

Abstract
Cancer is considered a high‐risk condition for severe illness resulting from COVID‐19. The interaction between severe acute respiratory syndrome coronavirus‐2 (SARS‐CoV‐2) and human metabolism is key to elucidating the risk posed by COVID‐19 for cancer patients and identifying effective treatments, yet it is largely uncharacterised on a mechanistic level. We present a genome‐scale map of short‐term metabolic alterations triggered by SARS‐CoV‐2 infection of cancer cells. Through transcriptomic‐ and proteomic‐informed genome‐scale metabolic modelling, we characterise the role of RNA and fatty acid biosynthesis in conjunction with a rewiring in energy production pathways and enhanced cytokine secretion. These findings link together complementary aspects of viral invasion of cancer cells, while providing mechanistic insights that can inform the development of treatment strategies.
Keywords: cancer, COVID‐19, flux balance analysis, genome‐scale metabolic modelling, multi‐omics, SARS‐CoV‐2
Abbreviations
AGP, alpha‐1 acid glycoprotein
COVID‐19, coronavirus disease 2019
DARs, differentially active reactions
DEGs, differentially expressed genes
DEPs, differentially expressed proteins
dUMP, deoxyuridine monophosphate
FBA, flux balance analysis
FVA, flux variability analysis
GO, gene ontology
GSMMs, genome‐scale metabolic models
hpi, hours post‐infection
IP10, interferon γ‐induced protein‐10
MIP‐1β, macrophage inflammatory protein‐1β
PPARalpha, peroxisome proliferator‐activated receptor alpha
SARS‐CoV‐2, severe acute respiratory syndrome coronavirus‐2
SDH, succinate dehydrogenase
VEGFA, vascular endothelial growth factor A
The rapid spread of severe acute respiratory syndrome coronavirus‐2 (SARS‐CoV‐2) has created a global public health emergency that is currently affecting countries across the globe [1, 2]. The coronavirus disease 2019 (COVID‐19) pandemic poses challenges to healthcare systems due to the lack of specific therapeutics for prevention or patient treatment. In turn, drug development requires scientists across biomedical disciplines to expand our knowledge on biomolecular mechanisms regulating SARS‐CoV‐2 infection and the associated cascade of cellular alterations.
Cancer is considered by the Centers for Disease Control and Prevention a high‐risk condition for severe illness resulting from COVID‐19. However, it is not clear to what extent this comorbidity affects the cellular mechanisms observed in both diseases. While cancer patients infected with SARS‐CoV‐2 are known to be at increased risk, the human metabolic response to SARS‐CoV‐2 infection in cancer cells remains largely uncharacterised to date [3, 4, 5, 6]. The severity of physiological symptoms experienced by individuals is largely dependent on the host response, which means understanding the metabolic changes caused by infection is crucial to identifying treatments for managing both the immediate and longer‐term effects on health [7].
Further, liver dysfunction has been reported to increase with increasing severity of COVID‐19 infection [8, 9, 10, 11, 12]. It has been reported that the ACE2 receptor that facilitates viral entry to the cell is highly expressed in cholangiocytes and is also expressed in hepatocytes to a lesser extent [13, 14]. Chu et al. [15] demonstrated that COVID‐19 can replicate within liver cancer cells such as Huh7. However, the precise mechanism underlying the viral effect on the liver has still to be determined [16, 17].
Recently, time‐course transcriptomic measurements of SARS‐CoV‐2‐infected Huh7 cells have been reported [18]. In this study, the ErbB, HIF‐1, mTOR and TNF signalling pathways were identified as significantly modulated during the course of the SARS‐CoV‐2 infection. The mTOR signalling pathway was thus suggested as a potential drug target to treat COVID‐19 patients. mTOR is known to be implicated in the metabolic control of glucose, nucleotides and lipids [19]. This study thus highlights the relevance of omics analyses for the consolidation of knowledge with potential for clinical translation. Studies based on gene and protein transcription levels, however, provide scattered information that may be difficult to unify and interpret.
Similarly, studies of circulating blood metabolites in COVID‐19 patients have yielded interesting biomarkers of infection, including hijacking of nucleic acid intermediates [20, 21, 22, 23], dysregulation of lipid metabolism [22, 24, 25], changes in amino acid metabolism [23, 25], alteration of energy metabolism [26], immune response [27], and indicators of hepatic cell damage [25]. However, a limitation of metabolomics alone is that it focuses on alterations of metabolites at a pathway level rather than identifying altered reaction/enzyme activity, which allows for more specific therapeutic targeting.
To unify and interpret heterogeneous omics data, systems biology offers powerful tools such as genome‐scale metabolic modelling [28]. This approach mathematically describes metabolic networks and their activity by unifying large corpuses of detailed biochemical knowledge, thus allowing the estimation of metabolic phenotypes on a single‐reaction resolution [29]. For instance, previous studies exploited it for predicting biomarkers in the context of cancer and other diseases [30, 31, 32]. In the context of SARS‐CoV‐2, there have been few studies to investigate the metabolic effects of viral infection at genome scale [33, 34] and to our knowledge, none that investigate the metabolic effects of infection in cancer cells, an aspect of special relevance in cancer [35].
To address these gaps, we apply a systems biology approach to explore how SARS‐CoV‐2 infection impacts human cancer cell metabolism and how distinct pathways are affected over time. To this end, we build genome‐scale metabolic models (GSMMs) that incorporate multiple transcriptional and protein expression states after infection and that reflect consecutive stages of immune response to viral invasion. Thanks to a flux balance analysis (FBA) framework, such models allowed us to explore in detail how the disease insurgence impacts single metabolic reactions. Through this approach, we identify altered pathway capabilities and reconstruct a global overview of metabolic rewiring under SARS‐CoV‐2 infection (Fig. 1 for the workflow).
Fig. 1.
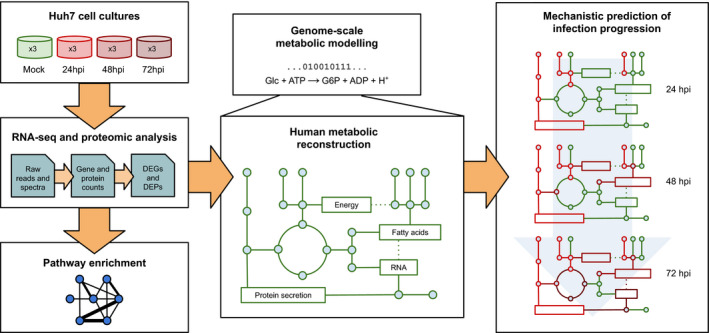
Workflow followed in the study. Time series RNA‐seq and proteomic data from infected Huh7 cells sampled at 24, 48 and 72 hours post‐infection (hpi) were analysed and used to identify DEGs, DEPs and dysregulated pathways compared with uninfected cultures. Results from RNA‐seq and proteomic analysis were used to inform a genome‐scale metabolic reconstruction of human metabolism, which was expanded to include translation and secretion pathways for a range of immune proteins alongside a SARS‐CoV‐2 biomass objective function. Gene and protein expression counts were thus incorporated in the baseline model to capture time‐dependent changes in metabolic activity by estimating DARs with respect to uninfected Huh7 cells.
Our analysis identified four main areas of metabolism affected by SARS‐CoV‐2 infection: RNA production, energy production, fatty acid metabolism and the secretome. Glycolysis showed upregulated production of ATP while the mitochondrial electron transport chain and oxidative phosphorylation were downregulated. This diversion of energy production through glycolysis is in addition to the upregulation of glycolysis already known to occur in cancer cells. The amplified effect of cancer metabolism may explain why cancer patients are at higher risk from SARS‐CoV‐2 infection. Synthesis of fatty acids for the viral cell envelope was increased, while unsaturated fatty acid synthesis was decreased. Secretion of interferon γ‐induced protein‐10 (IP10/CXCL10), which is associated with the cytokine storm, was highly upregulated. Finally, we suggest some biological processes that could be targeted by therapeutic treatments based on our findings.
Materials and methods
Data collection
RNA‐seq data were taken from the SRA database (accession PRJNA627100). Briefly, Huh7 cells were used to perform transcriptomic analysis in uninfected and 24, 48 and 72 hours post‐infection (hpi) by the SARS‐CoV‐2 virus. Three specimens for each time point were collected. Complete data production is extensively reported in the original paper of Appelberg et al. [18]. Preprocessed proteomic data for Huh7 cells at the same time points were obtained from the authors.
RNA‐seq and proteomic data processing and analysis
RNA read preprocessing was performed by using fastp v0.20.0 [36], applying specific parameters in order to remove residual adapter sequences and to keep only high‐quality data (qualified_quality_phred = 20, unqualified_percent_limit = 30, average_qual = 25, low_complexity_filter = True, complexity_threshold = 30). Then, passing filter reads were mapped to the human genome reference (version GRCh38) using star v2.7.0 [37] with standard parameters, except for sjdbOverhang option set to 74 due to read length. Genome and transcript annotation provided as input were downloaded from the Ensembl repository v99 (genome [38], annotation [39]). Alignments were then elaborated by rsem v1.3.3 [40] to estimate transcript and gene abundances. Subsequently, sample‐specific gene‐level abundances were merged into a single raw expression matrix applying a dedicated rsem command (rsem‐generate‐data‐matrix). Genes with less than 10 counts in all samples were then filtered out. Gene differential expression (pairwise comparisons) was computed by edger [41] from raw counts in each comparison, following the authors' instructions. A multiple testing correction was applied (estimateDisp, glmQLFit and glmQLFtest), and genes with a P‐value ≤ 0.05 were considered differentially expressed.
