

Key Points
Question
What is the clinical, histological, and pathophysiological significance of skin lesions in Vacuoles, E1 enzyme, X-linked, Autoinflammatory, Somatic (VEXAS) syndrome?
Findings
In this case series study of 8 men, VEXAS skin lesions included neutrophilic dermatosis lesions (tender red or violaceous papules, inflammatory papules, firm erythematous purpuric, or pigmented infiltrated plaques and nodules) and livedo racemosa, with mostly perivascular neutrophils and myeloid cell infiltration on histology. The skin infiltrate harbored the UBA1 clonal variation.
Meaning
This study’s findings suggest that the dermal infiltrates seen in VEXAS skin lesions are derived from the pathological myeloid clone.
Abstract
Importance
VEXAS (vacuoles, E1 enzyme, X-linked, autoinflammatory, somatic) is a recently described severe adult-onset autoinflammatory disease that is associated with myeloid lineage-restricted ubiquitin-activating enzyme 1 (UBA1) somatic variations that primarily affect the skin (Sweet syndrome), cartilage, and bone marrow. Skin symptoms have been poorly described.
Objective
To better describe clinical and pathological skin manifestations and their pathophysiology in VEXAS syndrome.
Design, Setting, and Participants
This multicenter retrospective case series study of clinical and histological features of 8 patients with VEXAS syndrome and skin involvement was conducted in France from December 2007 to March 2021, with molecular data obtained from March to April 2022. Any UBA1 variations were detected by Sanger or next-generation sequencing that was performed on bone marrow and formalin-fixed paraffin-embedded tissue sections of skin lesion biopsies.
Results
All 8 patients were men, and the median age at symptom onset was 65.5 years (interquartile range, 54-76 years). All patients had neutrophilic dermatosis skin lesions, including tender red or violaceous papules, sometimes edematous, without fever, arthralgia, recurrence or pathergy, inflammatory edematous papules on the neck and trunk (sometimes umbilicated), and firm erythematous purpuric or pigmented infiltrated plaques and nodules. Three patients had livedo racemosa. The infiltrates were perivascular and consisted of mature neutrophils with leukocytoclasia, which were admixed with myeloperoxidase-positive CD163-positive myeloid cells with indented nuclei and lymphoid cells in all cases. A sequencing analysis of paired bone marrow samples and skin lesion biopsies identified the same loss-of-function UBA1 variation in both samples for all patients.
Conclusions and Relevance
This case series study describes the different clinical presentations of skin lesions found in VEXAS syndrome, which is characterized histologically by neutrophilic dermatosis. The findings suggested that the dermal infiltrates seen in VEXAS skin lesions are derived from the pathological myeloid clone. This suggests that using therapies that target the pathological clone may be effective in the long-term management of the disease.
This case series study of 8 French men examines clinical and pathological skin manifestations and their pathophysiology in VEXAS syndrome.
Introduction
VEXAS syndrome is a recently described adult-onset autoinflammatory disease that is associated with myeloid lineage-restricted ubiquitin-activating enzyme 1 (UBA1) somatic variations. It is characterized by skin involvement (88% of patients), relapsing polychondritis (RP) (64%), and myelodysplastic syndrome (MDS) (24%).1 To our knowledge, little is known about VEXAS dermatological lesions, except that neutrophilic dermatoses (NDs) and cutaneous vasculitis may occur (respectively 32% met the criteria of Sweet syndrome and 12% met the criteria of polyarteritis nodosa).1 The pathophysiological association between hematological features and autoinflammatory symptoms encountered in VEXAS syndrome is unknown. Neutrophilic dermatoses are characterized by an accumulation of mature polymorphonuclear cells (PMNs) in the skin. Approximately 20% of NDs occur within myeloid neoplasm, and the PMNs found in these lesions have been shown to derive from clonal myeloid cells.2,3 We aimed to describe the dermatological lesions found in VEXAS syndrome and investigate the possible clonal origin of skin-infiltrating cells. We analyzed a retrospective series of patients using Sanger or next-generation sequencing (NGS) of paired hematopoietic and skin lesions samples.
Methods
We identified 8 patients with VEXAS syndrome and skin involvement (Saint Louis, Saint Antoine, Tenon, Avicenne, and René Dubos Hospitals; Paris, France; December 2007 to March 2021). Any UBA1 variations were initially searched for using Sanger sequencing of exons 2, 3, and exon-intron junctions, and the UBA1 gene was thereafter included in the NGS panel. Sequencing was performed on DNA extracted from bone marrow mononuclear cells and from formalin-fixed paraffin-embedded tissue sections of skin lesion biopsies. May Grunwald Giemsa–stained bone marrow aspirates of 6 patients were reviewed for cytomorphologic evaluation. This study received the agreement of the local ethics committee, and participants provided written informed consent.
