

 This work is licensed under a
This work is licensed under a Abstract
Single-cell profiling of circulating tumor cells (CTCs) as part of a minimally invasive liquid biopsy presents an opportunity to characterize and monitor tumor heterogeneity and evolution in individual patients. In this study, we aimed to compare single-cell copy number variation (CNV) data with tissue and define the degree of intra- and inter-patient genomic heterogeneity. We performed next-generation sequencing (NGS) whole-genome CNV analysis of 125 single CTCs derived from seven patients with neuroendocrine neoplasms (NEN) alongside matched white blood cells (WBC), formalin-fixed paraffin-embedded (FFPE), and fresh frozen (FF) samples. CTC CNV profiling demonstrated recurrent chromosomal alterations in previously reported NEN copy number hotspots, including the prognostically relevant loss of chromosome 18. Unsupervised hierarchical clustering revealed CTCs with distinct clonal lineages as well as significant intra- and inter-patient genomic heterogeneity, including subclonal alterations not detectable by bulk analysis and previously unreported in NEN. Notably, we also demonstrated the presence of genomically distinct CTCs according to the enrichment strategy utilized (EpCAM-dependent vs size-based). This work has significant implications for the identification of therapeutic targets, tracking of evolutionary change, and the implementation of CTC-biomarkers in cancer.
Keywords: neuroendocrine tumors, circulating tumor cells, single cell, copy number variation, whole-genome sequencing
Background
The molecular characterization of tumors has advanced our understanding of the major somatic driver mutations and informed the development of targeted therapies, which have transformed outcomes in selected patient populations (Vogel et al. 2002, Sharma et al. 2007, Sosman et al. 2012). Whilst tissue biopsy remains central to diagnostic work-up, it is invasive, limited by the overall percentage of tumor cells, and subject to heterogeneity exhibited in primary and metastatic tumors (Navin et al. 2010, Gerlinger et al. 2012, Walter et al. 2018). Furthermore, bulk genomic analysis cannot provide resolution at the single-cell level, which is required to fully define the extent of tumor heterogeneity.
Technological advances in whole-genome amplification (WGA) and next-generation sequencing (NGS) methods now permit genomic analysis at the single-cell level and are uniquely placed to unravel complex clonal structures and track tumor evolution over time. Furthermore, characterization of single-circulating tumor cells (CTCs) as part of a minimally invasive 'liquid biopsy' provides an opportunity to explore tumor biology and identify therapeutic targets.
The first clinical applications of CTCs focused on enumeration using the EpCAM-dependent CellSearch® platform, which has been shown to be both prognostic and predictive across a wide range of epithelial malignancies (Cristofanilli et al. 2004, Cohen et al. 2008, de Bono et al. 2008, Krebs et al. 2011, Poveda et al. 2011), including neuroendocrine neoplasms (Khan et al. 2011, 2013, 2016, Mandair et al. 2021). More recently, molecular analysis of single CTCs has been used to identify predictive biomarkers, such as the T790M resistance allele in NSCLC (Maheswaran et al. 2008). In SCLC, a pretreatment CTC-based biomarker has been shown to predict sensitivity to first-line chemotherapy (Carter et al. 2017).
Neuroendocrine neoplasms (NEN) represent a heterogeneous disease entity with diverse histology, clinical features, and prognosis (Dasari et al. 2017). They are characterized by a low mutational burden (Banck et al. 2013), but recurrent patterns of copy number variation (CNV) have been observed (Kulke et al. 2008, Cunningham et al. 2011, Capurso et al. 2012). CNVs affect a greater portion of the cancer genome than any other somatic genetic alteration (Heitzer et al. 2016), and CNV burden is prognostic for cancer-free and overall survival in multiple tumor types (Hieronymus et al. 2018) including NEN, where aneuploidy can be used to define distinct molecular subgroups of prognostic relevance (Karpathakis et al. 2016).
In this study, we perform CNV analysis of single NEN CTCs, aiming to define the extent of genomic heterogeneity both within and between patients and to compare single-cell CTC data with bulk tissue analysis. CTC enrichment in NEN patients has to date been confined to EpCAM-dependent methodologies, which may fail to capture the full diversity of CTCs seen in this disease (Gorges et al. 2012). Here, we utilize both the EpCAM-based CellSearch and epitope-independent Parsortix® systems in order to interrogate the full diversity of cells at the CNV level and investigate whether single-cell CTCs may differ at a genomic level, according to EpCAM expression.
Methods
Patients
NEN patients were recruited at the Royal Free Hospital, London, between September 2014 and February 2018. The study was approved by the Local Ethics Committee (NRES Committee London – Bromley, IRAS ref 13/LO/0376), and all participants were required to provide written informed consent. Eligible patients had a histologically confirmed diagnosis of metastatic NEN in the absence of any other active malignancy. Tumors were graded according to the European Neuroendocrine Tumor Society (ENETS) guidelines (Bosman et al. 2010).
CTC enrichment using CellSearch
Peripheral blood samples (7.5 mL) were collected into CellSave tubes (Veridex LLC), stored at room temperature, and processed within 96 h using the Celltracks Autoprep and Analyzer II platform for the semi-automated staining, enrichment, and the enumeration of CTCs as previously described (Cristofanilli et al. 2005, Riethdorf et al. 2007). CTCs were defined as cells with a DAPI positive nucleus and positive EpCAM and cytokeratin expression in the absence of CD45 staining. All evaluations regarding enumeration of CTCs were made by two independent operators without the knowledge of patient pathology. Enriched samples were re-suspended, aspirated from the CellSearch cartridge, and stored at −20°C in 50% glycerol.
CTC enrichment using Parsortix
Blood was collected in Streck tubes (10 mL) and incubated for 24–48 h prior to size-based enrichment with the Parsortix platform (ANGLE) using software protocols provided by the manufacturer. Following enrichment, samples were harvested in a total volume of 1.2 mL of HBS by applying a reverse flow to the separation cassette. Enriched samples were re-suspended in 200 μL of autoMACS running buffer and fixed and stained for further processing on a sterile transwell polycarbonate membrane insert placed within a 50mL Falcon tube. BSA of 3% (200 μL) was pipetted to entirely cover its surface for a 10 min incubation. The 50 mL tube was centrifuged at 500 g for 2 min to elute the BSA solution from the filter prior to transferring the enriched patient sample onto the insert surface. One hundred microliter of a 10% CD45 staining solution (10 μL anti-CD45-APC (Miltenyi Biotec) and 90 μL of running buffer) and 100 μL of a 10% CK staining solution (10 μL anti-CK-PE (Abcam) and 90 μL Inside Perm (Miltenyi Biotec)) were used to sequentially stain samples for CD45 and cytokeratin prior to staining for nuclear content using 100 μL of a 0.001 mg/mL solution of Hoechst 33342 (Sigma–Aldrich). After washing with SB115 buffer, the cell suspension was transferred into a sterile 1.5 mL tube prior to volume reduction and loading into the DEPArray™ cartridge.
