

Abstract
USUTU virus (USUV) is an arbovirus maintained in the environment through a bird–mosquito enzootic cycle. Previous surveillance plans highlighted the endemicity of USUV in North-eastern Italy. In this work, we sequenced 138 new USUV full genomes from mosquito pools (Culex pipiens) and wild birds collected in North-eastern Italy and we investigated the evolutionary processes (phylogenetic analysis, selection pressure and evolutionary time-scale analysis) and spatial spread of USUV strains circulating in the European context and in Italy, with a particular focus on North-eastern Italy. Our results confirmed the circulation of viruses belonging to four different lineages in Italy (EU1, EU2, EU3 and EU4), with the newly sequenced viruses from the North-eastern regions, Veneto and Friuli Venezia Giulia, belonging to the EU2 lineage and clustering into two different sub-lineages, EU2-A and EU2-B. Specific mutations characterize each European lineage and geographic location seem to have shaped their phylogenetic structure. By investigating the spatial spread in Europe, we were able to show that Italy acted mainly as donor of USUV to neighbouring countries. At a national level, we identified two geographical clusters mainly circulating in Northern and North-western Italy, spreading both northward and southward. Our analyses provide important information on the spatial and evolutionary dynamics of USUTU virus that can help to improve surveillance plans and control strategies for this virus of increasing concern for human health.
Keywords: USUTU virus, evolutionary analysis, diffusion dynamics, North-eastern, Italy
1. Introduction
The USUTU virus (USUV) is an arbovirus belonging to the Flaviviridae family, genus Flavivirus. It has a (+)-strand RNA genome of 11,064 nucleotides that encodes a single polyprotein. The polyprotein is cleaved by viral and host proteases into structural (C, prM, E) and non-structural (NS1, NS2a, NS2b, NS3, NS4a, NS4b and NS5) proteins (Bakonyi et al. 2004). USUV is maintained in the environment through an enzootic cycle involving mosquitoes as vectors (Culex pipiens complex above all) and wild birds as the main amplifying hosts (Roesch et al. 2019; Vilibic-Cavlek et al. 2019a). Several episodes of mass mortality have been reported in wild birds in Central Europe, with blackbirds (Turdus merula) as one of the most affected species (Weissenböck et al. 2003; Steinmetz et al. 2011; Becker et al. 2012; Benzarti et al. 2020; Weidinger et al. 2020). Although USUV has mostly been associated with disease in birds, it can also infect mammals, including humans, sporadically causing neuro-invasive disease (Pecorari et al. 2009; Santini et al. 2015; Grottola et al. 2017; Barzon 2018; Nagy et al. 2019; Pacenti et al. 2019; Simonin et al. 2018; Vilibic-Cavlek et al. 2019b).
The first known emergence of USUV in Europe dates back to 1996, when it was associated to deaths among Eurasian blackbirds in Italy (Weissenböck et al. 2013), although Engel et al. (2016) estimated three separate virus introductions in Western and Central Europe likely between 1950s and 1990s (Engel et al. 2016) through bird migration from Africa, followed by a rapid virus expansion. Phylogenetic studies performed on full-genome and partial NS5 and envelope (E) genes (Engel et al. 2016; Bakonyi et al. 2017; Cadar et al. 2017; Calzolari et al. 2017; Kemenesi et al. 2018; Roesch et al. 2019; Oude Munnink et al. 2020) identified distinct lineages, named after their geographical origin: three African (Africa 1–3) and five European (Europe 1–5) lineages. USUV has been reported in many European countries (Austria, Belgium, Croatia, the Czech Republic, France, Germany, Hungary, Italy, the Netherlands, Serbia, Spain, Switzerland) and the co-circulation of different lineages has been demonstrated in recent years: Europe 2–3, Europe 5 and Africa 2–3 lineages have been detected in Germany (Michel et al. 2019), Europe 3 and Africa 3 in the Netherlands (Oude Munnink et al. 2020), Europe 3 and Africa 2–3 in France (Eiden et al. 2018), Europe 1–3 and Africa 3 in the Czech Republic (Hönig et al. 2019).
In Italy, USUV was reported in chickens in North-eastern Italy, more specifically in Trentino Alto Adige and Emilia-Romagna regions and respectively in 2005 and 2007 (Rizzoli et al. 2007; Lelli et al. 2008); fatal cases in wild birds (owls and blackbirds) due to USUV were described in North-western Italy (Lombardy region) between 2006 and 2008 (Manarolla et al. 2010). In North-eastern Italy (Veneto region), USUV was identified as being the causative agent of a severe blackbird die-off between 2008 and 2009 (Savini et al. 2011). USUV and USUV antibodies were then reported in chickens and wild birds in North-eastern Italy, including Emilia-Romagna, Veneto and Friuli Venezia Giulia (FVG) regions (Calzolari et al. 2017; Pacenti et al. 2019). Since 2008, surveillance programs for arboviruses have been set up in Northern Italy mainly for West Nile virus (WNV) control, but the screening of mosquitoes (Culex pipiens) and birds provides evidence of the co-circulation of different USUV lineages in the surveyed territory (Calzolari et al. 2017, 2010; Mulatti et al. 2015): Europe 2 in the North-eastern and Central regions (Emilia-Romagna, Veneto, FVG, Marche, Tuscany, Lazio Regions), and Europe 4 in the North-western and Central regions (Lombardy, Piedmont, Marche) (Calzolari et al. 2017).
These monitoring plans have identified the Northeast of Italy as a hotspot for mosquito flaviviruses circulation with multiple flavivirus species detected, such as WNV and Marisma virus (Da Rold et al. 2015; Ravagnan et al. 2015; Calzolari et al. 2016) and a surge in USUV infection cases. Evidence of both asymptomatic and symptomatic USUV infections in humans has been reported in the last few years (Pacenti et al. 2019); nonetheless, little is known about the genetic diversity and spatial spread of USUV in this area.
The aim of this study was to determine the role played by North-eastern Italy in the diffusion dynamics and evolution of USUV strains not only in Italy but also in the European context. For this purpose, the whole-genome and NS5 sequences available in public databases were analysed along with the complete genome of 138 newly characterized viruses, identified from mosquitoes and wild birds in Veneto and FVG regions between 2011 and 2018.
2. Materials and methods
2.1 Sample collection
Samples were collected in the framework of a WNV entomological surveillance carried out from 2011 to 2018 in the plain area (below 300 m above sea level) of the Veneto and FVG Regions (North-eastern Italy). The plain area was divided into geographical units of 15 km2; CO2-CDC traps (IMT®—Italian Mosquito Trap, Cantù, Italy) were placed in each geographical unit to collect mosquitoes. The number of monitored sites changed according to WNV epidemiology, ranging from 35 to 72 per year. The entomological surveillance was carried out each year from the second week of May to the first week of November.
The trapped mosquitoes (Culex pipiens) were pooled including from 3 to 100 specimens per pool. From 2011 to 2018, we tested 23,371 pools and we found 481 USUV-positive pools (2%), 125 (26%) of which were selected for this study in order to cover the entire period of time and the different locations.
With regard to avian species, 3,946 clinical samples (brain, kidney, heart and spleen) were collected from dead birds in the Veneto and FVG regions in the framework of the 2011–18 national monitoring plan for arboviruses (passive surveillance). We identified USUV from seven different avian species (Alavola arvensis, Ciconia ciconia, Larus michahellis, Sturnus vulgaris, Turdus iliacus, Turdus merula, Turdus pilaris), with the most represented avian species being Turdus merula. All the USUTU positive birds (n = 13, 0.3%) were included in our analyses.
Detailed information for each virus (host, collection date and location) is provided in Table S1 in Supplementary File 1 and Supplementary File 2.
2.2 Genome sequencing and generation of consensus sequences
Total RNA was extracted from homogenate tissue samples using QIAamp Viral RNA mini kit (QIAGEN) following the manufacturer’s instructions. SuperScript™ III One-Step RT-PCR (Reverse Transcriptase Polymerase Chain Reaction) System with Platinum™ Taq High Fidelity DNA Polymerase (Invitrogen) was used to amplify whole USUV genome using primers listed in Supplementary Table S2 in Supplementary File 1. Five overlapping PCR products were obtained for each sample. Amplicons were purified using Agencourt AMPure XP (Beckman Coulter™), quantified with Qubit™ DNA HS Assay (Thermo Fisher Scientific), and mixed in equimolar proportion. Sequencing libraries were prepared using Illumina Nextera XT DNA Sample Preparation Kit (Illumina, San Diego, CA, USA) following the manufacturer’s instructions. Sequencing was performed using Illumina MiSeq (2×300 bp PE). The read quality was assessed by using FastQC v0.11.2. Raw data were filtered by removing reads with more than 10 per cent of undetermined (‘N’) bases, reads with more than 100 bases with Q score below seven, duplicated paired-end reads. Reads were clipped from Illumina Nextera XT adaptors with scythe v0.991 (https://github.com/vsbuffalo/scythe) and trimmed with sickle v1.33 (https://github.com/najoshi/sickle). Complete genomes were generated through a reference-based approach. High-quality reads were aligned against a reference genome using BWA v0.7.12 (Van Der Auwera et al. 2014), processing the alignments with Picard-tools v2.1.0 (http://picard.sourceforge.net) and GATK v3.5 (McKenna et al. 2010; Depristo et al. 2011; Van Der Auwera et al. 2014). Single nucleotide polymorphisms were called using LoFreq v2.1.2 (Wilm et al. 2012). Consensus sequences were generated with a minimum coverage of 10× and a 75 per cent base call threshold. Sequences were submitted to the GenBank database under the accession numbers MW164634 - MW164771.
