

Abstract
Activation of mostly quiescent hematopoietic stem cells (HSC) is a prerequisite for life-long blood production1, 2. This process requires major molecular adaptations to meet the regulatory and metabolic requirements for cell division3–8. The mechanisms governing cellular reprograming upon stem cell activation and their subsequent return to quiescence are still not fully characterized. Here, we describe a role for chaperone-mediated autophagy (CMA)9, a selective form of lysosomal protein degradation, in sustaining adult HSC function. CMA is required for stem cell protein quality control and upregulation of fatty acid metabolism upon HSC activation. We identify that CMA activity decreases with age in HSC and show that genetic or pharmacological activation of CMA can restore functionality of old HSC. Together, our findings provide mechanistic insights into a new role for CMA in sustaining quality control, appropriate energetics and overall long-term hematopoietic stem cell function. Our work supports that CMA may be a promising therapeutic target to enhance hematopoietic stem cell function in conditions such as aging or stem cell transplantation.
Keywords: autophagy, chaperones, lipid metabolism, lysosomes, protein degradation, protein quality control, stem cells
Hematopoietic stem cells (HSC) harbor extensive self-renewal and multilineage differentiation capabilities to sustain production of blood cells over a lifetime8, 10–12. These adult stem cells are mostly quiescent, but intrinsic and extrinsic cues signal HSC to enter cell cycle and self-renew or differentiate6, 7, 13–17. Maintenance of a fully functional proteome that can rapidly change to facilitate the transition of HSC from quiescence to activation and, again return to quiescence, is essential to prevent stem cell exhaustion and bone marrow failure18, 19.
Repeated activation of HSC and exposure to cellular insults results in compromised HSC function with age7, 20, 21 in the form of severely reduced self-renewal22 and multilineage repopulation capacities23, 24. This functional decline of HSC in aging has been attributed to increasing levels of reactive oxygen species (ROS), accumulation of DNA damage and declining proteostasis25–28. Macroautophagy, the most studied type of autophagy, contributes to maintenance of HSC quiescence through mitochondria degradation to limit oxidative phosphorylation18, 29, 30. Here, we address the contribution of chaperone-mediated autophagy (CMA)9, to HSC maintenance and function. CMA is a selective type of autophagy for proteins bearing a targeting motif31 that when recognized by heat shock cognate protein of 70 kDa (HSC70) targets them to lysosomes for degradation32. Substrate proteins bind lysosome-associated membrane protein type 2A (LAMP2A)33 at the lysosomal surface and are internalized through the translocation complex formed by LAMP2A multimerization9. The selectivity of CMA relies on the targeting motif that can be present in the amino acid sequence as a full motif (canonical) or completed through posttranslational modifications, such as phosphorylation34 or acetylation35. Basal CMA activity is cell-type specific but, in most cells, CMA can be induced in response to stressors (i.e. starvation36, oxidative stress37, lipotoxicity38–40, proteotoxicity41, or DNA damage42). CMA contributes to quality control, through degradation of damaged proteins. CMA also degrades non-damaged proteins to terminate their function and, in this way, it contributes to regulate a variety of intracellular functions (i.e. T-cell activation, cell cycle, cell metabolism, cell growth, and survival)9, 35, 39, 42–44. CMA activity declines with age in many cells and tissues9, 43, 45 and malfunctions in conditions such as metabolic disorders46, neurodegeneration41 and cancer47. The dual role of CMA in protein quality control and in the regulation of cellular functions, motivated us to explore a possible involvement of CMA in the regulation of HSC function.
Results
CMA is upregulated in activated HSC
Using transgenic mice expressing a CMA reporter (KFERQ-Dendra2) that highlights lysosomes as fluorescent puncta when CMA is activated (Extended Data Fig. 1a)48, we found reduced CMA activity in HSC isolated from the older age mice compared to young mice. Both, CMA activity per cell and fraction of HSCs active for CMA decreased with age, whereas CMA remained unchanged with age in granulocyte-monocyte progenitor (GMP) cells (Fig. 1a,b and Extended Data Fig. 1b,c). Dendra fluorescent puncta were not detected in CMA-incompetent mice (knock-out for LAMP-2A (L2A), L2AKO) at any age, confirming the specificity of the CMA reporter (Extended Data Fig. 1d). Reduced CMA in old mice HSC is not a consequence of overall lower lysosome abundance with age, as levels of the endo lysosomal marker LAMP1 were comparable in young and old mice HSC. In contrast, levels of L2A, the spliced variant of the lamp2 gene required for CMA, were significantly reduced in HSCs from old mice, further supporting impairment of CMA activity in HSC with age (Extended Data Fig. 1e). A similar decline of CMA with age may also occur in humans, as we found significantly higher abundance of proteins bearing CMA-targeting motifs31 among the proteins reported to accumulate with age in human HSC49 (Extended Data Fig. 1f).
Fig. 1. CMA activity is required for HSC self-renewal.
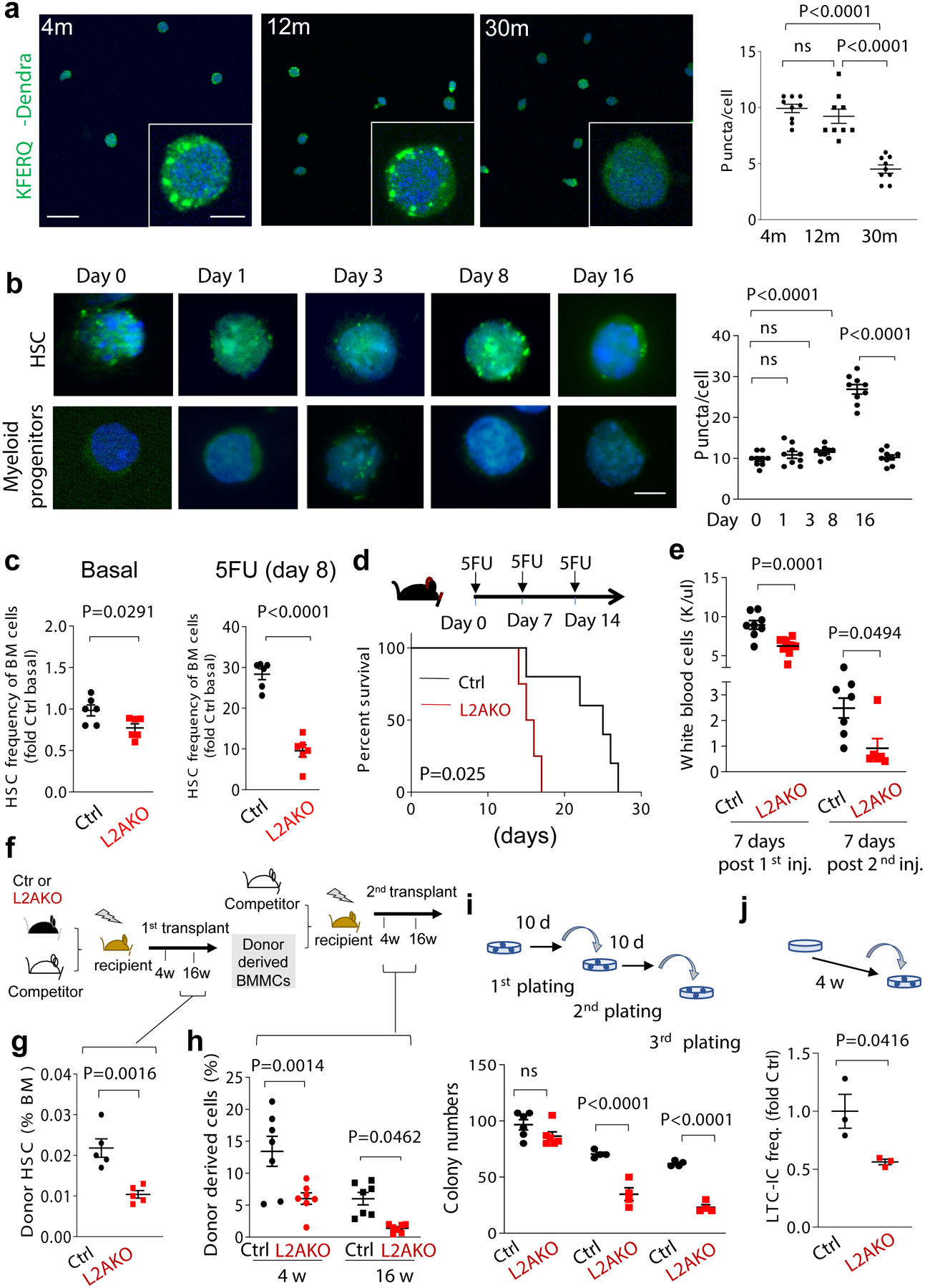
a, Dendra fluorescence in sorted HSC from 4m, 12m and 30m old KFERQ-Dendra mice. b, Quantification of Dendra puncta per cell in a. n=9 fields from 4 individual mice (individual points represent the average of approx. 7–10 cells per field). c, Dendra fluorescence in HSC and myeloid progenitors (Lin–cKit+Sca-1–) from KFERQ-Dendra mice before and at the indicated days after single injection of 5-fluorouracil (5FU). Right: quantification of Dendra+ puncta per cell in c. n=9 fields from 5 individual mice (individual points represent the average of 7–10 cells per field). d, HSC frequency in BM from Ctrl and L2AKO mice untreated (Basal) or 8 days after a single injection with 5-fluorouracil (5FU). n=6 mice. e, Survival curve of Ctrl and L2AKO mice after serial injections of 5FU 7 days apart. f, White blood cell counts 7 days after first (left) and second (right) 5FU injection. n=8 (Ctrl) and 10 (L2AKO) mice. g-i, Serial transplantation and competitive BM repopulation with Ctrl or L2AKO BM cells, experimental strategy (g); frequency of donor-derived HSC 16 weeks after the first competitive transplantation n=5 mice (h) and donor cell (from 3–4m CD45.2 mice) contribution in recipients’ peripheral blood 4 or 16 weeks after competitive secondary transplantation. n=7 mice (i). j, Serial colony formation assay with HSC from Ctrl or L2AKO mice. Number of colonies at day 10 after the indicated plating number is shown. n=6 (1st plating) and 4 (2nd and 3rd plating) mice. k, LTC-IC assay of LSK cells from 4m Ctrl and L2AKO mice. Fold change of LTC-IC frequency relative to Ctrl after 4 weeks of culture is shown, n=3 mice. One-way ANOVA test followed by Tukey’s multiple comparison post-hoc test (b,c) and two-way ANOVA test followed by Sidak’s multiple comparison post-hoc test (f,i,j), Log-rank (Mantel-Cox) test (e) and unpaired t-test (d,h,k) were used. P<0.05 (*), 0.01 (**), 0.0001(****). ns: no statistical significance.
To test if declining CMA contributes to aging-associated functional alterations of HSC, and because the role of CMA in hematopoietic stem cells has not been studied before, we first measured CMA activity in steady-state quiescent and activated HSC isolated from KFERQ-Dendra mice. Quiescent HSC showed higher basal CMA activity than myeloid progenitors, and further upregulate CMA upon in vivo activation of HSC following exposure to myeloablative agent 5-fluorouracil (5FU) (Fig. 1c and Extended Data Fig. 2a). Maximal CMA upregulation was observed by day 8 after 5FU injection, with CMA returning to basal levels by day 16 (Fig. 1c). Increase in CMA was mainly due to higher activity per cell as the fraction of HSC active for CMA remained unchanged (Extended Data Fig. 2a, right). We confirmed that lysosomal degradation of intracellular proteins and levels of LAMP2A, but not of other lysosomal markers, were also significantly higher at day 8 post activation (Extended Data Fig. 2b,c). In vivo treatment with polyI:C, which potently induces HSC cell cycle entry30, also upregulated CMA in these cells (Extended Data Fig. 2d, e), whereas none of these interventions resulted in changes in CMA activity in myeloid progenitor cells (Extended Data Fig. 2f–i). We conclude that HSC upregulate CMA upon activation.
CMA prevents HSC depletion upon activation
To determine the functional relevance of CMA upregulation upon HSC activation, we selectively eliminated CMA in hematopoietic cells by crossing LAMP2Af/f mice40 with Vav-iCre mice (Vav-iCre-LAMP2Af/f mice, hereafter termed L2AKO). L2AKO mice allow for hematopoietic cell-specific deletion of LAMP2A while preserving expression of the other Lamp2 splice variants (Extended Data Fig. 3a). Young L2AKO mice did not display differences in the total number of hematopoietic cells in the bone marrow (BM) or in the frequencies of T (CD3ε+), B (B220+), mature myeloid (CD11b+Gr1+) or of lineage-negative (Lin−) progenitor cell populations when compared to litter mate controls (Extended Data Fig. 3b–d), demonstrating that loss of CMA is compatible with multilineage hematopoiesis. However, we observed a moderate but significant decrease in the frequency and number of stem cell-enriched (Lin–Sca-1+cKit+, LSK) and rigorously defined phenotypical HSC (Lin–Sca-1+cKit+CD150+CD48–) populations at steady-state, which became markedly more pronounced following 5FU-mediated stem cell activation (Fig. 1d and Extended Data Fig. 3e,f). In fact, the expected expansion of the HSC pool, a prerequisite for restoring mature hematopoietic cell populations following 5FU-mediated myeloablation50–52, was also significantly attenuated in L2AKO mice (approximately 2.5-fold less expansion than control cells) (Fig. 1d and Extended Data Fig. 3f). The reduction in the steady-state HSC pool size and regeneration capabilities in L2AKO mice was attributable to a specific inhibition of the HSC compartment, as we did not find detectable changes in total BM cellularity in these mice (Extended data Fig. 3b).
Constitutive acute HSC activation using serial 5FU injections revealed that L2AKO mice could not recover their white blood cell counts and died of pre-mature bone marrow failure (Fig. 1e,f). Furthermore, serial transplantation and competitive BM repopulation, a more chronic paradigm of HSC activation (Fig. 1g), showed that CMA-deficient HSC were equally capable to engraft and reconstitute lethally irradiated congenic recipients upon primary transplantation but that, 16 weeks later, the number of L2AKO donor-derived HSC was reduced by 50%, while the total amount of immature Lin− cells was unaltered (Fig. 1h and Extended Data Fig. 3g,h). Secondary competitive bone marrow transplantation revealed a persistently reduced reconstitution with donor cells in all blood lineages in recipients of CMA-deficient cells compared to controls demonstrating a loss of functional stem cells (Fig. 1i and Extended Data Fig. 3i–k). Ex vivo studies also showed impaired self-renewal of L2AKO HSC (Fig. 1j,k and Extended data Fig. 3l), in further support of a cell intrinsic functional defect of CMA-deficient HSC.
Together, these findings demonstrate that albeit CMA is dispensable for multilineage blood cell differentiation, it is necessary to maintain functional HSC during steady state and, particularly, upon activation.
CMA deficiency impairs HSC activation
The reduced BM repopulation capability of CMA-deficient HSC originates, at least in part, from a marked reduction in the pool of actively cycling HSC 8 days after 5FU injection, as more than 50% of L2AKO HSC (vs. 15% of control HSC) were found in G0 at that time (Fig. 2a and Extended Data Fig. 4a–c). Interestingly, a higher fraction of L2AKO HSC was still cycling when most control HSC had fully returned to quiescence (Extended Data Fig. 4d,e). We did not find differences in cell cycle status between control and L2AKO myeloid progenitor cells (Extended Data Fig. 4f,g), supporting that CMA is important specifically for HSC cycling.
Fig. 2. Consequences of CMA blockage on HSC function.
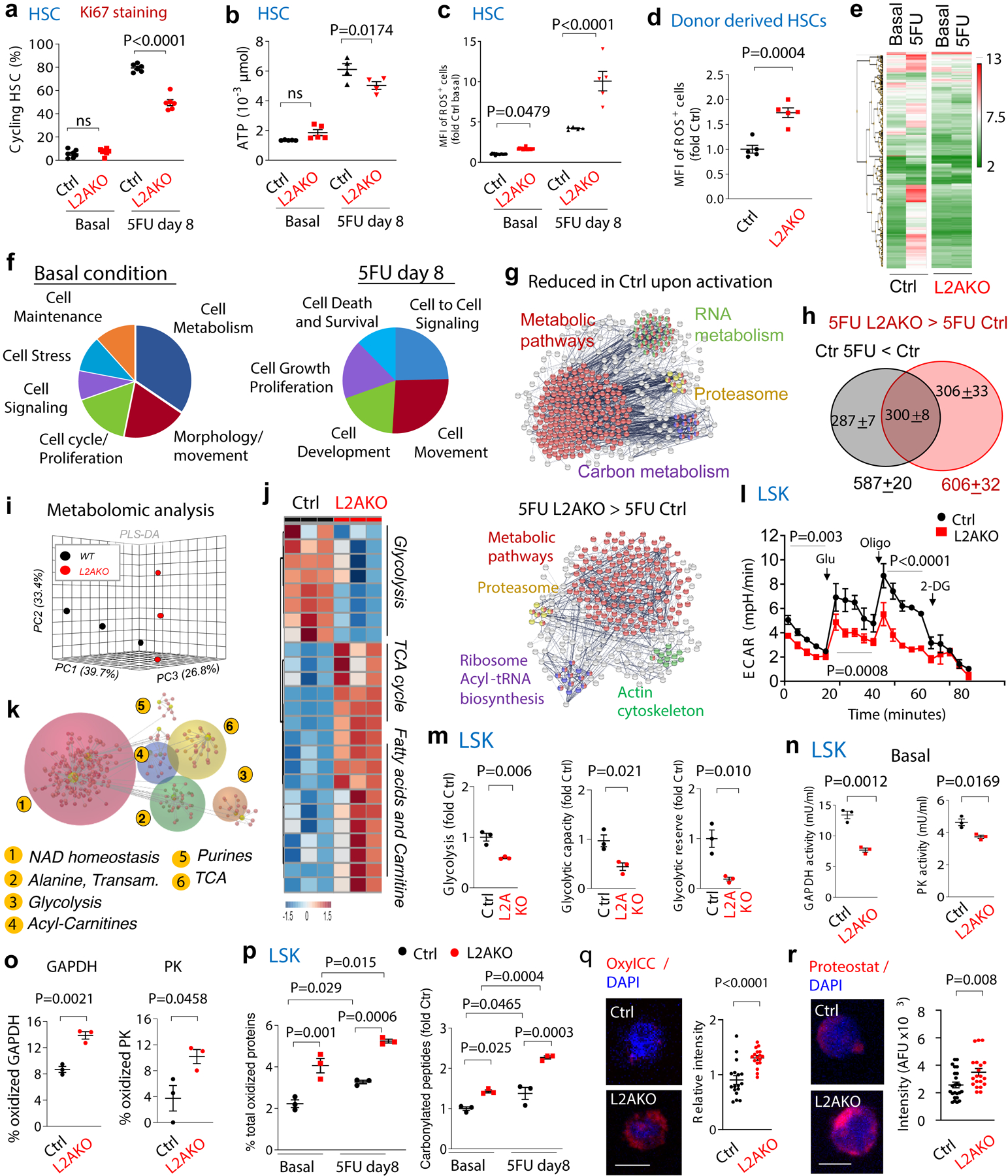
a-c, Percentage of stem cells in active cell cycle (not in G0 by Ki67 and Hoechst staining) (a), ATP levels (b) and median fluorescence intensity (MFI) for cellular ROS (reactive oxygen species) (c) in HSC from control (Ctrl) and L2AKO mice untreated (basal) or at day 8 after a single 5FU injection. n= 6–7 mice (a), 4–5 mice (b), 5–15 (c) mice. Representative examples of FACS plots for Fig. 2a and cycling analysis by Brdu incorporation are shown in Extended Data Fig. 4a-c. d, ROS levels in Ctrl and L2AKO bone marrow (BM) derived HSC in transplanted recipients. n= 5 mice . e,f, Heat map of gene expression (e) and enrichment pathway analysis (f) in HSC cells from Ctrl and L2AKO mice untreated or 8 days post 5FU injection. g, STRING analysis of proteins whose levels decrease in control cells upon activation (top) and those at higher levels in L2AKO LSK than Ctrl LSK 8 days post-5FU (bottom). h, Number of proteins different and overlapping in the same two groups as in g. n=3 proteomic experiments with pool of 3 mice per group. i-k, Metabolic phenotypes of LSK cells from Ctrl and L2AKO mice. Unsupervised principal component analysis of the two groups (i), hierarchical clustering analysis of the top 25 significant metabolites by two-tailed t test (j) and Omicsnet analysis of the most significantly affected pathways in L2AKO cells (k). n=3 metabolomic experiments with pool of 3 mice per group. l, Extracellular acidification rates (ECAR) in Ctrl and L2AKO LSK cells and changes upon addition of glucose (Glu), oligomycin (Oligo) and 2-Deoxy-D-glucose (2DG). n=3 independent experiments. m, Basal glycolysis (left), glycolytic capacity (middle) and glycolytic reserve (right) in L2AKO LSK cells relative to control cells. n=3 independent experiments. n. GAPDH (left) and pyruvate kinase (PK) (right) activity in Ctrl and L2AKO LSK cells. n=3 independent experiments. o, Percentage of cellular oxidized GAPDH and PK detected by mass spec in Ctrl and L2AKO LSK cells under basal conditions. n=9 mice in 3 different experiments. p, Percentage of total cellular proteins that are oxidized (left) and fold changes in the number of carbonylated peptides (right) in LSK from Ctrl and L2AKO mice under basal conditions or 8 days after 5FU injection. n=9 mice in 3 independent experiments. q, r Oxidized proteins (q) and protein inclusions (r) in basal Ctrl and L2AKO HSC detected by staining with OxylCC and Proteostat, respectively. Representative images (left) and quantification of staining intensity (right) are shown. n=15 fields from 5 individual mice (individual points represent the average of 5–10 cells per field). Nuclei are highlighted with DAPI. Full field images for q and r as shown in Extended Data Fig. 6g. Two-way ANOVA test followed by Sidak’s (a-c) or Tukey’s (p) multiple comparison post-hoc test, Chi-square test (k), multiple time point paired t-test (l) and unpaired t-test (d,m-o,q, r) were used for the statistics. P<0.05 (*), 0.01 (**), 0.001(***), 0.0001 (****). ns: no statistical significance.
