

Abstract
Arterial stiffness due to the vessel remodeling is closely linked to raised blood pressure, and its physiopathologic mechanism is still not fully understood. We here aimed to explore whether extracellular vesicle (EV) mediated intercellular communication between endothelium and smooth muscle cell contribute to the blood vessel remodeling under hypertension. We here revealed that the arterial endothelial cells robustly secreted EV, which in turn could be circulated and/or directly taken up by the subendothelial smooth muscle cells (SMC). Under hypertension, the EV secretion increased and the miRNA profile changed significantly mainly due to the raised mechanical force and subsequent enhanced reactive oxygen species generation. Among the miRNA cargos in the EV, miR-320d/423-5p were found increased most significantly. In vivo delivery of miR-320d/423-5p mimics via engineered EV increased their expression in arterial vessels, recapitulating the phenotype in hypertension. In contrast, therapeutic delivery of miR-320d/423-5p inhibitors via engineered EV alleviated the phenotype in spontaneous hypertension rat model. Together, we have found that the injured endothelium due to the raised mechanical force in hypertension contributes to the arterial wall remodeling via the secreted EV. Our study has not only provided novel insights on the mechanism of hypertension associated blood vessel wall remodeling, but also shed light on therapeutic intervention of hypertension associated vascular diseases.
Keywords: Hypertension, Extracellular vesicles, Endothelial cell, Smooth muscle cell, Phenotype switch, microRNA
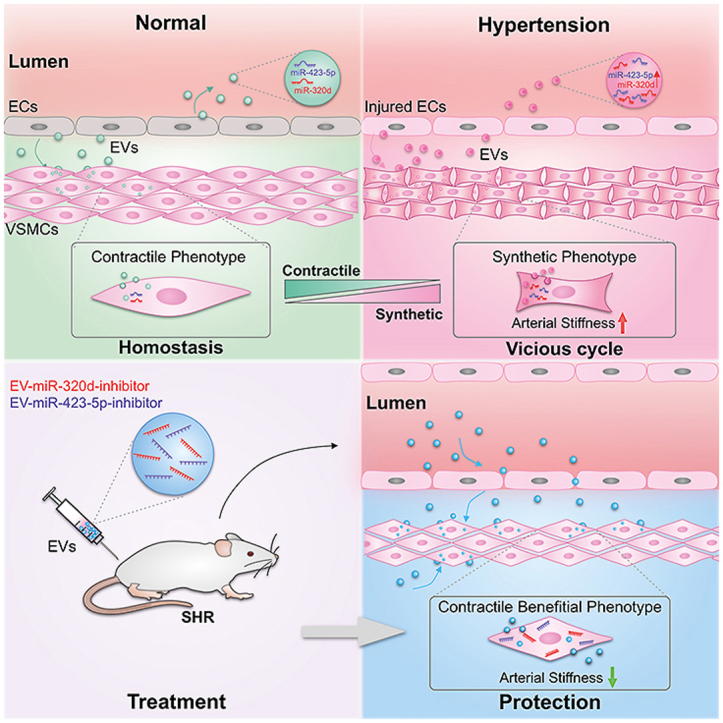
Graphical abstract
High blood pressure alters the EV secretion and the component profile. In turn, the altered EVs are taken up by the subendothelial smooth muscle cells and deliver the encapsulated cargos, such as miR-423 into the recipient cells, resulting in the phenotype switch and blood vessel wall structure remodeling. The remodeled blood vessel structure further increases the blood pressure, forming a vicious cycle. In vivo delivery of therapeutic EVs antagonizing miR-320/423 function in hypertension rat model partially reverses the disease phenotype.
Highlights
-
•
EVs derived from arterial endothelium alter significantly under hypertension.
-
•
EV-mediated transfer of miR-423/320 induces VSMC synthetic phenotype switch.
-
•
EVs from the injured endothelium promote arterial remodeling.
-
•
Therapeutic delivery of miR-423/320 inhibitors via EVs alleviates arterial stiffening.
1. Introduction
Hypertension, or elevated blood pressure, is a serious chronic disease that significantly increases the disease risks of heart, brain, kidney and other organs [[1], [2], [3]]. It is estimated that 1.13 billion people worldwide suffering from hypertension. Hypertension features as endothelial dysfunction and artery remodeling, resulting in increased vascular wall thickness and elevated arterial stiffness. Arterial remodeling in turn further accelerates the deteriorative progression of blood pressure and generates a vicious cycle ultimately [[4], [5], [6]]. The disturbed communication between endothelial cells (EC) and subendothelial smooth muscle cells (SMC) plays a crucial role in the arterial remodeling [5,[7], [8], [9]]. Besides the intensively studied hormone and cytokines in the process, extracellular vesicles should be also involved, though the details are largely unknown [[10], [11], [12]].
Extracellular vesicles (EVs), which are less than 2000 nm in diameter, are emerging as an important mediator for regulating intercellular communication and diagnostic marker for many diseases [[13], [14], [15]]. EVs typically contain proteins, nucleic acids, and lipids, reflecting their cell origins and mediating intercellular communication under normal and pathological conditions [[16], [17], [18]]. EVs have been extensively and intensively studied due to their promising potential in treating diseases [19,20]. Elevated EV level has been reported in a variety of diseases, especially the inflammatory and damaged vascular diseases [21,22]. All of these raise the possibility that EV from EC might alter the function of subendothelial SMC and thus arterial wall remodeling under hypertension.
In the present study, we first profiled the changes of the EV from the EC in hypertension by combinatorial use of human blood specimen and pressure load cell model. Then, we confirmed that hypertension derived EV could remodel the vessel wall at least partially via switching the SMC phenotype with miR-423. Our study has not only provided novel insights on the mechanism of hypertension associated blood vessel wall remodeling, but also shed light on therapeutic intervention of arterial stiffening.
2. Materials and methods
2.1. Recruitment of healthy subjects and hypertension patients
In the EV-miRNA screening experiment, 3 adult male healthy subjects and 3 age-matched patients with stage one hypertension were enrolled. Blood samples in the morning after 12 h fasting were collected. The blood samples of equal volume in each group were pooled and the EVs were isolated with the ExoQuick™ (ExoQ5TM-1/TMEXO-1, SBI, USA). miRNA abundance in EV were profiled by RNA-sequencing. In the validation experiment, eleven male volunteers were enrolled. The blood pressure was monitored in the morning. The mean arterial pressure was calculated as the sum of 1/3 systolic blood pressure and 2/3 diastolic blood pressure. The plasma EVs were harvested for candidate miRNAs analysis by qPCR. All the subjects were informed consent and the study was ethically approved by the institutional review board at the Fourth Military Medical University.
2.2. RNA-seq and bioinformatics
A total of 4 mL of plasma in each group was mixed with RiboTM EV Isolation Reagent (Ribobio, China). After the isolation, EV-RNA was extracted by HiPure Plasma miRNA Kit (Megan, China). EV-RNAs were used to prepare small RNA libraries by NEBNext® Multiplex Small RNA Library Prep Set for Illumina (NEB, USA) according to manufacturer's instructions. The libraries were sequenced by HiSeq 2500 (Illumina, USA) with single-end 50 bp at Ribobio Co. Ltd (Ribobio, China). The differentially expressed miRNAs were identified and shown by heatmap using Cluster 3.0 and Java Treeview.
2.3. Experimental animals
Eight-week-old male mice (C57BL/6) from Model Animal Research Center of Fourth Military Medical University were fed with normal chow diet (11%). Twelve-week-old male Wistar-Kyoto (WKY) and spontaneously hypertensive rats (SHR) from Beijing Vital River Laboratory Animal Technology Co. were fed with normal chow diet (11%).
For EV intervention, mice or rats were injected with EVs via tail vein once per week for 4 weeks. At the end of the experiment, all animals were sacrificed and the tissues were isolated for further analysis. All animal procedures were in accordance with the guidelines of the Animal Care and Use Committee of Fourth Military Medical University.