Results
Clinical characteristics of all 8 patients are summarized in the Table. All patients were men, and the median age at the onset of symptoms was 65.5 years (range, 54-76 years). All patients had ND skin lesions. The clinical description of skin lesions showed Sweet syndrome–like dermatosis (8 of 8 patients), including tender red or violaceous papules (sometimes edematous), without fever, arthralgia, recurrence or pathergy (Figure 1A), inflammatory edematous papules on the neck and trunk (Figure 1B) (sometimes umbilicated), and firm erythematous purpuric or pigmented infiltrated plaques and nodules (Figure 1C) and livedo racemosa (3 of 8 patients; Figure 1D). In 5 patients, skin involvement appeared before or at the time of other clinical features of VEXAS, with a median delay of 4 months (range, 0-72 months). Other symptoms included fever (3 of 8), RP (3 of 8), venous thromboembolism (4 of 8), and pulmonary involvement (2 of 8). Cytoplasmic vacuoles involving granulocytic precursors were found in all the bone marrow aspirates that were available for review (6 of 8) (eTable in the Supplement). Sequencing analysis of paired bone marrow samples and skin lesion biopsies identified the same loss-of-function UBA1 variation in both samples for all patients. Histopathological analysis of skin biopsies taken from all clinical types of lesions found ND with myeloid cells in all 8 patients (Figure 2A). Livedo racemosa lesions were not biopsied. The infiltrates consisted of mature PMNs with leukocytoclasia, which were admixed with myeloperoxidase-positive CD163-positive myeloid cells with indented nuclei (metamyelocytes and immature band neutrophils), and lymphoid cells in all cases (Figure 2, C and D). The infiltrates were typically perivascular and involved the entire dermis and subcutaneous fat in 2 cases. Infiltration of dermal vessel wall was seen in 2 patients, 1 with associated thrombosis without fibrinoid necrosis (Figure 2B). Patient 4 had in addition to ND a typical image of polyarteritis nodosa on the biopsy of a subcutaneous reddish-purple tender nodule. All 8 patients had either MDS (6 of 8) or clonal cytopenia of undetermined significance (2 of 8).4 Patients 1 and 6 also had monoclonal gammopathies, and patient 4 had essential thrombocytopenia. Three patients had abnormal karyotypes (del[6q], monosomy 7, and loss of Y). All bone marrow and skin lesions were analyzed by NGS of a panel of genes that had recurrently variated in myeloid malignant tumors: 3 patients harbored other somatic variations in addition to UBA1 (ie, ZRSR2, BRCA2, DNMT3A, TP53 and CALR) (eTable in the Supplement).5 The remaining patients showed no other hemopathy. No other somatic variation was detected in patients who were screened via bone marrow and/or skin for 80 genes involved in myeloid malignancies using NGS. Detailed hematological features are described in the eTable in the Supplement.
Table. Patient Characteristics.