Cell isolation from FFPE
FFPE tissue sections of 40–60 μm thickness were dissociated into single-cell suspensions and stained as previously described (Bolognesi et al. 2016). To enable visualization and identification of cells using the DEPArray, cytokeratin and vimentin were used as tumor and stromal cell markers, respectively. Cell suspensions were stained with anti-cytokeratin MNF116 (IgG1) (DAKO), anti-cytokeratin AE1/AE3 (IgG1) (Millipore–Chemicon), and anti-Vimentin 3B4 (IgG2A) (DAKO).
Dissociated FFPE samples were subjected to a DNA quality-control assay using the DEPArray FFPE QC kit (Silicon Biosystems). Each sample was given a QC score between 0 and 1 based on a qPCR-based assay. Samples with a sufficiently high DNA quality as determined by a QC score ≥ of 0.4 according to manufacturer’s guidelines were processed on the DEPArray platform for retrieval of single tumor cells.
Cell isolation from fresh tissue
Fresh tissue samples were collected in RPMI 1640 medium (Gibco) and processed within 3 h of collection. The tumor sample was placed in 1mL of dissociation solution (240 μL collagenases, 150 μL DNAse, and 13.85 mL of RPMI media) and processed in a gentleMacs dissociator for one cycle, followed by two consecutive 30 min incubations at 37°C. Single-cell suspensions were created using a 50 μL cell strainer and centrifuged and re-suspended in 5mL of RPMI prior to re-suspending in 1mL of freezing medium (10% DMSO in FBS) for storage at −80°C. Samples were fixed with 2% paraformaldehyde (Fluka) for 20 min at room temperature prior to staining for cytokeratin, vimentin, and DAPI performed as per FFPE samples.
DEPArray sorting and recovery
Both CellSearch- and Parsortix-enriched samples were imaged and sorted using the DEPArray system (Silicon Biosystems) as per the manufacturer’s instructions (Abonnenc et al. 2013). Image-based selection was used to identify and recover individual cells of interest as either single cells or pools of cells, based on their morphological features, DNA content, and fluorescence labeling; CTCs (CK-PE+/CD45-APC−/DAPI+) and white blood cells (WBC) (CK-PE−/CD45-APC+/DAPI+).
For analysis of FFPE samples with the DEPArray, between 5000 and 10000 stained cells were loaded into the cartridge, and cell sorting was executed according to DEPArray User’s Manual rev 1.1_sw 2.1.1. The cytokeratin+ vimentin- tumor cell population and cytokeratin-vimentin+ stromal cell population were gated separately to evaluate morphology and staining characteristics prior to selecting cells for recovery.
Whole-genome amplification of single-cell DNA and quality-control assay
WGA was performed on all recovered single-cells using the Ampli1™ WGA kit version 02 (Silicon Biosystems) as per the manufacturer’s instructions to generate a 50 μL WGA product. For single cells derived from blood (CTCs and WBC) and fresh tissue (tumor and stromal cells), the quality of the WGA product was determined using the Ampli1 QC Kit (Silicon Biosystems). A genomic integrity index (GII) was allocated for each sample, scored from 0 to 4. Only single cells with sufficiently good quality DNA as determined by a GII ≥ 2 were selected for downstream analysis.
Nucleic acid extraction
For bulk sequencing, DNA was extracted from 5 to 10 sections of 10 μm thickness from three FFPE blocks using the DNAstorm FFPE DNA Isolation Kit (CELLDATA) following the manufacturer's instructions. DNA was eluted into 75 μL of nuclease-free water and concentrations were measured using the NanoDrop-1000 Spectrophotometer (NanoDrop) and Qubit 2.0 Fluorometer (Invitrogen). Hematoxylin and eosin-stained sections were evaluated to ensure >80% purity of tumor specimens prior to processing.
Lowpass whole-genome sequencing and bioinformatics
Ampli1 LowPass kit for Illumina (Menarini Silicon Biosystems) was used for preparing low-pass whole-genome sequencing (WGS) libraries from single cells. For high-throughput processing, the manufacturer’s procedure was implemented in a fully automated workflow on a STARlet Liquid Handling Robot (Hamilton). Ampli1 LowPass libraries were normalized and sequenced by HiSeq 2500 instrument using 150 SR rapid-run mode. The obtained FASTQ files were aligned to the hg19 human reference sequence using Burrows–Wheeler Aligner version 0.7.12 (BWA). Copy number alterations in the data were identified using Control-FREEC software (version 11.0).
For bulk analysis of FFPE samples, genomic DNA was quantified using Qubit 3 fluorometer with dsDNA BR kit according to the manufacturer’s instructions. One microgram of genomic DNA was used to prepare whole-genome sequencing libraries using Nonacus Cell 3 Target: Library Preparation kit. Library preparation was done according to the manufacturer’s instructions. Enzymatic fragmentation was performed at 32°C for 14 min to obtain library fragments with an average size of 250 bp followed by ligation of UMI Adapters on both ends of the 5’-phosphorylated/3’-dA-tailed DNA fragments. Libraries were purified using Target Pure NGS clean-up beads, and minimal PCR amplification was carried out using four cycles of amplification. Libraries were quantified using Qubit 3 fluorometer with dsDNA BR kit and run on an Agilent Bioanalyzer DNA 1000 chip according to the manufacturer’s instructions. Average library fragment length was determined from the bioanalyzer trace. Library molar concentration was determined based on the average fragment size and the Qubit concentration. All libraries were normalized to 10 nM working concentration and pooled. The dual-indexed library pool was sequenced on Illumina Nextseq 500/550 platform to generate paired-end reads. The Nonacus Cell 3 Target: Library preparation protocol adds unique molecular identifiers (UMIs) to the sequencing libraries which were sequenced by additional nine cycles of sequencing added on to the i7 index read.
Bulk sequencing data were processed with the nextflow Sarek v2.3.FIX1 pipeline (https://github.com/UCL-BLIC/Sarek_v2.3.FIX1) following GATK best practices. Specifically, reads were aligned against hg38 with BWA v0.7.17, duplicated reads were marked, and reads were recalibrated with GATK v4.1.1.0. CNV profiles were obtained by running Control-FREEC v11.5 with WGS recommended parameters.
Statistics
All statistical analyses were performed in R. Pairwise Manhattan distances were calculated for all samples, using only copy number bins that were not NA for each pair. Hierarchical clustering of copy number profiles using these distances was performed with Ward’s minimum variance method.
When comparing bulk and CTC copy number profiles, the mean copy number across CTC copy number bins that overlapped a bulk bin was taken. Any bulk bin without an overlapping CTC bin was not given a copy number designation.
t-Distributed stochastic neighbor embedding (TSNE) analysis was performed using the R package Rtsne, using only the genomic bins that were non-missing for all samples analyzed, with a perplexity of 30.