L2AKO HSC, but not myeloid progenitor cells, showed a reduced ability to increase intracellular ATP content and a more pronounced increase in ROS levels in response to 5FU when compared to control cells (Fig. 2b,c and Extended Data Fig. 4h,i). Higher ROS levels - a common manifestation of metabolic deregulation and proven cause of impaired HSC self-renewal ability27, 28- were also observed in L2AKO bone marrow derived HSC upon transplantation (Fig. 2d). Overall, our data support that CMA is required to promote timely HSC cell cycle entry upon activation and that the poor energetic status and elevated ROS content of CMA-deficient HSC may be behind their compromised self-renewal capacity.
Although blockage of macroautophagy in HSC also increases intracellular ROS18, 53, the phenotype of CMA-deficient HSC was not secondary to changes in the status of macroautophagy, which was comparable between control and L2AKO HSC in both basal and cytokine starved conditions (Extended Data Fig. 5a–d show no differences in LC3 flux, the autophagy and lysosomal TFEB-dependent transcriptional program or in the number of autophagic compartments between both cell genotypes). Furthermore, blockage of macroautophagy in HSC increases proliferation rates and causes pronounced mitochondrial alterations3, 18, 53, which is in clear contrast with the reduced cycling and absence of noticeable changes in mitochondrial morphology and cellular and cytosolic areas of CMA-deficient HSC (Extended Data Fig. 5e,f).
CMA-dependent proteome remodeling in HSC
Comparative gene expression analysis of control and L2AKO HSC identified 405 genes differentially expressed during steady-state, and 855 genes following stem cell activation (Fig. 2e). Most transcriptional differences between resting control and L2AKO HSC were in genes related to cellular metabolism, cellular motility, cell cycle and proliferation; while upon HSC activation, differential expression occurred in genes with known roles in cell-to-cell signaling and interaction, cellular movement and cell proliferation (Fig. 2f and Extended Data Fig. 6a). Comparative quantitative proteomics also identified higher abundance of proteins related to metabolic pathways in L2AKO compared to control stem cells (Extended Data Fig. 6b). In an attempt to identify proteins degraded by CMA during stem cell activation, we focused on two groups of proteins: i) proteins whose levels decrease in CMA-competent control cells upon activation, and ii) proteins that accumulate in activated L2AKO cells (vs. activated control HSC). Notably, in the pool of proteins shared by both groups (300±8 proteins), the vast majority of them harbored CMA-targeting motifs (94.8±0.4% compared with the 84.7±8% in the total of 1,578±32 proteins (in average) detected in HSC by the proteomic analysis, p<0.05), further supporting that they are putative CMA substrates under these conditions (Fig. 2g,h). Proteins involved in metabolic pathways (mostly enzymes) were present in both groups of putative CMA substrates during stem cell activation (Extended data Fig. 6c). The fact that most changes in enzyme abundance during stem cell activation were only detected at the protein level (Fig. 2g) but not at the transcriptional level (Fig. 2f, right), suggests that CMA facilitates metabolic adaptation of activated HSC through direct degradation of metabolic enzymes.
Reduced glycolysis in CMA-deficient HSC
Metabolomics analyses of steady-state HSC revealed distinct metabolic phenotypes between L2AKO and control (partial least square-discriminant analysis and hierarchical clustering analysis in Fig. 2i, j). Pathway analysis of the top 25 metabolites significantly different between Ctrl and L2AKO HSC, highlighted a pronounced impairment in L2AKO cells of glycolysis and the glucose-alanine pathway, known energy sources critical for HSC quiescence27, 54 (Fig. 2k and Extended Data Fig. 6d). Extracellular metabolic flux analyses confirmed a decrease in glycolysis in L2AKO LSK cells compared to CMA-proficient controls, that was not observed for myeloid progenitor cells; thus, confirming that the defect in glycolysis upon CMA blockage is specific to stem cell-enriched cell populations (Fig. 2l,m and Extended Data Fig. 6e,f).
In agreement with the metabolomic and metabolic flux data, we found a significant decrease in the enzymatic activities of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and pyruvate kinase (PK) - two glycolytic enzymes previously identified as CMA substrates in other cell types40 - in L2AKO stem cells compared to control cells (Fig. 2n). We attributed the reduced enzymatic activity to the higher content of oxidized residues per protein detected by mass spectrometry in these enzymes in steady-state L2AKO stem cells (Fig. 2o). We conclude that failure to degrade oxidized glycolytic enzymes in CMA-deficient stem cells leads to accumulation of non-functional enzymes and impaired glycolytic capability in these cells. Reduced quality control in L2AKO HSC is not limited to glycolytic enzymes, as we found a general loss of proteostasis in these cells. Indeed, mass spectrometry analysis revealed that CMA-deficient stem cell enriched LSK have significantly higher levels of carbonylated peptides and oxidized proteins (including reversible and irreversible oxidation of sulfur-containing amino acids, methionine and cysteine – collectively plotted in Fig. 2p). Direct cellular staining also confirmed the higher content of oxidized proteins and protein inclusions in L2AKO HSC (Fig. 2q,r and Extended data Fig. 6g). In agreement with the fact that CMA exclusively degrades proteins, we found that oxidized products accumulating in L2AKO HSC seem to be preferentially proteins and peptides, while for example, lipid peroxidation products appeared unaltered (Extended Data Fig. 6g,h). Analysis of the subset of proteins bearing CMA-targeting motifs that display higher oxidation in L2AKO HSC identified besides glycolytic enzymes, other proteins related to cellular energetics, proteins involved in cytosolic and ER protein quality control (synthesis, folding, degradation and trafficking) – which could explain the dilated ER observed in L2AKO HSC cells (Extended Data Fig. 5c,g) - and proteins that contribute to cell cycle and cellular mobilization (Extended Data Fig. 6i,j).
Together, our results support that loss of CMA leads to defective protein quality control in quiescent HSC and reduces the ability to accommodate the metabolic requirements for appropriate stem cell activation.
CMA regulates lipid metabolism in activated HSC
Metabolomic analysis of 5FU-activated LSK cells revealed that glycolysis was also impaired in CMA-incompetent cells during activation, likely through similar mechanisms, since we also found higher rates of oxidation and of metabolic markers of oxidant stress (methionine sulfoxide, xanthine and spermine), higher levels of damage-related PTMs and reduced activity of glycolytic enzymes in L2AKO stem cells than in control upon activation (Fig. 2p and Extended Data Fig. 6k, l).
However, the most noticeable metabolic differences between CMA-competent and CMA-incompetent stem cells during activation occurred in pathways related to lipid metabolism, with linoleic acid metabolism and fatty acid biosynthesis and beta-oxidation among the top affected pathways (Fig. 3a–d and Extended Data Fig. 7a). Activation of control LSK cells associated with higher flux through linoleic and α-linolenic acid metabolism, whereas in activated L2AKO LSK we observed accumulation of the two precursors (α-linolenic and linoleic acid) and very pronounced decrease in all downstream metabolites (Fig. 3b–d). Blocking linoleic acid metabolism with an inhibitor of fatty acid desaturase 2 (FADS2), the rate limiting enzyme of this pathway55, 56, was sufficient to reduce self-renewal ability of control cells (Fig. 3e); while conversely, treatment of CMA-defective HSC with γ-linolenic acid (GLA), the direct product of FADS2, restored their self-renewal capacity ex vivo (Fig. 3f). Thus, the blockage in α-linolenic and linoleic acid metabolism is of functional relevance in L2AKO HSC.
Fig. 3. CMA increases linoleic acid metabolism upon HSC activation.
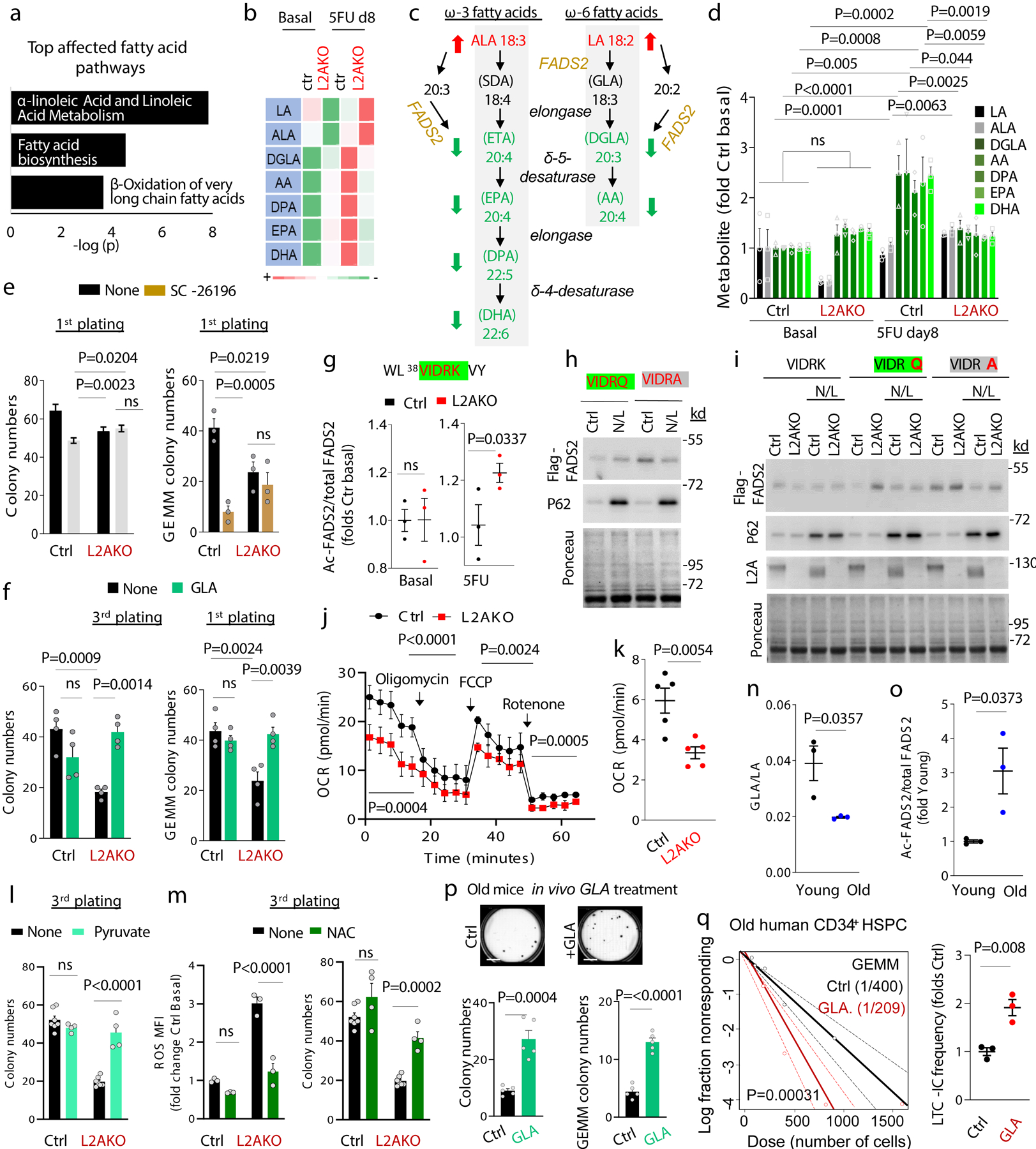
a, Top lipid metabolism pathways identified from enrichment analysis of metabolites as different between control and L2AKO LSK cells 8 days post 5FU injection. b, Heat map of substrate and metabolite abundance in the linoleic acid and α-linolenic metabolism in the indicated conditions. c, Scheme of linoleic and α-linolenic fatty acid metabolism pathway showing the increased (red arrow) and decreased (green arrow) metabolites in L2AKO LSK cells comparing with Ctrl cells in 5FU-activated conditions. d, Levels of substrates (black, gray) and downstream metabolites (green) of the linoleic acid and α-linolenic metabolism relative to those in unstimulated control cells. n= 9 mice in 3 different experiments. Significant differences with control untreated are marked with * and between L2AKO basal and 5FU treated with †. e, Total colony numbers (left) and number of immature, multipotent colony-forming unit-granulocyte/ erythroid/macrophage/megakaryocyte CFU-GEMM (right) in the first plating from Ctrl and L2AKO HSC treated or not with the FADS2 inhibitor SC-26196 (SC). n=3 independent experiments. f, Total colony numbers in the 3rd plating (left) or number of CFU-GEMM colonies in the first plating (right) from Ctrl and L2AKO HSC treated or not with γ-linolenic acid (GLA). n=4 independent experiments. g, Predicted CMA-targeting motif in FADS2 (green) generated by acetylation of 42K (g top) and analysis of the acetylation level of this peptide detected by mass spectrometry Ctrl and L2AKO LSK cells in basal and 5FU activated conditions (g bottom). n=3 mice. h,i, Immunoblot for Flag of Ctrl (h) and L2AKO (i) ex vivo expanded HSC expressing Flag-myc tagged FADS2 wild type (VIDRK) or mutated as indicated at the top (VIDRQ or VIDRA) treated or not with NH4Cl/Leupeptin (N/L). For gel source data, see supplementary Figure 1. j, Oxygen consumption rate (OCR) in Ctrl and L2AKO LSK cells at day 8 after 5FU injection. Responses to addition of oligomycin (Oligo), FCCP and rotenone (Roteno) are shown. n=5 independent experiments. k, Quantification of mitochondrial respiration from fatty acid β-oxidation (etomoxir-sensitive) in Ctrl and L2AKO LSK cells. n=5 independent experiments. l, Total colony numbers in 3rd plating from both Ctrl and L2AKO HSC treated or not with Methyl-pyruvate (5mM) starting from the 1st plating. n=7 (None) and 4 (Methyl-pyruvate) independent mice. m, Quantification of MFI of ROS+ cells (left) and total colony numbers (right) in the 3rd plating of Ctrl or L2AKO HSC untreated or supplemented with NAC from the 1st plating. n=7 (None) and 4 (NAC) mice. n, Ratio of γ-linolenic acid (GLA) and linoleic acid (LA) in LSK cells from young and old mice calculated from the metabolomics data. n= 9 mice in 3 different experiments. o, Ratio of K42 acetylated and total FADS2 peptide in LSK from young and old mice calculated from the mass spectrometry analysis. n= 9 mice in 3 different experiments. p, Total colony numbers (left) and number of CFU-GEMM colonies (right) in the colony formation assay with HSC cells from old (22m) mice daily injected with either saline or GLA (1mg/kg bw) for 7 weeks. Representative images of wells with Ctrl or GLA treated cells are shown on the top. n=5 mice per group. q, LTC-IC assay according to GEMM colonies from Ctrl and GLA treated human CD34+ cells from old (>65 year) multi-myeloma patients. n=2 patients. Two-way ANOVA test followed by Tukey’s (d,f) or Sidak (e,l,m) multiple comparison post-hoc test, unpaired t-tests (g,k,n,o,p), time point paired t-test (j), and Chi-square test (q) were used for the statistics. P<0.05 (*), 0.01 (**), 0.001(***), 0.0001(****). ns: no statistical significance.
To elucidate how CMA may regulate linoleic acid metabolism, we focused on FADS2 because the blockage in this pathway in CMA-deficient cells occurred at the step catalyzed by this enzyme (Fig. 3c shows the steps, metabolites and regulatory enzymes in linoleic and α-linolenic acid metabolism and Fig. 3b,d show accumulation of the precursors of the reaction catalyzed by FADS2 and reduction of the downstream products). FADS2 content increased upon 5FU activation at comparable levels in control and L2AKO LSK cells (Extended Data Fig. 7b,c), suggesting that qualitative rather than quantitative differences may be behind the lower activity of FADS2 in CMA-deficient cells. Among the different posttranslational modifications that could modulate FADS2 activity, we focused on acetylation because the following findings support an important role for acetylation in the CMA-dependent proteome remodeling of HSC and progenitor cells upon activation: i) FADS2 would acquire a CMA-targeting motif if lysine 42 becomes acetylated (38VIDRK42 changes to VIDRQ-like) (Fig. 3g); ii) CMA-targeting motifs that can be generated by acetylation were significantly enriched in the 300 proteins that decreased in control cells upon activation and remain elevated in L2AKO HSC (Extended Data Fig. 7d), iii) mass spectrometry demonstrated a trend toward higher acetylation in putative CMA substrates (KFERQ-like motif containing proteins) among the proteins that accumulate in L2AKO stem cells upon activation (Extended Data Fig. 7e); and iv) the pools of proteins with higher acetylation in L2AKO LSK cells and those with predicted acetylation generated CMA-targeting motifs in proteins that accumulate in L2AKO HSC and in human HSC from old individuals, were all enriched in proteins involved in cellular metabolism, including lipid metabolism (Extended Data Fig. 7d, e).
Mass spectrometry analysis identified higher acetylation of the FADS2 K42 residue in L2AKO LSK cells upon 5FU activation (Fig. 3g). Single point mutagenesis in this residue confirmed that an acetylation mimetic form of FADS2 (FADS2VIDRQ) when expressed in HSC was rapidly degraded in lysosomes (Fig. 3h) in a L2A-dependent manner (Fig. 3i), and that this rapid degradation of FADS2 by CMA was no longer observed in an acetylation-resistant mutant at that residue (FADS2VIDRA) (Fig. 3h,i). These results support that, as previously described for other enzymes35, acetylation triggers removal of inactive forms of FADS2 by CMA and that, by changing the active/inactive enzyme ratio, CMA positively regulates FADS2 activity and increases linoleic and α-linolenic fatty acid metabolism during HSC activation.