| Patient No. | First symptoms/delay between skin lesions and other symptoms | Initial diagnosis | Clinical presentation of skin involvements | Histopathological classification of skin involvements (clinical appearance) | Hematological disorder | UBA1 variation (VAF)/other variated genes/karyotype abnormalities | Fever/arthralgia | RP | Venous thromboembolism | Pulmonary involvement |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Skin/3 mo | Eosinophilic pustular folliculitis |
|
ND (1 tender red/violaceous papule, 1 firm erythematous purpuric plaque) |
|
c.T122C/NA/NK | No | Yes | No | Yes |
| 2 | Skin and venous thromboembolism | Cutaneous PAN |
|
|
MDS-SLD | c.T122C/ZRSR2, BRCA, DNMT3A/Del(6q) | Yes | No | Yes | No |
| 3 | Skin/4 mo | Insect bites | Inflammatory edematous papules on neck and trunk | ND (2 inflammatory edematous papules) | CCUS | c.A121C/NA/NK | No | Yes | No | No |
| 4 | Skin/3 y | Cutaneous localization of myelodysplastic syndrome |
|
ND (1 tender red/violaceous papule, 1 firm erythematous purpuric plaque) |
|
c.A121C/DNMT3A, CALR/-7 | No | No | No | No |
| 5 | Cytopenia/1 y | SS | Inflammatory edematous papules on neck and trunk | ND (2 inflammatory edematous papules) | MDS-MLD | c.118-1G>C/NA/−Y | No | Yes | Yes | No |
| 6 | Venous thromboembolism/3 y | SS |
|
ND (2 tender red/violaceous papules) |
|
c.T122C/NA/NK | No | No | Yes | No |
| 7 | Skin/6 y | SS | Tender red/violaceous papules | ND (3 tender red/violaceous papules) | MDS-MLD | c.A121C (58%)/NA/Not documented | Yes | No | No | No |
| 8 | Venous thromboembolism/1 y | Unspecified autoinflammatory disease | Firm erythematous purpuric plaques | ND (1 firm erythematous purpuric plaque) | MDS-EB | c.A121G (34%)/TP53/Not documented | Yes | No | Yes | Yes |
| Total |
|
|
|
|
Yes (3/8) | Yes (3/8) | Yes (4/8) | Yes (2/8) |
Abbreviations: CCUS, clonal cytopenia of undetermined significance; ET, essential thrombocytopenia; MDS-EB, myelodysplastic syndrome with excess blasts; MDS-MLD, MDS with multilineage dysplasia; MDS-SLD, MDS with single-lineage dysplasia; MGUS, monoclonal gammopathy of undetermined significance; MM, multiple myeloma; NA, not applicable; ND, neutrophilic dermatosis; NK, normal karyotype; PAN, polyarteritis nodosa; RP, relapsing polychondritis; VAF, variant allele frequency; Y, y chromosome.
