

ABSTRACT
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is the viral pathogen causing the coronavirus disease 2019 (COVID-19) global pandemic. No effective treatment for COVID-19 has been established yet. The serine protease transmembrane protease serine 2 (TMPRSS2) is essential for viral spread and pathogenicity by facilitating the entry of SARS-CoV-2 into host cells. The protease inhibitor camostat, an anticoagulant used in the clinic, has potential anti-inflammatory and antiviral activities against COVID-19. However, the potential mechanisms of viral resistance and antiviral activity of camostat are unclear. Herein, we demonstrate high inhibitory potencies of camostat for a panel of serine proteases, indicating that camostat is a broad-spectrum inhibitor of serine proteases. In addition, we determined the crystal structure of camostat in complex with a serine protease (uPA [urokinase-type plasminogen activator]), which reveals that camostat is inserted in the S1 pocket of uPA but is hydrolyzed by uPA, and the cleaved camostat covalently binds to Ser195. We also generated a homology model of the structure of the TMPRSS2 serine protease domain. The model shows that camostat uses the same inhibitory mechanism to inhibit the activity of TMPRSS2, subsequently preventing SARS-CoV-2 spread.
IMPORTANCE Serine proteases are a large family of enzymes critical for multiple physiological processes and proven diagnostic and therapeutic targets in several clinical indications. The serine protease transmembrane protease serine 2 (TMPRSS2) was recently found to mediate SARS-CoV-2 entry into the host. Camostat mesylate (FOY 305), a serine protease inhibitor active against TMPRSS2 and used for the treatment of oral squamous cell carcinoma and chronic pancreatitis, inhibits SARS-CoV-2 infection of human lung cells. However, the direct inhibition mechanism of camostat mesylate for TMPRSS2 is unclear. Herein, we demonstrate that camostat uses the same inhibitory mechanism to inhibit the activity of TMPRSS2 as uPA, subsequently preventing SARS-CoV-2 spread.
KEYWORDS: SARS-CoV-2, COVID-19, TMPRSS2, uPA, serine protease inhibitor, camostat
INTRODUCTION
The global coronavirus disease 2019 (COVID-19) pandemic, caused by severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2), has led to over 78.6 million diagnosed cases and over 1.7 million reported deaths as of 24 December 2020. Elucidating the mechanism of SARS-CoV-2 infection is necessary for the rational design of therapeutics, developing novel or repurposed therapeutics, and understanding the clinical course of COVID-19. It has been demonstrated that infections by the SARS-CoV-2 depend on the host cell receptor angiotensin-converting enzyme 2 (ACE2) receptor (1–3).
One key discovery in understanding the mechanism of SARS-CoV-2 infection involves the role of transmembrane serine protease 2 (TMPRSS2), a cell surface protein that is expressed by epithelial cells of specific tissues, e.g., in the aerodigestive tract (4, 5). Viral entry requires TMPRSS2 to prime the virus’s spike (S) protein by the cleavage of the S proteins at the S1/S2 and S2 sites (6, 7). Moreover, not only SARS-CoV-2 but also other types of coronaviruses and influenza viruses depend on TMPRSS2 for viral activation and cell entry, including SARS-CoV and H1N1 (8, 9). Therefore, TMPRSS2 plays a central and conserved role in the pathogenesis of the illnesses caused by coronaviruses and influenza viruses.
Recent works show that the serine protease inhibitor camostat mesylate (FOY 305), which has been used for the treatment of oral squamous cell carcinoma and chronic pancreatitis (10, 11), inhibits SARS-CoV-2 infection of human lung cells by acting against TMPRSS2 (12). Although Hoffmann et al. reported potential mechanisms of viral resistance as well as camostat mesylate metabolization and antiviral activity of metabolites (13), the direct inhibition mechanism of camostat mesylate for TMPRSS2 is unclear. In addition, there is no report of the atomic-resolution structure of TMPRSS2 so far, even only its serine protease domain. TMPRSS2 is a plasma membrane-anchored serine protease with one extracellular serine protease domain, including the canonical triad His57-Asp102-Ser195 (chymotrypsinogen numbering) and a canonical protease family S1 fold (14).
