

Abstract
Two-component signaling systems are a major strategy employed by bacteria, and to some extent, yeast and plants, to respond to environmental stress. The EnvZ/OmpR system in E. coli responds to osmotic and acid stress and is responsible for regulating the protein composition of the outer membrane. EnvZ is a histidine kinase located in the inner membrane. Upon activation, it is autophosphorylated by ATP and subsequently, it activates OmpR. Phosphorylated OmpR binds with high affinity to the regulatory regions of the ompF and ompC porin genes to regulate their transcription. We set out to visualize these two-components in single bacterial cells during different environmental stress conditions and to examine the subsequent modifications to the bacterial nucleoid as a result. We created a chromosomally-encoded, active, fluorescent OmpR–PAmCherry fusion protein and compared its expression levels with RNA polymerase. Quantitative western blotting had indicated that these two proteins were expressed at similar levels. From our images, it is evident that OmpR is significantly less abundant compared to RNA polymerase. In cross-sectional axial images, we observed OmpR molecules closely juxtaposed near the inner membrane during acidic and hyposomotic growth. In acidic conditions, the chromosome was compacted. Surprisingly, under acidic conditions, we also observed evidence of a spatial correlation between the DNA and the inner membrane, suggesting a mechanical link through an active DNA–OmpR–EnvZ complex. This work represents the first direct visualization of a response regulator with respect to the bacterial chromosome.
Introduction
All cells need to sense and respond to their environment in order to survive and bacteria use two-component signaling systems to perform this function. Environmental changes are sensed by histidine kinases (HKs) that in gram-negative bacteria are most often embedded in the inner membrane. Upon activation, HKs are autophosphorylated by intracellular ATP. The phosphoryl group is then transferred to an aspartic acid residue of the second component, the response regulator (RR). Most RRs are DNA binding proteins and phosphorylation enhances or represses transcription.
In the archetype EnvZ/OmpR system, bacteria respond to increasing extracellular osmolality by concentrating their cytoplasm. This intracellular signal drives a disordered region around the active site histidine of EnvZ to become ordered, activating phosphorylation.1,2 Phospho-OmpR binds to the regulatory regions of the porin genes, activating ompC transcription at high osmolality and repressing ompF. The two porins are reciprocally regulated3 and OmpF has a larger pore and a faster flow rate to allow for enhanced nutrient exchange in dilute environments.4 In Salmonella enterica, the EnvZ/OmpR system is essential to activate a second two-component system, SsrA/B, which drives expression of virulence factors that enable Salmonella to replicate in the acidic environment of the macrophage vacuole.5,6
In the EnvZ/OmpR system, we recently established that signals that were once believed to be extracellular, were actually intracellular.1,2,7 For example, at high external osmolality, the cytoplasm of E. coli becomes concentrated, up to 1.8 osmoles,8 altering EnvZ conformation.1 Likewise, when Salmonella is in a macrophage vacuole, its cytoplasm acidifies to pH 5.6.7 This acidification is dependent on the cytoplasmic domain of EnvZ (EnvZc), which contains both kinase and ATP-binding subdomains. Acidification works through EnvZ–OmpR, as OmpR represses the cadaverine transport system cadC/BA to prevent neutralization of the cytosol after acid stress. Furthermore, when E. coli and Salmonella are exposed to external osmolytes, they respond by acidifying the cytoplasm.9 EnvZ responds to these intracellular signals by increasing its autophosphorylation and activating OmpR via phosphotransfer.1 This may be in part a response to mechanical cues that arise as E. coli undergoes substantial volume changes during large alterations in osmolality. Recent studies used ChIP-seq to identify OmpR binding sites in response to external acid stress.10 Those studies employed a FLAG-tag fused to the C-terminus of OmpR. However, previous studies have shown that alterations to the OmpR C-terminal DNA binding region are deleterious.11 Our goal was to establish a photoactivatable construct of OmpR that was chromosomally-expressed under its native promoter and capable of porin gene regulation so that we could examine the OmpR distribution in single cells in response to various environmental stresses.
To investigate the EnvZ/OmpR system at the single-cell level, a fluorescent label can be attached to the protein of interest, which facilitates the microscopic study of its intracellular localization. Due to the small size of bacterial cells and the diffraction limit of light, conventional fluorescence microscopy is of limited use. Super-resolution microscopy allows visualization of biological structures at scales far below the diffraction limit of light.12 We applied single-molecule localization microscopy (SMLM)13 to visualize protein and chromosome structures at the nanometer scale. In SMLM, the fluorescence signal is separated in time using photoactivatable or photoswitchable fluorescent probes. By activating only a subset of fluorophores, single molecules can be detected and their position determined with a precision of a few nanometers.14 Next to imaging cells with subdiffraction resolution, photoactivated localization microscopy (PALM)15 can provide copy numbers of the target protein in single cells, an approach that was recently demonstrated for RNA polymerase in bacteria.16 Point accumulation for imaging in nanoscale topography (PAINT)17,18 and direct stochastic optical reconstruction microscopy (dSTORM)19 can be used to visualize the bacterial envelope or chromosomal DNA, respectively. Combining these techniques sequentially enabled us to investigate the localization of OmpR under different stress conditions, to determine its relative position within the cell cylinder, and its spatial relationship with the chromosome. In this study, we labeled OmpR with the photomodulatable proteins mEos20 and PAmCherry,21 while conserving its functionality.
In the case of two-component signaling systems, the HK is often not an abundant protein and it is usually embedded in the inner membrane, making it difficult to study. In the case of envZ, it is in an operon with ompR (the ompB operon), with the promoter upstream of ompR.22 The stop signal for ompR overlaps the start signal for envZ, resulting in substantially fewer copies of EnvZ compared to OmpR. This provides a significant imaging challenge, as the low copy prevents the generation of a suitable super-resolution image. We overcame this difficulty by over-expression of EnvZc-mEos2 using an arabinose inducible-plasmid and induction with 0.02% arabinose.2 This produced a level of EnvZ that was as functional as the wildtype and the protein was widely distributed throughout the cytoplasm. However, at copy numbers approaching in vivo levels, not enough molecules could be collected to produce a super-resolution image. To overcome these obstacles and examine the interaction of EnvZc with OmpR as a function of environmental stress, we turned to fluorescence correlation spectroscopy (FCS) to examine the EnvZc/OmpR interactions in vitro, in solution.
Imaging the distribution of OmpR within single cells using PALM indicated that OmpR was largely localized near the plasma membrane during growth in acidic conditions and in low osmolality medium. We also provide evidence of chromosomal compaction during acid stress. Our results suggest an in vivo linkage with EnvZ in the inner membrane, to OmpR to the nucleoid23 and explains why some dominant mutations in ompR require envZ for expression of their phenotype.24 Furthermore, the visible linkages observed stretching from the nucleoid to the inner membrane provide evidence of structural interactions between the chromosome and the membrane and suggest mechanical stress could be easily transmitted from the membrane to the DNA.
Methods
Construction of an active, chromosomally-expressed OmpR-photoactivatable fusion protein
In constructing an OmpR fluorescent fusion protein, we placed a flexible linker of GGSG repeats (encoded by 5ʹ-ggtggttctggt-3ʹ) in between ompR and the fluorophore. Plasmid pCAH63-PompR-ompR-GGSGx4-PAmCherry was constructed in two steps. First, a fragment with the ompR promoter and the ompR gene (PompR-ompR) was amplified from E. coli genomic DNA with primers PompR-EcoR1-F and FP-GGSGx4-ompR-R, and PAmCherry was amplified with primers ompR-GGSGx4-PAmC-F and PAmC-BamH1-R. These two fragments were equally mixed and used as a template to amplify PompR-ompR-GGSGx4-PAmCherry, which was flanked with EcoR1 and BamH1 restriction sites. The resulting fragment PompR-ompR-GGSGx4-PAmCherry was digested with EcoR1 and BamH1, and then inserted into pCAH63 that had been treated with the same restriction enzymes, generating pCAH63-PompR-ompR-GGSGx4-PAmCherry. The mEos2 plasmid (pCAH63-PompR-ompR-GGSGx4-mEos2) was created using a similar method except that mEos2 was amplified with primers ompR-GGSGx4-mEos2-F and mEos2-BamH1-R. PompR-ompR and mEos2 was fused together with primers PompR-EcoR1-F and mEos2-BamH1-R, yielding a PompR-ompR-GGSGx4-mEos2 fragment. PompR-ompR-GGSGx4-mEos2 and pCAH63 were digested with EcoR1 and BamH1, and ligated to generate pCAH63-PompR-ompR-GGSGx4-mEos2. Plasmid pCAH63-PompR-ompR-GGSGx10-mEos2 was randomly generated during the construction of pCAH63-PompR-ompR-GGSGx4-mEos2.
