

ABSTRACT
Hepatitis B virus (HBV) can integrate into the chromosomes of infected hepatocytes, creating potentially oncogenic lesions that can lead to hepatocellular carcinoma (HCC). However, our current understanding of integrated HBV DNA architecture, burden, and transcriptional activity is incomplete due to technical limitations. A combination of genomics approaches was used to describe HBV integrations and corresponding transcriptional signatures in three HCC cell lines: huH-1, PLC/PRF/5, and Hep3B. To generate high-coverage, long-read sequencing data, a custom panel of HBV-targeting biotinylated oligonucleotide probes was designed. Targeted long-read DNA sequencing captured entire HBV integration events within individual reads, revealing that integrations may include deletions and inversions of viral sequences. Surprisingly, all three HCC cell lines contain integrations that are associated with host chromosomal translocations. In addition, targeted long-read RNA sequencing allowed for the assignment of transcriptional activity to specific integrations and resolved the contribution of overlapping HBV transcripts. HBV transcripts chimeric with host sequences were resolved in their entirety and often included >1,000 bp of host sequence. This study provides the first comprehensive description of HBV integrations and associated transcriptional activity in three commonly utilized HCC-derived cell lines. The application of novel methods sheds new light on the complexity of these integrations, including HBV bidirectional transcription, nested transcripts, silent integrations, and host genomic rearrangements. The observation of multiple HBV-associated chromosomal translocations gives rise to the hypothesis that HBV is a driver of genetic instability and provides a potential new mechanism for HCC development.
IMPORTANCE HCC-derived cell lines have served as practical models to study HBV biology for decades. These cell lines harbor multiple HBV integrations and express only HBV surface antigen (HBsAg). To date, an accurate description of the integration burden, architecture, and transcriptional profile of these cell lines has been limited due to technical constraints. We have developed a targeted long-read sequencing assay that reveals the entire architecture of integrations in these cell lines. In addition, we identified five chromosomal translocations with integrated HBV DNA at the interchromosomal junctions. Incorporation of long-read transcriptome sequencing (RNA-Seq) data indicated that many integrations and translocations were transcriptionally silent. The observation of multiple HBV-associated translocations has strong implications regarding the potential mechanisms for the development of HBV-associated HCC.
KEYWORDS: chromosomal translocation, HBV, HCC, viral integration
INTRODUCTION
In chronic hepatitis B (CHB) patients, HBV integration is a risk factor that has been associated with HBV chronicity and hepatocellular carcinoma (HCC). HBV integration occurs early and can be detected even in pediatric patients (1, 2). Integrations often produce viral transcripts, presenting a major obstacle for complete clearance of HBV antigens (3, 4). Transcripts from integrations are often chimeric with host, which can lead to the synthesis of chimeric proteins. These chimeric transcripts and proteins have been associated with progression to HCC in CHB patients, and multiple HCC-derived cell lines harboring integrations have been described (5–9). PLC/PRF/5, huH-1, and Hep3B cells were first characterized in the 1970s and have been used extensively to study HBV and carcinogenesis (10). While these cell lines do not produce infectious viral particles, they do produce HBV surface antigen (HBsAg), presumably from integrated HBV DNA (11). Determination of the exact architecture, burden, and transcriptional activity of all integrations in these cell lines could provide insights into the transcriptional activity of integrated HBV and the mechanisms of HCC development.
Characterization of HBV integrations has largely relied on identifying virus-host junctions using inverse nested PCR (2, 12, 13). While this method can greatly amplify integrated HBV sequences, results may be biased by the presence, absence, and proximity of primer binding and restriction sites. More recent studies have used next-generation sequencing (NGS) for an unbiased approach to detection (14, 15). Using sequencing by synthesis (SBS), the most common NGS platform, millions of reads can be generated from a single sample. However, libraries for SBS sequencing have an average length of 300 bp, approximately 10 times shorter than the 3.2-kb length of the HBV genome (16). While it is possible to identify junctions between HBV and host genomes, resolution of the architecture of individual integrations has required validation by PCR (17). Third-generation sequencing platforms (e.g., PacBio and Oxford Nanopore) have made it possible to generate sequences significantly long enough to contain full-length HBV and flanking host regions within a single read (18). However, the human haploid genome (3.3Gbp) is one million times the size of the HBV genome (3.2 kbp), so most reads from shotgun DNA sequencing contain only human sequences.
Ascribing HBV transcripts to specific integrations has also proven challenging due to the shared sequence homology among integration events and the overlapping nature of transcripts produced. In addition, traditional transcriptome sequencing (RNA-Seq) cannot distinguish the origin of reads containing only HBV, whether from integrated or covalently closed circular DNA (cccDNA). Moreover, HBV produces multiple overlapping transcripts from cccDNA that cannot be easily differentiated with short reads (19). Isoform sequencing (Iso-Seq) is long-read sequencing applied to cDNA libraries and offers important advantages over short-read RNA-Seq. Iso-Seq has the potential to sequence entire transcripts, including kilobases of host chimeric sequence, to reveal the entire architecture of chimeric HBV-host reads. Iso-Seq reads made from multiple passes of individual molecules also have high enough accuracy that single-nucleotide polymorphisms (SNPs) can be phased (20).
In this study, we sought to resolve the architecture of integrated HBV DNA and assign corresponding transcriptional profiles to each event. A combination of genomic approaches was used to sequence integrated HBV and associated HBV RNAs from three HCC cell lines: huH-1, PLC/PRF/5, and Hep3B. We used a custom HBV-targeting panel of biotinylated oligonucleotides to enrich for HBV sequences followed by long-read sequencing of genomic DNA (targeted PacBio) or cDNA (targeted Iso-Seq). Targeted PacBio revealed multiple integrations in all cell lines, including several associated with host chromosomal translocations. Targeted Iso-Seq enabled the detection and differentiation of full-length transcripts generated from HBV integrations and assigned transcripts to specific integrations. Notably, many integrations appear transcriptionally silent. Lastly, we identified both chimeric and nonchimeric transcripts originating from integrations. The nonchimeric reads from integrations utilize a novel transcription stop site and can be differentiated from nonchimeric transcripts of cccDNA origin. This important observation allows accurate quantification of the transcriptional burden from integrations versus cccDNA.
RESULTS
Short-read sequencing reveals location and transcriptional activity of HBV integrations.
