

ABSTRACT
The majority of previously described Staphylococcus aureus bacteriophages belong to three major groups, namely, P68-like podophages, Twort-like or K-like myophages, and a more diverse group of temperate siphophages. Here, we present the following three novel S. aureus “jumbo” phages: MarsHill, Madawaska, and Machias. These phages were isolated from swine production environments in the United States and represent a novel clade of S. aureus myophage. The average genome size for these phages is ∼269 kb with each genome encoding ∼263 predicted protein-coding genes. Phage genome organization and content are similar to those of known jumbo phages of Bacillus sp., including AR9 and vB_BpuM-BpSp. All three phages possess genes encoding complete virion and nonvirion RNA polymerases, multiple homing endonucleases, and a retron-like reverse transcriptase. Like AR9, all of these phages are presumed to have uracil-substituted DNA which interferes with DNA sequencing. These phages are also able to transduce host plasmids, which is significant as these phages were found circulating in swine production environments and can also infect human S. aureus isolates.
IMPORTANCE This study describes the comparative genomics of the following three novel S. aureus jumbo phages: MarsHill, Madawaska, and Machias. These three S. aureus myophages represent an emerging class of S. aureus phage. These genomes contain abundant introns which show a pattern consistent with repeated acquisition rather than vertical inheritance, suggesting intron acquisition and loss are active processes in the evolution of these phages. These phages have presumably hypermodified DNA which inhibits sequencing by several different common platforms. Therefore, these phages also represent potential genomic diversity that has been missed due to the limitations of standard sequencing techniques. In particular, such hypermodified genomes may be missed by metagenomic studies due to their resistance to standard sequencing techniques. Phage MarsHill was found to be able to transduce host DNA at levels comparable to that found for other transducing S. aureus phages, making it a potential vector for horizontal gene transfer in the environment.
KEYWORDS: Staphylococcus aureus, bacteriophage, jumbo phage, MarsHill, Madawaska, Machias
INTRODUCTION
Staphylococcus aureus is an opportunistic pathogen of both humans and animals and is a leading cause of bacteremia and skin, soft tissue, and device-related infections in humans (1). The expansion and prevalence of methicillin-resistant S. aureus (MRSA) impose a significant burden to the health care system (2). S. aureus infections, particularly MRSA infections, can be difficult and costly to treat, with one study reporting the median cost for treatment of a MRSA surgical site infection as $92,363 (3).
Carriage of S. aureus in the general public in the continental United States ranges from 26% to 32% (4), with an estimated 1.3% of that S. aureus being MRSA (5). However, in individuals in the United States that are swine farmers, production workers, or veterinarians, carriage of multidrug-resistant S. aureus (MDRSA) is two to six times greater than that in individuals in the community or those who are not exposed to swine (6, 7). As with humans, S. aureus is considered to be part of the normal bacterial flora of swine (8). Certain activities on swine farms, such as pressure washing and tail docking, generate particle sizes capable of depositing primarily in human upper airways but also the primary and secondary bronchi as well as terminal bronchi and alveoli (9). Livestock-associated methicillin-resistant S. aureus (LA-MRSA) isolates have been found to have a half-life of 5 days in settled barn dust with an approximate die-off of 99.9% after 66 to 72 days (10). Therefore, a better understanding of the ecology of S. aureus in these environments and mitigation of MRSA in swine production facilities would be of benefit to farmers and workers for safety reasons, as MRSA isolates are able to persist and possibly spread throughout the environment and workers.
Bacteriophages (phages) are viruses that infect bacteria and are the most abundant organisms on Earth, with an estimated abundance of 1031 in the biosphere (11). However, despite their abundance and possible utility, there are still many unknowns about the basic biology of most phages and their interactions with their hosts in the environment. Previous investigations of S. aureus phages have described three main classes of phages, namely, small, virulent P68-like podophages with genomes of ∼18 to 20 kb; various temperate siphophages with genomes of ∼45 kb; and large, virulent Twort-like myophages with genomes of ∼130 kb (12, 13). A novel large S. aureus myophage that appears to be distinct from the K-like phages named S6 was reported by Uchiyama et al., in 2014 (14), and the large virulent siphophage vb_SauS_SA2, related to the S. epidermidis phage 6ec, was reported in 2019 (15).
While S6 was estimated to have a 270-kb genome by pulse-field gel electrophoresis and contain DNA in which thymine was replaced by uracil, no genomic sequence for S6 has been reported. Phages that have genomes over 200 kb are classified as “jumbo” phages (16). Most jumbo phages have been isolated from Gram-negative hosts, with the only known jumbo phages for Gram-positive hosts infecting Bacillus spp. Jumbo phage phylogeny and taxonomy are complicated by their often distant relationships to each other and to other known phage types; jumbo phage genomes typically encode large numbers of hypothetical proteins (16). This study describes three novel S. aureus jumbo phages isolated from swine environments: MarsHill, Machias, and Madawaska.
RESULTS AND DISCUSSION
Phage isolation and characterization.
Phages MarsHill, Madawaska, and Machias were isolated from S. aureus-enriched environmental samples collected from swine production facilities located in Texas and Minnesota (Table 1). MarsHill and Madawaska were isolated against a human clinical USA300 MRSA host (NRS384), and Machias was isolated on a sequence type 9 (ST9) methicillin-susceptible S. aureus (MSSA) isolate of swine origin (strain PD32). Phage enrichment and culture were routinely conducted at 30°C, and phages MarsHill, Madawaska, and Machias were found to be unable to form plaques or propagate in liquid culture at 37°C. Temperature sensitivity between 37°C and 30°C was also noted for the S. aureus jumbo phage S6 (14) and may be a limiting factor for recovery of this type of phage from the environment.
TABLE 1.
Summary of three S. aureus jumbo myophage genomes
Transmission electron microscopy of MarsHill revealed a large phage with typical myovirus morphology (Fig. 1). MarsHill was observed to have a mean head size of ∼115 nm (8 virions; ± 6 nm) and a tail length of ∼233 nm (6 virions; ± 4 nm). For comparison, the K-like S. aureus phage phi812 has a reported head diameter of 90 nm and a tail length of 240 nm (17). The measurements of the jumbo phage reported here are comparable to those of phage S6, with a reported head diameter of 118 nm and tail length of 237 nm (14). Although the genome sequence of S6 is not available as of this writing, the large size and temperature-dependent growth characteristics reported for this phage make it likely to be related to the jumbo phages reported here.
FIG 1.

Transmission electron micrograph of the jumbo S. aureus myophage MarsHill.
DNA sequencing and genomic characterization.
Genomic DNA extracted from all three phages was not able to be digested by EcoRI-HF (5′-GAATTC-3′) or DraI (5′-TTTAAA-3′), but it could be digested with MspI (5′ CCGG 3′) and Taq-αI (TaqI) (5′-TCGA-3′) (Fig. 2). TaqI was used as it has been shown to cut heavily modified phage DNA such as that of SPO1, which contains hydroxymethyluracil in place of thymine in its DNA (18). These results are consistent with phage DNA modifications to adenine or thymine bases, as MspI, which lacks A or T bases in its recognition sequence, was also able to digest this DNA.
FIG 2.
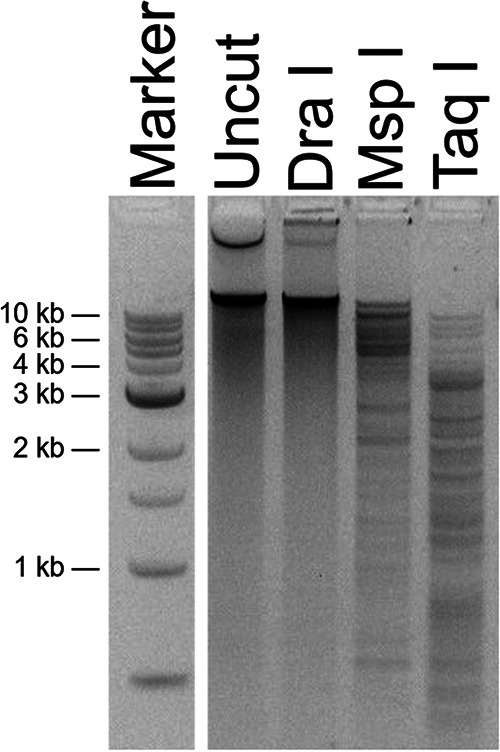
Restriction digests of MarsHill genomic DNA. DNA was not digestible with DraI but was cut with the enzymes MspI and TaqI. Marker is a 1-kb ladder (NEB).
