

ABSTRACT
Cytotoxic CD4 T lymphocytes (CD4-CTL) are important in antiviral immunity. For example, we have previously shown that in mice, CD4-CTL are important to control ectromelia virus (ECTV) infection. How viral infections induce CD4-CTL responses remains incompletely understood. We demonstrate here that not only ECTV but also vaccinia virus and lymphocytic choriomeningitis virus induce CD4-CTL, though the response to ECTV is stronger. Using ECTV, we also demonstrate that in contrast to CD8-CTL, CD4-CTL differentiation requires constant virus replication and ceases once the virus is controlled. We also show that major histocompatibility complex class II molecules on CD11c+ cells are required for CD4-CTL differentiation and for mousepox resistance. Transcriptional analysis indicated that antiviral CD4-CTL and noncytolytic T helper 1 (Th1) CD4 T cells have similar transcriptional profiles, suggesting that CD4-CTL are terminally differentiated classical Th1 cells. Interestingly, CD4-CTL and classical Th1 cells expressed similar mRNA levels of the transcription factors ThPOK and GATA-3, necessary for CD4 T cell linage commitment, and Runx3, required for CD8 T cell development and effector function. However, at the protein level, CD4-CTL had higher levels of the three transcription factors, suggesting that further posttranscriptional regulation is required for CD4-CTL differentiation. Finally, CRISPR/Cas9-mediated deletion of Runx3 in CD4 T cells inhibited CD4-CTL but not classical Th1 cell differentiation in response to ECTV infection. These results further our understanding of the mechanisms of CD4-CTL differentiation during viral infection and the role of posttranscriptionally regulated Runx3 in this process.
IMPORTANCE While it is well established that cytotoxic CD4 T cells (CD4-CTLs) directly contribute to viral clearance, it remains unclear how CD4-CTL are induced. We now show that CD4-CTLs require sustained antigen presentation and are induced by CD11c-expressing antigen-presenting cells. Moreover, we show that CD4-CTLs are derived from the terminal differentiation of classical T helper 1 (Th1) subset of CD4 cells. Compared to Th1 cells, CD4-CTLs upregulate protein levels of the transcription factors ThPOK, Runx3, and GATA-3 posttranscriptionally. Deletion of Runx3 in differentiated CD4 T cells prevents induction of CD4-CTLs but not classical Th1 cells. These results advance our knowledge of how CD4-CTLs are induced during viral infection.
KEYWORDS: CD4 T cells, Runx3, cytotoxic CD4 T cells, ectromelia virus, immune mechanisms, immune response
INTRODUCTION
Cytotoxic lymphocytes play a key role in eliminating virus-infected and tumor cells and in the pathogenesis of IgG4-related disease through direct cytolysis (1–5). The classically defined cytotoxic lymphocytes are CD8 T cells and natural killer (NK) cells, but CD4 T cells can also have cytolytic function. The underappreciated subset of CD4 cytotoxic T lymphocytes (CD4-CTLs) has been observed in human blood samples for a number of chronic viral infections, including human immunodeficiency virus (6, 7), hepatitis viruses (8), human cytomegalovirus (7, 9) and also following vaccinia virus immunization (10). CD4-CTL responses have also been shown in mice following virus infection with ectromelia virus (ECTV) (1), influenza A virus (IAV) (2), lymphocytic choriomeningitis virus (LCMV) (3), and gammaherpesvirus 68 (11), as well as in murine models of melanoma (4, 5).
We previously demonstrated that CD4-CTLs contribute to the direct killing of infected cells in vivo in mice infected with the DNA poxvirus ECTV, a natural mouse pathogen and causative agent of mousepox (1). CD4 T cells specifically required the cytolytic molecule, perforin, to help eliminate ECTV (1). Likewise, others demonstrated that CD4 T cells expressing perforin can protect mice from IAV lethality (2). Thus, similar to CD8 T cells (12), CD4-CTLs mediate apoptosis of target cells primarily through release of cytolytic granzymes and perforin. Interestingly, CD4-CTLs targeted major histocompatibility complex class II (MHC-II)-expressing cells that were infected (1), in contrast to CD8 T cells and NK cells, which utilize MHC class I for activation or inhibition, respectively, suggesting a unique target cell population for the CD4-CTL response. However, despite the important role of CD4-CTLs during viral infection, the mechanism for their induction in vivo remains unclear.
In the present study, we determine that multiple acute viral infections can induce CD4-CTLs, although the response is transient in comparison to the CD8-CTL response. We also show that MHC-II on CD11c+ cells, most likely dendritic cells (DCs), are required to for optimal total and cytotoxic CD4 T cell responses. We further determined that CD4-CTLs have a transcriptional profile similar to that of activated noncytotoxic T helper 1 (Th1) CD4 T cells (classical Th1 cells) but vastly different compared to naive CD4 T cells. These results indicate a singular differentiation pathway in which activated classical Th1 cells are further terminally differentiated to produce CD4-CTLs rather than arising from a separate unique pathway. We also demonstrate that CD4-CTLs posttranscriptionally upregulate protein levels of the transcription factors ThPOK and GATA-3, required for CD4 T cell differentiation, and Runx3, which is necessary for CD8 T cell commitment (13, 14). Finally, we also show that deletion of Runx3, known to be necessary for CD8 T cell maintenance (15), NK cell maturation (16), and intestinal epithelial CD4 T cell programing (17, 18), prevents CD4-CTL differentiation but does not alter the overall CD4 T cell response. These findings define several of the mechanisms involved in antiviral CD4-CTL differentiation.
RESULTS
CD4-CTLs are induced following viral infection and require the presence of live virus for optimal accumulation.
We have previously demonstrated that infection of C57BL/6 (B6) with ECTV elicits robust CD4-CTL responses in the liver and spleen following viral dissemination (1). The subset of CD4-CTLs can be readily identified via flow cytometry by their expression of activation marker CD44 and the cytolytic granule granzyme B (GzmB; Fig. 1A). In addition, a portion of CD4 T cells expressed perforin (Fig. 1A), which together with GzmB can mediate cytolysis. The frequency and total number of GzmB or perforin expressing CD4-CTLs peaked at 8 days postinfection (dpi) with ECTV in both the liver and the spleen (Fig. 1B and C). Of note, the expression of GzmB in CD4 T cells was transient. For example, in the liver, ∼15%, ∼39%, ∼20%, and ∼14% of CD4 T cells were GzmB+ at 7, 8, 9, and 10 dpi, respectively.
FIG 1.
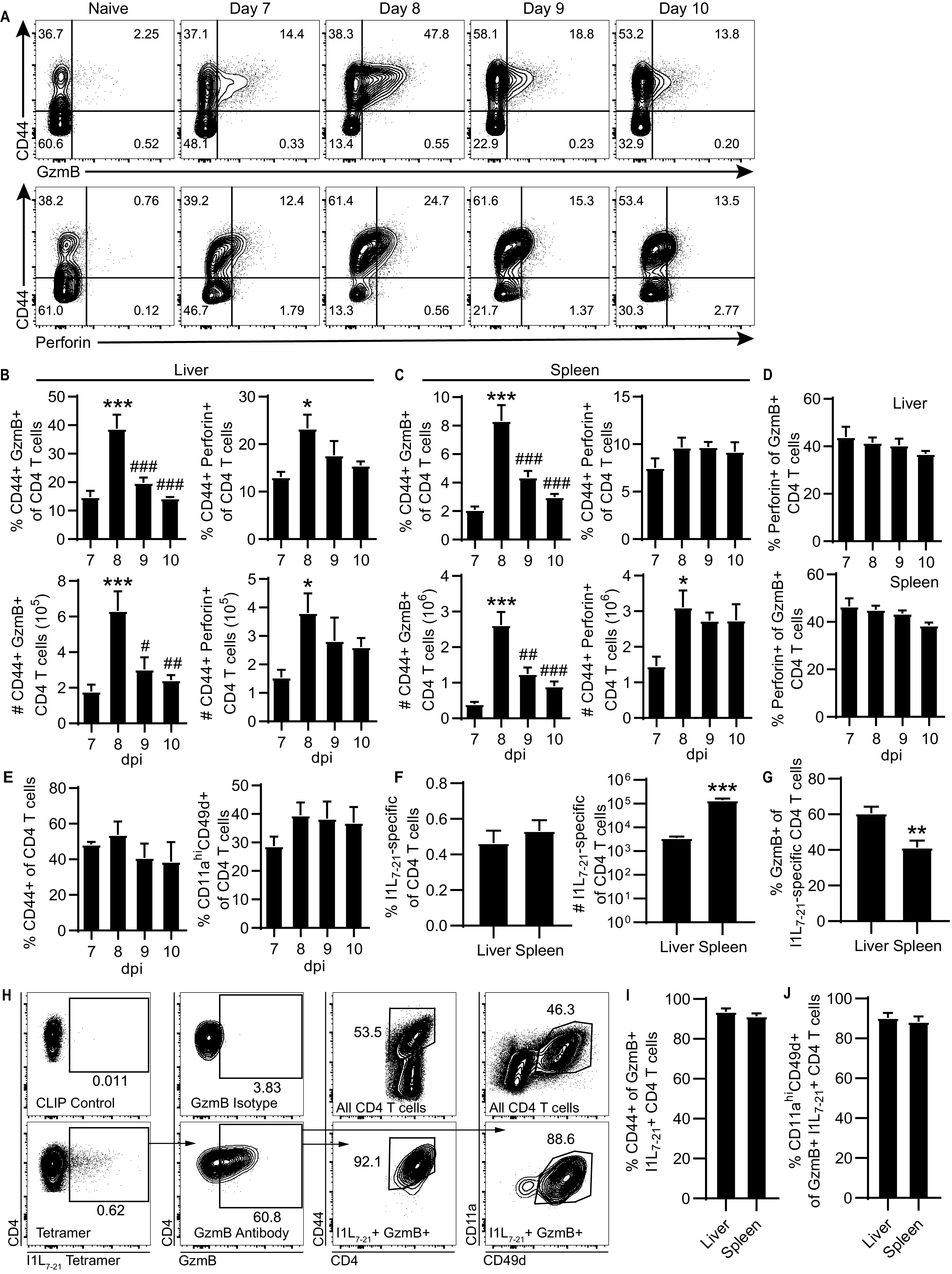
Transient CD4-CTL response induced following ECTV infection. B6 mice were infected with 3,000 PFU of ECTV in the footpad. (A) Representative flow plots of CD44 by GzmB (top panels) or perforin (bottom panels) gated on CD4 T cells from the liver at the indicated time points after infection. The percentages (top panels) and total numbers (bottom panels) of GzmB+ and perforin+ CD4 T cells in the liver (B) or spleen (C) are shown for the indicated dpi. (D) Frequency perforin+ cells of GzmB+ CD4 T cells. (E) Percentage activated CD44+ or CD11ahi CD49d+ of CD4 T cells. (F) Frequency and total number of I1L7-21-specific CD4 T cells in the liver and spleen at 8 dpi. (G) Percent GzmB+ of I1L7-21-specific CD4 T cells at 8 dpi in the liver and spleen. (H) Staining of I1L7-21 tetramer, GzmB, CD44, CD11a, and CD49d at 8 dpi in the liver. Flow plots are concatenated from four mice. The top panels show control stains for determination of gate placement, as indicated by text. (I and J) CD44+ (I) or CD11ahi CD49d+ (J) frequency of GzmB+I1L7-21-specific CD4 T cells at 8 dpi in the liver and spleen. Asterisks (*) denote statistical differences compared to 7 dpi, whereas number symbols (#) denote statistical differences compared to 8 dpi. Compiled data are displayed as means ± the standard errors of the mean (SEM) representing two independent experiments (A to E, n = 6; F to J, n = 9).
Approximately 40% of the CD4 T cells that expressed GzmB also coexpressed perforin (Fig. 1D). The magnitude of the CD4-CTL response for both frequency and total number was greater in the liver than in the spleen on all days analyzed. However, there was no significant change in the frequency of activated CD4 T cells from 7 to 10 dpi, as measured by CD44 expression or upregulation of integrins CD11a and CD49d, which denote antigen-experienced T cells (Fig. 1E) (19). Importantly, 40 to 60% of virus-specific CD4 T cells corresponding to the I1L7-21 epitope (QLIFNSISARALKAY) (1) expressed GzmB at 8 dpi (Fig. 1F to H). While the frequency of I1L7-21-specific CD4 T cells was similar between the liver and spleen, the total number was significantly greater in the spleen (Fig. 1F). The percentage of I1L7-21-specific CD4 T cells expressing GzmB at 8 dpi was significantly greater in the liver compared to the spleen (Fig. 1G). Furthermore, approximately 90% of I1L7-21-specific GzmB+ CD4 T cells were CD44+, CD11ahi, and CD49d+ (Fig. 1H to J). These results show that virus-specific CD4-CTL response correlates with the overall antigen-experienced CD4 T cell response against ECTV.