2.4. Cell culture
Human umbilical vein endothelial cells (HUVEC) (ATCC®, CRL-1730™) and mouse aortic smooth muscle cells (MOVAS) (ATCC®, CRL-2797™) were cultured in complete Dulbecco's modified Eagle Medium (DMEM, Gibco) medium supplemented with 10% fetal bovine serum (FBS, Gibco), 1% penicillin–streptomycin (Hyclone, GE), and maintained in a humidified atmosphere with 5% CO2 at 37 °C.
2.5. Transmission electron microscopy of blood vessel
To observe EV in the blood vessel, the aortas of mice were explanted after euthanasia using cervical dislocation. The samples were fixed with 2% paraformaldehyde and 2% glutaraldehyde in 500 μL PBS at room temperature. After incubating for 30 min at 4 °C, the aortas were fixed with 2% glutaraldehyde in PBS at 4 °C overnight. The samples were washed three times with PBS and post-fixed with 2% osmium tetroxide in PBS at 4 °C for 1 h. Then, the samples were dehydrated in graded ethanol solutions, embedded in Epon-812 resin (Sigma-Aldrich, USA) and polymerized at 60 °C for 48 h. The polymerized resins were sectioned using an ultramicrotome (Ultracut-UCT, Leica, Germany). The sections were mounted on copper grids, stained with 2% uranyl acetate at room temperature for 10 min, washed with distilled water, and stained with lead stain solution (Sigma-Aldrich, St. Louis, USA) at room temperature for 3 min. Then the aorta samples were analyzed by TEM (JEM-1400, JEOL, Japan).
2.6. Application of mechanical force on endothelial cells
HUVECs were seeded onto six-well plates and cultured in growth medium containing 10% EV-free FBS. After reaching 70% confluence, a cover slip was placed over the cell layer and the compressive force was conducted by laying the weight of 2 g on the cover slip [[23], [24], [25]]. The weight load was performed for 8 cycles, with 30 min on and 30 min off. The group without the weight served as control. The secreted EVs from the different culture were then isolated. To analysis the effect of ROS in the process, cells were additionally treated with N-acetylcysteine at 10 mM (NAC, Sigma, USA).
2.7. Pulse wave velocity measurement
Animal pulse wave velocity (PWV) was performed by experienced technicians using Vevo 2100 Imaging System (FUJIFILM VisualSonics, Canada). Mice or rats, with the hair around their chest and abdomen removed, were anesthetized by isoflurane (RDW, China) on a heating pad. The inhalation concentration of isoflurane was 3.5–4% for anesthesia induction and maintained at 2–2.5% during experiment. The heartbeats of mice or rats were kept around 350 or 400 beats per minute during the examination respectively. The velocity spectra were acquired at the ascending aorta and abdominal aorta for aortic PWV calculation. The time from the R wave of the electrocardiogram (ECG) to the start of pulse waveform for each measurement location was calculated by using a real-time signal acquisition and spectrum analyzer system. PWV was calculated as the distance divided by the time difference of the peak aortic flow at two distinct locations in the aorta.
2.8. EV isolation and characterization
For EV isolation from the blood plasma, blood was drawn into BD Vacutainer Blood Collection Tubes (BD, Bioscience) containing buffered sodium citrate as anticoagulant. Within 15 min of blood draw, blood plasma was isolated by centrifugation at 2000 g for 15 min, followed by a second round of centrifugation to remove any platelets remained. Then, plasma EVs were then isolated using the commercial kit ExoQuick™ (ExoQ5TM-1/TMEXO-1, SBI, USA).
For EV isolation from the culture medium, the supernatant was first centrifuged at 800 g for 10 min to remove cells, and then at 10,000 g for 20 min to eliminate the cellular debris. The resulting supernatant was regularly filtered through 0.22 μm filters and then subjected to ultracentrifugation for 3 h before EV harvesting. The protein concentration of EV was determined using the Pierce BCA Protein Assay Kit (Thermo Scientific, Waltham, MA, USA). Transmission electron microscopy (TEM) (HT7800, Hitachi) was used to determine EV size and morphology [[26], [27], [28]]. For biomarker analysis of EV, the EV pellet was dissolved in lysis buffer for Western blot of inclusive markers. The size distribution of EV was directly measured using the nanoparticle tracking analysis (NTA) with ZetaView PMX110 (Particle Metrix, Meerbusch, Germany).
2.9. EV tracking analysis
DiI and DiR (DiIC18(3) and DiIC18(7)), a family of fluorescent probe widely used lipophilic tracers for membrane labeling were included for EV labeling. Briefly, EVs (1 μg/μL) were incubated with DiI or DiR dye (1 mM, Invitrogen, China) at the ratio of (500:1 in volume) for 30 min, and the unbound dye was removed by another round of EV isolation as described above.
For in vivo fluorescence imaging, 200 μg of EVs were labeled with fluorescent dye DiR followed by tail vein injection for distribution analysis in vivo. Four hours later, the distribution of fluorescent-labeled EVs was observed under using in vivo imaging system (IVIS, PerkinElmer, Thermo Fisher, USA) [29]. Mice were anesthetized by isoflurane and had their thoracic and abdominal hair removed before imaging. After whole-body imaging, mice were sacrificed and the organs were taken out for subsequent fluorescence imaging.
For analysis of the EVs distribution in the blood vessel, DiI-labeled EVs were prepared similarly before tail vein injection. Four hours later, mice were sacrificed and isolated blood vessels were fixed by 4% paraformaldehyde before sectioning. Sections were incubated with rabbit α-SMA (Servicebio, GB111364, 1:1000) at 4 °C overnight, followed by incubation with goat anti-rabbit IgG H&L (Alexa Fluor 488) (Servicebio, GB25303, 1:300) secondary antibody. The nuclei were then counterstained with Hoechst. Distribution of DiI-labeled EVs was observed by laser scanning confocal microscope (ECLIPSE Ti, Nikon, Tokyo, Japan). The entire process was conducted in dark.
For in vitro tracing of EVs in vascular SMC, MOVAS cells were incubated with DiI-labeled EVs for 3 h. The cells were then washed with PBS three times and fixed with 4% paraformaldehyde for 10 min. Cell nuclei was counterstained with Hoechst (Invitrogen, H3570), and DiI labeled EVs in the cells were observed using a Nikon A1 Spectral Confocal Microscope (Nikon, Japan).
2.10. Measurement of reactive oxygen species (ROS)
Superoxide production was detected by DHE (Dihydroethidium, S0063, 1:1000, Beyotime, China) [30]. DHE is a cell-permeable fluorescent probe for the evaluation of ROS level. After being oxidized by superoxide, DHE can produce red fluorescence by binding to RNA or DNA. For DHE staining of the cells, 10 μM DHE was added in the medium and incubated with cells in a light-protected, humidified chamber at 37 °C for 30 min. After incubation, the cells were washed with PBS for 3 times. Then, cells were fixed for 15 min by 4% paraformaldehyde and the nuclei was counterstained with Hoechst (1:1000, Invitrogen, China). After washing 3 times with PBS, the fluorescence signal for DHE and the blue nuclei were viewed by laser scanning confocal microscope (ECLIPSE Ti, Nikon, Tokyo, Japan) and the intensities of the images were quantified with ImageJ software.
2.11. miRNA loading into EVs
The isolated EVs were loaded with miRNA-mimics, miRNA-inhibitor or negative control (NC) (Gene Pharma, China) via electroporation. Briefly, EVs and miRNA mimics (150 μg EV/0.5 OD mimics) were resuspended in 400 μL PBS and electroporated with Gene Pulser Xcell (Bio-Rad) at 700 V, 50 μF for 6 pulses in 4 mm Bio-Rad cuvettes (Bio-Rad). Electroporated EVs were then incubated in ice for 30 min and unloaded miRNAs were removed by RNase treatment at 37 °C for 20 min (0.5 μg/μl, Thermo Fischer Scientific, USA). After RNase treatment, EVs were then washed and additionally isolated with the ultracentrifugation method before further analysis or downstream application.