Obtaining the genomic DNA sequence of these phages was challenging. Multiple attempts at standard DNA sequencing approaches using library preparation by Illumina TruSeq Nano or Swift Accel-NGS 1S plus kits (Swift Biosciences) followed by a MiSeq V2 Nano 500-cycle sequencing kit produced no detectable phage DNA sequence. SMRTbell Express (Pacific Biosciences) failed to produce sequenceable DNA libraries. Sequencing by a MinION device (Oxford Nanopore) produced sequence data that could not be interpreted, even though the DNA sequence of coliphage Lambda could easily be recovered as the positive control for the sequencing run. Illumina library preparation by a PCR-free library protocol, which required significantly larger amounts of input DNA, produced DNA libraries sequenceable by the Illumina MiSeq platform. Modification or base substitution of phage DNA has been reported as an impediment to sequencing for other phages, such as the 5-hydroxymethyluracil substitution for thymine found in the Bacillus phage CP-51 (19) or uracil substitution for thymine in the Bacillus jumbo phage AR9 (20). Phage AR9 was sequenced by the Illumina platform following library amplification using a uracil-compatible DNA polymerase (20), and CP-51 was sequenceable only by a combination of PacBio and Sanger sequencing (19). The previously reported S. aureus jumbo phage S6 was also determined to contain DNA with thymine completely replaced by uracil (14). The behavior of the DNA of the MarsHill-like phages reported here, namely, resistance to digestion by restriction enzymes with A/T bases in the recognition site, sensitivity to digestion by TaqI, and an inability to be amplified by PCR using polymerases other than the uracil-insensitive PhusionU, is consistent with such a thymine-uracil base substitution as reported for phages AR9 and S6 (14, 20).
The substitution of nonstandard bases in phage DNA is likely an adaptation that provides broad protection of the phage chromosome from cleavage by host restriction systems (21). The glycosylated 5-hydroxymethylcytosine (glc-HMC) of coliphage T4 protects phage DNA from not only HMC-specific nucleases but also CRISPR-Cas9 (22). Substituting uracil for thymine is also a means of circumventing host defenses; B. subtilis phage SPO1 has hydroxymethyluracil in place of thymine, and replacement of this base with thymine renders the phage DNA more susceptible to restriction enzymes (23, 24). It is worth noting that the incompatibility of such hypermodified phage DNA with standard sequencing approaches likely impacts various metaviromic studies of natural phage communities, as this DNA will be rendered “invisible” to such approaches unless procedures are adapted to capture this sequence.
Genomic analysis of MarsHill-like phages, a clade of phages related to Bacillus jumbo phages.
Phages MarsHill, Machias, and Madawaska were sequenced by the Illumina MiSeq platform to final coverages of 271-, 151-, and 212-fold, respectively. These phages are jumbo phages with an average genome size of ∼269 kb (Table 1). The phages’ G+C content of ∼25% is lower than the median G+C content of 32.7% found in their S. aureus host. A comparison of phage DNA sequences by progressiveMauve (25) (Fig. 3) found MarsHill and Madawaska were more closely related (92.4% identity), with the Machias DNA sequence more diverged (61.5% identity with MarsHill, and 61.8% identity with Madawaska). In addition to being ∼8 kb longer than either MarsHill or Madawaska, Machias contains an inversion in its genome that corresponds to MarsHill genes 171 to 193. Differences between these genomes are primarily confined to a loss or replacement of individual genes, typically in predicted homing endonucleases or hypothetical genes (Fig. 3). A blastclust comparison of the 789 predicted proteins found across the 3 S. aureus jumbo phages (threshold of >50% identity aligned over >50% protein length) generated 311 protein clusters, with 197 of them conserved in all 3 phages. Sixty-two of these protein clusters were singletons found in only a single phage, with the majority (46/62, 74%) found only in Machias. The results of the protein cluster analysis are provided in Table S1 in the supplemental material.
FIG 3.

Comparison of three S. aureus jumbo phage genomes by progressiveMauve. The annotated genome map for phage MarsHill is added to scale at the top of the figure. The genomes are generally syntenic, with the exception of an inverted region in phage Machias. DNA sequence discontinuities are primarily located at homing endonuclease genes (light blue) or hypothetical genes (gray).
There is little detectable DNA sequence similarity between the MarsHill-like phages and any other organisms in the NCBI nucleotide database (<5% alignable DNA sequence identity by BLASTn). A comparison of the predicted MarsHill, Madawaska, and Machias proteomes to other phages by the Center for Phage Technology (CPT) Galaxy comparative genomics workflow (26) shows that these phages are related to previously described Bacillus jumbo phages AR9 (GenBank accession no. NC_031039 [27]), PBS1 (NC_043027), and vB_BpuM-BpSp (KT895374.1 [28]) (Table 2). These phages are more distantly related to jumbo phages phiR1-37 (which also contains hypermodified DNA [29]) and MS32 infecting Gram-negative hosts Yersinia enterocolitica and Vibrio mediterranei, respectively. Phage Machias shares a greater number of proteins with these other jumbo phages and also shares more proteins with the staphylococcal myophage Twort and other Twort-like phages (Table 2). Genes shared between the MarsHill-like and Twort-like phages are primarily those encoding homing endonucleases (14 of 21 shared proteins), ribonucleotide reductases (2 proteins), and a predicted amidase. The AR9-like jumbo phages are part of a larger clade of phiKZ-like jumbo phages that can be found infecting Gram-negative and Gram-positive hosts (30).
TABLE 2.
Predicted proteins shared between the MarsHill-like phages and other related phages, as determined by BLASTpa
| Related phage name | Accession no. | Host | No. of predicted proteins shared with phages in this study: |
||
|---|---|---|---|---|---|
| Mars Hill | Madawaska | Machias | |||
| AR9 | NC_031039 | B. subtilis | 77 | 76 | 81 |
| PBS1 | NC_043027 | B. subtilis | 76 | 75 | 81 |
| BpuM-BpSp | KT895374 | B. pumilus | 75 | 75 | 80 |
| phiR-37 | NC_016163 | Y. enterocolitica | 60 | 59 | 62 |
| MS32 | MK308677 | V. mediterranei | 20 | 19 | 19 |
| Twort | NC_007021 | S. aureus | 13 | 12 | 21 |
E < 10−5.
The genomes of phages MarsHill, Madawaska, and Machias were annotated using the CPT Galaxy-Apollo toolset (26). Due to the overall similarity of the three phages, they will be discussed primarily using phage MarsHill as a reference. The phage genomes were closed and then analyzed by PhageTerm (31) to determine genomic termini, which indicated phage chromosomes with pac-like circular permutation. The phage genomes were reopened immediately upstream of the genes encoding predicted nonvirion RNA polymerase (RNAP) β and β’ subunit N-terminal domains (Fig. 4). The MarsHill genome encodes 262 predicted proteins, of which 76 could be assigned functions. The genomes exhibit a general lack of clear modular organization, which is common in jumbo phages (16). Multiple structural components could be identified bioinformatically in the MarsHill genome, based on the presence of conserved domains or their similarity to proteins previously described in phages AR9 (20, 32) and vB_BpuM-BpSp (28, 33), including the major capsid protein, predicted tail fibers, tail sheath, baseplate wedge, prohead scaffold, and portal protein (Table S1). Two potential tape measure proteins, namely, gp86 and gp87, were identified; both proteins contain significant alpha-helical content as well as several predicted transmembrane domains which is indicative of tape measure proteins (34). However, one protein (MarsHill gp86; QQM14620) contains detectable peptidoglycan-degrading domains (PF18013 and PF00877) and thus was annotated as a potential tail lysin. However, peptidoglycan-degrading activity has been associated with multiple known phage tail tape measure proteins (35–37), and thus, either of these proteins could plausibly provide this function. All three phages contain dCMP deaminase genes, but other genes associated with modifications to uracil, such as dUMP hydroxymethylase or HMdUMP kinase, were not identified in the genomes of the S. aureus jumbo phages (24). A GroEL-like chaperonin protein was identified in all three S. aureus jumbo phage genomes; a similar GroEL-like protein has also been identified in phage AR9 (20). The GroEL-like protein in AR9 has been shown to possess chaperone activity without requiring a cochaperonin to function when purified and expressed in Escherichia coli (38). In coliphage T4, a GroES-like protein (T4 gp31) is essential for folding of the major capsid protein and aids in the assembly of phage particles (39), and this jumbo phage chaperonin likely plays a similar role in folding key viral proteins.
FIG 4.

Annotated genome map of the jumbo S. aureus phage MarsHill. Genes transcribed from the top DNA strand are drawn above the black line, and genes transcribed from the minus strand are drawn below. Genes are color coded based on their predicted functions as shown in the legend, and selected genes are annotated with their predicted functions. Open reading frames that are predicted to be part of intron-disrupted genes are joined by heavy black lines.