The CD8-CTL response also peaked at 8 dpi (Fig. 2A to C) and was larger than the CD4-CTL response (compare to Fig. 1B and C), with a significant increase in the frequency of activated CD44+ CD8 T cells at 8 and 9 dpi (Fig. 2D). Of note, in contrast to the transient response of CD4-CTLs (Fig. 1B-C), CD8-CTL maintained high frequencies from 7 to 10 dpi (Fig. 2A to C). Together, the data in Fig. 1 and 2 suggested that the presence of CD4-CTLs, but not of CD8-CTLs, may be driven by high virus loads.
FIG 2.

Robust CD8-CTL response following ECTV infection. B6 mice were infected with 3,000 PFU of ECTV in the footpad. (A) Representative flow plot CD44 by GzmB (top) or perforin (bottom) gated on CD8 T cells from the liver at the indicated time points after infection. (B and C) Percentages (top) and total numbers (bottom) of GzmB+ and perforin+ CD8 T cells in the liver (B) or spleen (C) at the indicated dpi. (D) Percent activated CD44+ or CD11ahi CD49d+ CD8 T cells. Asterisks (*) and number symbols (#) are used to denote statistical differences compared to 7 or 8 dpi, respectively. Compiled data are displayed as means ± the SEM representing two independent experiments (n = 6).
To test this hypothesis, infected mice were administered the antiviral drug cidofovir, which effectively reduces ECTV titers in mice, at 3 dpi (1, 20). Cidofovir treatment resulted in almost undetectable virus loads at 8 dpi (Fig. 3A and B) but did not reduce the total CD4 and CD8 T cell responses as indicated by CD44 expression (Fig. 3C and D). However, cidofovir-treated mice had a significant reduction (∼66% in liver and ∼41% in spleen) in frequency of GzmB+ CD4-CTLs (Fig. 3E). Conversely, the frequency of GzmB+ CD8-CTLs was not affected in the liver and spleen (Fig. 3F). These results suggest that, in contrast to noncytolytic CD4s and CD8-CTLs, CD4-CTLs require the presence of virus-infected cells and likely constant TCR signaling prior to the peak of the CD4 T cell response for optimal cytotoxic programming.
FIG 3.

CD4-CTL response requires late viral replication. ECTV-infected mice were treated with PBS or 600 μg of cidofovir intraperitoneally at 3 dpi. (A and B) Virus titers in the liver (A) and spleen (B) at 8 dpi. (C and D) Percentage of CD44+ in CD4 (C) or CD8 (D) T cells in the liver and spleen at 8 dpi. (E and F) Frequency of GzmB+ in CD4 T cells (E) and CD8 T cells (F) in the liver and spleen at day 8 after infection. Unpaired t tests were conducted on all graphs, and asterisks (*) denote statistical differences. Compiled data are displayed as means ± the SEM representing three independent experiments (n = 12 to 15).
We questioned whether, similar to ECTV, other viral infections could also induce large CD4-CTL responses. Mice were infected in the footpad with the RNA arenavirus LCMV, which has been previously shown to induce CD4 T cells capable of lysing target cells (3). We also evaluated another poxvirus, vaccinia virus (VACV), as well as the natural mouse pathogen murine cytomegalovirus (MCMV). At 8 dpi, all viruses except MCMV induced significantly increased CD4 T cell responses in the liver and spleen, as indicated by CD44 or CD11a and CD49d expression. However, ECTV infection resulted in a significantly higher CD4 T cell response compared to the other viruses (Fig. 4A and B). Accordingly, infection with ECTV also resulted in a higher frequency and total number of CD4-CTLs in both the liver and the spleen (Fig. 4C to E). Of note, mice infected with LCMV or VACV had detectable CD4-CTL responses in the liver, as indicated by frequency (Fig. 4D). While the total number of CD4-CTLs in the liver and spleen following LCMV or VACV infection was not increased compared to naive mice by one-way analysis of variance (ANOVA), a more direct unpaired t test comparison indicated significant increases in both groups (LCMV, P < 0.001; VACV, P < 0.01; Fig. 4E). In contrast, the frequency of CD8-CTLs in the liver and spleen was significantly greater in all infection groups compared to naive mice (Fig. 4D). ECTV also generated the greatest frequency and total number of CD8-CTLs in both the liver and the spleen compared to all other groups. Furthermore, the relative frequency of CD4-CTLs and CD8-CTLs in relation to total activated CD44hi T cells was the highest following ECTV infection and significantly greater than all other infections in both the liver and the spleen (Fig. 4F and G). To account for potentially altered kinetics and peak magnitude of the T cell response, we examined the ratio of percentage CD4-CTLs to CD8-CTLs at 8 dpi. Relative to the other viruses, ECTV infection induced the greatest ratio of CD4-CTLs in relation to the CD8-CTL response (Fig. 4H). These results indicate that different acute viral infections can induce CD4-CTLs. However, after footpad infection, ECTV promotes the largest total T cell and CTL responses (for both CD4 and CD8 T cells) when measured at 8 dpi. Whether the other viruses could induce stronger responses at other times postinfection was not tested. Nevertheless, our data indicate that ECTV is an excellent model to study the immune factors that induce CD4-CTLs.
FIG 4.
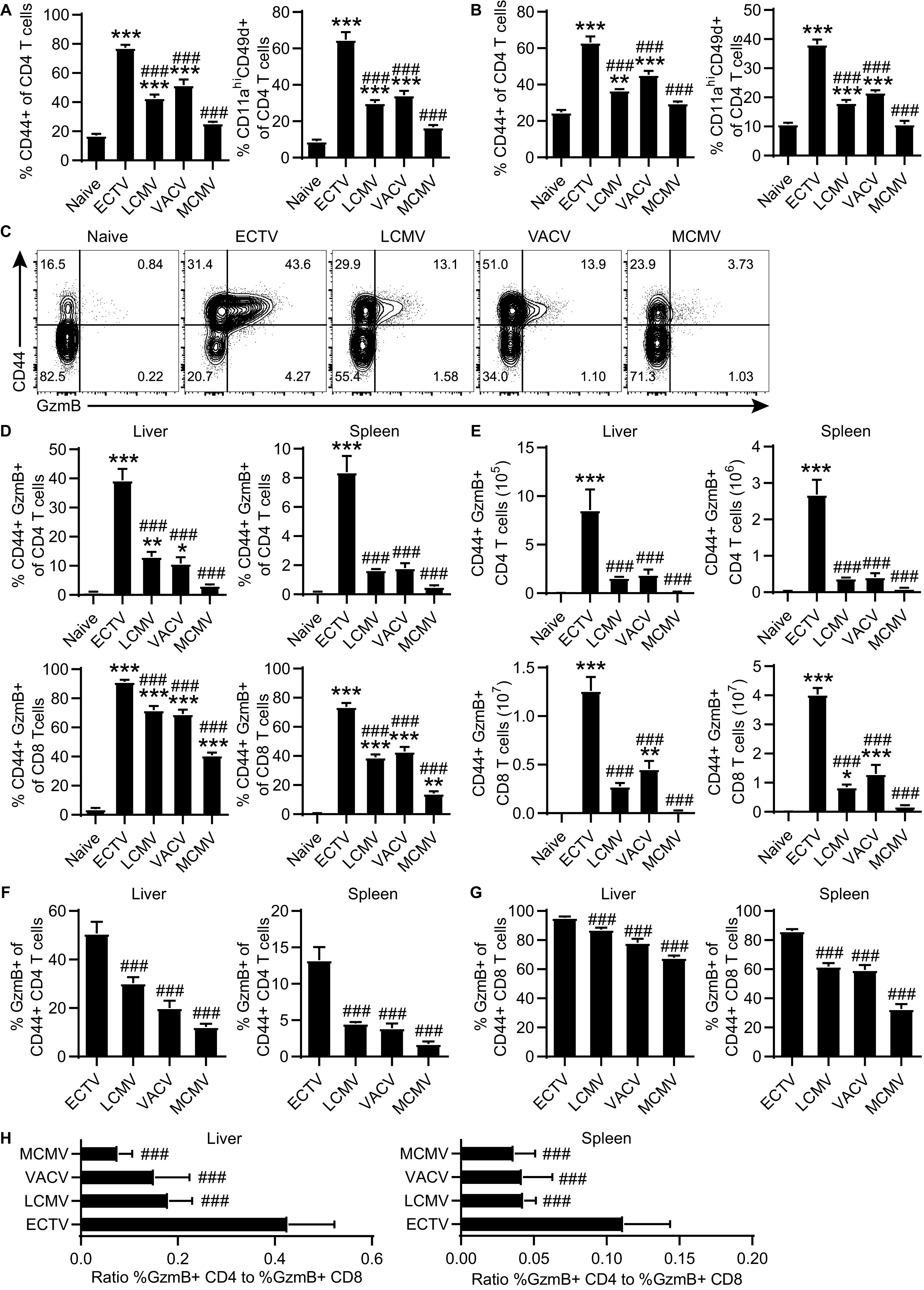
CD4-CTLs are induced by different infections. B6 mice were infected with ECTV, LCMV, VACV, or MCMV. (A and B) Frequency of CD44+ or CD11ahi CD49d+ of CD4 T cells in the liver (A) and spleen (B) at day 8 after infection. (C) Representative flow plots of CD44 by GzmB staining in liver at 8 dpi. (D) Frequency and (E) total number of GzmB+ CD4 T cells (top row) or CD8 T cells (bottom row) in the liver and spleen. (F and G) Percent of GzmB+ in CD44+ CD4 (F) and CD44+ CD8 (G) T cells in the liver and spleen at 8 dpi. (H) Ratio of frequency GzmB+ CD4 T cells to percentage GzmB+ CD8 T cells in the liver and spleen at 8 dpi. Asterisks (*) denote statistical differences compared to naive group, while number symbols (#) are comparisons to the ECTV-infected group. The data are displayed as means ± the SEM compiled from two independent experiments (n = 6).
MHC-II on CD11c+ cells is required for the CD4 T cell response and protection against mousepox.
MHC-II is primarily expressed by DCs, B cells, and monocytes/macrophages. Interestingly, when comparing the different viral infections, the number of DCs and monocytes/macrophages, but not B cells, directly correlated with the size of the CD4-CTL response in the liver (Fig. 4C and 5A). To determine whether MHC-II on specific subsets of antigen-presenting cells is required to induce CD4-CTLs, we crossed MHC-II floxed mice (MHC-IIfl/fl) with mouse strains expressing cre recombinase under specific promoters. Cre expressed from the Vav1 (Vav1-cre), CD11c (Itgax-cre), lysozyme 2 (Lyz2-cre), or CD79a (Mb1-cre) is known to, respectively, delete floxed genes from all hematopoietic cells, CD11c+ cells, monocytes/macrophages, or B cells (21–24). Only cre-expressing mice with a normal CD4/CD8 ratio of T cells in the blood were used for experimentation to ensure differences were not due to an absence of CD4 T cells. Analysis of the target antigen-presenting cell populations revealed almost complete ablation of MHC-II expression on CD11c+ cells in Itgax-cre MHC-IIfl/fl and B cells in Mb1-cre MHC-IIfl/fl mice (Fig. 5B and C). Lyz2-cre MHC-IIfl/fl mice had a greater, but incomplete deletion of MHC-II on monocytes and macrophages (Fig. 5C). In addition, we also observed leakiness of the cre-lox system across the three strains where the frequency of MHC-II-expressing cells was reduced in the nontargeted antigen-presenting cell subsets (Fig. 5C). However, MHC-II expression was not completely absent in other antigen-presenting cell subsets that were not being targeted by the gene expressing cre recombinase.
FIG 5.