Starting from quantile‐normalised protein abundance, proteomic profiles were processed as described in the original paper [18]. Proteomic‐transformed data were processed through linear models using the limma r package [42]. In the limma design matrix, separated coefficients were associated with time and samples in order to define the contrasts. For the pairwise comparisons, a single factorial design was implemented to fit models with a coefficient for each of four factors: uninfected, 24, 48 and 72 hpi. As a result, significant differentially expressed proteins were selected based on P‐values after a Benjamini–Hochberg (BH) adjustment. Proteins with an alpha value less than 0.05 were considered significant. For both proteomic and transcriptomic results, no threshold was applied on the log2 fold change.
Re‐annotation of differentially expressed genes (DEGs) and proteins was performed using the biomart package [43] in r 3.6, querying available Ensembl Gene IDs and retrieving Gene Names and HGNC gene IDs to allow mapping of genes onto the metabolic model. Then, GO (gene ontology) and pathway enrichment analyses were performed on the KEGG [44] and Reactome [45] public pathway databases, which consist of graphical diagrams of biochemical pathways including most of the known metabolic reactions.
Expansion of a human genome‐scale metabolic reconstruction
We modelled the hepatocyte‐derived cellular carcinoma cell line Huh7 metabolism based on the Recon 2.2 genome‐scale reconstruction [46]. This model incorporates a large number of biochemical pathways of human metabolism and is the result of joint efforts by many scientists and several rounds of curation. We expanded Recon by including translation and secretion pathways for relevant proteins using a recently introduced pipeline [47]. Secretory proteins were selected based on the secretomes of COVID‐19 [11, 12, 48, 49, 50], SARS [51, 52, 53], oxidative stress [54] and liver cells [55] (Table 1). Moreover, we introduced a SARS‐CoV‐2 biomass pseudoreaction accounting for viral metabolic requirements within host cells, to enable an integrated human virus metabolic modelling simulation [56]. Specifically, we employed a biomass stoichiometry defined in previous works by assembling molecular investments in SARS‐CoV‐2 biomass and that accounts for nucleotide, amino acid, lipid and energetic requirements [33, 34]. The resulting model spans 1838 genes, 9892 reactions and exchange pseudoreactions and 7386 metabolites across 10 cellular compartments.
Table 1.
Genome‐scale metabolic model secretory proteins. Table showing the name, gene and protein UniProt ID for the secretome added to the GSMM.
| Name | Gene | Protein |
|---|---|---|
| Alpha‐1 acid glycoprotein | ORM1 | P02763 |
| Apolipoprotein A1 | APOA1 | P02647 |
| Basic fibroblast growth factor | FGF2 | P09038 |
| Chemokine (C‐C motif) ligand 20 | CCL20 | P78556 |
| C‐reactive protein | CRP | P02741 |
| Eotaxin | CCL11 | P51671 |
| Fetuin | AHSG | P02765 |
| Granulocyte colony‐stimulating factor (GSCF) | CSF3 | P09919 |
| Haptoglobin | HP | P00738 |
| IFN‐γ | IFNG | P01579 |
| Proinflammatory interleukins IL‐1β, IL‐2, IL‐5, IL‐6, IL‐8, IL‐12A, IL‐12B, IL‐15, IL‐17A | IL1B, IL‐2, IL‐5, IL‐6, CXCL8, IL‐12A, IL‐12B, IL‐15, IL‐17RA | P01584, P60568, P05113, P10145, P29459, P29460, P40933, Q96F46 |
| Anti‐inflammatory interleukins IL1RA, IL‐4, IL‐7, IL‐9, IL‐10, IL‐13 | IL1RN, IL‐4, IL‐7, IL‐10, IL‐13, IL‐9 | P18510, P05112, P05231, P13232, P22301, P35225, P15248 |
| Interferon γ‐induced protein‐10 (IP10) | CXCL10 | P02778 |
| Macrophage inflammatory protein‐1α (MIP‐1α) | CCL3 | P10147 |
| Macrophage inflammatory protein‐1β (MIP‐1β) | CCL4 | P13236 |
| Monocyte chemoattractant protein‐1 (MCP1) | CCL2 | P13500 |
| Peroxiredoxin‐1 | PRDX1 | Q06830 |
| Peroxiredoxin‐2 | PRDX2 | P32119 |
| Platelet‐derived growth factor subunit B | PDGFB | P01127 |
| RANTES | CCL5 | Q9Y2Y9 |
| Thymic stromal lymphopoietin | TSLP | Q969D9 |
| Transferrin | TF | P02787 |
| Transthyretin | TTR | P02766 |
| Tumour necrosis factor‐α (TNF‐α) | TNF | P01375 |
| Vascular endothelial growth factor A (VEGFA) | VEGFA | P15692 |
| α1‐Antichymotrypsin | SERPINA3 | P01011 |
| α1‐Antitrypsin (TF‐α1‐AT) | SERPINA1 | P01009 |
Infection‐stage‐specific metabolic modelling
Flux balance analysis is a mathematical approach for analysing the flux of biochemical reactions through a genome‐scale reconstruction of metabolic networks [28, 29]. FBA requires a genome‐scale metabolic network reconstruction, represented as a stoichiometric matrix S where the rows correspond to metabolites and the columns represent reactions. Under a steady‐state assumption, there is no net change in mass in the system and the mass is conserved. Therefore, the rate of production of each internal metabolite equals its rate of consumption. A column vector v represents the flux through the system (reaction rate of each reaction). Under the steady‐state assumption, the matrix multiplication of the stoichiometric matrix S and column vector v provides the linear equations representing the constraints (Sv = 0).
Further constraints are added through RNA‐seq expression profiles, and specifically the lower and upper bounds of each metabolic flux lb ≤ v ≤ ub. These are vectors representing the lowest and highest reaction rate possible for each reaction. These constraints reduce the possible solution space and can be set for each condition, therefore creating condition‐specific metabolic models [29].
Temporal progression was modelled through the creation of condition‐specific models, based on the gene and protein expression fold change profiles over the three time points. Gene expression for the unaffected cells was used as a control when calculating the fold changes. Condition‐specific models were generated through a modified version of metrade [57] where flux bounds are a linear function of gene set expression values. This process requires, as input, expression fold changes, which are then converted into reaction flux bounds through gene‐protein‐reaction rules encoded in the model. The same process was repeated with proteomic data, therefore generating two sets of models, which will be denoted by transcriptomic‐informed and proteomic‐informed GSMMs. The flux bounds obtained after solving these models were then used in the simulations described below.
Simulating alterations in metabolic network capabilities
In addition to the transcriptional constraints described above, we utilised additional constraints devised based on the literature and our RNA‐seq analysis results. Regulation of lipid metabolism by peroxisome proliferator‐activated receptor alpha (PPARalpha), which increases the size and number of peroxisomes, was identified from the pathway analysis. PPARalpha is known to be involved in regulating fatty acid metabolism and the immune response in hepatocytes [58]. Acyl‐CoA oxidase is a rate‐limiting enzyme activated by PPARalpha [59]. Two further constraints were added from a review of the literature and their correlated increase in activity with the viral infection. Phosphogluconate dehydrogenase catalyses the conversion of 6‐phosphogluconate to ribulose‐5‐phosphate in the cytosol generating NADPH. Ribulose‐5‐phosphate is used for nucleotide biosynthesis. The reaction catalysed by phosphogluconate dehydrogenase is increased when there is a need for nucleotide and fatty acid synthesis, which uses the NADPH, such as for viral replication [60, 61]. The third and final reaction chosen occurs in peroxisomes, where lactate and NAD+ are converted to pyruvate and NADH by NAD+ oxidoreductase. The pyruvate/lactate ratio is linked with the NAD+/NADH ratio [62], potentially suggesting that the pyruvate is then shuttled back into the cytosol to be recycled into the citric acid cycle. Following the evidence described above, we set the lower bound of phosphogluconate dehydrogenase, acyl‐CoA oxidase and NAD+ oxidoreductase to 80% of their maximum value. Moreover, to account for cells growing in a culture, we constrained biomass production to be at least 50% of its maximum value. Analogously, we set a lower bound on viral biomass production to simulate the effects of intracellular viral replication, based on a total virion dry weight estimate of one‐third the host cell's at peak infection [63]. The viral biomass production was therefore constrained to be above a variable fraction of the maximum host biomass across the time points, in order to get a peak bound of one‐third of its value for the 72‐h time point. Only for the translation and secretion pathways, we used the same bound over all the time points, in order to best balance the impact of the viral biomass. Finally, oxygen uptake was set as unlimited, while oxygen production and superoxide consumption were blocked.
After having obtained a constrained model, flux variability analysis (FVA) was used to evaluate the minimum and maximum value of the flux that each reaction can carry, while still satisfying the given constraints. FVA was run by solving a double linear programming problem on each condition‐specific metabolic model. Estimation of metabolic potential on each time point was performed by FVA as implemented in the COBRA toolbox [64]. A metabolic network visualisation of flux fold changes was generated using ReconMap [65].