Correlations between copy number profiles were calculated with respect to a base copy number of 2, as described in Gao et al. (2017):
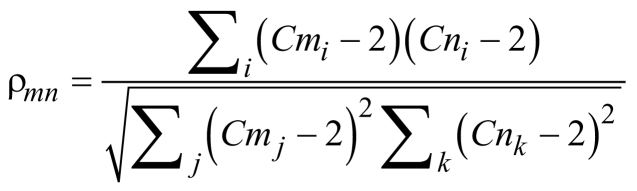
where ρmn is the correlation between samples m and n, while Cmi is the copy number for sample m at bin i.
To account for differences in ploidy, correlation was also calculated with respect to the average copy number across all bins for each sample:
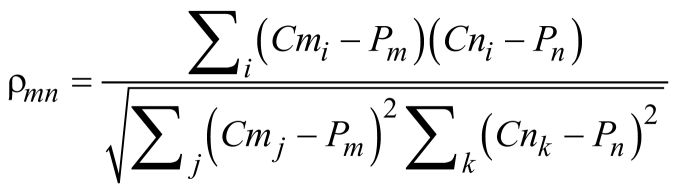
where Pm is the mean copy number for sample m across all bins.
Metrics chosen to investigate copy number dynamics within a sample were the proportion of genome altered (number of CN! = 2 bins divided by the total number of bins) and Shannon’s diversity index, 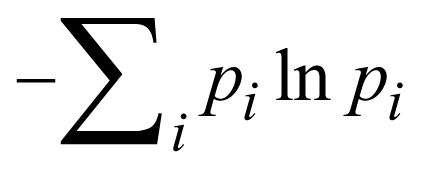 , where pi is the proportion of copy number bins with copy number state i, that is, CN = 2. Tests for statistical differences between distributions for these metrics were performed using the Kolgomorov–Smirnov test.
, where pi is the proportion of copy number bins with copy number state i, that is, CN = 2. Tests for statistical differences between distributions for these metrics were performed using the Kolgomorov–Smirnov test.
Copy number gains and losses were defined in relation to ploidy. Gains were defined as log2(CN/ploidy) > 0.9, while losses were defined as log2(CN/ploidy) < −0.9. The proportion of cells with a loss at a given genomic bin was used as a metric for a single patient. When combining multiple patients, the mean proportion of cells across all patients considered was used. A threshold for statistically significant recurrent gain or loss was determined by bootstrapping the original copy number data; for each patient, copy number states were sampled with replacement from every copy number state seen in the original data for that patient, this was performed for the same number of cells as were originally profiled for that patient. Gains and losses were defined as previously, and the proportion of simulated cells with a gain/loss at each genomic bin was calculated. This was repeated 1000 times per patient, and the threshold for determining recurrent gains/losses was set as 99.9th percentile value across all genomic bins for gains or losses separately. For a threshold where multiple patients are being considered, the same bootstrapping was performed for each patient, but the threshold was determined as the 99.9th percentile of the mean proportion of cells with gain/loss across the patients being evaluated.
Results
Patient characteristics and sample collection
Seven NEN patients were included with primary tumor sites comprising the small intestine (SINET) (n = 4), pancreas (n = 1), gastro-esophageal junction (GOJ) (n = 1), and kidney (n = 1). All patients had peripheral blood samples taken for CTC enrichment using the EpCAM-dependent CellSearch platform, and three patients had concomitant samples enriched using the size-based Parsortix device (Fig. 1). Blood samples were taken from new patients at the time of the first presentation to our clinic (patients 1, 3, 5, 6) or at the time of disease progression prior to commencing systemic therapy (patients 2, 4, 7). Matched WBC were analyzed as negative controls. A total of seven tissue samples (6 FFPE, 1 FF) from six patients were analyzed. Of the seven samples, four were primary tumor samples (3 small intestine, 1 GOJ) and three were metastatic sites (2 liver, 1 brain). One patient (patient 1) had no available tissue for analysis. The clinical and treatment characteristics as well as the samples analyzed per patient are summarized in Table 1.
Figure 1.

Experimental design of the study. Workflow used in the study to enrich for CTCs and CNV profiling using Ampli1 WGA and LowPass kit for Illumina. Following enrichment (EpCAM-dependent vs size-based platforms), single NEN CTCs and matched WBC are selectively recovered in dynamically controlled dielectrophoretic cages using the DEPArray Image-Assisted Digital Cell Sorter. CTC samples undergo WGA and QC prior to low-resolution whole-genome sequencing for CNV profiling. Where surgical resection or biopsy specimens are available, samples are processed for bulk LPWGS and single-cell LPWGS as per CTCs.
Table 1.
Summary of clinical characteristics.
| Patient ID | Sex | Age | Primary site | Grade | Treatment | CellSearch CTCs | Parsortix CTCs | WBC | Fresh tissue single cells | FFPE single cells | FFPE bulk samples |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | M | 74 | Small intestine | 2 | na | 4 | na | 4 | na | na | na |
| 2 | F | 45 | Small intestine | 3 | PEN-221 | 15 | 5 | na | na | 2 | Pituitary metastasis |
| 3 | F | 69 | Small intestine | 2 | na | 7 | 8 | na | 8 | na | Small bowel |
| 4 | M | 65 | Small intestine | 1 | SSA | 18 | na | na | na | na | Small bowel |
| 5 | M | 47 | Pancreas | 2 | na | 12 | na | 4 | na | na | na |
| 6 | M | 64 | GOJ | 3 | na | 11 | na | na | na | 1 | na |
| 7 | F | 33 | Renal | 2 | PEN-221 | 21 | 11 | 10 | na | 2 | na |
All tissue samples are FFPE unless specifically indicated otherwise.
F, female; FFPE, formalin-fixed paraffin-embedded; GOJ, gastro-esophageal junction; M, male; na, not applicable; SSA, somatostatin analogs; PEN-221, novel antibody-drug conjugate.
CTC sequencing
In total, 125 single CTCs were isolated from seven patients and successfully subjected to the whole-genome amplification (WGA), quality-control PCR, and low-pass whole-genome sequencing (LPWGS). Single CTCs displayed high-quality metrics, with only 3.5% failing to pass the quality checks for single-cell CNV. As a control, 17 single WBC (CD45 positive cells) were isolated and subjected to the same procedures. CD45 positive cells showed balanced copy number profiles (Supplementary Fig. 1, see section on supplementary materials given at the end of this article) whereas CTCs showed multiple gains and losses (Figs 2 and 3), confirming the aberrant nature of these tumor cells and the uniformity of single-cell WGA with the Ampli1 kit. The sensitivity and specificity of CTC identification and recovery by the DEPArray were assessed across all single cells subjected to LPWGS. Cells with CNV profiles demonstrating an overabundance of substantial chromosomal gains and losses were considered CTC, whilst cells demonstrating flat profiles were classified as WBC (Ferrarini et al. 2018, Mangano et al. 2019). Using CNV profiles as the ultimate classifier of cell status, DEPArray selection had a positive predictive value of 95% and a negative predictive value of 100% (P < 0.0001).
Figure 2.