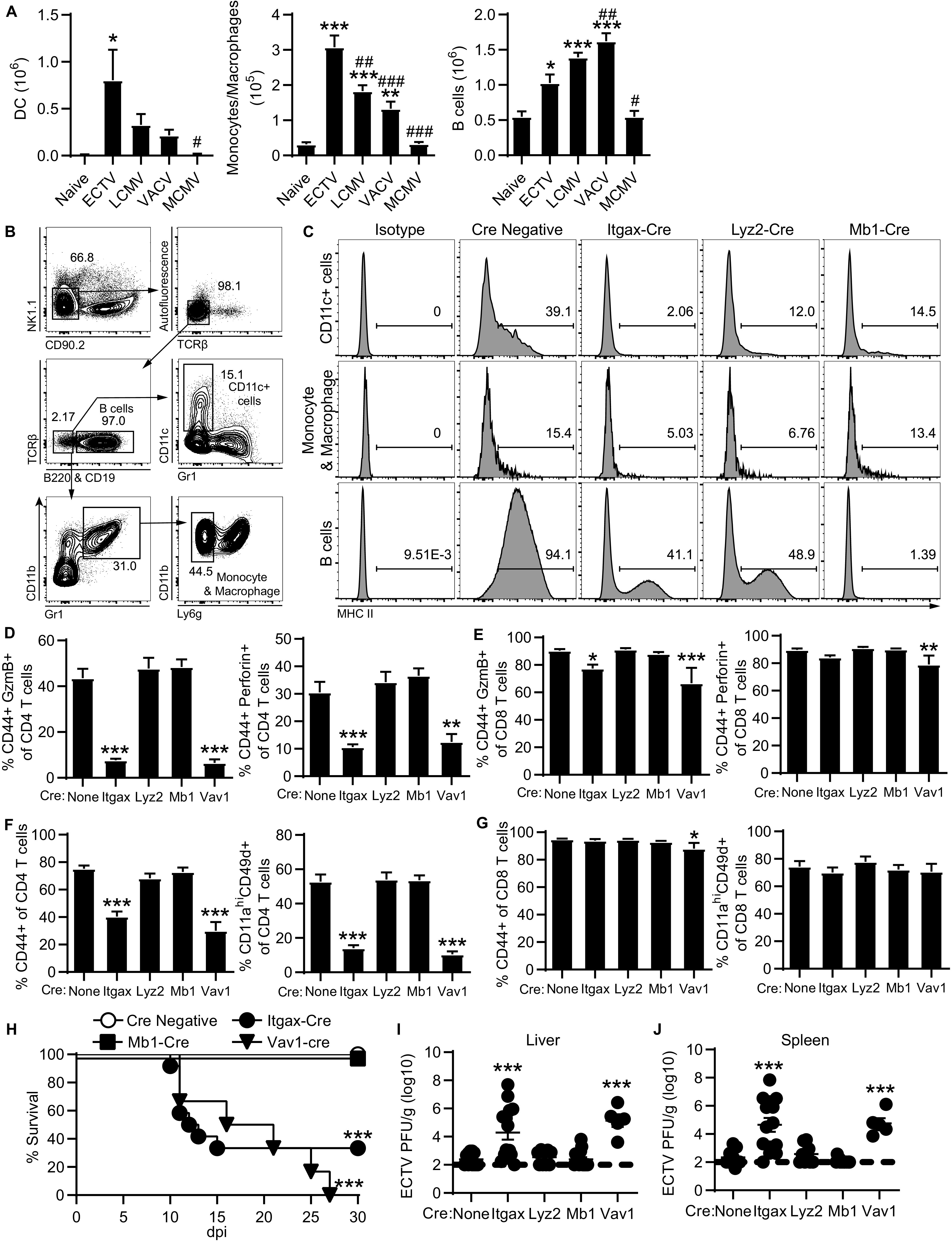
MHC-II on CD11c+ cells induces the overall CD4 T cell response and CD4-CTLs. (A) Total number of DCs, monocytes/macrophages, and B cells in the liver from mice infected with ECTV, LCMV, VACV, or MCMV. Asterisks (*) denote statistical differences compared to naive group, while number symbols (#) are comparisons to the ECTV-infected group. The data are displayed as means ± the SEM compiled from two independent experiments (n = 6). (B) Concatenated flow plot representing gating for antigen-presenting cell subsets from spleens of 4 naive MHC-IIfl/fl mice. MHC-IIfl/fl mice were bred with indicated cre-expressing strains. (C) Histograms of MHC-II expression on CD11c+ cells, monocytes and macrophages, or B cells in spleens from crossed naive mice. The data are concatenated from four mice in each group. (D to J) The crossed cre-lox mice were challenged with ECTV and analyzed at 8 dpi. Groups are compared to the MHC-IIfl/fl mice with no cre expression (none) for statistical analysis. (D and E) GzmB+ or perforin+ frequency of CD4 T cells (D) and CD8 T cells (E) in the liver. (F and G) Frequency of activated CD44+ or CD11ahi CD49d+ of CD4 T cells (F) and CD8 T cells (G). (H) Survival curve tracking mice for 30 days following infection (cre negative, n = 38; Itgax-cre, n = 12; Mb1-cre, n = 10; Vav1-cre, n = 6). (I and J) Virus titers in the livers (I) and spleens (J) of infected mice. Asterisks (*) denote statistical differences compared to naive mice, while number symbols (#) denote differences compared to the ECTV-infected group. Compiled data are displayed as means ± the SEM from two to three independent experiments (None, n = 12; Itgax, n = 14; Lyz2, n = 9; Mb1, n = 14; Vav1, n = 5).
After ECTV infection, both Itgax-cre MHC-IIfl/fl and Vav1-cre MHC-IIfl/fl mice had a significantly reduced CD4-CTL response in the liver at 8 dpi compared to control MHC-IIfl/fl mice (Fig. 5D). The CD4-CTL response was not altered in the Lyz2-cre- or Mb1-cre expressing strains. This result indicates that MHC-II on CD11c+ cells, most likely DCs, is necessary for the CD4-CTL response following ECTV infection. Itgax-cre MHC-IIfl/fl mice also had a statistically significant, albeit relatively small, reduction in the frequency of GzmB+ CD8-CTLs (Fig. 5E). However, deletion of MHC-II on CD11c+ cells also resulted in a significant decrease to the overall percentage of activated CD4 T cells (Fig. 5F). In contrast, there was no significant change to the overall CD8 T cell response of Itgax-cre MHC-IIfl/fl mice (Fig. 5G). These results suggest that CD11c+ cells induce the CD4 T cell response following ECTV infection, thereby affecting the overall magnitude of the CD4-CTL response. Despite having strong CD8 T cell responses, most Itgax-cre- and Vav1-cre-expressing mice succumbed to ECTV infection (66 and 100% mortality, respectively; Fig. 5H), whereas Mb1-cre MHC-IIfl/fl mice were resistant. Furthermore, Itgax-cre and Vav1-cre MHC-IIfl/fl mice had significantly increased virus titers in the livers and spleens at 8 dpi compared to MHC-IIfl/fl controls (Fig. 5I and J). Mortality is not likely to be mediated by altered antibody responses, since B cell-deficient mice only succumb to ECTV after approximately 2 months of infection (1). In addition, the deletion of MHC-II on nontargeted antigen-presenting cell populations of Lyz2-cre MHC-IIfl/fl or Mb1-cre MHCIIfl/fl mice was insufficient to significantly alter the CD4-CTL and overall CD4 T cell response. Only the near complete ablation of MHC-II on CD11c+ cells in Itgax-cre MHC-IIfl/fl mice caused impairment of the CD4 T cell response and mortality following ECTV infection. These data indicate that the total and cytotoxic CD4 T cell responses are induced by CD11c+ cells and that effector CD4 T cells are required for protection against ECTV infection.
CD4-CTLs and activated noncytolytic classical Th1 CD4 T cells have similar transcriptional profiles.
While the magnitude of the CD4-CTL response directly correlated with the overall CD4 T cell response, it was unclear whether CD4-CTLs represented a distinct subset of T helper cell differentiation. Therefore, we performed total RNA sequencing on nonactivated (CD44lo), activated noncytotoxic (CD44hi GzmB–), and cytotoxic (CD44hi GzmB+) CD4 T cells (Fig. 6A). To identify CTLs during live cell sorting, we used transgenic reporter mice in which GzmB is fused to the fluorescent protein tdTomato (GzmB-tdTomato) (25). For controls, CD44lo and CD44hi T cells from naive mice were included in the analysis along with CD8-CTLs from infected mice. The T cell subsets were sorted at 7 dpi to analyze a time point prior to the peak of the CD4-CTL response as CD4 T cells continue to acquire cytolytic function (Fig. 1). Heatmap cluster analysis of all sequenced genes revealed that noncytotoxic classical Th1 (CD44hi GzmB–) and cytotoxic (CD44hi GzmB+) CD4 T cells from infected mice were transcriptionally similar and that both of these groups were also comparable to cytotoxic CD8 T cells (Fig. 6B). The remaining CD4 and CD8 T cell groups from naive mice and non-activated CD44lo CD4 T cells from infected mice formed a separate cluster, suggesting that their transcriptional profiles were similar. These data suggest that a similar transcriptional circuitry is induced in all activated T cell subsets compared to resting T cells, whether antigen experienced or not.
FIG 6.
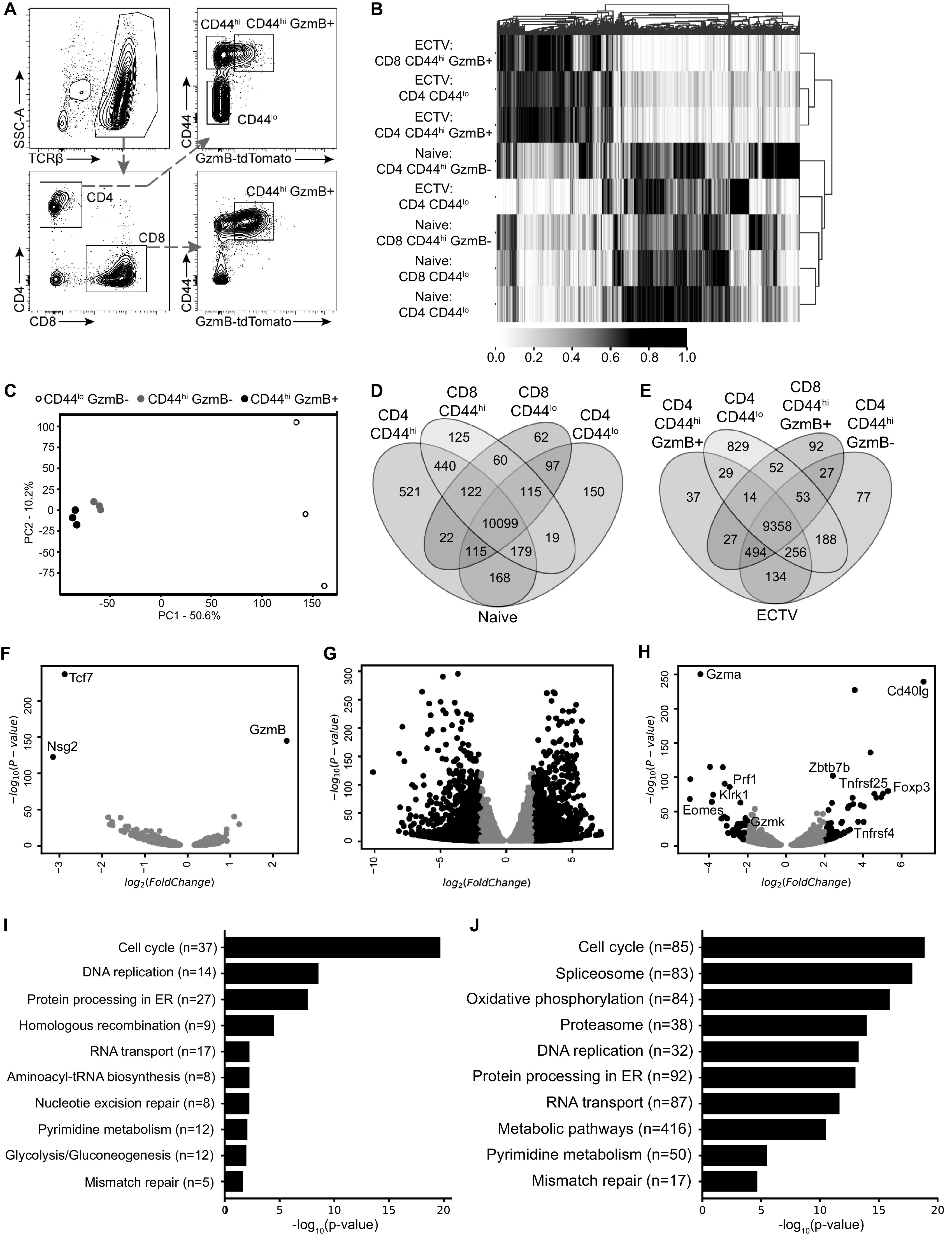
Activated noncytotoxic and cytotoxic CD4 T cells have similar transcriptional profiles. GzmB-tdTomato reporter mice were infected with ECTV and 10 livers were processed into a single cell suspension, followed by magnetic purification for T cells at 7 dpi. RNA samples for each cell population were collected a total of three times across independent experiments. (A) Representative flow plot illustrating gating strategy used to sort and isolate T cell populations based on CD44 and GzmB-tdTomato expression. (B) Heatmap with cluster analysis with normalized values for sequenced genes from sorted T cell populations. (C) PCA for sorted CD4 T cell populations from infected mice. The percentages indicate the total variance represented by each component. (D and E) Venn diagram analysis of the indicated T cell populations from naive mice (D) or ECTV-infected mice (E). (F to H) Volcano plots of differentially expressed genes comparing CD44hi GzmB+ and CD44hi GzmB– CD4 T cells (F), CD44hi GzmB+ and CD44lo CD4 T cells (G), and CD44hi GzmB+ CD4 T cells and CD44hi GzmB+ CD8 T cells (H). Comparisons were done on samples from infected mice. Genes of interest are shown as black dots using cutoffs of log2(FoldChange) = 2 and an adjusted P value of <0.05. Gray dots represent genes that did not meet cutoff values. (I and J) Analysis showing top 10 enriched pathways using KEGG database ranked by adjusted P value comparing CD44hi GzmB+ and CD44hi GzmB– CD4 T cells (I) and CD44hi GzmB+ and CD44lo CD4 T cells (J).