Statistical analysis of differentially active reactions
For each time point, we determined the most perturbed metabolic pathways by a one‐sided hypergeometric test with false discovery rate correction for multiple testing on the differentially active reactions (DARs). Overactive DARs were defined as those reactions whose maximal flux fold change was 1.5 or greater and above the 95th percentile of the fold change distribution. For underactivity, we considered reactions whose maximal flux fold change was 0.8 or lower and fell in the 5th fold change percentile. In this way, we could select metabolic reactions characterised by activity changes with both statistical relevance and meaningful effect size.
Results and Discussion
Reconstructing the metabolic evolution of SARS‐CoV‐2 infection
To evaluate how SARS‐CoV‐2 infection affects metabolic activity along short‐term post‐infection time points (0, 24, 48 and 72 h), we applied our condition‐specific modelling approach, as described in the Materials and methods section and illustrated in Fig. 1. Integrated with machine learning methods, such an approach has previously revealed insights in infectious and complex disorders [66, 67, 68, 69]. Our modelling and analyses steps were performed starting from the multi‐omics dataset recently published by Appelberg et al. [18].
As a result of transcriptomic analysis, we obtained a high percentage of uniquely mapped reads, ranging from 88.02 and 89.13, with a mean value of 88.5. Then, we calculated DEGs among all the possible time point combinations, focusing on the differences between uninfected cells and the most advanced stage of virus incubation (72 hpi), which is expected to disclose the most valuable and significant results. Analogously, we obtained DEPs for the same contrasts. At 72 hpi, we identified 3464 DEGs and 883 DEPs (Table S1), associated with a large number of biological pathways (Fig. 2), including upregulated glycolysis and inflammatory secretome as found previously [18]. In addition, we identified dysregulation in the biosynthesis of amino acids and fatty acid metabolism, biomarkers of hepatic cell injury and biomarkers of endoplasmic reticulum stress due to increased demand for protein folding [70]. The enrichment results are listed in Tables S2 and S3. DEGs and DEPs derived from the comparisons across time points were used to inform the genome‐scale model in order to specifically highlight metabolic alterations due to the SARS‐CoV‐2 infection.
Fig. 2.
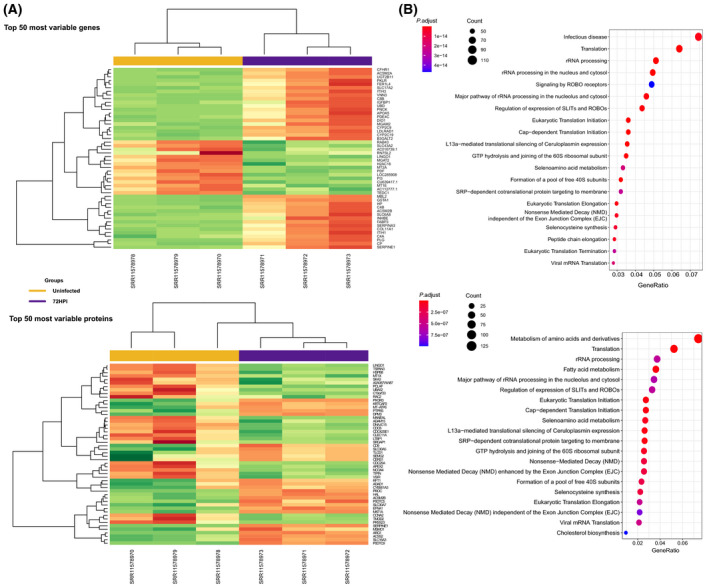
Dysregulated DEGs and DEPs identified by RNA‐seq and proteomic analysis. (A) Hierarchical clustering heatmap representing the most variable genes (|logFC| > 2) considering their transcript (top) and protein (bottom) expression in uninfected (yellow) and 72 hpi (violet) samples. Overexpression is red coloured, while underexpression is depicted in green. (B) Enrichment analysis showing the most relevant KEGG and Reactome pathways populated by differentially expressed genes (top) and proteins (bottom). Circle size indicates the number of genes involved in each pathway. Colour gradient represents statistical significance based on adjusted P‐value.
To obtain a mechanistic understanding of the alterations observed on a transcriptional level, we integrated the gene and protein expression profiles described above within Recon 2.2, a GSMM of human cells. The selected GSMM is among the most complete templates for the human metabolic network and was chosen based on its manually curated pathways and the validated representation of energy generation across various carbon sources [46]. As Recon 2.2 is a general‐purpose GSMM, it incorporates all the main metabolic pathways for a range of tissues and can be tailored to any target tissue through integration with omics data, as reviewed elsewhere [29, 71, 72]. Moreover, we expanded the GSMM by including a SARS‐CoV‐2 biomass accumulation pseudoreaction and secretory pathways for a range of relevant immune proteins (see Materials and methods). Our multi‐omics genome‐scale metabolic modelling approach thus allows us to investigate key dysregulated enzymes and reactions, giving a more holistic view of the metabolic phenotype of the infected cancer cell.
Our modelling approach assumes a metabolic steady state, which can be assumed by considering different stages of infection that are distant in time. This approach has previously been used to detect biomarkers and drug targets for a range of disorders [30, 31, 73]. The result is a global picture of metabolic capabilities associated with varying transcriptional activity at each time point (24, 48 and 72 h). Uninfected cells were used as a control to analyse the changes in metabolic capabilities (see Materials and methods). Figure 3A shows the average activity fold change across reactions for all the pathways in the human metabolic network, while detailed results can be found in Tables S4 and S5. Viral metabolic perturbations affect a range of pathways including amino acids, energy production and coenzymes, which appear directly linked to specific protein oversecretion. In Fig. 3C, we visualise the activity of main energy production pathways, providing a graphical account of local alterations. Further, Fig. 3B,D shows pathways enriched in DARs at each time point, respectively, based on RNA‐seq and proteomic data integration.
Fig. 3.
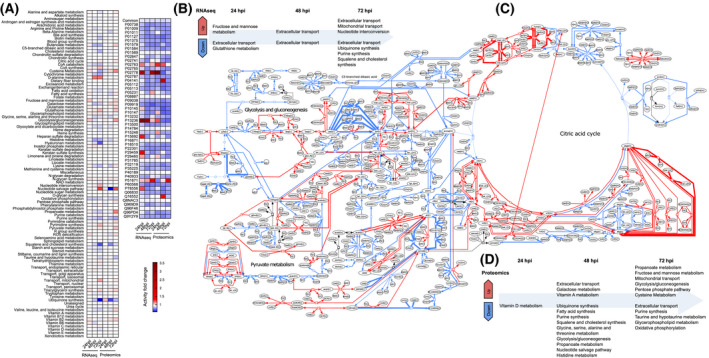
Genome‐scale metabolic modelling predicts alterations in multiple pathways following SARS‐CoV‐2 infection. (A) Pathway‐level temporal progression of Huh7 cell metabolic capabilities up to 72 h from infection. (B) Pathway enrichment of DARs identified at each time point in transcriptomic‐informed GSMMs. (C) Visualisation of alterations in central metabolism after 72 h from infection. (D) Pathway enrichment of DARs identified at each time point in proteomic‐informed GSMMs. In all the panels, red denotes increased activity potential for biochemical reactions and pathways, while blue indicates reduced activity.
To obtain a global overview of how SARS‐CoV‐2 affects cancer cell metabolism, we closely inspected altered pathways and individual reactions. In the following sections, we describe the main patterns identified and discuss their potential role in SARS‐CoV‐2 invasion inside host cells.
Genome‐scale modelling identifies RNA production upregulation
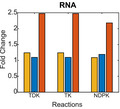
The nucleotide interconversion pathway shows evidence of increasing the uracil available to produce RNA by ensuring that the dUTP/dTTP ratio is low to minimise uracil incorporation into DNA [74]. The two top upregulated reactions at 72 hpi in the transcriptomic‐informed flux data are the catalysis of uridine monophosphate (UMP) to deoxyuridine monophosphate (dUMP) by deoxyuridine kinase and of thymidine to dTMP (deoxythymidine monophosphate) by thymidine kinase (see TDK and TK in Fig. 4A). dUMP is a precursor for de novo dTTP (thymidine triphosphate) synthesis. Thymidylate synthase converts dUMP to TMP and dTMP to dTDP. NDP (nucleoside diphosphate) kinase then converts dTDP into dTTP. This last reaction is among the second most highly upregulated reactions (see NDPK in Fig. 4A). Also upregulated is the production of deoxyguanosine, uracil, cytosine and adenosine triphosphates, which are the components of RNA. The nucleotide salvage pathway shows upregulated adenine recovery from RNA/DNA degradation by adenine phosphoribosyltransferase, which would coincide with an increased demand for adenine to bind to uracil. Within the pyrimidine synthesis pathway, three out of the top four upregulated pathways are involved in the production of dTTP. Proteomic‐informed flux data begin to show these same patterns at 72 hpi, apart from deoxyuridine kinase and thymidine kinase, which are upregulated at 48 hpi. Altogether, these results thus suggest increased viral RNA production.
Fig. 4.