Here, the inhibitory effects of camostat mesylate on different types of serine protease were investigated, and a homology structure model of the TMPRSS2 serine protease domain (TMPRSS2-SPD) was built, which was shown to be highly identical to the structure of another serine protease, urokinase-type plasminogen activator (uPA), with a root mean square deviation (RMSD) of 0.681 Å. Next, the crystal structure of uPA in complex with camostat was determined and showed that camostat had been hydrolyzed and that the cleaved product covalently binds to Ser195 of uPA, leading inhibition of the enzyme activity. Thus, camostat mesylate uses covalent binding to inhibit the activity of serine proteases, including TMPRSS2.
RESULTS AND DISCUSSION
Inhibition assays of camostat for different serine proteases.
Camostat is well-known as a synthetic serine protease inhibitor with anti-inflammatory, antifibrotic, and potential antiviral activities. We performed enzyme inhibition assays using purified soluble recombinant serine protease domains of uPA, murine uPA (muPA), plasma kallikrein (PK), factor XIa (FXIa), trypsin, and matriptase (Fig. 1A). These results showed that camostat significantly inhibits (at a nanomolar order of magnitude) the protease activity on the above-mentioned serine proteases. Among these serine proteases, camostat has the highest inhibitory potency for trypsin, with a 50% inhibitory concentration (IC50) of 9.3 ± 1.2 nM. These potencies rank as follows: trypsin, 9.3 ± 1.2 nM; PK, 10.4 ± 2.7 nM; matriptase, 21.1 ± 3.5 nM; FXIa, 44.1 ± 1.1 nM; uPA, 86.4 ± 9.4 nM; and muPA, 100.4 ± 41.9 nM (Fig. 1B to G). These inhibitory potencies are consistent with the reported IC50 for TMPRSS2 (4 to 6 nM) (15, 16), demonstrating that camostat is a broad-spectrum serine protease inhibitor.
FIG 1.

SDS-PAGE electrophoresis (15%) showing the purity of serine proteases (A) and the activity of camostat against a panel of serine proteases, including uPA (B), muPA (C), PK (D), FXIa (E), matriptase (F), and trypsin (G). Dose-response curves were visualized and curve fitting was performed using GraphPad Prism. The calculated concentrations required for 50% inhibition (IC50) are displayed, in nanomolar units. The average of two independent experiments, each performed with triplicate samples, is shown.
Sequence alignment of TMPRSS2 and other serine proteases.
The architectures of the serine protease domains are generally well conserved. Although serine proteases recognize their distinct substrates in various ways, they utilize a canonical catalytic triad for activity. The catalytic triad of the classic trypsin and subtilisin families is composed of three highly conserved residues, His57, Asp102, and Ser195 (chymotrypsinogen numbering), and exhibits similar spatial arrangements (17). Several other highly conserved residues in serine protease families have been linked to catalysis, such as the backbone of Ser214 in chymotrypsin-like proteases, which contributes to the S1 binding pocket (18). Alignment of the eight serine protease domains, including uPA, muPA, PK, FXIa, and matriptase, supports that the sequence in the active site of TMPRSS2-SPD is highly conserved with other serine proteases (Fig. 2) and also indicates that the secondary structure of TMPRSS2-SPD might adopt a folding type similar to those of other serine proteases.
FIG 2.

Sequence alignments of TMPRSS2 and other representative chymotrypsin family serine proteases, including human uPA, murine uPA, tPA, trypsin, plasma kallikrein, FXIa, and matriptase. The secondary structure elements shown at the bottom of the sequences are based on human uPA. Red triangles represent residues of the serine protease catalytic triad: His57, Asp102, and Ser195.
Homology modeling of the structure of TMPRSS2 serine protease domain.