Principal-component analysis (PCA) of only the CD4 T cell subsets from infected mice demonstrated a clear similarity between activated CD44hi CD4 T cells, regardless of GzmB expression (Fig. 6C). In contrast, the nonactivated CD44lo CD4 T cells clustered separately, suggesting a vastly different transcriptional profile compared to the virus-specific CD4 T cells. A Venn diagram of differentially expressed genes (P < 0.05) further corroborated the heatmap cluster analysis in which subsets of T cells from naive mice did not have a large number of unique genes (Fig. 6D). The CD44hi CD4 T cells were the most different group among the four T cell subsets from naive mice (521 unique genes), which may be due to a greater diversity of differentiation for CD4 T helper cells. We also found a greater number of unique genes differentially expressed by non-activated CD44lo CD4 T cells (829 genes) compared to virus-specific CD4 and CD8 T cells regardless of GzmB expression (only 37 to 92 genes; Fig. 6E).
When directly comparing the differentially expressed genes with a fold change of at least 2, only three genes met the requirements between GzmB– and GzmB+ CD4 T cells. As expected, Gzmb was significantly upregulated in the cytotoxic CD4 T cells, but both Tcf7 and Nsg2 were decreased compared to noncytotoxic classical Th1 CD44hi CD4 T cells (Fig. 6F). This was in stark contrast to GzmB+ versus nonactivated CD44lo CD4 T cells, which had 1,317 increased and 1,438 decreased genes (Fig. 6G). Furthermore, comparison of CD4-CTLs and CD8-CTLs revealed 66 upregulated and 43 downregulated genes (Fig. 6H). CD4-CTLs had increased expression of a number of molecules commonly associated with CD4 T cells, including Foxp3, Zbtb7b (ThPOK), CD40lg (CD40 ligand), Tnfrsf4 (OX40 receptor), and Tnfrsf25 (death receptor 3), compared to CD8-CTL (Fig. 6H). On the other hand, CD8-CTLs had greater expression of genes, including cytolytic proteins Prf1 (Perforin), Gzma, Gzmk, the activating receptor Klrk1 (NKG2D), and the transcription factor Eomes. Analysis using the Kyoto Encyclopedia of Genes and Genomes (KEGG) database revealed that cell cycle was the top ranked pathway upregulated in CD4-CTLs compared to CD44hi GzmB– Th1 (Fig. 6I) or CD44lo (Fig. 6J) CD4 T cells. DNA replication and repair pathways were also upregulated in CD4-CTLs, indicating that robust cellular division is required for clonal expansion. Furthermore, pathways related to energy metabolism were altered in CD4-CTLs. Gene expression pathways related to oxidative phosphorylation were enriched in CD4-CTLs compared to non-activated CD44lo CD4 T cells (Fig. 6J), but there was an increase in glycolysis-related pathways, contrasting the change from noncytolytic CD44hi CD4 T cells. These results demonstrate CD4-CTLs are highly similar to the noncytolytic but activated CD44hi CD4 T cells, suggesting a single pathway of development with further terminal differentiation.
CD4-CTLs express Runx3 and are type 1 T helper cells.
Given the similarity between cytolytic and noncytolytic activated CD4 T cells, we searched for any key transcription factors that would induce cytotoxic function in activated CD4 T cells. Previous work by Reis et al. demonstrated that a subset of CD4+ CD8αα+ intraepithelial lymphocytes (IELs), which have cytotoxic effector function, require expression of Runx3 and downregulation of ThPOK (18), the master transcriptional regulators that, respectively, determine CD8 and CD4 T cell lineage commitment (14, 26, 27). Peripheral naive T cells largely express only their respective lineage commitment transcription factor due to negative regulation of the opposing factor (Fig. 7A). Interestingly, we did observe minor Runx3 expression by naive splenic CD4 T cells above the isotype control staining (Fig. 7A). We examined the relative gene counts from RNA sequencing and observed that CD4 T cells expressed ThPOK with some minor transcript detected in CD8 T cells (Fig. 7B). Following ECTV infection, ThPOK gene transcript number was reduced in CD4 T cells but upregulated upon activation. Interestingly, Runx3 transcripts were detected in both CD4 and CD8 T cells, regardless of infection status (Fig. 7B). Activated CD4 T cells also had similar transcript copy number as CD8 T cells, but this did not appear to change in CD4-CTLs. The T helper type 1 (Th1)-associated transcription factor T-bet was upregulated in all CD44hi T cells. GATA-3, which is necessary for Th2 differentiation and important for CD4 lineage commitment, was expressed in all sorted CD4 T cell subsets and increased in CD44hi CD8 T cells. Transcription factor Eomes was only expressed in CD8 T cells and is known to contribute to CD8 T cell expansion and memory formation (28), as well as NK cell maturation (16). While Runx3 and T-bet gene expression was increased in activated CD4 T cells, no obvious differences were observed compared to CD4-CTLs.
FIG 7.
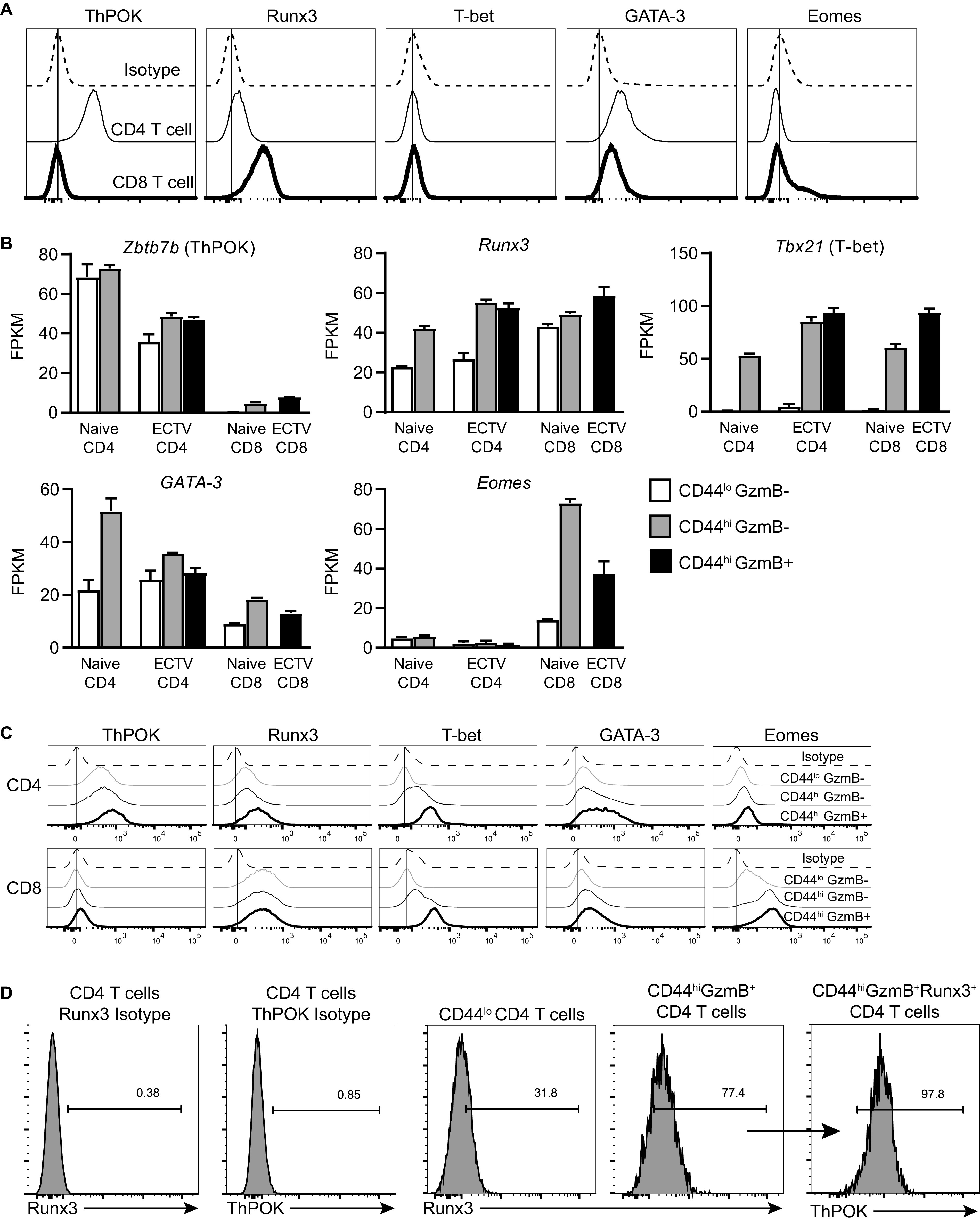
CD4-CTLs upregulate Runx3 and maintain ThPOK expression. (A) Splenocytes from naive mice were stained with antibodies targeting the indicated transcription factors. Histogram plots are shown for both CD4 and CD8 T cells compared to isotype antibody staining. (B) Relative transcription numbers, represented as fragments per kilobase of transcripts per million map reads (FPKM), for indicated genes for T cell populations analyzed as in Fig. 6. The data are presented as means ± the SEM from the three RNA samples collected. (C) Histograms of antibody staining for transcription factors for indicated splenic T cell populations from infected mice at 7 dpi. (D) Splenic CD4-CTLs at 7 dpi were first gated on Runx3-positive cells, followed by ThPOK gating. Isotype antibody staining on CD4 T cells was used to set gates. Concatenated or representative flow plots gated on three to four mice were used for histograms, and the results are representative of two independent experiments.
We next investigated the protein levels of the same transcription factors following infection using flow cytometry. Both CD4 and CD8 T cells from infected mice had antibody-staining profiles for ThPOK, T-bet, and Eomes that were consistent to what was observed with RNA sequencing (Fig. 7C). However, CD4 T cell protein expression of ThPOK was not greatly reduced following infection, as suggested from the RNA-seq analysis, but rather increased upon T cell activation compared to cells from naive mice (Fig. 7C). GATA-3 protein expression was increased in CD4-CTLs specifically in contrast to the transcript levels, which did not change compared to CD44hi GzmB– CD4 T cells. Interestingly, Runx3 expression was increased in CD4-CTLs compared to both CD44lo and classical Th1 CD44hi GzmB– CD4 T cell subsets. In addition, the level of Runx3 in CD4-CTLs was similar to what was observed in CD8 T cells. Furthermore, almost all Runx3+ CD4-CTLs still expressed the transcription factor ThPOK (Fig. 7D).
Since T-bet expression would suggest a largely Th1-biased CD4 T cell response, we also examined the phosphorylation status of signal transducer and activator of transcription (STAT) protein family members. Only phosphorylated STAT4 levels were increased in both activated CD44hi CD4 and CD8 T cells (Fig. 8A). This is consistent with previous results, as STAT4 also helps to promote Th1 differentiation of the CD4 T cell response (29, 30). Since CD4-CTLs had increased expression of both Runx3 and T-bet, we determined whether Runx3+ CD4 T cells could produce gamma interferon (IFN-γ). When we compared activated CD44hi CD4 T cells based on Runx3 expression (Fig. 8B), virtually all Runx3+ cells expressed IFN-γ at 7 dpi, and more than two-thirds were also positive for tumor necrosis factor alpha (TNF-α+) (Fig. 8C). In contrast, only ∼33% of CD44hi Runx3– CD4 T cells were IFN-γ+, and CD44lo cells expressed neither cytokine. Furthermore, almost 70% of Runx3+ CD4 T cells expressed CD107a, a marker of degranulation (Fig. 8D). Taken together, these data demonstrate CD4-CTLs are differentiated from Th1 cells and express the CD8 T cell lineage commitment transcription factor Runx3 but do not downregulate ThPOK at the protein level. Of note, the increase in Runx3 protein of CD4-CTLs was posttranscriptionally regulated.
FIG 8.
