

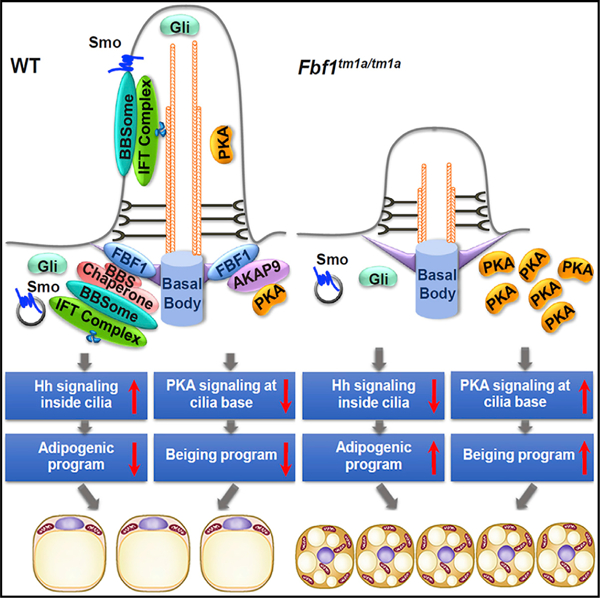
SUMMARY
Preadipocytes dynamically produce sensory cilia. However, the role of primary cilia in preadipocyte differentiation and adipose homeostasis remains poorly understood. We previously identified transition fiber component FBF1 as an essential player in controlling selective cilia import. Here, we establish Fbf1tm1a/tm1a mice and discover that Fbf1tm1a/tm1a mice develop severe obesity, but surprisingly, are not predisposed to adverse metabolic complications. Obese Fbf1tm1a/tm1a mice possess unexpectedly healthy white fat tissue characterized by spontaneous upregulated beiging, hyperplasia but not hypertrophy, and low inflammation along the lifetime. Mechanistically, FBF1 governs preadipocyte differentiation by constraining the beiging program through an AKAP9-dependent, cilia-regulated PKA signaling, while recruiting the BBS chaperonin to transition fibers to suppress the hedgehog signaling-dependent adipogenic program. Remarkably, obese Fbf1tm1a/tm1a mice further fed a high-fat diet are protected from diabetes and premature death. We reveal a central role for primary cilia in the fate determination of preadipocytes and the generation of metabolically healthy fat tissue.
Graphical abstract
In brief
Zhang et al. demonstrate that Fbf1tm1a/tm1a mice exhibit ‘‘healthy obesity’’ with healthy and beiging white fat tissue. Mechanistically, FBF1 governs preadipocyte differentiation by constraining the beiging program through cilia-regulated PKA signaling, while suppressing the Hh-dependent adipogenic program. Fbf1tm1a/tm1a mice fed a high-fat diet are protected from diabetes and premature death.
INTRODUCTION
Obesity and associated type 2 diabetes mellitus (T2DM) constitute a public health crisis. Factors that lead to adipose tissue dysfunction cause chronic inflammation in adipose tissue, insulin resistance, and the ‘‘spillover’’ of fat to other tissues, lipotoxicity, and systemic organ dysfunction in metabolic syndrome (Després et al., 2008; Galic et al., 2010). Unlike energy-storing white adipocytes, brown/beige adipocytes dissipate energy in the form of heat (Harms and Seale, 2013). Adult humans, especially obese or aged individuals, possess little or no active brown adipose tissue (BAT) (Harms and Seale, 2013). White adipose tissue (WAT) browning, or beiging, can be strongly induced by certain environmental stimuli such as cold exposure (Nedergaard and Cannon, 2013). WAT-to-BAT metabolic conversion of adipocytes is inversely associated with metabolic complications (Cohen et al., 2014), which makes the beige adipocyte an attractive target for therapeutic strategies to combat metabolic disorders. However, the intrinsic mechanism of WAT beiging remains as a challenging question.
Obesity is caused by genetics or the environment. Although monogenic obesities are rare, the elucidation of the physiological function of causal genes, such as leptin in ob/ob mutant mice, was transformative in understanding obesity physiopathology. Primary cilia are sensory organelles emanating from the surface of most eukaryotic cells (Goetz and Anderson, 2010; Singla and Reiter, 2006). Dozens of human rare diseases, such as Bardet-Biedl syndrome (BBS), Alström syndrome (ALMS), and Joubert syndrome, have been characterized as ciliopathies (Goetz and Anderson, 2010). Notably, a significant share of genetic obesity involves ciliopathies (Oh et al., 2015; Vaisse et al., 2017). In the prevailing view by studying ciliopathy models, abnormal satiety signaling mediated by neuronal cilia leads to hyperphagia and obesity (Vaisse et al., 2017). A few in vitro studies showed that the differentiating preadipocyte is transiently ciliated, and dysfunctional ciliopathy genes could promote or inhibit adipogenesis through unclear mechanisms (Oh et al., 2015). Intriguingly, morbid obesity and ciliopathy ALMS patients, but not some BBS patients, are predisposed to T2DM (Feuillan et al., 2011). This highlights that cilia-dysfunction-induced obesity is complex and could involve different ciliated organ/tissues, and alterations to particular cilia pathways may even be protective against metabolic disorders.
Unlike other membrane-enclosed organelles, the ciliary lumen is open to the cytoplasm. Tightly regulated cilia trafficking at cilia base (i.e., cilia gating) is required to maintain proper protein composition and make the cilium a distinct sensory antenna. Non-ciliary membrane proteins cannot pass through a lateral diffusion barrier at cilia base (Vieira et al., 2006). For soluble proteins, a size-dependent gate at cilia base controls selective import (Kee et al., 2012; Lin et al., 2013; Nachury et al., 2010). Transition fibers (TFs) (transformed from the distal appendages of the mother centriole; Anderson, 1972) may constitute a critical part of the proposed ciliary gate (Reiter et al., 2012; Rosenbaum and Witman, 2002). TFs anchor basal bodies to the apical membrane and constitute a visible physical barrier between the cytoplasm and the ciliary lumen (Anderson, 1972). Emerging evidence suggests that TF dysfunction leads to impaired cilia gating for both membrane and soluble proteins (Wei et al., 2015). Among identified TF components, Fas binding factor 1 (FBF1) is distinctive; its deficiency does not compromise TF integrity but specifically disrupts cilia gating (Wei et al., 2013, 2016).
In this study, we established an Fbf1tm1a/tm1a mouse model to assess the physiological consequences of FBF1 inactivation. Fbf1tm1a/tm1a adult mice develop progressive obesity, but surprisingly without hyperphagia. Despite greater adiposity, adult Fbf1tm1a/tm1a mice are robustly protected from glucose and insulin resistance. Unexpectedly, both subcutaneous WAT (sWAT) and visceral WAT (vWAT) of obese Fbf1tm1a/tm1a mice show spontaneous beiging, along with other healthy features, including hyperplasia and minimal inflammation along the lifetime. The FBF1 level is inversely correlated with the activation of beiging programs. Mechanistically, FBF1 associates with the A-kinase anchoring protein AKAP9 on TFs to regulate local homeostasis of protein kinase A (PKA) holoenzymes at cilia base. Ectopic PKA accumulation at cilia base in FBF1-deficient cells induces the beiging program in a p38-dependent manner. A cilia base-specific PKA inhibition effectively blunts the beiging program in FBF1-depleted cells. At the same time, other than its role in regulating beiging, FBF1 also recruits the BBS chaperone complex to TFs, a process ensuring proper BBSome assembly and BBSome-dependent Hh signaling. Impaired Hh signaling in FBF1-depleted adipose precursors results in enhanced adipogenesis and thus contributes to the healthy expansion of adipose tissue. We further demonstrated that the role of FBF1 in beiging and adipogenesis is highly conserved in primary isolated human adipocyte precursors. More exciting is that the Fbf1tm1a/tm1a mice are protected from insulin resistance and premature death upon a high-fat diet treatment, underscoring the preadipocyte cilium as an intriguing therapeutic target in the future treatment of metabolic disorders.
RESULTS
Fbf1tm1a/tm1a mice develop obesity without metabolic syndrome
To determine the in vivo physiological importance of Fbf1, we established and examined homozygous Fbf1tm1a/tm1a mutant mice. Fbf1tm1(KOMP)Vlcg embryonic stem cells (ESCs) were generated by a ‘‘knockout first strategy’’ (Skarnes et al., 2011) and obtained from the KIOMP Repository (University of California, Davis) (Figure S1A). We established and bred Fbf1tm1a/tm1a homozygous mice in C57BL/6 background (hereafter called Fbf1tm/tm) (Figures S1B and S1C). Approximately 50% of homozygous Fbf1tm/tm mice die before birth, and ~15% develop hydrocephalus, small size, and die within 4 weeks postnatally. Intriguingly, the surviving Fbf1tm/tm mice were otherwise healthy but developed progressive obesity on ad libitum feeding with regular chow (Figures 1A–1C). Analyses of whole Fbf1tm/tm embryos revealed that the inserted knockout first cassette does not completely block Fbf1 transcription, resulting in an ~5% leaky expression of Fbf1 mRNA and residual FBF1 protein translated in Fbf1tm/tm embryos (Figures S1D and S1E). This indicates that the Fbf1 tm/tm mouse line is a hypomorphic line and explains the incomplete penetrance of embryonic lethality, a phenotype commonly associated with other ciliopathy mice models. In adult mice, Fbf1 is expressed globally but shows higher expression levels in heart, muscle, and fat tissues (Figure S1F). Fbf1 expression is disrupted in various organs examined in adult Fbf1tm/tm mice (Figure S1G).
Figure 1. Fbf1tm/tm homozygous mice exhibit ‘‘healthy obesity’’.
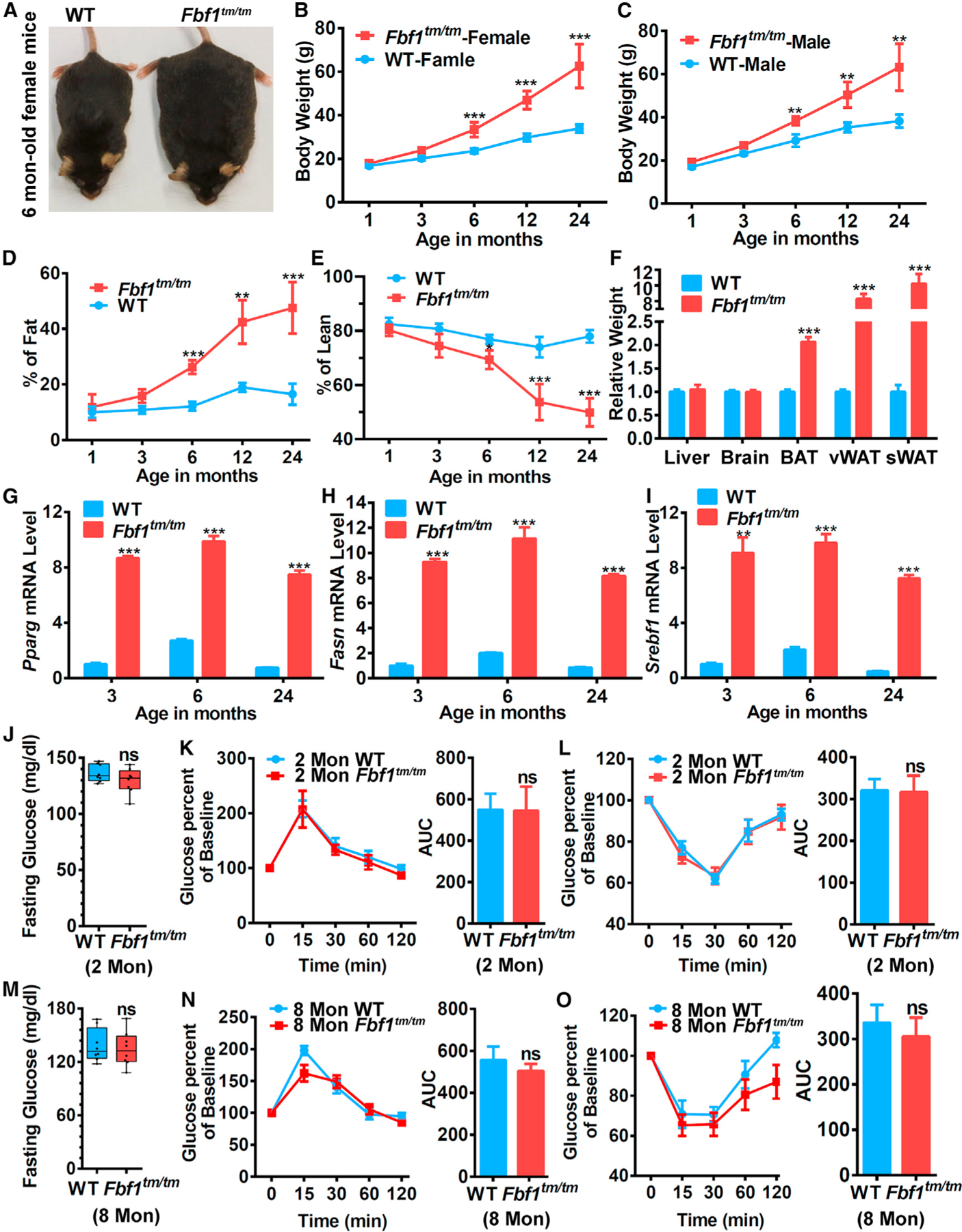
(A) A comparison between 6-month-old Fbf1tm/tm and WT littermate.
(B and C) Growth curve of (B) female and (C) male Fbf1tm/tm/WT littermates.
(D and E) Fat (D) and (E) lean mass percentage were measured by MRI in different ages.
(F) Relative tissue weight in 24-month-old Fbf1tm/tm/WT littermates.
(G–I) Adipogenic gene expression levels of vWAT in different ages.
(J–L) Fasting blood glucose (J), (K and L) glucose and insulin tolerance tests, with corresponding area under the curves in 2-month-old Fbf1tm/tm/WT littermates.
(M–O) Fasting blood glucose (M), (N and O) glucose and insulin tolerance tests, with corresponding area under the curves in 8-month-old Fbf1tm/tm/WT littermates.
Values are expressed as means ± SEMs, n = 8 in (B)–(E) and (J)–(O) in each group. p values were indicated as follows: *p < 0.05, **p < 0.01, and ***p < 0.001.