There is no experimental structure for TMPRSS2 yet, but it is possible to develop quality structural models for the serine protease domain using comparative modeling strategies, as such domains are highly structurally conserved. The three-dimensional structure of the TMPRSS2 serine protease domain (TMPRSS2-SPD) was built using the online server SWISS-MODEL (19) as shown in Fig. 3A. The modeling result shows that TMPRSS13 (PDB code 6KD5) (15) is the best template to model the structure of TMPRSS2-SPD (QMEAN, −1.15; sequence identity, 44.44%). The stereochemical analysis was performed using Procheck by considering the overall structural geometry of the respective protein (20). The results showed that 97.9% of the residues were in favorable and allowed regions of the Ramachandran plot, indicating that the structure is properly refined and can be used for further studies. TMPRSS2 belongs to the trypsin-like type; these enzymes usually cleave peptide bonds at Lys or Arg residues. This so-called P1 residue fits into a negatively charged S1 pocket. This pocket displays a conserved Asp residue at the bottom and as such tends to prefer a positively charged residue at the P1 position. In this study, we compared the modeled structure of TMPRSS2-SPD with that of a representative trypsin-like serine protease, urokinase-type plasminogen activator (uPA), and found that these structures had a high similarity (RMSD = 0.681 Å). Especially, the S1 pockets and the catalytic triads are identical to each other with a sequence identity of 34.4% (Fig. 3B).
FIG 3.

Homology modeling of the structure of TMPRSS2-SPD and comparison with the uPA structure. (A) Overview of the modeled structure of TMPRSS2-SPD (orange) by SWISS-MODEL. (B) Comparison of TMPRSS2-SPD (orange) and uPA-SPD (light blue) as a representative serine protease showing that these two structures had a high similarity (RMSD = 0.681 Å), which indicates that TMPRSS2-SPD might have a structure highly similar to that of other serine proteases.
Camostat was hydrolyzed by serine protease and irreversibly inhibited serine protease.
To study how camostat inhibits serine proteases, we determined the crystal structure of camostat in complex with uPA. The crystal structure of the uPA-camostat complex was determined to a resolution of 2.0 Å and was refined to good statistics (R factor = 19.2%, Rfree factor = 23.1% [Table 1]). We identified the strong electron density map (Fo − Fc = 3.0 σ) at the major substrate-binding pocket of uPA (or S1 pocket) and close to Ser195 (Fig. 4A). However, the electron density map was obviously small in volume and not large enough to accommodate intact camostat, suggesting that camostat might have been hydrolyzed by uPA. After modeling and refining the structure, the clear electron density map (2Fo − Fc = 1.5 σ) showed that one of the hydrolysates of camostat, 4-guanidinobenzoic acid (GBA), covalently bound to the Ser195 (Fig. 4B and C). Moreover, we did not find the primary metabolite of camostat [4-(4-guanidinobenzoyloxy)phenylacetic acid (GBPA; FOY-251)] and another metabolite (6-amidino-2-naphthol; PDB code 6A2N) in the complex structure. The mass spectrometric analysis also determined that the purified uPA-camostat sample contained the hydrolysates of camostat GBA (Fig. 5). Thus, we clearly determined that the GBA fragment binds to the active sites of uPA and the guanidine group of GBA points to the bottom of the S1 pocket, forming hydrogen bonds with the uPA residues Asp189, Ser190, and Gly219 (Fig. 6A and B). Additionally, interactions such as π-cation, π-π stacked, π-π T-shaped, amide-π stacked and π-alkyl, which largely involves charge transfer, helped in intercalating the GBA in the binding site of the enzyme. The combination of these covalent and noncovalent interactions with uPA made the GBA fragment very stable (B factor, 10.8 Å2) in the complex structure (B factor, 19.9 Å2). The current results demonstrate the possible antiviral mechanism of camostat as an inhibitor of TMPRSS2 by blocking the spike protein priming and then preventing SARS-CoV-2 entry into the host cell (Fig. 6C).
TABLE 1.