Genome integration was conducted following the method of Haldimann and Wanner.25 Briefly, strains MH225.101 and MH513.101 that were ompR null26 were transformed with plasmid pINT-ts by electroporation. After that, the recombinant strains MH225.101/pINT-ts and MH513.101/pINT-ts were made competent and transformed with pCAH63-PompR-ompR-GGSGx4-PAmCherry, pCAH63-PompR-ompR-GGSGx4-mEos2 and pCAH63-PompR-ompR-GGSGx10-mEos2 plasmids, and colonies were then recovered on plates using chloramphenicol selection. A single colony was re-streaked and the correct integration was confirmed by colony PCR with primers P1, P2, P3 and P4. The pINT-ts plasmid was eliminated by incubation at the restrictive temperature, as it possesses a temperature-sensitive origin of replication. Finally, we obtained the following strains MH225.101 attB:PompR-ompR-GGSGx4-PAmCherry, MH225.101 attB:PompR-ompR-GGSGx4-mEos2, MH225.101 attB:PompR-ompR-GGSGx10-mEos2, MH513.101 attB:PompR-ompR-GGSGx4-PAmCherry, MH513.101 attB:PompR-ompR-GGSGx4-mEos2 and MH513.101 attB:PompR-ompR-GGSGx10-mEos2.
| Primer | Sequence (5ʹ–3ʹ) |
|---|---|
| PompR-EcoR1-F | ccggaattcggtttgagttcatcttttcgg |
| FP-GGSGx4-ompR-R | accagaaccaccaccagaaccaccaccagaaccaccaccagaaccacctgctttagagccgtccggtac |
| ompR-GGSGx4-mEos-F | ggtggttctggtggtggttctggtggtggttctggtatgtcagcaatcaaaccggatatg |
| ompR-GGSGx4-PAmC-F | ggtggttctggtggtggttctggtggtggttctggtatggtgagcaagggcgaggaggataac |
| PAmC-BamH1-R | cgcggatccctacttgtacagctcgtccatgccgcc |
| mEos2-BamH1-R | cgcggatccttagcgacgggcgttatccggcag |
| P1 | GGCATCACGGCAATATAC |
| P2 | ACTTAACGGCTGACATGG |
| P3 | ACGAGTATCGAGATGGCA |
| P4 | TCTGGTCTGGTAGCAATG |
β-galactosidase assays
β-Galactosidase assays were performed as previously described.27
Protein purification and fluorescent-labeling
EnvZc and EnvZcM were sub-cloned into the pET28b vector with an N-terminal His-tag, over-expressed and purified as described previously.1 OmpR was over-expressed on a pET15b vector and purified using a similar method as EnvZc. The proteins were stored in 20 mM phosphate (pH 7.4), 200 mM NaCl (500 mM for OmpR), 5% glycerol (v/v) and 0.05% Tween-20. EnvZc purified as a dimer based on the elution time on the FPLC. Alexa dyes were added in five-fold molar excess for labeling of EnvZc, EnvZcM and OmpR. The reaction was allowed to proceed at 4 °C overnight with gentle rocking. The labeled protein was separated from the excess dye using two PD-10 desalting columns in tandem (GE Healthcare).
Native PAGE
EnvZc, EnvZcM and OmpR were incubated at final concentrations of 7 μM ina 10 ml volume reaction in 50 mM Tris-HCl, pH 8.0, 50 mM KCl and 5% glycerol buffer. The samples were loaded onto a native gel with 2× loading buffer (40 mM Tris-HCl, pH 7.5, 80 mM β-mercaptoethanol, 0.08% bromophenol blue, 8% glycerol). The composition of the stacking gel was 5% acrylamide/bis (37.5 : 1) in 63 mM Tris-HCl (pH 7.4) and the composition of the separation gel was 10% acrylamide/bis (37.5 : 1) in 188 mM Tris-HCl (pH 8.8), while the running buffer was 83 mM Tris-HCl (pH 8.9), 33 mM glycine. The gel was run at a constant current of 0.02A at 4 °C. The native gel was then imaged using a UV imager (BioRad, ChemiDoc MP) before staining with Coomassie based InstantBluet™ (Expedeon).
Fluorescence correlation spectroscopy (FCS) and fluorescence cross-correlation spectroscopy (FCCS) measurements
FCS and FCCS were performed on the LSM710-ConfoCor3 (Carl Zeiss) system with a 40×, NA 1.2 C-apochromat objective. Alexa Fluor 488 and 568 were excited using the 488 nm and 561 nm laser line, respectively. The protein samples were incubated in 20 mM phosphate (pH 7.4), 50 mM NaCl, and 0.05% Tween-20 at room temperature for 30 min before spotting (20 μl) onto a clean coverslip for measurements. The autocorrelations functions (ACFs) from the EnvZcM titrations FCS were fitted with either a single component (for the lower EnvZcM concentrations) or two-component 3D diffusion model (for the higher EnvZcM concentrations where there are two distinct diffusion times):28
where G(∞) is a convergence value for the ACF at long (infinite) delay times. Ftrip is the fraction of the particles that reside in the triplet state and τtrip is the triplet state relaxation time. G(0) is the amplitude of the ACF and is related to the average number of molecules N detected in the observation volume by:
D(τ) for a single component 3D diffusion is given by:
and D(τ) for a two-component 3D diffusion model is given by:
where Fbound is the fraction of bound molecules with a higher diffusion time of τD(bound). τD(free) is the diffusion time for the faster free molecule. K is the structural parameter defining a confocal laser spot obtained from calibrating the microscope with either Alexa 488 or 568 dyes.
In the FCCS measurements, as the concentration of EnvZc labeled with Alexa 568 is always in excess, cross-talk of Alexa 488 into the Alexa 568 detection channel can be neglected. Therefore, the fraction of Alexa 488-labeled OmpR (OmpR-A488) bound can be simply obtained from the ratio of the amplitude of the cross-correlation function (CCF) to the amplitude of the ACF of Alexa 568 labeled protein (cross-correlation ratio):
where Ncomplex is the number of molecules in complex and NTot,OmpR is the total number of molecules of OmpR-A488 (bound and free). The microscope was calibrated using known controls to obtain the range of cross-correlation ratio values obtainable (ESI,† Fig. S1A and B). The range of the cross-correlation ratio for the microscope system was determined to be between 0.02 and 0.99.
Super-resolution microscopy
Cell culture.
The OmpR–PAmCherry strain was inoculated from glycerol stocks and grown over night (ON) in the respective media, supplemented with 10 μM chloramphenicol, at 32 °C or 37 °C and 200 rpm shaking. Working cultures were inoculated 1 : 200 from ON cultures and OD600 was determined every 30 minutes to obtain the doubling time for each culture. For media composition and respective doubling times see ESI,† Table S1. EdU (Baseclick) was added at 10 μM concentration for 40 minutes to the LB culture at an OD600 of ~0.25. Due to overlapping replication cycles, EdU-incorporation should be incorporated throughout the entire chromosome. The mass-doubling times were significantly longer in the minimal media, therefore EdU was added for 1.5 mass-doubling times.
Fixation and immobilization.
Cultures were fixed in solution by adding sodium-phosphate-buffer pH 7.5 and methanol-free formaldehyde (Sigma) to a final concentration of 15 mM and 1%, respectively. After 30 minutes, the cells were pelleted for 3 minutes at 5000 × g and washed once with 100 mM sodium-phosphate-buffer pH 7.5 containing 50 mM ammonium-chloride to reduce excess formaldehyde. Resuspended bacteria were then immobilized on KOH-cleaned (3 M, 30 min), poly-l-lysine-coated 8-well-chamberslides (Sarstedt) and washed twice with phosphate buffer. The buffer was then removed and the sample was placed on ice 5 min before proceeding with gel matrix coating.
Coating and post-labeling.
To maintain the positions of immobilized bacteria and prohibit detachment from the poly-l-lysine surface, we added a hydrophilic gel matrix to the sample as described previously, but with shorter gel polymerization time (15 min).29 During the sequential workflow, we also followed the provided protocol for DNA post-labeling. In brief, immobilized bacteria were permeabilized with 0.5% Triton-X 100 in PBS (the osmolality was adjusted for each growth condition) for 60 min. After two washing steps, 300 μl click-reaction buffer (100 mM Tris pH 8.0, 1 mM CuSO4 10 μM Alexa Fluor 647 azide, 100 mM ascorbic acid, osmolality-adjustment with NaCl if required) was added to the sample for 60 minutes. Afterwards, the sample was washed 3 times for 5 min and further 2 times for 30 min with PBS (osmolality adjusted). The sample was then transferred onto the microscope for dSTORM imaging.
Super-resolution imaging.
Samples were imaged on a custom-built microscope.29 A mechanical stage (TI-S-ER, Nikon) and an adjustable TIRF mirror were used to relocate bacteria during the sequential imaging workflow and adjust the angle of the inclined laser for proper HILO imaging. μManager30 was used to control the microscope and camera. PALM imaging of RNAP–PAmCherry in fixed cells was performed using 568 nm for excitation (1.5 kW cm‒2) and 404 nm for activation (gradually increased up to 12 W cm‒2). Typically, between 5000 and 7000 frames were recorded at a camera frame-rate of 10 Hz in HILO mode until all PAmCherry-molecules were photoconverted and bleached. Afterwards, 200 pM Nile red in phosphate buffer was added to facilitate PAINT-imaging (10 000 frames at 33 Hz camera frame-rate) using 568 nm excitation light (0.7 kW cm‒2). dSTORM-imaging of Alexa Fluor 647 was performed in PBS containing 100 mM cysteamin (MEA, Sigma) pH 8.5 using excitation at 647 nm (2 kW cm‒2) and 488 nm for reactivation (up to 30 W cm‒2). Appropriate dichroic mirrors and filters (AHF, Germany) were used for each spectral channel. The data was analyzed with rapidSTORM31 and resulting images were post-processed with Fiji.32
Image processing.
For visualization, a Gaussian blur of the mean localization precision was applied to the SMLM images using Fiji. For the averaging analysis of OmpR distribution, cells were angular reorientated and normalized for cell length and width using custom-written ImageJ macros. In order to prevent orientation artifacts, each bacteria was mirrored along the length axis, the cross axis and both axes with subsequent averaging of the resulting four images. 13–20 images per condition were averaged to obtain ‘heat maps’ of OmpR localization. Cells of similar size were chosen in order to average bacteria in comparable cell cycle stages and to prevent normalization artifacts (LB: 3.75–4.25 μm, Tris: 2.0–2.5 μm, MES: 1.75–2.25 μm, 0.5× M9: 1.5 to 2.0 μm).