Fbf1tm/tm mice develop progressive obesity throughout their lives. The fat mass reached >200% of controls at 24 months, with lean mass reduced accordingly (Figures 1D and 1E). Intriguingly, both WAT and BAT expanded dramatically in adult Fbf1tm/tm mice (Figures 1F). Consistently, expression of the adipogenic markers Pparg, Fasn, and Srebf1, were robustly upregulated in Fbf1tm/tm adipose tissue throughout their lifespan (Figures 1G–1I). Young Fbf1tm/tm mice (2 months old) were not obese and possessed a healthy metabolic profile (Figures 1J–1L). We were surprised to find that although already severely obese, 8-month-old Fbf1tm/tm mice also showed no signs of glucose intolerance and insulin resistance (Figures 1M–1O).
Fbf1tm/tm mice show no hyperphagia
Hyperphagia drives obesity in reported ciliopathy rodent models (Vaisse et al., 2017). Intriguingly, Fbf1tm/tm mice showed completely normal food intake throughout their lives (Figures 2A, S2A, and S2B). In comparison with age-matched wild-type (WT) littermates under either fed or fast conditions, the total energy expenditure was similar in non-obese 2-month-old Fbf1tm/tm mice but reduced in obese 8-month-old Fbf1tm/tm mice (Figures 2B, S2C, and S2D). Of note, a subtle decrease in physical activities started to show up in 2-month-old Fbf1tm/tm mice under rest conditions and then expanded to both fed and fast conditions in 8-month-old animals (Figures 2C, 2D and S2E–S2H).
Figure 2. FBF1 depletion induces the in vivo beiging of WAT.
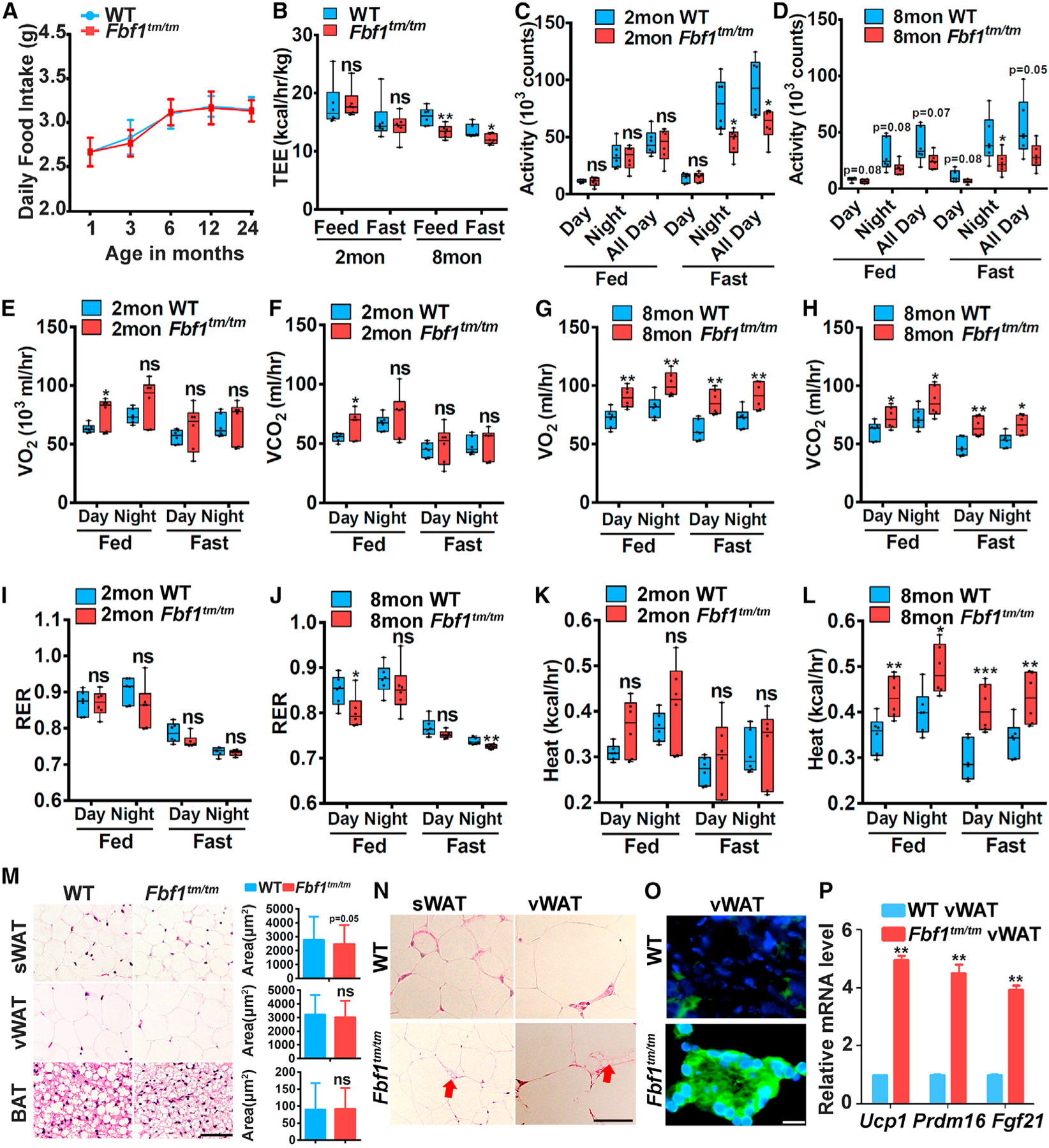
(A) Food intake of Fbf1tm/tm/WT littermates.
(B–L) Multiple metabolic parameters were monitored in Fbf1tm/tm/WT littermates at 2 and 8 months.
(B) Total energy expenditure was monitored by CLAMS in Fbf1tm/tm/WT littermates.
(C and D) Activity in 2- (C) and 8-month-old (D) mice.
(E and F) O2 consumption (E) and (F) CO2 production of 2-month-old mice.
(G and H) O2 consumption (F) and (H) CO2 production of 8-month-old mice.
(I–L) Average O2 consumption and CO2 production were measured by CLAMS and used to calculate RER (I and J) and heat production (K and L) in 2- and 8-month-old mice.
(M) H&E staining of WATs and BAT from 6-month-old WT/Fbf1tm/tm female mice. Scale bar, 150 µm. The adipocyte area was calculated from 200 adipocytes in each group.
(N) H&E staining of sections of WATs of Fbf1tm/tm and WT littermates. Arrow: multilocular beige adipocytes. Scale bar, 150 µm.
(O) Immunofluorescence staining of UCP1 in vWAT. Scale bar, 100 µm.
(P) Relative mRNA levels of beiging genes in vWAT.
Values are expressed as means ± SEMs, n = 8 in (A) and n = 6 in (B)–(I) in each group. p values were indicated as follows: *p < 0.05, **p < 0.01, and ***p < 0.001.
Obese Fbf1tm/tm mice show decreased respiratory exchange ratio (RER)
In a Comprehensive Lab Animal Monitoring System (CLAMS) analysis, Fbf1tm/tm mice showed a slightly but significantly higher O2 consumption and CO2 production than WT animals during normal chow feeding at 2 months old (Figures 2E and 2F). O2 consumption and CO2 production was further increased in obese 8-month-old Fbf1tm/tm mice with or without food (Figures 2G and 2H). RER data showed that the average RER of non-obese 2-month-old mice appears to be lower but not statistically different from controls (Figure 2I), while the changes become significant in obese 8-month-old mice (Figure 2J), signifying a preference for fat over carbohydrate as the metabolic substrate in obese Fbf1tm/tm mice. Consistently, when comparing thermogenesis in mice maintained at room temperature, the heat slightly increased in 2-month-old Fbf1tm/tm mice (but not statistically different from controls; Figure 2K) and exhibited a statistically sigificant increase in 8-month-old Fbf1tm/tm mice (Figure 2L).
FBF1 depletion induces WAT beiging
White-to-brown metabolic conversion of adipocytes is inversely associated with RER and insulin resistance (Cohen et al., 2014). We asked whether Fbf1 depletion upregulates the beiging program in adipocytes. Histological examination revealed no hypertrophy in Fbf1tm/tm fat tissue (including sWAT, vWAT, and BAT) (Figure 2M). Mouse white adipocytes contain fewer but larger lipid droplets, whereas brown adipocytes have more but smaller lipid droplets (Harms and Seale, 2013). Notably, multilocular beige-like adipocytes were frequently observed in the Fbf1tm/tm sWAT and vWAT (Figure 2N). The hallmark of adipocyte beiging is the upregulation of Uncoupling protein 1 (UCP1), a mitochondrial protein that, when active, uncouples the oxidation of fuel from ATP production to heat generation (Harms and Seale, 2013). The UCP1 level dramatically increased in Fbf1tm/tm vWAT (Figure 2O). Consistently, transcription signature of the beiging program was strongly upregulated in Fbf1tm/tm vWAT and BAT (Figures 2P, S2I, and S2J).
FBF1 level is inversely correlated with beiging activation
We isolated preadipocytes from Fbf1tm/tm vWAT and subjected them to in vitro adipogenesis induction. The morphology and size distribution of lipid droplets in Fbf1tm/tm adipocytes were typical of brown-like adipocytes on day 10 of differentiation (Figures 3A–3D). This is intriguing since adipogenesis induction could not induce the beiging program in WT controls. In vitro differentiated Fbf1tm/tm adipocytes were accompanied by elevated UCP1 expression and increased mitochondria content (Figures 3E and 3F). Knocking down Fbf1 in 3T3-L1 cells also upregulated UCP1 expression and beiging gene transcription upon adipogenesis induction (Figures S3A–S3C).
Figure 3. FBF1 level is inversely correlated with the activation of the beiging program.
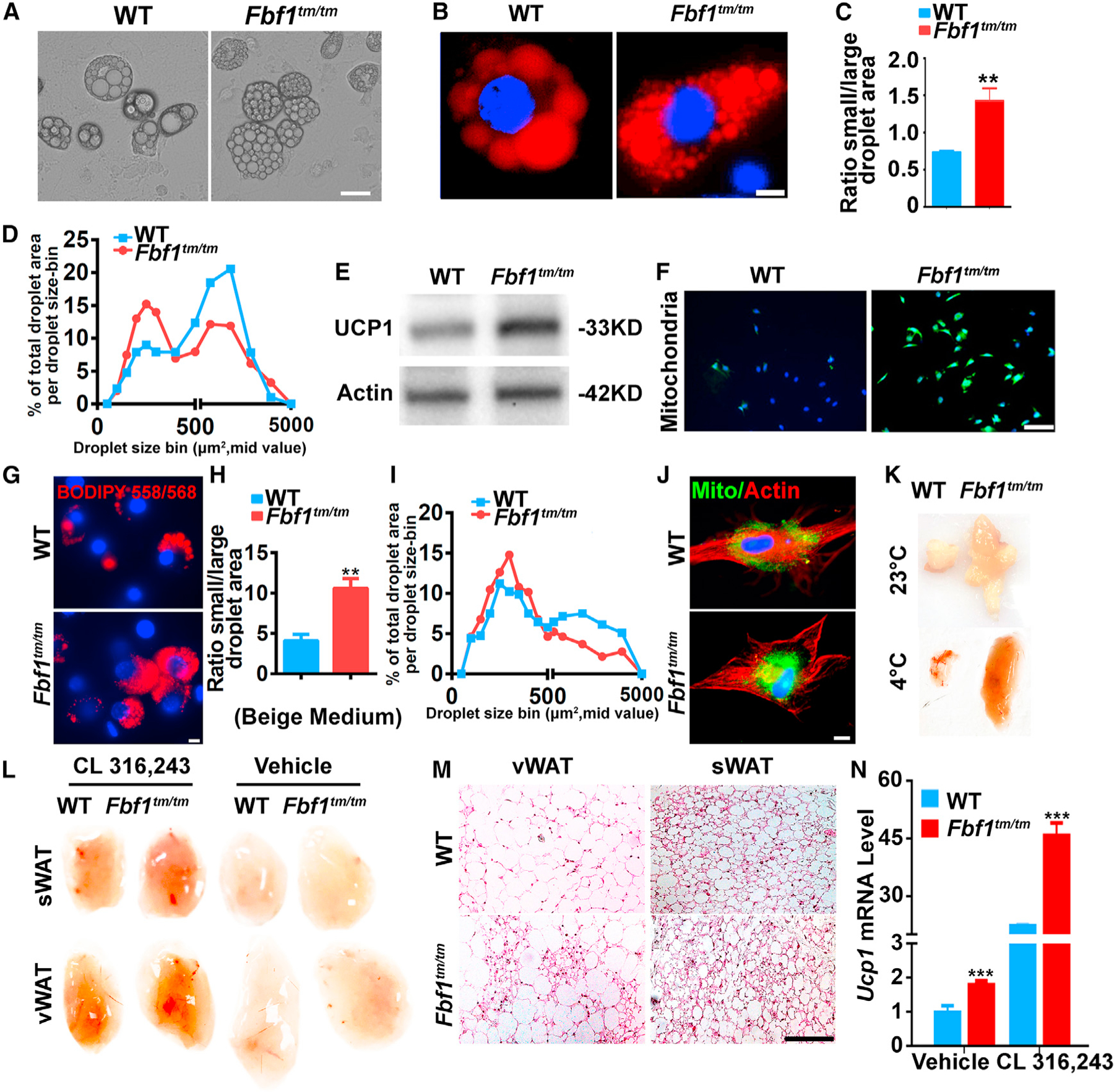
(A and B) Preadipocytes isolated from vWAT and differentiated. At day 10, (A) bright-field images were taken. Scale bar, 35 µm; (B) cells were fixed, stained with BIDIPY. Red: lipids, blue: nuclei. Scale bar, 50 µm.
(C) Bar graph illustrating the quantification of a brown-like lipid index, determined by calculating the ratio of total area for small (<1,000 µm2) versus large (>1,000 µm2) lipid droplets.
(D) Quantification of changes in lipid morphology is depicted as fraction of total lipid area per lipid droplet size.
(E) Immunoblotting on UCP-1 in preadipocytes from vWAT.
(F) Preadipocytes from vWAT were cultured and differentiated for 7 days, then labeled with mitochondria-GFP. At day 8, cells were fixed, stained, and imaged by fluorescence microscopy. Green: mitochondria, blue: nuclei. Scale bar, 200 µm.
(G–J) Preadipocytes from vWAT were cultured and induced to brown-like lipid morphology.
(G) At day 7, cells were fixed, stained with BIDIPY. Red: lipids, blue: nuclei. Scale bar, 50 µm.