X-ray data collection and model refinement statistics for the uPA-camostat crystal
| Compound | Value(s) for uPA-camostata |
|---|---|
| Data collection | |
| X-ray source wavelength (Å) | 1.54 |
| Resolution limits (Å) | 60.2–2.0 (2.03–2.00) |
| Space group | R3 |
| Temp of experiments (K) | 100 |
| Cell constants | |
| a, b, c (Å) | 120.317, 120.317, 42.451 |
| α = β; γ (°) | 90; 120 |
| Completeness (%) | 96.5 |
| Multiplicity | 3.04 |
| Rmergeb | 0.021 (0.064) |
| No. of observations | 46,862 |
| No. of unique reflections | 15,407 |
| Refinement data | |
| R | 0.192 |
| Rfree | 0.231 |
| Avg B factor (Å2) of uPA | 19.6 |
| Avg B factor (Å2) of camostat | 10.8 |
| RMSD of bond lengths (Å) | 0.006 |
| RMSD of angle (°) | 1.442 |
| Ramachandran analysis (%) | |
| Favored regions | 97.1 |
| Allowed regions | 2.5 |
| Disallowed regions | 0.4 |
The highest-resolution shell is shown in parentheses.
Rmerge = Σ|Ii – 〈I〉|/ΣIi, where Ii is the intensity of the ith observation and 〈I〉 is the mean intensity of the reflections.
FIG 4.
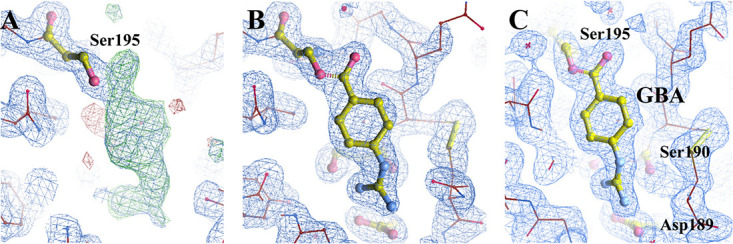
(A) A Fourier (2Fo − Fc) electron density map at a 1.5 σ cutoff showing the quality of the map and a difference (Fo − Fc) Fourier electron density map at a 3.0 σ cutoff showing the positive (green) and negative (red) electron densities, which indicates that camostat was hydrolyzed by uPA and one of the hydrolysates of camostat (GBA) covalently binds to the Ser195. (B) The simulated annealing 2Fo − Fc (1.5 σ) omit map of GBA in the active site of uPA. (C) Final (2Fo − Fc) electron density map at a 1.5 σ cutoff showing the well-refined position for GBA.
FIG 5.

Mass spectrometric analysis. (A) Mass spectra of the 2,5-dihydroxy benzoic acid (DHB)-only matrix. (B) Mass spectra of camostat bound to uPA. The peak at m/z 177 indicates that the hydrolysates of camostat and 4-guanidinobenzoic acid (GBA) are consistent with the m/z value predicated by ChemDraw.
FIG 6.

The overall complex structure of uPA-camostat. (A) Ribbon representation of the crystal structure of uPA (light blue) in complex with the hydrolyzed camostat (magenta stick model). The inset is a close-up view of the bound camostat inhibitor and the S1 pocket of uPA. The catalytic triad Ser195-His57-Asp102 and Asp189 in the bottom residue of the S1 pocket are shown in the stick model. The uPA and camostat are in orange and magenta, respectively. Dashed lines indicate hydrogen bonds. (B) Bound camostat with uPA and interactive site residues along with hydrogen bonds and other interactions. (C) The anti-SARS-CoV-2 mechanism of camostat is that camostat binds to TMPRSS2 and is hydrolyzed by it, and then the hydrolysate of camostat covalently binds to the Ser195 to deactivate TMPRSS2, sequentially block spike protein priming, and prevent SARS-CoV-2 entry into the host cell.
Conclusion.
In this study, we performed inhibitory assays of camostat for a series of serine proteases and provide evidence showing that camostat is a high-efficiency and broad-spectrum serine protease inhibitor. Moreover, we compared the modeled structure of TMPRSS2-SPD with that of uPA as a representative serine protease and found a high degree of similarity between them. Furthermore, the crystal structure of uPA in complex with camostat was determined and showed that camostat was hydrolyzed and cleaved to a fragment that was covalently bound to Ser195 of uPA. These results show that camostat might adopt the same inhibitory mechanism to inhibit the activity of TMPRSS2 as other serine proteases, thus preventing SARS-CoV-2 spread.
MATERIALS AND METHODS
Expression and purification of serine proteases.