Results
A chromosomally-expressed fluorescent fusion protein of OmpR is active
Initially, we constructed photoactivatable fusion proteins based on a previously published strategy33 that consisted of a four amino acid linker between the end of the ompR gene and the gene encoding the fluorophore. That study did not examine the functionality of the fusions that were constructed, and more recently, it was reported that the fluorescent fusion protein had significant effects on its distribution.34 To avoid this problem, we used a functional assay and examined the ability of our OmpR construct to activate its target genes, using ompF and ompC transcriptional fusions.26,35 These fusions contain the ompF/C promoter fused to the lacZ gene, encoding β-galactosidase. The original fusion was ~20% active (data not shown), similar to other OmpR chimeric fluorescent protein fusions36 that we have also measured. OmpR does not tolerate substitutions at its C-terminus, which disrupt its ability to bind to DNA.11 Increasing the linker length in four amino acid increments eventually led to an active chimera at sixteen amino acids (Fig. 1A and B, see also ref. 37). A linker length of 16 amino acids was sufficient for PAmCherry, but a longer linker length was required for mEos (Fig. 1A and B), and it was not as active as the PAmCherry chimera (79% compared to 93% at ompF and 60% compared to 71% at ompC). As we have also previously observed, ompC transcription was more sensitive than ompF to substitutions in OmpR that alter function.38 Our OmpR–PAmCherry was expressed from its native promoter and was integrated at the attB site of the E. coli chromosome, to avoid disrupting its downstream gene envZ (see Methods).
Fig. 1.
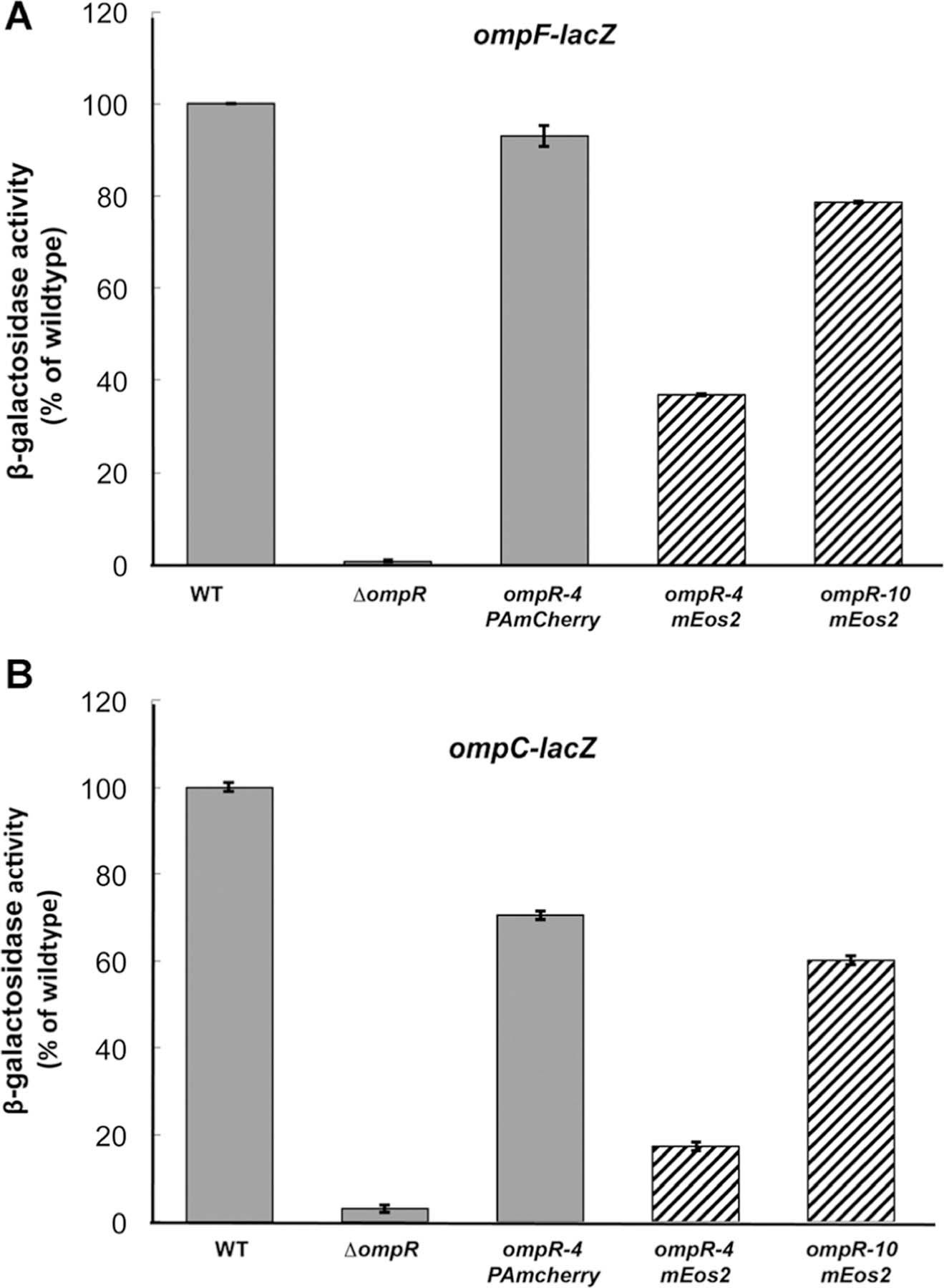
Comparison of OmpR–PAmCherry with OmpR-mEos and wildtype OmpR. MH513 and MH225 are bacterial strains containing a chromosomal ompF-lacZ (A) or ompC-lacZ (B) transcriptional fusion, respectively. Wildtype OmpR (column 1) represented 100% and the other backgrounds were normalized to it. In the ompR null strain (ompR101), there was no activation of ompF or ompC. The PAmCherry fusion was 93% as active as the wildtype, when a 16 amino acid linker, GGSGx4, was placed between the 3ʹ end of ompR and the beginning of PAmCherry. Its activity was higher than the mEos2 fusion that contained the same linker length (striped column 4, 37%), but a longer linker (40 amino acids, GGSGx10) improved activity to 79% of the wildtype (column 5). Similarly, at ompC, the PAmCherry fusion was 71% of the wildtype activity and better than the mEos2 fusion (17% with a 16 amino acid linker and 60% with a 40 amino acid linker). The photoactivatable fusion affected ompC activity more than ompF as we have observed with other ompR mutants38 as well as with envZ photoactivatable fusions.2
Correlating OmpR distribution with the bacterial nucleoid
We were next interested in visualizing the distribution of OmpR in single bacterial cells using super-resolution microscopy. In order to correlate the distribution of OmpR near the membrane or the bacterial chromosome, we applied a sequential workflow29 (Fig. 2). In medium at high osmolality (LB, 460 mOsm per kg), OmpR molecules were located close to the edge of the nucleoid, while the chromosomal core was not decorated by OmpR (Fig. 2A). This behavior was not readily apparent in low salt media (100 mM Tris pH 7.2, 210 mOsm per kg) and the nucleoid appeared to be more condensed (Fig. 2B, compare green circles outlining the chromosome in the third row panels). When cells were grown in acidic conditions (100 mM MES medium pH 5.6, 205 mOsm per kg) (Fig. 2C) or 0.5× M9 medium (115 mOsm per kg) (Fig. 2D), OmpR localized near the plasma membrane and appeared to be more clustered than in rich media at higher osmolality (LB, Fig. 2A). In order to quantify this distribution, we constructed average localization maps of OmpR, each consisting of 13–20 bacteria (bottom panel, Fig. 2). While OmpR shows a more uniform distribution in LB and Tris, the preferential localization near the membrane is evident for acidic and low osmolality 0.5× M9 medium. Furthermore, the bacterial nucleoids appeared to be more condensed during acidic growth compared to cells grown at neutral pH (Tris) (ESI,† Fig. S2).
Fig. 2.
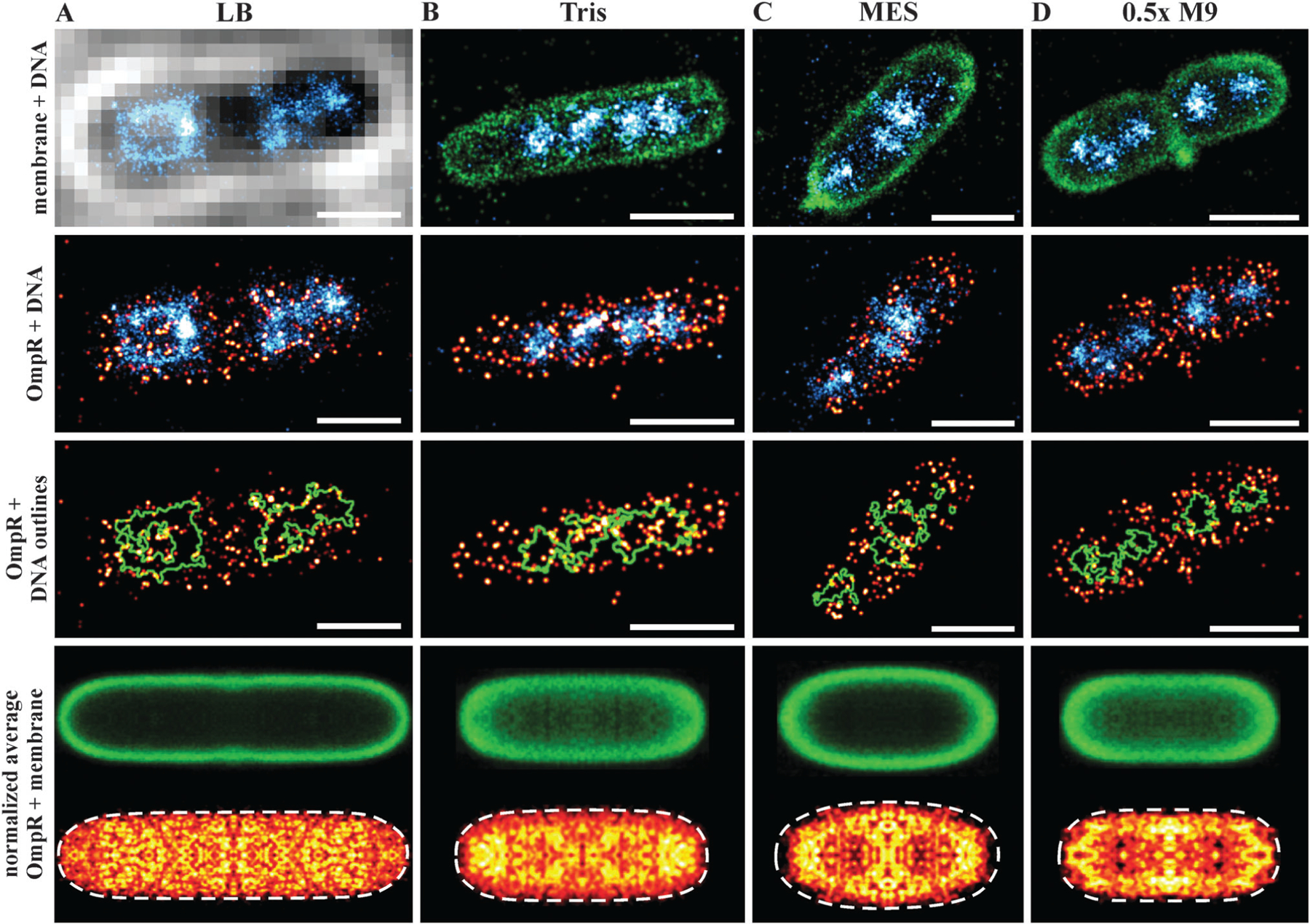
Sequential super-resolution imaging of OmpR–PAmCherry (red hot), bacterial cell membrane (green) and DNA (cyan) in various growth media. LB (A), 100 mM Tris pH 7.2 (B), 100 mM MES pH 5.6 (C) and 0.5× M9 buffer (D). Evidence for DNA compaction is noticeable in the third row panels, where the green outlines the nucleoid edges. OmpR was distributed around the nucleoid edges (outlined in green) in LB, whereas it was more uniformly distributed in Tris buffer. In acidic (MES, pH 5.6) and hypotonic (0.5× M9) conditions, OmpR was recruited to the plasma membrane (scale bar 1 μm). Images in the bottom row show the averaged distribution of the bacterial membrane (green) and OmpR (red hot). These average images indicate a localization of OmpR near the membrane in acid and low osmolality. The recruitment of OmpR to DNA in LB is averaged out because of the nucleoid heterogeneity and asymmetric segregation.74 Cells grown in Tris and 0.5× M9 medium exhibited a smaller cell diameter (~0.8 μm), resulting in projection effects for the membrane average image. The number of images used for averaging was: 19, cell length 3.75–4.25 μm (A), 20 cell length 2.0–2.5 μm (B), 15 cell length 1.75–2.25 μm (C) and 13 cell length 1.5–2.0 μm (D).