As expected from HSC unable to upregulate linoleic acid metabolism during activation, L2AKO cells showed lower fatty acid β-oxidation related mitochondrial respiration rates than control cells (Fig. 3j, k). Reduced lipid β-oxidation can explain the lower ATP levels in L2AKO stem cells upon activation (Fig. 2b) and their reduced self-renewal ability, as supplementation with methyl-pyruvate (to bypass reduced β-oxidation) significantly increased L2AKO HSC function ex vivo (Fig. 3l). Furthermore, a toxic effect of unmetabolized linoleic and α-linolenic fatty acids – known to increase oxidative and endoplasmic reticulum (ER) stress57–59 – also contributes to the reduced function of L2AKO HSC, as they display higher ROS levels (Fig. 2c,d) and abnormally dilated ER (Extended Data Fig. 5c,g), and addition of the ROS scavenger N-acetylcysteine (NAC) restored their ex vivo long-term colony initiation abilities (Fig. 3m). These data support that the compromised function of CMA-deficient HSC is likely a combination of the cells’ inability to meet the energetic requirements of activation and the toxic effects of accumulating unmetabolized, pro-oxidative lipid products.
Comparative proteomic analysis revealed that L2AKO HSC from young mice phenocopy in part the changes in the proteome of old control HSC. In fact, almost half of the proteins that we found that accumulated in HSC from 25m mice were also elevated under basal conditions in HSC from 4m L2AKO mice and they fell in similar functional family categories (Extended Data Fig. 7f). Similarly, analysis of the metabolic phenotypes and hierarchical clustering analysis of the top 50 metabolites significantly different between HSC from young and old mice demonstrated consistent changes in glycolysis and in metabolism of free fatty acids with those observed in HSC from young L2AKO mice (Extended Data Fig. 7g–i). Furthermore, the ratio of GLA to LA (metabolite and precursor in the step catalyzed by FADS2) was significantly reduced in LSK cells from old mice (Fig. 3n) and the acetylation state of FADS2 at the K42 residue in these cells was significantly elevated (Fig. 3o) when compared with LSK from young mice.
Overall, these findings support a possible similar molecular mechanism underlying the metabolic derangement of L2AKO HSC and old HSC. To test the possible contribution of the observed failure in linolenic and α-linolenic fatty acid metabolism to the functional deficiencies of old HSC, we subjected 22m mice to daily injections of GLA for 7 weeks. We observed that in vivo GLA supplementation improved HSC function as assessed by colony formation (Fig. 3p). Furthermore, ex vivo supplementation with GLA also increased the number of functional human stem cells in long-term cultures of CD34+ cells derived from old (>65 years) patients (Fig. 3q). These findings further support the relevance of linoleic and α-linolenic fatty acid metabolism for HSC activation both in mice and in humans, the contribution of defective flux through this pathway to HSC loss of function during aging and the similarity, at the molecular level, between the changes in FADS2 proteostasis in HSC in aging and in our model of deficient CMA.
CMA activation preserves HSC function in aging
We observed that the progressive depletion of functional HSC in absence of CMA accentuates with age as evidenced by: 1) reduced numbers of phenotypical HSC (Fig. 4a), 2) decreased HSC reconstitution ability noticeable in HSC from old L2AKO mice already upon the first round of transplantation (Fig. 4b,c), in contrast to young L2AKO mice that only displayed differences in reconstitution abilities after the second round of transplantation (Fig. 1i); and 3) more pronounced age-dependent increase in intracellular ROS levels in L2AKO mice HSC compared to wild type littermate mice (Fig. 4d).
Fig. 4. Modulation of CMA restores old HSC function.
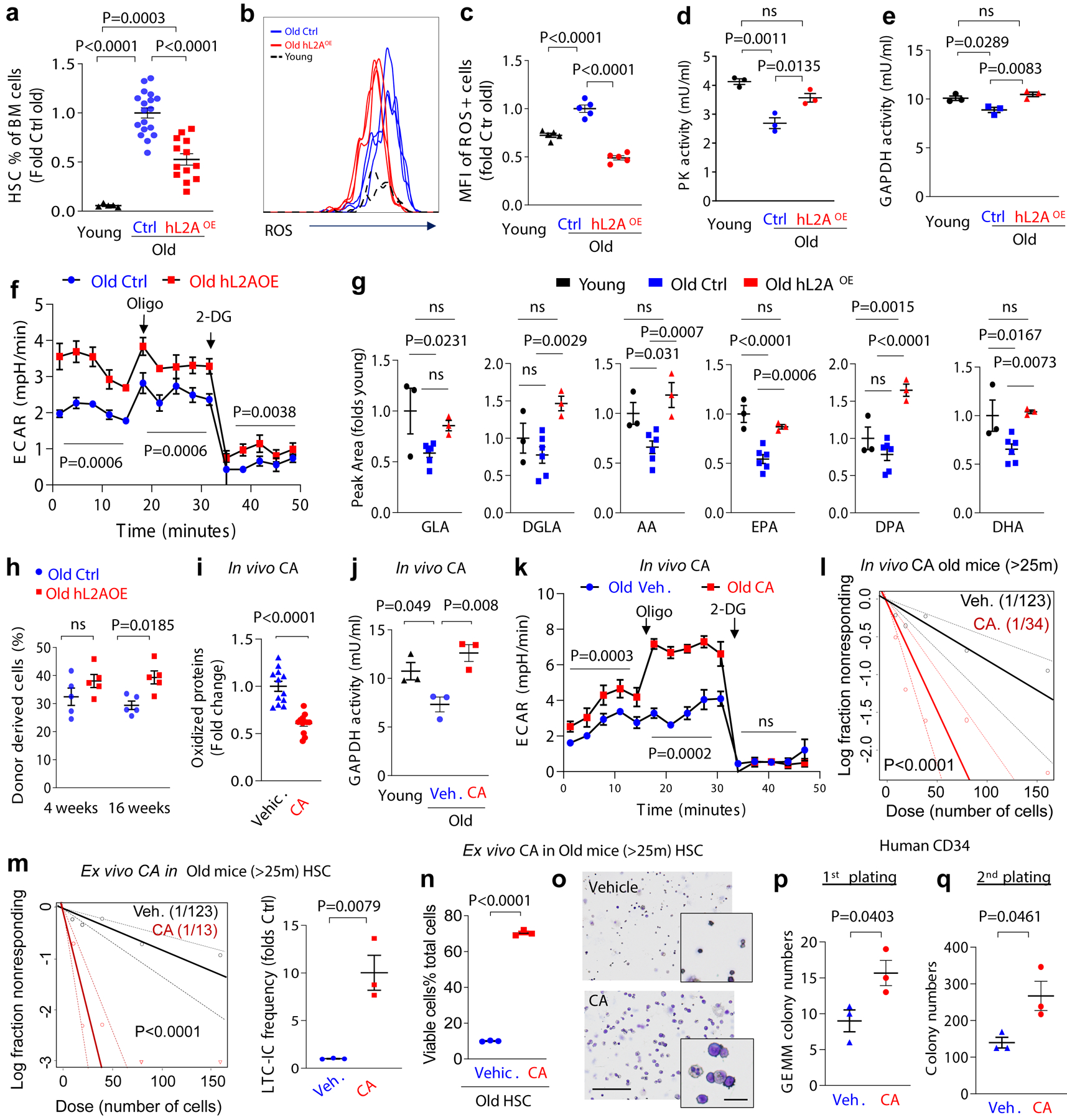
a, Changes in HSC numbers per femur and tibia in Ctrl and L2AKO mice with age. Values are relative to young control mice. n=9–11 mice. b, Donor chimerism at the indicated time points in peripheral blood of recipients after competitive transplantation of 25–30m Ctrl or L2AKO BM cells. n=5–6 mice. c, Donor lineage distribution in peripheral blood 24 weeks after second competitive transplantation of Ctrl or L2AKO BM cells. n=5–6 mice. d, ROS levels in HSC from 3–4m and >25m old Ctrl and L2AKO mice. Data is shown as median fluorescence intensity (MFI) per cell. n=5–6 mice. e, Percentage of HSC in BM of young, old control and old mice bearing an extra copy of human L2A (hL2AOE). n=5 (young), 18 (old ctrl) and 13 (old hL2AOE) mice. f,g, Representative FACS (f) and quantification (g) of ROS levels in HSC cells from young, old Ctrl and hL2AOE mice. Data is shown relative to old Ctrl mice. n=5 mice. h-j PK (h) and GAPDH (i) activity and extracellular acidification rates (ECAR) in basal conditions or after addition of oligomycin (Oligo) and 2-deoxy-D-glucose (2DG) (j) in LSK cells from old Ctrl and hL2AOE mice. n=3 mice. k. Levels of polyunsaturated fatty acids generated by FADS2 in old Ctrl and hL2AOE mice expressed as peak area relative to young mice. n= 9 (young), 18 (old), 9 (hL2AOE) mice in 3 independent experiments. l. Percentage of donor derived cells at the indicated times in mice transplanted with BM from old Ctrl and hL2AOE mice. n=5 mice. m-o, Levels of oxidized proteins (m), GAPDH activity (n) and ECAR in basal conditions or upon Oligo and 2DG addition (o) in HSC from old (>22m) mice 2 months after daily oral administration of a chemical activator of CMA (CA 20mg/Kg b.w.) or the corresponding vehicle (Veh.). Values in young mice are shown as reference in n. Representative images of cells used for quantification in m are in Extended Data Fig. 9n. n=12 fields from 4 and 3 mice for m and n, respectively. p. LTC-IC assay of LSK cells from BM of old mice administered daily for 2 months CA or vehicle. Scatter plot shows LTC-IC frequency for vehicle and CA treated old mice. n=4 mice. q,r LTC-IC assay of old LSK cells after ex vivo treatment with vehicle (DMSO) or a chemical activator of CMA (CA 10μM, daily for 4 weeks) (q) and viable cell percentage recovered from the colonies formed at the end of LTC-IC (r). Individual plot showing the fold change of LTC-IC frequency compared to old control cells (q left), scatter plot showing LTC-IC frequency for vehicle and CA treated old Ctrl cells (q right). n=3 mice. s. Representative images of Giemsa staining of the cells recovered at the end of the LTC-IC assay when cells were plated in presence of CA (10μM) or vehicle. n=3 experiments. t,u. Quantification of immature GEMM colonies in the 1st plating (t) and total colony numbers in the 2nd plating (u) of CD34+ enriched stem and progenitor cells from mobilized blood of multi-myeloma patients (59, 65, and 71 years old), when maintained in presence of CA (10μM) or vehicle starting from the 1st plating. n=3 patients. Multiple t-tests (a-c), unpaired t-tests (m,q left,r,t,u), time points paired t-test (j, o), two-way ANOVA test followed by Sidak’s multiple comparisons post-hoc test (d, l), one-way ANOVA tests followed by Tukey’s (e,g-i,n), Sidak’s (k) or Dunnett’s (n) multiple comparisons post-hoc test and Chi-square test (p,q right) were used for the statistics. P<0.05 (*), 0.01 (**), 0.001 (***), 0.001(****). ns: no statistical significance.
To gain a better understanding of the observed changes in CMA activity in HSC with age (Fig. 1a,b) and to place them in the context of the well-known functional heterogeneity of the aged HSC24, we next explored in more detail CMA activity in old HSC. Re-analysis of CMA activity in HSC from young and old KFERQ-Dendra mice, by plotting the absolute number of Dendra+ puncta per cell (instead of averaging number of Dendra+ puncta for all HSC), revealed the emergence of two groups of HSC with very different CMA activity (Extended Data Fig. 8a). Immunostaining images and FACS both demonstrated high levels of cytosolic KFERQ-Dendra protein in about 45% of cells, which corresponded to those with low number of fluorescent puncta (Extended Data Fig. 8b,c). Reduced number of Dendra+ puncta and increased cytosolic staining in some old cells was a direct consequence of reduced flux through CMA, that we measured upon blocking protein degradation in the lysosomal lumen with leupeptin (Extended Data Fig. 8d,e); notably, HSC cells from old mice with low cytosolic Dendra accumulation displayed CMA flux almost indistinguishable from young HSC (Extended Data Fig. 8d,e). In agreement with the observed disruption of proteostasis upon CMA failure, we found that the population of HSC from old mice with low CMA activity had significantly higher levels of oxidized proteins, higher cellular ROS levels and displayed lower GAPDH activity than HSC with preserved CMA from the same mice (Extended Data Fig. 8f–i). These data further support the contribution of reduced CMA with age to loss of HSC proteostasis.
To test whether preventing the age-dependent decline in CMA could preserve HSC function, we used ERCre-hL2A mice (hereafter hL2AOE) (Tasset et al., in preparation) in which expression of human L2A was induced at middle life (12 months of age) for the rest of their life to compensate for the decrease in endogenous L2A levels responsible for the decline in CMA activity with age60. We confirmed expression of hL2A, at the mRNA (one hL2A transcript for each mL2A) and protein level, without affecting levels of the endogenous mouse L2A (Extended Data Fig. 9a–c). We tested whether expression of hL2A was effective in preserving normal CMA activity until late in life by crossing KFERQ-Dendra mice with the hL2AOE mice. HSC from old (>22m) KFERQ-Dendra-hL2AOE mice displayed significantly higher CMA flux than control littermate and CMA activity was more homogeneous among old stem cells, to the point that the population with low CMA activity observed in control old mice was undetectable in hL2AOE mice (Extended Data Fig. 9d–h).
Contrary to young animals where the pool of stem cells is small but the potency of each stem cell is high, aging associates with a well-characterized expansion of the stem cell population but of restricted functionality at the single cell level22. Analysis of the BM of old hL2AOE mice revealed that the frequency of HSC in BM from old hL2AOE mice was significantly lower than in control old littermates (Fig. 4e). We predicted that the age-dependent expansion of the HSC pool was no longer occurring in these mice due to their preserved better functionality at the single cell level. To confirm this hypothesis, we directly analyzed quality and function of HSC and found that HSC cells from old hL2AOE mice displayed significantly lower intracellular ROS levels (Fig. 4f,g), PK and GAPDH activities closer to those observed in young mice (Fig. 4h,i) and higher glycolysis rates than control old littermates (Fig. 4j). Metabolomic analysis showed that maintenance of functional CMA in old mice also prevented the significant decrease in FADS2-generated polyunsaturated fatty acids observed in the old control group (Fig. 4k). The better proteostasis and preserved glucose and fatty acid metabolism observed in HSC from old hL2AOE mice is probably responsible for their improved functionality, detected as their higher efficiency in BM reconstitution upon transplantation (Fig. 4l).
Overall, contrary to CMA blockage which reduces HSC frequency and functionality, preservation of CMA activity until late in life, prevented the loss of HSC function with age and the expansion of poorly functional HSC observed in control old mice. Upregulation of L2A expression in young (4m) hL2AOE mice for 3m did not change HSC frequency in BM, HSC performance in the LTC-IC assay or ROS levels and PK activity of HSC (Extended Data Fig. 9 i–l), supporting a maximal beneficial effect of CMA upregulation in conditions with an already underlying CMA defect, such as aging.
Restoration of CMA rejuvenates aged HSC
Lastly, to explore if upregulation of CMA in already old HSC could still be a viable intervention to improve their function, we exposed HSC from old mice to a well-characterized pharmacological CMA activator (CA) optimized for in vivo use61. We confirmed that CA was still effective in activating CMA in HSC from old KFERQ-Dendra mice (Extended Data Fig. 9m), and demonstrated that daily oral administration of CA (20mg/kg b.w.) for only 2 months was sufficient to reduce levels of oxidized proteins, restore GAPDH activity and increase glycolytic flux (Fig. 4m–o and Extended Data Fig. 9n), which likely contributes to the remarkable increase in HSC function observed upon CA administration (shown as better performance in the LTC-IC assay) (Fig. 4p). To confirm that the functional improvement of HSC was due to direct activation of CMA in these cells and independent of any other systemic beneficial effect of CA when administered systemically in vivo, we exposed HSC from old mice to CA in vitro and found a similarly striking increase in the number of LTC-IC and higher ability of stem cells to generating viable mature cells of various lineages compared to vehicle treated controls (Fig. 4q–s and Extended Data Fig. 9o).
The connection between CMA and fatty acid metabolism through regulation of FADS2 identified in this study, along with the prevention of the age-dependent decline of this metabolic pathway in HSC of old mice with preserved CMA activity (Fig. 4k) and the fact that polyunsaturated fatty acids generated by FADS2 activity also decrease with age in human blood from 250 heathy blood donor volunteers62 (Extended Data Fig. 10a) motivated us to also test the effect of the CMA activator on human HSC cells. Addition of CA to CD34+ hematopoietic stem and progenitor cells (HSPC) from old donors (≥59 years) markedly increased multi-lineage potent HSPC and sustained overall cell output upon long term culture (Fig. 4t,u). These findings support that pharmacological activation of CMA could be an effective strategy to improve and restore function of aged HSC.
Discussion
In this study, we demonstrate how functional CMA is required for HSC maintenance and function. In quiescent steady-state HSC, CMA contributes to quality control and the defense against oxidative stress63–65 by removing oxidized proteins. Whereas upon HSC activation, timely degradation by CMA of acetylation-tagged proteins is required to stimulate long chain fatty acid metabolism and accommodate the metabolic requirements of a dividing stem cell66 (Extended Data Fig. 10b).
Other components of the proteostasis network have been reported to contribute to HSC homeostasis and function18, 53. Metabolic deregulation with massive accumulation of lipid droplets and increased glycolysis occurs upon macroautophagy blockage in neutrophil precursors67. This metabolic phenotype is in clear contrast with the reduced glycolysis observed in CMA-deficient HSC. In further support that both autophagic pathways are non-redundant, loss of CMA in HSC cannot be compensated for by macroautophagy in these cells. Different from other cell types (i.e. fibroblasts, hepatocytes) that respond to CMA blockage with an upregulation of macroautophagy activity to preserve cellular proteostasis40, 68, HSC fall in the group of cells (also including retinal cells or activated T cells43, 69) in which macroautophagy activity remains unaltered upon CMA blockage. This lack of compensatory upregulation is probably a determining factor for the early in life loss of proteostasis observed in HSC when CMA is non-functional. Differences in timing, signaling pathways and substrate recognition may be responsible for the inability of these proteolytic systems to compensate for each other in HSC. The sustained lower quality control of CMA-deficient HSC through life has likely a cumulative effect at the level of proteotoxicity, evidenced by the worsening of the L2AKO HSC phenotype with age.
As expected for a proteolytic system, the consequences of CMA failure in HSC may go beyond the changes in lipid metabolism reported in this work. Our comparative proteomic analysis demonstrate that CMA-deficient cells are unable to carry out the overall proteome remodeling required during the process of activation. Failure to timely modulate levels of proteins involved in processes such as cell cycle, cytoskeletal organization, or in the regulation of other proteostasis components, are likely to contribute to the functional impairment of HSC. The selectivity and timing of CMA degradation is likely behind the cell type-specific functions described for this type of autophagy. For example, CMA facilitates T cell activation by directly degrading proteins that actively repress T cell activation43, whereas here we uncovered that CMA partakes in regulation of the metabolic state of HSC upon activation. Interestingly, the mechanisms and metabolic pathways regulated by CMA appear also to be cell type and context specific as, for example, CMA participates in maintenance of pluripotency of embryonic stem cells also thorough metabolic changes but in that case by limiting α-ketoglutarate production, which ultimately affects activity of DNA demethylases involved in pluripotency70.