2.3 Datasets design
All USUV viral sequences belonging to the European lineages were downloaded from GenBank (available on 31 January 2020) and since most of them were partial genome sequences, we decided to use both complete genome sequences as well as the partial NS5 sequences covering at least the genomic region from nucleotide (nt) 8,950 to 9,210, as they represent the only sequences available for some geographic areas.
We generated four datasets that either contain full or partial genome sequences and are focused on local or European sequence data: dataset 1 consists of complete genome sequences (n = 138) collected in Veneto and FVG regions between 2011 and 2018 from mosquitoes (n = 125) and birds (n = 13); dataset 2 consists of complete genome sequences (n = 264) collected in Europe between 2001 and 2018 from mosquitoes (n = 149), birds (n = 115); dataset 3 consists of partial NS5 sequences (n = 324) collected in Europe between 2001 and 2018 from mosquitoes (n = 150), birds (n = 143) and humans (n = 31); dataset 4 consists of partial NS5 sequences (n = 205) collected in Italy between 2006 and 2018 from mosquitoes (n = 146), birds (n = 41) and humans (n = 18) (Table 1). Datasets 3 and 4 included both the NS5 deposited in GenBank as partial sequences and the NS5 contained in the full genome sequences.
Table 1.
Description of USUV sequence datasets used in this study.
| Dataset 1 | Dataset 2 | Dataset 3 | Dataset 4 | |
|---|---|---|---|---|
| Genome/gene | Complete genome | Complete genome | NS5 | NS5 |
| Collection area | North-eastern Italya | Europe | Europe | Italy |
| N. mosquitoes | 125 | 149 | 150 | 146 |
| N. birds | 13 | 115 | 143 | 41 |
| N. humans | – | – | 31 | 18 |
| N. total | 138 | 264 | 324 | 205 |
Veneto and FVG regions.
2.4 Phylogenetic analysis
Sequences were aligned using default parameters in MAFFT version 7 (Katoh and Standley 2013) (https://mafft.cbrc.jp/alignment/server/). A quartet likelihood mapping analysis was performed using the –lmap function in IQ-TREE version 1.6 in order to assess the phylogenetic information of our input alignments (Nguyen et al. 2015). Most of the quartets in dataset 1 (90.3%), dataset 2 (91.9%) and dataset 3 (73.6%) were equally distributed in the three corners of the triangle, representing well-resolved phylogeny. Differently, dataset 4 had a considerable number of quartets (46.1%) with uncertain topology, indicating a low phylogenetic signal and a star-like topology (Supplementary Fig. S1 in Supplementary File 4).
Maximum likelihood (ML) phylogenetic trees of dataset 2 and dataset 3 sequences were generated in IQTREE version 1.6, performing ultrafast bootstrap resampling analysis (1,000 replications) (Nguyen et al. 2015; Hoang et al. 2018). Phylogenetic analyses were limited to datasets 2 and 3 since these two datasets respectively include dataset 1 and dataset 4 sequences. African lineages were used as outgroup for tree rooting. ModelFinder (Kalyaanamoorthy et al. 2017), implemented in IQTREE, was used to select the best-fitted nucleotide substitution model for each dataset. In detail, according to Bayesian Information Criterion, the model GTR + F + I + G4 (general time reversible model with unequal rates and unequal base frequencies, base frequencies calculated empirically from the alignment, allowing for a proportion of invariable sites and a discrete Gamma model with 4 rate categories) was selected for dataset 2 and the model GTR + F + R3 (general time reversible model with unequal rates and unequal base frequencies, base frequencies calculated empirically from the alignment, three categories of rate heterogeneity, with rates and weights inferred by ML) was selected for dataset 3. Phylogenetic trees were visualized by using FigTree version 1.4.2 (http://tree.bio.ed.ac.uk/software/figtree/).
2.5 Amino acid signatures
We evaluated the presence of specific amino acids characterizing clusters of sequences along the phylogenetic tree. The amino acid substitutions fixed on specific groups of the ML tree were mapped to the phylogeny by using Mesquite v3.61 (Maddison and Maddison 2019).
2.6 Analysis of recombination and selection pressure
Recombination and selection pressure were investigated on the most comprehensive dataset, consisting of complete genome sequences collected in Europe between 2001 and 2018 (dataset 2). Genetic Algorithm for Recombination Detection (Kosakovsky Pond et al. 2006), available in the Datamonkey web application (Weaver et al. 2018), was used to screen the dataset alignment for recombination breakpoints and did not reveal any recombination events.
To estimate sites under positive selection, eleven nucleotide sequence datasets corresponding to the eleven USUV proteins were generated. For each dataset, we estimated the selection across individual sites applying different methods available in Datamonkey. We used a mixed-effects ML approach in MEME (Mixed Effects Model of Evolution) (Murrell et al. 2012) to test the hypothesis that individual sites have been subject to episodic positive or diversifying selections and to three different methods so as to determine whether codons may have been the results of adaptive evolution, estimated as a ratio of non-synonymous (dN) to synonymous (dS) nucleotide substitutions (dN/dS) per site: FEL (Fixed Effects Likelihood) (Kosakovsky Pond and Frost 2005), FUBAR (Fast, Unconstrained Bayesian AppRoximation) (Murrell et al. 2013) and SLAC (Single-Likelihood Ancestor Counting) (Kosakovsky Pond and Frost 2005). Positive selection at individual codon sites was considered significant with a P-value ≤0.1 for MEME, FEL and SLAC and a posterior probability (PP) ≥0.9 for FUBAR.
2.7 Codon usage analysis
The Codon Adaption Index (CAI) was determined in order to evaluate the relative adaptability of the codon usage of a gene towards that of highly expressed genes within a given host, for the sequences obtained in Northern Italy (Sharp and Li 1987). The CAI values of USUV genomes were analyzed in the context of the following: Homo sapiens as host; Culex pipiens as vector; Passer domesticus and Sturnus vulgaris as potential avian reservoirs. The codon usage was obtained from the codon usage database available at http://www.kazusa.or.jp/codon/ (codon usage for some hosts from which some sequences were obtained, such Parus caeruleus, Turdus merula, Cicocnia ciconia, Strix nebulosa, Alauda arvensis, Garrulus glandarius, Larus michahellis, were not available). DAMBE6 was then used to calculate the CAI values (Xia and Xie 2001).
2.8 Evolutionary analysis
The evolutionary rate and the time to the most common recent ancestor (tMRCA) were estimated using Bayesian inference for all the four datasets. Markov chain Monte Carlo (MCMC) sampling analyses were performed using BEAST v1.10.4 software (Suchard et al. 2018). We employed an uncorrelated lognormal relaxed molecular clock that allows for rate variation across lineages. The SRD06 substitution model (HKY85 + Γ4 with two partitions—1st + 2nd positions vs. 3rd position—base frequencies and Γ-rate heterogeneity unlinked across all codon positions) was used (Shapiro et al. 2006).
The performance of the marginal likelihood estimators as implemented in BEAST, specifically, path sampling (PS) and stepping-stone (SS) sampling marginal likelihood estimators (Baele et al. 2012a) were assessed to compare and select the best fitting molecular clock model and tree prior. Specifically, an uncorrelated lognormal relaxed molecular clock that allows for rate variation across lineages was tested against a strict clock, and a coalescent constant population size model was tested against a coalescent Gaussian Markov random field Bayesian Skyride model (Drummond et al. 2002; Minin et al. 2008).
MCMC chains were run for 50–200 million iterations until ESS of all selected probability densities and parameter values were higher than 200, and convergence was assessed using Tracer v1.7.1 (Rambaut et al. 2018). Maximum clade credibility (MCC) trees were summarized using TreeAnnotator v1.10.4 and visualized using FigTree v1.4.2 (http://tree.bio.ed.ac.uk/software/figtree/).
The association between phylogeny and the pattern of the geographical structure of USUV was assessed on dataset 2 using the software BaTS (Bayesian Tip-Association analysis) (Parker et al. 2008). Values of the association index (AI), parsimony score (PS) statistics and the level of clustering in six locations (Italy, Germany, Austria, Hungary, France, Belgium) using the monophyletic clade (MC) size statistic were calculated based on the posterior samples of trees produced by BEAST package. The null distribution for each statistic was estimated with 100 replicates of state randomization.
To explore the pattern of the spatial diffusion among a set of geographic regions, European countries, Italian regions or Italian provinces (depending on the dataset), we performed discrete phylogeographic analyses using the location as trait (Lemey et al. 2009). We assumed an asymmetric non-reversible transition model and incorporated Bayesian stochastic search variable selection (Lemey et al. 2009). Spread D3 v0.9.6 (Bielejec et al. 2016) was used to visualize the spatial dispersal of the viral strain through time and to assess Bayes factor (BF) supports for individual transitions between discrete locations. We interpreted the strength of statistical support as positive for 5 ≤ BF <20, strong for 20 ≤ BF <150 and very strong for BF ≥150.
As regards dataset 1, for which precise geographic coordinates were available, we also estimated the viral diffusion dynamics in continuous space (Lemey et al. 2010), as a complementary approach to discrete phylogeographic inference. A strict Brownian diffusion model was tested against relaxed random walk models and the best model was selected on the basis of the path sampling (PS) and SS sampling marginal likelihood estimators, implemented in BEAST (Baele et al. 2012a,b). The spatiotemporal dynamic of USUV in Veneto and FVG regions was visualized using Spread D3 v0.9.6 (Bielejec et al. 2016).