2.12. Cell transfection
MOVAS cells were plated in 6-well plates one day before transfection. Cells with indicated treatments were additionally transfected with 4 μg control, miRNA-mimics or inhibitors by Lipofectamine 2000 (Invitrogen, USA) according to the manufacturer's instructions. Cells were then incubated at 37 °C, 5% CO2 for 24 h followed by harvesting.
2.13. Quantitative real-time polymerase chain reaction analysis
Total RNAs were extracted from tissues, cultured cells or EVs using Tripure Isolation Reagent (Roche, Basel, Switzerland) according to manufacturer's protocol. miRNA was reversely transcribed by miRcute Plus miRNA First-strand cDNA Synthesis Kit (Tiangen, Beijing, China) and mRNA was reversely transcribed by First Strand cDNA Synthesis Kit (Genenode, Beijing, China) according to manufacturers' instructions. The qPCR reaction (in 20 μL system) was performed by FastStart Essential DNA Green Master (Roche, IN, USA) with specific forward primers. Relative miRNA expression was normalized to U6 or Gapdh levels and calculated with the 2-ΔΔCT method. The sequences of PCR primers were provided in Table S1.
2.14. Western blot analysis
The protein samples of EV from different groups were lysed using RIPA buffer (Beyotime, China). Protein concentrations were determined using BCA Protein Assay Kit (Thermo Scientific). Samples were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to nitrocellulose membranes (Immobilon P, Millipore, USA). The membranes were blocked with 5% milk in Tris-buffered saline containing 0.1% Tween-20 for 1 h at room temperature. Then, the membranes were incubated with primary antibodies at 4 °C overnight. The antibodies included anti-GM130 (Abcam, ab30637, 1:1000), anti-CD9 (Abcam, ab92726, 1:1000), anti-TSG101 (Abcam, ab125011, 1:1000). The membrane was then incubated with corresponding secondary antibodies for 1 h at room temperature. The bands were visualized using the ECL Prime Western Blotting Detection Reagent (GE, UK).
2.15. Histology
The blood vessels were harvested at the end of the experiments, fixed in 4% PFA, cleaned, embedded in OCT, and sectioned. Then, the serial cross-sections (10 μm) were stained with hematoxylin and eosin, Masson trichrome for histological analysis.
2.16. Statistical analysis
All data are expressed as the means ± SEM. Statistical significance was analyzed by GraphPad Prism 7.0 using the Student's t-test for two group comparison or ANOVA for more than three groups. Differences with P < 0.05 were considered statistically significant.
3. Results
3.1. Transfer of EVs from the endothelium into subendothelial SMC in artery
In order to explore whether EV involved in the intercellular communication between EC and subendothelial SMC, the ultrastructure of the artery was analyzed by transmission electron microscopy. As shown in Fig. 1A, there were amounts of vesicles in the basolateral side of the endothelium, while some of the vesicles were undergoing secretion. The vesicles were in the size range of 80–100 nm in diameter and displayed a typical ball-shaped morphology (Fig. 1A).
Fig. 1.

Transfer of EC derived EVs to the artery.
(A) Transmission electron microscopy of mouse aorta. Robust vesicles secretion could be seen in the basal side of the endothelium in the artery. Scale bars represents 1 μm or 200 nm. (B) Schematic illustration of the procedure how the HUVEC derived EVs were isolated. (C) Representative image of transmission electron microscopy of the EV released from HUVEC. Scale bars represents 100 nm. (D) Size distribution of EVs as analyzed by NTA. (E) Western blot analysis of EV inclusive and exclusive marker expression, including GM130, TSG101 and CD9. Representative data of at least three independent experiments. (F) Representative images of DiR-labeled EVs distributed in different tissues. About 150 μg (at protein level) in 100 μL of EV from EC labeled with DiR was injected via tail vein. About 4 h after the intravenous injection, in vivo and ex vivo fluorescence imaging were performed. Accumulation of EVs could be seen in the aorta, especially the branched region. n = 3 mice. (G) Quantification data of the relative fluorescence intensity of distributed EVs in different tissues. n = 3 mice.
To further confirm EV distribution in mice, EVs were harvest from the culture medium of HUVEC by ultracentrifugation (Fig. 1B). The particle size for EV was about 100 nm in diameter (Fig. 1C, D). In addition, the exosomal inclusive markers (TSG101, CD9) and exclusive marker GM130 [31] confirmed the EVs were mainly exosomes (Fig. 1E). In vivo analysis of the biodistribution of DiR labeled EVs revealed that the EVs mainly accumulated in the liver (Fig. 1F, left panel). Besides the main distribution in liver, EVs were also found in other organs (Fig. 1F, right panel, Fig. 1G). Notably, there were also EVs in the blood vessels, especially the branched area (Fig. 1F, G).
To mimicking the blood pressure force pushing the blood vessel wall, HUVECs were subjected to cyclic weight overloading (Fig. 2A). The EVs derived from control or 2 g weight loading cells were denoted as EVctrl and EV2g respectively. Weight loading didn't change the EV size, morphology and marker expression, while the yield under 2 g weight was moderately increased (Fig. 2B–D). Smooth muscle cells (MOVAS) were incubated with DiI labeled EVctrl and EV2g (Fig. S1A). Both EVctrl and EV2g could be efficiently taken up by MOVAS, while the uptake efficiency of EV2g was markedly higher than that of EVctrl (Fig. S1B, C). To explore whether these EVs could circulate into the subendothelial SMC in vivo, EVs were labeled with DiI and traced by confocal microscope after tail vein injection (Fig. 2E). As shown in Fig. 2E, EVctrl and EV2g were found in SMC of the artery 4 h after intravenous injection. Notably, there were more EV2g colocalized with the SMC in the blood vessel. To further confirm the finding, EVctrl and EV2g were loaded with cel-miR-54, which has no homolog in mouse, and cel-miR-54 expression in the aorta was analyzed by qPCR (Fig. S2A). Consistent with the fluorescence data, there were more cel-miR-54 delivered into the aorta in the EV2g group (Fig. S2B).
Fig. 2.

Uptake of the endothelial cell-derived EVs by the SMC.
(A) Schematic illustration of the setup of the cell model subjected to compressive forces and the procedure how the derived EVs were harvested. (B) Representative electron microscopy of the EV released from control or weight loaded HUVECs. Scale bars represents 100 nm. (C) Size distributions of EVctrl and EV2g as analyzed by NTA. (D) Western blot analysis of EV inclusive and exclusive marker expression, including GM130, TSG101 and CD9. Representative data of at least three independent experiments. (E) Schematic representation of the animal experiment procedure. EVs were labeled with DiI (Red) followed by tail vein injection. (F) EV distribution in the artery. SMCs were stained with anti-α-SMA (Green) and the nuclei were counterstained with Hoechst (Blue). Images were taken under confocal microscope. Arrow indicates the SMC distribution while arrowhead indicates the possible endothelium localization of the EVs. (G) Quantification data of the relative fluorescence intensity of DiI labeled EV distribution in the artery. Data are presented as mean ± SEM. n = 3 mice. *P < 0.05 as determined by t-test.
Besides efficient uptake by SMC, EC derived EVs could be also taken up by endothelium both in vitro and in vivo (Fig. 2F, G and Fig. S3A, B).
3.2. EVs from the injured endothelium recapitulate the phenotype in hypertension
Next, we explored whether the EVs from the injured endothelium conferred the functional information in vivo. EVs were injected into mice via tail vein once a week for 4 weeks, and the arterial stiffness was then assessed by PWV (Fig. 3A, B). The aorta PWV in mice of EV2g treatment showed a significant higher PWV in comparison with the control (Fig. 3C). Moreover, HE staining showed that the vascular wall thickness was markedly increased in EV2g group mice (Fig. 3D, E). And smooth muscle fibers were sparse and disordered with prominent gaps in the media (Fig. 3D). The lumen diameter had no significant change, while the wall and lumen ratio increased in the EV2g treatment group (Fig. 3E-G). Consistently, there was increased collagen deposition in vascular wall in EV2g mice group as revealed by Masson's trichrome staining (Fig. 3H). Moreover, the mRNA expression of Col3a1, Col1a1 and Spp1, which were the synthetic phenotype marker of SMC, was found significantly increased in EV2g treatment group, suggesting that EV2g treatment might promote SMC phenotype switch (Fig. 3I–K). Accordingly, EV2g treatment also promoted the synthetic phenotype switch of MOVAS, as shown by up-regulated expression of Col3a1, Col1a1 and Spp1 (Fig. S4A-D).