MarsHill gp047 was annotated as the phage holin due to the presence of conserved domains associated with staphylococcal and streptococcal phages (pfam04531) and a conserved protein domain for the holin of lactococcal bacteriophage phi LC3 (TIGR01598) (40). However, two MarsHill proteins (gp153 and gp203) possess characteristics of phage endolysins; thus, both proteins have been annotated simply as N-acetylmuramoyl-l-alanine amidases. MarsHill gp203 has similarity to amidases in phages AR9 (20) and PBS1 (NC_043027.1). However, this similarity is to AR9 gp272, not to AR9 gp194, which is noted to have a PlyB-like endolysin conserved domain (cd06523) and is not conserved in the MarsHill-like phages. The second MarsHill amidase gp153 is more closely related to bacterial amidases and does not share a homolog in AR9. It is not clear if one or both of the identified amidases function as the endolysin in the Mars-Hill-like phages; these phages may have “captured” a second amidase gene with redundant endolysin activity.
A large class of phiKZ-like jumbo phages are known to encode their own DNA-directed RNA polymerases (20). These jumbo phage RNA polymerases differ from the bacterial RNA polymerase in that they lack α and ω subunits, with only β and β’ subunits identified (41). These jumbo phages encode two RNA polymerase holoenzymes, of which one (the virion RNA polymerase) is packaged into the virion and presumably ejected into the host cell upon infection (42) and the other, nonvirion RNA polymerase is expressed during the phage lytic cycle (43). Identification and annotation of the jumbo phage RNA polymerase genes are complicated by their arrangement; each of the virion and nonvirion β and β’ RNA polymerase subunits are expressed from multiple loci, encoding subdomains of each of these subunits. The virion RNA polymerase is comprised of β and β’ subunits, with the β subunit expressed from two genes encoding N- and C-terminal domains and the β’ subunit encoded by three genes encoding N-terminal, midterminal, and recently discovered C-terminal domain (43). The nonvirion RNA polymerase is comprised of β and β’ subunits, with each encoded by two genes corresponding to N and C termini, and an additional, fifth protein subunit, which appears to be involved in phage-specific promoter recognition (44). The jumbo phage RNA polymerase subunit genes are also frequently disrupted by one or more introns (20). Genes encoding five virion RNA polymerase subunits (β-N, β-C, β’-N, β’-mid, and β’-C) and five nonvirion RNA polymerase subunits (β-N, β-C, β’-N, β’-C, and specificity determinant) were identified in the MarsHill-like jumbo phages based on the presence of conserved domains and similarity to homologs in related phages.
A comparison of the predicted MarsHill, Madawaska, and Machias proteins with their homologs in other phages indicated that multiple genes are interrupted by one or more intron sequences. The presence of intron-disrupted genes is well known in K-like S. aureus phages (45), in the Bacillus phages SPO1 and SP82 (46), and the Bacillus jumbo phage AR9 (20). As shown in Table 3, introns were detected in genes encoding multiple RNA polymerase subunits, the prohead scaffold/protease, DNA polymerase subunits, large terminase, and DNA gyrase B. A similar repertoire of genes is known to be disrupted by introns in AR9 (20). Based on protein sequence alignment to their intact homologs in AR9 and other related organisms, the number of exons in disrupted genes in MarsHill, Madawaska, and Machias was predicted (Table 3). This approach allowed for the prediction of the mature peptides for all intron-disrupted genes except for the large terminase subunit (MarsHill genes 70 and 71), which could not be confidently reconstructed based on alignment to any intact protein homolog. Some intron-disrupted genes identified in AR9, such as the nonvirion RNA polymerase β' N-terminal subunit, were found to be intact in MarsHill, while genes that are intact in AR9, such as the DNA polymerase I, were found to be disrupted in the MarsHill-like phages. The predicted number of exons and exon boundary positions was variable between the three S. aureus jumbo phages described here, which is consistent with the highly plastic nature of these regions (20).
TABLE 3.
Predicted intron content in the genomes of three S. aureus jumbo phages, in comparison with introns identified in the related Bacillus jumbo phage AR9a
| Gene product | Predicted content by phage genome |
|||||||
|---|---|---|---|---|---|---|---|---|
| MarsHill |
Madawaska |
Machias |
AR9 |
|||||
| Gene(s) | No. of predicted exons | Gene(s) | No. of predicted exons | Gene(s) | No. of exons | Gene | No. of exons | |
| nvRNAP β' N | 002 | 1 | 002 | 1 | 002 | 1 | g270 | 2 |
| scaffold/prohead protease | 065 | 2 | 064 | 3 | 065 | 3 | g114 | 2 |
| TerL | 070, 071 | ≥2 | 069, 071 | ≥2 | 070, 073 | ≥2 | g121 | 5 |
| NrdE | 077 | 1 | 077 | 1 | 077 | 3 | g223 | 1 |
| Virion protein/hypothetical protein | 085 | 1 | 085 | 1 | 090 | 2 | g205 | 3 |
| DNA pol catalytic subunit | 108 | 2 | 108 | 2 | 110 | 3 | g152 | 1 |
| vRNAP β' N | 113 | 3 | 113 | 2 | 117 | 3 | g145 | 3 |
| DNA pol exonuclease subunit | 138 | 1 | 137 | 2 | 137 | 3 | g132 | 1 |
| vRNAP β C2 | 161 | 2 | 160 | 2 | 170 | 4 | g189 | 6 |
| vRNAP β' C | 162 | 1 | 161 | 1 | 171 | 2 | g264 | 1 |
| DNA gyrase B | 216 | 2 | 218 | 2 | 225 | 2 | g024 | 1 |
| DNA gyrase A | 228 | 1 (intein) | 231 | 1 (intein) | 238 | 1 (intein) | g044 | 3 |
| vRNAP β C1 | 230 | 2 | 233 | 3 | 239 | 3 | g078 | 3 |
| nvRNAP β C | 248 | 4 | 254 | 3 | 266 | 6 | g089 | 1 |
Intron content in the S. aureus phages was predicted based on protein sequence alignment with intact coding sequences found in AR9 or other related organisms. Some genes, such as that encoding the nonvirion RNAP β' N-terminal domain, are intact in the MarsHill-like phages but disrupted by an intron in AR9; other genes, such as the DNA polymerase subunits, are intact in AR9 but disrupted in the MarsHill-like phages. The DNA gyrase A protein was found to be encoded by a single exon in the MarsHill-like phages but is disrupted by an intein.
Analysis of the protein sequence of the MarsHill DNA gyrase A (gp228) indicated that this gene is disrupted by an intein, based on the presence of intein-associated conserved domains (IPR006142, IPR003587, and IPR003586). The intein boundaries in MarsHill gp228 were predicted based on alignment with its intein-less homolog in AR9 (AR9 gp044) and the presence of a conserved cysteine at the N-terminal boundary and the conserved residues His-Asn at the C-terminal boundary (47). Based on this analysis, the intein is predicted to be inserted between residues Y117 and T492 of gp228. The gp228 intein is also predicted to contain a homing endonuclease, based on the presence of a LAGLIDADG endonuclease conserved domain (IPR004860); such domains are a common feature of intein sequences (48). The AR9 homolog of the MarsHill DNA gyrase A appears to lack this intein but is instead disrupted by two intron sequences (20), indicating this gene is a common target for mobile DNA elements.
Of 43 intron sequences that could be identified in the 3 phages, most (29 introns) were short (<250 bp) and contained no protein-coding genes. Eleven introns containing protein-coding genes were identified, ranging from 776 bp to 1,642 bp long. Phage Machias contains six such introns, MarsHill contains four, and Madawaska contains only one. Of the 12 proteins identified in these introns, 11 can be assigned functions based on conserved domain searches in InterProScan or HHpred (Table 4). Seven proteins contain very short patch repair (VSR)-like nuclease domains found in a class of homing endonucleases initially described in the Bacillus phage 0305phi8-36 (HHpred 3R3P_B; 97.9%) (49). Two were identified as HNH endonucleases, one was identified as a LAGLIDADG endonuclease, and one contains an IS605-like transposase domain that has been associated with intron-like elements in other Firmicutes hosts (50).
TABLE 4.