To better understand HBV integrations, we performed whole-genome sequencing (WGS) analysis on three HCC cell lines (huH-1, PLC/PRF/5, and Hep3B), known to contain HBV integrations (9, 10). These cell lines originated from individuals with HCC and have been used extensively to understand HBV-associated HCC. Each cell line contains more than one HBV integration and expresses HBsAg but no other viral antigens and does not produce infectious viral particles (9, 10, 21). Multiple reads containing both virus and host sequences were detected in all cell lines, indicating HBV integration into host chromosomes. Chimeric reads from all cell lines were mapped to both the HBV and human genomes, with arrows signifying the directionality of each read (Fig. 1). We were unable to resolve internal sequences from integrated HBV or describe potential open reading frames (ORFs), as reads containing only HBV sequences could not be mapped to specific integrations.
FIG 1.
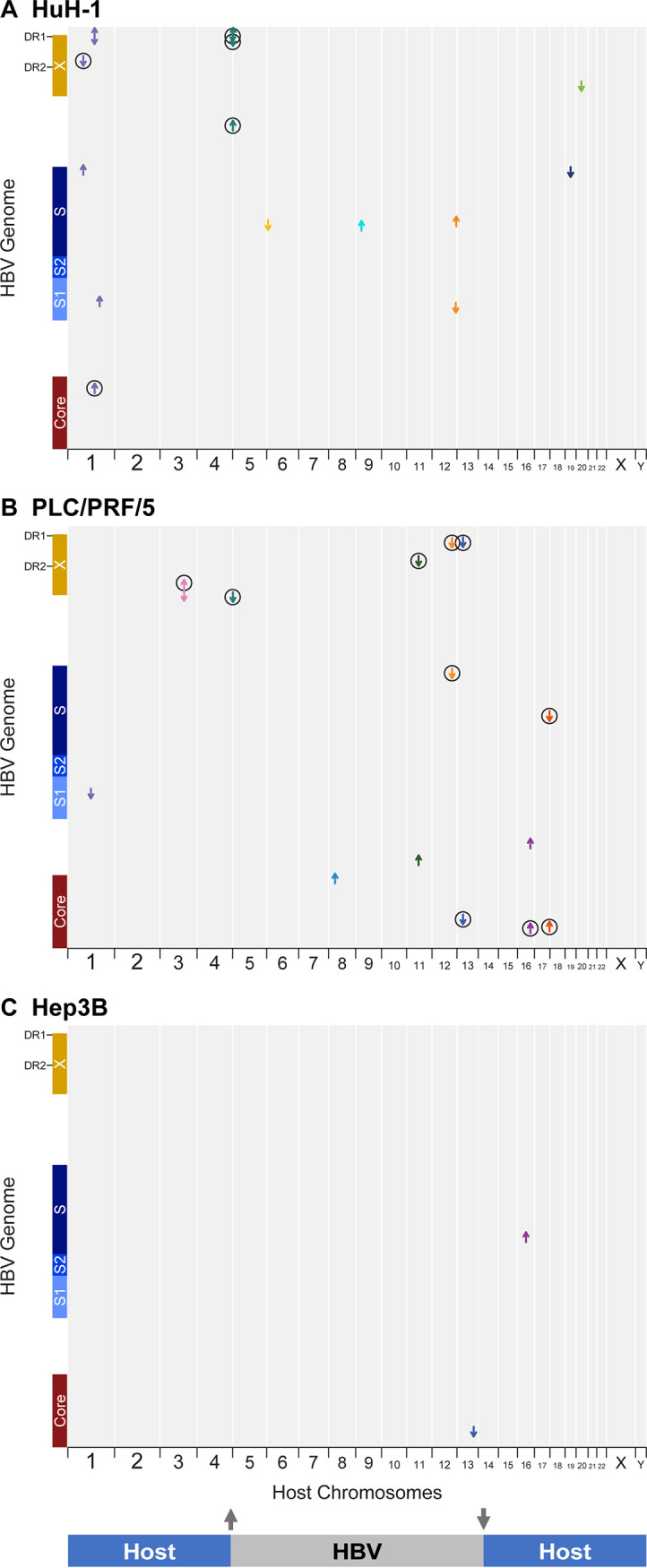
Short-read sequencing defines locations and transcriptional activity of integrated HBV in HCC cell lines. (A) huH-1, (B) PLC/PRF/5, and (C) Hep3B cell lines were analyzed by short-read RNA-Seq and WGS. HBV integrations were identified by chimeric HBV-host junctions and their locations mapped as arrows according to the aligned sequences to the HBV genome (y axis) and host genome (x axis). Each arrow color corresponds to an individual human chromosome, and its direction correlates to the orientation of the HBV sequence within the associated chimeric read. Circles indicate junctions that were also detected by RNA-Seq.
To reveal the transcriptional activity associated with each integration, we performed RNA-Seq from these same cell lines. Like WGS, RNA-Seq was able to detect chimeric reads in all samples. PLC/PRF/5 and huH-1 cells had high HBV transcriptional activity, with chimeric transcripts matching multiple integration events, as denoted by circled arrows on each plot (Fig. 1A and B). For Hep3B cells, RNA-Seq showed expression of chimeric transcripts derived from a single integration site in chromosome 4, although the site was not detected by WGS (Fig. 1C). HBV reads were >1,000-fold more abundant than WGS using RNA-Seq, allowing for deeper coverage of each expressed integration detected. Notably, WGS detected integrations in all three cell lines that were not detected by RNA-Seq, suggesting that these integrations are transcriptionally silent.
Targeted PacBio reveals the complete architecture of integrated HBV DNA.
To capture both transcriptionally active and silent integrations and map intervening sequences between chimeric junctions, we applied long-read sequencing, which allows for single reads to contain full-length HBV integrations and flanking host sequences (18). To enrich for HBV sequences, a panel of 37 biotinylated probes, each 120 bp in size, was designed to target HBV DNA and allow for facile detection of HBV integrations (Fig. 2A and B). This improved sequencing coverage by >2,000-fold compared to nonenriched samples, with HBV sequences representing 1 to 5% of total reads (see Fig. S1A in the supplemental material). In PLC/PRF/5 cells, short-read sequencing located two chimeric junctions on chromosome 17 (Fig. 2C). Given the proximity of these junctions in chromosome 17 and the orientation of the reads within HBV, the junctions were hypothesized to be associated with a single integration event (Fig. 2C). When the same sample was sequenced using targeted PacBio, multiple reads captured both junctions from the chromosome 17 integration, including intervening HBV sequences, confirming that these two junctions were associated with the same integration (Fig. 2D).
FIG 2.

Targeted PacBio platform established for HBV integrations and reveals complete architecture of integrated HBV DNA. (A) Schematic of target enrichment for PacBio method. (B) Targeting location of the 37 120-bp biotinylated oligonucleotides designed to enrich integrated HBV sequences. (C and D) Comparison between WGS (C) and targeted PacBio (D) data for the same integration on chromosome 17 in PLC/PRF/5 cells. WGS reads identify HBV-host junction ends, while targeted PacBio resolves the entire integration within single reads. Reads are plotted on circle aligning to the HBV genome. Orange circles indicated chimeric junctions with chromosome 17.