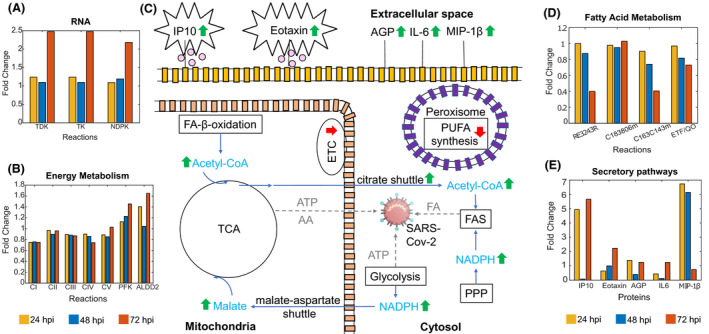
Summary of main metabolic changes in SARS‐CoV‐2‐infected Huh7 cells at 72 hpi. The mitochondrial ETC and oxidative phosphorylation are downregulated, reducing ATP production from the TCA cycle but potentially providing protection from apoptosis. The components of the TCA producing intermediates for viral amino acid synthesis are upregulated. While the production of ATP is reduced in the TCA, the energy‐producing reactions of glycolysis are upregulated, producing ATP for the viral production of amino acids and RNA. Fatty acid synthesis is also upregulated, producing fatty acids for the synthesis of the viral envelope. The production of anti‐inflammatory polyunsaturated fatty acids and the electron transport chain are both downregulated. Upregulation of inflammatory proteins IP10, eotaxin, MIP‐1β and IL‐6 has been linked with the cytokine storm. AGP is an acute‐phase protein involved in the inflammatory response. Green upward arrows represent upregulation. Red downward arrows represent downregulation. (A) Upregulated transcriptomic‐informed flux reactions involved in RNA production. (B) Dysregulated transcriptomic‐informed flux reactions involved in oxidative phosphorylation and glycolysis. (C) Visual overview of main metabolic changes. (D) Dysregulated transcriptomic‐informed flux reactions involved in fatty acid metabolism; RE3243R, production of polyunsaturated fatty acids, for example oleic acid; C183806m, final step of β‐oxidation producing octanyl‐CoA; C163C143m, first step of β‐oxidation; ETF/QO, electron transfer from octanoyl‐CoA to ubiquinone. (E) Upregulated transcriptomic‐informed secretory pathways involved in the immune response. PPP, pentose phosphate pathway; FAS, fatty acid synthesis; FA, fatty acid, TCA, citric acid cycle; AA, amino acid; FA‐β‐oxidation, fatty acid‐β‐oxidation; ETC, electron transport chain; PUFA, polyunsaturated fatty acid; IP10, interferon γ‐induced protein‐10; AGP, alpha‐1 acid glycoprotein; IL‐6, interleukin 6; MIP‐1β, macrophage inflammatory protein‐1β; TDK, deoxyuridine kinase; TK, thymidine kinase; NDPK, NDP kinase; C183806m, FAOXC183806m; C163C143m, FAOXC163C143m; ETF/QO, electron transfer flavoprotein/electron transfer flavoprotein–ubiquinone oxidoreductase, CI, NADH: ubiquinone oxidoreductase; CII, succinate dehydrogenase; CIII, cytochrome reductase; CIV, cytochrome c oxidase; CV, ATP synthase; PFK, phosphofructokinase; ALDD2, aldehyde dehydrogenase.
Altered energy metabolism for viral replication
For both sets of flux data, the components of the mitochondrial electron transport chain/oxidative phosphorylation are mostly downregulated through all the time points, indicating reduced ATP production from the citric acid cycle (TCA) [see transcriptomic‐informed results in (Fig. 4B) CI, CII, CIII, CIV, CV]. As shown in Fig. 3, the synthesis of ubiquinone, which is part of the mitochondrial electron transport chain, is progressively downregulated in the transcriptomic‐informed flux data and also downregulated in the proteomic‐informed flux data. Succinate dehydrogenase (SDH) is the only enzyme in the citric acid cycle that is also involved in the electron transport chain and is downregulated over all the time points in the transcriptomic‐informed flux data [75]. The resulting accumulation of succinate and fumarate led to an upsurge in mitochondrial transport of these metabolites into the cytosol (Fig. 3). Downregulation of the electron transport chain, including SDH, may indicate that COVID‐19 uses a similar mechanism to cancer cells to avoid oxidative stress initiating cell death while it is replicating [76, 77]. In the transcriptomic‐informed flux data, mitochondrial TCA reactions producing intermediates for amino acid synthesis are downregulated at 24 hpi, then become progressively upregulated for viral protein synthesis. Paradoxically, while citrate synthase is downregulated, the citrate shuttle and citrate lyase are upregulated providing acetyl‐CoA for fatty acid synthesis. The cytosolic reactions of the TCA are progressively upregulated through the time points, generating energy in the form of NADH/NADPH for fatty acid synthesis needed for the viral envelope. The NADPH producing enzymes in the pentose phosphate pathway, glucose 6‐phosphate dehydrogenase and phosphogluconate dehydrogenase, which also provide NADH/NADPH for fatty acid synthesis, are upregulated. These may have been influenced by maximising phosphogluconate dehydrogenase in the FVA. The proteomic‐informed flux data show very similar results for the TCA and pentose phosphate pathways.
In parallel, the majority of the reactions of glycolysis in both sets of flux data are upregulated or unchanged through all time points, with a dip in some upregulated reactions at 48 hpi. The main rate‐limiting enzyme of glycolysis, phosphofructokinase, is progressively upregulated through all time points in the transcriptomic‐informed flux data (see PFK in Fig. 4B). At the final 72‐h time point, the largest upregulation in the transcriptomic‐informed flux data occurs in the reaction catalysed by aldehyde dehydrogenase (see ALDD2 in Fig. 4B). Aldehyde dehydrogenase consumes the oxidative stressor acetaldehyde and produces energy in the form of NADH/NADPH. Interestingly, this reaction occurs in the cytosol, where viral replication takes place and is a known indicator of oxidative stress/lipid peroxidation [78]. The NADH from glycolysis is transported into the mitochondria to be used by the TCA. Transport is facilitated by both the malate‐aspartate shuttle, which is catalysed by the enzyme malate dehydrogenase, and the glycerol‐phosphate shuttle, which is catalysed by glycerol‐3‐phosphate dehydrogenase [79]. Here, in both sets of data, the flux of glycerol‐phosphate shuttle is unchanged, but the malate‐aspartate shuttle is upregulated, with the mitochondrial malate dehydrogenase reaction being the most highly upregulated in the transcriptomic‐informed TCA flux data. Within the glycolytic pathway for both sets of data, ATP production from pyruvate kinase is upregulated suggesting that glycolysis, rather than oxidative phosphorylation, is favoured by the virus for the generation of ATP for amino acid and RNA production [80].
Overall, proteomic‐informed flux data were in broad agreement with the transcriptomics‐informed flux data revealing greater influences on pathways at earlier time points. See Fig. 3B for a visual representation of the changes in the central energy metabolism.
Dysregulation of fatty acid metabolism
The largest perturbation in fatty acid synthesis occurs in the downregulation of desaturation of fatty acids in the endoplasmic reticulum, where polyunsaturated fatty acids such as oleic acid are produced (Fig. 4D, RE3243R). This downregulation is significantly more pronounced in the transcriptomics‐informed flux data as compared to the proteomic‐informed flux data. The downregulation of unsaturated fatty acid synthesis is a known effect of enveloped virus infection [81]. Yan et al. [82] found that oleic acid was upregulated in coronavirus HCoV‐229E infection in HuH7 cells, though this study did not include COVID‐19. This result could either be a result of the missing phospholipase A2 pathway enzymes or indicate the downregulation of unsaturated fatty acids involved in the host cell immune response [83, 84].
Moreover, the elongation stage of fatty acid synthesis in the cytosol is upregulated at 72 hpi, which could be linked to the production of the viral cell envelope. In the transcriptomic‐informed flux data, there is indeed an upregulation 4 of the 5 steps of the synthesis of CoA (Fig. 3), which is needed for the citric acid cycle and fatty acid metabolism [85], in conjunction with a decrease in the activity of the majority of enzymes associated with β‐oxidation of fatty acids and cholesterol synthesis. The breakdown of γ‐linolenoyl‐CoA to octanoyl‐CoA is the only β‐oxidation reaction that shows an increase in activity in both sets of the flux data (Fig. 4D, FAOXC183806m). This reaction is the final step of β‐oxidation in the mitochondria and provides acetyl‐CoA for the citric acid cycle. In the transcriptomic‐informed flux data, the transfer of electrons from octanoyl‐CoA to ubiquinone is progressively downregulated showing further evidence of the downregulation of the electron transport chain (Fig. 4D, ETF/QO) [86]. These perturbations are more pronounced in the transcriptomics‐informed flux data as compared to the proteomic‐informed flux data.
Secretome inflammatory biomarkers
As expected, protein secretion exhibited a widespread upregulation, though visible especially at 24 and 72 hpi, with 10 upregulated cytokine/chemokine secretory pathways. Macrophage inflammatory protein‐1β (MIP‐1β/CCL4), IP10/CXCL10, IL‐12B and eotaxin (CCL11) were found to be upregulated in the proteomic‐informed data, while in the transcriptomics‐informed data upregulation was found for IL‐6, IL‐8, IL‐12A, vascular endothelial growth factor A (VEGFA), chemokine (C‐C motif) ligand 20 (CCL20) and alpha‐1 acid glycoprotein (AGP). Analysis of the metabolic alterations further revealed high upregulation of IL‐17A, IL‐15 and IL‐7.