Characteristics of the proteins found within introns in phages MarsHill, Madawaska, and Machiasa
| Intron-encoded protein | Detected domainb | Domain accession no. |
|---|---|---|
| MarsHill gp64 | LAGLIDADG endonuclease | IPR004860 |
| MarsHill gp114 | VSR endonuclease | G3DSA:3.40.960.10 |
| MarsHill gp160 | VSR endonuclease | G3DSA:3.40.960.10 |
| MarsHill gp231 | VSR endonuclease | G3DSA:3.40.960.10 |
| Zinc finger RING | IPR013083 | |
| Madawaska gp114 | VSR endonuclease | G3DSA:3.40.960.10 |
| RE-like | IPR011335 | |
| Machias gp78 | HNH endonuclease | IPR003615 |
| Machias gp89 | Transposase | IPR001959 |
| IS605 transposase | IPR010095 | |
| Machias gp138 | HNH endonuclease | IPR003615 |
| Machias gp140 | VSR endonuclease | G3DSA:3.40.960.10 |
| Machias gp168 | VSR endonuclease | G3DSA:3.40.960.10 |
| Machias gp169 | None | |
| Machias gp268 | VSR endonuclease | G3DSA:3.40.960.10 |
Most proteins were identified as homing endonucleases, with one predicted to contain a transposase domain and one with no identifiable conserved domains.
Detected by InterProScan.
An analysis of all predicted intron sequences in Rfam (51) identified only three related sequences in the Rfam database. One of the two predicted introns interrupting MarsHill gene 113, encoding the virion RNAP β’N-terminal subunit, was identified as a group II D1D4-3 catalytic intron (RF02001); this sequence also contains a predicted VSR-like homing endonuclease. Introns interrupting MarsHill gene 248 and Madawaska gene 254, encoding the nonvirion RNAP β subunit C-terminal domain, were both identified as group I catalytic introns (RF00028). These two introns are relatively short (177 and 178 bp, respectively) and do not contain protein-coding genes.
Comparisons of the introns interrupting the same gene in the three phages showed a pattern dominated by independent acquisition with a limited amount of vertical inheritance. For example, MarsHill gene 65, encoding the prohead scaffold/protease, is disrupted by a single predicted intron that contains a homing endonuclease gene, while its homologs in Madawaska and Machias are each disrupted by two predicted introns with no other protein-coding genes (Table 3; Fig. 5). DNA sequence comparisons by BLASTn show the Madawaska gene 64 and Machias gene 65 introns are related to each other (47.5% to 93% overall sequence identity) but are not related to those found in MarsHill, indicating independent acquisition events, although the predicted N- and C-terminal intron boundaries are similar. In a second example, in the genes encoding the virion RNAP β’ N-terminal subunits of these phages, the MarsHill and Madawaska homologs are disrupted by unrelated introns containing different homing endonuclease genes, while the Machias homolog contains introns with no protein-coding genes (Fig. 5). In contrast, the inteins identified in the DNA gyrase A genes of all three phages are closely related (80% to 100% DNA sequence identity by BLASTn), suggesting vertical transmission following acquisition by a common ancestor.
FIG 5.

Examples of intron-disrupted genes found in three S. aureus jumbo phages. Exons part of the annotated function (scaffold/protease or RNAP) are shown in green, and intervening intron-encoded genes (e.g., homing endonucleases) are shown in light blue. DNA sequence similarity between gene regions is shown as gray boxes, as determined by BLASTn and generated by the tool EasyFig. These examples illustrate the frequent independent acquisition of introns in homologous phage genes.
A comparison of all predicted intron DNA sequences by blastclust (cutoff, 90%; identity, over 90% sequence length) identified only five cases where the intron sequences are shared between two phages and no cases where they are shared across all three phages. In the five cases where introns are shared, they are in the same position in homologous genes, suggesting acquisition by a common ancestor. Evidence of intragenomic spread of intron sequences was not observed in these phages, as no similar introns were identified within the same genome, even when blastclust cutoffs were relaxed to 50% identity over 50% of the sequence length.
A previous analysis of AR9 transcription signals identified a AT-rich early promoter that is recognized by the virion RNA polymerase (32). A DNA sequence 200 bp upstream of all protein-coding genes was extracted from the MarsHill genome and analyzed for sequence motifs using MEME (52). An AT-rich motif nearly identical to that observed in phage AR9, particularly the consensus motif 5′-TATATTAT-3′, was identified upstream of 29 genes in the MarsHill genome by MEME (Fig. 6). A second conserved signal corresponding to the phage late promoter was not identified by this approach. Transcriptomic profiling of AR9 has indicated that this signal is somewhat less conserved with a consensus motif of AACA(N6)TW (32) and thus is difficult to predict de novo without additional data from phage transcripts.
FIG 6.

Sequence logo of the predicted MarsHill early promoter sequence recognized by the phage virion RNA polymerase.
All three S. aureus jumbo phages encode a predicted RNA-dependent DNA polymerase (reverse transcriptase), exemplified by MarsHill gp206. This 501-residue protein contains a C-terminal retron-like reverse transcriptase domain (cd03487) and an ∼200-residue N-terminal domain of unknown function. Reverse transcriptases have been previously identified in phages as part of diversity-generating retroelements (DGRs) which selectively mutagenize the phage tail fiber to extend the phage host range (53, 54). The MarsHill gp206 reverse transcriptase does not appear to be a part of such a system, as the accessory variability determinant (Avd), template repeat, variable repeat, and initation of mutagenic homing sequences typically associated with DGRs were not identified in MarsHill. The MarsHill reverse transcriptase is more closely related to proteins associated with bacterial retroelements, with homologs found in Bacillus sp. (TQJ37342; E = 10−26) and Marinimicrobium sp. (ROQ19867; E = 10−21). The MarsHill reverse transcriptase is quite diverged from other sequences present in NCBI, with the closest bacterial homologs sharing only ∼30% sequence identity. Immediately upstream of MarsHill gp206 is a 1,200-bp noncoding region which is hypothesized to contain the retroelement-associated noncoding RNA (ncRNA) (55). Bacterial retroelements have recently been implicated as a type of bacterial antiphage defense mechanism which leads to abortive infection when an infecting phage inhibits RecBCD; however, the MarsHill reverse transcriptase does not appear to be associated with a potential effector protein as described in antiphage elements (55). If this element serves as an antiphage system (e.g., to exclude coinfecting phages), it is possible that the gene encoding such an effector has become unlinked in the MarsHill genome. It is also possible that the gp206 reverse transcriptase serves some other function or is simply present as a selfish genetic element (56).
There are no identified tRNAs in phage MarsHill, with Madawaska having one and Machias two pseudo-tRNA genes as identified by tRNAscan. The presence of tRNAs is positively associated with genome size in phages and is thought to be driven by the divergence of phage codon usage from that of the host (57). In these phages, tRNA genes appear to be degenerating, with aberrant pseudo-tRNA genes appearing in two of the phages and these genes being undetectable in MarsHill, indicating a lack of selection pressure to maintain these functions (58).
Phage MarsHill is able to transduce host DNA.
Phages AR9 of Bacillus sp. and S6 of S. aureus have been previously described as generalized transducing phages capable of aberrantly packaging host DNA and transferring it to a new cell (14, 20). The ability of MarsHill to transduce chromosomal and plasmid-borne antibiotic resistance marker genes was determined experimentally. MarsHill was able to transduce plasmid-borne kanamycin resistance from S. aureus strain Xen 36 to wild-type S. aureus isolate PD17 at an efficiency of 7 × 10−9 transductants/PFU, based on three replicate experiments. This transduction frequency is comparable to that of several different pac-type S. aureus Siphoviridae, which are able to transduce plasmids of various sizes at a frequency of 10−5 to 10−11 transductants/PFU (59). No transductants were recovered using lysates of the virulent S. aureus phage K or induced culture supernatants of Xen 36, indicating that any endogenous temperate phages residing in Xen 36 are not produced at high enough levels to account for the observed transduction. Phage K is not expected to be able to perform generalized transduction, as it has been reported to degrade the host chromosome shortly after infection (60), an activity which is associated with the lack of transducing capacity in other phages, such as the coliphage T4 (61). Transduction experiments conducted with the donor strain CA-347, a USA600 MRSA strain, did not yield any detectable transductants expressing mecA-mediated oxacillin resistance in the recipient cultures using either phage or culture supernatants. This observed inability to transduce chromosomal markers is not conclusive, as the measured transduction frequency for plasmid DNA was near the detection limit of the assay (2 × 10−9 transductants/PFU), and transduction of the mecA locus by temperate Siphoviridae phages has been reported ranging from 1 × 10−9 to 2 × 10−10 transductants/PFU (62). Phage S6 was reported to transduce plasmid pCU1 at an efficiency of 10−7 transductants per cell and a plasmid containing the mecA gene at an efficiency of 5.2 × 10−11 per cell into the restriction-deficient S. aureus strain RN4220. S6-mediated transduction of the pCU1 plasmid was also tested for several other staphylococci, including S. epidermidis, Staphylococcus pseudintermedius, Staphylococcus sciuri, and Staphylococcus felis, with reported efficiencies of 10−7 to 10−10 per cell (14). The transduction efficiency of MarsHill may be expected to be lower than that observed for S6, as the recipient strain used to measure MarsHill transduction is a wild-type S. aureus strain recently isolated from the environment which presumably has a functional restriction system, unlike the restrictionless S. aureus strain RN4220.