Fig. 3.

The effects of EVctrl and EV2g on arterial remodeling.
(A) Schematic representation of the experimental procedure. (B) Representative images showing how PWV was acquired. The pulse wave velocity spectra were acquired at the ascending aorta and abdominal aorta, with the simultaneous ECG. The time lagging of peak velocity in the two positions was used for PWV calculation. (C) The PWV of the EVctrl and EV2g treated mice. Data are means ± SEM. n = 5, **P < 0.01 by t-test. (D) Vascular change as revealed by Hematoxylin/eosin staining. (E-G) Wall thickness (E), internal diameter (F) and wall/lumen ratio (G) of the arteries of mice receiving EVctrl and EV2g treatment for 4 weeks. n = 5. n.s., no significance, **P < 0.01, ***P < 0.001 as determined by t-test. (H) Masson's trichrome staining of the aorta in mice with indicated treatment. Representative images of at least 3 mice of each group. Scale bars represent 50 μm. (I–K) Expression of Col3a1 (I), Col1a1 (J) and Spp1 (K) in the arteries of EVctrl and EV2g treated mice. Expression of mRNA candidates were normalized to Gapdh expression. Data are expressed as mean ± SEM of at least 3 biological replicates. *P < 0.05, **P < 0.01 by t-test.
3.3. Significant miRNA changes in the EVs derived from dysfunctional endothelium under hypertension
To identify the key molecules inside the EVs involved in the function, we focused on the miRNA components. Thus, the difference of miRNA profiles in plasma EVs between normal people and hypertension patients were profiled (Fig. 4A). The particle size for plasma-EV was 150–200 nm in diameter (Fig. 4B, C), expressing typical EV markers (including TSG101, CD9, and no GM130) (Fig. S5A). Then EVs were sequenced by HiSeq 2500 (Illumina, USA). Totally, 1228 EV-miRNAs were identified in samples of control and hypertension (Fig. S5B), with 730 miRNAs found in both groups (Fig. S5B). Moreover, there were 19 miRNAs with high abundance in either group showed significant difference between two groups, including 10 up-regulated and 9 down-regulated in hypertension group (Fig. 4D). Among these miRNAs, we were specifically interested in miR-320d and miR-423-5p for their most obvious fold change (Fig. 4D). To further explore the correlation between miR-320d/miR-423-5p abundance and the blood pressure, 11 more volunteers were recruited. Interestingly, we found a significant positive correlation between mean blood pressure and the abundance of the two EV-miRNAs (EV-miR-320d: R2 = 0.3845, P = 0.0418; EV-miR-423-5p: R2 = 0.3854, P = 0.0415) (Fig. 4E, F).
Fig. 4.

Profiling of the miRNA abundance in the EVs from hypertension patients.
(A) Schematic illustration of the procedure of blood harvesting and EV-miRNA sequencing in normal volunteers and hypertension patients. (B) Representative electron microscopy of the EVNBP and EVHBP. Scale bars represents 100 nm. (C) Size distributions of EVs from blood samples were compared by NTA. (D) Heat map analysis based on the 19 most significant miRNAs of EVs from control group or hypertension group. (E-F) Pearson correlations were performed to investigate the correlation between mean blood pressure and the expression levels of EV-miR-320d (E) or EV-miR-423-5p (F). Expression of miRNA candidates were normalized to U6 expression. Mean blood pressure represents 1/3SBP +2/3DBP.
3.4. EV miR-320d/423-5p from the dysfunctional endothelium contribute to the arterial remodeling
In view of the above data, we assumed that EVs derived from dysfunctional endothelial cells might participate in the arterial remodeling via transferring miRNA-320d/423-5p. As expected, EV2g treatment significantly increased expression of miRNA-320d/423-5p in the aorta (Fig. S6). To verify the effect of EV-miRNA-320d/423-5p, miR-320d-mimics and miR-423-5p-mimics were thus encapsulated into EV derived from control endothelial cells respectively by electroporation. qPCR analysis showed that miRNA-mimics were efficiently loaded into the EVs (Fig. S7A, B). Then, the EVs were tail vein injected once a week for 4 weeks, the arterial stiffness was measured by PWV (Fig. 5A). As expected, both of the EVmiR-320d-mimics group and EVmiR-423-5p-mimics group had significant higher PWV, compared with the negative control group (Fig. 5B). Moreover, the thickness of vascular wall and the wall/lumen ratio increased in the EVmiR-320d-mimics group and EVmiR-423-5p-mimics group (Fig. 5C-F). Histology analysis showed increased collagen deposition and disordered arrangement of SMC in the groups treated with the EVmiR-320d-mimics and EVmiR-423-5p-mimics (Fig. 5G). Accordingly, mRNA expression of Col3a1, Col1a1 and Spp1, was found increased in the two groups (Fig. 5H-J).
Fig. 5.

EVmiR-320d-mimics and EVmiR-423-5p-mimics treatments recapitulate the arterial remodeling of hypertension.
(A) Schematic representation of the experimental procedure. (B) The PWV of mice receiving the EVNC-mimics, EVmiR-320d-mimics and EVmiR-423-5p-mimics. Data are means ± SEM. n = 5, **P < 0.01 by one-way ANOVA. (C) Vascular change as revealed by Hematoxylin/eosin staining. (D-F) Wall thickness (D), internal diameter (E) and wall/lumen ratio (F) of the arteries of mice receiving EVNC-mimics, EVmiR-320d-mimics and EVmiR-423-5p-mimics treatment for 4 weeks. n = 5. n.s., no significance, *P < 0.05, **P < 0.01, ***P < 0.001 as determined by one-way ANOVA. (G) Masson's trichrome staining of the aorta in mice with indicated treatments. Representative images of at least 3 mice of each group. Scale bars represent 50 μm. (H-J) Expression of Col3a1 (H), Col1a1 (I) and Spp1 (J) in the arteries of mice receiving EVNC-mimics, EVmiR-320d-mimics and EVmiR-423-5p-mimics treatment. Expression of mRNA candidates were normalized to Gapdh expression. Data are expressed as mean ± SEM of at least 3 biological replicates. *P < 0.05, **P < 0.01 by one-way ANOVA.
3.5. Therapeutic delivery of miR-320d/miR-423-5p inhibitors alleviates the arterial remodeling in SHR model
The above data indicated that EV-mediated delivery of miRNA-320d/423-5p might contribute to the SMC phenotype switch and arterial remodeling. In the following experiments, we explored whether intervention of the intercellular communication would be therapeutic. SHR rat model was included. The PWV of SHR was much higher compared with the control Wistar-Kyoto (WKY) rats (Fig. S8A, B). Similarly, the thickness of vascular wall and wall/lumen ratio of SHR were much larger, compared with WKY (Fig. S8C–F). Moreover, more collagen deposition and prominent gaps increased as Masson's trichrome staining showed in SHR compared with WKY (Fig. S8C). qPCR revealed higher mRNA expression of Col3a1, Col1a1 and Spp1 in SHR rats (Fig. S8G-I). All of these suggested that high blood pressure aggravated the vascular stiffness and dysfunction in the SHR model, resembling the phenotype in hypertension patients.
miR-320d-inhibitor and miR-423-5p-inhibitor were thus encapsulated into EV derived from endothelial cells by electroporation respectively. Control EV (EVNC-inhibitor), EVmiR-320d-inhibitor and EVmiR-423-5p-inhibitor were injected via tail vein once a week for one month (Fig. 6A). The loading efficiency of miRNA-inhibitors was considerable by qPCR analysis (Fig. S7C, D). Compared with control group, EVmiR-320d-inhibitor group and EVmiR-423-5p-inhibitor treatment significantly decreased the aorta PWV in SHR rats (Fig. 6B). Consistent with the functional change, the thickness of vascular wall and the wall/lumen ratio was decreased after EVmiR-320d-inhibitor and EVmiR-423-5p-inhibitor treatment, while the diameters of the blood vessel showed no significant changes (Fig. 6C–F). In addition, EVmiR-320d-inhibitor and EVmiR-423-5p-inhibitor treatment reduced collagen deposition (Fig. 6G) and down-regulated the mRNA expression of Col3a1, Col1a1 and Spp1 in the blood vessels of SHR (Fig. 6H–J).