In preparing bacteria samples for microscopy, the cells were spatially fixed in a gel matrix. We found that some bacterial cells were oriented axially, which allowed cross-sectional imaging (Fig. 3). The membrane association and positioning around the chromosome became more obvious for cells visualized in this orientation, and the preferred localization of OmpR juxtaposed next to the cell membrane was clearly evident in both 0.5× M9 (Fig. 3A) and pH 5.6 (Fig. 3B). For cells grown in acidic conditions (MES, pH 5.6), we also observed a hollow-tube structure for DNA (Fig. 3B), from which thin DNA fibers appeared to reach towards the membrane (see arrows).
Fig. 3.
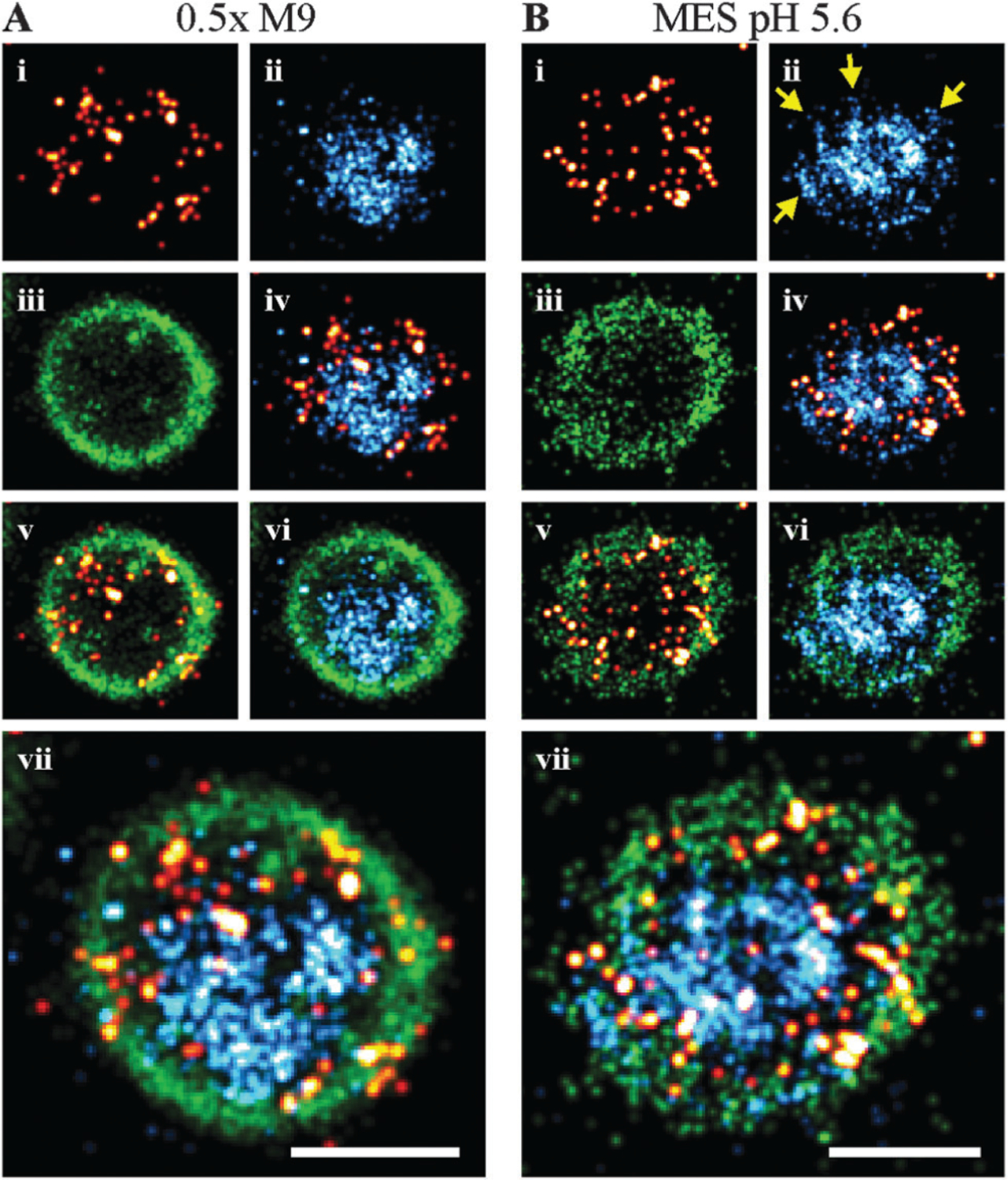
Cross-section of bacteria in 0.5× M9 (A) and MES pH 5.6 (B). Covering the bacterial cells with a hydrophilic gel matrix enabled some bacteria to orient axially, these provide cross-sectional images of the cells under various growth conditions. (i), (ii) and (iii) show the PALM, dSTORM and PAINT images of OmpR (red hot), DNA (cyan) and membrane (green), respectively. OmpR was distributed around the chromosome (iv) and along the plasma membrane (v), likely enabling recruitment to membrane-embedded EnvZ molecules during acidic and hypotonic shock. Interestingly, the chromosome seems to form a tube in the middle of the cell cylinder (vi) under acidic conditions, which exhibits small DNA fibers stretching from the tube towards the membrane (yellow arrows). (vii) Represents the composite image of all three SMLM channels (scale bar 0.5 μm).
We next investigated qualitative levels of OmpR expression in single bacterial cells. We recorded PALM images of bacterial cells expressing RNAP–PAmCherry endogenously (Fig. 4A) to compare with OmpR–PAmCherry (Fig. 4B). Quantitative western blotting reported that OmpR was present at 3500 copies per cell at high osmolality and 2000 copies at low osmolality.39 Copy numbers of RNAP were determined in a previous study using quantitative PALM.16 In rich media such as LB, an average of 3600 RNAP per cell were counted, i.e., similar to the copy number reported for OmpR by quantitative western blots. Compared to the PALM images of OmpR–PAmCherry recorded using the same experimental settings (Fig. 4B), it is immediately apparent that OmpR expression levels were significantly lower than those of RNAP. These observations question the accuracy of quantitative western blots in determining protein copy number (see Discussion).
Fig. 4.
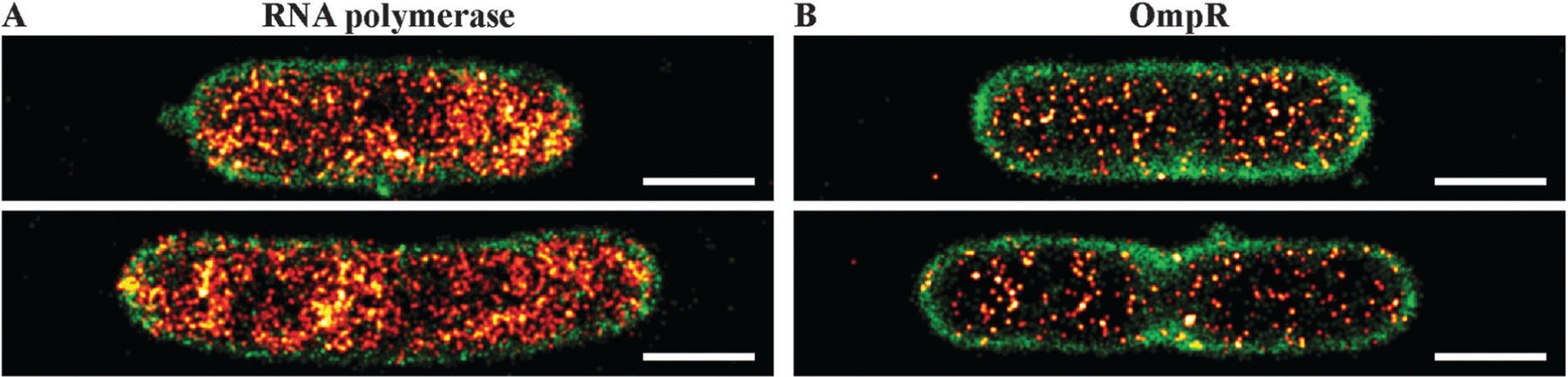
Representative super-resolution fluorescence images of OmpR (right panel, red hot) and RNAP (left panel, red hot) in E. coli cells grown in LB medium. The cell membrane was labeled with Nile red and visualized via PAINT imaging (green color). OmpR is clearly less abundant compared to RNAP (scale bar 1 μm).
