

Abstract
In this work, we investigated the redox transients of a number of water-soluble spin labels upon their interactions with Photosystem II (PS II) core complexes isolated from spinach leaves. We have found that the reactivity of nitroxide radicals, determined by the rate of their reduction upon illumination of PS II, depends on the chemical structure of radicals and the capability of their coming close to low-potential redox centers of photoactive PS II complexes. An enhanced capability of nitroxide radicals to accept electrons from PS II correlates with their chemical structure. Nitroxide radicals NTI (2,2,5,5-tetramethyl-4-nitromethylene-3-imidazolidine-N-oxyl) and Tacet (4-hydroxy-2,2,6,6-tetramethylpiperidine-1-oxyl-acetate), containing polar groups, appear to be most efficient acceptors of electrons donated by PS II compared to neutral (TEMPOL, 4-hydroxy-2,2,6,6-tetramethylpiperidine-1-oxyl) or positively charged (Tamine, 4-amino-2,2,6,6-tetramethylpiperidine-l-oxyl) spin labels. We assume that enhanced reactivities of polar nitroxide radicals, NTI and Tacet, are determined (1) by their relatively high redox potentials, providing the possibility to accept electrons from PS II, and (2) by their affinities to the closest binding sites on the surface of PS II in the vicinity of the primary plastoquinone acceptor PQA (12–14 Å) or/and in the intraprotein cavity for the secondary plastoquinone PQB (~ 22 Å).
Introduction
Water-soluble nitroxide radicals have found numerous applications in chemistry, biology, and medicine. The most interesting applications of nitroxide radicals in biology and medicine are associated with the development of drugs [1]. For example, recently the stable nitroxide 4-hydroxy-2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPOL) has been suggested as the reagent that can cause the oxidation of the Fe–S clusters of the RNA-dependent RNA polymerase (RdRp). These clusters serve as cofactors for the SARS-CoV-2 RdRp. The inhibition of the RdRp by TEMPOL and/or by related stable nitroxide radicals, blocking SARS-CoV-2 replication in cell culture, might be used for potential COVID-19 therapies during active viral infection [2].
In the current work, we have focused on applications of water-soluble nitroxide radicals for the study of electron transport processes in oxygenic photosynthesis. Photosynthetic apparatus of oxygenic organisms (cyanobacteria, alga, and higher plants) includes the pigment–protein complex of photosystem II (PS II), which is the light-driven water–plastoquinone oxidoreductase located in the thylakoid membranes. The structure of the dimeric form of PS II in thermophilic cyanobacteria was determined by X-ray analysis with the atomic resolution of 1.9 Å [3]. Each monomer of the PS II complex contains chlorophyll (Chl) dimer P680, monomeric Chl molecules ChlD1 and ChlD2, pheophytin molecules PheA and PheB, the primary (PQA) and the secondary (PQB) plastoquinone molecules, redox-active tyrosine YZ and the water-oxidizing complex (WOC). The WOC, consisting of the inorganic Mn4CaO5 cluster and surrounding protein matrix, is oriented towards the luminal side of the thylakoid membrane [3–7]. The energy of light quanta absorbed by the light-harvesting antenna migrates to the reaction center, where the charge separation occurs, forming the radical pair P680+PheA–. Reduced pheophytin donates an electron to the tightly bound PQA, which passes the electron further to PQB. After the consecutive two-electron reduction of PQB by PQA– and its protonation (PQB + 2e− + 2H+ → PQBH2), the fully reduced plastoquinone dissociates into the lipid phase of the thylakoid membrane in exchange for the PQ molecule from the PQ/PQH2 pool. The oxidized primary donor P680+ is reduced by YZ. The tyrosine YZ links P680 with the WOC (reviewed in [5–7]).
The light-induced charge transfer in PS II is coupled to the generation of the transmembrane electric potential difference [8, 9]. The search for organic and inorganic compounds capable of efficient accepting electrons from the acceptor side of PS II is relevant to better understanding of molecular mechanisms of the PS II functioning. This may be of practical application to create highly effective and ecologically friendly solar-electric energy converters [10–12]. In PS II, both PQA and PQB molecules are localized at a distance of ~ 10 Å from the protein–water boundary [3, 4]. It is conceivably, therefore, that they may transfer electrons to exogenous electron acceptors. Some of these acceptors may also serve as effective electron transfer intermediates between PS II and semiconductor electrodes [13]. In the presence of 3-(3,4-dichlorophenyl)-1,1-dimethylurea (DCMU)—an inhibitor of electron transfer from PQA– to PQB, PS II can donate electrons directly from PQA− to the oxidized form of cytochrome (cyt) c or the synthetic cationic complex Co(III). The commonly used mediators between PS II and the electrodes include 2,6-dichloro-p-benzoquinone, 2,6-dimethyl-p-benzoquinone, phenyl-p-benzoquinone [14–23] and the oxidized cyt c [24, 25]. The other potential intermediate electron acceptors from PQA− to semiconductors are paramagnetic molecules (spin labels) interacting with the acceptor side of PS II. The light-induced electron transfer from PS II to spin labels (nitroxide radicals) can be monitored by measuring the changes in the intensity of their electron paramagnetic resonance (EPR) signals [26].