Datasets 2 and 3 contain a higher number of sequences from Italy (N = 161 for dataset 2 and N = 190 for dataset 3) and Germany (N = 95 for dataset 2 and N = 96 for dataset 3) compared with the other countries (N = 12 for dataset 2 and N = 40 for dataset 3). Moreover, most of the German viruses were collected from birds (30.6% for dataset 2 and 25.5% for dataset 3), whereas the majority of the Italian viruses were identified in mosquitos (51.5% for dataset 2 and 40.8% for dataset 3). To mitigate sampling bias, we performed the phylogeographic analysis also on two further down-sampled datasets (balanced dataset 2 and balanced dataset 3) with a most balanced distribution of samples in terms of host and geographical origin. Balanced dataset 2 included twenty-two viruses from Italy, twenty-two viruses from Germany and eleven viruses from other European countries. Balanced dataset 3 was composed of thirty-three viruses collected in Italy, twenty-two viruses collected in Germany and thirty-nine viruses from other European countries (Supplementary File 2).
In order to mitigate sampling bias for dataset 4, which included a large number of NS5 sequences from the Veneto region compared to the other Italian regions (133 out of 205 sequences), we performed the phylogeographic analysis also on three different subsets obtained by randomly selecting the samples based on sampling location, in order to have roughly equitable numbers of sequences for each geographic region (Supplementary File 2).
3. Results
3.1 Phylogenetic relationship among USUV sequences reveals the circulation of a single genetic lineage in North-eastern Italy
ML phylogenetic trees of both complete genome and NS5 partial sequences show that the Italian USUV group within four distinct European (EU) lineages, namely EU1, EU2, EU3 and EU4 (Supplementary Figs. S2 and S3 in Supplementary File 4). Specifically, all viruses from North-eastern Italy, including the 138 complete genome sequences generated in this study, fall into two separate groups, here named A and B, within the EU2 lineage. Group A includes viruses collected over the period 2009–16 from mosquitoes (Culex pipiens) and birds mainly in the Veneto region, and one human virus collected in 2009 in the Emilia-Romagna region. Group B is more widespread and was detected between 2009 and 2018 from different hosts (mosquito, birds and humans) in the Northern and Central Italian regions, as well as in Austria and in Hungary (Supplementary Figs. S2 and S3 in Supplementary File 4).
Except for EU4, which includes only viruses collected in Italy (mainly in the Northwest), EU1 and EU3 are widespread in Europe. In particular, EU1 has been circulating mainly in the south and east of Europe, including Italy (Emilia-Romagna region), Austria, Hungary and Serbia, while EU3 has been detected mainly in Northern and Central Europe (Switzerland, Germany, Belgium and France).
USUV strains belonging to the African lineages detected in Northern and Central Europe have never been identified in Italy. For this reason, we decided to focus all our analyses on the EU lineages.
3.2 Fixed amino acid substitutions, selection pressure and codon usage analysis
Analysis of the complete genome of European USUV identified twenty-one fixed amino acid substitutions distinguishing the different EU lineages. Figure 1 shows the branches with the mutations, which are distributed over eleven proteins, more than a half in the non-structural proteins NS2a (NS2a74, NS2a91, NS2a108, NS2a128, NS2a179), NS4b (NS433, NS4174, NS4249) and NS5 (NS5116, NS5274, NS5834, NS5896) proteins (Table 2). The EU2 lineage is characterized by four amino acid signatures (E830G, I1324T, I1602V, N2304S at NS1, NS2a, NS3, NS4b proteins). In turn, subgroup EU2-A and EU2-B can be distinguished by two unique substitutions each: G595S, E3425D (at E and NS5 proteins) for EU2-A and K181R, P3363L (at prM and NS5 proteins) for EU2-B (Table 2, Fig. 1). Country-specific mutations N11S(C), L1549F(NS3), V1602I(NS3), K2445R(NS4b) have been observed for the German strains, while E830G(NS1), I1324T(NS2a), M2645I(NS5) are characteristics of the Italian strains. Since Italian and German viruses fall within distinct lineages, most of these mutations are also signatures of EU1, EU2 and EU4 (Fig. 1). Interestingly, just one lineage-specific mutation (G595S) has been found in the envelope protein, which was suggested to have a significant role for USUV evolution (Engel et al. 2016).
Figure 1.

Detail of the complete genome phylogenetic tree, showing fixed amino acid substitutions for each European lineage.
Table 2.
Fixed amino acid substitutions in supported phylogenetic clusters representing European USUV lineages EU1, EU2 (EU2-A and EU2-B), EU3, EU4.
| Codon | Protein and AA position | AA | EU1 | EU2 |
EU3 | EU4 | |
|---|---|---|---|---|---|---|---|
| EU2-A | EU2-B | ||||||
| 11 | C11 | N/S | N | N | N | S | N |
| 85 | C85 | K/R | K | K | K | R/K | K |
| 181 | prM55 | K/R | K | K | R | K | K |
| 262 | prM136 | F/L | F | L/F | F | F | F |
| 595 | E302 | S/G | G | S | G | G | G |
| 830 | NS137 | G/E | E | G | G | E | E |
| 1219 | NS2a74 | V/I | V | V | V | I | I |
| 1236 | NS2a91 | A/V | A | A | A | V | V/L |
| 1253 | NS2a118 | A/T | A | A/T | A | A | A |
| 1273 | NS2a128 | T/I | I | T/I | I | I | I/A |
| 1324 | NS2a173 | T/I | I/T | T | T | I | I |
| 1405 | NS2b173 | F/L | F | F/L | F | F/L | F |
| 1549 | NS333 | L/F | L | L/F | L | F | L |
| 1602 | NS399 | V/I | I | V | V | I | V |
| 2304 | NS4b33 | S/N | N | S | S | N | N |
| 2445 | NS4b174 | K/R | K | K | K | R | K |
| 2520 | NS4b249 | R/K | K | R/K | K | K | K |
| 2645 | NS5116 | I/M | M | I/M | I | M | M |
| 2803 | NS5274 | A/T | T | T/A | T | T | T |
| 3363 | NS5834 | L/P | P | P | L | P | P |
| 3425 | NS5896 | D/E | E | D | E | E | E |
The application of three selection pressure analysis methods (MEME, FUBAR, FEL) revealed that thirteen sites were under positive selection (MEME identified five sites under episodic positive selection; FUBAR and FEL identified nine and three sites, respectively, under pervasive positive selection), and most of them had occurred at internal branches. One of them was located at NS1 and NS4b, two at C and prM, three at NS3 and four at NS2A proteins (Table 3) (Supplementary Fig. S4 in Supplementary File 4). Particularly, four sites of the polyprotein (prM136, NS2a128, NS361 and NS3344) were identified under positive selection by two different selection pressure analysis methods (Table 3); two of which (NS361, NS3344) already reported by Engel et al. (2016). Further analyses need to be performed to investigate more in-depth the role of these sites and to determine their effect on the epidemiology and pathogenesis of USUV-related disease. Consistently with previous observations for other arboviruses replicating in alternate hosts, FUBAR and FEL found strong evidence of purifying selection at respectively 414 and 286 sites distributed in all the viral proteins (Holmes 2003; Lequime et al. 2016; Mavian et al. 2017). The majority of these sites were located in the E (sixty-four sites with FUBAR and thirty-nine with FEL), NS3 (seventy-seven sites with FUBAR and fifty-six with FEL), NS4b (thirty-five sites with FUBAR and thirty-six with FEL) and NS5 (186 sites with FUBAR and seventy-six with FEL) proteins.
Table 3.
Codons under positive selection using different selection pressure analysis methods (MEME, P < 0.1; FUBAR, PP > 0.9; FEL, P < 0.1). The analyses were performed on dataset 2.
| Codon | Protein and AA position | AA | MEME P-value |
FUBAR PP |
FEL P-value |
|---|---|---|---|---|---|
| 11 | C11 | N/S | ns | 0.964 | ns |
| 124 | C124 | V/A/I | ns | 0.900 | ns |
| 246 | prM120 | Y/N | 0.090 | ns | ns |
| 262 | prM136 | L/F | ns | 0.919 | 0.091 |
| 939 | NS1146 | V/A | ns | 0.928 | ns |
| 1147 | NS2a2 | R/Q | ns | 0.954 | ns |
| 1263 | NS2a118 | A/S | 0.030 | ns | ns |
| 1273 | NS2a128 | T/I | 0.020 | 0.920 | ns |
| 1318 | NS2a173 | T/I | ns | 0.906 | ns |
| 1549 | NS346 | L/F | ns | 0.928 | ns |
| 1564 | NS361 | G/S | 0.090 | ns | 0.071 |
| 1847 | NS3344 | A/V/P | ns | 0.908 | 0.098 |
| 2428 | NS4b157 | L/I | 0.000 | ns | ns |
Codons that resulted under positive selection for more than one method are marked in bold.
ns: not significant.
We compared complete genomes of USUV collected from birds and from mosquitoes. Five sites (C11, NS2a2, NS2a118, NS2a179, NS361) in the subset of sequences of avian origin and five sites (prM136, NS2a128, NS346, NS353, NS4b157) in the subset of sequences of mosquito origin resulted under positive selection (Table 4). Of note, viruses circulating in mosquito vectors and avian hosts show different sites under positive selection, although, in both cases, most of them are located in the NS2a and NS3 proteins. Anyway, as most of the sequences of avian origin come from Germany (EU3 lineage) and most of the sequences of mosquito origin were collected in Italy (EU2 and EU4 lineages), these results could be influenced by geographical origin rather than host/vector origin.