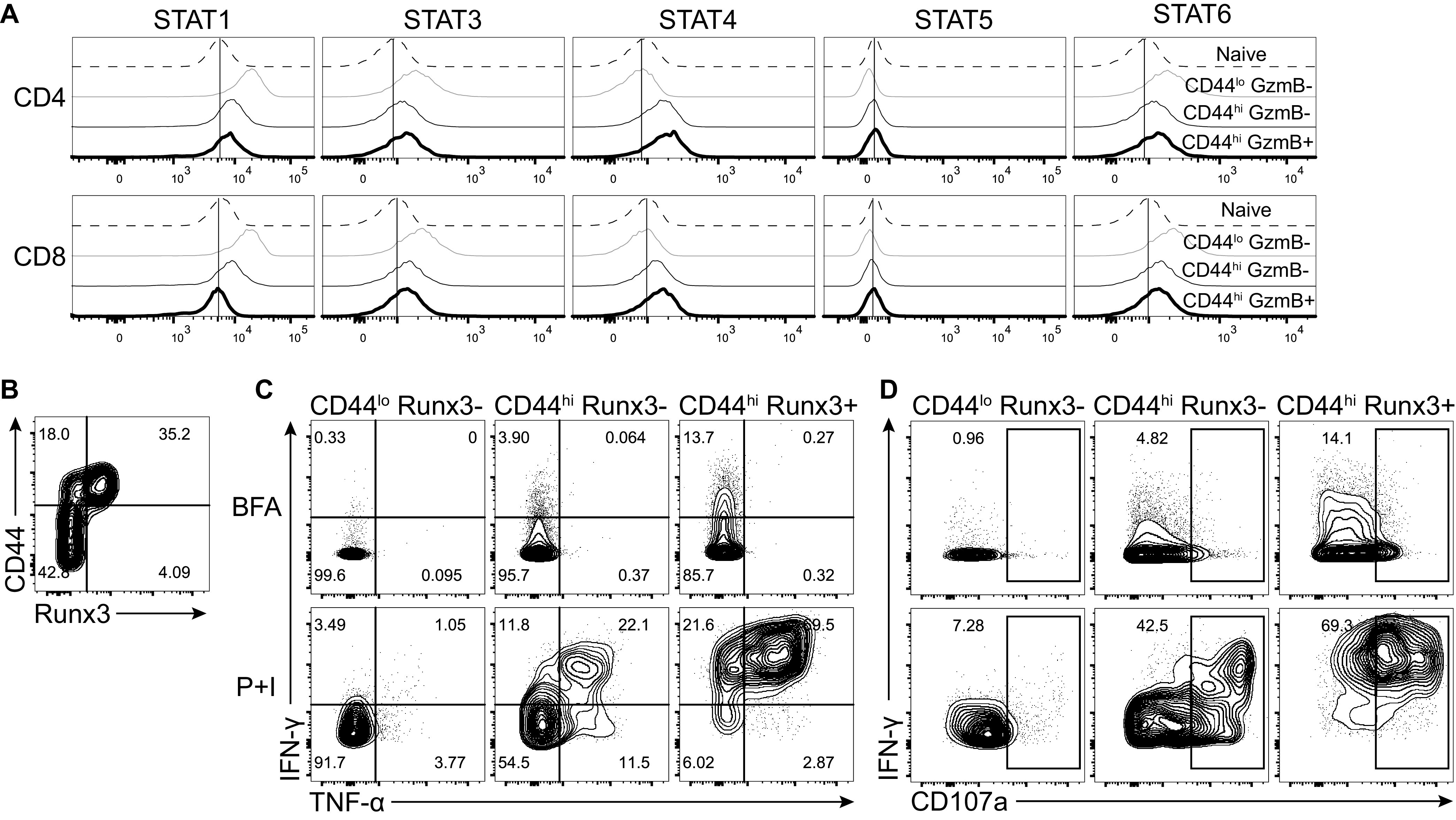
CD4-CTLs are terminally differentiation Th1 cells. (A) Histograms for staining of phosphorylated STATs on splenic T cell populations. (B to D) Splenocytes were restimulated with PMA and ionomycin (P+I) with brefeldin A (BFA) for 4 h on day 7 after ECTV infection (lower panels). As a control, splenocytes were only incubated with BFA (top panels). (B) Flow plot of CD44 by Runx3 expression used to gate CD4 subsets in panels C and D. (C and D) Representative flow plots of IFN-γ by TNF-α (C) or IFN-γ by CD107a (D) after gating on the indicated CD4 T cell populations. All flow plots are representative of the indicated group from two independent experiments.
Runx3 expression is required for CTL differentiation in CD4 T cells.
Given the important role of transcription factor Runx3 in NK maturation (16) and CD8 T cell lineage programming (27, 31), we hypothesized that Runx3 was also responsible for inducing the CD4-CTL response. To test this theory, we isolated CD4 T cells from spleens of naive CD45.1 homozygous and F1 CD45.1/2 heterozygous mice and electroporated cells with Cas9 protein and single-guide RNA (sgRNA) targeting either Runx3 or control nontargeting sgRNA, respectively (Fig. 9A). The electroporated CD4 T cells were then adoptively transferred into CD45.2 wild type B6 recipient mice at a 1:1 ratio and infected with ECTV. At 8 days after infection, we observed a significant reduction in the frequency of CD44hi GzmB+ CD4 T cells in the CD45.1 cells transfected with Runx3 sgRNA compared to the control treated CD45.1/2 cells and the endogenous CD45.2 response in both the liver and the spleen (Fig. 9B and C). There was no change in the frequency of activated CD44hi CD4 T cells in the liver, with a relatively small, but statistically significant decrease in the spleen for Runx3 sgRNA-treated cells compared to the control sgRNA group (Fig. 9D). In addition, the frequency of IFN-γ+ and IFN-γ+ TNF-α+ CD4 T cells was similar across all groups in the spleen and liver following restimulation (Fig. 9E). Taken together, these results highlight a role for Runx3 in the induction of CD4-CTL programming that is distinct from overall CD4 T cell activation and Th1 differentiation.
FIG 9.
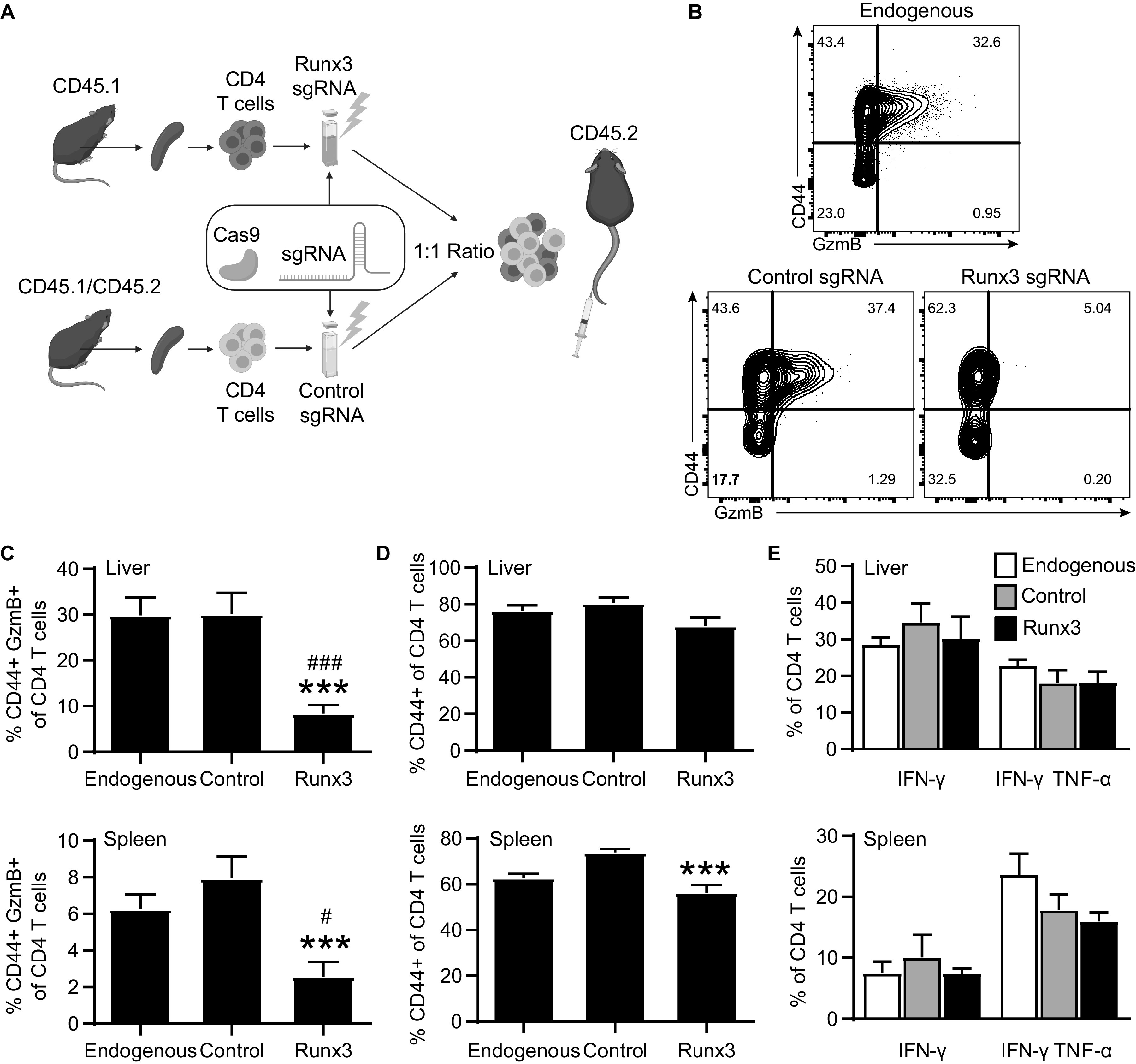
Runx3 is required for GzmB expression but not IFN-γ production. CD45.1 and CD45.1/2 splenocytes were magnetically purified for CD4 T cells and electroporated to incorporate Cas9 and Runx3 sgRNA or control nontargeting sgRNA. Totals of 2 × 106 to 4 × 106 electroporated cells were mixed at a 1:1 ratio, adoptively transferred into CD45.2 recipient mice, and infected with ECTV. The spleens and livers were collected at 8 dpi for analysis. (A) Depiction of experimental design. (B) Representative flow plot of CD44 by GzmB expression by CD4 T cell populations. Samples were concatenated across each cell population group. (C) Percentage of GzmB+ for CD4 T cell populations. (D) Frequency activated CD44+ of CD4 T cell populations. (E) Percentage IFN-γ+ or IFN-γ+ TNF-α+ for CD4 T cells for each CD4 T cell population. Asterisks (*) represent statistical differences compared to the control sgRNA group, whereas number symbols (#) denote differences compared to the endogenous group. Compiled data are displayed as means ± the SEM from three independent experiments (n = 14).
DISCUSSION
Previous work in our lab demonstrated that CD4-CTLs kill antigen-bearing cells in vivo and are necessary for survival to mousepox (1). A similar role was also observed with lethal influenza A virus infection (2). In both models, viral clearance required the cytolytic protein perforin in CD4 T cells. This suggested CD4-CTLs utilize granzymes and perforin to mediate cytolysis of target cells following viral infection. During ECTV infection, the CD4-CTL response targeted infected MHC-II+ cells (1), implying that CD4-CTLs may have a distinct role in killing infected cells presenting viral peptides on MHC-II, in contrast to NK cells, which recognize the loss of MHC-I on infected cells, and CD8 T cells, which recognize viral peptide bound to MHC-I. We show that CD4-CTLs are induced following murine infection with ECTV, LCMV, VACV, or MCMV, as has been previously observed, but not in direct comparison (1, 3, 32, 33). Footpad infection with ECTV results in the greatest overall proportion of CD4-CTLs even compared to a similar poxvirus, VACV (Fig. 4). The greater CD4-CTL response following ECTV infection compared to VACV has been previously demonstrated by Siciliano et al. (33).
Despite the important role of CD4-CTLs in mediating protection against ECTV infection, it remained unknown how CD4 T cells differentiated into CTLs. We show here that sustained viral replication and the posttranscriptional upregulation of Runx3 protein are required for the differentiation of classical noncytolytic Th1 cells into CD4-CTLs. Notably, the increase in Runx3 occurs despite high levels of ThPOK, which is a negative regulator of Runx3.
Our work demonstrates that, compared to the CD8 T cell response, the CD4-CTL response is more transient, peaking 1 day following the maximum virus titers in the spleen and liver during ECTV infection (34) and rapidly waning as the virus is eliminated. The transient response of CD4-CTLs may be due to the limited availability of MHC-II, which is largely found on antigen-presenting cells, whereas MHC-I is ubiquitously expressed. The continuous presence of live replicating virus, likely due to a need for high antigen load, is also important for CD4-CTLs, but not CD8-CTL differentiation because cidofovir treatment severely impaired only the CD4-CTL response. In addition, the inhibition of viral replication and subsequent loss of CD4-CTLs did not significantly alter the overall frequency of activated CD4 T cells. A possible explanation for the difference between CD4-CTLs and CD8-CTLs is that Runx3 is constitutively expressed in CD8 T cells but may require strong and continuous TCR signaling for posttranscriptional upregulation in CD4 T cells (14, 26). The transient kinetics and probable requirement for constant antigen exposure to maintain the CTL phenotype may explain why CD4-CTLs have been more commonly identified during chronic viral infections in humans. Whether the posttranscriptional upregulation of Runx3 is due to increased translation or protein half-life is not resolve by our studies.