FCS and FCCS measurements of EnvZ/OmpR interaction
Because OmpR appears to be more closely associated with the membrane where its cognate HK EnvZ is located (Fig. 3A and B), we attempted to determine whether OmpR interaction with EnvZ might be dependent on osmolality, pH or other activating signals. Unfortunately, the EnvZ copy number is too low to generate super-resolution images using photoactivatable fusions when envZ is expressed from its native chromosomal locus.2 Thus, we turned to in vitro methods to examine their interaction in solution in response to signals that are known to activate porin gene expression.
Fluorescence correlation spectroscopy (FCS)40,41 and its two-color variant, fluorescence cross-correlation spectroscopy (FCCS)42 are useful for determining protein–protein interactions at low concentrations (~ nanomolar), which is typically the physiological concentrations of many proteins. FCS is capable of determining the stoichiometry of ligand–receptor binding,43 while FCCS reports the strength of protein–protein interactions.44 For these experiments, we purified OmpR and the cytoplasmic domain of EnvZ (EnvZc), which is the osmolality-sensing domain.1,2 EnvZc consists of a four-helix bundle formed by a dimer that contains the phosphorylated histidine and an ATP-binding domain. EnvZc and OmpR were both labeled with Alexa 568 and Alexa 488 at their lone cysteines, respectively.
EnvZc was labeled at its lone cysteine C277 using the maleimide functional group attached to Alexa Fluor 568. We used FCCS to measure the EnvZ/OmpR interaction. OmpR was labeled with Alexa 488 at its lone cysteine at C67 (OmpR-A488, 90 nM), incubated with excess EnvZc-A568 (750 nM), and then FCCS measurements were performed (Fig. 5A). The cross-correlation ratio (the relative amplitude of the cross-correlation function to the ‘‘red’’ autocorrelation function of EnvZc-A568) is an indication of the fraction of OmpR-A488 molecules that were bound to EnvZc-A568. This value was 0.14 ± 0.02, which was very low, given that the range of the cross-correlation ratio for the microscope system is between 0.02 and 0.99 (ESI,† Fig. S1). The low level of complex formation determined by FCCS was also confirmed by native PAGE (ESI,† Fig. S3). This weak interaction was presumably due to interference from the label, as labeling at OmpRC67 is known to decrease phosphorylation.23
Fig. 5.
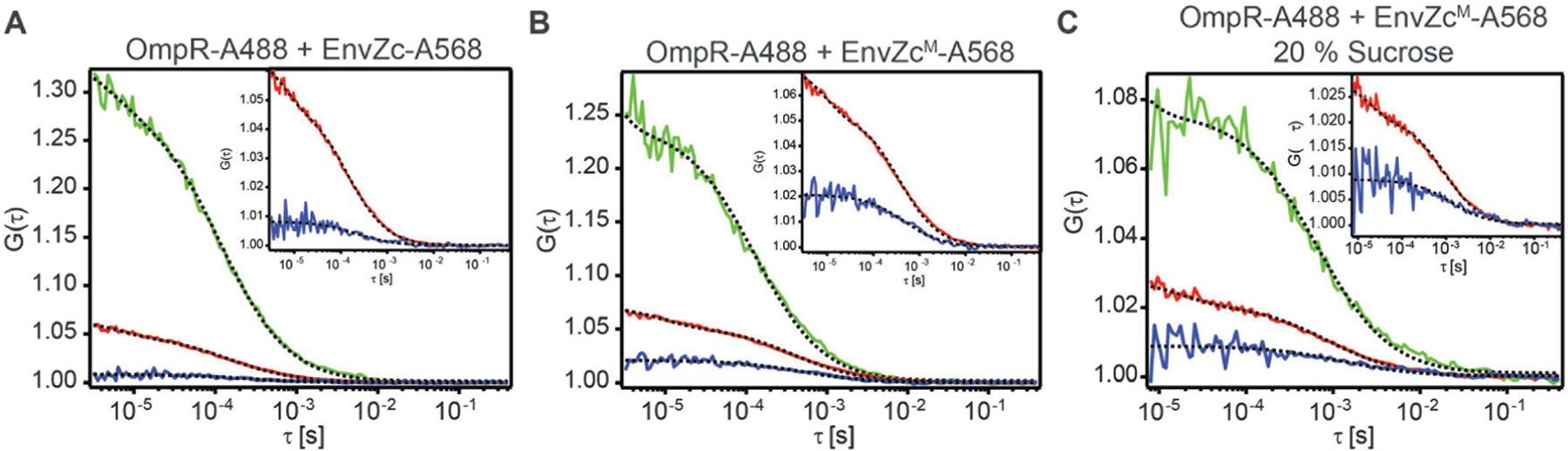
(A) FCCS measurement of EnvZc-A568 + OmpR-A488. Inset shows the close up of the relative amplitude of the CCF to the ACF of EnvZc-A568 (cross-correlation ratio). The fraction of OmpR-A488 bound to EnvZc-A568 is 0.14 ± 0.02. (B) FCCS measurement of EnvZcM-A568 + OmpR-A488. The fraction of OmpR-A488 bound to EnvZcM-A568 is 0.42 ± 0.08. (C) FCCS measurement of EnvZcM-A568 + OmpR-A488 in the presence of 20% sucrose. The fraction of OmpR-A488 bound to EnvZc-A568 is 0.37 ± 0.03.
In contrast, when the native cysteine of EnvZ was substituted with methionine45 and a lone cysteine was introduced in a new location between the four-helix bundle and the ATP binding subdomains (C277M/M294C), the labeled EnvZcM (EnvZcM-A568) was able to interact with OmpR. The FCCS measurement indicated that the fraction of OmpR that was bound to EnvZcM-A568 was three times higher (0.42 ± 0.08) than that of the EnvZc-A568/OmpR interaction (Fig. 5B). This result suggests that the four-helix bundle where C277 resides is important for interaction with OmpR, since labelling at C277 prevented OmpR interaction with EnvZc. This finding is consistent with previous studies analyzing conserved specificity determinants,46 co-variance,47 NMR titration experiments,48 and hydrogen : deuterium exchange mass spectrometry,1 which predicted that the four-helix bundle, and the second helix in particular, is the site of interaction for EnvZ with OmpR. Again, we confirmed our FCCS measurements by native PAGE (ESI,† Fig. S3).
Environmental conditions such as high osmolality that activate the EnvZ/OmpR system may do so by stimulating formation of an EnvZ–OmpR complex. We therefore examined the effect of high osmolality on the EnvZc interaction with OmpR (Fig. 5C). Similar FCCS measurements were performed in 20% sucrose and produced a fraction of OmpR bound to EnvZ that was 0.37 ± 0.03, i.e., similar to the result in low osmolality (Fig. 5B). Thus, the interaction of OmpR with EnvZ was not sensitive to increased osmolality and complex formation does not appear to play a role in activation during osmotic stress.
Two OmpR molecules bind to an EnvZ dimer
In order to determine the stoichiometry of the EnvZ/OmpR interaction, we performed an FCS experiment by titrating a fixed amount of OmpR-A488 (280 nM) with increasing amounts of unlabeled EnvZcM. The auto-correlation functions of OmpR-A488 provide information such as the average number of OmpR-A488 molecules and the change in diffusion time upon binding to the larger EnvZcM dimer (ESI,† Fig. S4). The number of OmpR-A488 molecules obtained from FCS at low EnvZcM concentrations was ~20 (Fig. 6A). At higher concentrations of EnvZcM (>100 nM), the number of OmpR-A488 molecules decreased by nearly 50% to ~11 (Fig. 6A). This observation indicates that two OmpR molecules bind to a single EnvZcM dimer. The reduction in the number of OmpR-A488 molecules was a result of detection of an EnvZc/OmpR complex as a single molecule instead of two OmpR individual molecules, halving N. As expected, the fluorescence count rate per molecule (cpm), obtained from normalizing the total fluorescence intensity by the number of molecules obtained from FCS at higher EnvZcM concentrations increased ~two fold compared to that obtained at low EnvZcM (Fig. 6B). The doubling of cpm values further supports the premise that two OmpR molecules are present in a single EnvZ/OmpR complex, resulting in a stoichiometry of 2EnvZ : 2OmpR. This configuration is presumably 1 OmpR bound to each EnvZ molecule of the dimer (see Discussion).
Fig. 6.