Comparison of low-resolution whole-genome copy number profiles for CTCs and bulk tissue reveals reproduction of the majority of the CNV from the formalin-fixed paraffin-embedded (FFPE) and fresh frozen (FF) tissue in CTC samples. Unsupervised hierarchical clustering heat map of each analyzed individual CTC and tissue sample based on CNV from three SINET patients. Each patient is depicted with one color as shown on the phenobar at the bottom of the heat map. Individual CTCs are categorized according to enrichment method and tissue into bulk vs single-cell FFPE (see key). Chromosomal CNV is shown from top to bottom for each individual cell or sample; copy number gains are depicted in blue, losses in orange.
Figure 3.

Individual CTC CNV data depicting complex intrapatient and interpatient genomic diversity. Unsupervised hierarchical clustering heat map of all analyzed CTCs based on CNV across seven patients. Each patient is depicted with one color as shown on the phenobar at the bottom of the heat map along with the cell sorting method and primary NET site.
Single tumor cells derived from FFPE surgical specimens/biopsies were also subjected to the same procedures as CTCs. DNA quality of single-cell suspensions was assessed using the Ampli1 QC Kit (Silicon Biosystems) prior to cell sorting. Four of the seven samples had QC values ≥ 0.4 indicating a sufficient DNA quality for single-cell CNV analysis, and eight to ten single cells from each sample were processed for CNV analysis. The majority of single tumor cells had high derivative log ratio spread values in keeping with low library quality and only 15% of recovered single cells yielded sufficient quality results for CNV analysis.
CTC vs tumor tissue CNV profiles
For the three patients with sufficient matched FFPE tissue available for bulk analysis, whole-genome CNV profiles were compared between CTCs and bulk FFPE samples (Fig. 2). The CNVs demonstrated in bulk tissue analysis were predominantly losses and these were also detectable in most CTCs. For example, in patient 2, losses in chromosomes 6, 9, and 18 are seen in bulk tissue and in 25, 80, and 65% of CTCs respectively, while patient 3, losses in chromosome 16 were observed in bulk tissue and 100% of CTCs (Fig. 2). The majority of these concordant genomic losses are located in regions of the genome previously described as altered in NENs, with loss of chromosomes 9 and 18 reported in 20% and 60–78% of SINETS, respectively. However, single CTC data demonstrated the presence of clones enriched in additional somatic copy number alterations not detectable at the bulk level, including the presence of a subclone of cells with evidence of whole-genome doubling, observed in patients 2 (10% of CTCs) and 4 (6%). These reproducible CNV patterns were not evident in bulk sequencing analysis and only detectable due to the resolution afforded by single-cell sequencing. Such subclonal copy number alterations were most pronounced in patient 4, where appreciable CNV gains or losses were only detectable at the single-cell level and not in the bulk tissue.
In patient 3, single tumor cells derived from a fresh frozen (FF) liver biopsy exhibited identical copy number profiles as CTCs and unsupervised hierarchical clustering of CTC and tumor copy number profiles demonstrated clustering of these cells together.
CTC analysis reveals significant inter-and intrapatient heterogeneity
To fully explore inter- and intrapatient CNV heterogeneity in NEN patients, the full set of 125 single CTCs from seven patient samples were further interrogated (Fig. 3). Copy number losses were seen more frequently than amplifications; however, whole-genome doubling was detected in all CTCs derived from two patients (patients 1 and 6). Despite the preponderance of losses, the CNV patterns of individual patients are dissimilar and this remains the case when considering only those patients of the small intestinal primary site (patients 1–4). These patient-specific patterns of CNV were confirmed using t-distributed stochastic neighbor embedding (TSNE; Fig. 4), which demonstrated clear clustering of individual patients, with no segregation according to the primary site. Conversely, all WBC clustered together regardless of patient of origin in keeping with their flat CNV profiles (Fig. 4B).
Figure 4.

Relationship between CTCs from all seven NEN patients is revealed through TSNE analysis. (A) Single CTCs from all seven patients are visualized and can be identified by color in the phenobar at the top of the figure. Cells are also depicted according to enrichment strategy (see key). (B) TSNE of all analyzed CTCs and WBC.
Within individual patients, there were observations of clonal CN alterations seen in 100% of CTCs, but also clear evidence of subclonal changes and individual cells with unique CNV profiles indicative of divergent evolution (Fig. 3). This intrapatient heterogeneity was only detectable at the single-cell level. The degree of intrapatient heterogeneity varied according to patient, with patients 3 and 6 demonstrating the highest average pairwise correlation of CTC CNV profiles, and hence the most homogenous copy number landscape across CTCs (Supplementary Fig. 2). However, the correlation of CNV profiles within patients remains higher than that observed between patients, underscoring the independent nature of CNV profiles originating in different patients, and the shared evolutionary history of CTCs, and thus CTC CNV profiles, within individual patients.
CNV profiles vary according to enrichment strategy
In patient 7, hierarchical clustering of CNV profiles demonstrated distinct clustering of CTCs enriched by the EpCAM-dependent CellSearch as compared to the epitope-independent, size-based Parsortix platform (Fig. 3). This is also demonstrated in Fig. 4 where Parsortix and CellSearch CTCs from patient 7 form largely separate groups. To investigate this further, we summarized single CTC profiles via two metrics; the proportion of the genome that is aberrant (copy number other than 2), and copy number diversity as enumerated by Shannon’s diversity index, and compared these metrics across cells according to the enrichment strategy utilized. There was a statistically significant difference in the distribution of both metrics between different enrichment strategies within patient 7 (Kolgomorov–Smirnov test, P < 0.01, Fig. 5A), where Parsortix CTCs demonstrate a larger range in both metrics as compared to CellSearch CTCs, indicating greater cell-to-cell variation. Interestingly, the difference seen in patient 7 was not found to be statistically significant across all patients (Fig. 5B), indicating that these differences may vary on a patient-to-patient basis. This data suggests that restricting the analysis of CTCs to only those that express EpCAM may exclude subsets of tumor cells that could be clinically relevant.
Figure 5.

Distribution plot describing the impact of enrichment strategy in patient 7 (A) and all patients (B) on the proportion of the genome that is aberrant and CNV diversity as quantified by Shannon Index. Each small line represents the described value for a single CTC. Large bars represent mean values.
CTC molecular characterization
In order to evaluate the clinical application of CTC CNV profiling as a surrogate for tissue biopsy, we interrogated CTC CNV profiles for prognostic or actionable copy number changes described in the NEN literature. Evaluation of the frequency of copy number amplifications and deletions within CTC CNV profiles from SINET patients revealed recurrent losses of chromosomes 9, 13q, 16q, and 18q (Fig. 6A). These have previously been described in SINETs supporting the technical reliability of our data and the potential use of CTCs as a tissue surrogate (Kulke et al. 2008, Banck et al. 2013, Hashemi et al. 2013, Karpathakis et al. 2016, Di Domenico et al. 2017). Of particular note is chromosome 18, loss of which is the most frequently reported genomic event in SINET, occurring in 60–78% of tumors and is of prognostic relevance (Karpathakis et al. 2016). Previously unreported alterations, including loss of chromosomes 2p and 7q22, were also identified. Although not reported in SINET, allelic losses in chromosome 2p are reported in colorectal, lung, and endometrial malignancies. The tumor suppressor gene CUX1 is located at chromosome 7q22, knockdown of which causes increased PI3K signaling and AKT phosphorylation (Ramdzan & Nepveu 2014). This may be relevant in this patient population as deregulation of the PI3K/Akt/mTOR pathway is well-established in NEN, supported by the clinical efficacy of the mTOR inhibitor everolimus (Pavel et al. 2011, Yao et al. 2011).