Table 4.
Codons under positive selection using different selection pressure analysis methods (MEME, P < 0.1 and FUBAR, PP > 0.9) identified 115 USUV complete genomes collected from birds and 149 USUV complete genomes collected from mosquitoes.
| Codon | Protein and AA position |
AA | Sequences of avian origin |
Sequences of mosquito origin |
||
|---|---|---|---|---|---|---|
| MEME P-value |
FUBAR PP |
MEME P-value |
FUBAR PP |
|||
| 11a | C11 | N/S | ns | 0.933 | ns | ns |
| 262b | prM136 | L/F | ns | ns | ns | 0.964 |
| 1147a | NS2a2 | R/Q | ns | 0.925 | ns | ns |
| 1263a | NS2a118 | A/S | 0.050 | ns | ns | ns |
| 1273b | NS2a128 | T/I | ns | ns | 0.030 | 0.957 |
| 1324a | NS2a179 | T/I | ns | 0.910 | ns | ns |
| 1549b | NS346 | L/F | ns | ns | ns | 0.943 |
| 1556b | NS353 | T/K | ns | ns | 0.090 | ns |
| 1564a | NS361 | G/S | ns | 0.936 | ns | ns |
| 2428b | NS4b157 | L/I | ns | ns | 0.000 | ns |
ns: not significant.
Codons characteristics of sequences of avian origin;.
Codons characteristics of sequences of mosquito origin.
In order to investigate potential differential adaptation of USUV isolates to mammalian hosts and arthropod vectors based on the origin of sampling, we compared Codon Adaptation Index estimates based on the codon usage bias of USUV full-genomes (Fig. 2). In the CAI analysis, full-genome sequences obtained in Northern Italy from mosquitoes and birds showed overlapping CAI values, therefore suggesting that USUV codon usage is conserved among strains circulating in different hosts (Fig. 7). USUV CAI values based on the NS5 gene sequences obtained in Italy and across Europe were overlapping. This finding indicates that the geographic origin of the USUV strains does not affect USUV codon usage. A wide range of CAI values was observed in the different hosts, and overall, USUV full genome sequences showed higher CAI values for the codon usage of Homo sapiens, thus suggesting that USUV in Europe has a high degree or better adaptability to humans. Although CAI values for Sturnus vulgaris and Passer domesticus were lower than the ones for humans, they suggest that these avian species are likely a reservoir host for USUV. Unfortunately, the codon usage for Turdus merula, which is the most abundant species in our datasets, was not available from the codon usage database (http://www.kazusa.or.jp/codon/), which prevented us from confirming the results for this species. Surprisingly, USUV showed low CAI values for Culex pipiens, suggesting low adaptability to this vector. These results highlighted that codon usage of USUV does not change among strains circulating in different hosts or different geographic regions, and that NS5 is a good proxy for full genome in terms of CAI analysis.
Figure 2.
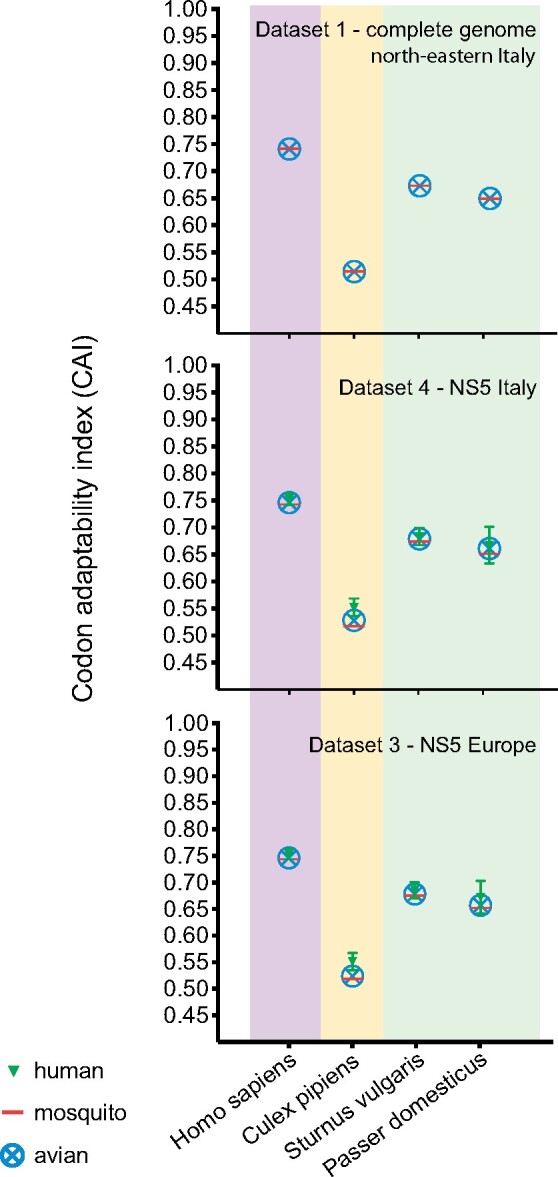
CAI: CAI analysis for USUV full-genome and NS5 gene sequences based on the codon usage of the most frequent genes of various hosts and vector species: Homo sapiens (pink shade), Culex pipiens (yellow shade) and avian hosts Sturnus vulgaris and Passer domensticus (green shade). Values for USUV from humans (green triangle symbol), mosquitoes (red horizontal line symbol) or avian samples (blue circle-cross symbol) are reported as mean ± standard deviation.
3.3 Evolutionary dynamics of USUV European lineages
To explore the evolutionary dynamics and the time of emergence of the EU lineages in Europe and North-eastern Italy, we estimated the tMRCA and the rate of nucleotide substitution of the complete USUV genomes belonging to the EU lineages (dataset 2) (n = 264) and of USUV genomes collected in North-eastern Italy (dataset 1) (n = 138).
For the complete genome sequences of the European USUV, we estimated a rate of nucleotide substitution of 3.41 × 10−4 sub/site/year with 95 per cent highest posterior density (HPD) of 3.04–3.77 × 10−4. The mean tMRCA of the root dates back to June 1993 (95% HPD, May 1988 to October 1997) (Fig. 3). This time point is three years prior the earliest identification of USUV in Europe (in 1996), when a USUV outbreak caused a mass mortality among wild birds (mostly Turdus merula) in the Tuscany region in Central Italy (Weissenböck et al. 2013). Among the EU lineages, the EU1 was the first one to emerge (mean tMRCA June 1999, 95% HPD, May 1997 to December 2000) while EU2, EU3 and EU4 appeared between October 1999 and December 2007. The mean tMRCA for the EU2 lineage was estimated to be May 2003 (95% HPD, October 1999 to February 2006), with EU2-A and EU2-B likely emerging in February 2007 (95% HPD, April 2005 to June 2008) and November 2007 (95% HPD, September 2005 to August 2009), respectively (Fig. 3, Supplementary Table S3 in Supplementary File 1).
Figure 3.

(a) tMRCA values of the USUV European complete genomes (EU) and of the EU sub-lineages EU1, EU2, EU3, EU4 and the EU2 sub-groups EU2-A and EU-B. (b) Skyride plot of dataset 1 (complete genome Northeast Italy) showing the viral population increase from 2010 to 2018 in North-eastern Italy.
Consistently with results from European USUV sequences, the complete genome of the North-eastern Italian viruses exhibited a rate of nucleotide substitution of 3.62 × 10−4 sub/site/year (95% HPD, 3.22–4.04 × 10−4) and since its introduction the virus has shown a constant increase in genetic diversity (Fig. 3).
3.4 Transmission dynamics of USUV in Europe—the role of Italy
Dataset 2 was also used to explore the gene flows and the pattern of geographical structure of USUV in the European and Italian scenario. Our Bayesian MCMC analysis of geographical association revealed an overall geographic clustering of USUV sequences by country of origin (P = 0 for both AI and PS). In particular, a significant subdivision of the population was observed for Italy, Germany and Austria (P = 0.01 for MC statistic), while the MC statistic indicated an extensive viral migration into Hungary, France and Belgium (Supplementary Table S4 in Supplementary File 1).