Figure 1. Clinical Presentations of Skin Lesions in Patients With VEXAS Syndrome.
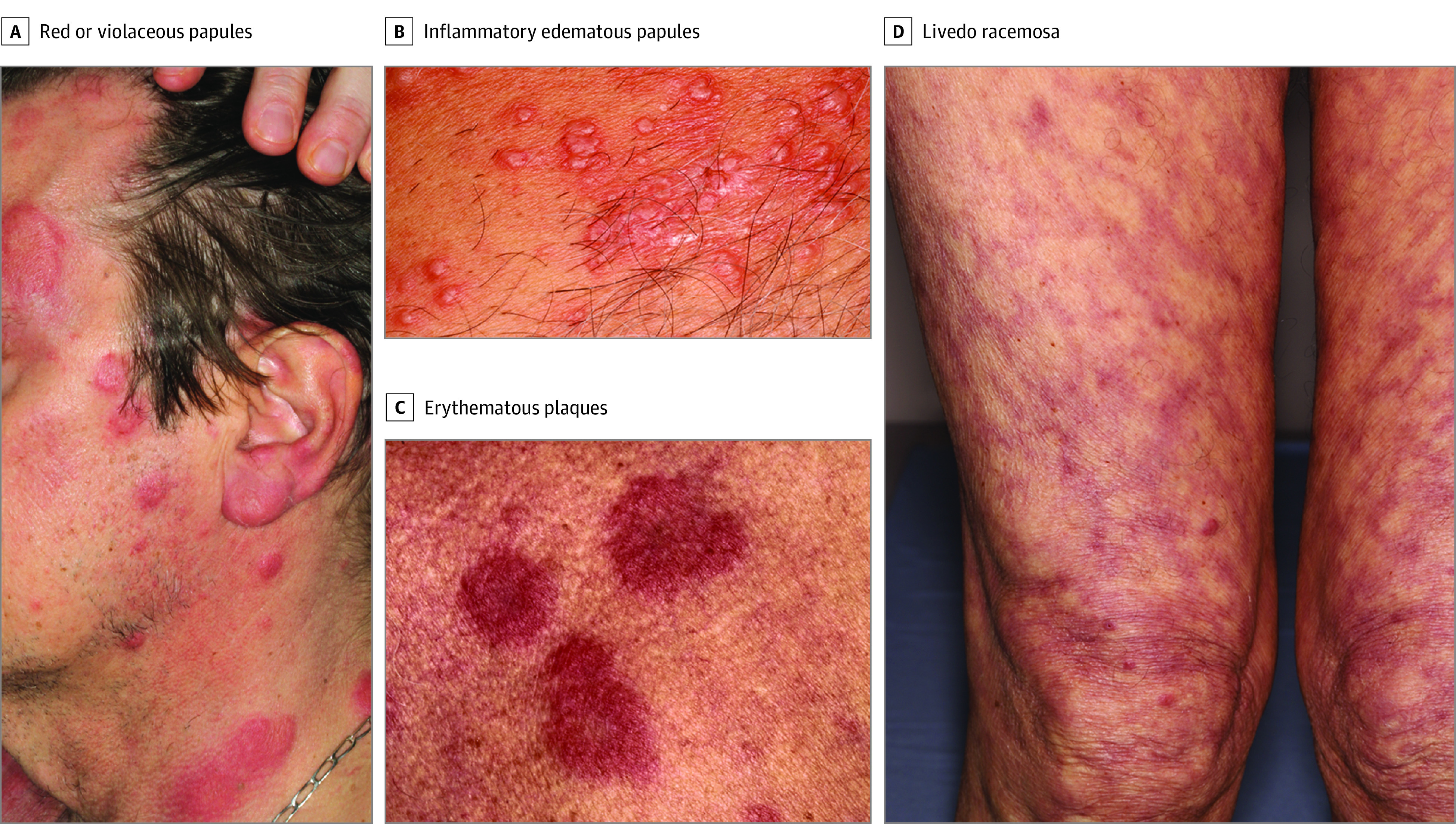
Skin lesions in patients with a diagnosis of Vacuoles, E1 enzyme, X-linked, Autoinflammatory, Somatic (VEXAS) syndrome included tender red or violaceous papules (A), inflammatory edematous papules on the neck and the trunk (B), firm erythematous purpuric or pigmented infiltrated plaques and nodules (C), and livedo racemosa (D).
Figure 2. Histopathological Features of Skin Lesions in Patients With VEXAS Syndrome.
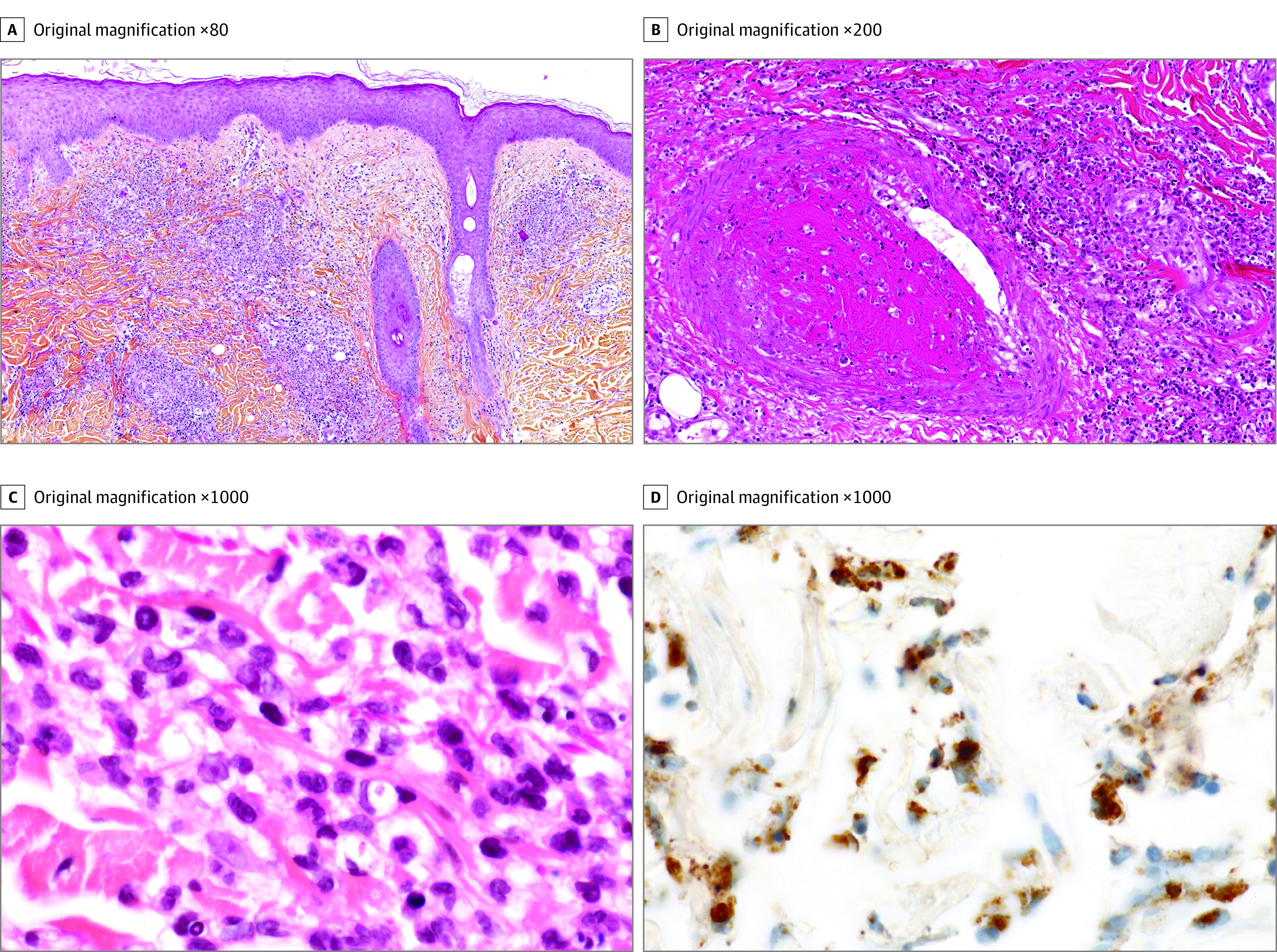
A, All patients had histopathological features of neutrophilic dermatosis (ND) with myeloid cells, with a perivascular infiltrate involving all dermis associated with moderate interstitial edema (hematoxylin-eosin). B, In 1 patient, medium-sized vein thrombosis was seen on the biopsy sample of ND, with infiltration of the vessel wall by mature polymorphonuclear cells (PMNs) and some myeloid cells with perivascular leukocytoclasia (hematoxylin-eosin). C, The infiltrate of ND consisted of rare mature PMNs and myeloid cells with indented nuclei that corresponded to metamyelocytes or band neutrophils (hematoxylin-eosin) admixed with lymphocytes. D, Myeloid cells expressed myeloperoxidase (immunostaining).
Discussion
Skin involvement in VEXAS syndrome has been poorly characterized.1 In this study, we showed the various skin clinical presentations of VEXAS syndrome and that the histopathological analysis of these lesions mainly shows ND with myeloid cells. Histiocytoid ND is a particular type of ND that is characterized by dermal infiltration of lymphocytes and myeloid myeloperoxidase–positive cells that is frequently associated with MDS.6 Histiocytoid ND may represent a form of cutaneous infiltration by MDS and show considerable overlap with myelodysplasia cutis.7,8 VEXAS syndrome was revealed by cutaneous manifestations in 5 of 8 patients, underlining the importance of an accurate description of these lesions to make an early diagnosis.