Targeted PacBio identifies chromosomal translocations associated with integrated HBV.
Several virus-host junctions detected by WGS appeared unpaired and could not be partnered with opposite junctions nearby on the same chromosome. For example, in huH-1 cells, virus-host junctions were observed in chromosome 9 and chromosome 20, but neither junction could be paired with a corresponding junction located nearby (Fig. 3A). When targeted PacBio was applied to huH-1 cells, we found read support linking chromosome 9 and 20 junctions to a single integration event. We observed 1,100 bp of HBV DNA at the interface of a novel chromosomal translocation joining chromosome 9 to chromosome 20 (Fig. 3B). We verified this translocation using spectral karyotyping (SKY), a method in which each chromosome is assigned a unique spectral signature and enables detection of chromosomal rearrangements (Fig. 3C). Spectral karyotyping detected two copies of the chromosome 9-to-chromosome 20 fusion event, validating the long-read sequencing results. Additionally, long-read sequencing detected a previously described chromosomal fusion event in PLC/PRF/5 cells between chromosome 1 and 8 (Fig. 3E to H) (22, 23). These data highlight a novel feature of integrated HBV DNA that is not sufficiently characterized using traditional NGS or nested PCR platforms. In total, we observed three chromosomal translocations in huH-1 cells, two in PLC/PRF/5 cells, and two in Hep3B cells (Fig. 4).
FIG 3.

Targeted PacBio identifies HBV-associated chromosomal translocations. WGS and targeted PacBio were performed on DNA from huH-1, PLC/PRF/5, and Hep3B cells. (A) WGS data from huH-1 cells found individual HBV-host junctions within chromosomes 9 (blue dots) and 20 (green dots), with no obvious pairing. (B) Targeted PacBio revealed that these junctions originated from an HBV-associated translocation. (C) Chromosome 9 to 20 translocation was confirmed by spectral karyotyping. (D) Alignment of all reads corresponding to this translocation generated a 12-kbp consensus sequence flanking ∼1,100 bp of HBV. (E) WGS data mapped individual HBV-host junctions within Chr1 (purple dots) and Chr8 (blue dots). (F) Targeted PacBio revealed that these junctions originated from an HBV-associated translocation. (G) This translocation event was confirmed by spectral karyotyping. (H) The consensus sequence revealed a segmented HBV with 722 bp adjoined to Chr1 and 2,238 bp adjoined to Chr8.
FIG 4.

Multiple HBV integrations are associated with chromosomal translocations. (A to C) Overview of HBV integration and translocation events in huH-1 (A), PLC/PRF/5 (B), and Hep3B (C) cell lines. Each chimeric junction is noted with an arrow mapped against the host genome on the x axis and HBV genome on the y axis. The direction of each arrow indicates the section of HBV included at the respective junction. Lines connecting two chimeric junctions indicate that reads support pairing of those junctions. Diagonal lines connecting two chimeric junctions indicate HBV-associated translocations. Major contigs from each cell line are shown on the right.
Resolution of integration architecture and transcriptional activity.
Generation of individual contigs for each integration event revealed a variety of structural changes to the HBV DNA, including large deletions and inversions (Fig. 5). None of the chromosomal translocations detected contained an intact full-length HBV sequence. The HBV sequences associated with chromosomal translocations either contained large truncations or consisted of two pieces of truncated and inverted HBV sequence joined together. These neighboring truncated sequences do not appear to generate novel viral open reading frames (ORFs); instead, each segment appears independently transcriptionally active (Fig. 5).
FIG 5.
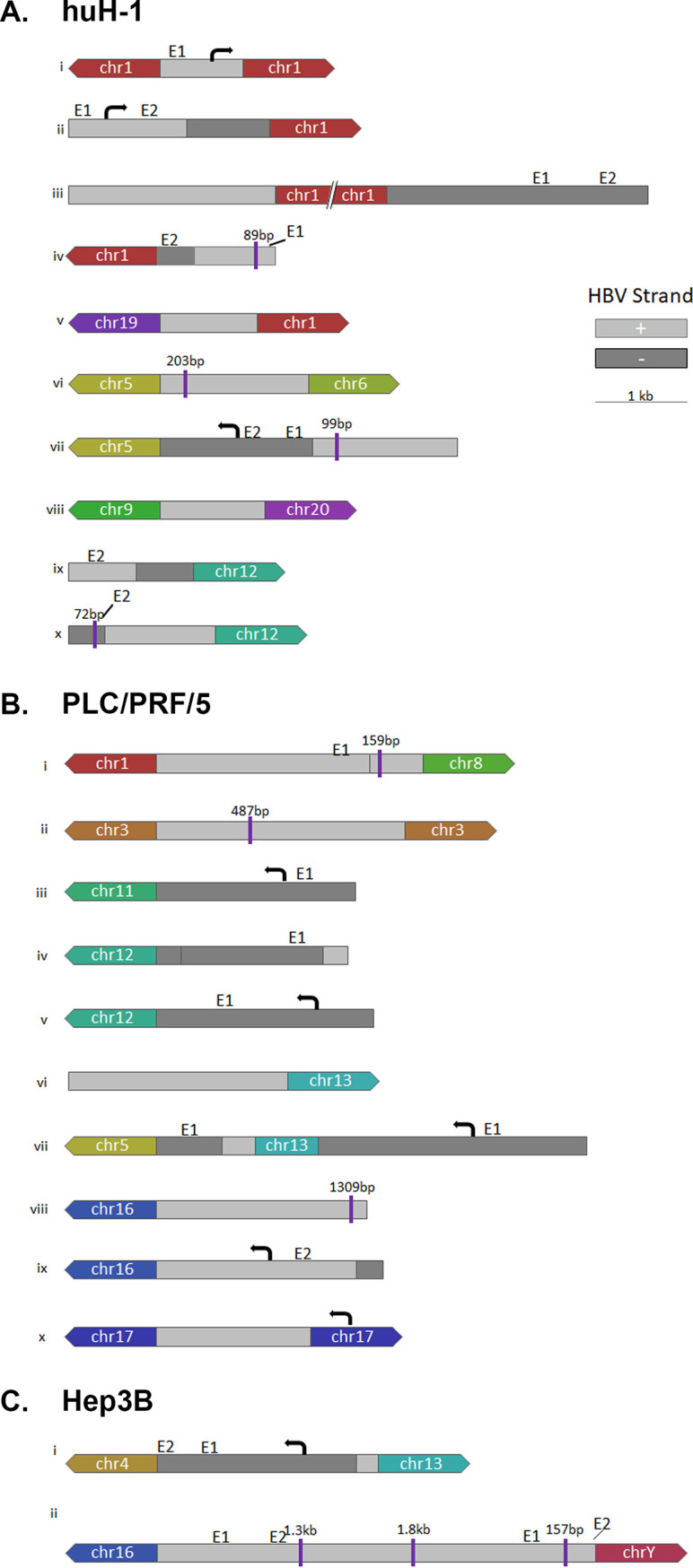
HBV architecture and transcriptional profiles of HBV integrations and translocations in huH-1 and PLC/PRF/5 cells. PacBio Reads from huH-1 (A), PLC/PRF/5 (B), and Hep3B (C) cells were used for de novo assembly of each major integration site. The resulting contigs delineated the sequence structure of integrated HBV sequences, including inversions and deletions. Chimeric reads generated by targeted Iso-Seq were used to assign a transcriptional activity, or lack thereof, to each integration event. E1 and E2 denote know enhancer regions (38, 39).