(H) Bar graph illustrating the quantification of a brown-like lipid index, determined by calculating the ratio of total area for small (<1,000 µm2) versus large (>1,000 µm2) lipid droplets.
(I) Quantification of changes in lipid morphology is depicted as fraction of total lipid area per lipid droplet size.
(J) Cells were fixed, stained, and imaged. Green: mitochondria, red: actin, blue: nuclei. Scale bar, 50 µm.
(K) Photograph of vWAT collected from Fbf1tm/tm/WT control exposed to 23°C or 4°C for 2 days.
(L) Photograph or WATs collected from Fbf1tm/tm/WT control treated with vehicle or CL-316,243.
(M and N) H&E staining (M) and (N) Ucp1 mRNA level of WATs collected from mice treated with vehicle or CL-316,243.
It is interesting that the beiging of WAT does not reduce adiposity in Fbf1tm/tm mice. One probable explanation is that the 5-fold increase in UCP1 expression could prevent individual adipocytes from hypertrophy, but is not sufficient to mediate significant thermogenesis to prevent Fbf1tm/tm mice from developing obesity. In the beiging mouse model that shows the lean phenotype, up to a 100-fold increase in UCP1 expression was observed (Nedergaard and Cannon, 2013). As expected, after treating cells with beiging induction medium, both Fbf1tm/tm adipocytes and siFbf1-treated 3T3-L1 cells showed more robust beiging phenotypes in the context of droplet size, UCP1 level, and mitochondria content (Figures 3G–3J and S3D). To confirm that Fbf1tm/tm mice were naturally predisposed to WAT beiging, we also performed cold acclimatization experiments. After exposing mice to 4°C for 2 days, we could visually detect stronger beiging of vWAT in Fbf1tm/tm (Figure 3K). When using β3-adrenergic agonist CL-316,243 to induce adaptive thermogenesis, we observed stronger beiging, more multilocular beige adipocytes, and upregulation of Ucp1 transcription in Fbf1tm/tm mice when compared with WT controls (Figures 3L–3N). Our data demonstrate that FBF1 deficiency produces healthier WAT tissues with the upregulated beiging program, which is not adequate to counteract adiposity but likely sufficient to protect adipocytes from hypertrophy and metabolic dysfunction.
FBF1 regulates adipose beiging through a cilia-specific PKA signaling
Adipose beiging is strongly induced by PKA-p38 activation (Harms and Seale, 2013). Cilia-specific PKA signaling was documented in various cell types (Hilgendorf et al., 2016). Cilia regulate cAMP concentration separately from the rest of the cell, thus affording localization-dependent regulation (Nachury and Mick, 2019). In CL-316,243-induced adaptive thermogenesis, PKA catalytic subunits PKACα accumulated significantly around cilia base in WT vWAT, which was further exaggerated in Fbf1tm/tm WAT (Figure S4A). A similar observation was also confirmed in mouse preadipocytes isolated from WT or Fbf1tm/tm vWAT and treated with CL-316,243 or another β3-adrenergic agonist, mirabegron (Wu et al., 2012; Figures S4B and S4C). To understand the molecular basis for the role of FBF1 in beiging, we used mouse embryonic fibroblasts (MEFs) and/or 3T3-L1 cells in an in vitro differentiation assay. We confirmed that endogenous PKA catalytic subunits PKACα localized at cilia base in WT MEFs or 3T3-L1 cells (Figures S4D and S4E). During induced beiging with β3-adrenergic agonists, PKACα accumulated significantly around cilia base in WT cells, which is further exaggerated in Fbf1tm/tm MEFs or siFbf1-treated 3T3-L1 cells (Figures 4A, 4B, and S4F). Biochemical analyses confirmed that vWAT from Fbf1tm/tm mice, Fbf1tm/tm MEFs, or siFbf1-treated 3T3-L1 cells showed increased phospho-p38, a signature for adipocyte beiging (Figures 4C and S4G).
Figure 4. FBF1 controls adipose beiging through a cilia-specific PKA signaling.
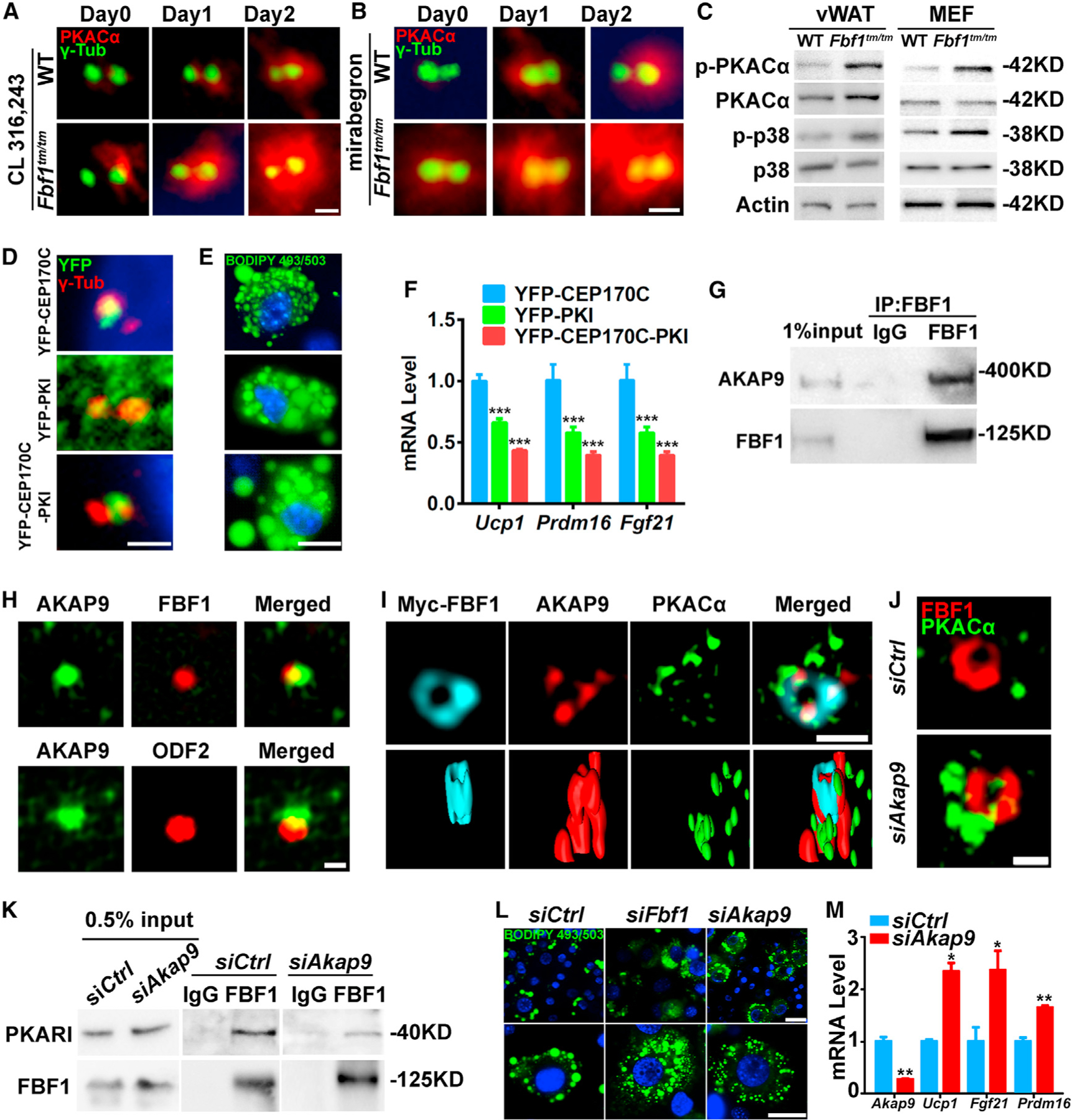
(A and B) PKACα localization in WT and Fbf1tm/tm MEFs before and after treated with (A) CL-316,243 or (B) mirabegron. Centrosomes were stained with γ-tubulin. Scale bar, 1 µm.
(C) Immunoblotting on vWAT or MEF extracts.
(D) WT vWAT preadipocytes labeled with yellow fluorescent protein (YFP)-tagged CEP170C-PKI, CEP170C, and PKI. Centrosomes were stained with γ-tubulin. Scale bar, 1 µm.
(E) WT vWAT preadipocytes overexpressed PKI, CEP170C, or CEP170C-PKI were treated with CL-316,243 and differentiated for 7 days in adipogenic medium (AM), then fixed, stained, and imaged. Green: lipids, blue: nuclei. Scale bar, 50 µm.
(F) Relative mRNA levels of beiging genes in WT vWAT preadipocytes overexpressed PKI, CEP170C, or CEP170C-PKI.
(G) Endogenous AKAP9 immunoprecipitates with FBF1 in 3T3-L1 cells.
(H) AKAP9 localization in 3T3-L1 cells. FBF1 or ODF2 labeled basal bodies. Scale bar, 1 µm.
(I) SIMS and 3-dimensional (3D) surface-rendering images of the basal bodies of 3T3-L1 cells stained for PKACα, AKAP9, and FBF1. Scale bar, 1 µm.
(J) SIMS images of the basal bodies of 3T3-L1 cells stained for PKACα and FBF1. Scale bar, 1 µm.
(K) Association between endogenous PKARI and FBF1 in 3T3-L1 cells treated with control or Akap9 siRNA.
(L) 3T3-L1 cells treated with control or Akap9 siRNA were differentiated with CL-316,243 inductions for 7 days, then fixed, stained, and imaged. Green: lipids, blue: nuclei. Scale bar, 100 µm.
(M) Relative mRNA levels of beiging genes in 3T3-L1 cells treated with control or Akap9 siRNA.
To directly assess the cilia-specific role of PKA in adipocyte beiging, we specifically targeted PKI, a peptide PKA inhibitor (Knighton et al., 1991), to cilia base by fusing it with the C terminus of (CEP170C). CEP170 is a subdistal appendage protein, and CEP170C faithfully targets PKI near the TFs (Figure 4D). Overexpression of either PKI or CEP170C-PKI, but not CEP170C alone, in WAT preadipocytes effectively blocked the transcriptional signature of the beiging program and changed the lipid morphology from a pattern of brown adipocytes to white adipocytes (Figures 4E and 4F). Of note, TF-enriched CEP170C-PKI show stronger inhibition of the beiging program (Figures 4E and 4F). Similarly, overexpressed CEP170C-PKI enriched around cilia base and effectively suppressed the beiging program in 3T3-L1 cells induced with CL-316,243 (Figures S4H–S4J). To further distinguish the impact of global versus ciliary PKI on the beiging program, we generated a chemically inducible trapping system (Hong et al., 2018) that can spatiotemporally recruit overexpressed PKI to the proximity of TFs once treated with rapamycin. Our data clearly support the conclusion that local PKA signaling around TFs is critical for activating the beiging program during preadipocyte differentiation (Figures S4K and S4L).
We further asked how FBF1 regulates PKA signaling at cilia base. Nascent PKA was confirmed inside cilia, but difficult to be detected because of its transient presence and/or the low sensitivity of the antibodies (Breslow et al., 2018). As a global kinase, PKA relies on specific A-kinase anchoring proteins (AKAPs) to localize to distinct subcellular locations (Chen and Kass, 2005). By using a proximity-labeling BioID assay (Roux et al., 2012), we profiled the FBF1 interactome in MEFs. AKAP9 was retrieved as one of the top FBF1 interactors. AKAP9 is enriched around the tip of mother centrioles in mammalian cells (Kolobova et al., 2017) and directly regulates local cyclic AMP (cAMP) signaling (Terrin et al., 2012). Immunoprecipitation confirmed that endogenous AKAP9 associates with FBF1 (Figure 4G). Endogenous AKAP9 localizes near TFs and overlaps with PKA in WT vWAT (Figures S4M and S4N), vWAT preadipocytes (Figure S4O), or 3T3-L1 cells (Figures 4H and 4I). FBF1 deficiency does not affect AKAP9 localization at cilia base (Figure S4P). It appears that AKAP9 is sorted to cilia base in an FBF1-independent manner, and FBF1 probably anchors AKAP9 to a distinct subcompartment so that AKAP9-associated PKA could be efficiently imported through TFs. Of note, the depletion of AKAP9 led to PKACα accumulation around cilia base (Figure 4J), compromised FBF1-PKARI association (Figure 4K), and induced the beiging of 3T3-L1 cells (Figures 4L, 4M, and S4Q).
Fbf1tm/tm mice possess healthy WAT with hyperplasia and low inflammation
Adipose tissue expands by hyperplasia or hypertrophy. Compared to hyperplasia, hypertrophic expansion of adipose tissue is metabolically unfavorable and associated with obesity-associated insulin resistance (Romieu et al., 2017; Rosen and Spiegelman, 2014). Consistent with the view that upregulated beiging and thermogenesis prevent adipocytes from developing hypertrophy, the size of adipocytes in sWAT, vWAT, and BAT from obese Fbf1tm/tm mice is either smaller or similar when compared to WT controls (Figure 2M). This suggests that the obesity of Fbf1tm/tm mice is very much driven by adipose hyperplasia or excessive adipogenesis. An EdU (5-ethynyl-2ʹ-deoxyuridine) incorporation assay confirmed that adipogenesis in Fbf1tm/tm vWAT is ~2-fold greater than those in WT animals (Figure 5A). Consistent with metabolism-favorable hyperplasia and beiging in WAT, Fbf1tm/tm mice possess healthier WAT characterized by a lower inflammation level (Figures 5B–5D) and decreased macrophages (Figure 5E) when compared with WT WAT.
Figure 5. FBF1 deficiency enhances adipogenic differentiation.