To examine more in detail the transmission dynamics of USUV EU lineages between Italy and the other European countries (Austria, Belgium, France, Germany, Hungary), as well as the role of the Italian regions (Emilia-Romagna, FVG, Lombardy, Marche, Piedmont, Veneto) (Fig. 4a, Supplementary Fig. S5a in Supplementary File 4), we performed discrete-trait phylogeographic analyses, considering either Italy as a whole or each Italian region separately. Results from both the analyses showed that Italy played an important role in the spread of EU lineages into Europe, acting as an important source of the virus. Our estimations indicated one single virus spread from Italy to Germany (BF = 27.86, PP = 0.86) in an undefined moment between 1999 and 2006, and two virus movements from Italy to Austria (BF = 274.21, PP = 0.98), which likely occurred in October 1995-June 1999 and in May 2012-October 2013 (Figs. 4a and 5a, Supplementary Fig. S6a in Supplementary File 4). A more detailed analysis that considers each Italian region separately, suggests Lombardy (North-western region) and Emilia-Romagna (North-eastern region) as the most likely sources of USUV for Germany (BF = 5, PP = 0.5) and Austria (BF = 56.48, PP = 0.86), respectively (Supplementary Figs. S5a and S7a in Supplementary File 4 and Supplementary File 3). Subsequently, EU lineages spread from Germany to France (BF = 38425.44, PP = 0.99) and Belgium (BF = 4265.70, PP = 0.99) and from Austria to Hungary (BF = 245.27, PP = 00.98) at various times (Figs. 4a and 5a, Supplementary Figs. S5a and S6a in Supplementary File 4). These findings are consistent with the results obtained from the analysis of the NS5 gene of the European viruses (n = 324, dataset 3) as well as with the migration patterns obtained from the down-sampled datasets 2 and 3 which have a more balanced distribution of samples among locations and hosts (Supplementary Fig. S11 in Supplementary File 4, Supplementary File 2 and Supplementary File 3). The availability in GenBank of NS5 sequences from a greater number of European countries (n = 8)—Italy, Austria, Belgium, France, Germany, Hungary, Switzerland and Serbia—as well as Italian regions (n = 8)—Emilia-Romagna, FVG, Lombardy, Marche, Piedmont, Veneto, Tuscany and Lazio—allowed us to better trace the virus spread throughout Europe. Specifically, an additional virus spread from Hungary to Serbia between May 2010 and September 2011 was identified (BF = 144.44, PP = 0.96) and showed that USUV had reached Germany from Italy crossing Switzerland (Italy to Switzerland BF = 25.72, PP = 0.80; Switzerland to Germany BF = 14.13, PP = 0.69) between August 2001 and October 2003 (Figs. 4b and 5b, Supplementary Figs. S5b and S6b in Supplementary File 4). This transmission pattern is not supported by the analyses based on the subdivision of Italy per region, which recognized Switzerland as a virus source for Italy (Switzerland to Tuscany region BF = 19.24, PP = 0.59), but this could be likely due to the limited sequence length (Supplementary Figs. S5b, Fig. S7b, Fig. S6, Fig. S7 in Supplementary File 4). Similarly, this analysis confirmed that Italy was most likely the country of origin of USUV detected in Austria since 2016 (BF = 456.99, PP = 0.99); however, in this case, North-eastern Veneto region was identified as the most likely virus source (BF = 270.23, PP = 0.95). These results need to be corroborated by complete genome analyses as soon as complete genomes of viruses collected in Switzerland, Tuscany and Austria are going to be available. Overall, our investigation reveals a general pattern in the virus spread, with the Western Italian regions as the major source of the virus for central EU countries and Eastern Italy as the most likely origin of the virus for central-eastern EU countries, although the specific role played by each Italian region as a source or sink of the virus for the other European countries may have been affected by the different number of available sequences.
Figure 4.

Spatial spread of USUVs detected in Europe. Maps showing statistically supported non-zero rates (BF > 5) of the EU lineages resulted from the analyses of two distinct datasets: (a) Dataset 2—complete genome, (b) Dataset 3—NS5. The location trait provides for a division of Europe on a country level. The thickness of the dashed lines representing the rates is proportional to the relative strength by which rates are supported: very strong (BF > 150, thick lines), strong (20 < BF < 150, medium lines) and positive (5 < BF < 20, dashed lines).
Figure 5.

(a) MCC trees of dataset 2—complete genome Europe; (b) MCC trees of dataset 3—NS5 Europe. PP values higher than 0.8 are indicated next to the nodes.
Indeed, the different availability of viral sequences from different countries can affect the results of the phylogeographic analyses. The more the complete genome sequences are made available, the more accurate the analyses will be.
3.5 Transmission dynamics of USUV in Italy—the role of North-eastern Italy
Throughout the whole Italian territory, the discrete-trait phylogeographic analysis of dataset 2 clearly suggests no or limited virus gene flows between Eastern and Western regions and suggests Emilia-Romagna (red in Supplementary Fig. S5 in Supplementary File 4) as the major source of USUV. Specifically, in the Eastern area, we evidenced a virus spread southward from Emilia-Romagna to Marche region (BF = 138.40, PP = 0.94), and northward from Emilia-Romagna to Veneto (BF = 83582.66, PP = 1) and from Veneto to FVG (BF = 83582.66, PP = 1) (Supplementary Fig. S5a in Supplementary File 4). At least three major dissemination events from Emilia-Romagna to Veneto were identified between 2003 and 2011, followed by a persistent circulation of the virus in Veneto (Supplementary Figs. S5a and S7a in Supplementary File 4). Differently, viruses identified in FVG originated from multiple independent virus movements from Veneto, which did not give rise to local clusters (Supplementary Figs. S5a and S7a in Supplementary File 4). On the other hand, in Western Italy, we identified a single virus gene flow from Lombardy to Piedmont (BF = 16.69, PP = 0.64).
Most of the inter-region geographic spreads were confirmed by the analyses of NS5 sequences (in datasets 3 and 4) (Supplementary Figs. S5b, S7b, S8, S9 in Supplementary File 4). These datasets contain 190 Italian sequences from eight different regions, including Lazio and Tuscany in Central Italy, for which only partial sequences were available. In order to assess the robustness of our analyses and mitigate sampling bias from the different Italian regions, we assembled three different down-sampled datasets for dataset 4, as described in the Section 2. All the results reported below were confirmed by all three down-sampled datasets, except for one transition (Veneto to FVG) which was confirmed in two out of three down-sampled datasets (Supplementary File 3).
Lazio appears to have played a marginal role in the virus diffusion within the country, with no supported virus spread identified between this region and the other Italian territories. The analyses of the NS5 datasets indicated a southward spread to the Marche region from the North-western Piedmont region and highlighted the role of Emilia-Romagna region in the viral spread southwards to the Marche and northwards to the Veneto and Piedmont regions (Supplementary Figs. S5b and S8 in Supplementary File 4). In Northwest Italy, we observed a marked link between the neighbouring Lombardy and Piedmont regions, which exhibited a strong virus exchange, although the spread direction from one region to the other is unclear (from Lombardy to Piedmont for dataset 2 and dataset 4 in Supplementary Figs. S5a and S8 in Supplementary File 4; from Piedmont to Lombardy for the NS5 European sequences of dataset 3 in Supplementary Fig. S5b in Supplementary File 4).
3.6 Transmission dynamics of USUV in North-eastern Italy
The discrete-trait phylogeographic analysis of the North-eastern Italian viruses suggests the Veneto provinces of Venice (VE), Treviso (TV) and Vicenza (VI) as the major sources of the virus, while the FVG provinces of Pordenone (PN) and Udine (UD) only received the virus from the Veneto region without spreading it further (Fig. 6a, Supplementary Fig. S10 in Supplementary File 4).
Figure 6.
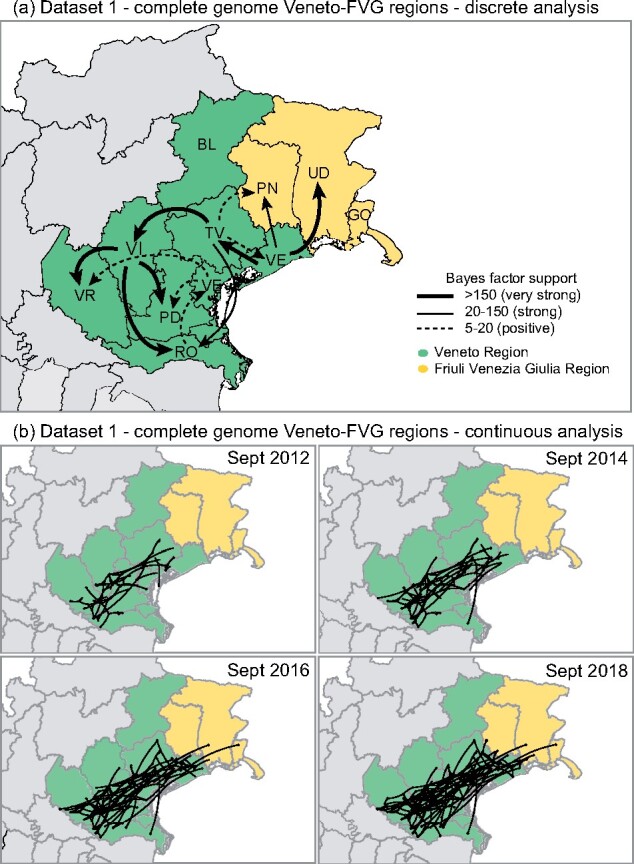
Dataset 1—USUV complete genome from Northeast Italy (Veneto and FVG regions). (a) Discrete analysis; (b) continuous analysis. Each window shows the viral movements in different time points (Sept 2012, Sept 2014, Sept 2016 and Sept 2018).
A more detailed phylogeographic reconstruction in continuous space corroborates this transmission dynamics, showing an extensive virus movement between 2012 and 2015 in the central flat area of the Veneto region (green in Fig. 6b), from the southern area of TV province to the southern part of VR province. Subsequently, since mid-2015 several independent North-eastern viral spreads from Veneto towards the flat area of FVG (yellow in Fig. 6b) were observed. None of these incursions in FVG appear to be responsible of further spread within the region.