As shown in Fig. 4E, MIP‐1β was the most highly expressed cytokine/chemokine at 24 and 48 hpi and was then replaced by IP10 at 72 hpi. IP10 has been reported as a good biomarker of SARS‐CoV‐2 disease progression and severity [87, 88, 89] and has been implicated in the cytokine storm with higher levels leading to more severe disease [90, 91]. Eotaxin was the second most highly upregulated protein at 72 hpi followed by AGP and IL‐6, respectively. In the proteomic‐informed flux data, only eotaxin was upregulated at 72 hpi. MIP‐1β is a proinflammatory chemokine known to induce cytokines such as IL‐6 and TNF‐α and could be an early predictor of severe disease [92]. Eotaxin has the potential to act as a marker to predict disease severity and outcome. Although it is seen to progressively elevate in patients with mild symptoms, eosinopenia has been detected in patients with severe disease with a subsequent increase being associated with a good outcome [93, 94, 95]. AGP is an acute‐phase protein, and plasma levels of AGP have previously been found to be correlated with influenza disease progression and could be a potential novel biomarker for SARS‐CoV‐2 disease progression [96]. IL‐6 has been implicated in the cytokine storm and has received a lot of attention as a potential therapeutic target with clinical trials of IL‐6 receptor blockers tocilizumab, siltuximab and sarilumab currently underway [97, 98, 99, 100]. IL‐17A induces inflammatory cytokines involved in the cytokine storm such as IL‐6 and IL‐8 and has been linked with severe disease [101, 102]. IL‐17A blockers such as secukinumab have been proposed as a treatment for COVID‐19, although their administration would have to be timed carefully [103, 104]. IL‐7 has a role in maintaining NKT (natural killer T cells) and T cells in liver cells [105] and promotes cell survival [106]. IL‐15 is an important regulator of the immune response and a known antiviral cytokine [107, 108, 109]. Increased levels of IL‐15 may also be a biomarker of severe lymphopenia requiring longer hospital care [110]. The decrease in the production of hyaluronan shown in the pathway‐level flux average (Fig. 3A) is a further indicator of liver damage [111]. Interestingly, the increase in d‐alanine metabolism also detected in the metabolic fluxes may be a mechanism for producing cytotoxic oxidative stress as part of the innate immune response [112] (Fig. 3A).
Significance for COVID‐19 treatments
Figure 4 shows a visual summary of the key metabolic changes identified at 72 hpi. Taken together, they can be utilised for hypothesising improved treatment strategies for COVID‐19 in cancer patients. The upregulated secretory proteins identified in this study provide some insights into therapeutic treatments for COVID‐19. Upregulation of IL‐7 is an indicator of T‐cell exhaustion and elevated inflammatory cytokine characteristics of COVID‐19 [113]. IL‐7 has a protective role in maintaining NKT (natural killer T cells) and T cells in liver cells [105] and promotes cell survival [106]. The levels of IL‐7 could be elevated in COVID‐19 patients by using IL7r (recombinant IL‐7) therapy, which has been used to treat HIV patients and was able to restore CD4+ T cells while being well‐tolerated [114]. Secondly, MitoQ (mitochondrial‐targeted ubiquinone) has powerful antioxidant effects that could be used as a treatment to protect against mitochondrial electron transport chain dysfunction/oxidative stress, compensating for the increased downregulation of ETC ubiquinone seen in COVID‐19 infection and restoring T‐cell homeostasis [115, 116]. Finally, our findings support dietary supplementation of unsaturated fatty acids administered orally or intravenously to counteract their downregulation by COVID‐19, which has been suggested as a treatment to suppress inflammation [83, 84, 117].
It should be noted that individuals suffering from cancer may already be taking medication. While the treatments suggested here do not interact with common liver cancer drugs according to the interactions listed in the Joint Formulary Committee [118] (Table S6), caution should be taken to ensure that any COVID‐19 treatments are compatible with existing treatment regimes.
Conclusions
While transcriptomic‐only approaches can provide insights into changes in gene expression caused by viral infection, translating these data into metabolic data can give deeper and mechanistic insights into the effects of viral infection on the host cell. Here, we use genome‐scale‐metabolic modelling of Huh7 cells with transcriptomic‐ and proteomic‐informed FVA to explore the metabolic effects of altered gene expression from COVID‐19‐infected cancer cells on cellular phenotype. This method allows the identification of dysregulation at the enzyme/reaction level, in contrast to previous studies of metabolism. Our models were constrained to represent the viral‐infected cell using evidence from literature and the pathway analysis of DEGs.
A pathway analysis revealed an upregulation of PPARalpha, known to be involved in fatty acid metabolism and regulation of the immune system [58]. The analysis of the resulting transcriptomic‐ and proteomic‐informed flux data revealed perturbations in four main areas of metabolism, RNA synthesis, energy production, fatty acid synthesis and the secretome. Proteomic‐informed flux data sometimes displayed a cascade effect where patterns in reactions/pathways appeared at a later time point than with flux data. Energy production showed an increased shift from the TCA to glycolysis, as evidenced by the upregulation of key glycolytic enzymes and the downregulation of the mitochondrial electron transport chain. This pseudohypoxic metabolic shift is in addition to the dysregulation already seen in cancer cells and may contribute to the increased risk seen in cancer patients infected with COVID‐19. Reactions involved in RNA production were found to be upregulated in accordance with viral replication needs. While the production of polyunsaturated acids was downregulated, the synthesis of fatty acids needed for the viral envelope was upregulated. Key inflammatory secretory proteins involved in the cytokine storm were found to be upregulated including IP10/CXCL10, eotaxin, MIP‐1β and IL‐6. Finally, we suggest therapeutic treatments based on mediating the inflammatory response and metabolic key changes that enable the virus to replicate in the host cancer cell. Future studies could enhance our understanding by investigating multicellular metabolic changes as well as how individual data such as gender, age and ethnicity affect the metabolic changes caused by COVID‐19 infection. This will facilitate the investigation of more personalised therapeutic interventions for cancer patients.
Author contributions
CA conceived and supervised the study; LB and GB performed the RNA‐seq data analyses; EY and GZ developed the software and performed the simulations; EY and GZ analysed the data; all authors contributed to drafting the manuscript; EY, GZ and CA produced the final version of the manuscript.
Supporting information
Table S1. Differentially expressed genes and proteins.
Table S2. Enrichment analysis of differentially expressed genes.
Table S3. Enrichment analysis of differentially expressed proteins.
Table S4. Transcriptomic‐informed flux rates for reactions and pathways.
Table S5. Proteomic‐informed flux rates for reactions and pathways.
Table S6. Common liver cancer drugs and their interactions.
Acknowledgements
We would like to thank Ujjwal Neogi and Ali Mirazimi, from the Karolinska Institute, Stockholm, for sharing the quantile‐normalised proteomics data. EY, GZ and CA would like to acknowledge funding from a Children's Liver Disease Foundation Research Grant and UKRI Research England's THYME project.
Elisabeth Yaneske and Guido Zampieri contributed equally to this article
Edited by Lukas Alfons Huber
Data accessibility
All software and data produced in the study are available at: https://github.com/Angione‐Lab/Metabolic‐modelling‐of‐SARS‐CoV‐2‐in‐cancer‐cells.