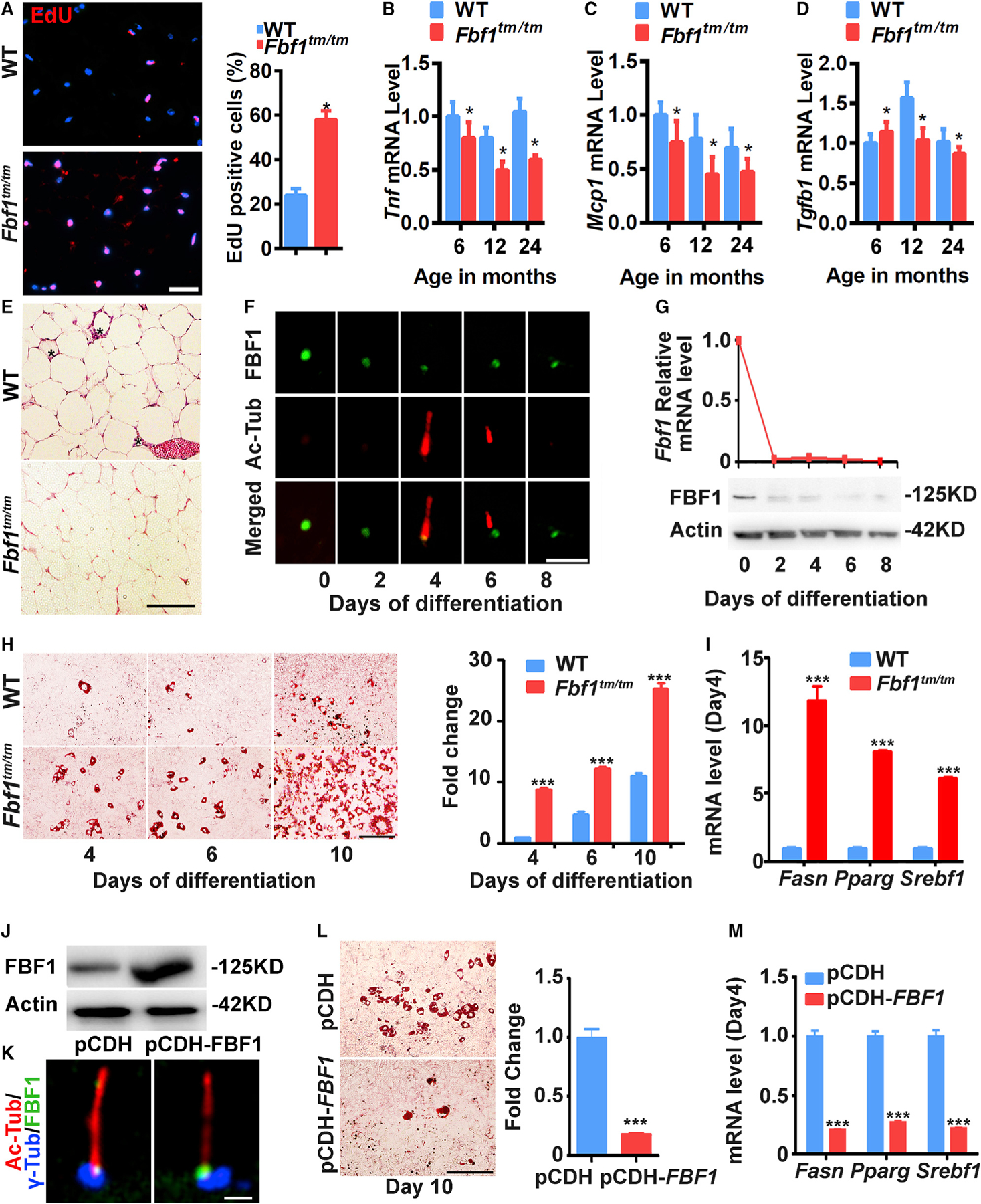
(A) Fluorescence images were obtained by the EdU incorporation assay for vWAT from 6-month-old female WT/Fbf1tm/tm mice. Red: EdU+ cells, blue: nuclei. Scale bar, 100 µm. Data were calculated from 300 cells in each group.
(B–D) Inflammatory genes expression levels of vWAT in different ages.
(E) H&E staining of sections of WATs of Fbf1tm/tm and WT littermates at 18 months. Macrophages surrounding adipocyte (asterisk). Scale bar, 200 µm.
(F and G) Immunofluorescence images of cilia (Ac-tubulin, red) and FBF1 staining during adipogenic differentiation (F) and (G) relative mRNA and protein levels of FBF1 of WT MEFs. Scale bars, 5 µm.
(H) WT/Fbf1tm/tm MEFs were induced to differentiate for 4, 6, and 8 days, then stained with oil red O. Images are representative of n = 6 replicates. Scale bar, 150 µm.
(I) Relative mRNA levels of adipogenic markers at day 4 of differentiation of MEFs.
(J) Protein levels of FBF1 in 3T3-L1 cells overexpressed with plasmid pCDH vector or pCDH-FBF1.
(K) Immunofluorescence assays in 3T3-L1 cells using antibodies against FBF1. Basal bodies and cilia were labeled with anti-γ-tubulin and anti-acetylated tubulin. Scale bar, 5 µm.
(L) 3T3-L1 cells were overexpressed with pCDH vehicle or pCDH-FBF1, differentiated for 10 days, and stained with oil red O. Scale bar, 150 µm.
(M) Relative mRNA levels of adipogenic markers at day 4 of differentiation of 3T3-L1 cells overexpressed vehicle or FBF1.
Values are expressed as means ± SEMs, n = 6 in (A)–(E) in each group. p values were indicated in figures as follows: *p < 0.05, **p < 0.01, and ***p < 0.001.
FBF1 acts as a negative regulator of adipogenesis
MEFs and 3T3-L1 cells show transient ciliogenesis during adipogenesis, with primary cilia appearing ~4 days after induction but disappearing in mature adipocytes at day 8 (Figures 5F and S5A–S5C). Intriguingly, upon adipogenesis induction, mRNA and protein levels of Fbf1 are tuned down immediately and kept at low levels throughout differentiation (Figure 5G). In agreement with in vivo observations, Fbf1tm/tm MEFs (Figure 5H), siFbf1-treated 3T3-L1 cells (Figure S5D), or Fbf1tm/tm vWAT preadipocytes (Figure S5F) showed greatly enhanced adipogenesis, accompanied by a robustly upregulated adipogenic program (Figures 5I, S5E, and S5G). Overexpressed FBF1 faithfully localizes to the endogenous site in 3T3-L1 cells (Figures 5J and 5K). Strikingly, FBF1 overexpression effectively blunts adipogenesis and suppresses the adipogenic program (Figures 5L and 5M).
FBF1 controls adipogenesis via an Hh signaling-dependent manner
Enhanced adipogenesis in Fbf1tm/tm MEFs or siFbf1-treated 3T3L-1 cells could be abolished by co-depleting Ift88 (Deren et al., 2016), a key player in ciliogenesis (Figures S6A–S6D). Among the signaling pathways implicated in regulating adipogenesis, Hh signaling inhibits adipogenesis and tightly correlates with primary cilia in different mammalian cell types (Kopinke et al., 2017; Rosen and MacDougald, 2006). Hh activation by SAG (smoothened agonist) could efficiently blunt induced adipogenesis and the transcription of adipogenic program in WT, but not in Fbf1tm/tm MEFs or Fbf1-depleted 3T3-L1 cells (Figures 6A, 6B, S6E, and S6F). SAG stimulation activates Hh signaling by promoting the ciliary localization of the G protein-coupled receptor (GPCR) Smoothened (Smo), zinc finger transcription factor Gli3, and the ciliary removal of GPCR GPR161. In SAG-treated Fbf1tm/tm MEF or siFbf1-treated 3T3-L1 cells, Smo and Gli3 were absent in cilia, but GPR161 was not removed from the cilium (Figures 6C–6H and S6G–S6J). Fbf1tm/tm MEFs show normal ciliation ratios, but with truncated cilia length (Figure 6G). As expected, SAG treatment could not activate the transcription of Hh target gene Gli1 in Fbf1tm/tm MEFs or siFbf1-treated 3T3-L1 cells, unlike WT control cells (Figures 6H and S6K). In vivo analyses in WT and Fbf1tm/tm vWAT or isolated vWAT preadipocytes confirmed that Hh signaling is blunted in Fbf1tm/tm WAT (Figures 6I–6K).
Figure 6. FBF1 controls Hh signaling by recruiting BBS chaperone complex to TFs.
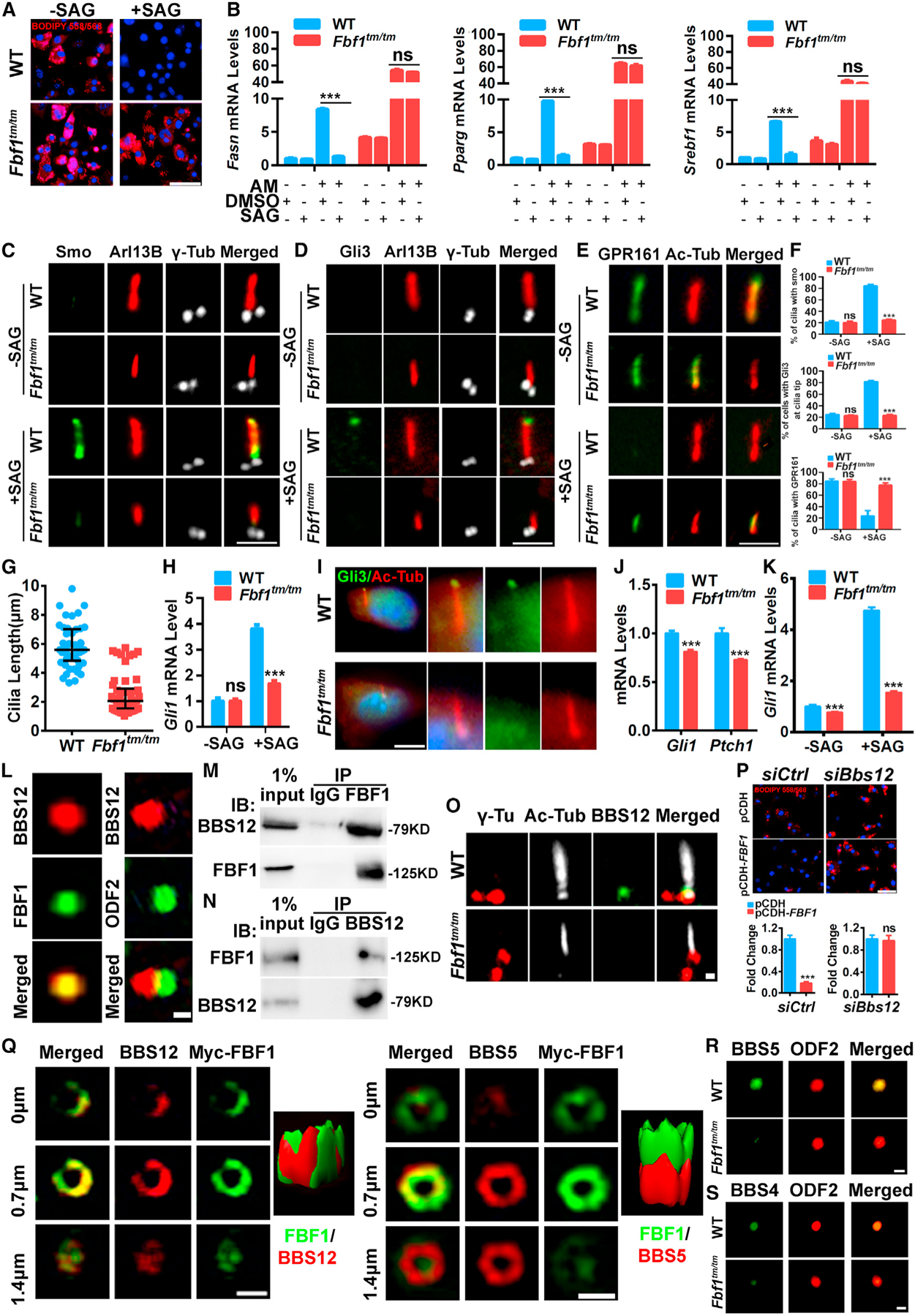
(A) WT/Fbf1tm/tm MEFs were treated with DMSO or SAG and differentiated for 7 days, then fixed and stained with BIDIPY. Red: lipids, blue: nuclei. Scale bar, 150 µm.
(B) Relative mRNA levels of adipogenic genes at day 4 of differentiation with or without AM in WT/Fbf1tm/tm MEFs treated with DMSO or SAG.
(C–E) Localization of Smo (C), (D) Gli3, and (E) GPR161 in primary cilia of WT/Fbf1tm/tm MEFs treated with DMSO or SAG. Basal bodies and cilia were labeled with anti-γ-tubulin and anti-acetylated tubulin. Scale bar, 5 µm.
(F) Statistical analysis of Smo+, Gli3+, and GPR161+ cilia. Data were calculated from 300 cilia in each group. Values are expressed as means ± SEMs. ***p < 0.001.
(G) Cilia length of WT and Fbf1tm/tm MEFs.
(H) Gli1 mRNA level in WT and Fbf1tm/tm MEFs before and after Hh activation.
(I) Localization of GLI3 in primary cilia of WT/Fbf1tm/tm vWAT tissues. Basal bodies and cilia were labeled with anti-γ-tubulin and anti-acetylated tubulin. Scale bar, 5 µm.
(J) Gli1 and Ptch1 mRNA levels in WT and Fbf1tm/tm vWAT tissues.
(K) Gli1 mRNA level in WT and Fbf1tm/tm vWAT preadipocytes before and after Hh activation.
(L) WT MEFs labeled with BBS12 and TF marker FBF1 or subdistal appendage marker ODF2, and analyzed with SIMS. Scale bar, 1 µm.
(M) Endogenous BBS12 immunoprecipitates with FBF1 in 3T3-L1 cells.
(N) Endogenous FBF1 immunoprecipitates with BBS12 in 3T3-L1 cells.
(O) The localization of BBS12 in WT/Fbf1tm/tm MEFs. Anti-γ-tubulin and anti-acetylated tubulin-labeled basal bodies and cilia. Scale bar, 5 µm.
(P) Control or Bbs12-knockdown 3T3-L1 cells were overexpressed vehicle or FBF1 and differentiated for 7 days, then fixed, stained, and imaged. Red: lipids, blue: nuclei. Scale bar, 150 µm.
(Q) SIM and 3D surface-rendering images of 3T3-L1 cells. Scale bar, 1 µm.
(R) BBSome component BBS5 localization in WT/Fbf1tm/tm MEFs. Basal bodies were stained with ODF2. Scale bar, 1 µm.
(S) BBSome component BBS4 localization in WT/Fbf1tm/tm MEFs. Basal bodies were stained with ODF2. Scale bar, 1 µm.