We show here that precluding the age-dependent decline of CMA in HSC is effective in preserving stem cell function and thus preventing the expansion of a functionally compromised HSC pool that observed in aging. We found particularly interesting that upregulation of CMA even later in life at a time when HSC function is already compromised, is efficient in restoring HSC function. These findings support that reactivation of CMA in HSC is possible and still has a beneficial effect at advanced age. The deficiencies in proteostasis maintenance and self-renewal capacity of HSC with defective CMA and the observed improvements in these two functions in old HSC upon genetic or pharmacological upregulation of CMA both in mouse and human HSC, unveils CMA as a possible target to restore hematopoietic stem cell function in aging.
Methods
Mice.
Hematopoietic system specific L2A KO mice were created by crossing C57BL/6 Vav-iCre mice with C57BL/6 L2Af/f mice. Wild type, Vav-iCre and L2Af/f40 male littermate mice were separately analyzed for each test and because no differences were detected among them, they were grouped in the results as “control” for the experimental group (Vav-iCreL2Af/f). For the aging studies, 3–4 months (labeled as 4m) and 25–30m (labeled as >25m) male mice were used in the young and old group, respectively. KFERQ-Dendra2 transgenic mice and KFERQ-Dendra mice defective in CMA (KFERQ-Dendra-L2AKO) were generated as described before48. Transgenic mouse models with targeted insertion of a human copy of L2A that can be induced by tamoxifen (hL2AOE) were generated by crossing hL2Af/f mice with TmxER-Cre71. hLAMP2A expression was induced with 3 intraperitoneal (i.p.) injections of tamoxifen (2mg/20g body weight) every other day either at 4m and analyzed at 7m of age or at 12m and analyzed at 27m of age. Young and old controls were similarly injected with tamoxifen. Injected animals did not show differences in HSC frequency with non-injected controls, thus discarding any possible direct effect of tamoxifen at the time that the animals were analyzed. KFERQ-Dendra mice with extra copy of human L2A (KFERQ-Dendra-hL2AOE) were generated by breeding KFERQ-Dendra male mice with hL2AOE mice. Where indicated, mice were i.p. injected with a single dose of 5-fluorouracil (5FU, 150mg/kg body weight, Invitrogen, sud-5fu). Blood cell count was analyzed in tail blood drawn at day 0, 1, 3, 6, 8 and 16 post 5FU injection using an Oxford Science Forcyte Blood Analysis Unit. Bone marrow was analyzed at the same times. Data is from the analysis 8 days post-injection, unless otherwise specified. For serial 5FU injections, mice were injected every week and 7 days post injection blood was taken and blood counts measured as above and dead mice and time of death were registered. For GLA supplementation, old mice (>25m) were injected with either saline or GLA (1mg/kg bw) for 40 days. The CMA activator (CA 20mg/Kg b.w. per mouse day for 2 months) was administered orally in the form of sucralose gelatin agar pills. The same delivery method was utilized in control mice but using drug-free pills. CA is a derivative from the previously generated first-in-class small molecules for selective activation of CMA in vitro61, that has been modified to make it suitable for in vivo administration (Gomez et al., in preparation). All mouse procedures were approved by the Institutional Animal Care and Use Committee of Albert Einstein College of Medicine.
Human hematopoietic stem and progenitor cells.
Mobilized peripheral blood from multiple myeloma patients (59–71 years old) was enriched for mononuclear cells by density gradient centrifugation using Ficoll-Paque PLUS (GE Healthcare). Mono-nuclear cells were further enriched for CD34+ cells using immunomagnetic bead sorting (CD34 MicroBead Kit, human; Miltenyi Biotec) according to the manufacturer’s protocol. The isolated CD34+ cells were then used for LTC-IC or colony formation assays.
Competitive bone marrow transplantation.
Total nucleated bone marrow (BM) cells (5×105) were injected retro-orbitally into lethally irradiated CD45.1 congenic mice in competition with the same amount of BM cells from CD45.1 mice. Transplanted mice were given antibiotic-containing water for 4 weeks post-irradiation. Reconstitution of donor derived (CD45.2) cells was monitored by staining blood cells with antibodies against CD45.2 and CD45.1 at different times as indicated. For serial transplantation, donor-derived BM cells were collected from recipient mice 24 weeks after first BM transplantation. Cells were then transplanted into recipient mice with fresh competitor cells (CD45.1).
Flow cytometry.
For flow cytometric analyses of mouse cells, the following monoclonal antibodies were used: APC-CD117(c-kit) (Biolegend 105812), PE/Cy7-Ly-6A/E (Sca-1) (Biolegend 108114), Pacific blue-CD48 (Biolegend 103418), PE-CD150 (Biolegend 115904), APC-CD3ε (Biolegend 100312), Percp/Cy5.5-B220 (Biolegend 103236), FITC-CD11b (Biolegend 101206), FITC-Ly-6G/Ly-6C (Gr-1) (Biolegend 108406), and FITC-CD34 (eBioscience, 11-0341-82), PE-CD16/32 (Biolegend 101308). Biotin-conjugated antibodies for lineage markers were as follows: CD3 (Biolegend 100244), CD4 (Biolegend 100404), CD8a (Biolegend 100704), IgM (Biolegend 406504), TER-119 (Biolegend 116204), B220 (Biolegend 103204), NK-1.1 (Biolegend 108704), CD19 (Biolegend, 115504). PerCP/Cy5.5 Streptavidin (Biolegend, 405214) was used against the biotin-conjugated antibodies. Before analysis, BM from tibias and femurs were depleted from red blood cells with ACK lysis buffer, then stained with HSC markers (Lin−/cKit+/Sca1+/CD48−/CD150+) or myeloid progenitor markers (Lin−/cKit+/Sca1−) or GMP markers (Lin−/cKit+/Sca1−/CD34hi/CD16/32hi) followed by either ROS staining or cell cycle analysis. All antibodies used in this study were from commercial sources and were validated following the multiple dilution method and where available through the use of cell lines or tissues from animals knock-out for the antigen. For ROS staining, cells were resuspended in staining buffer (2%FBS/2mM EDTA in PBS) with 5μM CellROX reagent (ThermoFisher, C10444) and incubated at 37°C for 30 min. Cells were washed 3 times with staining buffer, resuspended in the same buffer with 1μg/ml propidium iodide for dead cell exclusion and then subjected to FACS analysis. For cell cycle analysis, after staining for cell surface markers, cells were fixed with Cytofix/Cytoperm buffer (BD Bioscience, 554722) for 20 min at 4°C, washed twice with Perm/Wash buffer (BD Bioscience, 554723), re-suspended in Perm/Wash buffer containing Alexa Fluor 488 anti-mouse Ki-67 antibody (Biolegend, 652417) and incubated overnight at 4°C. After 3 washes with Perm/Wash buffer, cells were resuspended in cell staining buffer with Hoechst 33342 (ThermoFisher, H3570) for 5min at RT, and subjected to FACS analysis. For BrdU staining, mice were intraperitoneally injected with a single dose of 100mg/kg BrdU (Sigma-Aldrich, B5002) and provided with drinking water containing 0.8mg/ml BrdU and 5% glucose 24 hours before dissection. After staining with surface markers, cells were fixed, permeabilized, treated with DNase, and stained with anti-BrdU FITC antibody (Biolegend, 364103) following the BrdU labeling kit protocol (BD Pharmingen). Cells were then resuspended in staining buffer with Hoechst 33342. For sorting, BM cells from tibias, femurs and spine were centrifuged through Histopaque to isolate mononucleated cells. Cells were stained with HSC markers or LSK markers, resuspended in the staining buffer with 1μg/ml propidium iodide for dead cell exclusion and then subjected to sorting in a Beckman Moflo sorter with 70μm nozzle. Gating strategies used in the study are shown in Supplementary Figure 2. Data was acquired and analyzed with the BD FACSDiva™ software.
Colony formation and serial re-plating assays.
Sorted mouse HSC (180 cells Lin−/cKit+/Sca1+/CD48−/CD150+) or CD34-enriched human HSPC (5,000 cells) were plated into methylcellulose media (STEMCELL Technologies, MethoCult M 3434 for mouse or MethoCult H4434 for human) according to the manufacturer’s protocols. Colonies were scored after 10 to 12 days for mouse and 14 to 16 days for human using an Inverted Infinity and Phase Contrast Microscope (Fisher Scientific). For serial re-plating assays, methylcellulose medium from primary platings were dissolved in PBS to dissociate the colonies into a single-cell suspension, washed three times and 20,000 cells (mouse) or 40,000 cells (human) were re-plated in 1ml of MethoCult M3434 or MethoCult H4434 medium for mouse and human cells, respectively. Where indicated, media was supplemented with: 100 nM FADS2 inhibitor SC-26196 (Sigma, PZ0176), 100 μM γ-linolenic fatty acid (GLA) (Sigma, L2378), 100 μM NAC (Sigma, A7250) or 5 mM Methl-pyruvate (Sigma, 371173).
LTC-IC assay.
To assess LTC-IC frequencies, serial dilutions of FACS-sorted mouse Lin−Sca-1+c-Kit+ (LSK) cells or CD34+ enriched human HSPCs were plated in MyeloCult M5300 for mouse or MyeloCult H5100 for human (STEMCELL Technologies) mixed at 50:50 ratio with primary mouse stroma cell–conditioned medium or with HS5-conditioned medium for human72. For each cell dose (1:10, 1:20, 1:40, 1:80, 1:160 for mouse LSK cells, 1:50, 1:100, 1:200, 1:400, 1:800 for human HSPC), 10 technical replicates were performed. After 4 weeks (mouse) or 5 weeks (human) in culture with half change of the medium every week, cells in each well were used for the colony formation assay in MethoCult M3434 and MethoCult H4434 (STEMCELL technologies) for mouse and human, respectively. For CFU-C counting, in the case of mouse samples, wells containing any type of colonies were scored as positive, while in the case of human samples, only wells with GEMM colonies were scored as positive. Where indicated, the CMA activator (10μM) or the same volume of DMSO were added to the mouse LSK cells every 24 hours during the 4 weeks in culture. Supplementation of the γ-linolenic fatty acid (GLA) was done by adding 100μM GLA (Sigma, L2378) or the same volume of IMDM medium to the human HSPC cells every week for 5 weeks.
Long-term ex vivo expansion of mouse hematopoietic stem cells and electroporation.
The long-term ex vivo expansion of mouse HSC were conducted as previously described73. Briefly, Ctrl or L2AKO HSC were sorted into a well of 96-well plates (3 plates for each genotype) containing F-12 medium supplemented with 10 mM HEPES, 1× PSG, 1× ITSX, 1 mg/ml PVA, 100 ng/ml TPO, and 10 ng/ml SCF. Cells were cultured for one month with half of the medium changed twice a week. Cells were then collected and subjected to gradient centrifugation using Ficoll (Sigma, Histopaque-1083) to remove any dead cells. Electroporation was performed using the Amaxa Human CD34+ cell Nucleofector kit (Lonza, VPA-1003) according to the manufacturer’s instructions. Cells (2×106) were electroporated with 3 μg DNA coding for FADS2, FADS2-K42Q or FADS2-K42A together with 1.2 μg of the plasmid pmaxGFP. Neomycin (0.1 mg/ml) was used for selection after 48 hours of electroporation and cells were cultured for another 3 weeks following the protocol for long-term ex vivo expansion. Twenty-four hours before the experiment, cells were sorted for HSC (CD48−CD150+LSK) and then treated with leupeptin (100 μM) and NH4Cl (20 mM) for 16 hours and collected for SDS-PAGE and immunoblot.
Seahorse assay.
Oxygen consumption rates and extracellular acidification rates were measured using a 96-well Seahorse Bioanalyzer XF 96 according to the manufacturer’s instructions (Agilent Technologies). In brief, LSK cells were sorted and plated into 96-well plates pre-coated with CELL-TAK (CORNING, 354240). For extracellular acidification rates to measure glycolysis, cells (100,000 cells per well) were plated into 180μl base media (100nM SCF, 100nM TPO, 2mM L-Glutamine, 1mM Pyruvate), spin down at 80 g for 1 min and incubated within a CO2-free chamber at 37°C for 1 hour. Once in the reader, plates were sequentially injected with 30mM glucose, 2μM oligomycin and 100mM 2-DG or just oligomycin and 2-DG where is indicated in the related figures. To determine the fraction of oxygen consumption dependent on fatty acid β-oxidation, cells (200,000 cells/well) were plated into 180μl KHB media (111mM NaCl, 4.7mM KCl, 1.25mM CaCl2, 2mM MgSO4, 1.2mM NaH2PO4) and in half of the samples etomoxir (40μM, Sigma, E1905) was added for 30 min before the analysis. Glycolysis was calculated as ECAR rate after adding saturating amount of glucose, maximal glycolytic capacity as maximum ECAR rate reached upon addition of oligomycin and glycolytic reserve as the difference between glycolytic capacity and glycolysis rate. Once in the reader, plates were sequentially injected with 1μM oligomycin, 2μM FCCP and 0.5μM Rotenone. Fatty acid β-oxidation rate was calculated as the difference in oxygen consumption in presence or absence of etomoxir. Data were normalized to cell number using CyQuant (ThermoFisher, C7026).
Enzyme activity and ATP measurement.
Activity of Pyruvate Kinase or GAPDH were measured using Biovision kit (K709 for Pyruvate Kinase, K608 for GAPDH) according to the manufacturer’s instructions. Briefly, for Pyruvate Kinase, freshly sorted LSK cells (50,000) were collected by centrifugation, lysed with 50μl lysis buffer and after centrifugation, the supernatant was added to 96-well plate with clear bottom, followed by 50μl reaction mix. The plate was read using fluorescence Ex/Em=535/587 nm every 10 minutes. The Pyruvate Kinase activity was calculated by the two readings within a linear range. For the GAPDH activity, LSK cells (50,000) were lysed with 25μl GAPDH Assay buffer by incubation on ice for 10 min followed by centrifugation. The supernatant (20μl per well) adjusted to a final volume of 50μl with GAPDH Assay Buffer was incubated with 50μl of reaction mix and the plate measured at 450 nm in kinetic mode for 10–60 min at 37°C. ATP was measured using ATPlite luminescence ATP detection assay kit (PerkinElmer). Briefly, HSC (10,000) were plated in a white bottom 96-well plate with 100μl 2% FBS in PBS and 50μl mammalian cell lysis solution for 5 min with shaking, followed by additional 5 min incubation with 50μl substrate solution. Plates were measured in a luminometer.
Immunofluorescence staining.
HSC or myeloid progenitor cells were directly sorted into 16-well slides pre-coated with Cell-Tak Cell Tissue Adhesive (Corning, 354240) and then fixed with 4% PFA for 15 min at room temperature (RT). For LAMP2A, LAMP1, total human LAMP2 and FADS2 staining, slides were washed with PBS and incubated with blocking buffer (5% goat serum/0.3% TrionX-100 in PBS) for 1 hour at RT and incubated overnight at 4°C with the first antibody diluted in 1% BSA/0.3% TritonX-100 followed by 40 min incubation at RT with fluorescence-conjugated secondary antibodies. Cells were washed 3 times with PBS and mounted with mounting medium with DAPI (Southern biotech). For LC3 staining, after fixation, cells were permeabilized with 0.015% (v/v) digitonin in PBS (Sigma) and then incubated with blocking buffer (10% FBS in PBS) for 45 minutes. Both the 1st and 2nd antibody were diluted in blocking buffer and incubated for 30 minutes and 45 minutes at RT, respectively. Oxidized proteins were detected with OxyICC Oxidized Protein Detection Kit from Sigma (S7350) and protein inclusions were detected with PROTEOSTAT Aggresome detection kit from ENZO (ENZ-51035) following manufacturer’s instructions. For LysoTracker staining, cells were incubated with 50nM LysoTracker green (Invitrogen, L7526) for 30 min at 37°C, washed and fixed for 10 minutes with 4% PFA and mounted. For analysis of CMA activity using direct fluorescence and the KFERQ-Dendra CMA reporter, cells isolated from KFERQ-Dendra2 transgenic mice were fixed with 2% PFA for 5 min at RT and mounted for direct puncta counting or subjected for immunofluorescence as described above. Quantification was performed in TIFF converted images upon thresholding using the 3D object counter tool of Image J software (NIH). Average number of puncta per cell was determined for each of the cells in a field and at least 3 different fields per animal were counted. Where indicated, we also calculated the percentage of cells active for CMA, defined as those with at least two Dendra positive puncta. To determine CMA flux using the CMA reporter, cells were incubated or not in the presence of leupeptin (100 μM, 16 hours), fixed with 4% PFA for 15 minutes followed by blocking with 5% goat serum/0.01% tritonX-100 for 1 hour at RT, and then incubated with the dendra2 antibody and the corresponding secondary antibody in 1% BSA/0.01% TritonX-100 in PBS sequentially. CMA flux was calculated as the increase in number of Dendra positive puncta upon leupeptin treatment. The following primary antibodies were used: anti-mouse LAMP2A (Invitrogen, 512200), anti-mouse LAMP1 (Hybridoma Bank, 1D4B), total human LAMP2 (Hybridoma Bank, h4b4), FADS2 (Abclonal, A10270), LC3 (MBL, PM036), Dendra2 (antibodies, abin361314).The following secondary antibodies were used: Alexa Fluor 488 goat anti-rabbit IgG (H+L) (Invitrogen, A-11008) and Alexa Fluor 488 goat anti-rat IgG (H+L) (Invitrogen, A-11006), Alexa Fluor 555 goat anti-rabbit IgG (Invitrogen, A-21428), Alexa Fluor 555 goat anti-rat IgG (Invitrogen, A-21434), Alexa Fluor 635 goat anti-rabbit IgG (Invitrogen, A-31576). All the images were taken with a Confocal microscope (TCS SP8; Leica) using an HCX Plan Apo CS 63.0× 1.40 NA oil objective and the Leica Application Suite X (LAS X).
Transmission Electron microscopy.
Cells (100,000 per condition) were pelleted and fixed with 2% paraformaldehyde and 2.5% glutaraldehyde in 0.1M sodium cacodylate buffer, followed by sequential fixation in 1% osmium tetroxide followed and 2% uranyl acetate. Samples were dehydrated through a graded series of ethanol dilutions and embedded in LX112 resin (LADD Research Industries). Ultrathin (80nm) sections were cut on a Leica EM Ultracut UC7, stained with uranyl acetate followed by lead citrate and viewed on a JEOL 1200EX transmission electron microscope at 80kV.
RNA Purification, Amplification, and Microarray Analysis.
Cells (10,000) were sorted and total RNA was extracted using RNeasy Micro kit (Qiagen) according to the manufacturer’s protocols. All total RNA samples were quantified with the RNA Quantification Kit (ThermoFisher Scientific, 902905). Using the GeneChip WT Pico Kit (ThermoFisher Scientific, 902622), samples were amplified to cRNA from in vitro transcription of one round of linear amplification reaction and then converted to sense-strand single stranded cDNA followed fragmentation of biotinylated cDNA. 5.5 micrograms of cDNA from each sample was hybridized to ThermoFisher Scientific (Affymetrix) GeneChip Mouse Gene 2.0 ST Array (902118). Hybridization cocktail was made using the hybridization kit (ThermoFisher Scientific, 900454) and array scans were performed according to the manufacturer’s instructions using the high-resolution GeneArray Scanner 3000 7G (ThermoFisher Scientific, Affymetrix). The data were analyzed with the GeneChip Command Console Software from ThermoFisher Scientific (Affymetrix) default analysis settings.
Quantitative Proteomics and Protein Pathway Analysis.