Recombinant serine protease domains of uPA (21), FXIa (22), matriptase (23), plasma kallikrein (PK) (24), and tissue-type plasminogen activator (tPA) (25) were expressed from Pichia pastoris strain X-33 as described elsewhere. The serine protease domain of murine uPA (muPA) was expressed as inclusion bodies in Escherichia coli BL21(DE3); the subsequent refolding and purification of the serine protease domain of muPA were described previously (26). In addition, human trypsin was purchased from Sigma-Aldrich (St. Louis, MO, USA).
Determination of inhibitory potency of camostat mesylate for serine proteases.
For routine determination of IC50s for the inhibition of the various enzymes under steady-state inhibition conditions, 2 nM purified enzyme was preincubated in a total volume of 200 μl of 0.01 M HEPES, 0.15 M NaCl (pH 7.4) with 0.1% bovine serum albumin (BSA) at 37°C, with various concentrations of peptides for 15 min before the addition of the chromogenic substrate S-2444 (pyro-Glu-Gly-Arg-p-nitroanilide) for the uPA variants, S-2302 (H-d-Pro-Phe-Arg-p-nitroanilide) for PK, S-2288 (H-d-Ile-Pro-Arg-p-nitroanilide) for matriptase and tPA, and B-3133 (Nα-benzoyl-l-Arg-4-nitroanilide) for trypsin in concentrations approximately equal to the Km value for each particular variant. The initial reaction velocities were monitored at an absorbance of 405 nm. The nonlinear regression (sigmoidal) in GraphPad Prism 8 software was used to fit the IC50 of camostat for different serine proteases.
Homology modeling and validation of TMPRSS2 serine protease domain.
The three-dimensional structure of TMPRSS2 serine protease was built using homology modeling due to the unavailability of its structure in the Protein Data Bank (PDB). The protein sequence of the TMPRSS2 serine protease domain (TMPRSS2-SPD) was obtained at the UniProt website (entry O15393; https://www.uniprot.org/), which contains 236 amino acids. Then, the sequence was retrieved in the FASTA format and uploaded to the fully automated protein structure homology-modeling server SWISS-MODEL (https://swissmodel.expasy.org/) (27). The top model with the best QMEAN score and high percentage similarity was selected as the analytic structure.
Crystallization and data collection for the uPA-camostat complex.
The crystallization trials were carried out using the method of sitting drop vapor diffusion (28). The uPA protease domain (positions 159 to 411) was mixed with 500 μM camostat and crystallized with 2.0 M ammonium sulfate, 50 mM sodium citrate (pH 4.6), and 5% polyethylene glycol 400 (PEG400) at room temperature. The crystals appeared in about 3 days. A solution of 2.0 M ammonium sulfate, 50 mM sodium citrate (pH 4.6), 5% PEG400, and 20% (vol/vol) glycerol was used as a cryoprotectant for the crystals. X-ray diffraction data of the uPA-camostat complex were collected in a Bruker D8 Venture system at a wavelength of 1.54 Å. The space group of uPA-camostat was determined to be R3 by preliminary manipulation using the APEX3 suite program package, with the following unit cell parameters: a = 120.3 Å, b = 120.3 Å, c = 42.5 Å. The most probable Matthews coefficient was 2.11 Å3 Da−1 and corresponded to one protein molecule per asymmetric unit with a solvent content of 41.8%. The data set was 99.5% complete to 2.0 Å. The statistics of data collection are listed in Table 1.
Phasing and refinement.
The structure of the uPA-camostat complex was solved by the molecular replacement method using the MolRep program (29), which gave very strong and unambiguous solutions. A uPA was first positioned inside the crystal lattice using the uPA structure (PDB code 4DVA) (30) as a searching model and all the X-ray data up to 2.0 Å. The Fo − Fc electron density map calculated at this stage showed the presence of one compound at the active site of uPA. The molecular replacement model was subjected to iterative refinement and manual model rebuilding using Refmac (31) and Coot (32), alternately, giving a final R factor and Rfree factor of 0.192 and 0.231, respectively, at a resolution range of 60.2 to 2.0 Å (Table 1). The structure was validated with PROCHECK (33). None of these residues was in the disallowed region of the Ramachandran plot. The final results were analyzed and visualized by PyMOL (34) and LigPlot+ (35).