References
- 1. Dong E, Du H and Gardner L (2020) An interactive web‐based dashboard to track COVID‐19 in real time. Lancet Infect Dis 20, 533–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Skegg D, Gluckman P, Boulton G, Hackmann H, Karim SSA, Piot P and Woopen C (2021) Future scenarios for the COVID‐19 pandemic. Lancet 397, 777–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Fung M and Babik JM (2021) COVID‐19 in immunocompromised hosts: what we know so far. Clin Infect Dis 72, 340–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chin IS, Galavotti S, Yip KP, Curley H, Arnold R, Sharma‐Oates A, Chegwidden L, Lee SI, Lee LY, Pinato DJ et al. (2020) Influence of clinical characteristics and anticancer therapy on outcomes from SARS‐CoV‐2 infection: a systematic review and meta‐analysis of 5,678 cancer patients. medRxiv. [Google Scholar]
- 5. Guarneri V, Bassan F, Zagonel V, Milella M, Zaninelli M, Cattelan AM, Vianello A, Gori S, Aprile G, Azzarello G et al. (2021) Epidemiology and clinical course of severe acute respiratory syndrome coronavirus 2 infection in cancer patients in the Veneto Oncology Network: The Rete Oncologica Veneta covID19 study. Eur J Cancer 147, 120–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. van der Veldt AAM, Oosting SF, Dingemans A‐M, Fehrmann RSN, GeurtsvanKessel C, Jalving M, Rimmelzwaan GF, Kvistborg P, Blank CU, Smit EF et al. (2021) COVID‐19 vaccination: the VOICE for patients with cancer. Nat Med 27, 568–569. [DOI] [PubMed] [Google Scholar]
- 7. Ayres JS (2020) A metabolic handbook for the COVID‐19 pandemic. Nat Metab 2, 572–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Richardson S, Hirsch JS, Narasimhan M, Crawford JM, McGinn T, Davidson KW, Barnaby DP, Becker LB, Chelico JD, Cohen SL et al. (2020) Presenting characteristics, comorbidities, and outcomes among 5700 patients hospitalized with COVID‐19 in the New York City area. JAMA 323, 2052–2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Guan W‐J, Ni Z‐Y, Hu YU, Liang W‐H, Ou C‐Q, He J‐X, Liu L, Shan H, Lei C‐L, Hui DS et al. (2019) Clinical characteristics of coronavirus disease 2019 in China. N Engl J Med 382, 1708–1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhang C, Shi L and Wang FS (2020) Liver injury in COVID‐19: management and challenges. Lancet Gastroenterol Hepatol 5, 428–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Huang C, Wang Y, Li X, Ren L, Zhao J, Hu YI, Zhang LI, Fan G, Xu J, Gu X et al. (2020) Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 395, 497–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ding Z‐Y, Li G‐X, Chen L, Shu C, Song J, Wang W, Wang Y‐W, Chen Q, Jin G‐N, Liu T‐T et al. (2021) Association of liver abnormalities with in‐hospital mortality in patients with COVID‐19. J Hepatol 74, 1295–1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Xu L, Liu J, Lu M, Yang D and Zheng X (2020) Liver injury during highly pathogenic human coronavirus infections. Liver Int 40, 998–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chai X, Hu L, Zhang Y, Han W, Lu Z, Ke A, Zhou J, Shi G, Fang N, Fan J et al. (2020) Specific ACE2 expression in cholangiocytes may cause liver damage after 2019‐nCoV infection. bioRxiv [PREPRINT]. [Google Scholar]
- 15. Chu H, Chan J‐W, Yuen T‐T, Shuai H, Yuan S, Wang Y, Hu B, Yip C‐Y, Tsang J‐L, Huang X et al. (2020) Comparative tropism, replication kinetics, and cell damage profiling of SARS‐CoV‐2 and SARS‐CoV with implications for clinical manifestations, transmissibility, and laboratory studies of COVID‐19: an observational study. Lancet Microbe 1, e14–e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Marjot T, Webb GJ, Barritt AS, Moon AM, Stamataki Z, Wong VW and Barnes E (2021) COVID‐19 and liver disease: mechanistic and clinical perspectives. Nat Rev Gastroenterol Hepatol 18, 348–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Martinez MA and Franco S (2021) Impact of COVID‐19 in liver disease progression. Hepatol Commun 5, 1138–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Appelberg S, Gupta S, Svensson Akusjärvi S, Ambikan AT, Mikaeloff F, Saccon E, Végvári Á, Benfeitas R, Sperk M, Ståhlberg M et al. (2020) Dysregulation in Akt/mTOR/HIF‐1 signaling identified by proteo‐transcriptomics of SARS‐CoV‐2 infected cells. Emerg Microbes Infect 9, 1748–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Saxton RA and Sabatini DM (2017) mTOR signaling in growth, metabolism, and disease. Cell 169, 361–371. [DOI] [PubMed] [Google Scholar]
- 20. Delafiori J, Navarro LC, Siciliano RF, de Melo GC, Busanello ENB, Nicolau JC, Sales GM, de Oliveira AN, Val FFA, de Oliveira DN et al. (2021) Covid‐19 automated diagnosis and risk assessment through metabolomics and machine learning. Anal Chem 93, 2471–2479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wu D, Shu T, Yang X, Song J‐X, Zhang M, Yao C, Liu W, Huang M, Yu Y, Yang Q et al. (2020) Plasma metabolomic and lipidomic alterations associated with COVID‐19. Natl Sci Rev 7, 1157–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Shen BO, Yi X, Sun Y, Bi X, Du J, Zhang C, Quan S, Zhang F, Sun R, Qian L et al. (2020) Proteomic and metabolomic characterization of COVID‐19 patient sera. Cell 182, 59–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Blasco H, Bessy C, Plantier L, Lefevre A, Piver E, Bernard L, Marlet J, Stefic K, Benz‐de Bretagne I, Cannet P et al. (2020) The specific metabolome profiling of patients infected by SARS‐COV‐2 supports the key role of tryptophan‐nicotinamide pathway and cytosine metabolism. Sci Rep 10, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Song J‐W, Lam SM, Fan X, Cao W‐J, Wang S‐Y, Tian HE, Chua GH, Zhang C, Meng F‐P, Xu Z et al. (2020) Omics‐driven systems interrogation of metabolic dysregulation in COVID‐19 pathogenesis. Cell Metab 32, 188–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Thomas T, Stefanoni D, Reisz JA, Nemkov T, Bertolone L, Francis RO, Hudson KE, Zimring JC, Hansen KC, Hod EA et al. (2020) COVID‐19 infection alters kynurenine and fatty acid metabolism, correlating with IL‐6 levels and renal status. JCI Insight 5, e140327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ajaz S, McPhail MJ, Singh KK, Mujib S, Trovato FM, Napoli S and Agarwal K (2021) Mitochondrial metabolic manipulation by SARS‐CoV‐2 in peripheral blood mononuclear cells of patients with COVID‐19. Am J Physiol Cell Physiol 320, C57–C65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Heer CD, Sanderson DJ, Voth LS, Alhammad YMO, Schmidt MS, Trammell SAJ, Perlman S, Cohen MS, Fehr AR and Brenner C (2020) Coronavirus infection and PARP expression dysregulate the NAD metabolome: an actionable component of innate immunity. J Biol Chem 295, 17986–17996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Geng J and Nielsen J (2017) In silico analysis of human metabolism: Reconstruction, contextualization and application of genome‐scale models. Curr Opin Syst Biol 2, 29–38. [Google Scholar]
- 29. Vijayakumar S, Conway M, Lió P and Angione C (2018) Seeing the wood for the trees: a forest of methods for optimization and omic‐network integration in metabolic modelling. Brief Bioinform 19, 1218–1235. [DOI] [PubMed] [Google Scholar]
- 30. Moolamalla STR and Vinod PK (2020) Genome‐scale metabolic modelling predicts biomarkers and therapeutic targets for neuropsychiatric disorders. Comput Biol Med 125, 103994. [DOI] [PubMed] [Google Scholar]
- 31. Folger O, Jerby L, Frezza C, Gottlieb E, Ruppin E and Shlomi T (2011) Predicting selective drug targets in cancer through metabolic networks. Mol Syst Biol 7, 501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Angione C (2018) Integrating splice‐isoform expression into genome‐scale models characterizes breast cancer metabolism. Bioinformatics 34, 494–501. [DOI] [PubMed] [Google Scholar]
- 33. Renz A, Widerspick L and Dräger A (2020) FBA reveals guanylate kinase as a potential target for antiviral therapies against SARS‐CoV‐2. Bioinformatics 36 (Suppl 2), i813–i821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Renz A, Widerspick L and Dräger A (2021) Genome‐scale metabolic model of infection with SARS‐CoV‐2 mutants confirms guanylate kinase as robust potential antiviral target. Genes 12, 796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Seyfried TN, Flores RE, Poff AM and D'Agostino DP (2014) Cancer as a metabolic disease: implications for novel therapeutics. Carcinogenesis 35, 515–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chen S, Zhou Y, Chen Y and Gu J (2018) fastp: an ultra‐fast all‐in‐one FASTQ preprocessor. Bioinformatics 34, i884–i890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M and Gingeras TR (2013) STAR: ultrafast universal RNA‐seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yates AD, Achuthan P, Akanni W, Allen J, Allen J, Alvarez‐Jarreta J, Amode MR, Armean IM, Azov AG, Bennett R et al. (2020) Ensembl 2020. Oxford University Press. ftp://ftp.ensembl.org/pub/release‐99/fasta/homo_sapiens/dna/Homo_sapiens.GRCh38.dna.primary_assembly.fa.gz, version 99.
- 39. Yates AD, Achuthan P, Akanni W, Allen J, Allen J, Alvarez‐Jarreta J, Amode MR, Armean IM, Azov AG, Bennett R et al. (2020) Ensembl 2020. Oxford University Press. ftp://ftp.ensembl.org/pub/release‐99/gtf/homo_sapiens/Homo_sapiens.GRCh38.99.gtf.gz, version 99.