Conclusions.
Three novel S. aureus myophages isolated from swine environments across the United States presented unique challenges in both culturing and sequencing. Multiple commonly used approaches failed to produce usable sequences for these phages; however, using a PCR-free library preparation method produced libraries sequenceable by the Illumina platform. This study raises questions about the diversity of phages with modified DNA that are missed by metagenomic studies using traditional library preparations to identity phage sequences. Complete genomes were obtained for phages MarsHill, Madawaska, and Machias, with all phages having genomes well over 200 kb, classifying them as members of a new jumbo phage lineage in S. aureus (16). The S. aureus phages in this study were isolated from environmental samples at 30°C instead of the traditional 37°C, in hopes of more closely mimicking ambient barn temperatures as well as human nasopharynx temperatures which average around 34°C (63). Without this adjustment, these phages would not have been isolated, as they do not form observable plaques or propagate efficiently in liquid culture at 37°C. These three phages are distantly related to other known Bacillus phages, with the most similar being Bacillus phage vB_BpuM-BpSp (28) and Bacillus phage AR9 (20). All three phages carry identifiable virion and nonvirion RNA polymerase subunits, of which many are intron disrupted. Intron and intein disruption of multiple phage genes was identified, including the virion and nonvirion RNA polymerases, DNA polymerase, and DNA gyrase. Mature peptides were predicted for all intron-disrupted genes by alignment to homologs in AR9 and other organisms, except for the phages’ TerL. Unlike phage AR9, all three phages contain an RNA-directed DNA polymerase, and its function is unclear. MarsHill was able to transduce plasmid DNA at an efficiency of 7 × 10−9 transductants/PFU which indicates that these phages, in addition to temperate S. aureus Siphoviridae, are also possible vectors for horizontal gene exchange in S. aureus. This is particularly relevant, as these phages were found to be circulating in swine production environments and are able to infect both swine and human S. aureus isolates.
MATERIALS AND METHODS
Culture and maintenance of bacteria and phages.
S. aureus was routinely cultured on Bacto Trypticase soy broth (TSB; Difco) or Trypticase soy agar (TSA; TSB + 1.5% [wt/vol] Bacto agar; Difco) aerobically at 30°C. Phages were cultured using the double-layer overlay method (64) with 4 ml of top agar (10 g/liter Bacto tryptone [Difco], 10 g/liter NaCl, and 0.5% [wt/vol] Bacto agar [Difco]) supplemented with 5 mM each of CaCl2 and MgSO4 over TSA bottom plates. Lawns were inoculated with 0.1 ml of a mid-log S. aureus bacterial culture grown to an optical density at 550 nm (OD550) of ∼0.5. Phage stocks were produced by the confluent plate lysate method (65) using the original phage isolation host and harvested with 4 to 5 ml of lambda diluent (100 mM NaCl, 25 mM Tris-HCl [pH 7.4], 8 mM MgSO4, and 0.01% [wt/vol] gelatin). Harvested lysates were centrifuged at 10,000 × g, 10 min, and 4°C; sterilized by passage through a 0.2-μm syringe filter (Millipore, Burlington, MA); and stored in the dark at 4°C. Phages that could not be cultured to high titers using the double-layer overlay method were propagated in liquid culture by inoculating TSB 1:100 with an overnight culture of the appropriate S. aureus host strain and incubating at 30°C with shaking (180 rpm) until OD550 of ∼0.5 was reached. The culture was inoculated with phage at a multiplicity of infection (MOI) of 0.01 and incubated at 30°C with shaking (180 rpm) overnight. The next day, the lysate was harvested by centrifugation (10,000 × g, 10 min, and 4°C) and the supernatant was sterilized by passage through a 0.2-μm syringe filter (Millipore).
Phage isolation.
Phages were isolated from environmental swabs collected from 2015 to 2017 in swine production facilities in the United States. Five swabs per site were collected, targeting areas in production facilities with visible residue, such as floor slats or water lines. Swabs were collected dry or were wetted in Stuart’s medium (BD BBL CultureSwab) for transport. Swab heads were aseptically clipped into a 50-ml conical tube (Falcon Corning) and eluted in sterile TSB with shaking for 2 hours at room temperature. Swabs were pooled by collection site. The swab eluates were centrifuged at 8,000 × g, 10 min, and 4°C, and the supernatants were passed through 0.2-μm syringe filters (Millipore). Samples were enriched for phage using a mixed enrichment approach (66, 67) by mixing 12-ml sample supernatant with 8 ml TSB, inoculating with 0.2 ml of a mixed S. aureus culture, and incubating overnight at 30°C with aeration. Enrichment inocula were prepared by mixing equal volumes of overnight S. aureus cultures. An environmental sample collected from the O.D. Butler, Jr. Animal Science Complex (College Station, TX) yielded phage MarsHill which was enriched with a mixture of the four human S. aureus strains shown in Table 5. Samples collected from commercial swine operations in Minnesota and Texas yielded phages Machias and Madawaska, respectively. These sample elutes were divided and enriched using two different enrichment panels, namely, one panel of six swine isolates and one panel with four human isolates shown in Table 5. Enrichment cultures were centrifuged (8,000 × g, 10 min, and 4°C), and the supernatants were sterilized by passage through a 0.2-μm syringe filter (Millipore) and stored at 4°C. Phages were isolated from phage-positive samples by dilution and plating to lawns of individual S. aureus strains, followed by picking of well-isolated plaques. Each phage was subcultured three times to ensure clonality.
TABLE 5.
S. aureus strains used for phage isolation in this studya
| Strain no. | Strain name | ST | CC or PFGE type | SCCmec | Geographic origin | Isolation source |
|---|---|---|---|---|---|---|
| NRS384 | USA300-0114 | 8 | USA300 | IV | Mississippi, USA | Human |
| NRS255 | HT20020371 | 80 | 80 | IV | France | Human |
| NRS253 | HT20020354 | 398 | 398 | None | France | Human |
| NRS653 | CA-513 | 5 | USA800 | IV | California, USA | Human |
| N/A | PD6 | 9 | N/A | None | Illinois, USA | Swine |
| N/A | PD10 | 398 | N/A | Yes | Iowa, USA | Swine |
| N/A | PD17 | 398 | N/A | None | Texas, USA | Swine |
| N/A | PD18 | 9 | N/A | None | North Carolina, USA | Swine |
| N/A | PD19 | 5 | N/A | None | North Carolina, USA | Swine |
| N/A | PD32 | 9 | N/A | None | Alabama, USA | Swine |
ST, sequence type; CC, clonal complex; PFGE, pulsed-field gel electrophoresis type; SCCmec, the mecA allele that confers methicillin resistance; N/A, not available.
Phage DNA purification and sequencing.
High titer (>108 PFU/ml) lysates of each phage were produced from plate lysates or liquid cultures as described above, and genomic DNA (gDNA) was extracted from 10 to 20 ml of the phage lysate. Phage genomic DNA was extracted using the Wizard DNA cleanup kit (Promega) by a modified protocol as described previously (68, 69). Phage DNA was sheared to fragments of approximately 350 bp using a Bioruptor Pico device (Diagenode Diagnostics). One microgram of DNA for each phage isolate was used for Illumina Truseq library preparation using the manufacturer’s PCR-free library preparation protocol. Prepared libraries were quantified using the Kapa qPCR library quantification kit (Roche) and diluted to 4 nM. Sequencing of phage DNA was performed by Illumina MiSeq V2 Nano 500 cycle chemistry. Library preparation and sequencing were performed at The Institute for Genomics and Society core laboratory at Texas A&M University.
FastQC (70) and SPAdes 3.5.0 (71) were used for read quality control and read assembly, respectively. An analysis of phage contigs by PhageTerm (31) indicated a circularly permuted genome. MarsHill termini were determined by PCR using primers (forward, 5′-AGCAGGTATTACAGGCCATTT-3′; reverse, 5′-GAAGACGAAGTTAATGAGGCTAGA-3′) facing off the contig ends to produce an ∼1,000-bp product. DNA polymerase Phusion U (Thermo Fisher) was used in this end closure PCR, and the PCR product was sequenced by Sanger sequencing to close and correct the MarsHill contig. The Madawaska and Machias contigs were closed by reassembling the genomes with SPAdes (71) to produce contigs in which the original contig ends were internal to the genome with >40× coverage; this information was used to correct and close these contigs.
Restriction digests of phage DNA.