Fig. 6.

EVmiR-320d-inhibitor and EVmiR-423-5p-inhibitor treatments alleviate the arterial remodeling in SHR rats.
(A) Schematic illustration of the experimental procedure. EVs encapsulating miRNA inhibitors were injected into SHR rats once a week for 4 weeks. (B) The PWV of SHR rats receiving EVNC-inhibitor, EVmiR-320d-inhibitor and EVmiR-423-5p-inhibitor. Data were expressed as mean ± SEM. n = 5, *P < 0.05 by one-way ANOVA. (C) Vascular change as revealed by Hematoxylin/eosin staining. (D-F) Wall thickness (D), internal diameter (E) and wall/lumen ratio (F) of the arteries of rats receiving EVNC-inhibitor, EVmiR-320d-inhibitor and EVmiR-423-5p-inhibitor for 4 weeks. n = 5. n.s., no significance, *P < 0.05, **P < 0.01 as determined by one-way ANOVA. (G) Masson's trichrome staining of the aorta in rats with indicated treatments. Scale bars represent 50 μm. (H-J) Expression of Col3a1 (H), Col1a1 (I) and Spp1 (J) in the arteries of SHR rats receiving EVNC-inhibitor, EVmiR-320d-inhibitor and EVmiR-423-5p-inhibitor injection. Expression of mRNA candidates were normalized to Gapdh expression. Data were expressed as mean ± SEM of at least 3 biological replicates. *P < 0.05, **P < 0.01 by one-way ANOVA.
In the following experiments, we explored the downstream target of miR-320d and miR-423-5p. Inhibition of miR-423-5p with the miRNA inhibitor significantly blocked the effects of EV2g on the synthetic phenotype switch of vascular SMC in vitro (Figs. S9A–C), suggesting that miR-423-5p could regulate the synthetic phenotype switch of vascular SMC directly. Unexpectedly, inhibition of miR-320d with the inhibitor had no effect on EV2g induced synthetic phenotype switch of vascular SMC. The paradoxical roles of miR-320 in EV between in vivo animal experiment and the in vitro cell experiments suggested that miR-320 might regulate other cell types rather than directly on the SMC.
Previous study has found that miR-423-5p could regulate Mybl2, a putative regulator of synthetic phenotype switch of vascular SMC [32,33]. In order to further clarify the direct target of miR-423-5p, the mRNA level of Mybl2 in MOVAS treated with control or miR-423-5p-mimic transfection was analyzed by qPCR. As expected, miR-423-5p transfection significantly reduced the expression of Mybl2 (Fig. S10A). Accordingly, EV mediated delivery of miR-423-5p significantly reduced Mybl2 expression in the aorta (Fig. S10B). All the data suggested that miR-423-5p in EV2g or EV from hypertension might regulate SMC synthetic phenotype switch at least via Mybl2.
3.6. Increased ROS contribute to high abundance of miR-320d/miR-423-5p in EVs
In view of the above data, we asked whether the change of plasma EV-miR-320d/miR-423-5p from hypertension patients were mainly from the injured endothelium, the abundance of the two miRNAs level of EVctrl and EV2g and their donor cells were then analyzed. Consistent with the previous findings that mechanical force induces ROS generation [[34], [35], [36], [37]], ROS in the HUVEC with 2 g weight treatment were much higher than that in the control group (Fig. 7A, B). ROS scavenger N-acetylcysteine (NAC) treatment significantly reduced the ROS (Fig. 7A, B), together with the abundance of miR-320d and miR-423-5p in donor cells and EVs (Fig. 7C, D). The expression of miR-320d and miR-423-5p was significantly up-regulated in both the weight overload cells and the derived EV2g (Fig. 7C, D), suggesting that the increased abundance in EVs might be due to the enhanced expression of the two miRNAs in the donor cells. Together, these findings suggested that hypertension related mechanical force increased miR-320d and miR-423-5p abundance in EVs in an ROS dependent manner.
Fig. 7.

Mechanical force induces EV enrichment of miR-320d and miR-423-5p in an ROS dependent manner.
(A) Confocal microscope images of the DHE staining of the ROS (red) in HUVECs with different treatments. Nuclei were counterstained with Hoechst. Scale bars represent 30 μm. (B) Quantification of the DHE fluorescence intensity in Figure A. Data were presented as means ± SEM. ***P < 0.001 as determined by one-way ANOVA. (C-D) qPCR analysis of the abundance of miRNAs in cells with control or 2 g weight loading or 2 g weight loading plus NAC treatment (C) or the derived EVs (D). Data were expressed as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001 as determined by one-way ANOVA.
4. Discussion
Arterial remodeling is the key feature of hypertension, which in turn accelerates the deteriorative progression of blood pressure and generates a vicious cycle. In the present study, we have revealed that EV from the EC could be taken up by the subendothelial SMC in the artery. Upon hypertension, the EV from the injured EC could remodel the vessel wall at least partially via switching the SMC phenotype with miR-320/miR-423. In contrast, therapeutic delivery of miR-320d/423-5p inhibitors via engineered EVs alleviated the phenotype in spontaneous hypertension rat model.
Endothelium, the largest organ in the body, acts as an important regulator of vascular homeostasis, maintaining a balance between vasoconstriction, vasodilatation, and regulation of smooth muscle proliferation [38]. Endothelial dysfunction is a prominent feature in hypertension [39]. The pathophysiological mechanisms responsible for hypertension are complex, and the endothelium dysfunction could be at least partially attributed to up-regulated iNOS (inducible nitric oxide synthase) and down-regulated eNOS (endothelial nitric oxide synthase), resulting in disturbed release of NO and aberrant vasodilation [4]. It has also been reported that endothelial cells, exposed to hemodynamic forces of nonuniform and irregular flow, are easily injured [40,41]. The disturbed flow could also induce the expression of a number of atherogenic and thrombogenic genes, proliferation of EC, and modulation of synthetic SMC phenotype differentiation [42,43]. Here, we proposed that the blood pressure force pushing the vessel wall increased ROS generation in the endothelium, which in turn altered the EVs secretion and miRNA profile, resulting in the SMC phenotype switch and arterial stiffening when taken up by the SMC. Our study here not only provided another layer of the pathological mechanism of hypertension, but also revealed a relationship between mechanical force and EV yield in the model of endothelium. Our results suggested that high physical pressure favors EVs production in endothelial cells in vitro. Considering the promising future of EVs in drug delivery [[44], [45], [46], [47]], it is interesting to optimize the mechanical force in the culture condition toward increased yield of EVs for therapeutic purposes.