We next applied targeted Iso-Seq to characterize and quantify full-length sequences for all chimeric and nonchimeric transcripts from the HCC cell lines and assigned a transcriptional signature to each integration event (Fig. 5). Targeted Iso-Seq involves generation of full-length cDNA libraries followed by HBV target enrichment. Full-length chimeric transcripts could be mapped to three integrations in PLC/PRF/5 cells and one integration in huH-1 and Hep3B cells. In addition, we successfully differentiated PreS1 from PreS2/S transcripts. In PLC/PRF/5 cells, the integration on chromosome 11 expresses both PreS1 and PreS2/S transcripts, while the integration on chromosome 13 expresses only PreS2/S (Fig. 5A). Surprisingly, we obtained many nonchimeric transcripts in huH-1 cells. These nonchimeric reads from huH-1 cells appeared to have two distinct single-nucleotide variant (SNV) profiles that align to two separate integration sites (Fig. 6). These data suggest that detection of HBV mRNAs from integrations is underestimated by short-read RNA-Seq, which only quantifies chimeric sequences.
FIG 6.

SNVs resolve nonchimeric HBV RNAs from two unique integration events. (A) In addition to chimeric HBV RNAs, we identified a large number of nonchimeric HBV RNAs in huH-1 cells. The majority of these transcripts resolved into two unique SNV profiles. The first SNV aligned contig ix, shown in Fig. 5 (B) The second SNV aligned with contig iii, shown in Fig. 5.
Targeted Iso-Seq reveals HBV RNA transcripts that use multiple alternative poly(A) signals.
Targeted Iso-Seq generated from the HCC cell lines demonstrated that huH-1 and PLC/PRF/5 cells generate transcripts containing both the PreS1 and PreS2/S ORFs (Fig. 7A). Hep3B cells generate only PreS2/S transcripts that all originated from the same integration event and were chimeric with chromosome 4 (Fig. 7). We observed that all chimeric HBV transcripts utilize the host poly(A) signal, typically AAUAAA, and often contain over 1,000 bp of host sequence appended to their 3′ ends (Fig. 8C). The length of the host sequence was dependent on the proximity of the nearest poly(A) site within the host genome. Transcriptional readthrough into the host was independent of whether the integration occurred within a known coding region or in an intergenic region of the host chromosome. In addition to the chimeric reads, many huH-1 transcripts were nonchimeric despite originating from integrations (Fig. 7A and 8B). These nonchimeric transcripts start and stop within the HBV integration and do not contain any host sequences (Fig. 8D). All nonchimeric transcripts utilize a noncanonical poly(A) sequence, CAUAAA, located within the HBV integration, downstream of DR1 (24).
FIG 7.

Iso-Seq differentiates transcripts from integrations through unique 3′ termini. (A) Targeted Iso-Seq libraries were generated from huH-1, PLC/PRF/5, and Hep3B cells and AD38-infected PHHs. Gray lines indicate individual long reads. Colored dots indicate reads are chimeric with host chromosomes. (B) Quantification of each transcript was performed by reducing PCR duplicates using UMIs and then quantifying the number of unique reads assigned to each transcript type.
FIG 8.

Three novel transcripts types associated with HBV identified by Iso-Seq. Representative HBV RNA sequence reads from PHH (A), PLC/PRF/5 (B), and huH-1 (C) represent the three distinct transcript types identified. The poly(A) signal is in red, while the poly(A) tail is in green. Breakpoints between HBV sequence and host sequence is marked with a caret (^). In panel B, HBV is fused to the MVK gene and is in frame with MVK’s start codon (underlined). (D) Quantification of the host aligning sequence length from chimeric transcripts. A histogram of HBV aligning read length from huH-1, PLC/PRF/5, Hep3B, and AD38 infected PHH cells is plotted in the purple bar graphs. The corresponding total read length, which corresponds to HBV and host aligning sequence, is plotted in gray bar graphs. PLC/PRF/5 and Hep3B cells both contain many chimeric transcripts with >1,000 nucleotides of host sequence appended to the 3′ end of HBV reads.
Targeted Iso-Seq was also applied to HBV-infected PHHs to deconvolute overlapping transcripts from cccDNA. HBV transcripts from cccDNA overlap and terminate at the same 3′ poly(A) signal and cannot be resolved using traditional RNA-Seq. An alignment of all HBV RNAs from PHHs shows the differentiation of pgRNA, pre-Core RNA, preS1, and preS2/S transcripts (Fig. 7A and 8A). Quantitation of these reads suggests that the two highest expressed HBV RNAs are pgRNA and PreS2/S RNA, which is consistent with Northern blot analyses (Fig. 7B) (25). In contrast to the integrated transcripts, all HBV transcripts detected from cccDNA utilized the poly(A) signal, UAUAAA, located downstream of the X ORF (Fig. 8A). As this poly(A) signal embedded in the X ORF was absent from integrated transcripts, the site of HBV mRNA polyadenylation may sufficiently differentiate transcripts from integrations versus cccDNA. Altogether, our targeted Iso-Seq approach provides a high-resolution transcriptome that enables the detection of expression from integrations and cccDNA at the transcript isoform level.
DISCUSSION
We used a combination of genomic approaches to characterize the HBV integration architecture, burden, and transcriptional activity in three HCC cell lines, PLC/PRF/5, huH-1, and Hep3B. These cell lines have been extensively utilized for the understanding of HBV-associated HCC and are known to contain multiple HBV DNA integrations and produce HBsAg but not infectious virus (10). Although these cell lines have been characterized extensively, the comprehensive structure of their integrated HBV DNA had not previously been resolved. In addition, we observed orphaned junctions within our short-read sequencing data that prompted us to apply long-read sequencing to these samples first as a proof of concept for our method (Fig. 1). Targeted long-read sequencing identified HBV-associated interchromosomal translocations associated with the orphaned junctions. In addition, short-read RNA-Seq could not resolve individual HBV RNA isoforms associated with each integration event, such as differentiating the expression of PreS1 versus PreS2/S transcripts. Application of our target enrichment platform to long-read cDNA libraries resolved these HBV RNA isoforms.