- 40. Li B and Dewey CN (2011) RSEM: accurate transcript quantification from RNA‐Seq data with or without a reference genome. BMC Bioinformatics 12, 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. McCarthy DJ, Chen Y and Smyth GK (2012) Differential expression analysis of multifactor RNA‐Seq experiments with respect to biological variation. Nucleic Acids Res 40, 4288–4297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ritchie ME, Phipson B, Wu DI, Hu Y, Law CW, Shi W and Smyth GK (2015) limma powers differential expression analyses for RNAsequencing and microarray studies. Nucleic Acids Res 43, e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Durinck S, Spellman PT, Birney E and Huber W (2009) Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat Protoc 4, 1184–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kanehisa M, Sato Y, Kawashima M, Furumichi M and Tanabe M (2016) KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res 44, D457–D462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Fabregat A, Jupe S, Matthews L, Sidiropoulos K, Gillespie M, Garapati P, Haw R, Jassal B, Korninger F, May B et al. (2018) The reactome pathway knowledgebase. Nucleic Acids Res 46, D649–D655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Swainston N, Smallbone K, Hefzi H, Dobson PD, Brewer J, Hanscho M, Zielinski DC, Ang KS, Gardiner NJ, Gutierrez JM et al. (2016) Recon 2.2: from reconstruction to model of human metabolism. Metabolomics 12, 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Gutierrez JM, Feizi A, Li S, Kallehauge TB, Hefzi H, Grav LM, Ley D, Baycin Hizal D, Betenbaugh MJ, Voldborg B et al. (2020) Genome‐scale reconstructions of the mammalian secretory pathway predict metabolic costs and limitations of protein secretion. Nat Commun 11, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Liu K, Fang Y‐Y, Deng Y, Liu W, Wang M‐F, Ma J‐P, Xiao W, Wang Y‐N, Zhong M‐H, Li C‐H et al. (2020) Clinical characteristics of novel coronavirus cases in tertiary hospitals in Hubei Province. Chin Med J 133, 1025–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wang W, Liu X, Wu S, Chen S, Li Y, Nong L, Lie P, Huang L, Cheng L, Lin Y et al. (2020) Definition and risks of cytokine release syndrome in 11 critically ill COVID‐19 patients with pneumonia: analysis of disease characteristics. J Infect Dis 222, 1444–1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zhang H, Shen J, Xu H, Sun J, Yin W, Zuo YU, Wang Z, Xiong F, Zhang Y & Lin H et al. (2020) Activation of peroxiredoxin 1 by fluvastatin effectively protects from inflammation and SARS‐CoV‐2. Preprint on webpage at https://papers.ssrn.com/sol3/papers.cfm?abstract_id=3606782
- 51. Chen J‐H, Chang Y‐W, Yao C‐W, Chiueh T‐S, Huang S‐C, Chien K‐Y, Chen A, Chang F‐Y, Wong C‐H and Chen Y‐J (2004) Plasma proteome of severe acute respiratory syndrome analyzed by two‐dimensional gel electrophoresis and mass spectrometry. Proc Natl Acad Sci USA 101, 17039–17044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Bjerregaard A, Laing IA, Poulsen N, Backer V, Sverrild A, Fally M, Khoo S‐K, Barrett L, Baltic S, Thompson PJ et al. (2017) Characteristics associated with clinical severity and inflammatory phenotype of naturally occurring virus‐induced exacerbations of asthma in adults. Respir Med 123, 34–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wan J, Sun W, Li X, Ying W, Dai J, Kuai X, Wei H, Gao X, Zhu Y, Jiang Y et al. (2006) Inflammation inhibitors were remarkably up‐regulated in plasma of severe acute respiratory syndrome patients at progressive phase. Proteomics 6, 2886–2894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Perkins A, Nelson KJ, Parsonage D, Poole LB and Karplus PA (2015) Peroxiredoxins: guardians against oxidative stress and modulators of peroxide signaling. Trends Biochem Sci 40, 435–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Franko A, Hartwig S, Kotzka J, Ruoß M, Nüssler AK, Königsrainer A, Häring H‐U, Lehr S and Peter A (2019) Identification of the secreted proteins originated from primary human hepatocytes and HepG2 cells. Nutrients 11, 1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Aller S, Scott A, Sarkar‐Tyson M and Soyer OS (2018) Integrated human‐virus metabolic stoichiometric modelling predicts host‐based antiviral targets against Chikungunya, Dengue and Zika viruses. J R Soc Interface 15, 20180125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Angione C and Lió P (2015) Predictive analytics of environmental adaptability in multi‐omic network models. Sci Rep 5, 15147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Christofides A, Konstantinidou E, Jani C and Boussiotis VA (2020) The role of peroxisome proliferator‐activated receptors (PPAR) in immune responses. Metabolism 114, 154338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Yoon M, Lee H, Jeong S, Kim J‐J, Nicol CJ, Nam KW, Kim M, Cho BG and Oh GT (2003) Peroxisome proliferator‐activated receptor α is involved in the regulation of lipid metabolism by ginseng. Br J Pharmacol 138, 1295–1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Zuo T, Liu Q, Zhang F, Lui GCY, Tso EYK, Yeoh YK, Chen Z, Boon SS, Chan FKL, Chan PKS et al. (2020) Depicting SARS‐CoV‐2 faecal viral activity in association with gut microbiota composition in patients with COVID‐19. Gut 70, 276–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Abu‐Farha M, Thanaraj TA, Qaddoumi MG, Hashem A, Abubaker J and Al‐Mulla F (2020) The role of lipid metabolism in COVID‐19 virus infection and as a drug target. Int J Mol Sci 21, 3544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Chen X, Shang L, Deng S, Li P, Chen K, Gao T, Zhang X, Chen Z and Zeng J (2020) Peroxisomal oxidation of erucic acid suppresses mitochondrial fatty acid oxidation by stimulating malonyl‐CoA formation in the rat liver. J Biol Chem 295, 10168–10179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Sender R, Bar‐On YM, Gleizer S, Bernshtein B, Flamholz A, Phillips R and Milo R (2021) The total number and mass of SARS‐CoV‐2 virions. Proc Natl Acad Sci USA 118, e2024815118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Heirendt L, Arreckx S, Pfau T, Mendoza SN, Richelle A, Heinken A, Haraldsdóttir HS, Wachowiak J, Keating SM, Vlasov V et al. (2019) Creation and analysis of biochemical constraint based models using the COBRA Toolbox v. 3.0. Nat Protoc 14, 639–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Noronha A, Daníelsdóttir AD, Gawron P, Jóhannsson F, Jónsdóttir S, Jarlsson S, Gunnarsson JP, Brynjólfsson S, Schneider R, Thiele I et al. (2017) ReconMap: an interactive visualization of human metabolism. Bioinformatics 33, 605–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Yaneske E and Angione C (2018) Mechanistic effects of influenza in bronchial cells through poly‐omic genome‐scale modelling. http://www.iwbdaconf.org/2018/, iWBDA 2018: 10th International Workshop on Bio‐Design Automation; Conference date: 31‐07‐2018 Through 03‐08‐2018. Preprint on webpage at https://research.tees.ac.uk/en/publications/mechanistic‐effects‐of‐influenza‐in‐bronchial‐cells‐through‐poly‐
- 67. Lewis JE and Kemp ML (2021) Integration of machine learning and genome‐scale metabolic modeling identifies multi‐omics biomarkers for radiation resistance. Nat Commun 12, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Occhipinti A, Hamadi Y, Kugler H, Wintersteiger C, Yordanov B and Angione C (2020) Discovering essential multiple gene effects through large scale optimization: an application to human cancer metabolism. IEEE/ACM Trans Comput Biol Bioinform, 1. [DOI] [PubMed] [Google Scholar]
- 69. Zampieri G, Vijayakumar S, Yaneske E and Angione C (2019) Machine and deep learning meet genome‐scale metabolic modeling. PLoS Comput Biol 15, e1007084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Banerjee A, Czinn SJ, Reiter RJ and Blanchard TG (2020) Crosstalk between endoplasmic reticulum stress and anti‐viral activities: a novel therapeutic target for COVID‐19. Life Sci 255, 117842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Angione C (2019) Human systems biology and metabolic modelling: a review—from disease metabolism to precision medicine. Biomed Res Int 2019, 8304260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Opdam S, Richelle A, Kellman B, Li S, Zielinski DC and Lewis NE (2017) A systematic evaluation of methods for tailoring genome‐scale metabolic models. Cell Syst 4, 318–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Salazar DA, Rodríguez‐López A, Herreño A, Barbosa H, Herrera J, Ardila A, Barreto GE, González J and Alméciga‐Díaz CJ (2016) Systems biology study of mucopolysaccharidosis using a human metabolic reconstruction network. Mol Genet Metab 117, 129–139. [DOI] [PubMed] [Google Scholar]
- 74. Vértessy BG and Tóth J (2009) Keeping uracil out of DNA: physiological role, structure and catalytic mechanism of dUTPases. Acc Chem Res 42, 97–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Huang S and Millar AH (2013) Succinate dehydrogenase: the complex roles of a simple enzyme. Curr Opin Plant Biol 16, 344–349. [DOI] [PubMed] [Google Scholar]
- 76. Guzzo G, Sciacovelli M, Bernardi P and Rasola A (2014) Inhibition of succinate dehydrogenase by the mitochondrial chaperone TRAP1 has anti‐oxidant and anti‐apoptotic effects on tumor cells. Oncotarget 5, 11897–11908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. King A, Selak M and Gottlieb E (2006) Succinate dehydrogenase and fumarate hydratase: linking mitochondrial dysfunction and cancer. Oncogene 25, 4675–4682. [DOI] [PubMed] [Google Scholar]