Both of EC and SMC are the primary cell types regulating vascular homeostasis, and the information exchange between the two cell types is considered closely related to the vascular structure and function [16,48]. Coordinated functions of EC and SMC could be achieved by various mechanisms, such as direct physical contact and paracrine interaction [49,50]. Recently, EV-mediated intercellular communication has been intensively studied in different organs, including the cardiovascular system. For instance, Zheng et al. found that EVs derived from KLF5-overexpressing vascular SMCs could transfer miR-155 to ECs, which in turn inhibited EC proliferation and migration, eventually impairing the integrity of endothelial barriers [51]. In another study, Tong et al. has found that EVs derived from vascular adventitial fibroblasts can transfer angiotensin-converting enzyme to vascular SMCs [52]. In our study, we found that EVs derived from ECs could be taken up by vascular SMCs and significantly contribute to the vascular stiffness remodeling process during hypertension. All the studies indicate that an active and robust EV-based intercellular communication between EC and SMC. However, it remains to be determined that how EC derived EVs trafficking to the vascular SMC, for EV uptake involving more than one route [53]. Theoretically, the EVs could directly cross the basement membrane and endocytosed by the SMC, as EVs, mainly exosomes have been found to have the ability to cross types of biological barriers [[54], [55], [56]]. Alternatively, the EVs could be released into circulation and reach SMC via the vasa vasorum. Further real-time tracing of the in vivo labeled EVs with two-photon excitation microscopy might shed light on the interesting question. Notably, we here found that EVs derived from injured EC displayed higher efficiency in entering vascular SMC, with the details unknown. Future studies revealing the targeting moieties in the EVs could be harnessed for efficient targeting to the SMCs.
In our current study, we found dozens of miRNAs in the EVs changed significantly in hypertension, especially up-regulated miR-423-5p and miR-320d, two miRNAs involved in regulation of SMC phenotype switch and function [[57], [58], [59]]. Previously, miR-423 and miR-320 have been reported to be involved in various cardiovascular biological processes [32,60,61]. For example, miR-320 is involved in atherosclerosis via targeting SRF in endothelium [61], while miR-423 targets Mybl2 (a repressor of collagen genes) in multiple cells associated with cardiovascular development and cancer progression [32,62]. EV-miR-423 was reported being a promising biomarker for heart failure [63,64]. Our study here further revealed that EV-related miR-423-5p and miR-320d contribute to the arterial stiffening via SMC phenotype switch.
Notably, EV mediated delivery of either miR-423-5p-inhibitor or miR-320d-inhibitor can alleviate vascular stiffness, reverse the phenotypic switch of vascular SMC, and thus slow down the progression of hypertension in vivo. However, miR-423-5p, rather than miR-320d was found to have direct effects on the synthetic phenotype of vascular SMC in vitro. It is highly possible that miR-320d in EVs might be act on endothelial cells in vivo, which in turn affect the vascular SMCs. The explanation is supported by a recent study, which has revealed that overexpression of miR-320a in vivo attenuates endothelium cell function and promotes atherogenesis, together with significant increase in levels of plasma lipid and serum inflammatory cytokines [61]. The assumption is also supported by the observation that the EVs could also be taken up by endothelium in vivo.
In addition to the therapeutic effect, the potential of EVs as biomarkers has broad implications for personalized diagnosis, risk stratification, and prognosis of cardiovascular diseases [65,66]. Considering the causative role of miR-320d/423-5p in SMC phenotype switch, it is thus reasonable to speculate these two miRNAs may be valuable diagnostic biomarkers predicting the arterial remodeling.
5. Conclusion
In the present study, we have revealed that EVs from the ECs could be taken up by the subendothelial SMCs in the artery. Upon hypertension, the EVs from the injured ECs could remodel the vessel wall at least partially via switching the SMC phenotype with the miRNAs encapsulated. Our study has not only provided novel insights on the mechanism of hypertension associated blood vessel wall remodeling, but also shed light on therapeutic intervention of arterial stiffening.
CRediT authorship contribution statement
Chen Wang: Investigation, Writing - original draft, Writing - review & editing. Changyang Xing: Investigation, Writing - review & editing, Methodology, Funding acquisition. Zhelong Li: Investigation, Formal analysis. Yunnan Liu: Investigation. Qiaoying Li: Investigation. Yixiao Wang: Investigation. Jiao Hu: Investigation. Lijun Yuan: Conceptualization, Supervision, Funding acquisition. Guodong Yang: Conceptualization, Supervision, Funding acquisition, Writing - review & editing.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
This study was funded by NSFC 31771507 and 81970737 to Yang GD, NSFC 81871357 and 81671690 to Yuan LJ, NSFC 81901751 to Xing CY, and Provincial Scientific Foundation of Shaan’Xi (2020TD-038), Innovative Development Fund of Tangdu Hospital (2018QYTS007) and Clinical Trial Fund of Tangdu Hospital (2021LCYJ006) to Yuan LJ. This study was also funded by MOST (2016YFA0102100).
Footnotes
Peer review under responsibility of KeAi Communications Co., Ltd.
Supplementary data to this article can be found online at https://doi.org/10.1016/j.bioactmat.2021.06.005.
Contributor Information
Lijun Yuan, Email: yuanlj@fmmu.edu.cn.
Guodong Yang, Email: yanggd@fmmu.edu.cn.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Mills K.T., Bundy J.D., Kelly T.N., Reed J.E., Kearney P.M., Reynolds K. Global disparities of hypertension prevalence and control: a systematic analysis of population-based studies from 90 countries. Circulation. 2016;134:441–450. doi: 10.1161/CIRCULATIONAHA.115.018912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Forouzanfar M.H., Liu P., Roth G.A., Ng M., Biryukov S., Marczak L. Global burden of hypertension and systolic blood pressure of at least 110 to 115 mm Hg, 1990-2015. J. Am. Med. Assoc. 2017;317:165–182. doi: 10.1001/jama.2016.19043. [DOI] [PubMed] [Google Scholar]
- 3.Melgarejo J.D., Maestre G.E., Thijs L., Asayama K., Boggia J., Casiglia E. Prevalence, treatment, and control rates of conventional and ambulatory hypertension across 10 populations in 3 continents. Hypertension. 2017;70:50–58. doi: 10.1161/hypertensionaha.117.09188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Oparil S., Acelajado M.C., Bakris G.L., Berlowitz D.R., Cifkova R., Dominiczak A.F. Hypertension. Nat Rev Dis Primers. 2018;4:18014. doi: 10.1038/nrdp.2018.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Safar M.E. Arterial stiffness as a risk factor for clinical hypertension. Nat. Rev. Cardiol. 2017;15:97–105. doi: 10.1038/nrcardio.2017.155. [DOI] [PubMed] [Google Scholar]
- 6.Laurent S., Boutouyrie P. The structural factor of hypertension. Circ. Res. 2015;116:1007–1021. doi: 10.1161/circresaha.116.303596. [DOI] [PubMed] [Google Scholar]
- 7.Brandes R.P. Endothelial dysfunction and hypertension. Hypertension. 2014;64:924–928. doi: 10.1161/hypertensionaha.114.03575. [DOI] [PubMed] [Google Scholar]