We developed two long-read sequencing approaches: targeted PacBio and targeted Iso-Seq. Target enrichment increased sequencing coverage across integrated HBV DNA by >2,000-fold and resolved the comprehensive architecture of all integrations (Fig. 4 and 5). These integrations were highly diverse and contained inversions and deletions within the HBV sequences (Fig. 5). In addition, seven HBV-associated chromosomal translocations were detected, five of which were previously undescribed (17, 22). Four of these translocations were validated by spectral karyotyping (Fig. 3 and 4). One caveat of PacBio sequencing is that the SMRTBell adaptors are ligated by blunt ligation; therefore, there is a low level of ligation between library fragments during this step that leads to concatemers. Our bioinformatics pipeline resolves these concatemers by finding sample indexes in the middle of these reads and breaks them into the appropriate subreads. This step was critical to avoid miscalling HBV-associated chromosomal translocations. Target enrichment increased sequencing coverage across HBV RNA by >1,000-fold in all samples. We identified both chimeric and nonchimeric transcripts that originated from integrations and differentiated these from transcripts of cccDNA origin through the presence of unique 3′ termini (Fig. 7 and 8). In addition, we could assign each transcript to a specific integration and determine which integrations were transcriptionally silent (Fig. 5 and 6).
The HBV-associated chromosomal translocations identified in these cell lines all contained relatively short HBV sequences, and all appeared transcriptionally inactive. This observation leads to hypotheses on how integrations and translocations may arise in patients. Literature on HBV integration events supports a random mechanism for the initial HBV integration event in which double-stranded linear DNA (dslDNA) inserts into host chromosomes through nonhomologous end-joining reactions. Double-strand breaks occur randomly in the human genome, and this is supported by the observation that integrations in non-tumor tissue are found in all human chromosomes (26, 27). It may be possible for a translocation to occur if double-strand breaks in the human genome are synchronous with infection of a dslDNA-containing virion. These events would manifest as a full-length dslDNA insertion and may be rare. Alternatively, HBV-associated chromosomal translocations may arise from a double-strand break within the initial HBV integration and incorrect repair of that break. This mechanism is consistent with the short HBV sequences that we observe in these HCC cell lines (Fig. 3 and 4). Because of the proposed randomness of these events, it would be possible for HBV-associated chromosomal translocations to still express viral antigens such as HBsAg but would depend on how the translocation arose and the architecture of the inserted HBV DNA sequence. Finally, it is possible that the translocations identified here are unique to these cell lines. Now that the tools exist to probe for these events, it will be fascinating to explore if HBV-associated chromosomal translocations can be observed in patient samples and if they play a role in progression to HCC.
Targeted Iso-Seq allows resolution of all HBV RNA isoforms, including those from both integrations and cccDNA, including splice variants. Many integrations express both PreS1 and PreS2/S transcripts, while others only express PreS2/S (Fig. 7). In HBV-infected PHHs, we detected pre-Core RNA, pgRNA, PreS1, and PreS2/S RNAs. PreS2/S RNA and pgRNA were the most abundant species identified in PHHs. Notably, we did not observe HBx transcripts associated with the HCC cell lines or HBV-infected PHH, despite many integrations containing the X promoter and ORF sequences. The lack of HBx transcripts detected in this study may be related to the time point, as HBx has been shown to express early postinfection (25, 28). In addition to standard full-length HBV transcripts, spliced transcripts were found in the infected PHH sample, comprising approximately 7% of total HBV transcripts. Nearly 80% of spliced transcripts were SP1, but we also observed other previously characterized and novel splice variants that warrant further investigation. One notable splice variant was found associated with PLC/PRF/5 cells. The transcript contains the 5′ end of HBsAg fused, mid-HBs, to the mevalonate kinase gene (MVK). The MVK-aligning sequence appears to be spliced in the same manner as the traditional host MVK transcript. This fusion has been previously characterized and is known to encode an HBsAg-MVK fusion protein that has been shown to be oncogenic (29).
Targeted Iso-Seq revealed three transcript types associated with HBV, differentiated by their 3′ ends. The first type is nonchimeric transcripts originating from cccDNA, such as pgRNA, pre-Core RNA, PreS1 RNA, and PreS2/S RNA. These transcripts utilize the canonical HBV poly(A) sequence, UAUAAA, to terminate transcription (Fig. 8A). The second type is chimeric transcripts associated with integrations generated via readthrough into the host which are polyadenylated using host poly(A) cleavage signals, such as AAUAAA (Fig. 8C). The third transcript type is nonchimeric transcripts associated with integrations terminating at the viral noncanonical poly(A) sequence, CAUAAA (Fig. 8B) (24, 30). The ability to distinguish the transcript types is significant for two reasons. First, nonchimeric transcripts associated with HBV integrations represent a class of transcripts that should be used to assess the burden of integration. Measuring integration burden with short-read RNA-Seq relies on reads containing chimeric junctions and might otherwise miscount reads derived from these types of transcripts, leading to a significant underestimation of integration burden. Second, by taking advantage of the distinct polyadenylation signals utilized by each transcript, we may be able to differentiate HBV transcripts from cccDNA versus integrations in patient samples with mixed transcript origin.
PLC/PRF/5, huH-1, and Hep3B have been used extensively for decades to study HBV and carcinogenesis (10). In this study, we have generated the most comprehensive data set from these cell lines to date, describing integrations and their associated transcriptional activity. Importantly, HBV-associated chromosomal translocations were described in each cell line. Translocations have an important role in the initiation of carcinogenesis and have been associated with almost every tumor type (31). As these HCC cell lines are derived from livers from patients with HCC, it is possible that HBV-associated chromosomal translocations existed in these patients prior to development of a detectable neoplasm. However, we do not know if these specific translocations play any role in oncogenesis. If demonstrated, this could provide a novel mechanism for the role of HBV integrations in HCC progression. Application of these techniques to CHB human liver biopsy specimens may help understand the relationship between integrations and progression to HCC.
MATERIALS AND METHODS
Cell culture and nucleotide extraction.