Freshly sorted LSK cells (500,000) were pelleted and flash-frozen in liquid nitrogen for shipment to the Biological Mass Spectrometry Core Facility at University of Colorado Denver. For analysis, cells were lysed with RIPA buffer (ThermoFisher) and subjected to GeLC-MS74. Excised gel pieces were destained in ammonium bicarbonate in 50% acetonitrile and dehydrated in 100% acetonitrile, trypsin digested upon reduction and alkylation of unmodified cysteine residues, and analyzed by nano-UHPLC-MS/MS (Easy-nLC1000, QExactive HF-positive ion mode (ThermoFisher)). The peptide mixture was desalted and concentrated in a Thermo Scientific Pierce C18 Tip. Samples were analyzed on an Orbitrap Fusion mass spectrometer (ThermoFisher) coupled to an Easy-nLC 1200 system (ThermoFisher) through a nano-electrospray ion source according to manufacturer’s instructions. MS/MS spectra were extracted from raw data files and converted into mgf files using a Proteome Discoverer Software (ver. 2.1.0.62). The mgf files were then independently searched against the mouse database using an in-house Mascot server (Version 2.6, Matrix Science). Mass tolerances were +/− 10 ppm for MS peaks, and +/− 0.6 Da for MS/MS fragment ions. Trypsin specificity was used allowing for 1 missed cleavage. Met oxidation, protein N-terminal acetylation, and peptide N-terminal pyroglutamic acid formation were allowed as variable modifications while carbamidomethyl of Cys was set as a fixed modification. Scaffold (version 4.8, Proteome Software) was used to validate MS/MS based peptide and protein identifications. Peptide identifications were accepted if they could be established at greater than 95.0% probability as specified by the Peptide Prophet algorithm. Protein identifications were accepted if they could be established at greater than 99.0% probability and contained at least two identified unique peptides. Allocation of proteins to functional groups was done using the IPA software (Ingenuity Systems) and STRING database (https://string-db.org/). Protein oxidation state was performed by analyzing oxidation of methionine, carbonylation of proline to pyro-glutamate and various degrees of cysteine oxidation (disulfide, glutathionylation, sulfenic, sulfonic and beta-alanine). Total protein oxidation was determined by the total number and abundance of carbonylated residues via mass spectrometry. Oxidation of methionine and carbonylation were included in this work as they showed significant differences across genotypes and interventions. To determine the acetylation state of FADS2, integrated peak areas of the FADS2 acetylated intact peptides were obtained from their extracted ion chromatograms using Qual Browser of Xcalibur 2.2 (Thermo Fisher Scientific), while b and y series ions for the MS2 of those peptides were used for the assignment of the peptide sequence and position of the acetylation75.This approach allows identification and quantitation of both N-terminus acetylation as well as acetylation at specific amino acid residues, such as lysines – K. For the FADS2 peptides KVYNVTK and its di-acetylated form, the predicted m/z’s were: 426.2529 +/− 0.0043 and 468.2635 +/− 0.0046; and for the WLVIDRK peptide and its acetylated form 465.2820 +/− 0.0047 and 486.2873 +/− 0.0049 m/z’s.
Metabolomics.
Freshly sorted LSK cells (100,000) were centrifuged and the pellet was flash-frozen in liquid nitrogen and shipped to the Biological Mass Spectrometry Core Facility at University of Colorado Denver. Cell pellets were lysed with lysis solution (methanol:acetonitrile:water 5:3:2 v/v/v), before ice cold extraction by vortexing for 30 minutes at 4°C. Insoluble proteins were pelleted by centrifugation (15,000 g for 10 min at 4°C) and supernatants were analyzed using a UHPLC system (Vanquish, ThermoFisher) coupled online to a mass spectrometer (Q Exactive, ThermoFisher). Samples were resolved over a Kinetex C18 column (2.1 × 150 mm, 1.7 μm; Phenomenex) at 25°C using a 9‐minute gradient at 400 μl/min from 5% to 95% B (A: water/0.1% formic acid; B: acetonitrile/0.1% formic acid). MS analysis and data analysis were performed as described before76, 77. Metabolite assignments were performed using a metabolomics data analyzer (MAVEN). Metabolic pathway analysis was performed using the MetaboAnalyst software.
Measurement of intracellular protein degradation.
Freshly isolated LSK cells (25,000 cells) were labeled with [3H]Leucine (2μCi/ml) in stem span medium containing 100nmol mSCF for 24 hours in a 48-well plate pre-coated with CELL-TAK. After extensive washing, media (with 2.8mM unlabeled leucine) was added and cells were incubated at 37°C. Where indicated, 20mM NH4Cl and 100μM Leupeptin (Sigma) were added to the media. Aliquots of the medium taken at 12 hours were precipitated with trichloroacetic acid and proteolysis was calculated as the percentage of initial total acid precipitable radioactivity (protein) transformed to acid soluble (peptides and amino acids) at each time point78. Lysosomal proteolysis was determined as percentage of proteolysis sensitive to the combination of lysosomal inhibitors.
Other procedures.
For quantitative PCR, RNA was extracted using RNeasy Micro kit (Qiagen) according to the manufacturer’s protocols. Transcripts were reverse transcribed according to manufacturer protocols (Invitrogen), and qPCR was performed using SYBR Green (Applied Biosystems). The following primers were used: mouse FADS2, F-5’-gctctcagatcaccgaggac-3’, R-5’-agtgccgaagtacgagagga-3’. Immunoblot for different tissues (100μg total protein) was performed in nitrocellulose membranes after tissue sonication in RIPA buffer. The pCMV6-Entry-FADS2-Myc-DDK plasmid was purchased from Origene (MR207091), single point mutation of FADS2 42K to Q and A was performed at GenScript.
Statistics, sample size and software.
All data are presented both as individual values (symbols) and mean ± standard error of the mean (sem). Unpaired t-test, two-way ANOVA test, one-way ANOVA tests, chi-square test, log-rank (Mantel-Cox) test were used for the statistics as indicated in each figure legend. In all instances, “n” refers to individual experiments or animals and is indicated in the figure legends. The number of animals used per experiment was calculated through power analysis based in previous results. Animals were randomly attributed to control or treatment groups after separating them according to genotype. No mouse was excluded from the analysis unless there were technical reasons or the mouse was determined to be in very poor health by the veterinarian. Outliers were determined by the ROUT method (Q=1%). Investigators were blinded to the treatment during data collection and analysis and unblinding was done when the analysis was completed for plotting. All parameters were analyzed using independent sample tests and were tested for normal distribution using Shapiro-Wilk normality test. For software, image analysis was performed using Image J (NIH), pathway analysis using the Ingenuity Pathway Analysis (Ingenuity Systems) and STRING database (Https://string-db.org/), HSC frequency within the tested cell population was estimated using ELDA software, all graphical plots were made using GraphPad Prism software 7.04 (GraphPad) and images were assembled with Adobe Photoshop CC software (Adobe).
Extended Data
Extended Data Fig. 1. CMA changes in HSC with age.
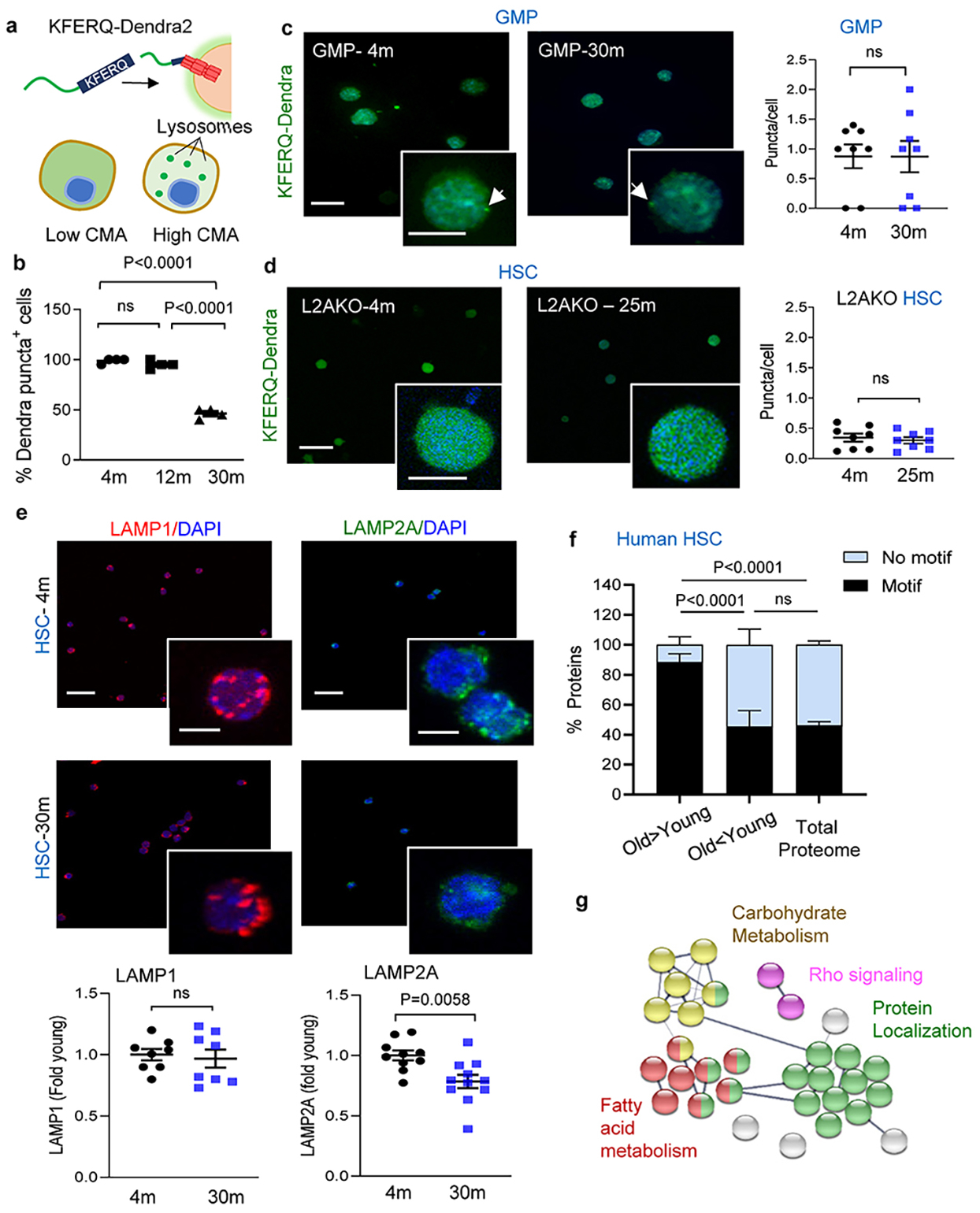
a, Scheme of the KFERQ-Dendra2 fluorescent CMA reporter. b, Percentage of HSC positive for Dendra puncta at the indicated ages. n=4 mice per group, individual points are fields . CMA+ cells = cells with >2 Dendra+ puncta. c, Dendra fluorescence in GMP cells from young and old KFERQ-Dendra mice. Left: Representative images and magnified cells. Right: Puncta per cell. n=8 fields from 3 mice per group, individual points show average puncta/field. d, Dendra fluorescence in HSC from young and old L2AKO-KFERQ-Dendra mice. Left: Representative images and magnified cells. Right: Puncta per cell. n=8 fields from 4 mice per group. e, Immunostaining for LAMP1 and LAMP2A in HSC from young and old mice. Top: Representative images and magnified cells. Bottom: Fluorescence intensity of each protein. n=8 (LAMP1), 11 (LAMP2A) fields from 3 mice per group, individual points shows average intensity/field. f, Proteins bearing KFERQ-like motifs in those at higher (O>Y) and lower (O<Y) abundance in old human BM HSC compared to young ones (data set in ref 31). Motif frequency in the total proteome is shown as reference. n=59 human subjects (45 male and 14 female) with age range 20–60 years (with median of 33.2 years). Only proteins displaying statistically significant changes with age (p<0.01) were included. g, STRING analysis of the proteins bearing CMA-targeting motifs that accumulate in old human BM HSC. Data shows individual values and mean ± SEM. One-way (b) or two-way (f) ANOVA with Tukey’s post-hoc test and unpaired two-tailed t test (c,d,e) were used. P values are shown and statistical analysis is in data source. ns: no statistical significance.
Extended Data Fig. 2. CMA upregulation during HSC activation.
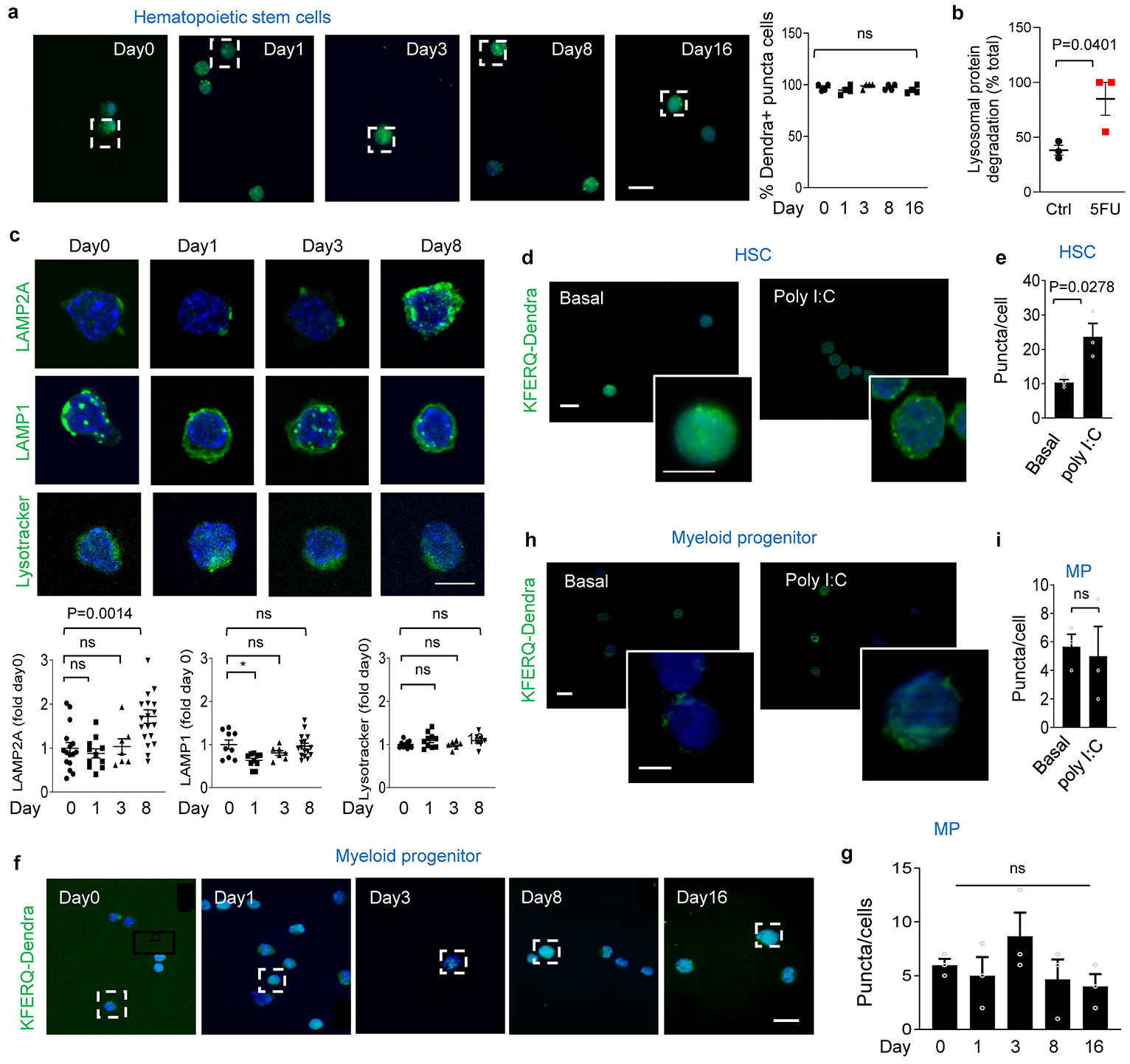
a, Left: Full fields of the images shown at higher magnification in Fig. 1b. Right: HSC positive for Dendra puncta. n=4 mice per group. CMA+ cells = cells with >2 Dendra+ puncta. b, Lysosomal proteolysis of long-lived proteins in HSC untreated (Ctrl) and 8 days post 5FU injection. n=3 independent experiments. c, Immunostaining for LAMP2A, LAMP1 and LysoTracker in HSC from mice before and 1, 3 and 8 days after single 5FU injection. Top: Representative images. Bottom: Fluorescence intensity. n=16, 11, 7, 19 fields (LAMP2A), 9, 10, 7, 16 fields (LAMP1) and 10, 10, 6, 7 fields (LysoTracker) from 3 mice per group, individual points show average intensity/field. d,e, Dendra fluorescence in HSC from KFERQ-Dendra mice 48h post PolyI:C or vehicle injection (Basal). Representative images and magnified cells (d) and puncta per cell (e), n=3 mice. Individual points show average puncta/field. f,g, Full fields of the images shown at higher magnification in Fig. 1b (f) and number of Dendra+ puncta per cell (g) in myeloid progenitors from KFERQ-Dendra mice before and at the indicated days post 5FU injection. n=3 mice. h,i, Dendra fluorescence in myeloid progenitors from KFERQ-Dendra mice 48h post PolyI:C or vehicle injection (Basal). Representative images and magnified cells (h) and puncta per cell (i), n=3 mice. Data shows individual values and mean ± SEM. Unpaired two tailed t tests (b,e,i) and one-way ANOVA with Tukey’s post-hoc test (a,c,g) were used. P values are shown and statistical analysis is in data source. ns: no statistical significance.
Extended Data Fig. 3. Effect of CMA blockage on HSC and myeloid progenitor cells.