Figure 6.

Frequency of genomic amplifications and deletions across all CTCs. Profiles demonstrated for SINET patients (A), patient 5; pancreatic NEN (B), patient 6; GOJ NEN (C), and patient 7; renal NEN (D).
Whole chromosome and arm gains at chromosome 4 have previously been described in SINET. We did not observe such large-scale gains, instead, we observed focal gains in the TEC gene on chromosome 4p12, which encodes a protein belonging to the Tec family of non-receptor protein-tyrosine kinases involved in the T-lymphocyte activation pathway and implicated in myelodysplastic syndrome.
CTCs from patient 7 (renal NET) demonstrated recurrent chromosomal alterations of likely clinical significance. Loss of chromosome 3p was observed in a high proportion of CTCs and harbors several tumor suppressor genes including the VHL gene at 3p25. Loss of heterozygosity (LOH) of 3p has been reported in the limited renal NET sequencing data available and is also found in over 90% of clear cell renal carcinoma (el-Naggar et al. 1995, Alimov et al. 2000). Loss of chromosomes 10q and 13q was also observed, the former of which encodes the tumor suppressor gene PTEN and is of prognostic relevance in renal cell carcinoma (Velickovic et al. 2002). Finally, as with SINET, chromosome 16q loss was frequently identified across patient 7 CTCs. Deletion of 16q is demonstrated across multiple malignancies, and LOH has been indicated as an early event in the development of breast and hepatocellular cancer with possible prognostic implications (Sakai et al. 1992, Hansen et al. 1998).
Discussion
Copy number analysis of NEN CTCs confirmed a wide range of genomic aberrations making them readily distinguishable from WBC. All cells classified as WBC using the pre-determined DEPArray criteria demonstrated balanced copy number profiles, confirming the specificity and reproducibility of these criteria and accuracy of DEPArray sorting.
In this study, we show for the first time that somatic CNVs of NEN CTCs mirror those seen in FFPE tissue, validating these CTC enrichment and isolation technologies in NEN and confirming their potential use as a surrogate for tissue biopsy. The clinical applications of this finding have been demonstrated in other tumor types such as NSCLC, where good concordance between ALK-rearranged CTCs and ALK-positive tumor biopsies has been demonstrated (Pailler et al. 2013). This finding is particularly relevant in tumor types where tissue biopsy is not readily available or as in NEN, where the relatively good prognosis of patients with low-grade disease means surgical specimens or biopsies may have been taken several years previously and, therefore, not be representative of the current genomic landscape of the disease after multiple lines of systemic therapy. CTCs have the additional benefit of being non-invasive and, therefore, easily repeatable, thus allowing the monitoring of genomic change in real-time. Beyond this, serial CTC monitoring may also enable the detection of mechanisms of resistance (Pailler et al. 2015). Importantly, subclonal CNVs not discernible in bulk tissue analysis were detectable in single CTC samples thus allowing the identification of intrapatient genomic heterogeneity.
Unsupervised hierarchical clustering identified intrapatient genomic heterogeneity in NEN patients, with diverse single CTC CNV traces observed in some patients. The intrapatient CNV heterogeneity demonstrated in this study has also been observed in other tumor types such as prostate (Dago et al. 2014, Lambros et al. 2018) and colorectal (Heitzer et al. 2013) cancer. This is in contrast to lung adenocarcinoma, SCLC, breast, and gastric cancer, where more homogeneous CNVs have been observed in CTCs from individual patients (Ni et al. 2013, Heidary et al. 2014, Gao et al. 2017). Intrapatient heterogeneity is of clinical relevance as it may impact prognosis, response to treatment, and biomarker development. High intratumoral heterogeneity in tissue samples has been associated with a worse overall survival across different tumor types (Seol et al. 2012, Mroz et al. 2013). This relationship has not yet been examined with regards to the genomic profiling of CTCs, but low phenotypic diversity of prostate cancer CTCs has been shown to correlate with improved OS in patients treated with androgen receptor signaling inhibitors (ARSI), whereas high heterogeneity was associated with increased risk of death on ARSI relative to taxanes. Considerable heterogeneity was also demonstrated in CNV patterns between patients. This appears to be cancer-type specific. Ni et al. observed almost identical global CNV patterns in five different patients with lung adenocarcinoma with 78% of the gain and loss regions shared between any two patients (Ni et al. 2013), and similar findings have been reported in gastric cancer (Gao et al. 2017). However, increased inter-patient heterogeneity is seen in other tumor types, such as SCLC and breast cancer (Ni et al. 2013, Gao et al. 2017). The inter-patient heterogeneity in CNV profiles demonstrated in this study persists even when the analysis is confined to those patients with small intestinal primaries.
Epitope-dependent enrichment technologies such as the CellSearch platform limit recovery of CTCs to an EpCAM-positive subpopulation. In this study, we performed the first direct comparison of CTC CNV profiles using identical blood draws between the epitope-independent size-based Parsortix and EpCAM-based CellSearch. In patient 7, CTCs enriched using the CellSearch platform demonstrate reproducible CNV with high inter-cell concordance. However, CTCs enriched using Parsortix appear genomically distinct, lacking the conserved CNV demonstrated in CellSearch CTCs and displaying a wider range of inter-cell heterogeneity. Different methods of enrichment may, therefore, impact the results of single-cell genomic analysis and have implications for serial monitoring of CNV profiles. This finding is clinically significant as it may impact biomarker development. For example, a CNV-based classifier of CTCs has been shown to predict chemosensitivity in SCLC patients (Carter et al. 2017). In that study, all CTCs were enriched using CellSearch, and the classifier was less effective in those patients demonstrating intrapatient heterogeneity. The data presented in our study suggest that the efficacy of CNV-based classifiers such as this may be affected by the form of enrichment used and could not be directly extrapolated to CTCs enriched using alternative technologies. Furthermore, it suggests combining epitope-independent enrichment strategies with CellSearch may allow sampling of a wider population of CTCs with greater potential to fully capture CTC diversity.
SINET are characterized by a low mutational burden, with the most frequent mutation occurring in the cell cycle regulator CDKN1B (cyclin-dependent kinase inhibitor 1B) in only 8% of tumors (Francis et al. 2013, Crona et al. 2015). In this study, we identify recurrent loss of chromosome 18, the most common genomic event in SINET and predictive of PFS in SINET. Karpathakis et al. have previously demonstrated that CNV analysis of SINET primary tissue can be used to divide patients into three molecular subtypes with significant impact on PFS (Karpathakis et al. 2016). We also demonstrated novel and potentially targetable alterations such as focal gains in chromosome 4p12, which encodes the TEC gene (Yu & Smith 2011). Further work is required to validate this finding in a larger cohort of patients.