We determined that CD4-CTLs were not a unique CD4 T cell subset, but rather Th1 cells that acquired CTL function. Activated noncytotoxic CD4 T cells and CD4-CTLs had transcriptional profiles that were very similar. Only three genes exhibited more than a 2-fold change, with Gzmb being increased and both Tcf7 (TCF-1) and Nsg2 reduced in CD4-CTLs (Fig. 10A to C). Nsg2 is a neuron-associated endosomal protein that modulates trafficking of AMPA receptors in the brain (35, 36). It is unclear what role it would be playing in T cells, but Nsg2 gene transcripts are expressed in both naive CD4 and CD8 T cells. Interestingly, transcription factor TCF-1 is required for CD4 lineage commitment, and its absence redirects CD4 T cells into the CD8 lineage (37). In addition, it has been previously suggested that Blimp1-mediated downregulation of both BCL6 and TCF-1 may be important for CD4-CTL differentiation during infection by an adenovirus 5-based vector (38). While TCF-1 gene expression was greatly reduced in CD4-CTLs during ECTV infection, there was no increase in CD8a transcript levels (Fig. 10D). TCF-1 is also suppressed in CD8 T cells to promote CD8 T cell effector function by IL-12 and STAT4 signaling (39). Thus, TCF-1 may also be downregulated in activated CD4 T cells to help promote cytotoxic function.
FIG 10.

Very few genes were differentially expressed when comparing CD4-CTLs and noncytolytic CD4 T cells. The relative gene transcript numbers from RNA-seq for T cell populations are presented in Fig. 7. (A to F) Gene copy numbers for (A) Gzmb, (B) Tcf7, (C) Nsg2, (D) Cd8a, (E) Socs1, and (F) Socs3. The data are shown as means ± the SEM for three replicates collected over three independent experiments.
Pathway analysis further highlighted significant increases in cell cycle, DNA replication, and DNA repair transcripts in CD4-CTLs. This could be due to CD4-CTLs undergoing constant cellular division during clonal expansion in response to the infection and to sustained TCR signaling. Furthermore, compared to activated noncytolytic CD4 T cells, CD4-CTLs upregulate glycolysis to help fulfill the energy demand from cellular division, differentiation, and effector functions (40). Thus, CD4-CTLs appear to undergo extensive terminal differentiation to ultimately acquire cytotoxic capabilities.
Several notable genes were differentially expressed in CD4- and CD8-CTLs (Fig. 6H). CD4-CTLs had higher expression of genes commonly associated with CD4 T cells, including regulatory transcription factor Foxp3, costimulatory ligands for CD40 and OX40, and death receptor 3, which has been shown to contribute to autoimmunity and inflammatory disease (41, 42). CD8-CTLs had greater expression of cytolytic molecules granzyme A, granzyme K, and perforin. This supported our previous data showing a greater magnitude of CD8-CTLs and a higher frequency of perforin-expressing CD8 compared to CD4 T cells (Fig. 1 and 2). CD8-CTLs also express more of the NK cell-associated activating receptor, NKG2D. Although NKG2D expression by NK cells is important for protection against ECTV (43), its expression can also improve CD8 T cell activation and effector function, serving as a costimulatory signal (44, 45). The transcription factor Eomes was largely upregulated by activated CD8 T cells, but minimal protein expression was observed in CD4 T cells (Fig. 7B and C). Previously, Eomes was demonstrated to contribute to CD8 T cell expansion and memory transition (28). These results indicate that whereas both CD4- and CD8-CTLs have a similar function to directly kill infected cells, they still have distinct gene profiles.
CD4-CTLs exhibited increases in the transcription factor protein levels of ThPOK, Runx3, T-bet, and GATA-3. T-bet expression was upregulated in activated noncytolytic CD4 T cells and had greater expression in CD4-CTLs. Paired with the elevated phosphorylated STAT4 levels, IFN-γ production following restimulation, and the similar transcriptional profile between GzmB– and GzmB+ CD44hi CD4 T cells, our data indicate that CD4-CTLs are a subset of Th1 cells. It was surprising to find that both Runx3 and ThPOK were highly expressed in CD4-CTLs at the same time. These opposing thymocyte lineage commitment, master transcription factors are almost exclusively expressed in their respective T cell subsets and have mechanisms to inhibit expression of one another (14, 26, 27, 46). Interestingly, despite being a Th2-associated transcription factor, GATA-3 protein was also upregulated in CD4-CTLs (47, 48). GATA-3 is known to induce ThPOK expression during thymocyte lineage commitment (49). Thus, the elevated GATA-3 protein expression may help explain why ThPOK is also increased in CD4-CTLs despite elevated Runx3. It is possible that GATA-3 in CD4-CTLs promotes ThPOK expression to help maintain their CD4 identity. A possible explanation for how Runx3 is expressed in the presence of ThPOK is that the levels of suppressor of cytokine signaling 1 (SOCS1) and SOCS3 gene transcripts were drastically lower in CD4-CTLs than in nonactivated CD44lo CD4 T cells (Fig. 10E and F). During thymocyte development, ThPOK induces SOCS proteins to repress expression of Runx3 (46). Therefore, the lack of SOCS1 and SOCS3 in activated CD4 T cells may allow CD4 T cells to express Runx3.
While Runx3 is the lineage commitment transcription factor for CD8 T cells, previous work has identified a population of CD4+ CD8αα+ IELs that express Runx3 and downregulate ThPOK to acquire cytotoxic programming (18, 50). In contrast, CD4-CTLs did not lose ThPOK expression or upregulate CD8a following viral challenge (Fig. 7D). This suggests CD4-CTLs programmed under drastically different environmental cues may have altered transcriptional profiles. We demonstrate that CD4 T cells deleted of Runx3 mount poor CD4-CTL responses following ECTV infection but produce IFN-γ normally. This contrasts with the findings of a previous study by Djuretic et al. (51), showing that CD4 T cells deficient in Runx3 produce reduced amounts of IFN-γ. These contrasting findings may be due to differences in mice, since Djuretic et al. used outbred strains, and all the experiments were done using in vitro cultured cells following anti-CD3/CD28 stimulation. Thus, our data imply that CD4-CTLs are generated from terminal differentiation of Th1 cells that eventually acquire Runx3 expression. Taken together, these results highlight a previously unknown role for Runx3 in the induction of cytotoxic programming in CD4 T cells during acute viral infection.
MATERIALS AND METHODS
Mice.
All experiments were approved by the Thomas Jefferson University Institutional Animal Care and Use Committee (IACUC). Wild type C57BL/6N (B6; CD45.2) mice and B6.SJL-Ptprca Pepcb/BoyJ (CD45.1) mice were purchased from Charles River and bred with B6 mice to generate F1 CD45.1/2 heterozygous mice. C57BL/6-Gzmbtm1Asmv/Orl (Gzmb-tdTomato) mice were purchased through the European Mutant Mouse Archive (EMMA), and heterozygous mice were used in experiments.
B6.129 × 1-H2-Ab1tm1Koni/J (MHC-IIfl/fl), B6.Cg-Tg(Itgax-cre)1-1Reiz/J (Itgax-cre), B6.129P2-Lyz2tm1(cre)Ifo/J (Lyz2-cre), and B6.Cg-Tg(Vav1-icre)A2Kio/J (Vav1-cre) mice were purchased from The Jackson Laboratory. The B6.C(Cg)-Cd79atm1(cre)Reth/EhobJ (Mb1-cre) strain was kindly provided by Kishore Alugupalli (Thomas Jefferson University, Philadelphia, PA). Strains generated from crossing MHC-IIfl/fl and cre-expressing mice were screened by flow cytometry to have normal levels of CD4 T cells in the periphery prior to use. cre-negative littermates were used as controls. All mice were bred and maintained in-house. Both females and males 6 to 12 weeks of age were used, and no gender biased results were observed.
Viruses and infection.
ECTV strain Moscow was obtained from ATCC (VR-1374) and propagated as previously described (20). Titers for ECTV were determined by plaque assay as previously outlined (52) with slight modifications. Briefly, BS-C-1 cells (ATCC CCL-26) were grown in 24-well tissue culture plates to 80 to 90% confluence in Dulbecco modified Eagle medium (DMEM) tissue culture medium (Invitrogen Life Technologies) supplemented with 10% fetal calf serum (FCS; Sigma-Aldrich), 4.5 g/liter glucose, 4.5 g/liter l-glutamine, 4.5 g/liter sodium pyruvate, 1× nonessential amino acids, and 100 IU/ml penicillin and streptomycin (complete DMEM). BS-C-1 monolayers were infected with 10-fold dilutions of organ homogenate for 1.5 h at 37°C with 5% CO2 in complete DMEM containing 2% FCS. Organ homogenate was obtained from processing samples for flow cytometry or by whole organ mechanical disruption using a TissueLyser (Qiagen) on frequency of 30/s for 2 min. After incubation, virus was removed, and monolayers were overlaid with 1:1 mixture of 2% carboxymethyl cellulose and complete DMEM containing 5% FCS. After 5 to 6 days of incubation at 37°C with 5% CO2, monolayers were fixed for 20 min at room temperature in 1% crystal violet in 20% ethanol solution containing 4% paraformaldehyde. Excess crystal violet was washed off in a pool of water and plaques were quantified. LCMV strain Armstrong was kindly provided by E. John Wherry (University of Pennsylvania, Philadelphia, PA) and propagated as previously described (53). VACV strain Western Reserve was initially obtained from Bernard Moss (National Institute of Allergy and Infectious Diseases [NIAID]) and amplified according to previous literature (54). MCMV (strain K181) was propagated as previously described (55). Mice were infected with 30 μl in the hind footpad with 3,000 PFU of ECTV, 1 × 105 PFU of LCMV, 1 × 103 PFU of VACV, or 1 × 105 PFU of MCMV. Virus doses for infection were based on previous studies using footpad infection route (1, 33, 56–58). Where indicated, mice were administered 600 μg of cidofovir in a 200-μl volume via i.p. route on day 3 after ECTV infection.
Flow cytometry.
Spleens were processed into single cell suspensions by gentle tissue dissociation using frosted microscope slides (Fisher Scientific). Livers were carefully manipulated through stainless steel wire mesh (88 T316 0.0035″ diameter; TWC, Inc.) in a 1.5″ × 1.25″ polyvinyl chloride female trap adapter (catalog no. 4804; Nibco). Hepatocytes were removed after resuspension in 37% Percoll (GE Healthcare Life Sciences) and centrifugation for 20 min at 930 relative centrifugal force at room temperature. The resulting liver cell pellet and splenocytes were treated with ammonium chloride potassium (ACK) buffer (155 mM NH4Cl, 1 mM KHCO3, 0.1 mM EDTA) for 5 min and washed with RPMI 1640 medium. For T cell restimulation experiments, up to 2 × 106 cells were incubated with brefeldin A (BFA; 10 μg/ml), phorbol myristate acetate (PMA; 50 ng/ml), and ionomycin (500 ng/ml) or BFA alone as a control for 4 h at 37°C and 5% CO2 in complete RPMI 1640 medium. For some experiments, CD107a antibody was added during the stimulation incubation. Stimulated samples were washed once in fluorescence-activated cell sorting (FACS) buffer (phosphate-buffered saline [PBS] with 3% Gibco newborn calf serum and 0.04% sodium azide) and further stained for other markers. To prevent nonspecific Fc receptor binding to antibodies, cells were stained with anti-CD16/32 (Fc-Block; 2.4G2 ATCC). For extracellular staining of surface molecules, single cell suspensions were incubated with antibodies in FACS buffer for 30 min at 4°C. In experiments requiring tetramer staining, I-Ab-QLIFNSISARALKAY tetramer conjugated to Alexa Fluor 647 (obtained from the NIH Tetramer Core Facility) was incubated with cells for 30 min at 4°C prior to staining with other antibodies. As negative control, cells were incubated with tetramer loaded with CLIP peptide (obtained from the NIH Tetramer Core Facility). For intracellular staining, samples were stained as described above and then fixed for 10 min in 1% paraformaldehyde in PBS. Cells were then incubated in 1× Perm/Wash buffer (BD Biosciences) for 5 min at 4°C and stained for 30 min with antibodies in 1× Perm/Wash buffer. Transcription factors were stained using an eBioscience transcription factor staining buffer kit (Thermo Fisher Scientific). Most transcription factors were stained after a 40-min incubation in fixation and permeabilization buffer for 30 min with antibodies in the 1× permeabilization buffer. Phospho-STAT antibodies required pretreatment of fixed cells with 100% ice-cold methanol for 30 min at 4°C. Samples were then stained for phospho-STAT molecules, along with other intracellular staining antibodies, in FACS buffer for 30 min at 4°C.