To distinguish different phage types, approximately 300 ng of extracted gDNA was digested with both DraI (5′-TTTAAA-3′) and EcoRI-HF (5′-GAATTC-3′) (New England BioLabs Inc., Ipswich, MA). gDNA samples that showed poor banding patterns or could not be digested by the enzymes listed above were then digested with TaqαI (5′-TCGA-3′) and MspI (5′-CCGG-3′). For DraI, EcoRI-HF, and MspI, 300 ng of gDNA from each phage was incubated with each enzyme and CutSmart buffer at 37°C overnight. For gDNA treated with TaqαI, 300 ng of gDNA was incubated with TaqαI and CutSmart buffer at 65°C for 2 hours. After incubation, 4 μl of loading dye was then added, and the total 24 μl for each sample was run on a 1% agarose gel at 90 V for 2.5 h.
Phage genome annotation.
Genome annotation was conducted on the Center for Phage Technology Galaxy-Apollo genome annotation platform using the Galaxy pipelines described in reference 26. Briefly, genes were identified using GLIMMER v3 (72) and MetaGeneAnnotator v1.0 (73), and tRNAs were identified using ARAGORN v2.36 (74). Protein function annotations were assigned based on results from BLAST v2.9.0 against the nonredundant (nr) and Swiss-Prot databases (75, 76), InterProScan v5.33 (27), HHpred (77), and TMHMM v2.0 (78). Each genomic DNA sequence was compared with that of other phages using BLASTn against the nucleotide database, progressiveMauve v2.4 (25), and Easyfig (79).
Transduction experiments.
The ability of phage MarsHill to transduce host DNA into a recipient cell was determined using methods based on those described in Stanczak-Mrozek et al. (80). S. aureus strains Xen 36 (PerkinElmer; containing a plasmid-borne kanamycin resistance marker) and CA-347 (NRS648; a USA 600 MRSA strain with a chromosomal mecA marker) were used as donor S. aureus strains, and the swine-associated strain PD17 was used as the recipient. Phage MarsHill was propagated on donor strains in liquid culture as described above. PD17 was grown in 10 ml TSB supplemented with 5 mm MgCl2 at 37°C to ∼5 × 108 CFU/ml. A MarsHill lysate propagated on a donor strain was added to the recipient culture at an MOI of 0.1 and incubated for 20 minutes at 30°C; culture supernatants of the donor strains cultured without phages were used as negative controls. The culture was centrifuged at 13,000 × g for 2 minutes, and the cell pellet was resuspended in 1 ml TSB with 50 mM sodium citrate. This culture was then incubated at 37°C for 1 h, washed twice in TSB by pelleting and resuspension, and resuspended in 10 ml TSB. This culture was plated in 1-ml aliquots onto TSA plates supplemented with either 150 μg/ml kanamycin (for lysates propagated on Xen 36) or 5 μg/ml oxacillin (for lysates propagated on CA-347) and incubated overnight at 37°C. Transduction efficiency was calculated as the number of transductants (CFU)/number of input phage particles (PFU), with a detection limit of 2 × 10−9.
Transmission electron microscopy.
Transmission electron micrographs of each phage were obtained by adhering the phage to a carbon film by the Valentine method (30) and staining with 2% uranyl acetate. Phages were viewed in a Jeol 1200EX transmission electron microscope (TEM) at 100-kV accelerating voltage.
ACKNOWLEDGMENTS
This work was supported by the National Pork Board, the National Science Foundation (DBI-1565146), NIH-NIAID (AI121689), and Texas AgriLife Research.
We thank H. Morgan Scott for their generosity and time in assisting in the sequencing of these phages. We also thank Peter Davies for providing swine S. aureus isolates and for his help in locating phage sampling sites. We acknowledge the support of the Network on Antimicrobial Resistance in Staphylococcus aureus (NARSA) for provision of bacterial isolates.
We declare no conflicts of interest.
Footnotes
Supplemental material is available online only.
Contributor Information
Jason J. Gill, Email: jason.gill@tamu.edu.
Julie K. Pfeiffer, University of Texas Southwestern Medical Center
REFERENCES
- 1.Tong SY, Davis JS, Eichenberger E, Holland TL, Fowler VG, Jr.. 2015. Staphylococcus aureus infections: epidemiology, pathophysiology, clinical manifestations, and management. Clin Microbiol Rev 28:603–661. 10.1128/CMR.00134-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Valiquette L, Chakra CN, Laupland KB. 2014. Financial impact of health care-associated infections: when money talks. Can J Infect Dis Med Microbiol 25:71–74. 10.1155/2014/279794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Engemann JJ, Carmeli Y, Cosgrove SE, Fowler VG, Bronstein MZ, Trivette SL, Briggs JP, Sexton DJ, Kaye KS. 2003. Adverse clinical and economic outcomes attributable to methicillin resistance among patients with Staphylococcus aureus surgical site infection. Clin Infect Dis 36:592–598. 10.1086/367653. [DOI] [PubMed] [Google Scholar]
- 4.Sivaraman K, Venkataraman N, Cole AM. 2009. Staphylococcus aureus nasal carriage and its contributing factors. Future Microbiol 4:999–1008. 10.2217/fmb.09.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Salgado CD, Farr BM, Calfee DP. 2003. Community-acquired methicillin-resistant Staphylococcus aureus: a meta-analysis of prevalence and risk factors. Clin Infect Dis 36:131–139. 10.1086/345436. [DOI] [PubMed] [Google Scholar]
- 6.Neyra RC, Frisancho JA, Rinsky JL, Resnick C, Carroll KC, Rule AM, Ross T, You Y, Price LB, Silbergeld EK. 2014. Multidrug-resistant and methicillin-resistant Staphylococcus aureus (MRSA) in hog slaughter and processing plant workers and their community in North Carolina (USA). Environ Health Perspect 122:471–477. 10.1289/ehp.1306741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wardyn SE, Forshey BM, Farina SA, Kates AE, Nair R, Quick MK, Wu JY, Hanson BM, O'Malley SM, Shows HW, Heywood EM, Beane-Freeman LE, Lynch CF, Carrel M, Smith TC. 2015. Swine farming is a risk factor for infection with and high prevalence of carriage of multidrug-resistant Staphylococcus aureus. Clin Infect Dis 61:59–66. 10.1093/cid/civ234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Frana TS. 2012. Staphylococcosis, p 834–840. In Zimmerman JJ, Karriker LA, Ramirez A, Stevenson GW, Schwartz KJ (ed), Diseases of swine, 10th ed. Wiley-Blackwell, Hoboken, NJ. [Google Scholar]
- 9.Madsen AM, Kurdi I, Feld L, Tendal K. 2018. Airborne MRSA and total Staphylococcus aureus as associated with particles of different sizes on pig farms. Ann Work Expo Health 62:966–977. 10.1093/annweh/wxy065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Feld L, Bay H, Angen Ø, Larsen AR, Madsen AM. 2018. Survival of LA-MRSA in dust from swine farms. Ann Work Expo Health 62:147–156. 10.1093/annweh/wxx108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Comeau AM, Hatfull GF, Krisch HM, Lindell D, Mann NH, Prangishvili D. 2008. Exploring the prokaryotic virosphere. Res Microbiol 159:306–313. 10.1016/j.resmic.2008.05.001. [DOI] [PubMed] [Google Scholar]
- 12.Łobocka M, Hejnowicz MS, Dąbrowski K, Gozdek A, Kosakowski J, Witkowska M, Ulatowska MI, Weber-Dąbrowska B, Kwiatek M, Parasion S, Gawor J, Kosowska H, Głowacka A. 2012. Genomics of staphylococcal Twort-like phages–potential therapeutics of the post-antibiotic era. Adv Virus Res 83:143–216. 10.1016/B978-0-12-394438-2.00005-0. [DOI] [PubMed] [Google Scholar]
- 13.Xia G, Wolz C. 2014. Phages of Staphylococcus aureus and their impact on host evolution. Infect Genet Evol 21:593–601. 10.1016/j.meegid.2013.04.022. [DOI] [PubMed] [Google Scholar]