Mass spectrometry.
Mass-spectrometric analysis of uPA preincubated with camostat under crystal growth conditions was carried out. After being purified by SDS-PAGE and trypsin digestion, the samples were analyzed on a Bruker Autoflex II mass spectrometer (Bruker Daltonics, Germany) using Flex Analysis 3.4 software.
Data availability.
The coordinate of the uPA-camostat complex was deposited in the Protein Data Bank (PDB code 7DZD).
ACKNOWLEDGMENTS
Our research work is financially supported by grants from the National Key R&D Program of China (no. 2017YFE0103200), the National Natural Science Foundation of China (no. 82070142 and 22077016), the Natural Science Foundation of Fujian Province (no. 2018J01897 and 2018J01729).
L.J. and M.H. designed the study. G.S., Y.S., Y.Z., and J.Y. performed research. G.S., Y.S. Y.Z., J.Y., C.Y., and L.J. analyzed the data. L.J. wrote the manuscript. All authors revised the manuscript.
We declare no competing financial interests.
Contributor Information
Longguang Jiang, Email: jianglg@fzu.edu.cn.
Mingdong Huang, Email: hmd_lab@fzu.edu.cn.
Rebecca Ellis Dutch, University of Kentucky College of Medicine.
REFERENCES
- 1.Zhou P, Yang XL, Wang XG, Hu B, Zhang L, Zhang W, Si HR, Zhu Y, Li B, Huang CL, Chen HD, Chen J, Luo Y, Guo H, Jiang RD, Liu MQ, Chen Y, Shen XR, Wang X, Zheng XS, Zhao K, Chen QJ, Deng F, Liu LL, Yan B, Zhan FX, Wang YY, Xiao GF, Shi ZL. 2020. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 579:270–273. 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lu R, Zhao X, Li J, Niu P, Yang B, Wu H, Wang W, Song H, Huang B, Zhu N, Bi Y, Ma X, Zhan F, Wang L, Hu T, Zhou H, Hu Z, Zhou W, Zhao L, Chen J, Meng Y, Wang J, Lin Y, Yuan J, Xie Z, Ma J, Liu WJ, Wang D, Xu W, Holmes EC, Gao GF, Wu G, Chen W, Shi W, Tan W. 2020. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet 395:565–574. 10.1016/S0140-6736(20)30251-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yan R, Zhang Y, Li Y, Xia L, Guo Y, Zhou Q. 2020. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 367:1444–1448. 10.1126/science.abb2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li F, Li W, Farzan M, Harrison SC. 2005. Structure of SARS coronavirus spike receptor-binding domain complexed with receptor. Science 309:1864–1868. 10.1126/science.1116480. [DOI] [PubMed] [Google Scholar]
- 5.Stopsack KH, Mucci LA, Antonarakis ES, Nelson PS, Kantoff PW. 2020. TMPRSS2 and COVID-19: serendipity or opportunity for intervention? Cancer Discov 10:779–782. 10.1158/2159-8290.CD-20-0451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ziegler CGK, Allon SJ, Nyquist SK, Mbano IM, Miao VN, Tzouanas CN, Cao Y, Yousif AS, Bals J, Hauser BM, Feldman J, Muus C, Wadsworth MH, II, Kazer SW, Hughes TK, Doran B, Gatter GJ, Vukovic M, Taliaferro F, Mead BE, Guo Z, Wang JP, Gras D, Plaisant M, Ansari M, Angelidis I, Adler H, Sucre JMS, Taylor CJ, Lin B, Waghray A, Mitsialis V, Dwyer DF, Buchheit KM, Boyce JA, Barrett NA, Laidlaw TM, Carroll SL, Colonna L, Tkachev V, Peterson CW, Yu A, Zheng HB, Gideon HP, Winchell CG, Lin PL, Bingle CD, Snapper SB, Kropski JA, Theis FJ, HCA Lung Biological Network, et al. 2020. SARS-CoV-2 receptor ACE2 is an interferon-stimulated gene in human airway epithelial cells and is detected in specific cell subsets across tissues. Cell 181:1016–1035.E19. 10.1016/j.cell.2020.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hoffmann M, Kleine-Weber H, Pohlmann S. 2020. A multibasic cleavage site in the spike protein of SARS-CoV-2 is essential for infection of human lung cells. Mol Cell 78:779–784.E5. 