4. Discussion
This study describes the characterization of 138 newly sequenced USUV full genomes from mosquito pools and wild birds collected in North-eastern Italy and investigates their evolutionary history within the European context. Our results confirm the circulation of viruses belonging to four different lineages in Italy (EU1-4), with the newly sequenced viruses from Veneto and FVG belonging to the EU2 lineage and clustering into two different sub-lineages, EU2-A and EU2-B that respectively emerged in February 2007 and November 2007, likely indicating a local evolution. EU2-B includes viruses collected between 2009 and 2016 mainly in the Veneto region, whereas EU2-A is more widespread and includes the most recent samples collected over the period 2009–18 from Northern and Central Italian regions, Austria and Hungary. Specific mutations characterize each European lineage (Fig. 1, Table 2) as well as the two different sub-lineages. One of these, G595S (E302), is located within the E protein and is distinctive of the EU2-A sub-lineage. The E protein was suggested to have a significant role for USUV evolution (Engel et al. 2016), it mediates the initial steps of host infection (cell binding and entry) and it is the main target of immune responses, thus influencing antigenic selection (Roehrig 2003). Mutation G595S, which characterizes the EU2-A sub-lineage, is located in the DIII domain of the E protein, and might have played a role in determining the neuroinvasive capacity of USUV towards the human host (Gaibani et al. 2013). The DIII domain in the E protein of flaviviruses is important for the interaction with cellular receptors in both human and mosquito cells; mutations in this region have been demonstrated to influence WNV infectivity, antigenicity and escape from neutralizing antibodies (Chu et al. 2005; Chin et al. 2007). Being this mutation fixed in a genetic group which includes the most recent sequences of the EU lineage currently circulating in Europe (EU2-A), it may pose a threat for humans. This mutation may also explain the higher number of human cases reported for this sub-lineage. Unfortunately, for most of these cases, NS5 is the only available gene, which prevents us from confirming the presence of this specific mutation for all the viruses.
The phylogenetic structure seems to be shaped by the geographic locations, in particular for the Italian and German clusters, suggesting an in situ evolution of this virus, as previously suggested also by other authors (Añez et al. 2013; Di Giallonardo et al. 2016) (Fig. 1, Supplementary Fig. S2 in Supplementary File 4). This finding is further supported by the identification of a single lineage in the area sampled in our study. Four mutations observed at C, NS3 and NS4b proteins are specific for German strains, while three mutations at NS1, NS2a and NS5 proteins characterize the Italian strains, indicating that mutations in different proteins have been involved in the formation of the lineages EU3 and EU2, respectively.
We identified thirteen sites under positive selection, most of which (70%) in the non-structural proteins (NS1, NS2a, NS3, NS4b). It has been shown that anti-NS1 antibodies are protective against severe diseases in flavivirus-borne infections and T-cell epitopes are localized in non-structural proteins of other flaviviruses; accordingly, the selective pressure at the sites located on non-structural proteins seems to be exerted by the host adaptive immune system (Chung et al. 2006; Lee et al. 2012; Mathew et al. 2014). Other works confirming the evidence for positive selection in WNV in mosquitoes demonstrated that WNV may undergo to convergent evolution in mosquitoes; however, the transmission of potentially mosquito-adaptive WNV variants is highly influenced by genetic drift in mosquitoes, and the genetic diversity is removed by strong purifying selection following the transmission to birds (Grubaugh and Ebel 2016; Grubaugh et al. 2017).
This is in line with the low CAI values for Culex pipiens, suggesting reduced adaptability of USUV to the vector rather than to its avian host. Our findings are supported by the transmission experiments demonstrating that WNV genetic diversity accumulates during mosquito infection within mosquitoes, although it is selectively purified during transmission from mosquitoes to birds (Grubaugh et al. 2017). Fifty percent (50%) of synonymous mutations is conserved during transmission, while 90 per cent of nonsynonymous mutations is lost, meaning that the strong purifying selection conserves virus population structure and removes deleterious mutations. The high level of purifying selection detected for WNV reflects what we have observed for USUV.
Overall, our investigations of the spatial spread in the European context show that Italy acted mainly as donor of USUV, which spread from North-eastern Italy to Austria and from North-western Italy to Switzerland. The direction of viral exchanges between Austria and Italy is controversial. While several authors speculated a presumptive viral movement from Austria to Italy (Engel et al. 2016; Calzolari et al. 2017; Roesch et al. 2019), our analyses revealed a USUV spread from Italy to Austria on two different occasions. The first dates back to the period 1995–99 and is consistent with the first identification of USUV in Europe in samples collected from Italian blackbirds in 1996 (Weissenböck et al. 2013); the second occurred in 2012–13. Although we cannot completely exclude it could actually represent a bias due to the large amount of Italian sequences available in our study, results from the balanced datasets support these observations. In between the two Austrian waves, from 1999 to 2006 Italy also exported USUV to Germany through Switzerland. USUV also spread westwards from Germany to France and Belgium (in 2011–15) and to the east from Austria to Hungary (in 1999–2011 and in 2013–16) and from Hungary to Serbia (in 2011). These findings are consistent with similar viral flows identified in previous works (Engel et al. 2016; Cadar et al. 2017; Roesch et al. 2019) and with the results we obtained by the analyses performed from down-sampled balanced datasets.
Despite our attempt to mitigate the effect of sampling bias, we cannot exclude that our results could be affected by the different surveillance and sequencing efforts implemented in countries and regions. Following our analyses, other USUV European sequences were added to public databases. As an example, Oude Munnink et al. (2020) (Oude Munnink et al. 2020) described 112 USUV viruses detected in the Netherlands between 2016 and 2018; most of them, 89 out of 112, belong to the African lineage (Africa3) while the remaining 23 strains group together with the German viruses within the EU3 lineage. As new sequences are going to be published, future analyses will help to fill the current gaps and provide a better understanding of the USUV diffusion dynamics in Europe.
Viral movements within Europe may have been shaped by geographic barriers, such as vegetation and climatic conditions affecting mosquito and bird populations, or by short or long-distance bird migrations. To give an example, the Turdus spp, and in particular blackbirds (Turdus merula), which are the most represented avian species in our datasets, can either be resident, partially migratory or fully migratory (Franchini et al. 2017). Turdus spp may therefore have contributed to the spread of the virus to neighbouring countries or in countries encountered along the migratory route. For the Turdus spp, the main direction of migration into Europe is North-East/South-West (Hasle, 2013; Höfle et al. 2013), which is consistent with the USUV spread from Germany to Belgium and France.
The strong geographical clustering we observed from the phylogenetic and BaTS analyses might reflect the emergence of local virus variants, which acquired changes during the adaptation to the local ecological conditions and were maintained in specific territories through an in situ evolution, as observed in North-eastern Italy, where a single lineage has been circulating. A similar scenario was observed for WNV in the United States (Añez et al. 2013; Di Giallonardo et al. 2016). The structure of bird populations changes depending on the environment (e.g. humid areas, forest areas), the availability of food, the vegetation in which they nest; based on these variations the vector population structure changes as well.
Among the elements that may have influenced our results we must also consider the viral transmission between migratory and indigenous bird populations and, lastly, the spread through short local movements and/or ship/airplane-borne transportation of USUV infected mosquitoes (Ibáñez-Justicia et al. 2020; Nielsen et al. 2020).
At a regional level, the Emilia-Romagna region acted as the major source of the virus. The spread between bordering regions of the Northern area of the country could have been favoured by the mosquitoes and wild birds abundance in the Po Valley, which expands from Piedmont to Lombardy, Emilia-Romagna, Veneto and FVG. In Northeast Italy, the Veneto provinces of Vicenza, Venice and Treviso resulted to be the main spreaders of the virus. Both USUV and WNV have been circulating in this area since 2008 and have become endemic. These viruses found the ideal conditions for their maintenance on site, with a warm and humid climate which creates favourable conditions for mosquito populations (Veronesi et al. 2012; Carrieri et al. 2014). USUV and WNV are characterized by a similar genetic structure and have in common clinical manifestations, besides presenting overlaps in geographic prevalence, host and vector species (Clé et al. 2019; Zannoli and Sambri 2019; Riccetti et al. 2020). We still do not know if the two viruses compete or co-exist, but our study revealed that the province of Vicenza, which was marginally affected by WNV (Busani et al. 2011), was strongly affected by the circulation of USUV in the time span considered (2011–18), suggesting that USUV was able to expand in these areas precisely because WNV was sparsely present. Overall, our Skyride plot analysis indicated a constant population growth of USUV in North-eastern Italy since 2010; whether this corresponds to a parallel decrease in the circulation of the WNV should be further explored.
Even though till now the limited circulation of USUV in humans has restrained the general consideration about this virus, our analyses rise concern for the massive USUV circulation in Northern Italy in recent years. Two sub-lineages (EU2-A and EU2-B) of the Europe 2 lineage were circulating between 2009 and 2018, with EU2-A still to be found throughout the Italian peninsula. Surveillance plans and research efforts are needed at a national and European levels to estimate USUV prevalence more accurately, identifying molecular markers associated with virulence or adaptation to new hosts and vectors. The development of control strategies at the vector/host interface will help to prevent the possible emergence of new outbreaks and reduce the impact this may have in the future.
Supplementary Material
Acknowledgments
We wish to thank Francesca Ellero for her excellent technical assistance. We thank the two anonymous reviewers for their careful and critical reading of our manuscript and their insightful comments and suggestions.
Data availability
Accession numbers of the sequences generated in this study have been provided in the Supplementary Data.
Funding
This study was made possible through the funding provided by the Italian Ministry of Health—Ricerca Corrente IZSVE 03/2017 “USUTU virus evolutionary route in endemic areas of north-eastern Italy and risk assessment of transmission to humans through blood transfusion”. LB acknowledge funding from the European Union’s Horizon 2020 research and innovation programme, under grant agreement no. 874735 (project VEO).