- 8.Miyagawa K., Shi M., Chen P.I., Hennigs J.K., Zhao Z., Wang M. Smooth muscle contact drives endothelial regeneration by BMPR2-notch1-mediated metabolic and epigenetic changes. Circ. Res. 2019;124:211–224. doi: 10.1161/CIRCRESAHA.118.313374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Van den Bergh G., Opdebeeck B., D'Haese P.C., Verhulst A. The vicious cycle of arterial stiffness and arterial media calcification. Trends Mol. Med. 2019;25:1133–1146. doi: 10.1016/j.molmed.2019.08.006. [DOI] [PubMed] [Google Scholar]
- 10.Nguyen H., Chiasson V.L., Chatterjee P., Kopriva S.E., Young K.J., Mitchell B.M. Interleukin-17 causes Rho-kinase-mediated endothelial dysfunction and hypertension. Cardiovasc. Res. 2013;97:696–704. doi: 10.1093/cvr/cvs422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fafian-Labora J.A., O'Loghlen A. Classical and nonclassical intercellular communication in senescence and ageing. Trends Cell Biol. 2020;30:628–639. doi: 10.1016/j.tcb.2020.05.003. [DOI] [PubMed] [Google Scholar]
- 12.Mathieu M., Martin-Jaular L., Lavieu G., Thery C. Specificities of secretion and uptake of exosomes and other extracellular vesicles for cell-to-cell communication. Nat. Cell Biol. 2019;21:9–17. doi: 10.1038/s41556-018-0250-9. [DOI] [PubMed] [Google Scholar]
- 13.Kalluri R., LeBleu V.S. The biology, function, and biomedical applications of exosomes. Science. 2020;367 doi: 10.1126/science.aau6977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jeppesen D.K., Fenix A.M., Franklin J.L., Higginbotham J.N., Zhang Q., Zimmerman L.J. Reassessment of exosome composition. Cell. 2019;177:428–445. doi: 10.1016/j.cell.2019.02.029. e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gao X., Ran N., Dong X., Zuo B., Yang R., Zhou Q. Anchor peptide captures, targets, and loads exosomes of diverse origins for diagnostics and therapy. Sci. Transl. Med. 2018;10 doi: 10.1126/scitranslmed.aat0195. [DOI] [PubMed] [Google Scholar]
- 16.Hergenreider E., Heydt S., Treguer K., Boettger T., Horrevoets A.J., Zeiher A.M. Atheroprotective communication between endothelial cells and smooth muscle cells through miRNAs. Nat. Cell Biol. 2012;14:249–256. doi: 10.1038/ncb2441. [DOI] [PubMed] [Google Scholar]
- 17.Yanez-Mo M., Siljander P.R., Andreu Z., Zavec A.B., Borras F.E., Buzas E.I. Biological properties of extracellular vesicles and their physiological functions. J. Extracell. Vesicles. 2015;4:27066. doi: 10.3402/jev.v4.27066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gutierrez-Vazquez C., Villarroya-Beltri C., Mittelbrunn M., Sanchez-Madrid F. Transfer of extracellular vesicles during immune cell-cell interactions. Immunol. Rev. 2013;251:125–142. doi: 10.1111/imr.12013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang P., Wang L., Li Q., Tian X., Xu J., Xu J. Atorvastatin enhances the therapeutic efficacy of mesenchymal stem cells-derived exosomes in acute myocardial infarction via up-regulating long non-coding RNA H19. Cardiovasc. Res. 2020;116:353–367. doi: 10.1093/cvr/cvz139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hu S., Li Z., Lutz H., Huang K., Su T., Cores J. Dermal exosomes containing miR-218-5p promote hair regeneration by regulating beta-catenin signaling. Adv. Sci. 2020;6 doi: 10.1126/sciadv.aba1685. eaba1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yuana Y., Sturk A., Nieuwland R. Extracellular vesicles in physiological and pathological conditions. Blood Rev. 2013;27:31–39. doi: 10.1016/j.blre.2012.12.002. [DOI] [PubMed] [Google Scholar]
- 22.de Jong O.G., Verhaar M.C., Chen Y., Vader P., Gremmels H., Posthuma G. Cellular stress conditions are reflected in the protein and RNA content of endothelial cell-derived exosomes. J. Extracell. Vesicles. 2012;1 doi: 10.3402/jev.v1i0.18396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kanzaki H., Chiba M., Shimizu Y., Mitani H. Periodontal ligament cells under mechanical stress induce osteoclastogenesis by receptor activator of nuclear factor kappaB ligand up-regulation via prostaglandin E2 synthesis. J. Bone Miner. Res. 2002;17:210–220. doi: 10.1359/jbmr.2002.17.2.210. [DOI] [PubMed] [Google Scholar]
- 24.Kirschneck C., Batschkus S., Proff P., Kostler J., Spanier G., Schroder A. Valid gene expression normalization by RT-qPCR in studies on hPDL fibroblasts with focus on orthodontic tooth movement and periodontitis. Sci. Rep. 2017;7:14751. doi: 10.1038/s41598-017-15281-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen L., Mo S., Hua Y. Compressive force-induced autophagy in periodontal ligament cells downregulates osteoclastogenesis during tooth movement. J. Periodontol. 2019;90:1170–1181. doi: 10.1002/JPER.19-0049. [DOI] [PubMed] [Google Scholar]
- 26.Zhu J., Liu B., Wang Z., Wang D., Ni H., Zhang L. Exosomes from nicotine-stimulated macrophages accelerate atherosclerosis through miR-21-3p/PTEN-mediated VSMC migration and proliferation. Theranostics. 2019;9:6901–6919. doi: 10.7150/thno.37357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kapustin A.N., Chatrou M.L., Drozdov I., Zheng Y., Davidson S.M., Soong D. Vascular smooth muscle cell calcification is mediated by regulated exosome secretion. Circ. Res. 2015;116:1312–1323. doi: 10.1161/CIRCRESAHA.116.305012. [DOI] [PubMed] [Google Scholar]
- 28.Tian T., Zhang H.X., He C.P., Fan S., Zhu Y.L., Qi C. Surface functionalized exosomes as targeted drug delivery vehicles for cerebral ischemia therapy. Biomaterials. 2018;150:137–149. doi: 10.1016/j.biomaterials.2017.10.012. [DOI] [PubMed] [Google Scholar]
- 29.de Couto G., Gallet R., Cambier L., Jaghatspanyan E., Makkar N., Dawkins J.F. Exosomal MicroRNA transfer into macrophages mediates cellular postconditioning. Circulation. 2017;136:200–214. doi: 10.1161/CIRCULATIONAHA.116.024590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hervera A., De Virgiliis F., Palmisano I., Zhou L., Tantardini E., Kong G. Reactive oxygen species regulate axonal regeneration through the release of exosomal NADPH oxidase 2 complexes into injured axons. Nat. Cell Biol. 2018;20:307–319. doi: 10.1038/s41556-018-0039-x. [DOI] [PubMed] [Google Scholar]
- 31.Thery C., Witwer K.W., Aikawa E., Alcaraz M.J., Anderson J.D., Andriantsitohaina R. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles. 2018;7:1535750. doi: 10.1080/20013078.2018.1535750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhu X., Lu X. MiR-423-5p inhibition alleviates cardiomyocyte apoptosis and mitochondrial dysfunction caused by hypoxia/reoxygenation through activation of the wnt/beta-catenin signaling pathway via targeting MYBL2. J. Cell. Physiol. 2019;234:22034–22043. doi: 10.1002/jcp.28766. [DOI] [PubMed] [Google Scholar]
- 33.Hofmann C.S., Sullivan C.P., Jiang H.Y., Stone P.J., Toselli P., Reis E.D. B-Myb represses vascular smooth muscle cell collagen gene expression and inhibits neointima formation after arterial injury. Arterioscler. Thromb. Vasc. Biol. 2004;24:1608–1613. doi: 10.1161/01.ATV.0000139010.71779.f3. [DOI] [PubMed] [Google Scholar]
- 34.Leopold J.A., Loscalzo J. Cyclic strain modulates resistance to oxidant stress by increasing G6PDH expression in smooth muscle cells. Am. J. Physiol. Heart Circ. Physiol. 2000;279:H2477–H2485. doi: 10.1152/ajpheart.2000.279.5.H2477. [DOI] [PubMed] [Google Scholar]
- 35.Chatterjee S., Fisher A.B. Mechanotransduction: forces, sensors, and redox signaling. Antioxidants Redox Signal. 2014;20:868–871. doi: 10.1089/ars.2013.5753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zeller K.S., Riaz A., Sarve H., Li J., Tengholm A., Johansson S. The role of mechanical force and ROS in integrin-dependent signals. PloS One. 2013;8 doi: 10.1371/journal.pone.0064897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang C., Lin S., Li T., Jiang Y., Huang Z., Wen J. Mechanical force-mediated pathological cartilage thinning is regulated by necroptosis and apoptosis. Osteoarthritis Cartilage. 2017;25:1324–1334. doi: 10.1016/j.joca.2017.03.018. [DOI] [PubMed] [Google Scholar]