FBF1 controls Hh signaling by recruiting BBS proteins to TFs
We next asked how FBF1 controls the ciliary localization of Hh signaling molecules in ciliated adipose precursors. Unlike most obesity-associated ciliopathies, a significant number of obese BBS patients retain a relatively normal metabolism profile (Marion et al., 2012), similar to that of obese Fbf1tm/tm mice. We thus reasoned FBF1 and BBS proteins may regulate obesity in the same pathway. BBS is a ciliopathy with >20 genes identified, of which BBS1, −10, and −12 are mutated in >50% of the patients (Nachury et al., 2007). BBS6, −10, and −12 form a chaperone complex to facilitate the assembly of the octameric BBSome complex, whose impaired assembly compromises cilia trafficking of ciliary proteins, including Hh signaling molecules, which contributes to the etiology of BBS phenotypes (Nachury et al., 2007; Seo et al., 2010). How and where BBS chaperone regulates BBSome assembly remains unknown.
Deficiency of BBS12 in mice enhances adipogenesis, but paradoxically causes greater insulin sensitivity (Marion et al., 2012). Interestingly, a significant part of endogenous BBS12 localizes to the cilia base in MEFs or 3T3-L1 cells (Figure S6L). Super resolution fluorescence structured illumination microscope, or SIM, revealed the complete colocalization of endogenous BBS12 and TFs (Figure 6L). Immunoprecipitation confirmed the association of endogenous BBS12 and FBF1 in 3T3-L1 cells (Figures 6M and 6N). FBF1 deficiency disrupted the TF localization of BBS12 in Fbf1tm/tm MEFs or siFbf1-treated 3T3-L1 cells without affecting the protein stability of BBS12 (Figures 6M, S6M, and S6N). In contrast, the loss of BBS12 does not affect the localization or expression of FBF1 (Figures S6O–S6Q). Reminiscent of what happened in FBF1-deficient cells, depleting BBS12 promoted the adipogenesis and transcription of adipogenic program in 3T3-L1 cells (Figures S6R and S6S). Remarkably, FBF1 overexpression effectively inhibits adipogenesis, which was abolished in siBbs12-treated 3T3-L1 cells (Figures 6O, S6P, and S6U). This strongly suggests that BBS12 acts downstream of FBF1 in regulating adipogenesis.
We then examined the BBSome. Serial SIMS analyses showed that the BBSome component BBS5 enriches below the TFs and only partially colocalizes with BBS12 and FBF1 (Figure 6Q). The spatial relationship among TFs, the BBS chaperone complex, and the BBSome suggests that the BBS chaperone complex is recruited to TFs by FBF1 and then regulates BBSome assembly in close proximity to TFs. The properly assembled BBSome then facilitates ciliary trafficking of Hh molecules and affects downstream adipogenesis. Consistent with our hypothesis, depleting Fbf1 or Bbs12 resulted in significantly reduced BBSome component BBS5 or BBS4 at cilia base (Figures 6R, 6S, S6V, and S6W).
FBF1 depletion in human preadipocytes promotes beiging and adipogenic programs during adipocyte differentiation
We next determined whether human preadipocyte cilia also regulate beiging and adipogenic programs. Small interfering RNA (siRNA) effectively knocked down FBF1 genes in primary human preadipocytes collected from healthy donors (Figures S7A and S7B). Consistent with the phenotypes in Fbf1tm/tm mice and Fbf1-deficient mouse cells, siFBF1-treated preadipocytes also showed classical beiging phenotypes with smaller lipid droplets, increased UCP1 expression, increased mitochondria content, and an upregulated transcription signature of the beiging program (Figures 7A–7G). Upon adipogenesis induction, siFBF1-treated human preadipocytes showed a greatly enhanced adipogenic program (Figures 7H, 7I, and S7C); disrupted localization of BBS12, Smo, and Gli3 (Figures 7J–7M, S7D, and S7E); and compromised Hh activation (Figure S7F). These data suggest that our discoveries made in the mouse model are highly relevant in human preadipocytes.
Figure 7. FBF1 negatively regulates beiging and adipogenic programs in human preadipocytes and protects mice from HFD-induced diabetes and premature death.
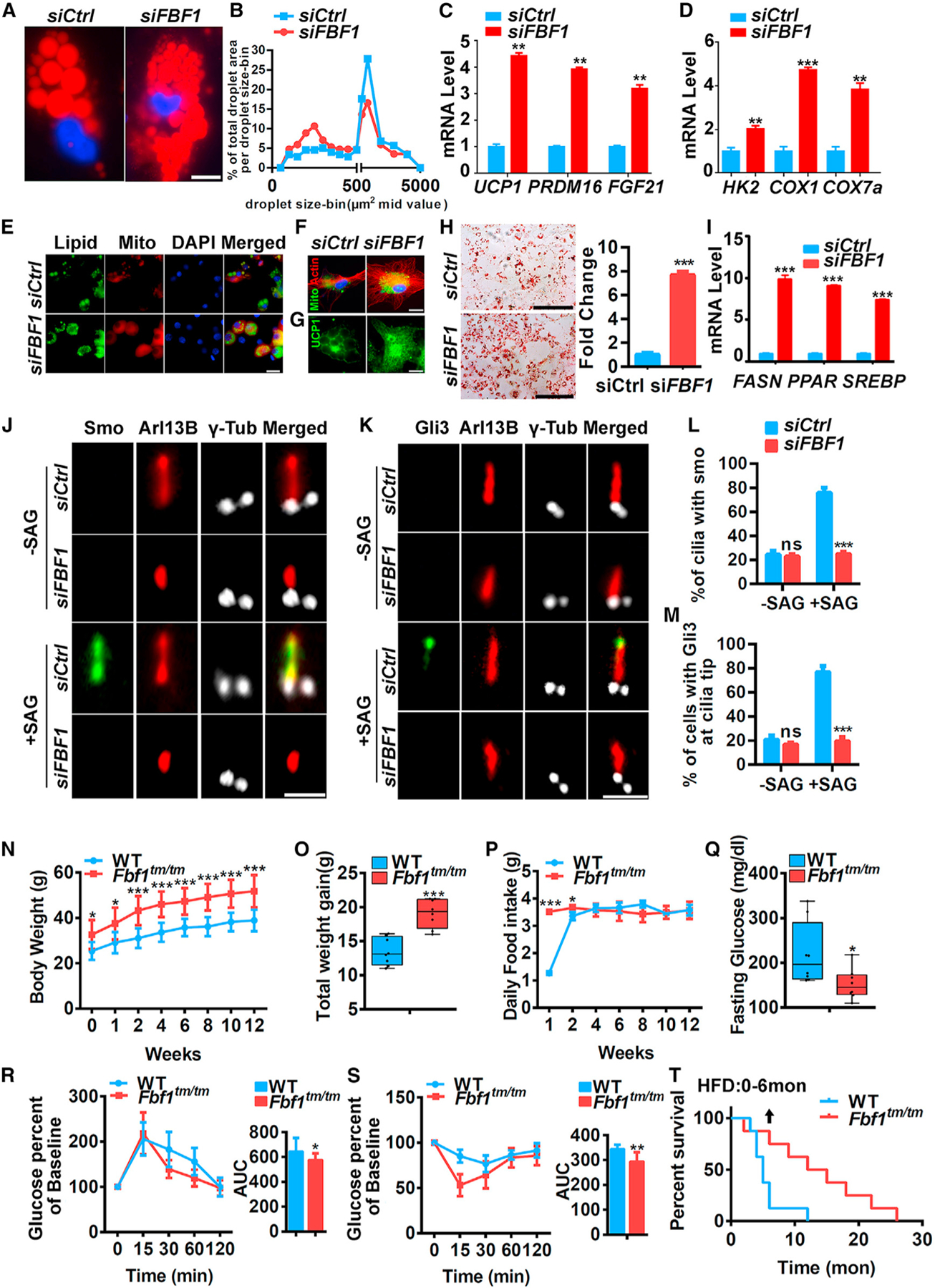
(A) Human adipocyte progenitors treated with control or FBF1 siRNA were induced to differentiate for 14 days, then fixed, stained, and imaged. Red: lipids, blue: nuclei. Scale bars, 50 µm.
(B) Quantification of changes in lipid morphology is depicted as fraction of total lipid area per lipid droplet size.
(C and D) Relative mRNA levels of beiging genes in human adipocyte progenitors treated with control or FBF1 siRNAs.
(E) Human adipocyte progenitors treated with control or FBF1 siRNA and differentiated. At day 14, cells were fixed, stained and imaged. Red: mitochondria, green: lipids, blue: nuclei. Scale bar, 100 µm.
(F) Human adipocyte progenitors treated with control or FBF1 siRNA and differentiated. At day 7, cells were fixed, stained, and imaged. Green: mitochondria, red: actin, blue: nuclei. Scale bar, 50 µm.
(G) Human adipocyte progenitors treated with control or FBF1 siRNA and differentiated. At day 14, cells were fixed, stained with anti-UCP1 (green), and imaged. Scale bar, 50 µm.
(H) Human adipocyte progenitors treated with control or FBF1 siRNA were induced to differentiate for 14 days, and cells were stained with oil red O. Scale bar, 150 µm.
(I) Relative mRNA levels of adipogenic genes at day 4 of differentiation of human adipocyte progenitors treated with control or FBF1 siRNA.
(J–M) The localization of SMO (J) and Gli3 (K) in primary cilia of control or FBF1-knockdown human adipocyte progenitors treated with SAG. Primary cilia were stained with Ac-tubulin. Centrosomes/basal bodies were stained with γ-tubulin. Scale bar, 5 µm. Statistical analysis of (L) SMO+ cilia and (M) Gli3+ cilia tips. Data were calculated from 300 cilia in each group. Values are expressed as means ± SEMs, ***p < 0.001.
(N–S) Growth curve (N), total weight gain (O), food intake (P), fasting glucose (Q), and glucose (R) and insulin (S) tolerance tests, with corresponding area under the curves and in 15-month-old Fbf1tm/tm and WT littermates that had been treated with HFD for 12 weeks. n = 6 in each group. *p < 0.05, **p < 0.01, and ***p < 0.001.
(T) Kaplan-Meier survival curve of Fbf1tm/tm and WT littermates treated with HFD; 12-month-old mice treated with HFD for 6 months. n = 8 in each group.
FBF1 deficiency protects animals from high-fat diet (HFD)-induced diabetes and premature death
Obesity and T2DM adversely affect health span and lifespan. Paradoxically, healthy adipocytes provide a safe storage place to prevent lipid spill-out from damaging the liver, muscle, and cardiovascular system (Rosen and MacDougald, 2006). To determine whether the healthier adipocytes of Fbf1tm/tm mice protect animal organs from HFD-induced metabolic anomalies, we subjected 2-month-old mice to an HFD. After a 12-week treatment, HFD induced progressively enlarged vacuoles, suggesting hepatic fat deposition, in the liver of WT mice, but not the Fbf1tm/tm littermates (Figure S7G). Consistently, HFD-fed WT mice show significantly higher inflammation levels in the liver (Figure S7H). To further assess the impact of FBF1 deficiency on health span and lifespan, we treated older 12-month-old mice to an HFD. After a 12-week treatment, HFD induced a significant increase in body mass in both WT and Fbf1tm/tm mice (Figure 7N), with the significantly larger increase occurring in the Fbf1tm/tm mice (Figure 7O). Although HFD-treated Fbf1tm/tm mice gained more weight, they had much lower fasting glucose (Figure 7N) and were protected from HFD-induced glucose intolerance and insulin resistance (Figures 7R and 7S). Amazingly, after fed HFD continuously for 6 months, Fbf1tm/tm mice had significantly better survival rates than WT littermates (Figure 7T). The protection of Fbf1tm/tm mice from HFD-induced diabetes and premature death strongly suggests that altering particular ciliary pathways could be an avenue for fighting metabolic disorders. Our data also indicate that the ciliary function of FBF1 protects against the development of morbid obesity, but to the detriment of metabolic function and lifespan, highlighting that a ciliary program exists to reduce fat tissues expansion in the setting of normal and excessive caloric intake.
DISCUSSION
Although the primary cilium is well accepted as ‘‘sensory antenna’’ for eukaryotic cells, its physiological importance in most cell types remains poorly defined. Here, we demonstrate the essential role of primary cilia in preadipocyte differentiation by controlling both beiging and adipogenic programs. Deficiency of FBF1, a key TF component that regulates the cilia gating for various signaling molecules, leads to an intriguing paradox that although progressively obese, Fbf1tm/tm mice are not predisposed to adverse metabolic complications along their lifetime. Lack of FBF1 simultaneously upregulates the beiging program in WAT through a cilia-specific AKAP9-PKA signaling and robustly enhances adipogenesis through BBS-regulated Hh signaling. The combined impact of WAT beiging and enhanced adipose expansion underscores plausible causes of ‘‘healthy obesity’’ in Fbf1tm/tm mice. Combined with the fact that HFD-fed Fbf1tm/tm mice are protected from diabetes and premature death, our study suggests that targeting preadipocyte cilium could be a promising strategy to combat metabolic disorders.
To date, the only ciliopathy protein implicated in WAT beiging is the zinc finger transcription factor ZFP423. Genetic ablation of Zfp423 in mice induces browning of vWAT (Shao et al., 2016). Mutations of the human ortholog ZNF423 have been identified in patients with the ciliopathy Joubert syndrome (Chaki et al., 2012). ZFP423 directly binds ciliary genes and alters the expression of key ciliary components (Chaki et al., 2012). ZFP423 is a key co-activator of RXR (a known regulator of adipose differentiation) in certain cells (Huang et al., 2009). Interestingly, the transcription of Fbf1 is immediately suppressed upon adipogenesis induction. Our bioinformatics analysis also revealed that there are multiple RXR binding sites in the Fbf1 promoter (data not shown). It is thus worthwhile to investigate whether ZFP423 function and FBF1 transcription converge on WAT adipogenesis and beiging.
One central question in understanding how cilia dysfunction affects metabolism regulation is which ciliated organs/tissues are involved. Conditional deletion of genes required for cilia biogenesis in a subset of hypothalamic neurons showed that neuronal cilia control feeding behavior (Davenport et al., 2007). Since most cilia-related mouse obesity models exhibit hyperphagia, previous studies focused on hypothalamic cilia and satiety signaling (Vaisse et al., 2017). In drastic contrast, Fbf1tm/tm mice retain normal satiety signaling but develop obesity due to the combined effects of reduced energy expenditure and enhanced adipose expansion, which provides an opportunity to examine the physiological importance of non-neuronal cilia in metabolism regulation. Our discoveries propose an intriguing model of cilia-regulated energy homeostasis, whereby primary cilia govern the differentiation of adipose precursors in both adipogenic and, surprisingly, beiging programs. Enhanced adipogenic and beiging programs in Fbf1tm/tm mice contribute to healthier adipose tissue, preventing adipocyte hypertrophy and avoiding metabolic anomalies. Our study differentiates the complicated contributions of primary cilia in whole-body energy homeostasis coordinated by a multitude of pathways, including satiety signaling, locomotion behavior, and adipose differentiation. The phenotypes of Fbf1tm/tm mice suggest that the indispensable role of preadipocyte cilia in energy homeostasis is equally important as that of neuronal cilia. With the beneficial influence of FBF1 depletion on metabolism regulation, mechanistically delineating the FBF1 pathway is of paramount importance to metabolism research.