10.1016/j.molcel.2020.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chaipan C, Kobasa D, Bertram S, Glowacka I, Steffen I, Tsegaye TS, Takeda M, Bugge TH, Kim S, Park Y, Marzi A, Pohlmann S. 2009. Proteolytic activation of the 1918 influenza virus hemagglutinin. J Virol 83:3200–3211. 10.1128/JVI.02205-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Matsuyama S, Nagata N, Shirato K, Kawase M, Takeda M, Taguchi F. 2010. Efficient activation of the severe acute respiratory syndrome coronavirus spike protein by the transmembrane protease TMPRSS2. J Virol 84:12658–12664. 10.1128/JVI.01542-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sai JK, Suyama M, Kubokawa Y, Matsumura Y, Inami K, Watanabe S. 2010. Efficacy of camostat mesilate against dyspepsia associated with non-alcoholic mild pancreatic disease. J Gastroenterol 45:335–341. 10.1007/s00535-009-0148-1. [DOI] [PubMed] [Google Scholar]
- 11.Ohkoshi M, Fujii S. 1983. Effect of the synthetic protease inhibitor [N,N-dimethylcarbamoyl-methyl 4-(4-guanidinobenzoyloxy)-phenylacetate] methanesulfate on carcinogenesis by 3-methylcholanthrene in mouse skin. J Natl Cancer Inst 71:1053–1057. [PubMed] [Google Scholar]
- 12.Hoffmann M, Kleine-Weber H, Schroeder S, Kruger N, Herrler T, Erichsen S, Schiergens TS, Herrler G, Wu NH, Nitsche A, Muller MA, Drosten C, Pohlmann S. 2020. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 181:271–280.E8. 10.1016/j.cell.2020.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hoffmann M, Hofmann-Winkler H, Smith JC, Kruger N, Sorensen LK, Sogaard OS, Hasselstrom JB, Winkler M, Hempel T, Raich L, Olsson S, Yamazoe T, Yamatsuta K, Mizuno H, Ludwig S, Noe F, Sheltzer JM, Kjolby M, Pohlmann S. 2020. Camostat mesylate inhibits SARS-CoV-2 activation by TMPRSS2-related proteases and its metabolite GBPA exerts antiviral activity. bioRxiv 10.1101/2020.08.05.237651. [DOI] [PMC free article] [PubMed]
- 14.Hedstrom L. 2002. Serine protease mechanism and specificity. Chem Rev 102:4501–4524. 10.1021/cr000033x. [DOI] [PubMed] [Google Scholar]
- 15.Ohno A, Maita N, Tabata T, Nagano H, Arita K, Ariyoshi M, Uchida T, Nakao R, Ulla A, Sugiura K, Kishimoto K, Teshima-Kondo S, Nikawa T, Okumura Y. 2020. Cystal structure of inhibitor-bound human MSPL/TMPRSS13 that can activate high pathogenic avian influenza. bioRxiv 10.1101/2020.06.12.149229:2020.06.12.149229. [DOI] [PMC free article] [PubMed]
- 16.Shrimp JH, Kales SC, Sanderson PE, Simeonov A, Shen M, Hall MD. 2020. An enzymatic TMPRSS2 assay for assessment of clinical candidates and discovery of inhibitors as potential treatment of COVID-19. ACS Pharmacol Transl Sci 3:997–1007. 10.1021/acsptsci.0c00106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jiang L, Yuan C, Huang M. 2021. A general strategy to inhibit serine protease by targeting its autolysis loop. FASEB J 35:e21259. 10.1096/fj.202002139RR. [DOI] [PubMed] [Google Scholar]
- 18.Krem MM, Di Cera E. 2001. Molecular markers of serine protease evolution. EMBO J 20:3036–3045. 10.1093/emboj/20.12.3036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R, Heer FT, de Beer TAP, Rempfer C, Bordoli L, Lepore R, Schwede T. 2018. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res 46:W296–W303. 10.1093/nar/gky427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Laskowski RA, Rullmannn JA, MacArthur MW, Kaptein R, Thornton JM. 1996. AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J Biomol NMR 8:477–486. 10.1007/BF00228148. [DOI] [PubMed] [Google Scholar]