Despite the novel findings reported, we acknowledge some limitations; namely, the relatively low number of patients involved, as well as their heterogeneity in terms of grade and primary site. However, limiting the analysis to a smaller patient cohort allowed assessment of multiple CTCs per patient in order to better characterize intrapatient heterogeneity, whilst the overall large number of single cells analyzed allowed comparison with bulk tissue data and of cell enrichment techniques at the molecular level.
In conclusion, this is the first study to demonstrate that CNV analysis of single CTCs in NEN patients is feasible. We have demonstrated significant intra- and inter-patient genomic heterogeneity undetected by bulk tissue analysis. Additionally, we demonstrate for the first time, the presence of genomically distinct CTCs according to the enrichment strategy utilized, which has implications for the study of CTCs across all tumor types.
Supplementary Material
Declaration of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
Funding
This work was supported by the CRUK & EPSRC Comprehensive Cancer Imaging Center, European Neuroendocrine Tumor Society Fellowship, University College London (UCL) CRUK, and NIHR Experimental Cancer Medicine Center Grant No. C12125/A15576, MRC award MR/M009033/1 and the UCL Hospitals NIHR Biomedical Research Center.
References
- Abonnenc M, Manaresi N, Borgatti M, Medoro G, Fabbri E, Romani A, Altomare L, Tartagni M, Rizzo R, Baricordi Oet al. 2013Programmable interactions of functionalized single bioparticles in a dielectrophoresis-based microarray chip. Analytical Chemistry 858219–8224. ( 10.1021/ac401296m) [DOI] [PubMed] [Google Scholar]
- Alimov A, Kost-Alimova M, Liu J, Li C, Bergerheim U, Imreh S, Klein G, Zabarovsky ER.2000Combined LOH/CGH analysis proves the existence of interstitial 3p deletions in renal cell carcinoma. Oncogene 191392–1399. ( 10.1038/sj.onc.1203449) [DOI] [PubMed] [Google Scholar]
- Banck MS, Kanwar R, Kulkarni AA, Boora GK, Metge F, Kipp BR, Zhang L, Thorland EC, Minn KT, Tentu Ret al. 2013The genomic landscape of small intestine neuroendocrine tumors. Journal of Clinical Investigation 1232502–2508. ( 10.1172/JCI67963) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolognesi C, Forcato C, Buson G, Fontana F, Mangano C, Doffini A, Sero V, Lanzellotto R, Signorini G, Calanca Aet al. 2016Digital sorting of pure cell populations enables unambiguous genetic analysis of heterogeneous formalin-fixed paraffin-embedded tumors by next generation sequencing. Scientific Reports 620944. ( 10.1038/srep20944) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosman FT, Carneiro F, Hruban RH, Theise ND.2010WHO Classification of Tumours. IARC. [Google Scholar]
- Capurso G, Festa S, Valente R, Piciucchi M, Panzuto F, Jensen RT, Dellefave G.2012Molecular pathology and genetics of pancreatic endocrine tumours. Journal of Molecular Endocrinology 49R37–R50. ( 10.1530/JME-12-0069) [DOI] [PubMed] [Google Scholar]
- Carter L, Rothwell DG, Mesquita B, Smowton C, Leong HS, Fernandez-Gutierrez F, Li Y, Burt DJ, Antonello J, Morrow CJet al. 2017Molecular analysis of circulating tumor cells identifies distinct copy-number profiles in patients with chemosensitive and chemorefractory small-cell lung cancer. Nature Medicine 23114–119. ( 10.1038/nm.4239) [DOI] [PubMed] [Google Scholar]
- Cohen SJ, Punt CJ, Iannotti N, Saidman BH, Sabbath KD, Gabrail NY, Picus J, Morse M, Mitchell E, Miller MCet al. 2008Relationship of circulating tumor cells to tumor response, progression-free survival, and overall survival in patients with metastatic colorectal cancer. Journal of Clinical Oncology 263213–3221. ( 10.1200/JCO.2007.15.8923) [DOI] [PubMed] [Google Scholar]
- Cristofanilli M, Budd GT, Ellis MJ, Stopeck A, Matera J, Miller MC, Reuben JM, Doyle GV, Allard WJ, Terstappen LWet al. 2004Circulating tumor cells, disease progression, and survival in metastatic breast cancer. New England Journal of Medicine 351781–791. ( 10.1056/NEJMoa040766) [DOI] [PubMed] [Google Scholar]
- Cristofanilli M, Hayes DF, Budd GT, Ellis MJ, Stopeck A, Reuben JM, Doyle GV, Matera J, Allard WJ, Miller MCet al. 2005Circulating tumor cells: a novel prognostic factor for newly diagnosed metastatic breast cancer. Journal of Clinical Oncology 231420–1430. ( 10.1200/JCO.2005.08.140) [DOI] [PubMed] [Google Scholar]
- Crona J, Gustavsson T, Norlen O, Edfeldt K, Akerstrom T, Westin G, Hellman P, Bjorklund P, Stalberg P.2015Somatic mutations and genetic heterogeneity at the CDKN1B locus in small intestinal neuroendocrine tumors. Annals of Surgical Oncology 22 (Supplement 3) S1428–S1435. ( 10.1245/s10434-014-4351-9) [DOI] [PubMed] [Google Scholar]
- Cunningham JL, Diaz de stahl T, Sjoblom T, Westin G, Dumanski JP, Janson ET.2011Common pathogenetic mechanism involving human chromosome 18 in familial and sporadic ileal carcinoid tumors. Genes, Chromosomes and Cancer 5082–94. ( 10.1002/gcc.20834) [DOI] [PubMed] [Google Scholar]
- Dago AE, Stepansky A, Carlsson A, Luttgen M, Kendall J, Baslan T, Kolatkar A, Wigler M, Bethel K, Gross MEet al. 2014Rapid phenotypic and genomic change in response to therapeutic pressure in prostate cancer inferred by high content analysis of single circulating tumor cells. PLoS ONE 9 e101777. ( 10.1371/journal.pone.0101777) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dasari A, Shen C, Halperin D, Zhao B, Zhou S, Xu Y, Shih T, Yao JC.2017Trends in the incidence, prevalence, and survival outcomes in patients With neuroendocrine tumors in the United States. JAMA Oncology 31335–1342. ( 10.1001/jamaoncol.2017.0589) [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Bono JS, Scher HI, Montgomery RB, Parker C, Miller MC, Tissing H, Doyle GV, Terstappen LW, Pienta KJ, Raghavan D.2008Circulating tumor cells predict survival benefit from treatment in metastatic castration-resistant prostate cancer. Clinical Cancer Research 146302–6309. ( 10.1158/1078-0432.CCR-08-0872) [DOI] [PubMed] [Google Scholar]
