, 2020 (95% HPD: October 07−December 16), whereas the alpha lineage could have been introduced around December 30, 2020 (95% HPD: December 27−31) (Figure 2). The Bayesian Skyline analysis indicates an increase of effective population size (Ne) of lambda lineage between January and February 2021, stabilized in February, and decreased in April, coincidently with the evolution of the “second wave.” The gamma lineage follows a similar evolution but presents a smaller size of its effective population, remaining below the effective population of the lambda lineage in the study period (Figure 2), we observed the same pattern for alpha lineage.
Figure 2.
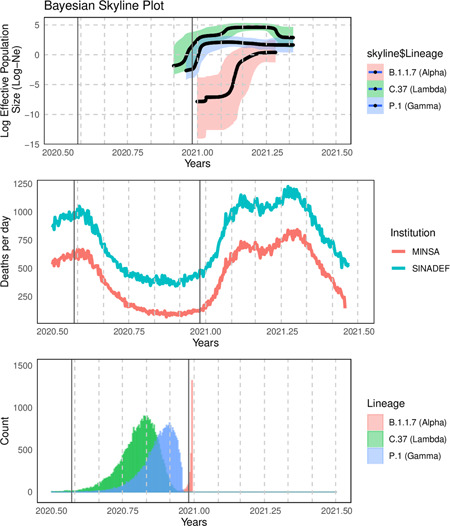
Upper: Bayesian Skyline analysis of the main lineages detected in Peru. The vertical line represents 25 December (Christmas holidays). Middle: Number of deaths from COVID‐19 reported by the Peruvian Ministry of Health (MINSA) and total deaths reported by the national death system (SINADEF). Down: Bayesian calculation of the mean divergence time of the alpha, gamma and lambda lineage
4. DISCUSSION
South America has been an important epicenter all throughout the pandemic and is now one of the still prevailing at a global level. Peru, in particular, has been considered to have the highest number of deaths per capita in the world. Our assessment shows that the lambda variant has become predominant at the country level during the second wave, despite the parallel circulation of two other variants of concern: alpha and gamma, which have not been let to gain preponderance.
We consider that this variant must have played an important role in producing such a high burden of morbidity and mortality in Peru during this second wave, together with the lack of regard to standard prevention measures of our population during the Christmas holidays. Due to the epidemiologic increases we have seen through this year, we believe it could have phenotypic characteristics that can even provide advantages in comparison to these other variants of concern, in particular to the alpha variant. Currently, it is extending throughout South America and is acquiring predominance in Chile and Argentina according to GISAID. 5 This variant has recently been labeled as a VOI by the World Health Organization, 10 as further data is needed to consider it a variant of concern.
The lambda variant was detected in more than 70% of analyzed samples in all the studied regions of Peru except for the Amazon ones, where the gamma variant was more prevalent. There could be an ancestral effect involved. The skyline analysis suggests that the increase in the proportion of the lambda lineage from January to April could reflect increased transmissibility. This should be confirmed by the comparison of its household transmission rate in relation to the other variants. Bayesian Skyline plots for both lambda and gamma lineages estimated most recent common ancestors (TMRCAs) as older as 2 and 1 month before the first reported case, respectively. The recent TMRCA estimated for gamma lineage was around November 15, 2020 (95% HPD: October 6–November 24, 2020) between 3 and 4 weeks before the first case 11 and perfectly agrees with our estimation for the same lineage (November 16). This close estimation could be explained by the dynamic commercial activities between the cities of Manaos and Iquitos in Brazil and Peru, respectively, through the Amazon basin.