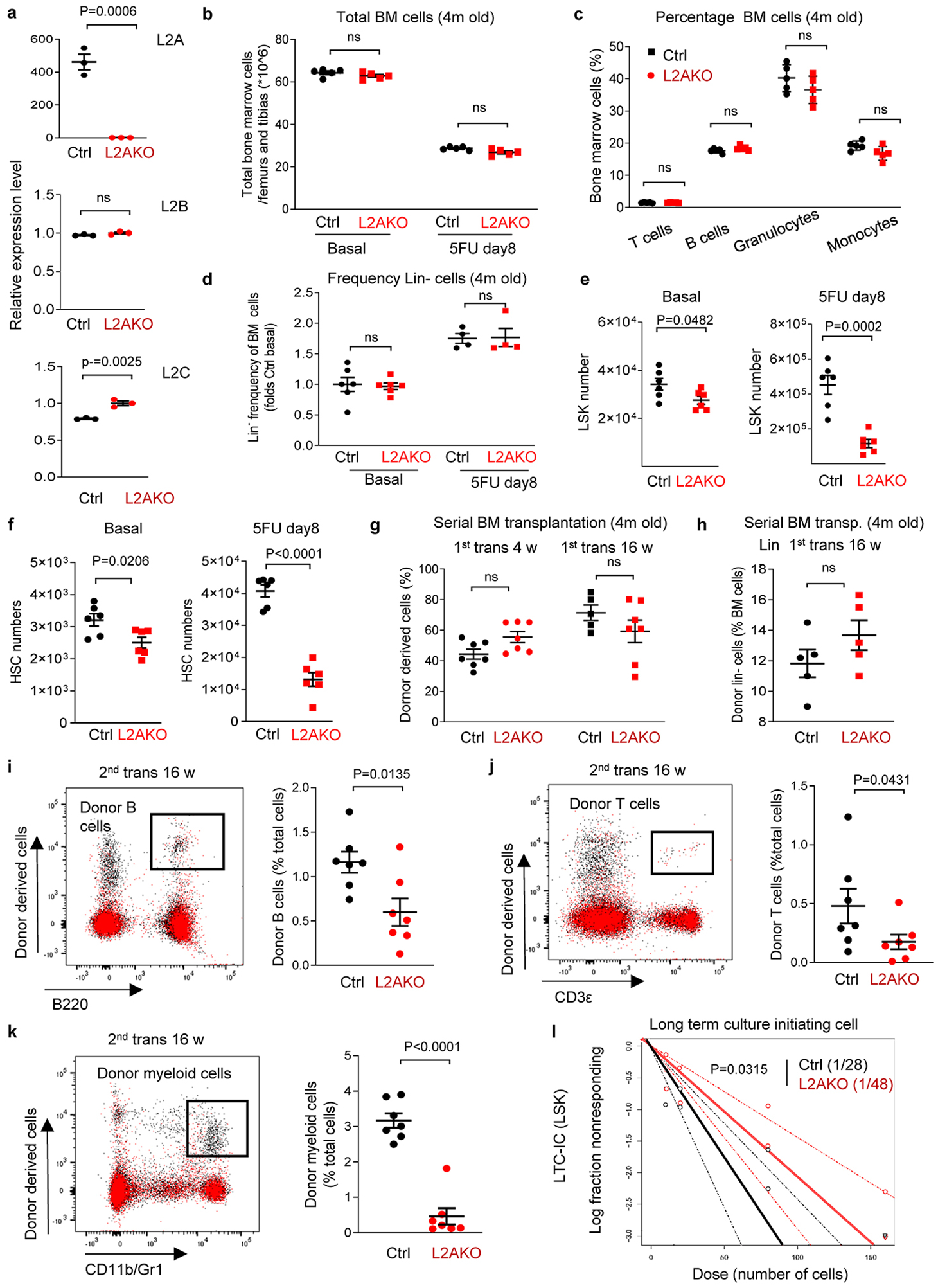
a, Quantitative PCR for the three lamp2 spliced variants in BM from Vav-iCreL2Af/f (L2AKO) and VaviCre (Ctrl) mice. n=3 independent experiments. b, Number of total BM cells of 2 femurs and tibias from Ctrl and L2AKO mice in basal and at day 8 post 5FU injection. n=5 mice. c, Percentage of T cells (CD3ε+), B cells (B220+), granulocytes (CD11b+ Gr1high) and monocytes (CD11b+ Gr1middle) in Ctrl and L2AKO mice BM in basal conditions. n=5 mice. d, Frequency of Lin- cells of 2 femurs and tibias from Ctrl and L2AKO mice in basal and day 8 post 5FU injection. n=6 (basal) and 4 (5FU) mice. e,f, Number of LSK cells (e) and HSC cells (f) in BM of Ctrl and L2AKO mice in basal and 5FU activated conditions. n=6 mice. g,h, Donor chimerism in peripheral blood (g) and donor derived Lin- population (h) in BM of recipients at the indicated weeks after the primary competitive transplantation using BM from 4m Ctrl and L2AKO mice. n=7 mice (g), n=5 mice (h). i-k, Donor lineage distribution in peripheral blood of recipients in the secondary competitive transplantation of Ctrl and L2AKO BM. Representative FACS plots (left) and quantification (right) are shown. n=7 mice. l, LTC-IC frequency for both Ctrl and L2AKO cells in the LTC-IC assay of Lin−Sca-1+c-Kit+ (LSK) cells from Ctrl and L2AKO mice. n=3 mice. Data shows individual values and mean ± SEM. Two way ANOVA with Sidak’s post-hoc test (b-d,g), unpaired two-tailed t tests (a,e,f,h-k) and Chi-square test (l) were used. P values are shown and statistical analysis is in data source. ns: no statistical significance.
Extended data Fig. 4. Impact of CMA blockage in the cell cycle of HSC and myeloid progenitor cells.
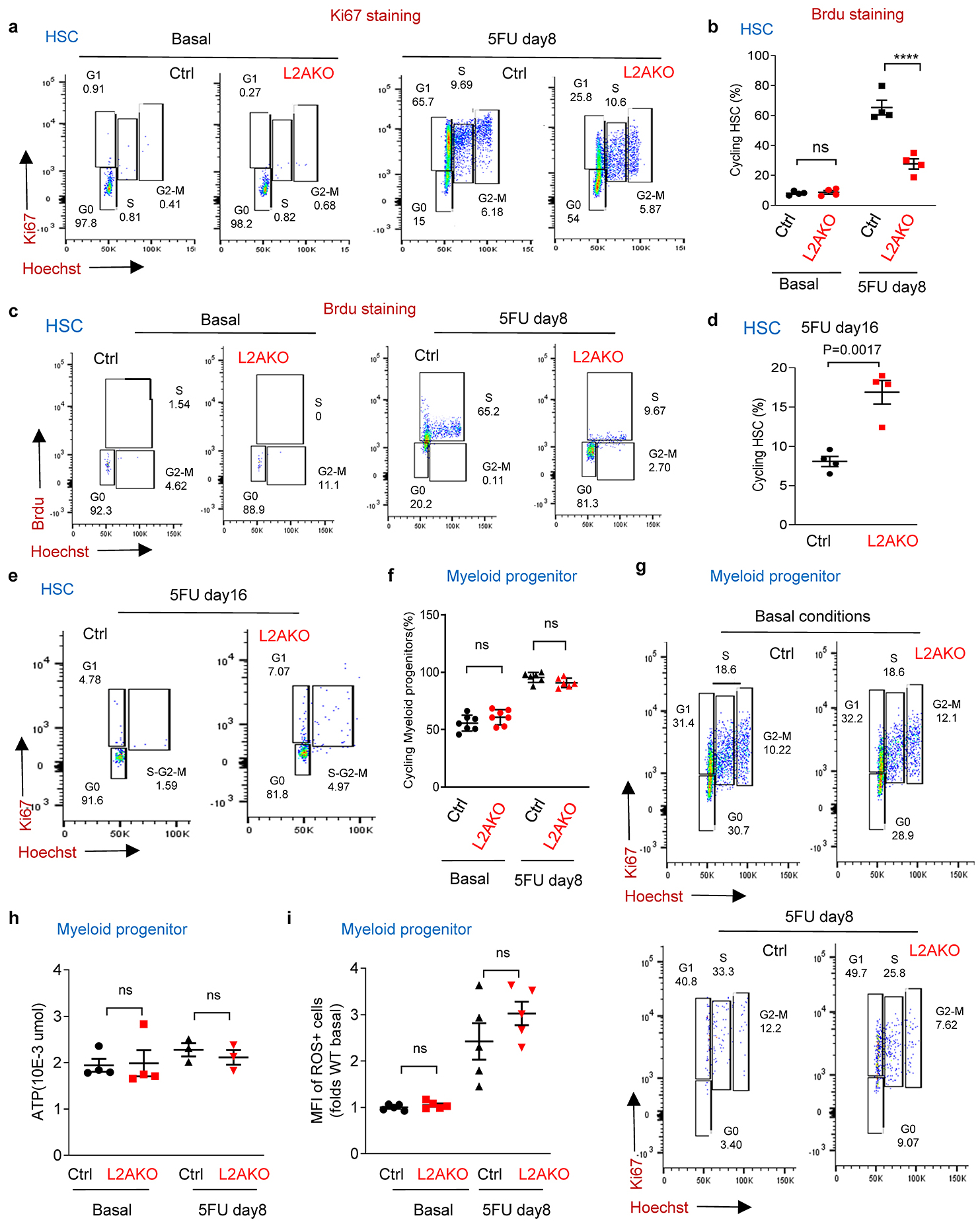
a-c, Representative FACS plots of Hoechst and Ki67 staining (a) or Hoechst and BrdU staining (c) in HSC from control (Ctrl) and L2AKO mice untreated (basal) or 8 days post single 5FU injection. Quantification of FACS in a is shown in main Fig. 2a and quantification of FACS in c is shown in b. n=4 mice. d,e, Representative FACS plots of Ki67 and Hoechst staining (e) and quantification (d) of cycling HSC from Ctrl and L2AKO mice untreated (basal) or 16 days post single 5FU injection. n=4 mice. f,g, Percentage of cycling cells (f) and representative flow cytometry plots (g) of Ki67 and Hoechst stained myeloid progenitors from Ctrl and L2AKO mice in basal conditions (top) and 8 days after a single 5FU injection (bottom). n=7 mice per condition. h,i, ATP (h) and ROS levels (i) in myeloid progenitors from Ctrl and L2AKO mice in basal or after single 5FU injection. n=4 (basal) and 3 (5FU) mice for h, n=5 mice for i. Data shows individual values and mean ± SEM. Two-way ANOVA with Sidak’s post-hoc test (b,f,h,i) and unpaired two tailed t-test (d) were used. P values are shown and statistical analysis is in data source. ns: no statistical significance.
Extended data Fig. 5. Characterization of macroautophagy in CMA-deficient HSC cells.
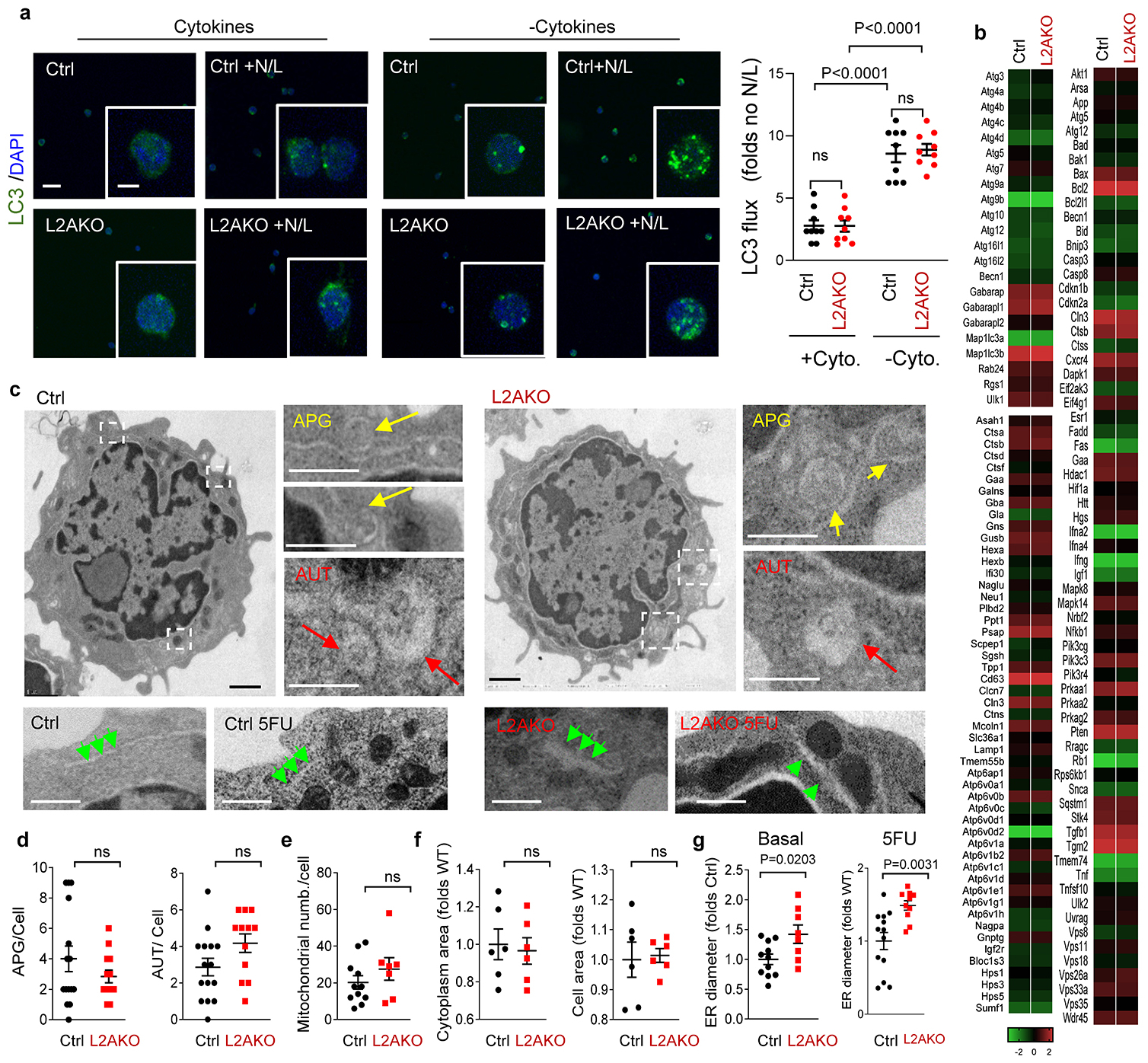
a, Immunostaining for endogenous LC3 in HSC from control (Ctr) and L2AKO mice in basal and cytokine starved conditions in presence or not of lysosomal inhibitors NH4Cl and leupeptin (N/L). Representative images of full fields and higher magnification cells (left) and LC3 flux (right) calculated as the increase in LC3+ puncta upon addition of N/L. n=9 fields from 3 mice, individual points show average puncta/field. b, Expression of genes in the TFEB-regulated transcriptional program in Ctrl and L2AKO LSK cells. Genes are grouped as: components of the macroautophagy machinery (top left), regulators of macroautophagy initiation (right) and genes involved in lysosome biogenesis (bottom left). c, Representative electron microscopy images of HSC (from 3 independent repeats). Top: whole fields and boxed areas at higher magnification to show examples of autophagosomes (APG, yellow arrows) and autolysosomes (AUT, red arrows). Bottom: representative images of ultrastructure of the endoplasmic reticulum (green arrows) in HSC under basal and 5FU (day8) activated conditions. d-f, Average number of APG, AUT (d) and mitochondrial number per cell (e) as well as cytoplasm and total cell area (f) from electron microscopy images in c. n=15 (Ctrl APG/AUT), 13 (L2AKO APG) and 12 (L2AKO AUT) fields (d), 11 fields (e) and 6 fields (f) from 3 independent samples. g, Average ER diameter in HSC under basal and 5FU (day8) activated conditions. n=11 (basal ctrl), 8 (basal L2AKO), 13 (5FU ctrl) and 10 (5FU L2AKO) fields. Data shows individual values and mean ± SEM. Two-way ANOVA with Tukey’s multiple post-hoc test (a) and unpaired two-tailed t test (d-g) were used. P values are shown and statistical analysis is in data source. ns: no statistical significance.
Extended data Fig. 6. Metabolism-related changes in the transcriptome and proteome of CMA-deficient HSC.
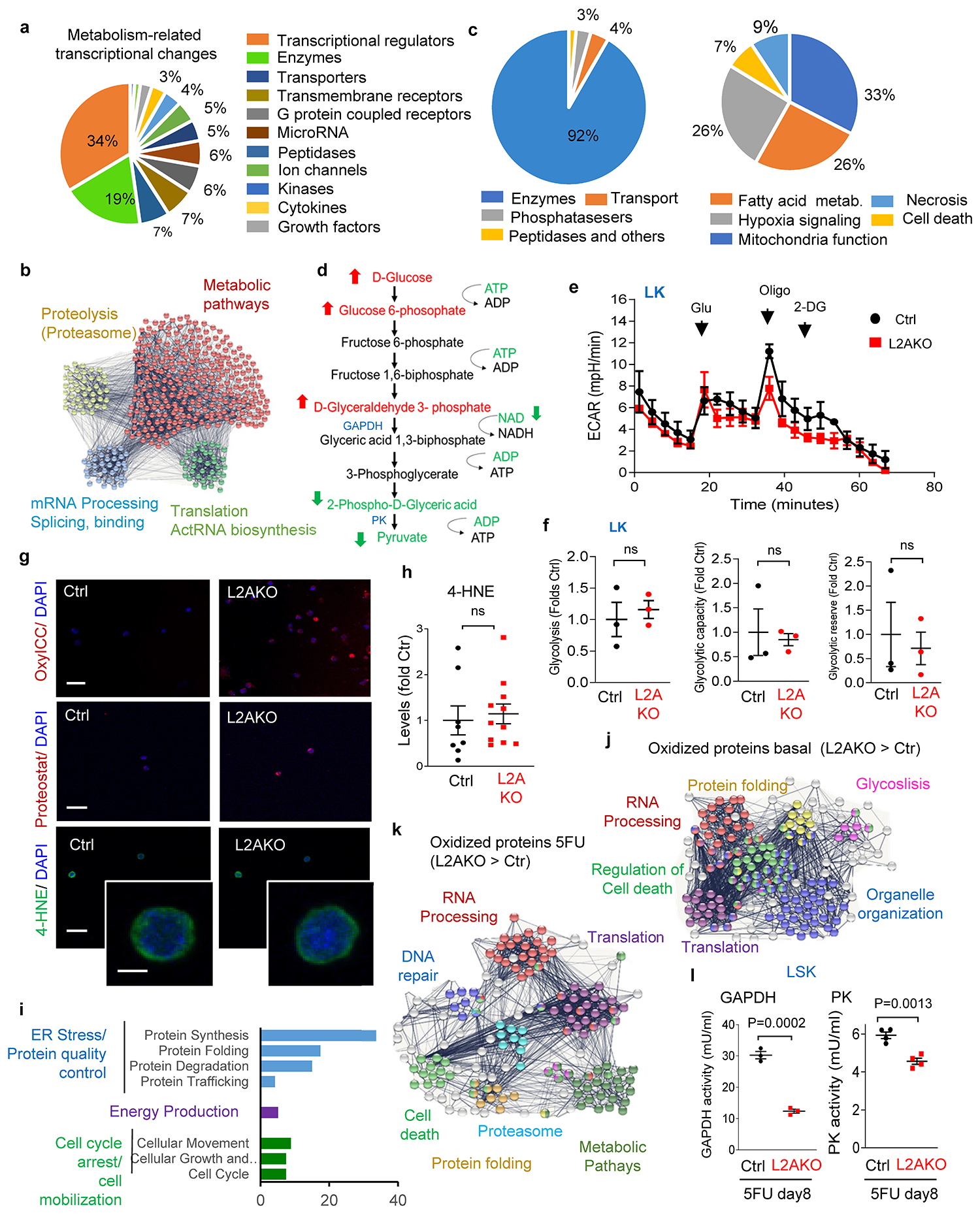
a, Categories of metabolism-related top transcriptional changes in L2AKO HSC cells in basal conditions. b, STRING analysis of proteins more abundant in L2AKO LSK in basal conditions. c, Top categories of proteins related with overall metabolism (left) and break down of the proteins under enzyme category (right) found in the overlapped proteins in Fig. 2h. d, Scheme of glycolysis-related substrates and metabolites to highlight those significantly increased (red) or decreased (green) in L2AKO LSK. e,f, Extracellular acidification rate (ECAR) (e) and basal glycolysis, glycolytic capacity and glycolytic reserve (f) in myeloid progenitors. Glu: Glucose, Oligo: oligomycin, 2DG: 2-Deoxy-D-glucose, n=3 independent experiments. g, Full field images of HSC straining for OxyICC or Proteostat shown in Fig. 2q,r. Bottom panel shows full field and higher magnification inset of HSC 4-HNE staining. h, Intensity of 4-HNE staining shown in g. n=8 (Ctrl) and 11 (L2AKO) fields from 3 mice, individual points represent average intensity per field. i, Ingenuity pathway analysis of the top proteins with higher percentage of oxidation in activated L2AKO LSK cells compared with control. j,k, STRING analysis of proteins with higher percentage of oxidation in basal (j) and 5FU activated conditions (k). l, GAPDH and PK activity in 5FU-activated Ctrl and L2AKO LSK cells. n=3 independent experiments. Data shows individual values and mean ± SEM. Multiple t test by time segment (e) and unpaired two-tailed t tests (f,h,l) were used. P values are shown and statistical analysis is in data source. ns: no statistical significance.