Myeloid cells represented around 10% to 30% of the skin cell population (which is a mixture of keratinocytes, fibroblasts, and other nonmyeloid immune cells). The sensitivity of the Sanger technique for detecting heterozygous variation is generally considered to be approximately 20%. In this case of variations affecting the UBA1 gene located on the X chromosome in male patients, one can extrapolate that at least 10% of the cells in the skin tissue section should be variated to detect the variation. In this case, the height of peaks for variated bases on Sanger sequencing (eFigure in the Supplement) as well as the high allelic frequencies on NGS (58% and 34% for patients 7 and 8, respectively) observed for UBA1 variation analyses of skin lesions rule out the possibility that our results are because of sample contamination by circulating blood myeloid cells, and rather suggested a clonal infiltration.
Associations of MDS with RP are well-documented in the literature and estimated to occur in around 10% of RP in retrospective studies. A strong and significant association between male sex, MDS, RP, and dermatological lesions (cutaneous vasculitis, livedo, and Sweet syndrome) was first reported in 2001 and could potentially retrospectively be reclassified as VEXAS syndrome.9,10
Our findings suggest that the inflammatory skin manifestations found in VEXAS syndrome are directly associated with clonal infiltration of the skin rather than a general proinflammatory activation state. This creates a better understanding of the VEXAS autoinflammatory symptoms seen in other organs (eg, cartilage, joints, lungs). It suggests that therapies that target the pathological clone ,as well as the proinflammatory milieu (eg, corticosteroids, Janus kinase inhibitors, tumor necrosis factor–α, and interleukin 6 antibodies), may be useful, as previously described in MDS.11,12,13
Limitations
The main limitation of our study is the limited number of patients who were analyzed, which may underestimate the diversity of the clinical and histopathological spectrum of VEXAS-associated skin lesions. There was also limited ethnic diversity among the patient cohort, as all patients were White, limiting generalizability.