The assumption of the increased transmissibility is aligned to some mutations seen in this variant. The lambda lineage contains a seven‐amino‐acid deletion in the S gene, which is probably a regression as this deletion is present in predecessor coronaviruses that infect other animal hosts. 12 Noteworthy, the L452Q mutation uniquely present in this variant is very similar to the L452R present in the delta variant and in the epsilon variants, which together with the D614G mutation, are considered to increase the transmissibility. 13 , 14 And most importantly, it has the T859N mutation in lambda, also present in variant B.1.526.1 (New York), which has been associated with a reduced neutralization by monoclonal antibodies, and by convalescent and postvaccination sera. 15
The biological significance of other mutations present in the lambda lineage is unknown, so our next step will be to perform studies devoted to assess neutralization of sera from patients with previous infections and vaccinated, as well as observational studies of the clinical profile of these patients including epidemiological studies, to measure transmissibility and impact on re‐infections to complete the characterization of this variant, which, in face of the results of these studies, could be upgraded to VOC.
Being Peru a country with very high transmission, and having reported the first case of this lambda variant, it is highly likely that it emerged here. Unfortunately, the circumstances that allowed its emergence have not varied greatly, and the vaccination coverage is currently less than 10%, highlighting the possibility of the emergence of new variants. Urgent action is needed to prevent this, focused on greater and quicker access to vaccines to prevent a new wave bringing a new variant.
CONFLICT OF INTERESTS
The authors declare that there are no conflict of interests.
ETHICS STATEMENT
This study was conducted in accordance with the Declaration of Helsinki. The data was generated as part of National Genomic Surveillance.
AUTHOR CONTRIBUTIONS
Carlos Padilla‐Rojas, Veronica Hurtado, Orson Mestanza, Iris Silva, Luis Barcena, Sandra Morales, Steve Acedo, and Wendy Lizarraga processed samples and obtained data. Carlos Padilla‐Rojas, Víctor Jimenez, Veronica Hurtado, Orson Mestanza, Iris Silva, Luis Barcena, Sandra Morales, Steve Acedo, Wendy Lizarraga, Henri Bailon, Omar Cáceres, Marco Galarza, Nancy Rojas‐Serrano, Natalia Vargas, Priscila Lope‐Pari, Joseph Huayra, and Lely Solari analyzed and interpreted data. All authors revised the article critically and approved the final version.
Supporting information
Supporting information.
ACKNOWLEDGMENTS
The authors wish to thank the COVID diagnostic team of the National Institute of Health of Peru for their hard contribution in confirming the cases of COVID‐19. We also want to thank the staff of laboratory technicians and assistants for their contribution to this article. We also wish to thank the various research groups that have contributed by sharing the SARS‐COV‐2 genome information in international databases.
Padilla‐Rojas C, Jimenez‐Vasquez V, Hurtado V, et al. Genomic analysis reveals a rapid spread and predominance of lambda (C.37) SARS‐COV‐2 lineage in Peru despite circulation of variants of concern. J Med Virol. 2021;93:6845‐6849. 10.1002/jmv.27261
Contributor Information
Carlos Padilla‐Rojas, Email: cpadilla@ins.gob.pe.
Orson Mestanza, Email: orsomm@gmail.com.
Sandra Morales Ruiz, Email: smr_sandra@yahoo.es.
DATA AVAILABLITY STATEMENT
The genomes characterized in this study have been shared in the international GISAID database are openly available at https://www.gisaid.org/, reference numbers EPI_ISL_2536701 to EPI_ISL_2844791, EPI_ISL_2921267 to EPI_ISL_2921399, EPI_ISL_2921401 to EPI_ISL_2921532, and EPI_ISL_3023377 to EPI_ISL_3023647, and EPI_ISL_3375862 to EPI_ISL_3376048.
REFERENCES
- 1. Padilla‐Rojas C, Lope‐Pari P, Vega‐Chozo K, et al. Near‐complete genome sequence of a 2019 novel Coronavirus (SARS‐CoV‐2) strain causing a COVID‐19 case in Peru. Microbiol Resour Announc. 2020;9(19):e00303‐e00320. 10.1128/MRA.00303-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Padilla‐Rojas C, Vega‐Chozo K, Galarza‐Perez M, et al. Genomic analysis reveals local transmission of SARS‐CoV‐2 in early pandemic phase in Peru. BioRvix. 10.1101/2020.09.05.284604 [DOI] [Google Scholar]
- 3. Dirección General de Epidemiologia, Ministerio de Salud Peru Sala COVID‐19. 2021. https://www.dge.gob.pe/covid19.html. Accessed July 22, 2021.