PLC/PRF/5 and Hep3B cell lines were purchased from the ATCC, and huH-1 (JCRB0199) was purchased from JCRB (7–9). All three cell lines were confirmed to be HBsAg positive and HBeAg negative using Meso Scale Discovery assays (25). PLC/PRF/5 and huH-1 cells were cultured in Dulbecco’s modified Eagle medium (DMEM; Thermo Fisher Scientific) supplemented with 10% fetal bovine serum (FBS; HyClone), 2 mM l-glutamine (Thermo Fisher Scientific), 100 U/ml penicillin, and 100 μg/ml streptomycin (Thermo Fisher Scientific) according to cell bank recommendations (Thermo Fisher Scientific). huH-1 cells had 1 μM dexamethasone (Thermo Fisher Scientific) added to the culture media to increase HBsAg production. Hep3B cells were cultured in Eagle’s minimum essential medium (EMEM; Thermo Fisher Scientific) supplemented with 10% FBS. Primary human hepatocytes (PHHs) were obtained from BioIVT and were cultured in William’s Medium E (Thermo Fisher Scientific) with 2% fetal bovine serum and 1.5% dimethyl sulfoxide (DMSO; Sigma-Aldrich) and hepatocyte maintenance supplement pack number CM4000 (Life Technologies). Cells were infected with in vitro HBV inocula derived from HepAD38 cells. At 1 day postinfection, cells were washed with William’s Medium E and cultured in William’s Medium E (Thermo Fisher Scientific) with 2% fetal bovine serum and 1.5% DMSO (Sigma-Aldrich) and hepatocyte maintenance supplement pack number CM4000 (Life Technologies).
DNA samples were harvested from PLC/PRF/5, huH-1, and Hep3B cells following 3 days in culture. RNA samples were harvested after 3 days in culture for PLC/PRF/5, after 9 days in culture for Hep3B, and after 10 days in culture for huH-1 to correlate with maximum HBsAg expression levels. Infected PHHs were harvested 1 week postinfection. Cells were harvested into RLT buffer (Qiagen) to preserve nucleic acids. RNA was isolated by HD Bioscience (Shanghai, China) or using the All-Prep kit (Qiagen). DNA was isolated using the MasterPure DNA and RNA purification kit (Epicentre) or using the All-Prep kit (Qiagen).
SKY.
Spectral karyotyping (SKY) was performed at the Molecular Cytogenetic Core at Albert Einstein College of Medicine (New York, USA). SKY was performed as previously described (32). Briefly, human SKYPaint probes (Applied Spectral Imaging) were hybridized to metaphase slides aged at 37°C for 5 days. After in situ hybridization, slides were counterstained with 4′,6-diamidino-2-phenylindole (DAPI), and images were acquired using an epifluorescence microscope (Olympus BX61) connected to an imaging interferometer (SD200; Applied Spectral Imaging, Migdal HaEmek, Israel).
Short-read sequencing.
Whole-genome sequencing and RNA-Seq were performed by WuXi (Jiangyin, China) on the Illumina HiSeq platform. Briefly, DNA was prepared for sequencing using the Illumina DNA prep kit and sequenced to at least 60× average coverage using 150-bp paired-end reads. RNA was prepared for sequencing using a TruSeq library preparation kit (Illumina). RNA-Seq had at least 75 million 150-bp paired-end reads per sample. DNA and RNA sequences were aligned to human genome reference hg38 using BWA (33).
Targeted PacBio and targeted Iso-Seq.
Genomic DNA from PLC/PRF/5, huH-1, and Hep3B cells was sheared to ∼7.5 kbp using the G-tube (Covaris) and purified using the AMPure PB DNA beads (Pacific Biosciences). Sheared DNA was barcoded using preannealed sample indexes (IDT) using the Kapa Hyper prep library kit (Kapa Biosystems). Barcoded DNA was amplified using the TaKaRa LA polymerase (Clontech). Thirty-seven 120-bp biotinylated probes were designed to be compatible with the xGen Lockdown platform (IDT) (see Fig. S2B in the supplemental material). Alternatively, RNA was reverse transcribed using the SMARTer cDNA synthesis kit (TaKaRa Bio) and a custom-designed poly(T) primer that incorporates a unique molecular index (UMI) for reduction of PCR duplicates. cDNA was amplified using indexing primers for multiplexed sequencing and the Kapa HiFi PCR polymerase (Kapa Biosystems). DNA and cDNA libraries were run on a Bioanalyzer 2100 (Agilent) prior to target enrichment.
DNA and cDNA libraries were enriched for HBV sequences by incubating with our custom probe pools at 65°C for 4 h (Fig. 2A). Targeted DNA fragments were captured using streptavidin Dynabeads (Invitrogen) at 65°C for 45 min and then washing using the xGen hybridization and wash kit (IDT). Captured DNA sequences were amplified using the TaKaRa LA polymerase (TaKaRa BioSciences) and the universal PacBio primer. Enriched DNA libraries were sequenced on the Sequel II (Pacific Biosciences) at the University of Arizona Genomics Institute (Tucson, AZ). To quantify HBV sequence enrichment, precapture and postcapture DNA libraries were compared by qPCR. In brief, 50 ng of each library was amplified for the HBx gene using the following primers: F (5′ CCG TCT GTG CCT TCT CAT CTG 3′), R (5′ AGT CCA AGA GTY CTC TTATGY AAG ACC TT 3′), and probe (5′ 6-carboxyfluorescein CCG TGT GCA CTT CGC TTC ACC TCT GC black hole quencher 1 3′). Fold enrichment is the ratio of postcapture DNA copies to precapture DNA copies.
HBV consensus sequence refinement.
Sequencing reads from each sample were aligned to HBV genome sequence KY003230.1 using minimap2 and then converted to a consensus sequence using bcftools (34). Each consensus sequence was aligned to a set of genotypes A through H using pairwise alignment (35). The genotype of each sample was inferred based on the highest mean similarity score. Genotype data were confirmed across sequencing methods to check for consistency.
minimap2 and bcftools were used as described above to create a second consensus sequence based on alignment to each cell line’s respective genotype. Data from all sequencing methods was combined in R with multiple-sequence alignment (MSA) (36). As the full 3.2-kb HBV sequence is not present in all cell lines, missing fragments in the consensus sequences were replaced with reference genotype sequences. A 2× length sequence was created by concatenating two copies of each consensus sequence into FASTA format, starting with the ATG of the core open reading frame.
Viral read analysis.
Reads were realigned to their respective cell line’s 2× consensus sequence combined with human genome reference hg38 using BWA or minimap2 for short or long reads, respectively. Reads with split alignment were used to infer chimerism between HBV and host. Potential concatemer artifacts were removed by searching for sequence fragments >25 bp between HBV and host alignments that aligned to barcodes, adapter sequences, or poly(A/T) tails using Biostrings (35). Iso-Seq reads with duplicate UMIs were filtered to only include one read per UMI.