Conclusions
The study data describe the different clinical presentations of skin lesions found in VEXAS syndrome, which are characterized histologically by ND with myeloid cells. The results suggest that the dermal infiltrate seen in VEXAS skin lesions is associated with the pathological myeloid clone. Clinicians should raise the hypothesis of a VEXAS syndrome in Sweet syndrome–like dermatosis that occurs in men older than 45 years, especially when associated with macrocytic anemia.
eTable. Haematological features of patients
eFigure. Sanger profiles of the UBA1 gene in bone marrow (BM) and skin lesion for two patients
References
- 1.Beck DB, Ferrada MA, Sikora KA, et al. Somatic mutations in UBA1 and severe adult-onset autoinflammatory disease. N Engl J Med. 2020;383(27):2628-2638. doi: 10.1056/NEJMoa2026834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chavan RN, Cappel MA, Ketterling RP, et al. Histiocytoid Sweet syndrome may indicate leukemia cutis: a novel application of fluorescence in situ hybridization. J Am Acad Dermatol. 2014;70(6):1021-1027. doi: 10.1016/j.jaad.2014.01.874 [DOI] [PubMed] [Google Scholar]
- 3.Passet M, Lepelletier C, Vignon-Pennamen MD, et al. Next-generation sequencing in myeloid neoplasm-associated Sweet’s syndrome demonstrates clonal relation between malignant cells and skin-infiltrating neutrophils. J Invest Dermatol. 2020;140(9):1873-1876.e5. doi: 10.1016/j.jid.2019.12.040 [DOI] [PubMed] [Google Scholar]
- 4.Steensma DP, Bejar R, Jaiswal S, et al. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood. 2015;126(1):9-16. doi: 10.1182/blood-2015-03-631747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ogawa S. Genetics of MDS. Blood. 2019;133(10):1049-1059. doi: 10.1182/blood-2018-10-844621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ghoufi L, Ortonne N, Ingen-Housz-Oro S, et al. Histiocytoid Sweet syndrome is more frequently associated with myelodysplastic syndromes than the classical neutrophilic variant: a comparative series of 62 patients. Medicine (Baltimore). 2016;95(15):e3033. doi: 10.1097/MD.0000000000003033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Osio A, Battistella M, Feugeas JP, et al. Myelodysplasia cutis versus leukemia cutis. J Invest Dermatol. 2015;135(9):2321-2324. doi: 10.1038/jid.2015.146 [DOI] [PubMed] [Google Scholar]
- 8.Vignon-Pennamen MD, Osio A, Battistella M. Histiocytoid sweet syndrome and myelodysplastic syndrome. JAMA Dermatol. 2017;153(8):835-836. doi: 10.1001/jamadermatol.2017.1669 [DOI] [PubMed] [Google Scholar]
- 9.Francès C, el Rassi R, Laporte JL, Rybojad M, Papo T, Piette JC. Dermatologic manifestations of relapsing polychondritis: a study of 200 cases at a single center. Medicine (Baltimore). 2001;80(3):173-179. doi: 10.1097/00005792-200105000-00003 [DOI] [PubMed] [Google Scholar]
- 10.Dion J, Costedoat-Chalumeau N, Sène D, et al. Relapsing polychondritis can be characterized by three different clinical phenotypes: analysis of a recent series of 142 patients. Arthritis Rheumatol. 2016;68(12):2992-3001. doi: 10.1002/art.39790 [DOI] [PubMed] [Google Scholar]
- 11.Delaleu J, Battistella M, Rathana K, et al. Identification of clonal skin myeloid cells by next-generation sequencing in myelodysplasia cutis. Br J Dermatol. 2021;184(2):367-369. doi: 10.1111/bjd.19547 [DOI] [PubMed] [Google Scholar]
- 12.Fraison J-B, Mekinian A, Grignano E, et al. Efficacy of azacytidine in autoimmune and inflammatory disorders associated with myelodysplastic syndromes and chronic myelomonocytic leukemia. Leuk Res. 2016;43:13-17. doi: 10.1016/j.leukres.2016.02.005 [DOI] [PubMed] [Google Scholar]
- 13.Bourbon E, Heiblig M, Gerfaud Valentin M, et al. Therapeutic options in VEXAS syndrome: insights from a retrospective series. Blood. 2021;137(26):3682-3684. doi: 10.1182/blood.2020010177 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
eTable. Haematological features of patients
eFigure. Sanger profiles of the UBA1 gene in bone marrow (BM) and skin lesion for two patients