Most adipogenic signaling, including anti-adipogenic (Wnt, Hh, transforming growth factor-β [TGFβ]) and pro-adipogenic (bone morphogenic proteins and insulin-like growth factor 1 [IGF1] signaling), correlate with cilia in non-fat cell types (Oh et al., 2015). Recently, omega-3 fatty acids were found to directly activate ciliary free fatty acid receptor 4 (FFAR4) to control adipogenesis (Hilgendorf et al., 2019). Thus, the central role of FBF1 in cilia gating could theoretically affect diverse cilia signaling to promote or inhibit adipogenesis. Although the enhanced adipogenic program in Fbf1tm/tm mice suggests that the dominant role of FBF1 in adipogenesis is inhibitory, it would be interesting to reveal the identities of other cilia-related signaling in the context of preadipocyte differentiation, as well as their synergies and co-ordination. For example, PKA is also a validated negative regulator of Hh pathway activity via regulation of the Gli3 repressor (Wang et al., 1999), and especially in the context of cilia (Truong et al., 2021). It is thus possible that the excessive PKA accumulation at cilia base in FBF1-deficient cells may collaterally affect Hh activity in preadipocytes.
Enhanced adipogenesis does not cause T2DM. Paradoxically, healthy adipocytes provide a safe storage place to prevent damage from lipid spill-out to liver, muscle, and the cardiovascular system (Rosen and MacDougald, 2006). Preadipocyte differentiation capacity declines during aging or in diabetes patients (Kirkland and Dobson, 1997), which may be a major cause for metabolic disorders. The combined effects of upregulated adipogeneses and beiging in Fbf1tm/tm WAT counteract adipose hypotrophy and result in healthier adipose tissue. Amazingly, although HFD-treated Fbf1tm/tm mice become extremely obese, they are still protected from HFD-induced glucose/insulin resistance and premature death. To date, multiple therapeutic options have been developed to treat obesity and T2DM, but unfortunately, many of these treatments are associated with significant adverse health risks. For example, the metabolism improvements of obese Fbf1tm/tm mice mimic the effect of the widely prescribed TZD family of antidiabetic drugs, which cause adipose hyperplasia, WAT beiging, and increased insulin sensitivity (Nissen and Wolski, 2007). However, TZD drugs are associated with a significant increase in the risk of myocardial infarction and, likely, death (Nissen and Wolski, 2007). Consequently, there is an unmet need for different antidiabetic therapies for rapidly increasing global obesity prevalence. Insights gained from studying preadipocyte cilia should facilitate the discovery of therapeutic targets/pathways/agents that can be targeted to promote the development of healthier adipose tissue, especially during high calorie input or aging scenarios.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Jinghua Hu (hu.jinghua@mayo.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
This study did not generate any unique datasets.
This study did not generate any unique code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
All animal experiments were performed according to protocols approved by the Institutional Animal Care and Use Committee (IACUC) at Mayo Clinic (protocol A00004472–19). C57BL/6J mice were purchased from Jackson Laboratory. The Fbf1tm1a/tm1a mice were obtained from Trust Sanger Institute with C57BL/6N genetic background and refreshed the genetic background10 generations by backcrossing to the C57BL/6J mice. Mice were maintained in a pathogen-free facility at 23–24 °C under a 12-h light, 12-h dark regimen with free access to a NCD (standard mouse diet with 20% protein, 5% fat (13.2% fat by calories), and 6% fiber; Lab Diet 5053, St. Louis, MO) and water. Quarterly testing was negative for the following pathogens: Clostridium piliforme, Mycoplasma pulmonis, cilia-associated respiratory (CAR) bacillus, ectromelia, rotavirus (EDIM), Hantaan virus, K virus, lymphocytic choriomeningitis virus (LCMV), lactate dehydrogenase–elevating virus (LDEV), mouse adenovirus 1 and 2, mouse cytomegalovirus (MCMV), mouse hepatitis virus (MHV), minute virus of mice (MVM), mouse parvovirus (MPV), mouse thymic virus (MTV), polyomavirus, pneumonia virus of mice (PVM), REO-3 virus, Sendai virus, Myocoptes, Theiler’s murine encephalomyelitis virus (TMEV), Encephalitozoon cuniculi, Aspiculuris tetraptera, Radfordia/Myobia, and Syphacia obvelata. All mice were fed normal chow unless otherwise indicated. For high-fat feeding, a 60% (by calories) fat diet (D12492, irradiated; Research Diets, New Brunswick, NJ) was used. All mice were housed in static autoclaved HEPA-ventilated microisolator cages (27 × 16.5 × 15.5 cm) with autoclaved Enrich-o’Cobs (The Andersons Incorporated) for bedding. Cages and bedding were changed biweekly. Cages were opened only in class II biosafety cabinets.
Both male and mice were used for the study, and the age was indicated in the figure legends.
CL-316,243-treated mice were injected intraperitoneally (i.p.) with 1 mg/kg CL-316, 243 in 0.9% NaCl for 5 days every week for 2 weeks; 0.9% NaCl was used in the control group instead of CL 316,243.
Tissues from mice sacrificed at the indicated time points were snap-frozen in liquid nitrogen for biochemical studies or fixed in 4% Paraformaldehyde (PFA) for 24 hours prior to processing and paraffin embedding. Paraffin-embedded tissues were cut at 5 µm intervals. For frozen sections, samples were snap frozen in liquid nitrogen, placed in OCT and cut into 7 µm sections.
Cell culture
Primary human fat progenitors were isolated from subcutaneous fat collected from healthy, lean (BMI 26.6 ± 0.9 kg/m2) kidney donors aged 39 ± 3.3 years as previously described (Tchkonia et al., 2005). The protocol (10–005236) was approved by the Mayo Clinic Foundation Institutional Review Board for Human Research.
3T3-L1 cells were cultured in DMEM medium containing 10% Bovine Calf Serum, 1% Pen/Strep and switched to DMEM containing 10% FBS, 1% Pen/Strep, and 1% GlutaMAX during adipogenesis.
Murine primary preadipocytes were isolated from visceral or subcutaneous white adipose tissue from wild-type and Fbf1tm1a/tm1a mice using mouse protocols approved by the Institutional Animal Care and Use Committee (IACUC) at Mayo Clinic. Briefly, preadipocytes were isolated from mice by following a tissue extraction and collagenase digestion procedure by 20min shaking (250rpm) at 37C. Digested samples were strained through a 100 µm filter and cells were collected by centrifugation (1300rpm, 5min) then cultured in DMEM containing 10% FBS. Primary preadipocytes were maintained and differentiated in DMEM medium containing 10% FBS, 1% Pen/Strep, and 1% GlutaMAX at 37°C in a humidified 7% CO2 incubator.
Mouse Embryo Fibroblasts (MEFS) were isolated from the embryos of a female that is at day 13.5 of her pregnancy. Embryos were removed and placed in PBS. Heads and internal organs were removed, and blood was washed away from the remaining carcasses with PBS. Tissue was minced in TrypLE Express Enzyme, followed by incubation with stirring for 30 minutes. TrypLE was neutralized with heat inactivated FBS, and cells were collected by centrifugation (1300rpm, 5min) then cultured in DMEM containing 10% FBS. Media switched to DMEM containing 10% FBS, 1% Pen/Strep during adipogenesis 24 hours after plating.
Cell line generation
Retroviral vectors carrying the gene of interest were co-transfected with psPAX2 and pMD2.G into 293T cells using Fugene6 (Promega). Media was replaced after 24 h and virus was harvested 48 and 72 h post-transfection. Virus was filtered with a 0.45 µm OVDF filter (Millipore) and used to infect cell lines with 10 µg/mL polybrene (Millipore). Media was replaced after 24 h and cells were subjected to 500 µg/ml G418 selection at 48 h after post-infection and fed with fresh G418 every 2 days.
METHOD DETAILS
In vitro adipogenic and beiging induction
3T3-L1 cells were grown to confluency in DMEM containing 10% Bovine Calf Serum, followed by another 2 days at confluency in DMEM containing 10% Bovine Calf Serum. Adipogenesis was then induced using DMEM containing 10% FBS and differentiation cocktail (1 µg/mL insulin, 1 µM dexamethasone, 0.5mM IBMX) for differentiation. After 2 days of differentiation, media was changed to DMEM containing 10% FBS and 1 µg/mL insulin. Maintenance media was changed every 2–3 days for a total differentiation time of 4–8 days.
Mouse Embryo Fibroblasts (MEFS) and mouse primary preadipocytes were grown to confluency in DMEM containing 10% FBS. Cells were not grown for another 2 days at confluency. Adipogenesis was induced using DMEM containing 10% FBS and differentiation cocktail (1 µg/mL insulin, 1 µM dexamethasone, 0.5mM IBMX). After 3 days of differentiation cocktail, media was changed to DMEM containing 10% FBS and 1 µg/mL insulin. Maintenance media was changed every 2–3 days for a total differentiation time of 4–8 days.
Human primary preadipocytes treated with differentiation medium containing DMEM/F12, 15nM HEPES, 15mM NaHCO3, 2mM glutamine, 10mg/L transferrin, 33 µM biotin, 0.5 µM insulin, 17 µM pantothenate, 0.1 µM dexamethasone, 2nM triiodo-L-thyronine (T3), 540 µM IBMX, 1 µM ciglitazone, 1mg/ml fetuin, and penicillin/streptomycin for 15 days unless indicated otherwise.
For brown/beige adipocyte differentiation, the cells were induced with induction medium contains DMEM, 10% FBS, 5 µg ml−1 insulin, 0.5 mM IBMX, 1 µM dexamethasone, 50 nM T3 and 5 µM troglitazone for 48 h, and further in growth medium supplemented with insulin, T3 and troglitazone for 6 days followed by 10 µM forskolin treatment for another 4 h.
Body composition
Lean and fat masses of individual mice were determined by quantitative nuclear magnetic resonance using an EchoMRI analyzer (Houston, TX) and expressed as a function of body weight. Un-anesthetized animals were placed in a plastic tube that was introduced into the EchoMRI instrument. Body composition, comprising fat mass and lean mass, was determined in approximately 90 s per animal.
Real-time RT-PCR
At predetermined endpoints, animals were sacrificed and organ samples were weighted and frozen in liquid nitrogen until further processing. Differential expression of genes of interest was quantified by real-time reverse transcriptase polymerase chain reaction (RT-qPCR). Total RNA was isolated from tissue or cell homogenates using TRIzol (ThermoFisher). Reverse transcription was performed using 0.5 µg of total RNA, random hexamer primers and Multiscribe reverse transcriptase (High-Capacity cDNA Reverse Transcription Kit, Applied Biosystems), according to the manufacturers protocol. SYBR Green (Bio-Rad) and cDNA was used as a master mix in a final volume of 20 µL containing primers for each candidate gene, as provided by the supplier. CFX Real-Time PCR Detection Systems (Bio-Rad) was used to assess RT-qPCR amplification. Expression of target genes in all samples was normalized to that of the 18S rRNA housekeeping gene.
EdU experiments
For EdU experiments, mice were injected with EdU (ThermoFisher) at a dose 100 ug/g twice a day for 2 days with the final concentration of 10mg/ml, dissolved in sterile PBS (pH 7.4), The animals were deeply anesthetized with 90mg/kg ketamine and 10mg/kg of xylazine in sterile PBS prior perfusion. Perfuse mice with PBS 200 mL and 4% PFA 200 ml. Adipose tissues were harvested and post-fixed overnight in 4% PFA for 24 hours prior to processing and paraffin embedding. Paraffin-embedded tissues were cut at 5 µm and incubated with Alexa 594-azide (Life Technologies). All preparations were mounted with fluorescent mounting medium (DAKO) and coverslipped.
Metabolic parameter measurement
For glucose tolerance testing, mice were fasted for 6 hours, and glucose (2g/kg body weight) was injected intraperitoneally. For insulin tolerance testing, mice were fasted for 4 hours and insulin (0.6 unit/kg body weight) was injected intraperitoneally. Glucose was measured using a handheld glucometer (Bayer) in blood from the tail vein.
Western blotting
Cells or tissues were homogenized in cell lysis buffer (50 mM Tris HCl (pH 7.4), 150 mM NaCl, 0.1 mM EDTA, 0.1 mM EGTA, 1% NP-40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate, 2 mM sodium orthovanadate, 10 mM β-glycerophosphate, 5 mM sodium fluoride) with protease inhibitors (Roche). Protein concentration in each sample was determined by the BCA method (ThermoFisher). Proteins were loaded on SDS-PAGE gels and transferred to immuno-blot PVDF membranes (Millipore), then blocked with 5% nonfat dry milk and sequentially incubated with primary and secondary antibodies. Horseradish peroxidase (HRP) substrates (ThermoFisher) were used to develop signals.
Immunoprecipitation
Cells were lysed in lysis buffer (25 mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.5% NP-40, 1 mM EDTA) with complete protease inhibitor cocktail (Roche). The lysate was centrifuged for 20 min at 12 000 g at 4 °C and then the supernatant was precleared with protein G Sepharose beads for 4 h. After removal of protein G, the supernatant was incubated with protein G beads and 2 µg of either primary antibody or control IgG overnight at 4 °C. After washing the beads five times, the immunocomplexes were separated by SDS–PAGE gels and analyzed.
Immunofluorescence
For centrosome staining, cells were fixed with −20 °C methanol for 20 min, then blocked with 3% BSA and sequentially incubated with primary and secondary antibodies. For cilia staining, cells were typically fixed with 4% paraformaldehyde for 20 min at room temperature, followed by permeabilization with 0.2% Triton X-100 for 10 min. Cells were then blocked with 3% BSA and sequentially incubated with primary and secondary antibodies. To stain cilia using the GPR161 antibody, we used an enhanced immunofluorescence protocol. Cells were prefixed with 0.4% paraformaldehyde for 5 min at 37 °C, extracted with 0.5% Triton X-100 in PHEM (50 mM PIPES, 50 mM HEPES, 10 mM EGTA and 10 mM MgCl2, pH 6.9) for 2 min at 37 °C and then stained following the immunofluorescence procedure described above.