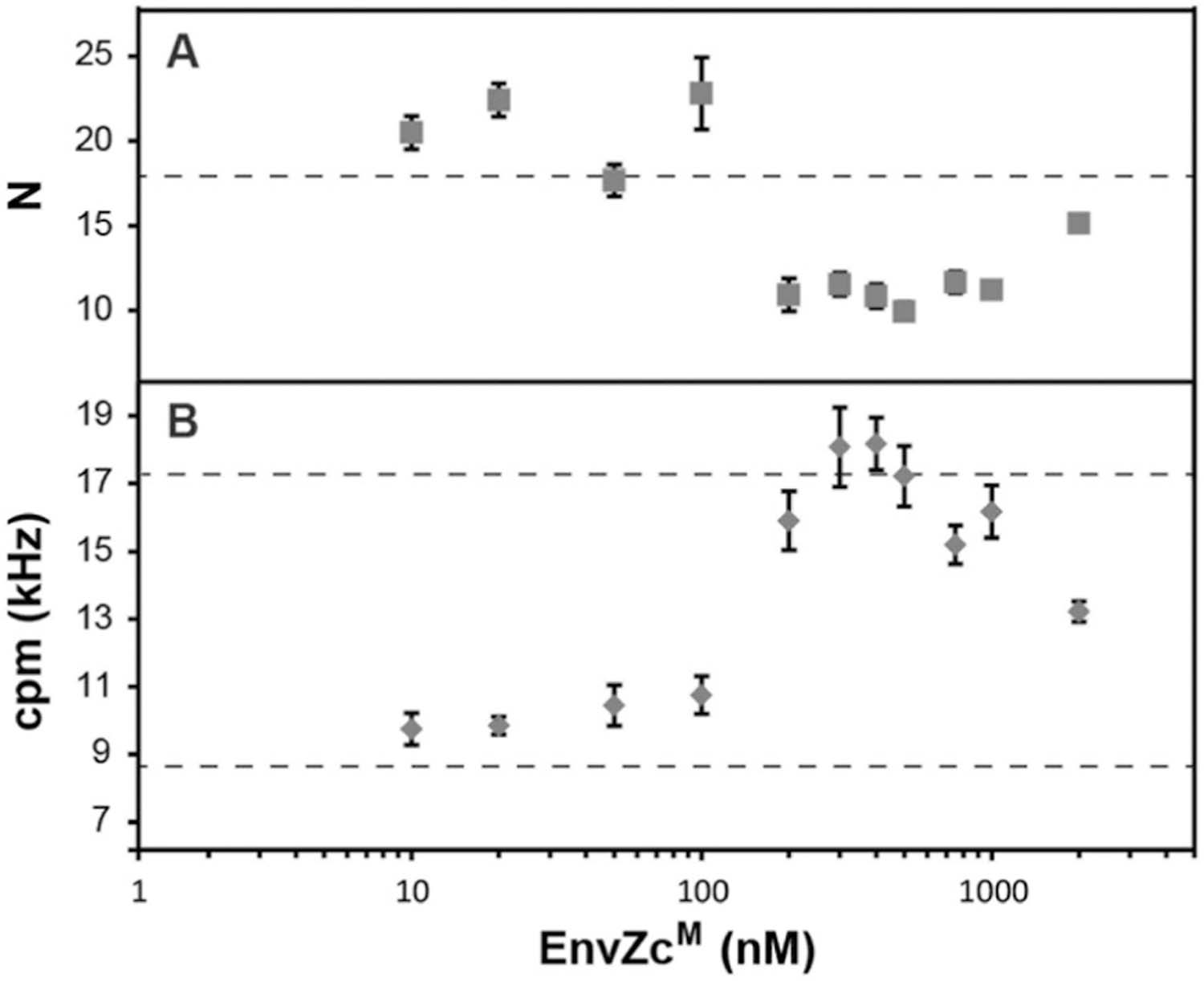
Titration of EnvZcM with 280 nM OmpR-A488. (A) The average number of OmpR-A488 (N). The dotted line indicates the value of N for the sample with only OmpR-A488. (B) Count rate per molecule (cpm) values were obtained by normalizing the fluorescence intensity by N. The lower dotted line indicates the cpm for the sample with only OmpR-A488. The upper dotted line indicates the expected cpm value (i.e., twice that of a monomer) of an OmpR-A488 dimer.
Discussion
In the present study, we constructed a photoactivatable fusion of OmpR that was expressed under its native promoter on the chromosome. The PAmCherry fusion was nearly as active as the wildtype protein, especially at ompF (93%), and it was ~70% as active as wildtype at ompC (Fig. 1). For reasons that were not obvious, the mEos2 construct containing identical linkers was not as active as the PAmCherry fusion (37% and 17% at ompF and ompC, respectively). Increasing the linker length to 40 amino acids improved the activity, but it was still lower compared to OmpR–PAmCherry. The active PAmCherry fusion enabled us to image OmpR in single cells under different environmental stress conditions, which provided new insights into the EnvZ/OmpR two-component system.
OmpR localizes to the edges of the nucleoid and near the inner membrane
Super-resolution microscopy allowed us to visualize OmpR, the bacterial chromosome, and the cell membrane in single cells. PALM imaging revealed that OmpR localized preferentially at the nucleoid edges (Fig. 2A) and close to the cell membrane under low salt and acidic conditions (Fig. 2C and D). We anticipate that OmpR would be bound to EnvZ under these conditions, but we were unable to generate a super-resolution image of EnvZ due to its low copy number. Visualizing EnvZc by super-resolution required over-expression.2
Interestingly, bacterial nucleoids appeared to be more condensed under acidic conditions (ESI,† Fig. S2). Nucleoid compaction was also evident in Fig. 2C, by comparing the green outline of the chromosome between samples (compare panels in the third row). This compaction could be an acid stress response, reducing acid-induced damage by reducing the exposed chromosome surface. Nucleoid-associated proteins such as Dps, contribute to acid stress tolerance in E. coli O157:H7.49,50 Further experiments are needed to provide more insight into the underlying mechanisms of acid-induced DNA compaction and the role of nucleoid-associated proteins in this process.
Chromosomal linkages to the inner membrane
One of the elements of the replicon model was that the chromosome was attached to the inner membrane.51 The attachment was postulated to play a role in the initiation of replication and to enable effective chromosome partitioning.52 Other studies reported an attachment at 20–30 membrane sites in addition to oriC.53 Recently, dynamic interactions of DNA with membrane proteins or associated pathways have been identified.54,55 Alternately, this linkage might result from an interaction with DNA coupling transcription, translation and insertion of membrane proteins56,57 i.e. a DNA–RNAP–mRNA–membrane interaction (transertion). We observed linkages stretching from the chromosome to the membrane that were evident in axial cross-sections (Fig. 3B). Understanding the molecular basis of these linkages requires further experimentation (e.g. a means of generating a high number of axially orientated bacteria) and the ability to identify EnvZ and other candidate molecules at low copy number.
OmpR localization near the membrane
It was interesting that in the cross-sectional images (Fig. 3), OmpR was located very close to the membrane. We attempted to determine whether it was in a complex with EnvZ, which would be expected. Unfortunately, we could not generate a super-resolution image because of the low in vivo levels of EnvZ. Other transcription factors are known to localize to the inner membrane as a regulatory strategy, including PutA,58 Mlc,59 MalT,60 TraR61 and NifL,62 but this strategy has not been shown to be involved in the EnvZ/OmpR signaling system. Experiments with artificially tethered BglG and LacI proteins have demonstrated that membrane tethering (or interaction) doesn’t necessarily lead to inactivation,63,64 as these tethered chimeras were still capable of binding to DNA. There are also examples of response regulators that lack the receiver domain, i.e. ToxR65 and CadC,66,67 but instead they are embedded in the inner membrane and they are clearly capable of binding to DNA. This brings us to the question of whether a complex of DNA–OmpR–EnvZ exists in vivo?23 Because of the difficulties of two-color PALM imaging in bacteria, addressing this question will require the development of new fluorescent proteins, a task that is underway currently in our laboratory.
The discrepancy between molecule counting by PALM and quantitative western blots
Comparing PALM images of OmpR with those of RNAP allowed us to assess the relative expression level of OmpR. In rich medium such as LB, RNAP copy numbers were determined to be B3600 copies per cell.16 OmpR copy numbers were significantly lower compared to RNA polymerase (Fig. 4). This result raises doubts as to the levels of OmpR that were previously determined by quantitative western blotting to be 2000–3500 copies per cell.39 This discrepancy between copy numbers determined from super-resolution imaging and western blots has recently been reported for numerous other proteins.34,39,68 If OmpR levels are indeed substantially lower than previously reported, this explains why OmpR~P levels were barely detectible using Phos-tag labeling, except in the presence of certain envZ mutations.69
Numbers for the detection efficiency of photoactivatable proteins vary widely. Copy numbers obtained by PALM-imaging compared to quantitative western blots yielded only 3.6% detection efficiency for PAmCherry,34 while the use of a nanotemplate resulted in 45–50% detectable proteins.68 Our previous study determined RNAP copy numbers using quantitative PALM resulted in ~3600 RNAP molecules per cell, in good agreement with quantitative western blots (~2600 molecules per cell).70 Even if a fraction of PAmCherry molecules does not photoactivate or fold properly, the number of active molecules can be compared, since both OmpR and RNAP fusions carry the same fluorescent protein with similar biological and photophysical properties, further emphasizing that the number of OmpR molecules are substantially lower than the 3500 copies reported from quantitative western blotting.39
OmpR binding to EnvZ is cooperative
When the EnvZc concentration exceeded 280 nM, i.e., when the stoichiometry of EnvZc to OmpR was more than 1 : 1, our data showed that the number of OmpR molecules in the EnvZc/OmpR complex remained at ~2, at least up to ~2 μM EnvZcM (Fig. 6). This would be expected if OmpR binds to EnvZ cooperatively. At an excess of EnvZcM (>280 nM), a non-cooperative reaction would ensure that most EnvZcM dimers would bind only one OmpR molecule. This would result in a decrease of cpm in the FCCS experiment back to the value of a OmpR monomer (i.e. cpm of ~17). If the reaction is cooperative, the first OmpR molecule that binds to an EnvZcM dimer will recruit the second OmpR molecule due to the higher affinity for OmpR compared to the EnvZcM dimer that does not have OmpR bound to it. Our results are consistent with similar findings using native PAGE.71 It clearly is not possible for OmpR to already be a dimer in solution before binding an EnvZcM dimer, as that result would not halve the number of OmpR molecules during the EnvZcM titration. This finding is also consistent with reports of monomeric OmpR in solution72 and our finding that OmpR~P (which does form dimers in solution) has a lower affinity for EnvZ compared to OmpR.73
In summary, we created a chromosomally-encoded, active OmpR–PAmCherry fusion protein. Using super-resolution microscopy, we showed that OmpR was localized near the membrane in low salt and acidic conditions and that its expression level was substantially lower than RNA polymerase, even though quantitative western blots had suggested they were expressed at similar levels. A linkage from the DNA to the inner membrane was observed, suggesting the possibility of a mechanical link from membrane-bound EnvZ to OmpR to the chromosome.