- Di Domenico A, Wiedmer T, Marinoni I, Perren A.2017Genetic and epigenetic drivers of neuroendocrine tumours (NET). Endocrine-Related Cancer 24 R315–R334. ( 10.1530/ERC-17-0012) [DOI] [PubMed] [Google Scholar]
- el-Naggar AK, Troncoso P, Ordonez NG.1995Primary renal carcinoid tumor with molecular abnormality characteristic of conventional renal cell neoplasms. Diagnostic Molecular Pathology 448–53. ( 10.1097/00019606-199503000-00009) [DOI] [PubMed] [Google Scholar]
- Ferrarini A, Forcato C, Buson G, Tononi P, Del Monaco V, Terracciano M, Bolognesi C, Fontana F, Medoro G, Neves Ret al. 2018A streamlined workflow for single-cells genome-wide copy-number profiling by low-pass sequencing of LM-PCR whole-genome amplification products. PLoS ONE 13 e0193689. ( 10.1371/journal.pone.0193689) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis JM, Kiezun A, Ramos AH, Serra S, Pedamallu CS, Qian ZR, Banck MS, Kanwar R, Kulkarni AA, Karpathakis Aet al. 2013Somatic mutation of CDKN1B in small intestine neuroendocrine tumors. Nature Genetics 451483–1486. ( 10.1038/ng.2821) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Ni X, Guo H, Su Z, Ba Y, Tong Z, Guo Z, Yao X, Chen X, Yin Jet al. 2017Single-cell sequencing deciphers a convergent evolution of copy number alterations from primary to circulating tumor cells. Genome Research 271312–1322. ( 10.1101/gr.216788.116) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerlinger M, Rowan AJ, Horswell S, Math M, Larkin J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Stewart Aet al. 2012Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. New England Journal of Medicine 366883–892. ( 10.1056/NEJMoa1113205) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorges TM, Tinhofer I, Drosch M, Rose L, Zollner TM, Krahn T, Von Ahsen O.2012Circulating tumour cells escape from EpCAM-based detection due to epithelial-to-mesenchymal transition. BMC Cancer 12 178. ( 10.1186/1471-2407-12-178) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen LL, Yilmaz M, Overgaard J, Andersen J, Kruse TA.1998Allelic loss of 16q23.2-24.2 is an independent marker of good prognosis in primary breast cancer. Cancer Research 582166–2169. [PubMed] [Google Scholar]
- Hashemi J, Fotouhi O, Sulaiman L, Kjellman M, Hoog A, Zedenius J, Larsson C.2013Copy number alterations in small intestinal neuroendocrine tumors determined by array comparative genomic hybridization. BMC Cancer 13 505. ( 10.1186/1471-2407-13-505) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidary M, Auer M, Ulz P, Heitzer E, Petru E, Gasch C, Riethdorf S, Mauermann O, Lafer I, Pristauz Get al. 2014The dynamic range of circulating tumor DNA in metastatic breast cancer. Breast Cancer Research 16 421. ( 10.1186/s13058-014-0421-y) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heitzer E, Auer M, Gasch C, Pichler M, Ulz P, Hoffmann EM, Lax S, Waldispuehl-Geigl J, Mauermann O, Lackner Cet al. 2013Complex tumor genomes inferred from single circulating tumor cells by array-CGH and next-generation sequencing. Cancer Research 732965–2975. ( 10.1158/0008-5472.CAN-12-4140) [DOI] [PubMed] [Google Scholar]
- Heitzer E, Ulz P, Geigl JB, Speicher MR.2016Non-invasive detection of genome-wide somatic copy number alterations by liquid biopsies. Molecular Oncology 10494–502. ( 10.1016/j.molonc.2015.12.004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hieronymus H, Murali R, Tin A, Yadav K, Abida W, Moller H, Berney D, Scher H, Carver B, Scardino Pet al. 2018Tumor copy number alteration burden is a pan-cancer prognostic factor associated with recurrence and death. eLife 7e37294. ( 10.7554/eLife.37294) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpathakis A, Dibra H, Pipinikas C, Feber A, Morris T, Francis J, Oukrif D, Mandair D, Pericleous M, Mohmaduvesh Met al. 2016Prognostic impact of novel molecular subtypes of small intestinal neuroendocrine tumor. Clinical Cancer Research 22250–258. ( 10.1158/1078-0432.CCR-15-0373) [DOI] [PubMed] [Google Scholar]
- Khan MS, Tsigani T, Rashid M, Rabouhans JS, Yu D, Luong TV, Caplin M, Meyer T.2011Circulating tumor cells and EpCAM expression in neuroendocrine tumors. Clinical Cancer Research 17337–345. ( 10.1158/1078-0432.CCR-10-1776) [DOI] [PubMed] [Google Scholar]
- Khan MS, Kirkwood A, Tsigani T, Garcia-Hernandez J, Hartley JA, Caplin ME, Meyer T.2013Circulating tumor cells as prognostic markers in neuroendocrine tumors. Journal of Clinical Oncology 31365–372. ( 10.1200/JCO.2012.44.2905) [DOI] [PubMed] [Google Scholar]
- Khan MS, Kirkwood AA, Tsigani T, Lowe H, Goldstein R, Hartley JA, Caplin ME, Meyer T.2016Early changes in circulating tumor cells are associated with response and survival following treatment of metastatic neuroendocrine neoplasms. Clinical Cancer Research 2279–85. ( 10.1158/1078-0432.CCR-15-1008) [DOI] [PubMed] [Google Scholar]
- Krebs MG, Sloane R, Priest L, Lancashire L, Hou JM, Greystoke A, Ward TH, Ferraldeschi R, Hughes A, Clack Get al. 2011Evaluation and prognostic significance of circulating tumor cells in patients with non-small-cell lung cancer. Journal of Clinical Oncology 291556–1563. ( 10.1200/JCO.2010.28.7045) [DOI] [PubMed] [Google Scholar]
- Kulke MH, Freed E, Chiang DY, Philips J, Zahrieh D, Glickman JN, Shivdasani RA.2008High-resolution analysis of genetic alterations in small bowel carcinoid tumors reveals areas of recurrent amplification and loss. Genes, Chromosomes and Cancer 47591–603. ( 10.1002/gcc.20561) [DOI] [PubMed] [Google Scholar]
- Lambros MB, Seed G, Sumanasuriya S, Gil V, Crespo M, Fontes M, Chandler R, Mehra N, Fowler G, Ebbs Bet al. 2018Single-cell analyses of prostate cancer liquid biopsies acquired by apheresis. Clinical Cancer Research 245635–5644. ( 10.1158/1078-0432.CCR-18-0862) [DOI] [PubMed] [Google Scholar]