- 4. Padilla‐Rojas C, Barcena‐Flores L, Vega‐Chozo K, et al. Near‐complete genome sequence of a SARS‐CoV‐2 VOC 202012/01 strain in Peru. Microbiol Resour Announc. 2021;10(12):e00069‐21. 10.1128/MRA.00069-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Shu Y, McCauley J. GISAID: global initiative on sharing all influenza data—from vision to reality. Euro Surveill. 2017;22(13):30494. 10.2807/1560-7917.ES.2017.22.13.30494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Romero PE, Dávila‐Barclay A & Gonzáles L et al. C.37: novel lineage expanding in Peru and Chile, with a convergent deletion in the ORF1a gene (Δ3675‐3677) and a novel deletion in the Spike gene (Δ246‐252, G75V, T76I, L452Q, F490S, T859N). 2021. https://virological.org/t/novel-sublineage-within-b-1-1-1-currently-expanding-in-peru-and-chile-with-a-convergent-deletion-in-the-orf1a-gene-3675-3677-and-a-novel-deletion-in-the-spike-gene-246-252-g75v-t76i-l452q-f490s-t859n/685. Accessed July 22, 2021.
- 7. Illumina COVIDSeq RUO Kits Reference guide. 2021. https://support.illumina.com//downloads/illumina-covidseq-test-reference-guide-1000000126053.html. Accessed July 22, 2021.
- 8. Drummond AJ, Rambaut A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol. 2007;7:214. 10.1186/1471-2148-7-214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30(4):772‐780. 10.1093/molbev/mst010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. World Health Organization Tracking SARS‐CoV‐2 variants. 2021. https://www.who.int/en/activities/tracking-SARS-CoV-2-variants/. Accessed July 22, 2021.
- 11. Faria NR, Mellan TA, Whittaker C, et al. Genomics and epidemiology of the P.1 SARS‐CoV‐2 lineage in Manaus, Brazil. Science. 2021;372(6544):815‐821. 10.1126/science.abh2644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Guruprasad L. Evolutionary relationships and sequence‐structure determinants in human SARS coronavirus‐2 spike proteins for host receptor recognition. Proteins. 2020;88(11):1387‐1393. 10.1002/prot.25967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Li Q, Wu J, Nie J, et al. The impact of mutations in SARS‐CoV‐2 spike on viral infectivity and antigenicity. Cell. 2020;182(5):1284‐1294. 10.1016/j.cell.2020.07.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Liu Z, VanBlargan LA, Bloyet LM, et al. Identification of SARS‐CoV‐2 spike mutations that attenuate monoclonal and serum antibody neutralization. Cell Host Microbe. 2021;29(3):477‐488. 10.1016/j.chom.2021.01.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhou H, Dcosta BM, Samanovic MI, Mulligan MJ, Landau NR, Tada T. B.1.526 SARS‐CoV‐2 variants identified in New York City are neutralized by vaccine‐elicited and therapeutic monoclonal antibodies. bioRxiv. 2021:436620. 10.1101/2021.03.24.436620 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information.
Data Availability Statement
The genomes characterized in this study have been shared in the international GISAID database are openly available at https://www.gisaid.org/, reference numbers EPI_ISL_2536701 to EPI_ISL_2844791, EPI_ISL_2921267 to EPI_ISL_2921399, EPI_ISL_2921401 to EPI_ISL_2921532, and EPI_ISL_3023377 to EPI_ISL_3023647, and EPI_ISL_3375862 to EPI_ISL_3376048.