Supplementary data
Supplementary data are available at Virus Evolution online.
Conflict of interest: None declared.
References
- Añez G.et al. (2013) ‘Evolutionary Dynamics of West Nile Virus in the United States, 1999–2011: Phylogeny, Selection Pressure and Evolutionary Time-Scale Analysis’, PLoS Neglected Tropical Diseases, 7: e2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baele G.et al. (2012a) ‘Improving the Accuracy of Demographic and Molecular Clock Model Comparison While Accommodating Phylogenetic Uncertainty’, Molecular Biology and Evolution, 29: 2157–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baele G.et al. (2012b) ‘Accurate Model Selection of Relaxed Molecular Clocks in Bayesian Phylogenetics’, Molecular Biology and Evolution, 30: 239–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakonyi T.et al. (2004) ‘Complete Genome Analysis and Molecular Characterization of Usutu Virus That Emerged in Austria in 2001: Comparison with the South African Strain SAAR-1776 and Other Flaviviruses’, Virology, 328: 301–10. [DOI] [PubMed] [Google Scholar]
- Bakonyi T.et al. (2017) ‘Usutu Virus Infections among Blood Donors, Austria, July and August 2017 – Raising Awareness for Diagnostic Challenges’, Eurosurveillance, 22: 17–00644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barzon L. (2018) ‘Ongoing and Emerging Arbovirus Threats in Europe’, Journal of Clinical Virology, 107: 38–47. [DOI] [PubMed] [Google Scholar]
- Becker N.et al. (2012) ‘Epizootic Emergence of Usutu Virus in Wild and Captive Birds in Germany’, PLoS One, 7: e32604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benzarti E.et al. (2020) ‘Usutu Virus Epizootic in Belgium in 2017 and 2018: Evidence of Virus Endemization and Ongoing Introduction Events’, Vector-Borne and Zoonotic Diseases, 20: 43–50. [DOI] [PubMed] [Google Scholar]
- Bielejec F.et al. (2016) ‘SpreaD3: Interactive Visualization of Spatiotemporal History and Trait Evolutionary Processes’, Molecular Biology and Evolution, 33: 2167–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busani L.et al. (2011) ‘West Nile Virus Circulation in Veneto Region in 2008-2009’, Epidemiology and Infection, 139: 818–25. [DOI] [PubMed] [Google Scholar]
- Cadar D.et al. (2017) ‘Widespread Activity of Multiple Lineages of Usutu Virus, Western Europe, 2016’, Eurosurveillance, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calzolari M.et al. (2010) ‘Evidence of Simultaneous Circulation of West Nile and Usutu Viruses in Mosquitoes Sampled in Emilia-Romagna Region (Italy) in 2009’, PLoS One, 5: e14324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calzolari M.et al. (2017) ‘Co-Circulation of Two Usutu Virus Strains in Northern Italy between 2009 and 2014’, Infection, Genetics and Evolution : Journal of Molecular Epidemiology and Evolutionary Genetics in Infectious Diseases, 51: 255–62. [DOI] [PubMed] [Google Scholar]
- Calzolari M.et al. (2016) ‘Insect-Specific Flaviviruses, a Worldwide Widespread Group of Viruses Only Detected in Insects’, Infection, Genetics and Evolution : Journal of Molecular Epidemiology and Evolutionary Genetics in Infectious Diseases, 40: 381–8. [DOI] [PubMed] [Google Scholar]
- Carrieri M.et al. (2014) ‘Weather Factors Influencing the Population Dynamics of Culex pipiens (Diptera: Culicidae) in the Po Plain Valley, Italy (1997-2011)’. Environmental Entomology, 43: 482–90. [DOI] [PubMed] [Google Scholar]
- Chin J. F. L., Chu J. J. H., Ng M. L. (2007) ‘The Envelope Glycoprotein Domain III of Dengue Virus Serotypes 1 and 2 Inhibit Virus Entry’, Microbes and Infection, 9: 1–6. [DOI] [PubMed] [Google Scholar]
- Chu J. J. H.et al. (2005) ‘Inhibition of West Nile Virus Entry by Using a Recombinant Domain III from the Envelope Glycoprotein’, The Journal of General Virology, 86: 405–12. [DOI] [PubMed] [Google Scholar]
- Chung K. M.et al. (2006) ‘Antibodies against West Nile Virus Nonstructural Protein NS1 Prevent Lethal Infection through Fc γ Receptor-Dependent and -Independent Mechanisms’, Journal of Virology, 80: 1340–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clé M.et al. (2019) ‘Usutu Virus: A New Threat?’, Epidemiology and Infection, 147: e232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Da Rold G.et al. 2015. ‘OP-D04. Detection of Marisma Virus and Characterization of Other Mosquito Flaviviruses In North-Eastern Italy’, in Second Conference on Neglected Vectors and Vector-Borne Diseases (EURNEGVEC) Abstract Book, p. 31.
- Depristo M. A.et al. (2011) ‘A Framework for Variation Discovery and Genotyping Using Next-Generation DNA Sequencing Data’, Nature Genetics, 43: 491–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Giallonardo F.et al. (2016) ‘Fluid Spatial Dynamics of West Nile Virus in the United States: Rapid Spread in a Permissive Host Environment’, Journal of Virology, 90: 862–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond A. J.et al. (2002) ‘Estimating Mutation Parameters, Population History and Genealogy Simultaneously from Temporally Spaced Sequence Data’, Genetics, 161: 1307–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eiden M.et al. (2018) ‘Emergence of Two Usutu Virus Lineages in Culex pipiens Mosquitoes in the Camargue, France, 2015’, Infection, Genetics and Evolution : Journal of Molecular Epidemiology and Evolutionary Genetics in Infectious Diseases, 61: 151–4. [DOI] [PubMed] [Google Scholar]
- Engel D.et al. (2016) ‘Reconstruction of the Evolutionary History and Dispersal of Usutu Virus, a Neglected Emerging Arbovirus in Europe and Africa’, mBio, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franchini P.et al. (2017) ‘Animal Tracking Meets Migration Genomics: Transcriptomic Analysis of a Partially Migratory Bird Species’, Molecular Ecology, 26: 3204–16. [DOI] [PubMed] [Google Scholar]
- Gaibani P.et al. (2013) ‘Comparative Genomic and Phylogenetic Analysis of the First Usutu Virus Isolate from a Human Patient Presenting with Neurological Symptoms’, PLoS One, 8: e64761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grottola A.et al. (2017) ‘Usutu Virus Infections in Humans: A Retrospective Analysis in the Municipality of Modena’, Clinical Microbiology and Infection, 23: 33–7. [DOI] [PubMed] [Google Scholar]
- Grubaugh N. D., Ebel G. D. (2016) ‘Dynamics of West Nile Virus Evolution in Mosquito Vectors’, Current Opinion in Virology, 21: 132–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grubaugh N. D.et al. (2017) ‘Mosquitoes Transmit Unique West Nile Virus Populations during Each Feeding Episode’, Cell Reports, 19: 709–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasle G. (2013) ‘Transport of Ixodid Ticks and Tick-Borne Pathogens by Migratory Birds’, Frontiers in Cellular and Infection Microbiology, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoang D. T.et al. (2018) ‘UFBoot2: Improving the Ultrafast Bootstrap Approximation’, Molecular Biology and Evolution, 35: 518–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Höfle U.et al. (2013) ‘Usutu Virus in Migratory Song Thrushes, Spain’, Emerging Infectious Diseases, 19: 1173–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes E. C. (2003) ‘Patterns of Intra- and Interhost Nonsynonymous Variation Reveal Strong Purifying Selection in Dengue Virus’, Journal of Virology, 77: 11296–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hönig V.et al. (2019) ‘Multiple Lineages of Usutu Virus (Flaviviridae, Flavivirus) in Blackbirds (Turdus merula) and Mosquitoes (Culex pipiens, Cx. modestus) in the Czech Republic (2016–2019)’. Microorganisms, 7: 568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibáñez-Justicia A.et al. (2020) ‘Detection of Exotic Mosquito Species (Diptera: Culicidae) at International Airports in Europe’, International Journal of Environmental Research and Public Health, 17: 3450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalyaanamoorthy S.et al. (2017) ‘ModelFinder: Fast Model Selection for Accurate Phylogenetic Estimates’, Nature Methods, 14: 587–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K., Standley D. M. (2013) ‘MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability’, Molecular Biology and Evolution, 30: 772–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemenesi G.et al. (2018) ‘First Genetic Characterization of Usutu Virus from Culex pipiens Mosquitoes Serbia, 2014’, Infection, Genetics and Evolution: Journal of Molecular Epidemiology and Evolutionary Genetics in Infectious Diseases, 63: 58–61. [DOI] [PubMed] [Google Scholar]
- Kosakovsky Pond S. L., Frost S. D. W. (2005) ‘Not so Different after All: A Comparison of Methods for Detecting Amino Acid Sites under Selection’, Molecular Biology and Evolution, 22: 1208–22. [DOI] [PubMed] [Google Scholar]
- Kosakovsky Pond S. L.et al. (2006) ‘Automated Phylogenetic Detection of Recombination Using a Genetic Algorithm’, Molecular Biology and Evolution, 23: 1891–901. [DOI] [PubMed] [Google Scholar]
- Lee T. H.et al. (2012) ‘A Cross-Protective mAb Recognizes a Novel Epitope within the Flavivirus NS1 Protein’, The Journal of General Virology, 93: 20–6. [DOI] [PubMed] [Google Scholar]