Extended Data Fig. 7. Regulation of lipid metabolism by CMA during HSC activation.
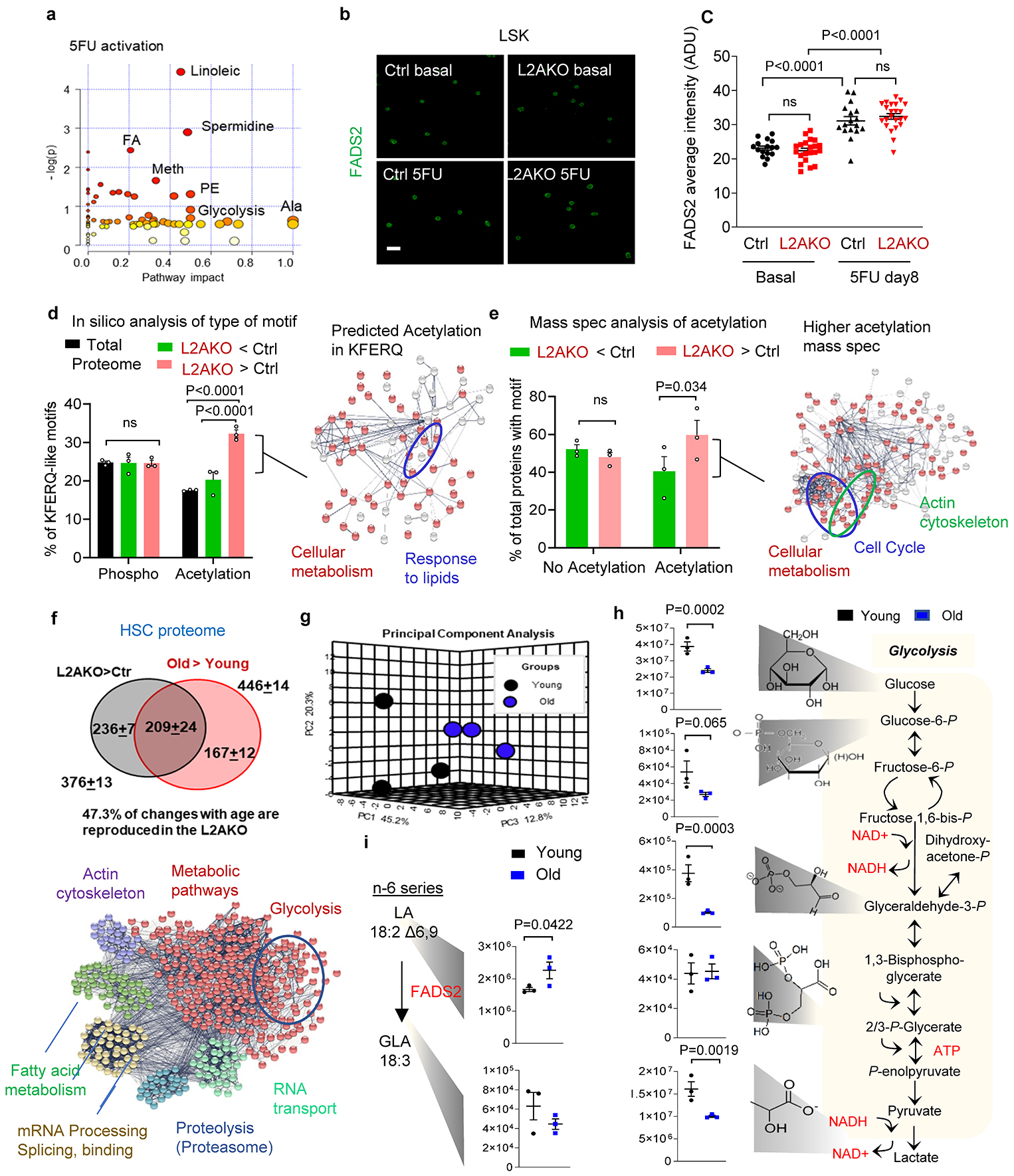
a, Metabolic pathways different between Ctrl and L2AKO LSK 8 days post-5FU injection. n=9 mice in 3 independent experiments (ie). b,c, Immunostaining for FADS2 in LSK cells. Representative images (b) and average of intensity per cell (c). n=16 (basal Ctrl), 21 (basal L2AKO), 17 (5FU Ctrl) and 23 (5FU L2AKO) fields from 3 ie. d, In silico analysis of proteins bearing phosphorylation- or acetylation-generated KFERQ-like motifs in the total proteome, and in the group of proteins less/more abundant in L2AKO LSK cells post-5FU. Left: Percentage of proteins with KFERQ motif. Right: STRING analysis of proteins bearing acetylation-generated KFERQ motif in the L2AKO>Ctrl group. n=3 mice in 3 ie. e, Mass spectrometry analysis of acetylation-generated KFERQ motifs in proteins with lower and higher acetylation in L2AKO LSK post-5FU. Left: Percentage of proteins with or without acetylation generated KFERQ motifs. Right: STRING analysis of proteins with acetylation-generated motifs in the group of proteins with higher acetylation in L2AKO. n=9 mice in 3 ie. f, Venn diagram of proteins more abundant in L2AKO LSK than Ctrl and in old than young LSK. n=9 mice in 3 ie. Bottom: STRING analysis of proteins that accumulate in both L2AKO and old mice LSK. g-i. Metabolomic analysis in young (4m) and old (>25m) mice LSK in basal conditions. Unsupervised principal component analysis (g). Comparison of metabolites in glycolysis (h) and in the FADS2-catalyzed step in linoleic acid metabolism (i). Values are integrated peak areas (arbitrary units) for each metabolite. n=9 mice in 3 ie. Data shows individual values and mean ± SEM. Two-way ANOVA with Tukey’s (d,e) or Sidak’s (c) post-hoc test and unpaired two-tailed t tests (h,i) were used. P values are shown and statistical analysis is in data source. ns: no statistical significance.
Extended Data Fig. 8. CMA activity in hematopoietic stem cells from old mice.
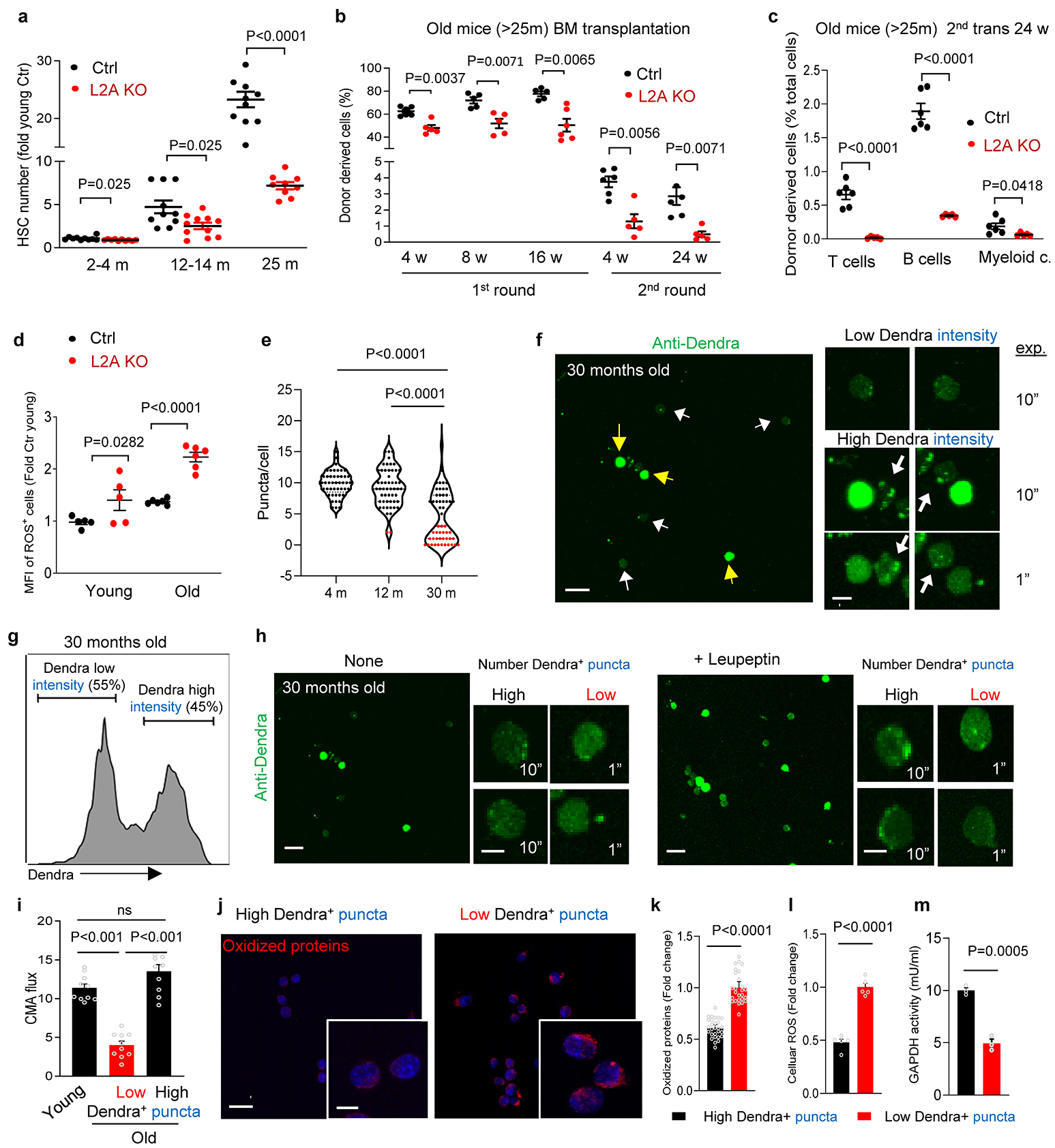
a, HSC numbers in control (Ctrl) and L2AKO mice with age, n=10,10,11 (Ctrl) and 10,11,9 (L2AKO) mice per age. b, Donor chimerism in recipients’ peripheral blood after old Ctrl and L2AKO mice BM competitive transplantation. n= 6,5,5,6,6 (Ctrl) and 5,5,6,5,5 (L2AKO) mice per time point. c, Donor lineage distribution in peripheral blood after Ctrl and L2AKO mice BM second competitive transplantation, n=6 (Ctrl) and 5 (L2AKO) mice. d, HSC ROS levels (MFI: median fluorescence intensity). n=5 (young) and 6 (old) mice. e, Fluorescence puncta in HSC from KFERQ-Dendra mice. Each individual point represents a cell. n=45 (4m), 52 (12m) and 53 (30m) cells from 4 mice per group. Red points: cells with <5 puncta. f, Representative full-field and higher magnification images (from 3 independent experiments (ie)) of dendra immunostained HSC from 30m old mice. Exposure times are indicated. Arrows: high (yellow) and low (white) cytosolic Dendra intensity. g, Flow cytometry analysis of direct Dendra fluorescence of HSC from 30m old mice. Representative plot from n=4 mice. h, Representative full-field and higher magnification images (from 3 ie) of dendra immunostained LSK cells from 30m old mice incubated or not with leupeptin. Exposure time is shown. i, CMA flux (increase in puncta number after leupeptin), n=9 fields from 4 mice per group. j,k, Oxidized protein staining in 30m old mice HSC FACS sorted according to their cytosolic Dendra fluorescence intensity. Representative full-field and higher magnification images (j) and intensity of oxidized proteins (k), n=26 (high intensity) and 28 (low intensity) fields from 4 mice. l,m, Cellular ROS levels (l) and GAPDH activity (m) in the same FACS sorted HSC populations as in j. n=5 fields from 3 mice (l) and n=3 mice (m). Data shows individual values and mean ± SEM. Multiple two tailed t-test (a-c), two-way ANOVA with Sidak’s post-hoc test (d), one-way ANOVA with Bonferroni post-hoc test (e, i) and unpaired two-tailed t tests (k-m) were used. P values are shown and statistical analysis is in data source. ns: no statistical significance.
Extended Data Fig. 9. Characterization of HSC with genetic or pharmacological activation of CMA.
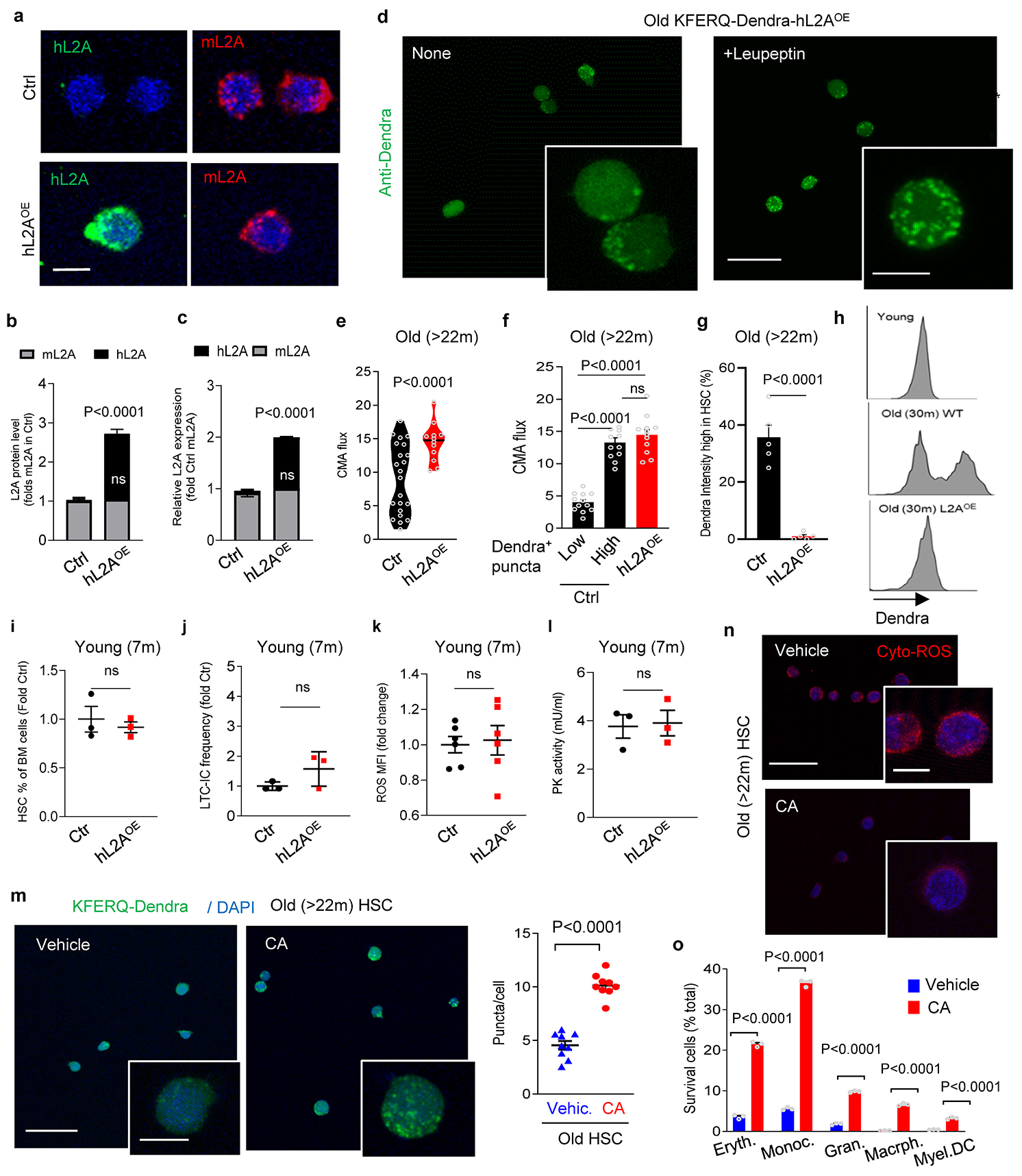
a,b, Immunostaining images for human and mouse L2A in HSC from Ctrl and hL2AOE mice (a) and average fluorescence intensity per cell relative to Ctrl (b), n=9 fields from 3 mice. c, Human and mouse L2A mRNA in HSC expressed as fold mL2A mRNA in Ctrl cells, n=3 independent experiments (ie). d, Images of Dendra immunostained HSC from 22–25m old KFERQ-Dendra-hL2AOE mice incubated or not with leupeptin, n=3 ie. e-g, CMA flux (increase in fluorescent puncta upon leupeptin) in HSC from old Ctrl (from Extended Fig. 8h) and KFERQ-Dendra-hL2AOE mice averaged (e) or separated by Dendra fluorescence intensity (f); fraction of cells with high cytosolic KFERQ-Dendra staining (g), n=12 fields from 4 mice. h, Flow cytometry plots (from 3 ie) of HSC sorted by their cytosolic KFERQ-Dendra fluorescence intensity. i-l, HSC frequency (i), LTC-IC frequency (j), levels of intracellular ROS (k) and PK activity (l) in HSC from 7m hL2AOE mice with upregulated L2A expression since 4m of age. n=3 (i,j,l) and 6 (k) mice/group. m, Direct fluorescence images (from 3 ie) of HSC from 22–25 months old KFERQ-Dendra mice ex vivo treated with vehicle or CMA activator (10 μM CA) for 4 weeks (left) and fluorescent puncta per cell (right). n=9 fields from 3 mice. n, Oxidized protein staining in HSC cells from 22–25 months old mice after 7 days of culture in presence of vehicle or CA (10 μM). Nuclei are stained with DAPI. Quantification is shown in Fig. 4i. o, Viability of cells recovered at the end of LTC-IC from LSK cells of 25m mice treated ex vivo with vehicle or CA. Data shows individual values and mean ± SEM. Two-way ANOVA with Sidak’s (b,o) or Tukey’s (c) post-hoc test, unpaired two-tailed t test (e,g, i-m) and one-way ANOVA with Tukey’s post-hoc test (f) were used. P values are shown and statistical analysis is in data source. ns: no statistical significance.
Extended Data Fig. 10. The effect of CMA on polyunsaturated fatty acid metabolism in HSC is the basis for the role of CMA in HSC function.