Supplementary Material
Insight, innovation, integration.
When bacteria encounter certain environmental stresses, the EnvZ/OmpR two-component system alters gene transcription patterns which enables adaptation. We constructed chromosomally-encoded, active, fluorescent fusion proteins and used super-resolution microscopy to visualize the EnvZ/OmpR two-component regulatory system for the first time. Technical innovation enabled us to serially image the chromosome, OmpR and the membrane and then overlay these in a combined image. During environmental stresses such as low osmolality and acid pH, OmpR was juxtaposed next to the membrane and the bacterial chromosome was condensed. In axial cross-sectional images, a linkage stretching from the DNA to the membrane was observed, suggesting a mechanism for mechanical coupling.
Acknowledgements
We are grateful to James Wiesshaar (University of Wisconsin-Madison) for the suggestion of increasing linker lengths between the end of ompR and the PAmCherrry to improve function. MH and CKS acknowledge funding by the German Science Foundation (DFG, grant EXC 115) and by Goethe University Frankfurt. LJK received support from Research Centre of Excellence Grant in Mechanobiology from the Ministry of Education, Singapore and VA 5IOBX000372.
Abbreviations
- FCS
Fluorescence correlation spectroscopy
- FCCS
Fluorescence cross-correlation spectroscopy
- PAmCherry
Photoactivatable mCherry
- Min
Minutes
- RT
Room temperature
- PCR
Polymerase chain reaction
- ACF
Autocorrelation function
- CCF
Cross-correlation function
- PAGE
Polyacrylamide gel electrophoresis
- RNAP
RNA polymerase
Footnotes
Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ib00077g
References
- 1.Wang LC, Morgan LK, Godakumbura P, Kenney LJ and Anand GS, The inner membrane histidine kinase EnvZ senses osmolality via helix-coil transitions in the cytoplasm, EMBO J, 2012, 31, 2648–2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Foo YH, Gao Y, Zhang H and Kenney LJ, Cytoplasmic sensing by the inner membrane histidine kinase EnvZ, Prog. Biophys. Mol. Biol, 2015, DOI: 10.1016/j.pbiomolbio.2015.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alphen WV and Lugtenberg B, Influence of osmolarity of the growth medium on the outer membrane protein pattern of Escherichia coli, J. Bacteriol, 1977, 131, 623–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nikaido H and Rosenberg EY, Porin channels in Escherichia coli: studies with liposomes reconstituted from purified proteins, J. Bacteriol, 1983, 153, 241–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Feng X, Oropeza R and Kenney LJ, Dual regulation by phospho-OmpR of ssrA/B gene expression in Salmonella pathogenicity island 2, Mol. Microbiol, 2003, 48, 1131–1143. [DOI] [PubMed] [Google Scholar]
- 6.Feng X, Walthers D, Oropeza R and Kenney LJ, The response regulator SsrB activates transcription and binds to a region overlapping OmpR binding sites at Salmonella pathogenicity island 2, Mol. Microbiol, 2004, 54, 823–835. [DOI] [PubMed] [Google Scholar]
- 7.Chakraborty S, Mizusaki H and Kenney LJ, A FRET-Based DNA Biosensor Tracks OmpR-Dependent Acidification of Salmonella during Macrophage Infection, PLoS Biol, 2015, 13, e1002116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cayley DS, Guttman HJ and Record MT Jr., Biophysical characterization of changes in amounts and activity of Escherichia coli cell and compartment water and turgor pressure in response to osmotic stress, Biophys. J, 2000, 78, 1748–1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chakraborty S, Kenney LJ, Osmolytes Stimulate Cytoplasmic Acidification in E. coli and S. Typhimurium, 2015, in preparation. [Google Scholar]
- 10.Quinn HJ, Cameron AD and Dorman CJ, Bacterial regulon evolution: distinct responses and roles for the identical OmpR proteins of Salmonella typhimurium and Escherichia coli in the acid stress response, PLoS Genet, 2014, 10, e1004215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Slauch JM, Garrett S, Jackson DE and Silhavy TJ, EnvZ functions through OmpR to control porin gene expression in Escherichia coli K-12, J. Bacteriol, 1988, 170, 439–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Heilemann M, Fluorescence microscopy beyond the diffraction limit, J. Biotechnol, 2010, 149, 243–251. [DOI] [PubMed] [Google Scholar]
- 13.Klein T, Proppert S and Sauer M, Eight years of single-molecule localization microscopy, Histochem. Cell Biol, 2014, 141, 561–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Furstenberg A and Heilemann M, Single-molecule localization microscopy-near-molecular spatial resolution in light microscopy with photoswitchable fluorophores, Phys. Chem. Chem. Phys, 2013, 15, 14919–14930. [DOI] [PubMed] [Google Scholar]
- 15.Betzig E, Patterson GH, Sougrat R, Lindwasser OW, Olenych S, Bonifacino JS, Davidson MW, Lippincott-Schwartz J and Hess HF, Imaging intracellular fluorescent proteins at nanometer resolution, Science, 2006, 313, 1642–1645. [DOI] [PubMed] [Google Scholar]
- 16.Endesfelder U, Finan K, Holden SJ, Cook PR, Kapanidis AN and Heilemann M, Multiscale Spatial Organization of RNA Polymerase in Escherichia coli, Biophys. J, 2013, 105, 172–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lew MD, Lee SF, Ptacin JL, Lee MK, Twieg RJ, Shapiro L and Moerner WE, Three-dimensional super-resolution colocalization of intracellular protein superstructures and the cell surface in live Caulobacter crescentus, Proc. Natl. Acad. Sci. U. S. A, 2011, 108, E1102–E1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sharonov A and Hochstrasser RM, Wide-field subdiffraction imaging by accumulated binding of diffusing probes, Proc. Natl. Acad. Sci. U. S. A, 2006, 103, 18911–18916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Heilemann M, van de Linde S, Schuttpelz M, Kasper R, Seefeldt B, Mukherjee A, Tinnefeld P and Sauer M, Subdiffraction-resolution fluorescence imaging with conventional fluorescent probes, Angew. Chem., Int. Ed. Engl, 2008, 47, 6172–6176. [DOI] [PubMed] [Google Scholar]
- 20.Zhang M, Chang H, Zhang Y, Yu J, Wu L, Ji W, Chen J, Liu B, Lu J, Liu Y, Zhang J, Xu P and Xu T, Rational design of true monomeric and bright photoactivatable fluorescent proteins, Nat. Methods, 2012, 9, 727–729. [DOI] [PubMed] [Google Scholar]
- 21.Subach FV, Patterson GH, Manley S, Gillette JM, Lippincott-Schwartz J and Verkhusha VV, Photoactivatable mCherry for high-resolution two-color fluorescence microscopy, Nat. Methods, 2009, 6, 153–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wurtzel ET, Chou MY and Inouye M, Osmoregulation of gene expression. I. DNA sequence of the ompR gene of the ompB operon of Escherichia coli and characterization of its gene product, J. Biol. Chem, 1982, 257, 13685–13691. [PubMed] [Google Scholar]
- 23.Ames SK, Frankema J and Kenney LJ, C-terminal DNA binding stimulates N-terminal phosphorylation by the outer membrane protein regulator OmpR from Escherichia coli, Proc. Natl. Acad. Sci. U. S. A, 1999, 96, 11792–11797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brissette RE, Tsung K and Inouye M, Mutations in a central highly conserved non-DNA-binding region of OmpR, an Escherichia coli transcriptional activator, influence its DNA-binding ability, J. Bacteriol, 1992, 174, 4907–4912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Haldimann A and Wanner BL, Conditional-replication, integration, excision, and retrieval plasmid-host systems for gene structure-function studies of bacteria, J. Bacteriol, 2001, 183, 6384–6393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hall M and Silhavy TJ, Genetic analysis of the ompB locus in Escherichia coli, J. Mol. Biol, 1981, 151, 1–15. [DOI] [PubMed] [Google Scholar]
- 27.Rhee JE, Sheng W, Morgan LK, Nolet R, Liao X and Kenney LJ, Amino acids important for DNA recognition by the response regulator OmpR, J. Biol. Chem, 2008, 283, 8664–8677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thompson NL, Fluorescence correlation spectroscopy, in Topics in Fluorescence Spectroscopy, ed. Lakowicz JR, Plenum Press, New York, 1991, pp. 337–378. [Google Scholar]
- 29.Spahn C, Cella-Zannacchi F, Endesfelder U and Heilemann M, Correlative super-resolution imagine of RNA polymerase distribution and dynamics, bacterial membrane and chromosomal structure in Escherichia coli, Methods Appl. Fluoresc, 2015, 3, DOI: 10.1088/2050-6120/3/1/014005. [DOI] [PubMed] [Google Scholar]
- 30.Edelstein A, Amodaj N, Hoover K, Vale R and Stuurman N, Computer control of microscopes using microManager, Current Protocols in Cell Biology, 2010, ch. 14, unit 14.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wolter S, Loschberger A, Holm T, Aufmkolk S, Dabauvalle MC, van de Linde S and Sauer M, rapidSTORM: accurate, fast open-source software for localization microscopy, Nat. Methods, 2012, 9, 1040–1041. [DOI] [PubMed] [Google Scholar]