PacBio reads from each junction were used for de novo assembly of integration sites using MSA followed by indel adjustment (37). If any reads encompassed two junctions, reads from either junction were combined to create a single contiguous sequence (contig). Reads were realigned to the new contigs to check accuracy. If the realigned reads showed discrepancies in single-nucleotide variant (SNV) profiles or significant soft clipping, reads were split by phased variants to replace with new contigs. Reads were assessed to determine if there were upstream and downstream junctions with the host within individual reads. Reads were filtered to remove low-quality sequences based on high indel frequency and then grouped by either junction positions or SNVs. Groups of reads were used to create individual contig sequences. Nonchimeric Iso-Seq reads were clustered based on SNVs identified in at least 5% of reads.
ACKNOWLEDGMENTS
We acknowledge Jidong Shan (Albert Einstein College of Medicine) for expertise in spectral karyotyping, Ryan Demeter (Integrated DNA Technologies) for help with designing our enrichment platform, and Dave Kudrna (University of Arizona) for expertise in PacBio sequencing.
All authors were employees and stock owners at Gilead Sciences, Inc.
The study was funded in full by Gilead Sciences, Inc.
Performed experiments, L.G., L.M., N.V.B., M.Y., N.B., and J.C.; bioinformatics, R.R. and C.S.; concept and design of experiments, L.G., R.R., N.V.B., N.B., L.L., R.C., and G.C.; writing of article, L.G., R.R., N.V.B., B.F., L.L., R.M., H.M., and W.D.
Contributor Information
Li Li, Email: li.li@gilead.com.
J.-H. James Ou, University of Southern California
REFERENCES
- 1.Furuta M, Tanaka H, Shiraishi Y, Unida T, Imamura M, Fujimoto A, Fujita M, Sasaki-Oku A, Maejima K, Nakano K, Kawakami Y, Arihiro K, Aikata H, Ueno M, Hayami S, Ariizumi SI, Yamamoto M, Gotoh K, Ohdan H, Yamaue H, Miyano S, Chayama K, Nakagawa H. 2018. Characterization of HBV integration patterns and timing in liver cancer and HBV-infected livers. Oncotarget 9:25075–25088. 10.18632/oncotarget.25308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mason WS, Gill US, Litwin S, Zhou Y, Peri S, Pop O, Hong ML, Naik S, Quaglia A, Bertoletti A, Kennedy PT. 2016. HBV DNA integration and clonal hepatocyte expansion in chronic hepatitis B patients considered immune tolerant. Gastroenterology 151:986–998. 10.1053/j.gastro.2016.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tu T, Budzinska MA, Shackel NA, Urban S. 2017. HBV DNA integration: molecular mechanisms and clinical implications. Viruses 9:75. 10.3390/v9040075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Summers J, Mason WS. 2004. Residual integrated viral DNA after hepadnavirus clearance by nucleoside analog therapy. Proc Natl Acad Sci U S A 101:638–640. 10.1073/pnas.0307422100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ruan P, Dai X, Sun J, He C, Huang C, Zhou R, Chemin I. 2020. Integration of hepatitis B virus DNA into p21-activated kinase 3 (PAK3) gene in HepG2.2.15 cells. Virus Genes 56:168–173. 10.1007/s11262-019-01725-4. [DOI] [PubMed] [Google Scholar]
- 6.Lau CC, Sun T, Ching AK, He M, Li JW, Wong AM, Co NN, Chan AW, Li PS, Lung RW, Tong JH, Lai PB, Chan HL, To KF, Chan TF, Wong N. 2014. Viral-human chimeric transcript predisposes risk to liver cancer development and progression. Cancer Cell 25:335–349. 10.1016/j.ccr.2014.01.030. [DOI] [PubMed] [Google Scholar]
- 7.Huh N, Utakoji T. 1981. Production of HBs-antigen by two new human hepatoma cell lines and its enhancement by dexamethasone. Gan 72:178–179. [PubMed] [Google Scholar]
- 8.Aden DP, Fogel A, Plotkin S, Damjanov I, Knowles BB. 1979. Controlled synthesis of HBsAg in a differentiated human liver carcinoma-derived cell line. Nature 282:615–616. 10.1038/282615a0. [DOI] [PubMed] [Google Scholar]
- 9.Alexander JJ, Bey EM, Geddes EW, Lecatsas G. 1976. Establishment of a continuously growing cell line from primary carcinoma of the liver. S Afr Med J 50:2124–2128. [PubMed] [Google Scholar]
- 10.Alexander JJ. 1987. Human hepatoma cell lines, p 47–56. In OKaI KG (ed), Neoplasms of the liver. Springer, Tokyo, Japan. [Google Scholar]
- 11.Kekule AS, Lauer U, Meyer M, Caselmann WH, Hofschneider PH, Koshy R. 1990. The preS2/S region of integrated hepatitis B virus DNA encodes a transcriptional transactivator. Nature 343:457–461. 10.1038/343457a0. [DOI] [PubMed] [Google Scholar]
- 12.Bill CA, Summers J. 2004. Genomic DNA double-strand breaks are targets for hepadnaviral DNA integration. Proc Natl Acad Sci U S A 101:11135–11140. 10.1073/pnas.0403925101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang W, Summers J. 1999. Integration of hepadnavirus DNA in infected liver: evidence for a linear precursor. J Virol 73:9710–9717. 10.1128/JVI.73.12.9710-9717.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Toh ST, Jin Y, Liu L, Wang J, Babrzadeh F, Gharizadeh B, Ronaghi M, Toh HC, Chow PK, Chung AY, Ooi LL, Lee CG. 2013. Deep sequencing of the hepatitis B virus in hepatocellular carcinoma patients reveals enriched integration events, structural alterations and sequence variations. Carcinogenesis 34:787–798. 10.1093/carcin/bgs406. [DOI] [PubMed] [Google Scholar]