- 14.Uchiyama J, Takemura-Uchiyama I, Sakaguchi Y, Gamoh K, Kato S, Daibata M, Ujihara T, Misawa N, Matsuzaki S. 2014. Intragenus generalized transduction in Staphylococcus spp. by a novel giant phage. ISME J 8:1949–1952. 10.1038/ismej.2014.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang J, Zhao F, Sun H, Wang Q, Zhang C, Liu W, Zou L, Pan Q, Ren H. 2019. Isolation and characterization of the Staphylococcus aureus bacteriophage vB_SauS_SA2. AIMS Microbiol 5:285–307. 10.3934/microbiol.2019.3.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yuan Y, Gao M. 2017. Jumbo bacteriophages: an overview. Front Microbiol 8:403. 10.3389/fmicb.2017.00403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nováček J, Šiborová M, Benešík M, Pantůček R, Doškař J, Plevka P. 2016. Structure and genome release of Twort-like Myoviridae phage with a double-layered baseplate. Proc Natl Acad Sci U S A 113:9351–9356. 10.1073/pnas.1605883113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang LH, Farnet CM, Ehrlich KC, Ehrlich M. 1982. Digestion of highly modified bacteriophage DNA by restriction endonucleases. Nucleic Acids Res 10:1579–1591. 10.1093/nar/10.5.1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Klumpp J, Schmuki M, Sozhamannan S, Beyer W, Fouts DE, Bernbach V, Calendar R, Loessner MJ. 2014. The odd one out: bacillus ACT bacteriophage CP-51 exhibits unusual properties compared to related Spounavirinae W.Ph. and Bastille. Virology 462-463:299–308. 10.1016/j.virol.2014.06.012. [DOI] [PubMed] [Google Scholar]
- 20.Lavysh D, Sokolova M, Minakhin L, Yakunina M, Artamonova T, Kozyavkin S, Makarova KS, Koonin EV, Severinov K. 2016. The genome of AR9, a giant transducing Bacillus phage encoding two multisubunit RNA polymerases. Virology 495:185–196. 10.1016/j.virol.2016.04.030. [DOI] [PubMed] [Google Scholar]
- 21.Loenen WA, Raleigh EA. 2014. The other face of restriction: modification-dependent enzymes. Nucleic Acids Res 42:56–69. 10.1093/nar/gkt747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bryson AL, Hwang Y, Sherrill-Mix S, Wu GD, Lewis JD, Black L, Clark TA, Bushman FD. 2015. Covalent modification of bacteriophage T4 DNA inhibits CRISPR-Cas9. mBio 6:e00648. 10.1128/mBio.00648-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hoet PP, Coene MM, Cocito CG. 1992. Replication cycle of Bacillus subtilis hydroxymethyluracil-containing phages. Annu Rev Microbiol 46:95–116. 10.1146/annurev.mi.46.100192.000523. [DOI] [PubMed] [Google Scholar]
- 24.Stewart CR, Casjens SR, Cresawn SG, Houtz JM, Smith AL, Ford ME, Peebles CL, Hatfull GF, Hendrix RW, Huang WM, Pedulla ML. 2009. The genome of Bacillus subtilis bacteriophage SPO1. J Mol Biol 388:48–70. 10.1016/j.jmb.2009.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Darling AE, Mau B, Perna NT. 2010. progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One 5:e11147. 10.1371/journal.pone.0011147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ramsey J, Rasche H, Maughmer C, Criscione A, Mijalis E, Liu M, Hu JC, Young R, Gill JJ. 2020. Galaxy and Apollo as a biologist-friendly interface for high-quality cooperative phage genome annotation. PLoS Comput Biol 16:e1008214. 10.1371/journal.pcbi.1008214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jones P, Binns D, Chang HY, Fraser M, Li W, McAnulla C, McWilliam H, Maslen J, Mitchell A, Nuka G, Pesseat S, Quinn AF, Sangrador-Vegas A, Scheremetjew M, Yong SY, Lopez R, Hunter S. 2014. InterProScan 5: genome-scale protein function classification. Bioinformatics 30:1236–1240. 10.1093/bioinformatics/btu031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yuan Y, Gao M. 2016. Characteristics and complete genome analysis of a novel jumbo phage infecting pathogenic Bacillus pumilus causing ginger rhizome rot disease. Arch Virol 161:3597–3600. 10.1007/s00705-016-3053-y. [DOI] [PubMed] [Google Scholar]
- 29.Kiljunen S, Hakala K, Pinta E, Huttunen S, Pluta P, Gador A, Lonnberg H, Skurnik M. 2005. Yersiniophage phiR1-37 is a tailed bacteriophage having a 270 kb DNA genome with thymidine replaced by deoxyuridine. Microbiology (Reading) 151:4093–4102. 10.1099/mic.0.28265-0. [DOI] [PubMed] [Google Scholar]
- 30.Valentine RC, Shapiro BM, Stadtman ER. 1968. Regulation of glutamine synthetase. XII. Electron microscopy of the enzyme from Escherichia coli. Biochemistry 7:2143–2152. 10.1021/bi00846a017. [DOI] [PubMed] [Google Scholar]
- 31.Garneau JR, Depardieu F, Fortier L-C, Bikard D, Monot M. 2017. PhageTerm: a tool for fast and accurate determination of phage termini and packaging mechanism using next-generation sequencing data. Sci Rep 7:8292. 10.1038/s41598-017-07910-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lavysh D, Sokolova M, Slashcheva M, Förstner KU, Severinov K. 2017. Transcription profiling of bacillus subtilis cells infected with AR9, a giant phage encoding two multisubunit RNA polymerases. mBio 8:e02041-16. 10.1128/mBio.02041-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yuan Y, Gao M. 2016. Proteomic analysis of a novel bacillus jumbo phage revealing glycoside hydrolase as structural component. Front Microbiol 7:745–745. 10.3389/fmicb.2016.00745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mahony J, Alqarni M, Stockdale S, Spinelli S, Feyereisen M, Cambillau C, Sinderen D. 2016. Functional and structural dissection of the tape measure protein of lactococcal phage TP901-1. Sci Rep 6:36667. 10.1038/srep36667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Piuri M, Hatfull GF. 2006. A peptidoglycan hydrolase motif within the mycobacteriophage TM4 tape measure protein promotes efficient infection of stationary phase cells. Mol Microbiol 62:1569–1585. 10.1111/j.1365-2958.2006.05473.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rodriguez-Rubio L, Gutierrez D, Martinez B, Rodriguez A, Gotz F, Garcia P. 2012. The tape measure protein of the Staphylococcus aureus bacteriophage vB_SauS-phiIPLA35 has an active muramidase domain. Appl Environ Microbiol 78:6369–6371. 10.1128/AEM.01236-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Boulanger P, Jacquot P, Plancon L, Chami M, Engel A, Parquet C, Herbeuval C, Letellier L. 2008. Phage T5 straight tail fiber is a multifunctional protein acting as a tape measure and carrying fusogenic and muralytic activities. J Biol Chem 283:13556–13564. 10.1074/jbc.M800052200. [DOI] [PubMed] [Google Scholar]
- 38.Semenyuk PI, Moiseenko AV, Sokolova OS, Muronetz VI, Kurochkina LP. 2020. Structural and functional diversity of novel and known bacteriophage-encoded chaperonins. Int J Biol Macromol 157:544–552. 10.1016/j.ijbiomac.2020.04.189. [DOI] [PubMed] [Google Scholar]
- 39.Linder CH, Carlson K, Albertioni F, Söderström J, Påhlson C. 1994. A late exclusion of bacteriophage T4 can be suppressed by Escherichia coli GroEL or Rho. Genetics 137:613–625. 10.1093/genetics/137.3.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Birkeland NK. 1994. Cloning, molecular characterization, and expression of the genes encoding the lytic functions of lactococcal bacteriophage phi LC3: a dual lysis system of modular design. Can J Microbiol 40:658–665. 10.1139/m94-104. [DOI] [PubMed] [Google Scholar]
- 41.Sokolova ML, Misovetc I, V Severinov K. 2020. Multisubunit RNA polymerases of jumbo bacteriophages. Viruses 12:1064. 10.3390/v12101064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wu W, Thomas JA, Cheng N, Black LW, Steven AC. 2012. Bubblegrams reveal the inner body of bacteriophage ϕKZ. Science 335:182. 10.1126/science.1214120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Thomas JA, Benítez Quintana AD, Bosch MA, Coll De Peña A, Aguilera E, Coulibaly A, Wu W, Osier MV, Hudson AO, Weintraub ST, Black LW. 2016. Identification of essential genes in the Salmonella phage SPN3US reveals novel insights into giant phage head structure and assembly. J Virol 90:10284–10298. 10.1128/JVI.01492-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sokolova M, Borukhov S, Lavysh D, Artamonova T, Khodorkovskii M, Severinov K. 2017. A non-canonical multisubunit RNA polymerase encoded by the AR9 phage recognizes the template strand of its uracil-containing promoters. Nucleic Acids Res 45:5958–5967. 10.1093/nar/gkx264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.O'Flaherty S, Coffey A, Edwards R, Meaney W, Fitzgerald GF, Ross RP. 2004. Genome of Staphylococcal Phage K: a new lineage of Myoviridae infecting Gram-positive bacteria with a low G+C content. J Bacteriol 186:2862–2871. 10.1128/JB.186.9.2862-2871.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Belfort M, Bonocora RP. 2014. Homing endonucleases: from genetic anomalies to programmable genomic clippers. Methods Mol Biol 1123:1–26. 