- Lelli R.et al. (2008) ‘Serological Evidence of USUTU Virus Occurrence in North-Eastern Italy’, Zoonoses and Public Health, 55: 361–7. [DOI] [PubMed] [Google Scholar]
- Lemey P.et al. (2009) ‘Bayesian Phylogeography Finds Its Roots’, PLoS Computational Biology, 5: e1000520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemey P.et al. (2010) ‘Phylogeography Takes a Relaxed Random Walk in Continuous Space and Time’, Molecular Biology and Evolution, 27: 1877–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lequime S.et al. (2016) ‘Genetic Drift, Purifying Selection and Vector Genotype Shape Dengue Virus Intra-Host Genetic Diversity in Mosquitoes’, PLoS Genetics, 12: e1006111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddison W., Maddison D. (2019) ‘Mesquite: A Modular System for Evolutionary Analysis. Version 3.61’. http://www.mesquiteproject.org
- Manarolla G.et al. (2010) ‘Usutu Virus in Wild Birds in Northern Italy’, Veterinary Microbiology, 141: 159–63. [DOI] [PubMed] [Google Scholar]
- Mathew A., Townsley E., Ennis F. A. (2014) ‘Elucidating the Role of T Cells in Protection against and Pathogenesis of Dengue Virus Infections’, Future Microbiology, 9(3), 411–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mavian C.et al. (2017) ‘Emergence of Recombinant Mayaro Virus Strains from the Amazon Basin’, Science Report, 7: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A.et al. (2010) ‘The Genome Analysis Toolkit: A MapReduce Framework for Analyzing Next-Generation DNA Sequencing Data’, Genome Research, 20: 1297–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel F.et al. (2019) ‘Evidence for West Nile Virus and Usutu Virus Infections in Wild and Resident Birds in Germany, 2017 and 2018’, Viruses, 11: 674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minin V. N., Bloomquist E. W., Suchard M. A. (2008) ‘Smooth Skyride through a Rough Skyline: Bayesian Coalescent-Based Inference of Population Dynamics’, Molecular Biology and Evolution, 25: 1459–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulatti P.et al. (2015) Retrospective Space-time Analysis Methods to Support West Nile Virus Surveillance Activities’, Epidemiology and Infection, 143: 202–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murrell B.et al. (2013) ‘FUBAR: A Fast, Unconstrained Bayesian Approximation for Inferring Selection’, Molecular Biology and Evolution, 30: 1196–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murrell B.et al. (2012) ‘Detecting Individual Sites Subject to Episodic Diversifying Selection’, PLoS Genetics, 8: e1002764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy A., Mettenleiter T. C., Abdelwhab E. M. (2019) ‘A Brief Summary of the Epidemiology and Genetic Relatedness of Avian Influenza H9N2 Virus in Birds and Mammals in the Middle East and North Africa’. Epidemiology and Infection. 145: 3320–3333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen L.-T.et al. (2015) ‘IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies’, Molecular Biology and Evolution, 32: 268–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen S. S.et al. (2020) ‘Rift Valley Fever: Risk of Persistence, Spread and Impact in Mayotte (France)’, EFSA J, 18: 1–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oude Munnink B. B.et al. (2020) ‘Genomic Monitoring to Understand the Emergence and Spread of Usutu Virus in The Netherlands, 2016-2018’, Scientific Reports, 10: 2798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacenti M.et al. (2019) ‘Clinical and Virological Findings in Patients with Usutu Virus Infection, Northern Italy, 2018’, Eurosurveillance, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker J., Rambaut A., Pybus O. G. (2008) ‘Correlating Viral Phenotypes with Phylogeny: Accounting for Phylogenetic Uncertainty’, Infection, Genetics and Evolution : Journal of Molecular Epidemiology and Evolutionary Genetics in Infectious Diseases, 8: 239–46. [DOI] [PubMed] [Google Scholar]
- Pecorari M.et al. (2009) ‘First Human Case of Usutu Virus Neuroinvasive Infection, Italy, August-September 2009’, Euro Surveill, 14: 19446. [PubMed] [Google Scholar]
- Rambaut A.et al. (2018) ‘Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7’, Systematic Biology, 67: 901–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravagnan S.et al. (2015) ‘OP-D05. Co-Circulation of Lineages and Strains of West Nile Virus in the Mosquito Vector Culex Pipiens of North-Eastern Italy’, in Second Conference on Neglected Vectors and Vector-Borne Diseases (EURNEGVEC) Abstract Book, p. 32.
- Riccetti S.et al. (2020) ‘Modelling West Nile Virus and Usutu Virus Pathogenicity in Human Neural Stem Cells’, Viruses, 12: 882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzoli A.et al. (2007) ‘West Nile Virus Circulation Detected in Northern Italy in Sentinel Chickens’, Vector-Borne and Zoonotic Diseases, 7: 411–7. [DOI] [PubMed] [Google Scholar]
- Roehrig J. T. (2003) ‘Antigenic Structure of Flavivirus Proteins’, Advances in Virus Research, 59: 141–75. [DOI] [PubMed] [Google Scholar]
- Roesch F.et al. (2019) ‘Usutu Virus: An Arbovirus on the Rise’, Viruses, 11: 640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santini M.et al. (2015) ‘First Cases of Human Usutu Virus Neuroinvasive Infection in Croatia, August–September 2013: Clinical and Laboratory Features’, Journal of Neurovirology, 21: 92–7. [DOI] [PubMed] [Google Scholar]
- Savini G.et al. (2011) ‘Usutu Virus in Italy: An Emergence or a Silent Infection? ’, Veterinary Microbiology, 151: 264–74. [DOI] [PubMed] [Google Scholar]
- Shapiro B., Rambaut A., Drummond A. J. (2006) ‘Choosing Appropriate Substitution Models for the Phylogenetic Analysis of Protein-Coding Sequences’, Molecular Biology and Evolution, 23: 7–9. [DOI] [PubMed] [Google Scholar]
- Sharp P. M., Li W.-H. (1987) ‘The Codon Adaptation Index-a Measure of Directional Synonymous Codon Usage Bias, and Its Potential Applications’, Nucleic Acids Research, 15: 1281–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonin Y.et al. (2018) ‘Human Usutu Virus Infection with Atypical Neurologic Presentation, Montpellier, France, 2016’, Emerging Infectious Diseases, 24: 875–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinmetz H. W.et al. (2011) ‘Emergence and Establishment of Usutu Virus Infection in Wild and Captive Avian Species in and around Zurich, Switzerland-Genomic and Pathologic Comparison to Other Central European Outbreaks’, Veterinary Microbiology, 148: 207–12. [DOI] [PubMed] [Google Scholar]
- Suchard M. A.et al. (2018) ‘Bayesian Phylogenetic and Phylodynamic Data Integration Using BEAST 1.10’, Virus Evolution, 4: vey016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Der Auwera G. A.et al. (2014) ‘From FastQ Data to High Confidence Variant Calls: The Genome Analysis Toolkit Best Practices Pipeline’, Current Protocols in Bioinformatics, 11: 11.10.1–11.10.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veronesi R.et al. (2012) ‘Seasonal Pattern of Daily Activity of Aedes caspius, Aedes detritus, Culex modestus, and Culex pipiens in the Po Delta of Northern Italy and Significance for Vector-Borne Disease Risk Assessment’, Journal of Vector Ecology : Journal of the Society for Vector Ecology, 37: 49–61. [DOI] [PubMed] [Google Scholar]
- Vilibic-Cavlek T.et al. (2019a) ‘Emerging Trends in the Epidemiology of West Nile and Usutu Virus Infections in Southern Europe’, Frontiers in Veterinary Science, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilibic-Cavlek T.et al. (2019b) ‘Prevalence and Molecular Epidemiology of West Nile and Usutu Virus Infections in Croatia in the ‘One Health’ Context, 2018’, Transboundary and Emerging Diseases, 66: 1946–57. [DOI] [PubMed] [Google Scholar]
- Weaver S.et al. (2018) ‘Datamonkey 2.0: A Modern Web Application for Characterizing Selective and Other Evolutionary Processes’, Molecular Biology and Evolution, 35: 773–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weidinger P.et al. (2020) ‘Different Dynamics of Usutu Virus Infections in Austria and Hungary, 2017–2018’, Transboundary and Emerging Diseases, 67: 298–307. 10.1111/tbed.13351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissenböck H.et al. (2013) ‘Usutu Virus, Italy, 1996’, Emerging Infectious Diseases, 19: 274–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissenböck H.et al. (2003) ‘Usutu Virus Activity in Austria, 2001-2002’, Microbes and Infection, 5: 1132–6. [DOI] [PubMed] [Google Scholar]
- Wilm A.et al. (2012) ‘LoFreq: A Sequence-Quality Aware, Ultra-Sensitive Variant Caller for Uncovering Cell-Population Heterogeneity from High-Throughput Sequencing Datasets’, Nucleic Acids Research, 40: 11189–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia X., Xie Z. (2001) ‘DAMBE: Software Package for Data Analysis in Molecular Biology and Evolution’, The Journal of Heredity, 92: 371–3. [DOI] [PubMed] [Google Scholar]
- Zannoli S., Sambri V. (2019) ‘West Nile Virus and Usutu Virus Co-Circulation in Europe: Epidemiology and Implications’, Microorganisms, 7:184. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Accession numbers of the sequences generated in this study have been provided in the Supplementary Data.