- 38.Watson T., Goon P.K., Lip G.Y. Endothelial progenitor cells, endothelial dysfunction, inflammation, and oxidative stress in hypertension. Antioxidants Redox Signal. 2008;10:1079–1088. doi: 10.1089/ars.2007.1998. [DOI] [PubMed] [Google Scholar]
- 39.Shimbo D., Muntner P., Mann D., Viera A.J., Homma S., Polak J.F. Endothelial dysfunction and the risk of hypertension. Hypertension. 2010;55:1210–1216. doi: 10.1161/hypertensionaha.109.143123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kruger-Genge A., Blocki A., Franke R.P., Jung F. Vascular endothelial cell biology: an update. Int. J. Mol. Sci. 2019;20 doi: 10.3390/ijms20184411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Davies P.F., Civelek M., Fang Y., Fleming I. The atherosusceptible endothelium: endothelial phenotypes in complex haemodynamic shear stress regions in vivo. Cardiovasc. Res. 2013;99:315–327. doi: 10.1093/cvr/cvt101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chiu J.J., Chien S. Effects of disturbed flow on vascular endothelium: pathophysiological basis and clinical perspectives. Physiol. Rev. 2011;91:327–387. doi: 10.1152/physrev.00047.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhou J., Li Y.S., Chien S. Shear stress-initiated signaling and its regulation of endothelial function. Arterioscler. Thromb. Vasc. Biol. 2014;34:2191–2198. doi: 10.1161/ATVBAHA.114.303422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Alvarez-Erviti L., Seow Y., Yin H., Betts C., Lakhal S., Wood M.J. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat. Biotechnol. 2011;29:341–345. doi: 10.1038/nbt.1807. [DOI] [PubMed] [Google Scholar]
- 45.Garcia-Manrique P., Matos M., Gutierrez G., Pazos C., Blanco-Lopez M.C. Therapeutic biomaterials based on extracellular vesicles: classification of bio-engineering and mimetic preparation routes. J. Extracell. Vesicles. 2018;7:1422676. doi: 10.1080/20013078.2017.1422676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wiklander O.P.B., Brennan M.A., Lotvall J., Breakefield X.O., El Andaloussi S. Advances in therapeutic applications of extracellular vesicles. Sci. Transl. Med. 2019;11 doi: 10.1126/scitranslmed.aav8521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tian Y., Li S., Song J., Ji T., Zhu M., Anderson G.J. A doxorubicin delivery platform using engineered natural membrane vesicle exosomes for targeted tumor therapy. Biomaterials. 2014;35:2383–2390. doi: 10.1016/j.biomaterials.2013.11.083. [DOI] [PubMed] [Google Scholar]
- 48.Harper E., Rochfort K.D., Forde H., Davenport C., Smith D., Cummins P.M. Activation of the non-canonical NF-kappaB/p52 pathway in vascular endothelial cells by RANKL elicits pro-calcific signalling in co-cultured smooth muscle cells. Cell. Signal. 2018;47:142–150. doi: 10.1016/j.cellsig.2018.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sun L., Zhang T., Yu X., Xin W., Lan X., Zhang D. Asymmetric dimethylarginine confers the communication between endothelial and smooth muscle cells and leads to VSMC migration through p38 and ERK1/2 signaling cascade. FEBS Lett. 2011;585:2727–2734. doi: 10.1016/j.febslet.2011.07.032. [DOI] [PubMed] [Google Scholar]
- 50.Chang S.F., Chen L.J., Lee P.L., Lee D.Y., Chien S., Chiu J.J. Different modes of endothelial-smooth muscle cell interaction elicit differential beta-catenin phosphorylations and endothelial functions. Proc. Natl. Acad. Sci. U.S.A. 2014;111:1855–1860. doi: 10.1073/pnas.1323761111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zheng B., Yin W.N., Suzuki T., Zhang X.H., Zhang Y., Song L.L. Exosome-mediated miR-155 transfer from smooth muscle cells to endothelial cells induces endothelial injury and promotes atherosclerosis. Mol. Ther. 2017;25:1279–1294. doi: 10.1016/j.ymthe.2017.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 52.Tong Y., Ye C., Ren X.S., Qiu Y., Zang Y.H., Xiong X.Q. Exosome-mediated transfer of ACE (Angiotensin-Converting enzyme) from adventitial fibroblasts of spontaneously hypertensive rats promotes vascular smooth muscle cell migration. Hypertension. 2018;72:881–888. doi: 10.1161/HYPERTENSIONAHA.118.11375. [DOI] [PubMed] [Google Scholar]
- 53.Mulcahy L.A., Pink R.C., Carter D.R. Routes and mechanisms of extracellular vesicle uptake. J. Extracell. Vesicles. 2014;3 doi: 10.3402/jev.v3.24641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.El Andaloussi S., Lakhal S., Mager I., Wood M.J. Exosomes for targeted siRNA delivery across biological barriers. Adv. Drug Deliv. Rev. 2013;65:391–397. doi: 10.1016/j.addr.2012.08.008. [DOI] [PubMed] [Google Scholar]
- 55.Yuan D., Zhao Y., Banks W.A., Bullock K.M., Haney M., Batrakova E. Macrophage exosomes as natural nanocarriers for protein delivery to inflamed brain. Biomaterials. 2017;142:1–12. doi: 10.1016/j.biomaterials.2017.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Das C.K., Jena B.C., Banerjee I., Das S., Parekh A., Bhutia S.K. Exosome as a novel shuttle for delivery of therapeutics across biological barriers. Mol. Pharm. 2019;16:24–40. doi: 10.1021/acs.molpharmaceut.8b00901. [DOI] [PubMed] [Google Scholar]
- 57.Ling S., Nanhwan M., Qian J., Kodakandla M., Castillo A.C., Thomas B. Modulation of microRNAs in hypertension-induced arterial remodeling through the beta 1 and beta3-adrenoreceptor pathways. J. Mol. Cell. Cardiol. 2013;65:127–136. doi: 10.1016/j.yjmcc.2013.10.003. [DOI] [PubMed] [Google Scholar]
- 58.Ren W., Liang L., Li Y., Wei F.Y., Mu N., Zhang L. Upregulation of miR 423 improves autologous vein graft restenosis via targeting ADAMTS7. Int. J. Mol. Med. 2020;45:532–542. doi: 10.3892/ijmm.2019.4419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Li H., Zhao J., Liu B., Luo J., Li Z., Qin X. MicroRNA-320 targeting neuropilin 1 inhibits proliferation and migration of vascular smooth muscle cells and neointimal formation. Int. J. Med. Sci. 2019;16:106–114. doi: 10.7150/ijms.28093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Xu F., Xiang Q., Huang J., Chen Q., Yu N., Long X. Exosomal miR-423-5p mediates the proangiogenic activity of human adipose-derived stem cells by targeting Sufu. Stem Cell Res. Ther. 2019;10:106. doi: 10.1186/s13287-019-1196-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chen C., Wang Y., Yang S., Li H., Zhao G., Wang F. MiR-320a contributes to atherogenesis by augmenting multiple risk factors and down-regulating SRF. J. Cell Mol. Med. 2015;19:970–985. doi: 10.1111/jcmm.12483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wu X., Gong Z., Ma L., Wang Q. lncRNA RPSAP52 induced the development of tongue squamous cell carcinomas via miR-423-5p/MYBL2. J. Cell Mol. Med. 2021 doi: 10.1111/jcmm.16442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Luo H., Li X., Li T., Zhao L., He J., Zha L. microRNA-423-3p exosomes derived from cardiac fibroblasts mediates the cardioprotective effects of ischaemic post-conditioning. Cardiovasc. Res. 2019;115:1189–1204. doi: 10.1093/cvr/cvy231. [DOI] [PubMed] [Google Scholar]
- 64.Viereck J., Thum T. Circulating noncoding RNAs as biomarkers of cardiovascular disease and injury. Circ. Res. 2017;120:381–399. doi: 10.1161/CIRCRESAHA.116.308434. [DOI] [PubMed] [Google Scholar]
- 65.Lu M., Yuan S., Li S., Li L., Liu M., Wan S. The exosome-derived biomarker in atherosclerosis and its clinical application. J. Cardiovasc. Transl. Res. 2019;12:68–74. doi: 10.1007/s12265-018-9796-y. [DOI] [PubMed] [Google Scholar]
- 66.Yellon D.M., Davidson S.M. Exosomes: nanoparticles involved in cardioprotection? Circ. Res. 2014;114:325–332. doi: 10.1161/CIRCRESAHA.113.300636. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.