- 32.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P and Cardona A, Fiji: an open-source platform for biological-image analysis, Nat. Methods, 2012, 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang W, Li GW, Chen C, Xie XS and Zhuang X, Chromosome organization by a nucleoid-associated protein in live bacteria, Science, 2011, 333, 1445–1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang S, Moffitt JR, Dempsey GT, Xie XS and Zhuang X, Characterization and development of photoactivatable fluorescent proteins for single-molecule-based superresolution imaging, Proc. Natl. Acad. Sci. U. S. A, 2014, 111, 8452–8457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hall MN and Silhavy TJ, Transcriptional regulation of Escherichia coli K-12 major outer membrane protein 1b, J. Bacteriol, 1979, 140, 342–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Batchelor E and Goulian M, Imaging OmpR localization in Escherichia coli, Mol. Microbiol, 2006, 59, 1767–1778. [DOI] [PubMed] [Google Scholar]
- 37.Snapp E, Design and use of fluorescent fusion proteins in cell biology, Current Protocols in Cell Biology, 2005, ch. 21, unit 21.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Maris AE, Walthers D, Mattison K, Byers N and Kenney LJ, The response regulator OmpR oligomerizes via beta-sheets to form head-to-head dimers, J. Mol. Biol, 2005, 350, 843–856. [DOI] [PubMed] [Google Scholar]
- 39.Cai SJ and Inouye M, EnvZ-OmpR interaction and osmoregulation in Escherichia coli, J. Biol. Chem, 2002, 277, 24155–24161. [DOI] [PubMed] [Google Scholar]
- 40.Elson EL and Magde D, Fluorescence correlation spectroscopy. I. Conceptual basis and theory, Biopolymers, 1974, 13, 1–27. [DOI] [PubMed] [Google Scholar]
- 41.Magde D, Elson EL and Webb WW, Fluorescence correlation spectroscopy. II. An experimental realization, Biopolymers, 1974, 13, 29–61. [DOI] [PubMed] [Google Scholar]
- 42.Schwille P, Meyer-Almes FJ and Rigler R, Dual-color fluorescence cross-correlation spectroscopy for multi-component diffusional analysis in solution, Biophys. J, 1997, 72, 1878–1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wohland T, Friedrich K, Hovius R and Vogel H, Study of ligand-receptor interactions by fluorescence correlation spectroscopy with different fluorophores: evidence that the homopentameric 5-hydroxytryptamine type 3As receptor binds only one ligand, Biochemistry, 1999, 38, 8671–8681. [DOI] [PubMed] [Google Scholar]
- 44.Shi X, Foo YH, Sudhaharan T, Chong SW, Korzh V, Ahmed S and Wohland T, Determination of dissociation constants in living zebrafish embryos with single wavelength fluorescence cross-correlation spectroscopy, Biophys. J, 2009, 97, 678–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Oropeza R and Calva E, The cysteine 354 and 277 residues of Salmonella enterica serovar Typhi EnvZ are determinants of autophosphorylation and OmpR phosphorylation, FEMS Microbiol. Lett, 2009, 292, 282–290. [DOI] [PubMed] [Google Scholar]
- 46.Skerker JM, Perchuk BS, Siryaporn A, Lubin EA, Ashenberg O, Goulian M and Laub MT, Rewiring the specificity of two-component signal transduction systems, Cell, 2008, 133, 1043–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Szurmant H and Hoch JA, Interaction fidelity in two-component signaling, Curr. Opin. Microbiol, 2010, 13, 190–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tomomori C, Tanaka T, Dutta R, Park H, Saha SK, Zhu Y, Ishima R, Liu D, Tong KI, Kurokawa H, Qian H, Inouye M and Ikura M, Solution structure of the homodimeric core domain of Escherichia coli histidine kinase EnvZ, Nat. Struct. Biol, 1999, 6, 729–734. [DOI] [PubMed] [Google Scholar]
- 49.Choi SH, Baumler DJ and Kaspar CW, Contribution of dps to acid stress tolerance and oxidative stress tolerance in Escherichia coli O157:H7, Appl. Environ. Microbiol, 2000, 66, 3911–3916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jeong KC, Hung KF, Baumler DJ, Byrd JJ and Kaspar CW, Acid stress damage of DNA is prevented by Dps binding in Escherichia coli O157:H7, BMC Microbiol, 2008, 8, 181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hendrickson WG, Kusano T, Yamaki H, Balakrishnan R, King M, Murchie J and Schaechter M, Binding of the origin of replication of Escherichia coli to the outer membrane, Cell, 1982, 30, 915–923. [DOI] [PubMed] [Google Scholar]
- 52.Leibowitz PJ and Schaechter M, The attachment of the bacterial chromosome to the cell membrane, Int. Rev. Cytol, 1975, 41, 1–28. [DOI] [PubMed] [Google Scholar]
- 53.Dworsky P and Schaechter M, Effect of rifampin on the structure and membrane attachment of the nucleoid of Escherichia coli, J. Bacteriol, 1973, 116, 1364–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Adachi S, Murakawa Y and Hiraga S, SecA defects are accompanied by dysregulation of MukB, DNA gyrase, chromosome partitioning and DNA superhelicity in Escherichia coli, Microbiology, 2014, 160, 1648–1658. [DOI] [PubMed] [Google Scholar]
- 55.Buss J, Coltharp C, Shtengel G, Yang X, Hess H and Xiao J, A multi-layered protein network stabilizes the Escherichia coli FtsZ-ring and modulates constriction dynamics, PLoS Genet, 2015, 11, e1005128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bakshi S, Choi H, Mondal J and Weisshaar JC, Time-dependent effects of transcription- and translation-halting drugs on the spatial distributions of the Escherichia coli chromosome and ribosomes, Mol. Microbiol, 2014, 94, 871–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Woldringh CL, The role of co-transcriptional translation and protein translocation (transertion) in bacterial chromosome segregation, Mol. Microbiol, 2002, 45, 17–29. [DOI] [PubMed] [Google Scholar]
- 58.Ostrovsky de Spicer P and Maloy S, PutA protein, a membrane-associated flavin dehydrogenase, acts as a redox-dependent transcriptional regulator, Proc. Natl. Acad. Sci. U. S. A, 1993, 90, 4295–4298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tanaka Y, Itoh F, Kimata K and Aiba H, Membrane localization itself but not binding to IICB is directly responsible for the inactivation of the global repressor Mlc in Escherichia coli, Mol. Microbiol, 2004, 53, 941–951. [DOI] [PubMed] [Google Scholar]
- 60.Schlegel A, Bohm A, Lee SJ, Peist R, Decker K and Boos W, Network regulation of the Escherichia coli maltose system, J. Mol. Microbiol. Biotechnol, 2002, 4, 301–307. [PubMed] [Google Scholar]
- 61.Qin Y, Luo ZQ, Smyth AJ, Gao P, Beck von Bodman S and Farrand SK, Quorum-sensing signal binding results in dimerization of TraR and its release from membranes into the cytoplasm, EMBO J, 2000, 19, 5212–5221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Klopprogge K, Grabbe R, Hoppert M and Schmitz RA, Membrane association of Klebsiella pneumoniae NifL is affected by molecular oxygen and combined nitrogen, Arch. Microbiol, 2002, 177, 223–234. [DOI] [PubMed] [Google Scholar]
- 63.Gorke B and Rak B, Efficient transcriptional antitermination from the Escherichia coli cytoplasmic membrane, J. Mol. Biol, 2001, 308, 131–145. [DOI] [PubMed] [Google Scholar]
- 64.Gorke B, Reinhardt J and Rak B, Activity of Lac repressor anchored to the Escherichia coli inner membrane, Nucleic Acids Res, 2005, 33, 2504–2511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Haas BL, Matson JS, DiRita VJ and Biteen JS, Single-molecule tracking in live Vibrio cholerae reveals that ToxR recruits the membrane-bound virulence regulator TcpP to the toxT promoter, Mol. Microbiol, 2015, 96, 4–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Miller VL, Taylor RK and Mekalanos JJ, Cholera toxin transcriptional activator toxR is a transmembrane DNA binding protein, Cell, 1987, 48, 271–279. [DOI] [PubMed] [Google Scholar]
- 67.Rhee JE, Rhee JH, Ryu PY and Choi SH, Identification of the cadBA operon from Vibrio vulnificus and its influence on survival to acid stress, FEMS Microbiol. Lett, 2002, 208, 245–251. [DOI] [PubMed] [Google Scholar]
- 68.Durisic N, Laparra-Cuervo L, Sandoval-Alvarez A, Borbely JS and Lakadamyali M, Single-molecule evaluation of fluorescent protein photoactivation efficiency using an in vivo nanotemplate, Nat. Methods, 2014, 11, 156–162. [DOI] [PubMed] [Google Scholar]
- 69.Adediran J, Leatham-Jensen MP, Mokszycki ME, Frimodt-Moller J, Krogfelt KA, Kazmierczak K, Kenney LJ, Conway T and Cohen PS, An Escherichia coli Nissle 1917 missense mutant colonizes the streptomycin-treated mouse intestine better than the wild type but is not a better probiotic, Infect. Immun, 2014, 82, 670–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Piper SE, Mitchell JE, Lee DJ and Busby SJ, A global view of Escherichia coli Rsd protein and its interactions, Mol. BioSyst, 2009, 5, 1943–1947. [DOI] [PubMed] [Google Scholar]
- 71.Yoshida T, Qin L and Inouye M, Formation of the stoichiometric complex of EnvZ, a histidine kinase, with its response regulator, OmpR, Mol. Microbiol, 2002, 46, 1273–1282. [DOI] [PubMed] [Google Scholar]
- 72.Jo YL, Nara F, Ichihara S, Mizuno T and Mizushima S, Purification and characterization of the OmpR protein, a positive regulator involved in osmoregulatory expression of the ompF and ompC genes in Escherichia coli, J. Biol. Chem, 1986, 261, 15252–15256. [PubMed] [Google Scholar]
- 73.King ST and Kenney LJ, Application of fluorescence resonance energy transfer to examine EnvZ/OmpR interactions, Methods Enzymol, 2007, 422, 352–360. [DOI] [PubMed] [Google Scholar]
- 74.Spahn C, Endesfelder U and Heilemann M, Super-resolution imaging of Escherichia coli nucleoids reveals highly structured and asymmetric segregation during fast growth, J. Struct. Biol, 2014, 185, 243–249. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.