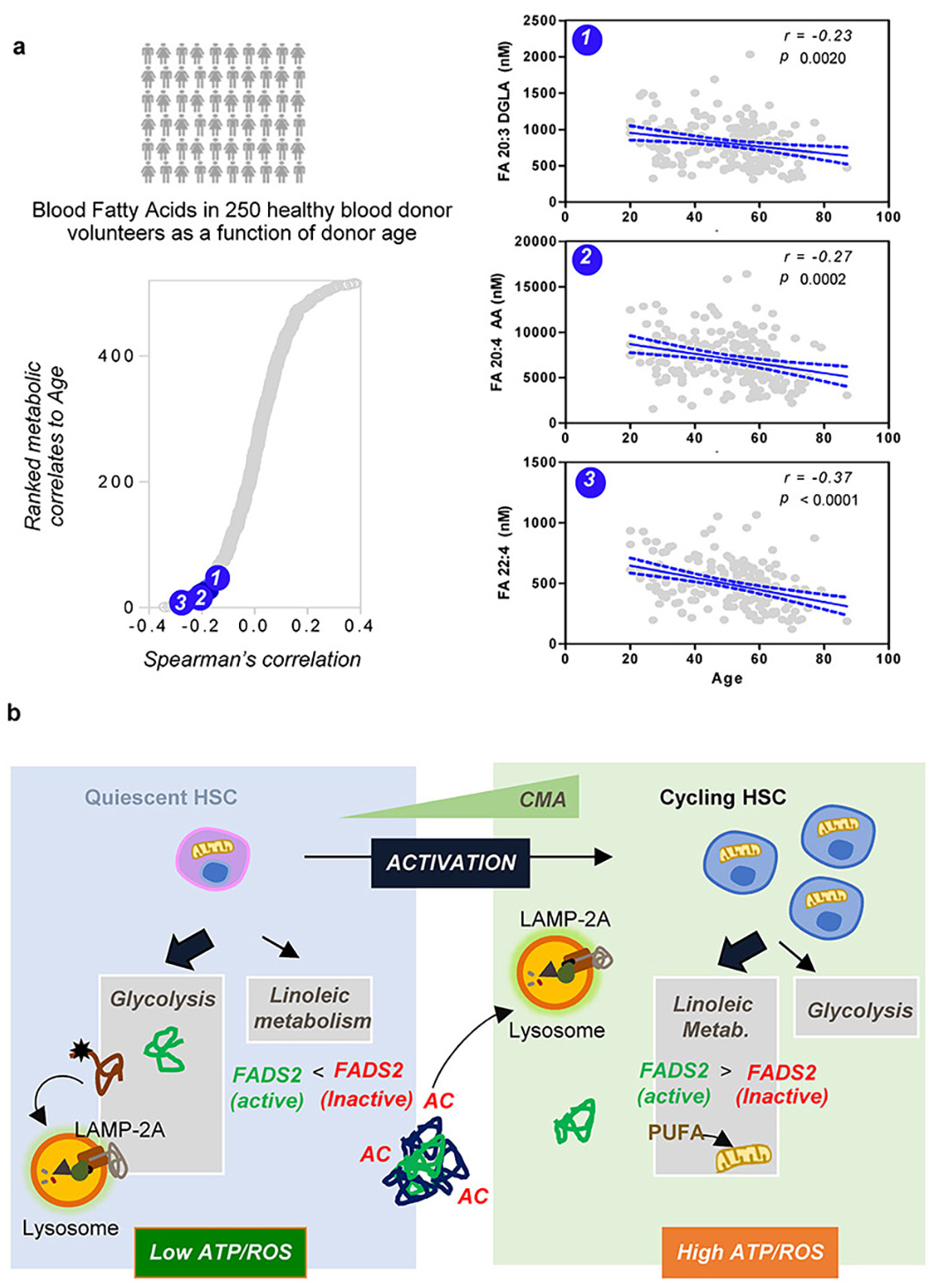
a, Polyunsaturated fatty acids that are generated by FADS2 activity decrease with age in human blood from healthy donor volunteers (n=250)40. Top metabolic changes positively and negatively correlating (Spearman’s correlation) with aging were determined (left) and placement of 3 products of FADS2 activity in the graph is marked by the blue circles as (1) for DGLA (Dihomo-γ-linolenic acid), (2) for AA (arachidonic acid) and (3) for DCA (Docosatetraenoic acid). Right shows levels of each of the three metabolites in 250 healthy individuals with age range 20–90 years. Pearson correlation was used for statistics. b, Scheme of the role of CMA in HSC function. Left: under basal conditions, functional CMA is required in HSC for protein quality control including that of enzymes involved in glucose metabolism. Failure of CMA leads to persistence of damaged enzymes and reduced glycolytic activity. Right: during HSC activation, CMA is required for increasing FADS2 activity the limiting enzyme in linoleic and α-linolenic metabolism, to activate this pathway and thus facilitate the metabolic switch from glucose to lipid metabolism. CMA changes the active/inactive enzyme ratio by selective removal of the inactive forms of FADS2. Acetylation of inactive forms of FADS2 during HSC activation completes a KFERQ-like motif in FADS2 that allows its recognition by hsc70 and subsequent targeting to lysosomes for degradation.
Supplementary Material
Acknowledgements
We thank Jinghang Zhang at the Cell Sorting Core Facility, Daqian Sun at the Stem Cell Isolation and Xenotransplantation facility and experts at the Analytical Image Facilities at Einstein (NCI P30CA013330) for technical assistance with different experiments in this work. We are grateful to Dr. Amit Verma and Mr. Richard Elkind at Montefiore Medical Center for providing human hematopoietic stem cell specimen. This work was supported by grants from the National Institutes of Health AG021904, AG038072 (to AMC), AG031782 (to AMC and EG), DK105134 and CA230756 (to BW), R01HL146442, R01HL149714, R01HL148151 and R21HL150032 (to AD) and the generous support of the Rainwaters Foundation (to AMC and EG), the JPB foundation and of Robert and Renée Belfer. YRK is the recipient of a T32 Training in aging research grant (PI: Barzilai, AG023475).
Footnotes
Competing interest
AMC and EG are cofounders of Selphagy Therapeutics (Boston, MA, USA).
Supplementary information is available for this paper
Data availability
There are not restrictions on data availability in this manuscript. All the information is included in the manuscript. All Main and Extended Figures have associated raw data that is provided as an Excel worksheet organized by figures. Transcriptomic data was deposited in GEO with accession number GSE142632. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD021340.
References
- 1.Suda T, Suda J & Ogawa M Single-cell origin of mouse hemopoietic colonies expressing multiple lineages in variable combinations. Proceedings of the National Academy of Sciences of the United States of America 80, 6689–6693 (1983). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jordan CT & Lemischka IR Clonal and systemic analysis of long-term hematopoiesis in the mouse. Genes Dev 4, 220–232 (1990). [DOI] [PubMed] [Google Scholar]
- 3.Ito K et al. Self-renewal of a purified Tie2+ hematopoietic stem cell population relies on mitochondrial clearance. Science (New York, N.Y.) 354, 1156–1160 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cabezas-Wallscheid N et al. Identification of regulatory networks in HSCs and their immediate progeny via integrated proteome, transcriptome, and DNA methylome analysis. Cell Stem Cell 15, 507–522 (2014). [DOI] [PubMed] [Google Scholar]
- 5.Arai F & Suda T Maintenance of quiescent hematopoietic stem cells in the osteoblastic niche. Ann N Y Acad Sci 1106, 41–53 (2007). [DOI] [PubMed] [Google Scholar]
- 6.Wilson A et al. c-Myc controls the balance between hematopoietic stem cell self-renewal and differentiation. Genes Dev 18, 2747–2763 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cheng T et al. Hematopoietic stem cell quiescence maintained by p21cip1/waf1. Science (New York, N.Y.) 287, 1804–1808 (2000). [DOI] [PubMed] [Google Scholar]
- 8.Morrison SJ & Weissman IL The long-term repopulating subset of hematopoietic stem cells is deterministic and isolatable by phenotype. Immunity 1, 661–673 (1994). [DOI] [PubMed] [Google Scholar]
- 9.Kaushik S & Cuervo AM The coming of age of chaperone-mediated autophagy. Nature Reviews Molecular Cell Biology 19, 365–381 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baum CM, Weissman IL, Tsukamoto AS, Buckle AM & Peault B Isolation of a candidate human hematopoietic stem-cell population. Proceedings of the National Academy of Sciences of the United States of America 89, 2804–2808 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Spangrude GJ, Heimfeld S & Weissman IL Purification and characterization of mouse hematopoietic stem cells. Science (New York, N.Y.) 241, 58–62 (1988). [DOI] [PubMed] [Google Scholar]
- 12.McCulloch EA & Till JE The radiation sensitivity of normal mouse bone marrow cells, determined by quantitative marrow transplantation into irradiated mice. Radiat Res 13, 115–125 (1960). [PubMed] [Google Scholar]
- 13.Adams GB et al. Stem cell engraftment at the endosteal niche is specified by the calcium-sensing receptor. Nature 439, 599–603 (2006). [DOI] [PubMed] [Google Scholar]
- 14.Arai F et al. Tie2/angiopoietin-1 signaling regulates hematopoietic stem cell quiescence in the bone marrow niche. Cell 118, 149–161 (2004). [DOI] [PubMed] [Google Scholar]
- 15.Calvi LM et al. Osteoblastic cells regulate the haematopoietic stem cell niche. Nature 425, 841–846 (2003). [DOI] [PubMed] [Google Scholar]
- 16.Katayama Y et al. Signals from the sympathetic nervous system regulate hematopoietic stem cell egress from bone marrow. Cell 124, 407–421 (2006). [DOI] [PubMed] [Google Scholar]
- 17.Lacorazza HD et al. The transcription factor MEF/ELF4 regulates the quiescence of primitive hematopoietic cells. Cancer Cell 9, 175–187 (2006). [DOI] [PubMed] [Google Scholar]
- 18.Mortensen M et al. The autophagy protein Atg7 is essential for hematopoietic stem cell maintenance. J Exp Med 208, 455–467 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Signer RA, Magee JA, Salic A & Morrison SJ Haematopoietic stem cells require a highly regulated protein synthesis rate. Nature 509, 49–54 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Walter D et al. Exit from dormancy provokes DNA-damage-induced attrition in haematopoietic stem cells. Nature 520, 549–552 (2015). [DOI] [PubMed] [Google Scholar]
- 21.Bernitz JM, Kim HS, MacArthur B, Sieburg H & Moore K Hematopoietic Stem Cells Count and Remember Self-Renewal Divisions. Cell 167, 1296–1309.e1210 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dykstra B, Olthof S, Schreuder J, Ritsema M & de Haan G Clonal analysis reveals multiple functional defects of aged murine hematopoietic stem cells. J Exp Med 208, 2691–2703 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sudo K, Ema H, Morita Y & Nakauchi H Age-associated characteristics of murine hematopoietic stem cells. J Exp Med 192, 1273–1280 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Beerman I et al. Functionally distinct hematopoietic stem cells modulate hematopoietic lineage potential during aging by a mechanism of clonal expansion. Proceedings of the National Academy of Sciences of the United States of America 107, 5465–5470 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rossi DJ et al. Deficiencies in DNA damage repair limit the function of haematopoietic stem cells with age. Nature 447, 725–729 (2007). [DOI] [PubMed] [Google Scholar]
- 26.Yahata T et al. Accumulation of oxidative DNA damage restricts the self-renewal capacity of human hematopoietic stem cells. Blood 118, 2941–2950 (2011). [DOI] [PubMed] [Google Scholar]
- 27.Suda T, Takubo K & Semenza GL Metabolic regulation of hematopoietic stem cells in the hypoxic niche. Cell Stem Cell 9, 298–310 (2011). [DOI] [PubMed] [Google Scholar]
- 28.Mohrin M et al. Stem cell aging. A mitochondrial UPR-mediated metabolic checkpoint regulates hematopoietic stem cell aging. Science (New York, N.Y.) 347, 1374–1377 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Warr MR et al. FOXO3A directs a protective autophagy program in haematopoietic stem cells. Nature 494, 323–327 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xie SZ et al. Sphingolipid Modulation Activates Proteostasis Programs to Govern Human Hematopoietic Stem Cell Self-Renewal. Cell Stem Cell 25, 639–653.e637 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kirchner P et al. Proteome-wide analysis of chaperone-mediated autophagy targeting motifs. PLoS Biol 17, e3000301 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chiang H, Terlecky S, Plant C & Dice JF A role for a 70-kilodalton heat shock protein in lysosomal degradation of intracellular proteins. Science (New York, N.Y.) 246, 382–385 (1989). [DOI] [PubMed] [Google Scholar]
- 33.Cuervo AM & Dice JF A receptor for the selective uptake and degradation of proteins by lysosomes. Science (New York, N.Y.) 273, 501–503 (1996). [DOI] [PubMed] [Google Scholar]
- 34.Thompson LM et al. IKK phosphorylates Huntingtin and targets it for degradation by the proteasome and lysosome. The Journal of cell biology 187, 1083–1099 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lv L et al. Acetylation targets the M2 isoform of pyruvate kinase for degradation through chaperone-mediated autophagy and promotes tumor growth. Molecular cell 42, 719–730 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cuervo AM, Knecht E, Terlecky SR & Dice JF Activation of a selective pathway of lysosomal proteolysis in rat liver by prolonged starvation. The American journal of physiology 269, C1200–1208 (1995). [DOI] [PubMed] [Google Scholar]
- 37.Kiffin R, Christian C, Knecht E & Cuervo AM Activation of chaperone-mediated autophagy during oxidative stress. Molecular biology of the cell 15, 4829–4840 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rodriguez-Navarro JA et al. Inhibitory effect of dietary lipids on chaperone-mediated autophagy. Proceedings of the National Academy of Sciences of the United States of America 109, E705–714 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kaushik S & Cuervo AM Degradation of lipid droplet-associated proteins by chaperone-mediated autophagy facilitates lipolysis. Nature cell biology 17, 759–770 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schneider JL, Suh Y & Cuervo AM Deficient chaperone-mediated autophagy in liver leads to metabolic dysregulation. Cell Metab 20, 417–432 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT & Sulzer D Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science (New York, N.Y.) 305, 1292–1295 (2004). [DOI] [PubMed] [Google Scholar]
- 42.Park C, Suh Y & Cuervo AM Regulated degradation of Chk1 by chaperone-mediated autophagy in response to DNA damage. Nature communications 6, 6823 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Valdor R et al. Chaperone-mediated autophagy regulates T cell responses through targeted degradation of negative regulators of T cell activation. Nature immunology 15, 1046–1054 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gao L et al. Oxidation of survival factor MEF2D in neuronal death and Parkinson’s disease. Antioxidants & redox signaling 20, 2936–2948 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cuervo AM & Dice JF Age-related decline in chaperone-mediated autophagy. The Journal of biological chemistry 275, 31505–31513 (2000). [DOI] [PubMed] [Google Scholar]
- 46.Sooparb S, Price SR, Shaoguang J & Franch HA Suppression of chaperone-mediated autophagy in the renal cortex during acute diabetes mellitus. Kidney Int 65, 2135–2144 (2004). [DOI] [PubMed] [Google Scholar]
- 47.Kon M et al. Chaperone-Mediated Autophagy Is Required for Tumor Growth. Science Translational Medicine 3, 109ra117–109ra117 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dong S et al. A novel mouse model for spatio-temporal analysis of chaperone-mediated autophagy in vivo. Nat. Comm e-pub ahead of printing (2019). [Google Scholar]
- 49.Hennrich ML et al. Cell-specific proteome analyses of human bone marrow reveal molecular features of age-dependent functional decline. Nature communications 9, 4004 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Harrison DE & Lerner CP Most primitive hematopoietic stem cells are stimulated to cycle rapidly after treatment with 5-fluorouracil. Blood 78, 1237–1240 (1991). [PubMed] [Google Scholar]
- 51.Randall TD & Weissman IL Phenotypic and functional changes induced at the clonal level in hematopoietic stem cells after 5-fluorouracil treatment. Blood 89, 3596–3606 (1997). [PubMed] [Google Scholar]
- 52.Wilson A et al. Hematopoietic stem cells reversibly switch from dormancy to self-renewal during homeostasis and repair. Cell 135, 1118–1129 (2008). [DOI] [PubMed] [Google Scholar]
- 53.Ho TT et al. Autophagy maintains the metabolism and function of young and old stem cells. Nature 543, 205–210 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Simsek T et al. The distinct metabolic profile of hematopoietic stem cells reflects their location in a hypoxic niche. Cell Stem Cell 7, 380–390 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vagner M & Santigosa E Characterization and modulation of gene expression and enzymatic activity of delta-6 desaturase in teleosts: A review. Aquaculture 315, 131–143 (2011). [Google Scholar]
- 56.Geay F et al. Characteristics of fads2 gene expression and putative promoter in European sea bass (Dicentrarchus labrax): comparison with salmonid species and analysis of CpG methylation. Mar Genomics 5, 7–13 (2012). [DOI] [PubMed] [Google Scholar]
- 57.Yuan T et al. Linoleic acid induces red blood cells and hemoglobin damage via oxidative mechanism. Int J Clin Exp Pathol 8, 5044–5052 (2015). [PMC free article] [PubMed] [Google Scholar]
- 58.Fernanda Cury-Boaventura M, Cristine Kanunfre C, Gorjao R, Martins de Lima T & Curi R Mechanisms involved in Jurkat cell death induced by oleic and linoleic acids. Clin Nutr 25, 1004–1014 (2006). [DOI] [PubMed] [Google Scholar]
- 59.de Kok TM et al. Analysis of oxidative DNA damage after human dietary supplementation with linoleic acid. Food Chem Toxicol 41, 351–358 (2003). [DOI] [PubMed] [Google Scholar]
- 60.Zhang C & Cuervo AM Restoration of chaperone-mediated autophagy in aging liver improves cellular maintenance and hepatic function. Nat Med 14, 959–965 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Anguiano J et al. Chemical modulation of chaperone-mediated autophagy by retinoic acid derivatives. Nat Chem Biol 9, 374–382 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.D’Alessandro A et al. Donor sex, age and ethnicity impact stored red blood cell antioxidant metabolism through mechanisms in part explained by glucose 6-phosphate dehydrogenase levels and activity. Haematologica (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Akunuru S & Geiger H Aging, Clonality, and Rejuvenation of Hematopoietic Stem Cells. Trends in molecular medicine 22, 701–712 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Urao N & Ushio-Fukai M Redox regulation of stem/progenitor cells and bone marrow niche. Free Radic Biol Med 54, 26–39 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hamilton ML et al. Does oxidative damage to DNA increase with age? Proceedings of the National Academy of Sciences of the United States of America 98, 10469–10474 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Vander Heiden MG, Cantley LC & Thompson CB Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science (New York, N.Y.) 324, 1029–1033 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Riffelmacher T et al. Autophagy-Dependent Generation of Free Fatty Acids Is Critical for Normal Neutrophil Differentiation. Immunity 47, 466–480.e465 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Massey AC, Kaushik S, Sovak G, Kiffin R & Cuervo AM Consequences of the selective blockage of chaperone-mediated autophagy. Proc Nat Acad Sci USA 103, 5905–5910 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rodriguez-Muela N et al. Balance between autophagic pathways preserves retinal homeostasis. Aging cell 12, 478–488 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Xu Y et al. Chaperone-mediated autophagy regulates the pluripotency of embryonic stem cells. Science (New York, N.Y.) 369, 397–403 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tasset I et al. Anti-aging effects of systemic restoration of chaperone-mediated autophagy. (in preparation). [Google Scholar]
- 72.Kao YR et al. Thrombopoietin receptor-independent stimulation of hematopoietic stem cells by eltrombopag. Sci Transl Med 10 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wilkinson AC, Ishida R, Nakauchi H & Yamazaki S Long-term ex vivo expansion of mouse hematopoietic stem cells. Nature Protocols 15, 628–648 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dzieciatkowska M, Hill R & Hansen KC GeLC-MS/MS analysis of complex protein mixtures. Methods Mol Biol 1156, 53–66 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Das C, Lucia MS, Hansen KC & Tyler JK CBP/p300-mediated acetylation of histone H3 on lysine 56. Nature 459, 113–117 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Reisz JA, Zheng C, D’Alessandro A & Nemkov T Untargeted and Semi-targeted Lipid Analysis of Biological Samples Using Mass Spectrometry-Based Metabolomics. Methods Mol Biol 1978, 121–135 (2019). [DOI] [PubMed] [Google Scholar]
- 77.Nemkov T, Reisz JA, Gehrke S, Hansen KC & D’Alessandro A High-Throughput Metabolomics: Isocratic and Gradient Mass Spectrometry-Based Methods. Methods Mol Biol 1978, 13–26 (2019). [DOI] [PubMed] [Google Scholar]
- 78.Auteri JS, Okada A, Bochaki V & Dice JF Regulation of intracellular protein degradation in IMR- 90 human diploid fibroblasts. J Cell Physiol 115, 159–166 (1983). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
There are not restrictions on data availability in this manuscript. All the information is included in the manuscript. All Main and Extended Figures have associated raw data that is provided as an Excel worksheet organized by figures. Transcriptomic data was deposited in GEO with accession number GSE142632. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD021340.