Antibodies targeting the following molecules were used in this study: CD3 (clone 17A2; BV785), CD4 (clone M4-5; BV605, BV785), CD8a (clone 53-6.7; FITC, BV711, PE), CD11a (clone 2D7; FITC BioLegend; BUV395 BD Biosciences), CD11b (clone M1/70; BV605), CD11c (clone N418; APC, PE/Cy7), CD19 (clone 6D5; PerCP/Cy5.5), CD44 (clone IM7; APC BioLegend, BUV395 BD Biosciences), CD45 (clone 30-F11; FITC), CD45.1 (clone A20; PE/Cy7), CD45.2 (clone 104; FITC), CD49d (clone 9C10 PE; clone R1-2 PerCP/Cy5.5), CD90.2 (clone 53-2.1; BV605), CD107a (clone 1D4B; PE), B220 (clone RA3-6B2; PerCP/Cy5.5), Eomes (clone Dan11mag; eBioscience Thermo Fisher Scientific; Alexa Fluor 488, PE/Cy7), F4/80 (clone BM8; FITC BioLegend, BUV395; BD Biosciences), GATA-3 (clone TWAJ; eBioscience/Thermo Fisher Scientific; PE), Gr-1 (clone RB6-8C5; APC/Fire750; Pacific Blue), granzyme B (clone GB11; Alexa Fluor 647, FITC; Pacific Blue), IFN-γ (clone XMG1.2; PE/Cy7, BV785), Ly6G (clone 1A8; PE), NK1.1 (clone PK136; APC, BV605), perforin (clone S16009A; PE), Runx3 (clone R3-5G4 purified BD Biosciences; conjugated to APC using an Abcam conjugation kit ab201807), pSTAT1 (clone A15151B; Alexa Fluor 488), pSTAT3 (clone 4/P-STAT3; BD Biosciences; Alexa Fluor 488), pSTAT4 (clone 38/p-Stat4; BD Biosciences; PE), pSTAT5 (clone SRBCZX; eBioscience/Thermo Fisher Scientific; PE/Cy7), pSTAT6 (clone CHI2S4N; eBioscience Thermo Fisher Scientific; PE), T-bet (clone 4B10; FITC, APC), TCRβ (clone H57-597; BV605; Pacific Blue), ThPOK (clone 11H11A14; PE), and TNF-α (clone MP6-XT22; Alexa Fluor 647). All antibodies, including any isotype control antibodies, were purchased from BioLegend unless otherwise stated. The following expression patterns were used to identify specific leukocyte populations: T cells, TCRβ+ CD90.2+; B cells, B220+ CD19+; DCs, TCRβ– CD3– B220– CD19– NK1.1– Gr1– CD11c+; and monocytes/macrophages, TCRβ– CD3– B220– CD19– NK1.1– Ly6g– CD11b+ Gr1+F4/80lo/hi. Data were acquired using the BD LSRFortessa or BD LSR II cytometers (BD Biosciences) and analyzed with FlowJo software (BD Biosciences).
Total RNA sequencing.
Livers from infected mice at 7 dpi were processed as described above and pooled across 10 mice. T cells were initially purified using the BioLegend magnetic cell sorting system targeting CD90.2 for positive selection (catalog no. 480102). Splenocytes from noninfected mice were used to select naive T cell populations due to lower T cell numbers in naive livers. The indicated populations of T cells were then sorted on a BD FACSAria II cytometer into PBS supplemented with 2% FCS for up to 1 × 106 cells. Cells were pelleted by centrifugation, and total RNA was extracted with DNase I treatment using RNA Clean and Concentrator-5 kit (Zymo Research). RNA for each T cell population was isolated three separate times over different experiments. RNA samples were submitted to Novogene for quality control analysis (All RIN values were >7) and sequencing on the Illumina NovaSeq 6000 system. Differentially expressed genes were identified using DESeq2 R package (59) where genes had an adjusted P value of <0.05. Heatmap cluster analysis, PCA, and volcano plots were generated using python packages matplotlib, seaborn, and pandas. KEGG pathway analysis was performed on differentially expressed genes that were significantly altered between groups, as indicated, and the top 10 pathways ranked by P value were shown (60–62).
CRISPR/Cas9 gene editing of CD4 T cells.
Spleens from CD45.1 and F1 CD45.1/2 mice were processed into single cell suspensions, and red blood cells were lysed as indicated above. CD4 T cells were purified using BioLegend Mojosort magnetic separation kits for total CD4 T cells by negative selection (catalog no. 480033). Approximately 2 × 106 to 4 × 106 CD4 T cells were electroporated with 180 pmol of Cas9 protein (from S. pyogenes; PNA Bio) and 540 pmol of sgRNA via program X-001 on the Amaxa Nucleofector II apparatus using the mouse T cell Nucleofector kit (Lonza; VPA-1006) according to the manufacturer’s instructions, with slight modifications. The Cas9 protein and sgRNA were mixed at a 3:1 ratio, followed by incubation for 2 min at room temperature, and then added to the CD4 T cells resuspended in the nucleofection solution as previously described (63). CD4 T cells were electroporated with a mixture containing 180 pmol of each of three sgRNA constructs (540 pmol total; Synthego)—5′-GGGGGGAGUGAAGCGGCGGC, 5′-CGCAGGGGAAGGCCGUGGAG, and 5′-ACGGCCUUCCCCUGCGGCGG—targeting exon 2 of Runx3. As a negative control, sgRNA that was designed through bioinformatic approaches to not recognize any sequence in the murine genome was used (540 pmol; Synthego Negative nontargeting control sgRNA 1: 5′-GCACUACCAGAGCUAACUCA). Electroporated CD4 T cells were rested at 37°C with 5% CO2 for 4 h prior to counting, mixed at a 1:1 ratio, and adoptively transferred into B6 mice through tail vein injection. Recipient mice were then immediately infected with ECTV. A representative model figure was generated with BioRender.com.
Statistical analysis.
Data were analyzed using Prism (GraphPad) software. A parametric unpaired t test was used to compare two groups, while one-way ANOVA with the Tukey’s multiple-comparison test was used for more than two groups. Survival curves were analyzed using the log-rank Mantel-Cox test. All experiments were done a minimum of two times. For all figures, P values are represented in the figures by symbols: */#, P < 0.05; **/##, P < 0.01; and ***/###, P < 0.001.
Data availability.
Raw and processed mRNA-Seq data are available in the Gene Expression Omnibus (GEO) under series entry GSE179289.
ACKNOWLEDGMENTS
This study was supported by grants R01AI110457 and R01AI065544 from the National Institute of Allergy and Infectious Diseases (NIAID) and AG048602 from the National Institute on Aging (NIA) to L.J.S. C.J.K. was supported by T32 AI134646 from NIAID. Research reported in this publication utilized the Flow Cytometry and Laboratory Animal facilities at the Sidney Kimmel Cancer Center at Jefferson Health and was supported by National Cancer Institute/National Institutes of Health award P30CA056036.
Contributor Information
Luis J. Sigal, Email: luis.sigal@jefferson.edu.
Rozanne M. Sandri-Goldin, University of California, Irvine
REFERENCES
- 1.Fang M, Siciliano NA, Hersperger AR, Roscoe F, Hu A, Ma X, Shamsedeen AR, Eisenlohr LC, Sigal LJ. 2012. Perforin-dependent CD4+ T-cell cytotoxicity contributes to control a murine poxvirus infection. Proc Natl Acad Sci U S A 109:9983–9988. doi: 10.1073/pnas.1202143109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brown DM, Lee S, Garcia-Hernandez ML, Swain SL. 2012. Multifunctional CD4 cells expressing gamma interferon and perforin mediate protection against lethal influenza virus infection. J Virol 86:6792–6803. doi: 10.1128/JVI.07172-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zajac AJ, Quinn DG, Cohen PL, Frelinger JA. 1996. Fas-dependent CD4+ cytotoxic T-cell-mediated pathogenesis during virus infection. Proc Natl Acad Sci U S A 93:14730–14735. doi: 10.1073/pnas.93.25.14730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Quezada SA, Simpson TR, Peggs KS, Merghoub T, Vider J, Fan X, Blasberg R, Yagita H, Muranski P, Antony PA, Restifo NP, Allison JP. 2010. Tumor-reactive CD4+ T cells develop cytotoxic activity and eradicate large established melanoma after transfer into lymphopenic hosts. J Exp Med 207:637–650. doi: 10.1084/jem.20091918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xie Y, Akpinarli A, Maris C, Hipkiss EL, Lane M, Kwon EK, Muranski P, Restifo NP, Antony PA. 2010. Naive tumor-specific CD4+ T cells differentiated in vivo eradicate established melanoma. J Exp Med 207:651–667. doi: 10.1084/jem.20091921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Appay V, Zaunders JJ, Papagno L, Sutton J, Jaramillo A, Waters A, Easterbrook P, Grey P, Smith D, McMichael AJ, Cooper DA, Rowland-Jones SL, Kelleher AD. 2002. Characterization of CD4+ CTLs ex vivo. J Immunol 168:5954–5958. doi: 10.4049/jimmunol.168.11.5954. [DOI] [PubMed] [Google Scholar]
- 7.Zaunders JJ, Dyer WB, Wang B, Munier ML, Miranda-Saksena M, Newton R, Moore J, Mackay CR, Cooper DA, Saksena NK, Kelleher AD. 2004. Identification of circulating antigen-specific CD4+ T lymphocytes with a CCR5+, cytotoxic phenotype in an HIV-1 long-term nonprogressor and in CMV infection. Blood 103:2238–2247. doi: 10.1182/blood-2003-08-2765. [DOI] [PubMed] [Google Scholar]
- 8.Aslan N, Yurdaydin C, Wiegand J, Greten T, Ciner A, Meyer MF, Heiken H, Kuhlmann B, Kaiser T, Bozkaya H, Tillmann HL, Bozdayi AM, Manns MP, Wedemeyer H. 2006. Cytotoxic CD4 T cells in viral hepatitis. J Viral Hepat 13:505–514. doi: 10.1111/j.1365-2893.2006.00723.x. [DOI] [PubMed] [Google Scholar]
- 9.van Leeuwen EM, Remmerswaal EB, Vossen MT, Rowshani AT, Wertheim-van Dillen PM, van Lier RA, ten Berge IJ. 2004. Emergence of a CD4+ CD28– granzyme B+, cytomegalovirus-specific T cell subset after recovery of primary cytomegalovirus infection. J Immunol 173:1834–1841. doi: 10.4049/jimmunol.173.3.1834. [DOI] [PubMed] [Google Scholar]
- 10.Munier CML, van Bockel D, Bailey M, Ip S, Xu Y, Alcantara S, Liu SM, Denyer G, Kaplan W, Group PS, Suzuki K, Croft N, Purcell A, Tscharke D, Cooper DA, Kent SJ, Zaunders JJ, Kelleher AD, PHIIDO Study Group. 2016. The primary immune response to vaccinia virus vaccination includes cells with a distinct cytotoxic effector CD4 T-cell phenotype. Vaccine 34:5251–5261. doi: 10.1016/j.vaccine.2016.09.009. [DOI] [PubMed] [Google Scholar]
- 11.Stuller KA, Flano E. 2009. CD4 T cells mediate killing during persistent gammaherpesvirus 68 infection. J Virol 83:4700–4703. doi: 10.1128/JVI.02240-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lowin B, Peitsch MC, Tschopp J. 1995. Perforin and granzymes: crucial effector molecules in cytolytic T lymphocyte and natural killer cell-mediated cytotoxicity. Curr Top Microbiol Immunol 198:1–24. doi: 10.1007/978-3-642-79414-8_1. [DOI] [PubMed] [Google Scholar]
- 13.Wang L, Bosselut R. 2009. CD4-CD8 lineage differentiation: Thpok-ing into the nucleus. J Immunol 183:2903–2910. doi: 10.4049/jimmunol.0901041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.He X, He X, Dave VP, Zhang Y, Hua X, Nicolas E, Xu W, Roe BA, Kappes DJ. 2005. The zinc finger transcription factor Th-POK regulates CD4 versus CD8 T-cell lineage commitment. Nature 433:826–833. doi: 10.1038/nature03338. [DOI] [PubMed] [Google Scholar]
- 15.Shan Q, Zeng Z, Xing S, Li F, Hartwig SM, Gullicksrud JA, Kurup SP, Van Braeckel-Budimir N, Su Y, Martin MD, Varga SM, Taniuchi I, Harty JT, Peng W, Badovinac VP, Xue HH. 2017. The transcription factor Runx3 guards cytotoxic CD8+ effector T cells against deviation towards follicular helper T cell lineage. Nat Immunol 18:931–939. doi: 10.1038/ni.3773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gordon SM, Chaix J, Rupp LJ, Wu J, Madera S, Sun JC, Lindsten T, Reiner SL. 2012. The transcription factors T-bet and Eomes control key checkpoints of natural killer cell maturation. Immunity 36:55–67. doi: 10.1016/j.immuni.2011.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Reis BS, Hoytema van Konijnenburg DP, Grivennikov SI, Mucida D. 2014. Transcription factor T-bet regulates intraepithelial lymphocyte functional maturation. Immunity 41:244–256. doi: 10.1016/j.immuni.2014.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reis BS, Rogoz A, Costa-Pinto FA, Taniuchi I, Mucida D. 2013. Mutual expression of the transcription factors Runx3 and ThPOK regulates intestinal CD4+ T cell immunity. Nat Immunol 14:271–280. doi: 10.1038/ni.2518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McDermott DS, Varga SM. 2011. Quantifying antigen-specific CD4 T cells during a viral infection: CD4 T cell responses are larger than we think. J Immunol 187:5568–5576. doi: 10.4049/jimmunol.1102104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fang M, Roscoe F, Sigal LJ. 2010. Age-dependent susceptibility to a viral disease due to decreased natural killer cell numbers and trafficking. J Exp Med 207:2369–2381. doi: 10.1084/jem.20100282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.de Boer J, Williams A, Skavdis G, Harker N, Coles M, Tolaini M, Norton T, Williams K, Roderick K, Potocnik AJ, Kioussis D. 2003. Transgenic mice with hematopoietic and lymphoid specific expression of Cre. Eur J Immunol 33:314–325. doi: 10.1002/immu.200310005. [DOI] [PubMed] [Google Scholar]