10.1007/978-1-62703-968-0_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tori K, Dassa B, Johnson MA, Southworth MW, Brace LE, Ishino Y, Pietrokovski S, Perler FB. 2010. Splicing of the mycobacteriophage Bethlehem DnaB intein: identification of a new mechanistic class of inteins that contain an obligate block F nucleophile. J Biol Chem 285:2515–2526. 10.1074/jbc.M109.069567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Elleuche S, Pöggeler S. 2010. Inteins, valuable genetic elements in molecular biology and biotechnology. Appl Microbiol Biotechnol 87:479–489. 10.1007/s00253-010-2628-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Taylor GK, Heiter DF, Pietrokovski S, Stoddard BL. 2011. Activity, specificity and structure of I-Bth0305I: a representative of a new homing endonuclease family. Nucleic Acids Res 39:9705–9719. 10.1093/nar/gkr669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.He S, Corneloup A, Guynet C, Lavatine L, Caumont-Sarcos A, Siguier P, Marty B, Dyda F, Chandler M, Ton Hoang B. 2015. The IS200/IS605 family and “peel and paste” single-strand transposition mechanism. Microbiol Spectr 3. 10.1128/microbiolspec.MDNA3-0039-2014. [DOI] [PubMed] [Google Scholar]
- 51.Kalvari I, Nawrocki EP, Ontiveros-Palacios N, Argasinska J, Lamkiewicz K, Marz M, Griffiths-Jones S, Toffano-Nioche C, Gautheret D, Weinberg Z, Rivas E, Eddy SR, Finn Robert D, Bateman A, Petrov AI. 2021. Rfam 14: expanded coverage of metagenomic, viral and microRNA families. Nucleic Acids Res 49:D192–D200. 10.1093/nar/gkaa1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW, Noble WS. 2009. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res 37:W202–W208. 10.1093/nar/gkp335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dai W, Hodes A, Hui WH, Gingery M, Miller JF, Zhou ZH. 2010. Three-dimensional structure of tropism-switching Bordetella bacteriophage. Proc Natl Acad Sci U S A 107:4347–4352. 10.1073/pnas.0915008107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liu M, Deora R, Doulatov SR, Gingery M, Eiserling FA, Preston A, Maskell DJ, Simons RW, Cotter PA, Parkhill J, Miller JF. 2002. Reverse transcriptase-mediated tropism switching in Bordetella bacteriophage. Science 295:2091–2094. 10.1126/science.1067467. [DOI] [PubMed] [Google Scholar]
- 55.Millman A, Bernheim A, Stokar-Avihail A, Fedorenko T, Voichek M, Leavitt A, Oppenheimer-Shaanan Y, Sorek R. 2020. Bacterial retrons function in anti-phage defense. Cell 183:1551–1561.e12. 10.1016/j.cell.2020.09.065. [DOI] [PubMed] [Google Scholar]
- 56.Wang C, Villion M, Semper C, Coros C, Moineau S, Zimmerly S. 2011. A reverse transcriptase-related protein mediates phage resistance and polymerizes untemplated DNA in vitro. Nucleic Acids Res 39:7620–7629. 10.1093/nar/gkr397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bailly-Bechet M, Vergassola M, Rocha E. 2007. Causes for the intriguing presence of tRNAs in phages. Genome Res 17:1486–1495. 10.1101/gr.6649807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mira A, Ochman H, Moran NA. 2001. Deletional bias and the evolution of bacterial genomes. Trends Genet 17:589–596. 10.1016/s0168-9525(01)02447-7. [DOI] [PubMed] [Google Scholar]
- 59.Mašlaňová I, Stříbná S, Doškař J, Pantůček R. 2016. Efficient plasmid transduction to Staphylococcus aureus strains insensitive to the lytic action of transducing phage. FEMS Microbiology Lett 363:fnw211. 10.1093/femsle/fnw211. [DOI] [PubMed] [Google Scholar]
- 60.Rees PJ, Fry BA. 1981. The morphology of staphylococcal bacteriophage K and DNA metabolism in infected Staphylococcus aureus. J Gen Virol 53:293–307. 10.1099/0022-1317-53-2-293. [DOI] [PubMed] [Google Scholar]
- 61.Fineran PC, Petty NK, Salmond GPC. 2009. Transduction: host DNA Transfer by Bacteriophages, p 666–679. In Schaechter M (ed), Encyclopedia of microbiology (third edition). Academic Press, Oxford, UK. [Google Scholar]
- 62.Scharn CR, Tenover FC, Goering RV. 2013. Transduction of staphylococcal cassette chromosome mec elements between strains of Staphylococcus aureus. Antimicrob Agents Chemother 57:5233–5238. 10.1128/AAC.01058-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Keck T, Leiacker R, Riechelmann H, Rettinger G. 2000. Temperature profile in the nasal cavity. Laryngoscope 110:651–654. 10.1097/00005537-200004000-00021. [DOI] [PubMed] [Google Scholar]
- 64.Kropinski AM, Mazzocco A, Waddell TE, Lingohr E, Johnson RP. 2009. Enumeration of bacteriophages by double agar overlay plaque assay. Methods Mol Biol 501:69–76. 10.1007/978-1-60327-164-6_7. [DOI] [PubMed] [Google Scholar]
- 65.Adams MH. 1959. Bacteriophages. Interscience Publishers, New York, NY. [Google Scholar]
- 66.Gill JJ, Svircev AM, Smith R, Castle AJ. 2003. Bacteriophages of Erwinia amylovora. Appl Environ Microbiol 69:2133–2138. 10.1128/AEM.69.4.2133-2138.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Xie Y, Wahab L, Gill JJ. 2018. Development and validation of a microtiter plate-based assay for determination of bacteriophage host range and virulence. Viruses 10:189. 10.3390/v10040189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Summer EJ. 2009. Preparation of a phage DNA fragment library for whole genome shotgun sequencing. Methods Mol Biol 502:27–46. 10.1007/978-1-60327-565-1_4. [DOI] [PubMed] [Google Scholar]
- 69.Gill JJ, Berry JD, Russell WK, Lessor L, Escobar-Garcia DA, Hernandez D, Kane A, Keene J, Maddox M, Martin R, Mohan S, Thorn AM, Russell DH, Young R. 2012. The Caulobacter crescentus phage phiCbK: genomics of a canonical phage. BMC Genomics 13:542. 10.1186/1471-2164-13-542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Andrew S. 2010. FastQC: a quality control tool for high throughput sequence data. http://www.bioinformatics.babraham.ac.uk/projects/fastqc.
- 71.Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, Pyshkin AV, Sirotkin AV, Vyahhi N, Tesler G, Alekseyev MA, Pevzner PA. 2012. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol 19:455–477. 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Delcher AL, Harmon D, Kasif S, White O, Salzberg SL. 1999. Improved microbial gene identification with GLIMMER. Nucleic Acids Res 27:4636–4641. 10.1093/nar/27.23.4636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Noguchi H, Taniguchi T, Itoh T. 2008. MetaGeneAnnotator: detecting species-specific patterns of ribosomal binding site for precise gene prediction in anonymous prokaryotic and phage genomes. DNA Res 15:387–396. 10.1093/dnares/dsn027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Laslett D, Canback B. 2004. ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res 32:11–16. 10.1093/nar/gkh152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.The UniProt Consortium. 2019. UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res 47:D506–D515. 10.1093/nar/gky1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL. 2009. BLAST+: architecture and applications. BMC Bioinformatics 10:421. 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zimmermann L, Stephens A, Nam S-Z, Rau D, Kübler J, Lozajic M, Gabler F, Söding J, Lupas AN, Alva V. 2018. A completely reimplemented MPI bioinformatics toolkit with a new HHpred server at its core. J Mol Biol 430:2237–2243. 10.1016/j.jmb.2017.12.007. [DOI] [PubMed] [Google Scholar]
- 78.Krogh A, Larsson B, von Heijne G, Sonnhammer EL. 2001. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol 305:567–580. 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- 79.Sullivan MJ, Petty NK, Beatson SA. 2011. Easyfig: a genome comparison visualizer. Bioinformatics 27:1009–1010. 10.1093/bioinformatics/btr039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Stanczak-Mrozek KI, Manne A, Knight GM, Gould K, Witney AA, Lindsay JA. 2015. Within-host diversity of MRSA antimicrobial resistances. J Antimicrob Chemother 70:2191–2198. 10.1093/jac/dkv119. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1<br>. Download JVI.02391-20-s0001.xlsx, XLSX file, 0.02 MB (25.1KB, xlsx)