- Maheswaran S, Sequist LV, Nagrath S, Ulkus L, Brannigan B, Collura CV, Inserra E, Diederichs S, Iafrate AJ, Bell DWet al. 2008Detection of mutations in EGFR in circulating lung-cancer cells. New England Journal of Medicine 359366–377. ( 10.1056/NEJMoa0800668) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandair D, Khan MS, Lopes A, Furtado O’Mahony L, Ensell L, Lowe H, Hartley JA, Toumpanakis C, Caplin M, Meyer T.2021Prognostic threshold for circulating tumor cells in patients with pancreatic and midgut neuroendocrine tumors. Journal of Clinical Endocrinology and Metabolism 106872–882. ( 10.1210/clinem/dgaa822) [DOI] [PubMed] [Google Scholar]
- Mangano C, Ferrarini A, Forcato C, Garonzi M, Tononi P, Lanzellotto R, Raspadori A, Bolognesi C, Buson G, Medoro Get al. 2019Precise detection of genomic imbalances at single-cell resolution reveals intra-patient heterogeneity in Hodgkin’s lymphoma. Blood Cancer Journal 9 92. ( 10.1038/s41408-019-0256-y) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mroz EA, Tward AD, Pickering CR, Myers JN, Ferris RL, Rocco JW.2013High intratumor genetic heterogeneity is related to worse outcome in patients with head and neck squamous cell carcinoma. Cancer 1193034–3042. ( 10.1002/cncr.28150) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navin N, Krasnitz A, Rodgers L, Cook K, Meth J, Kendall J, Riggs M, Eberling Y, Troge J, Grubor Vet al. 2010Inferring tumor progression from genomic heterogeneity. Genome Research 2068–80. ( 10.1101/gr.099622.109) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni X, Zhuo M, Su Z, Duan J, Gao Y, Wang Z, Zong C, Bai H, Chapman AR, Zhao Jet al. 2013Reproducible copy number variation patterns among single circulating tumor cells of lung cancer patients. PNAS 11021083–21088. ( 10.1073/pnas.1320659110) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pailler E, Adam J, Barthelemy A, Oulhen M, Auger N, Valent A, Borget I, Planchard D, Taylor M, Andre Fet al. 2013Detection of circulating tumor cells harboring a unique ALK rearrangement in ALK-positive non-small-cell lung cancer. Journal of Clinical Oncology 312273–2281. ( 10.1200/JCO.2012.44.5932) [DOI] [PubMed] [Google Scholar]
- Pailler E, Auger N, Lindsay CR, Vielh P, Islas-Morris-Hernandez A, Borget I, Ngo-Camus M, Planchard D, Soria JC, Besse Bet al. 2015High level of chromosomal instability in circulating tumor cells of ROS1-rearranged non-small-cell lung cancer. Annals of Oncology 261408–1415. ( 10.1093/annonc/mdv165) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavel ME, Hainsworth JD, Baudin E, Peeters M, Horsch D, Winkler RE, Klimovsky J, Lebwohl D, Jehl V, Wolin EMet al. 2011Everolimus plus octreotide long-acting repeatable for the treatment of advanced neuroendocrine tumours associated with carcinoid syndrome (RADIANT-2): a randomised, placebo-controlled, phase 3 study. Lancet 3782005–2012. ( 10.1016/S0140-6736(1161742-X) [DOI] [PubMed] [Google Scholar]
- Poveda A, Kaye SB, Mccormack R, Wang S, Parekh T, Ricci D, Lebedinsky CA, Tercero JC, Zintl P, Monk BJ.2011Circulating tumor cells predict progression free survival and overall survival in patients with relapsed/recurrent advanced ovarian cancer. Gynecologic Oncology 122567–572. ( 10.1016/j.ygyno.2011.05.028) [DOI] [PubMed] [Google Scholar]
- Ramdzan ZM, Nepveu A.2014CUX1, a haploinsufficient tumour suppressor gene overexpressed in advanced cancers. Nature Reviews: Cancer 14673–682. ( 10.1038/nrc3805) [DOI] [PubMed] [Google Scholar]
- Riethdorf S, Fritsche H, Muller V, Rau T, Schindlbeck C, Rack B, Janni W, Coith C, Beck K, Janicke Fet al. 2007Detection of circulating tumor cells in peripheral blood of patients with metastatic breast cancer: a validation study of the CellSearch system. Clinical Cancer Research 13920–928. ( 10.1158/1078-0432.CCR-06-1695) [DOI] [PubMed] [Google Scholar]
- Sakai K, Nagahara H, Abe K, Obata H.1992Loss of heterozygosity on chromosome 16 in hepatocellular carcinoma. Journal of Gastroenterology and Hepatology 7288–292. ( 10.1111/j.1440-1746.1992.tb00982.x) [DOI] [PubMed] [Google Scholar]
- Seol H, Lee HJ, Choi Y, Lee HE, Kim YJ, Kim JH, Kang E, Kim SW, Park SY.2012Intratumoral heterogeneity of HER2 gene amplification in breast cancer: its clinicopathological significance. Modern Pathology 25938–948. ( 10.1038/modpathol.2012.36) [DOI] [PubMed] [Google Scholar]
- Sharma SV, Bell DW, Settleman J, Haber DA.2007Epidermal growth factor receptor mutations in lung cancer. Nature Reviews: Cancer 7169–181. ( 10.1038/nrc2088) [DOI] [PubMed] [Google Scholar]
- Sosman JA, Kim KB, Schuchter L, Gonzalez R, Pavlick AC, Weber JS, Mcarthur GA, Hutson TE, Moschos SJ, Flaherty KTet al. 2012Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. New England Journal of Medicine 366707–714. ( 10.1056/NEJMoa1112302) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velickovic M, Delahunt B, Mciver B, Grebe SK.2002Intragenic PTEN/MMAC1 loss of heterozygosity in conventional (clear-cell) renal cell carcinoma is associated with poor patient prognosis. Modern Pathology 15479–485. ( 10.1038/modpathol.3880551) [DOI] [PubMed] [Google Scholar]
- Vogel CL, Cobleigh MA, Tripathy D, Gutheil JC, Harris LN, Fehrenbacher L, Slamon DJ, Murphy M, Novotny WF, Burchmore Met al. 2002Efficacy and safety of trastuzumab as a single agent in first-line treatment of HER2-overexpressing metastatic breast cancer. Journal of Clinical Oncology 20719–726. ( 10.1200/JCO.2002.20.3.719) [DOI] [PubMed] [Google Scholar]
- Walter D, Harter PN, Battke F, Winkelmann R, Schneider M, Holzer K, Koch C, Bojunga J, Zeuzem S, Hansmann ML,et al. 2018Genetic heterogeneity of primary lesion and metastasis in small intestine neuroendocrine tumors. Scientific Reports 83811. ( 10.1038/s41598-018-22115-0) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao JC, Shah MH, Ito T, Bohas CL, Wolin EM, Van Cutsem E, Hobday TJ, Okusaka T, Capdevila J, De Vries EGet al. 2011Everolimus for advanced pancreatic neuroendocrine tumors. New England Journal of Medicine 364514–523. ( 10.1056/NEJMoa1009290) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu L, Smith CI.2011Tec family kinases. FEBS Journal 2781969. ( 10.1111/j.1742-4658.2011.08135.x) [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.