- 78. Singh S, Brocker C, Koppaka V, Chen Y, Jackson BC, Matsumoto A, Thompson DC and Vasiliou V (2013) Aldehyde dehydrogenases in cellular responses to oxidative/electrophilicstress. Free Radic Biol Med 56, 89–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Singh G (2014) Mitochondrial FAD‐linked glycerol‐3‐phosphate dehydrogenase: a target for cancer therapeutics. Pharmaceuticals 7, 192–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Codo AC, Davanzo GG, Monteiro LDB, de Souza GF, Muraro SP, Virgilio‐da‐Silva JV, Prodonoff JS, Carregari VC, de Biagi Junior CAO, Crunfli F et al. (2020) Elevated glucose levels favor SARS‐CoV‐2 infection and monocyte response through a HIF‐1α/glycolysis‐dependent axis. Cell Metab 32, 437–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Kohn A, Gitelman J and Inbar M (1980) Unsaturated free fatty acids inactivate animal enveloped viruses. Adv Virol 66, 301–307. [DOI] [PubMed] [Google Scholar]
- 82. Yan B, Chu H, Yang D, Sze K‐H, Lai P‐M, Yuan S, Shuai H, Wang Y, Kao R‐T, Chan J‐W et al. (2019) Characterization of the lipidomic profile of human coronavirus infected cells: implications for lipid metabolism remodeling upon coronavirus replication. Viruses 11, 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Ghaffari S, Roshanravan N, Tutunchi H, Ostadrahimi A, Pouraghaei M and Kafil B (2020) Oleoylethanolamide, a bioactive lipid amide, as a promising treatment strategy for coronavirus/COVID‐19. Arch Med Res 51, 464–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Das UN (2021) Bioactive lipids in COVID‐19‐further evidence. Arch Med Res 52, 107–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Leonardi R and Jackowski S (2007) Biosynthesis of pantothenic acid and coenzyme A. EcoSal Plus 2, 1–28. 10.1128/ecosalplus.3.6.3.4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Watmough NJ and Frerman FE (2010) The electron transfer flavoprotein: ubiquinone oxidoreductases. Biochim Biophys Acta 1797, 1910–1916. [DOI] [PubMed] [Google Scholar]
- 87. Yang Y, Shen C, Li J, Yuan J, Wei J, Huang F, Wang F, Li G, Li Y, Xing LI et al. (2020) Plasma IP‐10 and MCP‐3 levels are highly associated with disease severity and predict the progression of COVID‐19. J Allergy Clin Immunol 146, 119–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Haroun RAH, Osman WH and Eessa AM (2021) Interferon‐γ‐induced protein 10 (IP‐10) and serum amyloid A (SAA) are excellent biomarkers for the prediction of COVID‐19 progression and severity. Life Sci 269, 119019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Lev S, Gottesman T, Sahaf Levin G, Lederfein D, Berkov E, Diker D, Zaidman A, Nutman A, Ilan Ber T, Angel A et al. (2021) Observational cohort study of IP‐10's potential as a biomarker to aid in inflammation regulation within a clinical decision support protocol for patients with severe COVID‐19. PLoS One 16, e0245296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Blot M, Jacquier M, Aho Glele L‐S, Beltramo G, Nguyen M, Bonniaud P, Prin S, Andreu P, Bouhemad B, Bour J‐B et al. (2020) CXCL10 could drive longer duration of mechanical ventilation during COVID‐19 ARDS. Crit Care 24, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Coperchini F, Chiovato L and Rotondi M (2021) Interleukin‐6, CXCL10 and infiltrating macrophages in COVID‐19‐related cytokine storm: not one for all but all for one! Front Immunol 12, 668507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Codina H, Vieitez I, Gutierrez‐Valencia A, Skouridou V, Martínez C, Patiño L, Botero‐Gallego M, Trujillo‐Rodríguez M, Serna‐Gallego A, Muñoz‐Muela E et al. (2021) Elevated anti‐SARS‐CoV‐2 antibodies and IL‐6, IL‐8, MIP‐1β, early predictors of severe COVID‐19. bioRxiv [PREPRINT]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Burgos‐Blasco B, Güemes‐Villahoz N, Santiago JL, Fernandez‐Vigo JI, Espino‐Paisán L, Sarriá B, García‐Feijoo J and Martinez‐de‐la‐Casa JM (2020) Hypercytokinemia in COVID‐19: tear cytokine profile in hospitalized COVID‐19 patients. Exp Eye Res 200, 108253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Cazzaniga M, Fumagalli LAM, D'angelo L, Cerino M, Bonfanti G, Fumagalli RM, Schiavo G, Lorini C, Lainu E, Terragni S et al. (2021) Eosinopenia is a reliable marker of severe disease and unfavourable outcome in patients with COVID‐19 pneumonia. Int J Clin Pract 75, e14047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Soni M (2021) Evaluation of eosinopenia as a diagnostic and prognostic indicator in COVID‐19 infection. Int J Lab Hematol 43 (Suppl 1), 137–141. [DOI] [PubMed] [Google Scholar]
- 96. Sidwell RW, Wong MH, Bailey KW, Barnard DL, Jackson MK and Smee DF (2001) Utilization of alpha‐1‐acid glycoprotein levels in the serum as a parameter for in vivo assay of influenza virus inhibitors. Antivir Chem Chemother 12, 359–365. [DOI] [PubMed] [Google Scholar]
- 97. Zhang C, Wu Z, Li J‐W, Zhao H and Wang G‐Q (2020) Cytokine release syndrome in severe COVID‐19: interleukin‐6 receptor antagonist tocilizumab may be the key to reduce mortality. Int J Antimicrob Agents 55, 105954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Salama C, Han J, Yau L, Reiss WG, Kramer B, Neidhart JD, Criner GJ, Kaplan‐Lewis E, Baden R, Pandit L et al. (2021) Tocilizumab in patients hospitalized with Covid‐19 pneumonia. N Engl J Med 384, 20–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Benucci M, Giannasi G, Cecchini P, Gobbi FL, Damiani A, Grossi V, Infantino M and Manfredi M (2020) COVID‐19 pneumonia treated with sarilumab: a clinical series of eight patients. J Med Virol 92, 2368–2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Gritti G, Raimondi F, RipamontiD RI, Landi F, Alborghetti L, Frigeni M, Damiani M, Micò C, Fagiuoli S, Cosentini R et al. (2020) IL‐6 signalling pathway inactivation with siltuximab in patients with COVID‐19 respiratory failure: an observational cohort study. MedRxiv. [Google Scholar]
- 101. Megna M, Napolitano M and Fabbrocini G (2020) May IL‐17 have a role in COVID‐19 infection? Med Hypotheses 140, 109749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Casillo GM, Mansour AA, Raucci F, Saviano A, Mascolo N, Iqbal AJ and Maione F (2020) Could IL‐17 represent a new therapeutic target for the treatment and/or management of COVID‐19‐related respiratory syndrome? Pharmacol Res 156, 104791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Pacha O, Sallman MA and Evans SE (2020) COVID‐19: a case for inhibiting IL‐17? Nat Rev Immunol 20, 345–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Bulat V, Situm M, Azdajic MD and Likic R (2021) Potential role of IL‐17 blocking agents in the treatment of severe COVID‐19? Br J Clin Pharmacol 87, 1578–1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Liang B, Hara T, Wagatsuma K, Zhang J, Maki K, Miyachi H, Kitano S, Yabe‐Nishimura C, Tani‐ichi S and Ikuta K (2012) Role of hepatocyte‐derived IL‐7 in maintenance of intrahepatic NKT cells and T cells and development of B cells in fetal liver. J Immunol 189, 4444–4450. [DOI] [PubMed] [Google Scholar]
- 106. Rathmell JC, Farkash EA, Gao W and Thompson CB (2001) IL‐7 enhances the survival and maintains the size of naive T cells. J Immunol 167, 6869–6876. [DOI] [PubMed] [Google Scholar]
- 107. Purton JF, Tan JT, Rubinstein MP, Kim DM, Sprent J and Surh CD (2007) Antiviral CD4+ memory T cells are IL‐15 dependent. J Exp Med 204, 951–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Verbist KC and Klonowski KD (2012) Functions of IL‐15 in anti‐viral immunity: multiplicity and variety. Cytokine 59, 467–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Guo Y, Luan L, Patil NK and Sherwood ER (2017) Immunobiology of the IL‐15/IL‐15Rα complex as an antitumor and antiviral agent. Cytokine Growth Factor Rev 38, 10–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Singh AK, Kasarpalkar NJ, Bhowmick SDK, Paradkar G, Talreja M, Shah K, Tiwari A, Palav H, Kaginkar S, Kulkarni R et al. (2021) IL‐15 and sMAdCAM: novel roles in COVID‐19 pathogenesis. medRxiv. [Google Scholar]
- 111. Kaul A, Singampalli KL, Parikh UM, Yu L, Keswani SG and Wang X (2021) Hyaluronan, a double‐edged sword in kidney diseases. Pediatr Nephrol, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Sasabe J and Suzuki M (2018) Emerging role of d‐amino acid metabolism in the innate defense. Front Microbiol 9, 933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Vardhana SA and Wolchok JD (2020) The many faces of the anti‐COVID immune response. J Exp Med 217, e20200678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Thiébaut R, Jarne A, Routy J‐P, Sereti I, Fischl M, Ive P, Speck RF, D'Offizi G, Casari S, Commenges D et al. (2016) Repeated cycles of recombinant human interleukin 7 in HIV‐infected patients with low CD4 T‐cell reconstitution on antiretroviral therapy: results of 2 phase II multicenter studies. Clin Infect Dis 62, 1178–1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Ouyang L and Gong J (2020) Mitochondrial‐targeted ubiquinone: a potential treatment for COVID‐19. Med Hypotheses 144, 110161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Israel A, Schäffer AA, Cicurel A, Feldhamer I, Tal A, Cheng K, Sinha S, Schiff E, Lavie G and Ruppin E (2020) Large population study identifies drugs associated with reduced COVID‐19 severity. medRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Das UN (2020) Can bioactive lipid arachidonic acid prevent and ameliorate COVID‐19? Medicina 56, 418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Digital Medicines Information Suite (2021) British National Formulary. https://about.medicinescomplete.com/
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Differentially expressed genes and proteins.
Table S2. Enrichment analysis of differentially expressed genes.
Table S3. Enrichment analysis of differentially expressed proteins.
Table S4. Transcriptomic‐informed flux rates for reactions and pathways.
Table S5. Proteomic‐informed flux rates for reactions and pathways.
Table S6. Common liver cancer drugs and their interactions.
Data Availability Statement
All software and data produced in the study are available at: https://github.com/Angione‐Lab/Metabolic‐modelling‐of‐SARS‐CoV‐2‐in‐cancer‐cells.