Oil red O staining for lipid visualization
Cells were typically fixed with 4% PFA/PBS for 10min at room temperature, followed by 3 rinses in PBS. Samples were incubated in 60% isopropanol for 5min at room temperature and then allowed to dry completely. Samples are then incubated in freshly diluted 60% Oil Red O staining solution in water (stock is 0.5% Oil Red O (Sigma) in isopropanol) for 20min at room temperature, followed by 3 rinses in water. Samples were allowed to dry completely and imaged.
Histological analysis
Adipose tissue was fixed in formalin overnight at RT, paraffin embedded and sectioned. Paraffin blocks were cut into 5 µM sections and stained with hematoxylin and eosin (H&E) or used for immunohistofluorescence (IHF) staining. Tissue sections were de-waxed and rehydrated before undergoing standard in H&E staining. To assess the area of individual adipocytes, H&E-stained WAT sections were thresholded and adipocyte area quantified using Analyze Particle function in ImageJ. For IHF, rehydrated tissue sections were blocked with 3% BSA for 1h at room temperature and sequentially incubated with primary and secondary antibodies. For IHF performed with frozen sections, OCT sections were washed with PBS and incubated in blocking buffer containing 5% (vol/vol) sheep serum, 1% (vol/vol) FBS, and 0.1% (vol/vol) Triton X-100 in PBS then stained following the immunofluorescence procedure described above.
Chemically inducible trapping system and beiging induction
The basis of the technology is chemically inducible dimerization, in which rapamycin induces the dimerization of FK506 binding protein (FKBP) and FKBP–rapamycin-binding domain (FRB). To anchor a fusion protein of PKI-FRB to the ciliary base, we employed C terminus of (CEP170C). Transfected cells at 80%–90% confluency was serum-starved for 24 h and then incubated with 100 nM rapamycin for 30 min. Washout of rapamycin after 30 min did not affect protein dimerization in cells owing to the irreversible nature of the system.
During beige differentiation, the cells were induced with induction medium contains DMEM, 10% FBS, 5 µg ml−1 insulin, 0.5 mM IBMX, 1 µM dexamethasone, 50 nM T3 and 5 µM troglitazone for 48 h, and further in growth medium supplemented with insulin, T3 and troglitazone for 6 days, then incubated with 100 nM rapamycin for 30 min to induce dimerization and then washout rapamycin, followed by 10 µM forskolin treatment for another 4 h.
Staining of lipid droplets
Cells were incubated with 20 µg/ml BODIPY 558/568 or BODIPY 493/503 (ThermoFisher) for 45 min at 37°C in PBS, rinsed to remove excess stain followed by a further incubation of 60 min at 37°C in cell culture media. Cells were either viewed live or fixed with 4% paraformaldehyde at 4°C for 30 min. Fixed cells were washed with PBS prior to mounting fluorescent mounting medium (DAKO) and coverslipped.
LacZ reporter gene detection
Embryos were isolated female that is at day 15 of her pregnancy. Embryos were removed and placed in PBS and fixed by immersion in 4% PFA for a further 60 min. Embryos were incubated in staining solution containing 0.1% X-Gal (5-bromo-4-chloro-3-indolyl-β-d-galactoside (ThermoFisher) at 4°C for 48 h followed by a post-fixation with 4% PFA overnight at 4°C. Clearing of the tissues was performed in glycerol, and the resulting resource was stored in the dark at room temperature.
Comprehensive laboratory animal monitoring system
Indirect calorimetry was performed during light and dark cycles to determine the extent to which treatment affected metabolic parameters during conditions of rest (light cycle) and activity (dark cycle). In addition, the effects of treatment on metabolic flexibility were assessed by measuring metabolic parameters under fed conditions (provision of a predominantly carbohydrate-based fuel source) and then fasted conditions (reliance on endogenous lipid-based energy stores). On the day of the experiment, mice were weighed and acclimated overnight. In a subset of eight mice per group, the habitual ambulatory, rearing, and total activity, oxygen consumption (VO2), and carbon dioxide production (VCO2) of individual mice were monitored over a 24h period (12h light/12h dark) using a Comprehensive Laboratory Animal Monitoring System (CLAMS) equipped with an Oxymax Open Circuit Calorimeter System (Columbus Instruments). The VO2 and VCO2 values were used to calculate the respiratory exchange ratio (RER) and VO2. RER values were used to determine the basal metabolic rate (in kilocalories per kilogram per hour). Data were analyzed using the CLAX software from Columbus Instruments, exported into Excel and plotted in GraphPad.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical parameters including the statistical test used, exact value of n, what n represents, and the distribution and deviation are reported in the figures and corresponding figure legends. Most data are represented as the mean ± standard error of the mean and the p value was determined using two-tailed Student’s t tests. p values were indicated in figures as follows: *p < 0.05, **p < 0.01 and ***p < 0.001. Unless otherwise stated, statistical analyses were performed in Microsoft Excel and GraphPad Prism.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
|
| ||
| Anti-UCP1, Rabbit polyclonal | Abcam | Cat# ab10983; RRID: AB_2241462 |
| Anti-ODF2, mouse monoclonal | Abnova | Cat# H00004957-M01; RRID: AB_1146453 |
| Anti-BBS12, rabbit polyclonal | Bioss | Cat# bs-11505R |
| Anti-PKACα, Rabbit polyclonal | Cell signaling | Cat# 4782; RRID: AB_2170170 |
| Anti-Phospho-PKAC (Thr197), Rabbit polyclonal | Cell signaling | Cat# 9624; RRID: AB_331817 |
| Anti-p38, Rabbit polyclonal | Cell signaling | Cat# 9212; RRID: AB_330713 |
| Anti-phospho-p38 (Thr180/Tyr182), Rabbit polyclonal | Cell signaling | Cat# 9211; RRID: AB_331641 |
| Anti-AKAP9, mouse monoclonal | GeneTex | Cat# GTX79473; RRID: AB_11174025 |
| Anti-FBF1, rabbit polyclonal | Proteintech | Cat# 11531–1-AP; RRID: AB_2246683 |
| Anti-Arl13B, rabbit polyclonal | Proteintech | Cat# 17711–1-AP; RRID: AB_2060867 |
| Anti-GPR161, rabbit polyclonal | Proteintech | Cat# 13398–1-AP; RRID: AB_2113965 |
| Anti-BBS5, rabbit polyclonal | Proteintech | Cat# 14569–1-AP; RRID: AB_10597551 |
| Anti-BBS4, rabbit polyclonal | Proteintech | Cat# 12766–1-AP; RRID: AB_10596774 |
| IgG Control Antibody, rabbit polyclonal | Proteintech | Cat# 30000–0-AP; RRID: AB_2819035 |
| Anti-Gli3, goat polyclonal | R&D Systems | Cat# AF3690; RRID: AB_2232499 |
| Anti-Smo, mouse monoclonal | Santa Cruz | Cat# sc-166685; RRID: AB_2239686 |
| normal mouse IgG | Santa Cruz | Cat# sc-2025; RRID: AB_737182 |
| Anti-Acetylated Tubulin, mouse monoclonal | Sigma | Cat# T7451; RRID: AB_609894 |
| Anti-γ-Tubulin, mouse monoclonal | Sigma | Cat# T5326; RRID: AB_532292 |
| Anti-β-Actin, Mouse monoclonal | Sigma | Cat# A1978; RRID: AB_476692 |
| Anti-Myc, mouse monoclonal | ThermoFisher | Cat# MA1–980; RRID: AB_558470 |
| Peroxidase AffiniPure Goat Anti-mouse IgG (H+L) | Jackson ImmunoResearch | Cat# 115–005-166; RRID: AB_2338459 |
| Peroxidase AffiniPure Goat Anti-Rabbit IgG (H+L) | Jackson ImmunoResearch | Cat# 111–035-144; RRID: AB_2307391 |
| Peroxidase AffiniPure Donkey Anti-Goat IgG (H+L) | Jackson ImmunoResearch | Cat# 705–035-003; RRID: AB_2340390 |
| Goat anti-rabbit conjugated to Alexa Fluor 488 | ThermoFisher | Cat# A32731; RRID: AB_2633280 |
| Goat anti-rabbit conjugated to Alexa Fluor 555 | ThermoFisher | Cat# A32732; RRID: AB_2633281 |
| Goat anti-rabbit conjugated to Alexa Fluor 647 | ThermoFisher | Cat# A32733; RRID: AB_2633282 |
| Donkey anti-rabbit conjugated to Alexa Fluor 555 | ThermoFisher | Cat# A32794; RRID: AB_2762834 |
| Goat anti-mouse IgG1 conjugated to Alexa Fluor 488 | ThermoFisher | Cat# A-21121; RRID: AB_2535764 |
| Goat anti-mouse IgG1 conjugated to Alexa Fluor 555 | ThermoFisher | Cat# A-21127; RRID: AB_2535769 |
| Goat anti-mouse IgG1 conjugated to Alexa Fluor 647 | ThermoFisher | Cat# A-21240; RRID: AB_2535809 |
| Goat anti-mouse IgG2a conjugated to Alexa Fluor 555 | ThermoFisher | Cat# A-21137; RRID: AB_2535776 |
| Goat anti-mouse IgG2b conjugated to Alexa Fluor 555 | ThermoFisher | Cat# A-21147; RRID: AB_2535783 |
| Goat anti-mouse IgG2b conjugated to Alexa Fluor 647 | ThermoFisher | Cat# A-21242; RRID: AB_2535811 |
| Donkey anti-mouse conjugated to Alexa Fluor 647 | ThermoFisher | Cat# A32787; RRID: AB_2762830 |
| Donkey anti-goat conjugated to Alexa Fluor 488 | ThermoFisher | Cat# A32814; RRID: RRID:AB_2762838 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| SYBR Green | Bio-Rad | Cat# 172–5270 |
| Mirabegron | Cayman | Cat# 17319 |
| Mounting media | Dako | Cat# C056330–2 |
| Humulin R U-100 | Eli Lilly and Company | Cat# 0002–8215 |
| TrypLE Express Enzyme | GIBCO | Cat# 12604013 |
| Bovine Calf Serum | GIBCO | Cat# 16170–078 |
| Fetal Bovine Serum | Life Technologies | Cat# 10–013-CV |
| Alexa 594-azide | Life Technologies | Cat# C10270 |
| DMEM/F-12 | Life Technologies | Cat# 31330 |
| SAG | Millipore | Cat# 566660 |
| Fugene6 Transfection Reagent | Promega | Cat# E2691 |
| Rodent Diet With 60 kcal% Fat | Research Diets | Cat# D12492 |
| cOmplete-Mini protease inhibitor cocktail | Roche | Cat# 11836153001 |
| D-(+)-Glucose | Sigma | Cat# G-6152 |
| 3-isobutyl-1-methylxanthine (IBMX) | Sigma | Cat# I7018 |
| Dexamethazone | Sigma | Cat# D4902 |
| Oil Red O | Sigma | Cat# 00625 |
| Collagenase | Sigma | Cat# C6885 |
| NP40 | Sigma | Cat# 11332473001 |
| Transferrin | Sigma | Cat# T3309 |
| Biotin | Sigma | Cat# B4501 |
| Pantothenate | Sigma | Cat# C8731 |
| Triiodo-L-thyronine (T3) | Sigma | Cat# T2877 |
| Ciglitazone | Sigma | Cat# 230950 |
| Fetuin | Sigma | Cat# F2379 |
| Forskolin | Sigma | Cat# 115116–37-5 |
| X-tremeGENE 9 DNA Transfection Reagent | Sigma | Cat# 6365779001 |
| TRIzol | ThermoFisher | Cat# 15596026 |
| Beta-galactosidase | ThermoFisher | Cat# 15520034 |
| BODIPY 558/568 | ThermoFisher | Cat# D-3835 |
| BODIPY 493/503 | ThermoFisher | Cat# D-3922 |
| EdU | ThermoFisher | Cat# A10044 |
| G418 | ThermoFisher | Cat# 11811023 |
| Mitochondria-GFP | ThermoFisher | Cat# C10508 |
| Pen/Strep | ThermoFisher | Cat# 15140163 |
| DMEM | ThermoFisher | Cat# 11995065 |
| Lipofectamine RNAiMAX Transfection Reagent | ThermoFisher | Cat# 13778500 |
| CL-316,243 | Tocris | Cat# 1499 |
|
| ||
| Critical commercial assays | ||
|
| ||
| Blood glucose test strips (Bayer Breeze2) | Bayer | Cat# 1468A |
| High-Capacity cDNA Reverse Transcription Kit | Applied Biosystems | Cat# 4368814 |
| Pierce BCA Protein Assay Kit | ThermoFisher | Cat# 23227 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| 3T3-L1 | ATCC | Cat# CL-173 |
| Mouse Embryonic Fibroblasts (MEFs) | This work | N/A |
| Primary mouse preadipocytes | This work | N/A |
| Primary human preadipocytes | This work | N/A |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| Mouse: C57BL/6J | Jackson Laboratories | Cat# 000664 |
| Mouse: C57BL/6J; Fbf1tm1a/tm1a | This work | N/A |
|
| ||
| Oligonucleotides | ||
|
| ||
| pCDH-CMV-MCS-EF1-Neo | System Biosciences | Cat# CD533A-2 |
| pCDH-CMV-MCS-EF1-Neo-FBF1 | This work | N/A |
| pCDH-CMV-MCS-EF1-Neo-Myc-FBF1 | This work | N/A |
| pCDH-CMV-MCS-EF1-Neo-Myc-BirA-FBF1 | This work | N/A |
| pCDH-CMV-MCS-EF1-YFP-CEP170C | This work | N/A |
| pCDH-CMV-MCS-EF1-Neo-YFP-CEP170C-PKI | This work | N/A |
| pCDH-CMV-MCS-EF1-Neo-YFP-PKI | This work | N/A |
| pCDH-CMV-MCS-EF1-Neo-CFP-FRB-CEP170C | This work | N/A |
| pCDH-CMV-MCS-EF1-Neo-YFP-FKBP-PKI | This work | N/A |
| Stealth RNAi siRNA Negative Control Med GC | ThermoFisher | Cat# 12935–300 |
| Primers for genotyping | ThermoFisher | See Table S1 |
| Primers for Real-time RT-PCR | ThermoFisher | See Table S1 |
| si RNAs | ThermoFisher | See Table S2 |
|
| ||
| Software and algorithms | ||
|
| ||
| Graphpad Prism | Graphpad Software | https://www.graphpad.com/scientific-software/prism/ |
| Image Lab | Bio-Rad | https://www.bio-rad.com/ |
| CFX Real-Time PCR Detection Systems | Bio-Rad | https://www.bio-rad.com/ |
| ZEN - ZEISS Efficient Navigation | ZEISS | https://www.zeiss.com/corporate/int/home |
| IMARIS | Oxford Instruments | https://imaris.oxinst.com/ |
| NIS-Elements Viewer | Nikon | https://www.microscope.healthcare.nikon.com/ |
| CLAMS Examination Tool | Columbus Instruments | https://www.colinst.com |
| IMARIS | Oxford Instruments | https://imaris.oxinst.com/ |
| NIS-Elements Viewer | Nikon | https://www.microscope.healthcare.nikon.com/ |
| CLAMS Examination Tool | Columbus Instruments | https://www.colinst.com |
Highlights.