- 21.Zhao G, Yuan C, Wind T, Huang Z, Andreasen PA, Huang M. 2007. Structural basis of specificity of a peptidyl urokinase inhibitor, upain-1. J Struct Biol 160:1–10. 10.1016/j.jsb.2007.06.003. [DOI] [PubMed] [Google Scholar]
- 22.Long-Guang J, Cai Y, Hong-Wei C, Yu W, Bao-Yu Z, Xu Z, Ming-Dong H. 2011. Preparation and structure of a new coagulation factor XI catalytic domain for drug discovery. Chin J Struct Chem 30:1021–1029. [Google Scholar]
- 23.Yuan C, Chen LQ, Meehan EJ, Daly N, Craik DJ, Huang MD, Ngo JC. 2011. Structure of catalytic domain of Matriptase in complex with Sunflower trypsin inhibitor-1. BMC Struct Biol 11:30. 10.1186/1472-6807-11-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xu M, Chen Y, Xu P, Andreasen PA, Jiang L, Li J, Huang M. 2018. Crystal structure of plasma kallikrein reveals the unusual flexibility of the S1 pocket triggered by Glu217. FEBS Lett 592:2658–2667. 10.1002/1873-3468.13191. [DOI] [PubMed] [Google Scholar]
- 25.Gong L, Liu M, Zeng T, Shi X, Yuan C, Andreasen PA, Huang M. 2015. Crystal structure of the Michaelis complex between tissue-type plasminogen activator and plasminogen activators inhibitor-1. J Biol Chem 290:25795–25804. 10.1074/jbc.M115.677567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kromann-Hansen T, Louise Lange E, Peter Sorensen H, Hassanzadeh-Ghassabeh G, Huang M, Jensen JK, Muyldermans S, Declerck PJ, Komives EA, Andreasen PA. 2017. Discovery of a novel conformational equilibrium in urokinase-type plasminogen activator. Sci Rep 7:3385. 10.1038/s41598-017-03457-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schwede T, Kopp J, Guex N, Peitsch MC. 2003. SWISS-MODEL: an automated protein homology-modeling server. Nucleic Acids Res 31:3381–3385. 10.1093/nar/gkg520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Benvenuti M, Mangani S. 2007. Crystallization of soluble proteins in vapor diffusion for x-ray crystallography. Nat Protoc 2:1633–1651. 10.1038/nprot.2007.198. [DOI] [PubMed] [Google Scholar]
- 29.Vagin A, Teplyakov A. 1997. MOLREP: an automated program for molecular replacement. J Appl Crystallogr 30:1022–1025. 10.1107/S0021889897006766. [DOI] [Google Scholar]
- 30.Jiang L, Botkjaer KA, Andersen LM, Yuan C, Andreasen PA, Huang M. 2013. Rezymogenation of active urokinase induced by an inhibitory antibody. Biochem J 449:161–166. 10.1042/BJ20121132. [DOI] [PubMed] [Google Scholar]
- 31.Murshudov GN, Vagin AA, Dodson EJ. 1997. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr 53:240–255. 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 32.Emsley P, Cowtan K. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60:2126–2132. 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 33.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. 1993. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr 26:283–291. 10.1107/S0021889892009944. [DOI] [Google Scholar]
- 34.DeLano WL. 2002. The PyMOL molecular graphics system. https://pymol.org/2/. [Google Scholar]
- 35.Wallace AC, Laskowski RA, Thornton JM. 1995. LIGPLOT: a program to generate schematic diagrams of protein-ligand interactions. Protein Eng 8:127–134. 10.1093/protein/8.2.127. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The coordinate of the uPA-camostat complex was deposited in the Protein Data Bank (PDB code 7DZD).