- 15.Jiang Z, Jhunjhunwala S, Liu J, Haverty PM, Kennemer MI, Guan Y, Lee W, Carnevali P, Stinson J, Johnson S, Diao J, Yeung S, Jubb A, Ye W, Wu TD, Kapadia SB, de Sauvage FJ, Gentleman RC, Stern HM, Seshagiri S, Pant KP, Modrusan Z, Ballinger DG, Zhang Z. 2012. The effects of hepatitis B virus integration into the genomes of hepatocellular carcinoma patients. Genome Res 22:593–601. 10.1101/gr.133926.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Quail MA, Swerdlow H, Turner DJ. 2009. Improved protocols for the Illumina genome analyzer sequencing system. Curr Protoc Hum Genet Chapter 18:Unit 18.2. 10.1002/0471142905.hg1802s62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ishii T, Tamura A, Shibata T, Kuroda K, Kanda T, Sugiyama M, Mizokami M, Moriyama M. 2020. Analysis of HBV genomes integrated into the genomes of human hepatoma PLC/PRF/5 cells by HBV sequence capture-based next-generation sequencing. Genes 11:661. 10.3390/genes11060661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kingan SB, Urban J, Lambert CC, Baybayan P, Childers AK, Coates B, Scheffler B, Hackett K, Korlach J, Geib SM. 2019. A high-quality genome assembly from a single, field-collected spotted lanternfly (Lycorma delicatula) using the PacBio Sequel II system. Gigascience 8:giz122. 10.1093/gigascience/giz122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Seeger CZF, Mason WS. 2014. Hepadnaviruses, p 3376–3436. Field's virology, 6th ed. Wolters Kluwer/Lippincott Williams & Wilkins Health, Philadelphia, PA. [Google Scholar]
- 20.Poplin R, Chang PC, Alexander D, Schwartz S, Colthurst T, Ku A, Newburger D, Dijamco J, Nguyen N, Afshar PT, Gross SS, Dorfman L, McLean CY, DePristo MA. 2018. A universal SNP and small-indel variant caller using deep neural networks. Nat Biotechnol 36:983–987. 10.1038/nbt.4235. [DOI] [PubMed] [Google Scholar]
- 21.Morishita A, Iwama H, Fujihara S, Sakamoto T, Fujita K, Tani J, Miyoshi H, Yoneyama H, Himoto T, Masaki T. 2016. MicroRNA profiles in various hepatocellular carcinoma cell lines. Oncol Lett 12:1687–1692. 10.3892/ol.2016.4853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Meng G, Tan Y, Fan Y, Wang Y, Yang G, Fanning G, Qiu Y. 2019. TSD: a computational tool to study the complex structural variants using PacBio targeted sequencing data. G3 9:1371–1376. 10.1534/g3.118.200900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wong N, Lai P, Pang E, Leung TW, Lau JW, Johnson PJ. 2000. A comprehensive karyotypic study on human hepatocellular carcinoma by spectral karyotyping. Hepatology 32:1060–1068. 10.1053/jhep.2000.19349. [DOI] [PubMed] [Google Scholar]
- 24.Hilger C, Velhagen I, Zentgraf H, Schroder CH. 1991. Diversity of hepatitis B virus X gene-related transcripts in hepatocellular carcinoma: a novel polyadenylation site on viral DNA. J Virol 65:4284–4291. 10.1128/JVI.65.8.4284-4291.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Niu C, Livingston CM, Li L, Beran RK, Daffis S, Ramakrishnan D, Burdette D, Peiser L, Salas E, Ramos H, Yu M, Cheng G, Strubin M, Delaney WI, Fletcher SP. 2017. The Smc5/6 complex restricts HBV when localized to ND10 without inducing an innate immune response and is counteracted by the HBV X protein shortly after infection. PLoS One 12:e0169648. 10.1371/journal.pone.0169648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Podlaha O, Wu G, Downie B, Ramamurthy R, Gaggar A, Subramanian M, Ye Z, Jiang Z. 2019. Genomic modeling of hepatitis B virus integration frequency in the human genome. PLoS One 14:e0220376. 10.1371/journal.pone.0220376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Svicher V, Salpini R, Piermatteo L, Carioti L, Battisti A, Colagrossi L, Scutari R, Surdo M, Cacciafesta V, Nuccitelli A, Hansi N, Ceccherini Silberstein F, Perno CF, Gill US, Kennedy PTF. 2020. Whole exome HBV DNA integration is independent of the intrahepatic HBV reservoir in HBeAg-negative chronic hepatitis B. Gut 10.1136/gutjnl-2020-323300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kornyeyev D, Ramakrishnan D, Voitenleitner C, Livingston CM, Xing W, Hung M, Kwon HJ, Fletcher SP, Beran RK. 2019. Spatiotemporal analysis of hepatitis B virus X protein in primary human hepatocytes. J Virol 93:e00248-19. 10.1128/JVI.00248-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Graef E, Caselmann WH, Hofschneider PH, Koshy R. 1995. Enzymatic properties of overexpressed HBV-mevalonate kinase fusion proteins and mevalonate kinase proteins in the human hepatoma cell line PLC/PRF/5. Virology 208:696–703. 10.1006/viro.1995.1201. [DOI] [PubMed] [Google Scholar]
- 30.Su Q, Wang SF, Chang TE, Breitkreutz R, Hennig H, Takegoshi K, Edler L, Schroder CH. 2001. Circulating hepatitis B virus nucleic acids in chronic infection: representation of differently polyadenylated viral transcripts during progression to nonreplicative stages. Clin Cancer Res 7:2005–2015. [PubMed] [Google Scholar]
- 31.Mitelman F, Johansson B, Mertens F. 2007. The impact of translocations and gene fusions on cancer causation. Nat Rev Cancer 7:233–245. 10.1038/nrc2091. [DOI] [PubMed] [Google Scholar]
- 32.Montagna C, Lyu MS, Hunter K, Lukes L, Lowther W, Reppert T, Hissong B, Weaver Z, Ried T. 2003. The Septin 9 (MSF) gene is amplified and overexpressed in mouse mammary gland adenocarcinomas and human breast cancer cell lines. Cancer Res 63:2179–2187. [PubMed] [Google Scholar]
- 33.Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25:1754–1760. 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li H. 2018. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34:3094–3100. 10.1093/bioinformatics/bty191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ha Pages PA, Gentleman R, DebRoy S. 2019. Biostrings: efficient manipulation of biological strings. https://bioconductor.org/packages/Biostrings. [Google Scholar]
- 36.Wright ES. 2016. Using DECIPHER v2.0 to analyze big biological sequence data in R. R J 8:352–359. 10.32614/RJ-2016-025. [DOI] [Google Scholar]
- 37.Bodenhofer U, Bonatesta E, Horejs-Kainrath C, Hochreiter S. 2015. msa: an R package for multiple sequence alignment. Bioinformatics 31:3997–3999. 10.1093/bioinformatics/btv494. [DOI] [PubMed] [Google Scholar]
- 38.Qin Y, Zhou X, Jia H, Chen C, Zhao W, Zhang J, Tong S. 2016. Stronger enhancer II/core promoter activities of hepatitis B virus isolates of B2 subgenotype than those of C2 subgenotype. Sci Rep 6:30374. 10.1038/srep30374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shamay M, Agami R, Shaul Y. 2001. HBV integrants of hepatocellular carcinoma cell lines contain an active enhancer. Oncogene 20:6811–6819. 10.1038/sj.onc.1204879. [DOI] [PubMed] [Google Scholar]