- 22.Caton ML, Smith-Raska MR, Reizis B. 2007. Notch-RBP-J signaling controls the homeostasis of CD8– dendritic cells in the spleen. J Exp Med 204:1653–1664. doi: 10.1084/jem.20062648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Clausen BE, Burkhardt C, Reith W, Renkawitz R, Forster I. 1999. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res 8:265–277. doi: 10.1023/a:1008942828960. [DOI] [PubMed] [Google Scholar]
- 24.Hobeika E, Thiemann S, Storch B, Jumaa H, Nielsen PJ, Pelanda R, Reth M. 2006. Testing gene function early in the B cell lineage in mb1-cre mice. Proc Natl Acad Sci U S A 103:13789–13794. doi: 10.1073/pnas.0605944103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mouchacca P, Schmitt-Verhulst AM, Boyer C. 2013. Visualization of cytolytic T cell differentiation and granule exocytosis with T cells from mice expressing active fluorescent granzyme B. PLoS One 8:e67239. doi: 10.1371/journal.pone.0067239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Egawa T, Littman DR. 2008. ThPOK acts late in specification of the helper T cell lineage and suppresses Runx-mediated commitment to the cytotoxic T cell lineage. Nat Immunol 9:1131–1139. doi: 10.1038/ni.1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Taniuchi I, Osato M, Egawa T, Sunshine MJ, Bae SC, Komori T, Ito Y, Littman DR. 2002. Differential requirements for Runx proteins in CD4 repression and epigenetic silencing during T lymphocyte development. Cell 111:621–633. doi: 10.1016/S0092-8674(02)01111-X. [DOI] [PubMed] [Google Scholar]
- 28.Banerjee A, Gordon SM, Intlekofer AM, Paley MA, Mooney EC, Lindsten T, Wherry EJ, Reiner SL. 2010. Cutting edge: the transcription factor eomesodermin enables CD8+ T cells to compete for the memory cell niche. J Immunol 185:4988–4992. doi: 10.4049/jimmunol.1002042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thieu VT, Yu Q, Chang HC, Yeh N, Nguyen ET, Sehra S, Kaplan MH. 2008. Signal transducer and activator of transcription 4 is required for the transcription factor T-bet to promote T helper 1 cell-fate determination. Immunity 29:679–690. doi: 10.1016/j.immuni.2008.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jacobson NG, Szabo SJ, Weber-Nordt RM, Zhong Z, Schreiber RD, Darnell JE, Jr, Murphy KM. 1995. Interleukin 12 signaling in T helper type 1 (Th1) cells involves tyrosine phosphorylation of signal transducer and activator of transcription 3 (Stat3) and Stat4. J Exp Med 181:1755–1762. doi: 10.1084/jem.181.5.1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cruz-Guilloty F, Pipkin ME, Djuretic IM, Levanon D, Lotem J, Lichtenheld MG, Groner Y, Rao A. 2009. Runx3 and T-box proteins cooperate to establish the transcriptional program of effector CTLs. J Exp Med 206:51–59. doi: 10.1084/jem.20081242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Verma S, Weiskopf D, Gupta A, McDonald B, Peters B, Sette A, Benedict CA. 2016. Cytomegalovirus-specific CD4 T cells are cytolytic and mediate vaccine protection. J Virol 90:650–658. doi: 10.1128/JVI.02123-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Siciliano NA, Hersperger AR, Lacuanan AM, Xu RH, Sidney J, Sette A, Sigal LJ, Eisenlohr LC. 2014. Impact of distinct poxvirus infections on the specificities and functionalities of CD4+ T cell responses. J Virol 88:10078–10091. doi: 10.1128/JVI.01150-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chaudhri G, Panchanathan V, Buller RM, van den Eertwegh AJ, Claassen E, Zhou J, de Chazal R, Laman JD, Karupiah G. 2004. Polarized type 1 cytokine response and cell-mediated immunity determine genetic resistance to mousepox. Proc Natl Acad Sci U S A 101:9057–9062. doi: 10.1073/pnas.0402949101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Saberan-Djoneidi D, Marey-Semper I, Picart R, Studler JM, Tougard C, Glowinski J, Levi-Strauss M. 1995. A 19-kDa protein belonging to a new family is expressed in the Golgi apparatus of neural cells. J Biol Chem 270:1888–1893. doi: 10.1074/jbc.270.4.1888. [DOI] [PubMed] [Google Scholar]
- 36.Chander P, Kennedy MJ, Winckler B, Weick JP. 2019. Neuron-specific gene 2 (NSG2) encodes an AMPA receptor interacting protein that modulates excitatory neurotransmission. eNeuro 6:ENEURO.0292-18.2018. doi: 10.1523/ENEURO.0292-18.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Steinke FC, Yu S, Zhou X, He B, Yang W, Zhou B, Kawamoto H, Zhu J, Tan K, Xue HH. 2014. TCF-1 and LEF-1 act upstream of Th-POK to promote the CD4+ T cell fate and interact with Runx3 to silence Cd4 in CD8+ T cells. Nat Immunol 15:646–656. doi: 10.1038/ni.2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Donnarumma T, Young GR, Merkenschlager J, Eksmond U, Bongard N, Nutt SL, Boyer C, Dittmer U, Le-Trilling VT, Trilling M, Bayer W, Kassiotis G. 2016. Opposing development of cytotoxic and follicular helper CD4 T cells controlled by the TCF-1-Bcl6 nexus. Cell Rep 17:1571–1583. doi: 10.1016/j.celrep.2016.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Danilo M, Chennupati V, Silva JG, Siegert S, Held W. 2018. Suppression of Tcf1 by inflammatory cytokines facilitates effector CD8 T cell differentiation. Cell Rep 22:2107–2117. doi: 10.1016/j.celrep.2018.01.072. [DOI] [PubMed] [Google Scholar]
- 40.Roos D, Loos JA. 1973. Changes in the carbohydrate metabolism of mitogenically stimulated human peripheral lymphocytes. II. Relative importance of glycolysis and oxidative phosphorylation on phytohaemagglutinin stimulation. Exp Cell Res 77:127–135. doi: 10.1016/0014-4827(73)90561-2. [DOI] [PubMed] [Google Scholar]
- 41.Aiba Y, Nakamura M. 2013. The role of TL1A and DR3 in autoimmune and inflammatory diseases. Mediators Inflamm 2013:258164. doi: 10.1155/2013/258164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Meylan F, Davidson TS, Kahle E, Kinder M, Acharya K, Jankovic D, Bundoc V, Hodges M, Shevach EM, Keane-Myers A, Wang EC, Siegel RM. 2008. The TNF-family receptor DR3 is essential for diverse T cell-mediated inflammatory diseases. Immunity 29:79–89. doi: 10.1016/j.immuni.2008.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fang M, Lanier LL, Sigal LJ. 2008. A role for NKG2D in NK cell-mediated resistance to poxvirus disease. PLoS Pathog 4:e30. doi: 10.1371/journal.ppat.0040030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bauer S, Groh V, Wu J, Steinle A, Phillips JH, Lanier LL, Spies T. 1999. Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible MICA. Science 285:727–729. doi: 10.1126/science.285.5428.727. [DOI] [PubMed] [Google Scholar]
- 45.Jamieson AM, Diefenbach A, McMahon CW, Xiong N, Carlyle JR, Raulet DH. 2002. The role of the NKG2D immunoreceptor in immune cell activation and natural killing. Immunity 17:19–29. doi: 10.1016/S1074-7613(02)00333-3. [DOI] [PubMed] [Google Scholar]
- 46.Luckey MA, Kimura MY, Waickman AT, Feigenbaum L, Singer A, Park JH. 2014. The transcription factor ThPOK suppresses Runx3 and imposes CD4+ lineage fate by inducing the SOCS suppressors of cytokine signaling. Nat Immunol 15:638–645. doi: 10.1038/ni.2917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang DH, Cohn L, Ray P, Bottomly K, Ray A. 1997. Transcription factor GATA-3 is differentially expressed in murine Th1 and Th2 cells and controls Th2-specific expression of the interleukin-5 gene. J Biol Chem 272:21597–21603. doi: 10.1074/jbc.272.34.21597. [DOI] [PubMed] [Google Scholar]
- 48.Lee HJ, Takemoto N, Kurata H, Kamogawa Y, Miyatake S, O’Garra A, Arai N. 2000. GATA-3 induces T helper cell type 2 (Th2) cytokine expression and chromatin remodeling in committed Th1 cells. J Exp Med 192:105–115. doi: 10.1084/jem.192.1.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang L, Wildt KF, Zhu J, Zhang X, Feigenbaum L, Tessarollo L, Paul WE, Fowlkes BJ, Bosselut R. 2008. Distinct functions for the transcription factors GATA-3 and ThPOK during intrathymic differentiation of CD4+ T cells. Nat Immunol 9:1122–1130. doi: 10.1038/ni.1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mucida D, Husain MM, Muroi S, van Wijk F, Shinnakasu R, Naoe Y, Reis BS, Huang Y, Lambolez F, Docherty M, Attinger A, Shui JW, Kim G, Lena CJ, Sakaguchi S, Miyamoto C, Wang P, Atarashi K, Park Y, Nakayama T, Honda K, Ellmeier W, Kronenberg M, Taniuchi I, Cheroutre H. 2013. Transcriptional reprogramming of mature CD4+ helper T cells generates distinct MHC class II-restricted cytotoxic T lymphocytes. Nat Immunol 14:281–289. doi: 10.1038/ni.2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Djuretic IM, Levanon D, Negreanu V, Groner Y, Rao A, Ansel KM. 2007. Transcription factors T-bet and Runx3 cooperate to activate Ifng and silence Il4 in T helper type 1 cells. Nat Immunol 8:145–153. doi: 10.1038/ni1424. [DOI] [PubMed] [Google Scholar]
- 52.Xu RH, Cohen M, Tang Y, Lazear E, Whitbeck JC, Eisenberg RJ, Cohen GH, Sigal LJ. 2008. The orthopoxvirus type I IFN binding protein is essential for virulence and an effective target for vaccination. J Exp Med 205:981–992. doi: 10.1084/jem.20071854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Welsh RM, Seedhom MO. 2008. Lymphocytic choriomeningitis virus (LCMV): propagation, quantitation, and storage. Curr Protoc Microbiol Chapter 15:Unit 15A11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Elroy-Stein O, Moss B. 2001. Gene expression using the vaccinia virus/T7 RNA polymerase hybrid system. Curr Protoc Protein Sci Chapter 5:Unit 5-15. [DOI] [PubMed] [Google Scholar]
- 55.Zurbach KA, Moghbeli T, Snyder CM. 2014. Resolving the titer of murine cytomegalovirus by plaque assay using the M2-10B4 cell line and a low viscosity overlay. Virol J 11:71. doi: 10.1186/1743-422X-11-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sinickas VG, Ashman RB, Blanden RV. 1985. The cytotoxic response to murine cytomegalovirus. I. Parameters in vivo. J Gen Virol 66:747–755. doi: 10.1099/0022-1317-66-4-747. [DOI] [PubMed] [Google Scholar]
- 57.Puhach O, Ostermann E, Krisp C, Frascaroli G, Schluter H, Brinkmann MM, Brune W. 2020. Murine cytomegaloviruses m139 targets DDX3 to curtail interferon production and promote viral replication. PLoS Pathog 16:e1008546. doi: 10.1371/journal.ppat.1008546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zinkernagel RM, Haenseler E, Leist T, Cerny A, Hengartner H, Althage A. 1986. T cell-mediated hepatitis in mice infected with lymphocytic choriomeningitis virus. Liver cell destruction by H-2 class I-restricted virus-specific cytotoxic T cells as a physiological correlate of the 51Cr-release assay? J Exp Med 164:1075–1092. doi: 10.1084/jem.164.4.1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Love MI, Huber W, Anders S. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kanehisa M. 2019. Toward understanding the origin and evolution of cellular organisms. Protein Sci 28:1947–1951. doi: 10.1002/pro.3715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kanehisa M, Furumichi M, Sato Y, Ishiguro-Watanabe M, Tanabe M. 2020. KEGG: integrating viruses and cellular organisms. Nucleic Acids Res 49:D545–D551. doi: 10.1093/nar/gkaa970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kanehisa M, Goto S. 2000. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res 28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Seki A, Rutz S. 2018. Optimized RNP transfection for highly efficient CRISPR/Cas9-mediated gene knockout in primary T cells. J Exp Med 215:985–997. doi: 10.1084/jem.20171626. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Raw and processed mRNA-Seq data are available in the Gene Expression Omnibus (GEO) under series entry GSE179289.