Fbf1tm1a/tm1a mice exhibit ‘‘healthy obesity’’ with healthy and beiging white fat tissue
FBF1 controls the beiging program via a cilia-specific, AKAP9-dependent, PKA signaling
FBF1 governs the adipogenic program via Hh signaling
Fbf1tm1a/tm1a mice are protected from diabetes and show reduced risk of dying prematurely
ACKNOWLEDGMENTS
The authors acknowledge support from the National Institutes of Health (NIH) research grants R01DK090038 and R01DK099160 and P30 center grant P30DK90728 to J. Hu and R01 AG058812 to E.N.C.; a Department of Defense grant W81XWH2010214 to K.L.; and grants from the NIH (R37 AG013925, P01 AG062413, and R33 AG061456, Translational Geroscience Network), the Connor Fund, Robert J. and Theresa W. Ryan, and the Noaber Foundation to J.K.; a Mayo Center for Biomedical Discovery Award to J. Hu, E.N.C., and J.K.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109481.
DECLARATION OF INTERESTS
The authors declare no competing interests.
INCLUSION AND DIVERSITY
We worked to ensure sex balance in the selection of non-human subjects. We worked to ensure diversity in experimental samples through the selection of the cell lines.
REFERENCES
- Anderson RG (1972). The three-dimensional structure of the basal body from the rhesus monkey oviduct. J. Cell Biol 54, 246–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breslow DK, Hoogendoorn S, Kopp AR, Morgens DW, Vu BK, Kennedy MC, Han K, Li A, Hess GT, Bassik MC, et al. (2018). A CRISPR-based screen for Hedgehog signaling provides insights into ciliary function and ciliopathies. Nat. Genet 50, 460–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaki M, Airik R, Ghosh AK, Giles RH, Chen R, Slaats GG, Wang H, Hurd TW, Zhou W, Cluckey A., et al. (2012). Exome capture reveals ZNF423 and CEP164 mutations, linking renal ciliopathies to DNA damage response signaling. Cell 150, 533–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, and Kass RS (2005). A-kinase anchoring proteins: different partners, different dance. Nat. Cell Biol 7, 1050–1051. [DOI] [PubMed] [Google Scholar]
- Cohen P, Levy JD, Zhang Y, Frontini A, Kolodin DP, Svensson KJ, Lo JC, Zeng X, Ye L, Khandekar MJ, et al. (2014). Ablation of PRDM16 and beige adipose causes metabolic dysfunction and a subcutaneous to visceral fat switch. Cell 156, 304–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davenport JR, Watts AJ, Roper VC, Croyle MJ, van Groen T, Wyss JM, Nagy TR, Kesterson RA, and Yoder BK (2007). Disruption of intraflagellar transport in adult mice leads to obesity and slow-onset cystic kidney disease. Curr. Biol 17, 1586–1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deren ME, Yang X, Guan Y, and Chen Q (2016). Biological and Chemical Removal of Primary Cilia Affects Mechanical Activation of Chondrogenesis Markers in Chondroprogenitors and Hypertrophic Chondrocytes. Int. J. Mol. Sci 17, 188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Després JP, Lemieux I, Bergeron J, Pibarot P, Mathieu P, Larose E, Rodés-Cabau J, Bertrand OF, and Poirier P (2008). Abdominal obesity and the metabolic syndrome: contribution to global cardiometabolic risk. Arterioscler. Thromb. Vasc. Biol 28, 1039–1049. [DOI] [PubMed] [Google Scholar]
- Feuillan PP, Ng D, Han JC, Sapp JC, Wetsch K, Spaulding E, Zheng YC, Caruso RC, Brooks BP, Johnston JJ, et al. (2011). Patients with Bardet-Biedl syndrome have hyperleptinemia suggestive of leptin resistance. J. Clin. Endocrinol. Metab 96, E528–E535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galic S, Oakhill JS, and Steinberg GR (2010). Adipose tissue as an endocrine organ. Mol. Cell. Endocrinol 316, 129–139. [DOI] [PubMed] [Google Scholar]
- Goetz SC, and Anderson KV (2010). The primary cilium: a signalling centre during vertebrate development. Nat. Rev. Genet 11, 331–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harms M, and Seale P (2013). Brown and beige fat: development, function and therapeutic potential. Nat. Med 19, 1252–1263. [DOI] [PubMed] [Google Scholar]
- Hilgendorf KI, Johnson CT, and Jackson PK (2016). The primary cilium as a cellular receiver: organizing ciliary GPCR signaling. Curr. Opin. Cell Biol 39, 84–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilgendorf KI, Johnson CT, Mezger A, Rice SL, Norris AM, Demeter J, Greenleaf WJ, Reiter JF, Kopinke D, and Jackson PK (2019). Omega-3 Fatty Acids Activate Ciliary FFAR4 to Control Adipogenesis. Cell 179, 1289–1305.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong SR, Wang CL, Huang YS, Chang YC, Chang YC, Pusapati GV, Lin CY, Hsu N, Cheng HC, Chiang YC, et al. (2018). Spatiotemporal manipulation of ciliary glutamylation reveals its roles in intraciliary trafficking and Hedgehog signaling. Nat. Commun 9, 1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S, Laoukili J, Epping MT, Koster J, Hölzel M, Westerman BA, Nijkamp W, Hata A, Asgharzadeh S, Seeger RC, et al. (2009). ZNF423 is critically required for retinoic acid-induced differentiation and is a marker of neuroblastoma outcome. Cancer Cell 15, 328–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kee HL, Dishinger JF, Blasius TL, Liu CJ, Margolis B, and Verhey KJ (2012). A size-exclusion permeability barrier and nucleoporins characterize a ciliary pore complex that regulates transport into cilia. Nat. Cell Biol 14, 431–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkland JL, and Dobson DE (1997). Preadipocyte function and aging: links between age-related changes in cell dynamics and altered fat tissue function. J. Am. Geriatr. Soc 45, 959–967. [DOI] [PubMed] [Google Scholar]
- Knighton DR, Zheng JH, Ten Eyck LF, Xuong NH, Taylor SS, and Sowadski JM (1991). Structure of a peptide inhibitor bound to the catalytic subunit of cyclic adenosine monophosphate-dependent protein kinase. Science 253, 414–420. [DOI] [PubMed] [Google Scholar]
- Kolobova E, Roland JT, Lapierre LA, Williams JA, Mason TA, and Goldenring JR (2017). The C-terminal region of A-kinase anchor protein 350 (AKAP350A) enables formation of microtubule-nucleation centers and interacts with pericentriolar proteins. J. Biol. Chem 292, 20394–20409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopinke D, Roberson EC, and Reiter JF (2017). Ciliary Hedgehog Signaling Restricts Injury-Induced Adipogenesis. Cell 170, 340–351.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin YC, Niewiadomski P, Lin B, Nakamura H, Phua SC, Jiao J, Levchenko A, Inoue T, Rohatgi R, and Inoue T (2013). Chemically inducible diffusion trap at cilia reveals molecular sieve-like barrier. Nat. Chem. Biol 9, 437–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marion V, Mockel A, De Melo C, Obringer C, Claussmann A, Simon A, Messaddeq N, Durand M, Dupuis L, Loeffler J-P, et al. (2012). BBS-induced ciliary defect enhances adipogenesis, causing paradoxical higher-insulin sensitivity, glucose usage, and decreased inflammatory response. Cell Metab 16, 363–377. [DOI] [PubMed] [Google Scholar]
- Nachury MV, and Mick DU (2019). Establishing and regulating the composition of cilia for signal transduction. Nat. Rev. Mol. Cell Biol 20, 389–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nachury MV, Loktev AV, Zhang Q, Westlake CJ, Peränen J, Merdes A, Slusarski DC, Scheller RH, Bazan JF, Sheffield VC, and Jackson PK (2007). A core complex of BBS proteins cooperates with the GTPase Rab8 to promote ciliary membrane biogenesis. Cell 129, 1201–1213. [DOI] [PubMed] [Google Scholar]
- Nachury MV, Seeley ES, and Jin H (2010). Trafficking to the ciliary membrane: how to get across the periciliary diffusion barrier? Annu. Rev. Cell Dev. Biol 26, 59–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nedergaard J, and Cannon B (2013). How brown is brown fat? It depends where you look. Nat. Med 19, 540–541. [DOI] [PubMed] [Google Scholar]
- Nissen SE, and Wolski K (2007). Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. N. Engl. J. Med 356, 2457–2471. [DOI] [PubMed] [Google Scholar]
- Oh EC, Vasanth S, and Katsanis N (2015). Metabolic regulation and energy homeostasis through the primary cilium. Cell Metab 21, 21–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiter JF, Blacque OE, and Leroux MR (2012). The base of the cilium: roles for transition fibres and the transition zone in ciliary formation, maintenance and compartmentalization. EMBO Rep 13, 608–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romieu I, Dossus L, Barquera S, Blottière HM, Franks PW, Gunter M, Hwalla N, Hursting SD, Leitzmann M, Margetts B., et al. ; IARC working group on Energy Balance and Obesity (2017). Energy balance and obesity: what are the main drivers? Cancer Causes Control 28, 247–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen ED, and MacDougald OA (2006). Adipocyte differentiation from the inside out. Nat. Rev. Mol. Cell Biol 7, 885–896. [DOI] [PubMed] [Google Scholar]
- Rosen ED, and Spiegelman BM (2014). What we talk about when we talk about fat. Cell 156, 20–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbaum JL, and Witman GB (2002). Intraflagellar transport. Nat. Rev. Mol. Cell Biol 3, 813–825. [DOI] [PubMed] [Google Scholar]
- Roux KJ, Kim DI, Raida M, and Burke B (2012). A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J. Cell Biol 196, 801–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo S, Baye LM, Schulz NP, Beck JS, Zhang Q, Slusarski DC, and Sheffield VC (2010). BBS6, BBS10, and BBS12 form a complex with CCT/TRiC family chaperonins and mediate BBSome assembly. Proc. Natl. Acad. Sci. USA 107, 1488–1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao M, Ishibashi J, Kusminski CM, Wang QA, Hepler C, Vishvanath L, MacPherson KA, Spurgin SB, Sun K, Holland WL, et al. (2016). Zfp423 Maintains White Adipocyte Identity through Suppression of the Beige Cell Thermogenic Gene Program. Cell Metab 23, 1167–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singla V, and Reiter JF (2006). The primary cilium as the cell’s antenna: signaling at a sensory organelle. Science 313, 629–633. [DOI] [PubMed] [Google Scholar]
- Skarnes WC, Rosen B, West AP, Koutsourakis M, Bushell W, Iyer V, Mujica AO, Thomas M, Harrow J, Cox T., et al. (2011). A conditional knockout resource for the genome-wide study of mouse gene function. Nature 474, 337–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tchkonia T, Tchoukalova YD, Giorgadze N, Pirtskhalava T, Karagiannides I, Forse RA, Koo A, Stevenson M, Chinnappan D, Cartwright A., et al. (2005). Abundance of two human preadipocyte subtypes with distinct capacities for replication, adipogenesis, and apoptosis varies among fat depots. Am. J. Physiol. Endocrinol. Metab 288, E267–E277. [DOI] [PubMed] [Google Scholar]
- Terrin A, Monterisi S, Stangherlin A, Zoccarato A, Koschinski A, Surdo NC, Mongillo M, Sawa A, Jordanides NE, Mountford JC, and Zaccolo M (2012). PKA and PDE4D3 anchoring to AKAP9 provides distinct regulation of cAMP signals at the centrosome. J. Cell Biol 198, 607–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truong ME, Bilekova S, Choksi SP, Li W, Bugaj LJ, Xu K, and Reiter JF (2021). Vertebrate cells differentially interpret ciliary and extraciliary cAMP. Cell 184, 2911–2926.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaisse C, Reiter JF, and Berbari NF (2017). Cilia and Obesity. Cold Spring Harb. Perspect. Biol 9, a028217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieira OV, Gaus K, Verkade P, Fullekrug J, Vaz WL, and Simons K (2006). FAPP2, cilium formation, and compartmentalization of the apical membrane in polarized Madin-Darby canine kidney (MDCK) cells. Proc. Natl. Acad. Sci. USA 103, 18556–18561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Wang B, and Jiang J (1999). Protein kinase A antagonizes Hedgehog signaling by regulating both the activator and repressor forms of Cubitus interruptus. Genes Dev 13, 2828–2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Q, Xu Q, Zhang Y, Li Y, Zhang Q, Hu Z, Harris PC, Torres VE, Ling K, and Hu J (2013). Transition fibre protein FBF1 is required for the ciliary entry of assembled intraflagellar transport complexes. Nat. Commun 4, 2750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Q, Ling K, and Hu J (2015). The essential roles of transition fibers in the context of cilia. Curr. Opin. Cell Biol 35, 98–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Q, Zhang Y, Schouteden C, Zhang Y, Zhang Q, Dong J, Wonesch V, Ling K, Dammermann A, and Hu J (2016). The hydrolethalus syndrome protein HYLS-1 regulates formation of the ciliary gate. Nat. Commun 7, 12437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Boström P, Sparks LM, Ye L, Choi JH, Giang AH, Khandekar M, Virtanen KA, Nuutila P, Schaart G., et al. (2012). Beige adipocytes are a distinct type of thermogenic fat cell in mouse and human. Cell 150, 366–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate any unique datasets.
This study did not generate any unique code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.