Water-soluble nitroxide radicals have found numerous applications in chemistry and biology (for references, see [27]). In particular, the imidazoline- and imidazolidine-based nitroxides are widely used as molecular probes for measuring the proton activity inside the thylakoid lumen (for review, see [28]). EPR spectra of pH-sensitive spin labels have distinct features that allow to distinguish between the signals given by other paramagnetic species. In our earlier studies, we investigated the interaction of a number of water-soluble imidazoline- and imidazolidine-based nitroxide radicals with isolated chloroplasts [29]. It was demonstrated that the light-induced changes in the EPR signals of nitroxide radicals are determined by several factors: (1) the ΔpH-driven accumulation of radicals inside the thylakoids and concomitant changes in the shape of their EPR spectra, (2) the irreversible loss of paramagnetism due to spin label interactions with the chloroplast electron transport chain, and (3) specific effects caused by changes in the partial pressure of molecular oxygen (O2) in the result of the light-induced evolution (or consumption) of paramagnetic molecules 3O2. The loss of paramagnetism (the “reactivity” of spin labels) may cause the problems in their applications, e.g., for probing structural changes in biological systems and physical–chemical properties of spin label surroundings. This property of nitroxide radicals can be used, however, for the analysis of redox activity of biological systems. Having spin labels capable of redox transformations, one could use them as the indicators of electron transport in bioenergetic processes [27–29].
The main purpose of this work is to investigate redox transients of a number of water-soluble spin labels that can occur due to their interactions with isolated PS II. We have found that the reactivity of nitroxide radicals, determined by the rate of their reduction upon illumination of PS II, depends on the chemical structure of radicals and the capability of their approaching to PQA molecules on the acceptor side of PS II.
Materials and Methods
Isolation of Photosystem II Complexes
Spinach PS II core complexes were isolated from commercial spinach plants (Spinacia oleracea) as described in [30] by treatment of the membrane fragments with dodecyl-β-d-maltoside (DM, 10:1, detergent/Chl) for 1 h followed by sucrose density centrifugation (20–40%) (Beckman VTi 50 vertical rotor) for 7 h at 4 °C at 210,000 g.
A stock suspension of PS II core particles in 25 mM Mes-NaOH buffer (pH 6.5), containing 0.33 M sucrose, 15 mM NaCl, 10 mM CaCl2 and 0.03% (w/v) DM, was frozen in liquid nitrogen as small aliquots with a Chla concentration of 2.5 mg mL−1 and stored at − 80 °C.
To prepare manganese-depleted apo-WOC–PSII complexes, thawed intact PS II particles (0.5 mg Chl per ml) were incubated in the presence of 0.8 M Tris–HCl buffer (pH 8.3) for 30 min at 23 °C, followed by 3 times washing with the buffer (50 mM Mes-NaOH, pH 6.5) [31].
Final concentrations of P680 centers in the samples used for EPR measurements were 10 μM for PS II and 25 μM for apo-WOC–PSII, respectively. PS II and apo-WOC–PSII complexes were suspended in 30 mM Mes-NaOH (pH 7.5).
Nitroxides
Chemical structures of nitroxide radicals and their brief names used in this work are shown in Fig. 1. Spin labels 4-amino-2,2,6,6-tetramethylpiperidine-l-oxyl (Tamine) and 4-hydroxy-2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPOL) were purchased from Sigma (USA). Spin labels 4-amino-2,2,5,5-tetramethyl-3-imidazoline-1-oxyl (ATI) and 2,2,5,5-tetramethyl-4-nitromethylene-3-imidazolidine-N-oxyl (NTI) were synthesized by I.A. Grigor’ev and I.A. Kirilyuk (Institute of Organic Chemistry, Novosibirsk) as described in [29]. Spin label 4-hydroxy-2,2,6,6-tetramethylpiperidine-1-oxyl-acetate (TEMPOLacetate or Tacet) was a gift of professor E.K. Ruuge (Faculty of Physics, Moscow State University).
Fig. 1.

Chemical structures of spin labels used in this work
Concentrations of spin labels in the suspensions of PS II and apo-WOC–PSII used for EPR measurements were about 30 times higher than the concentrations of P680 in the samples, i.e., 300 and 750 μM, respectively.
EPR Measurements
The conventional first-derivative EPR spectra (first harmonic absorption in phase, 100 kHz field modulation) were measured with a Varian E-4 X-band spectrometer equipped with a variable temperature controller [32]. EPR spectra were recorded at a modulation amplitude of 0.5G and microwave power 1, 10, or 50 mW, as indicated. Samples were placed into a thin-walled gas-permeable capillaries made of optically transparent methylpentane polymer (TFE tubes from Zeus Inc.; internal diameter, 0.635 mm; wall thickness, 0.051 mm) as described in [29]. Measurements were performed at room temperature (~ 20–22 °C). Samples were illuminated by white light from a 100 W tungsten lamp (photon flux about 10 µEinstein m−2 s−1 at the sample surface); infrared light was cut-off with a 5 cm layer of water. The control experiments showed that illumination of water suspensions of spin labels (without PS II complexes) did not cause changes in the EPR spectra that might be caused by the light-induced destruction of the nitroxides.
Molecular Docking
Potential sites of spin label binding on the surface of PS II have been found with Autodock Vina software [33]. The docking sites were visualized and analyzed with VMD molecular viewer [34].
Results and Discussion
EPR Study of Spin Label Reduction by Photosystem II
In this section, we describe the results of the study of spin label interactions with intact core PS II complexes and the samples depleted of water-oxidizing complex (apo-WOC–PSII). In the case of apo-WOC–PSII, electron transport through PS II was supported by the addition of 10 mM NH2OH. The following nitroxide radicals were used (Fig. 1): ATI (pKa = 6.2), NTI (pKa = 8.0), TEMPOamine (Tamine, pKa = 8.9), TEMPOL, and TEMPOacetate (Tacet). All the radicals dissolved in aqueous solutions give triplet EPR signals with narrow lines, typical of nitroxide radicals rapidly tumbling in the water bulk phase (see, for example, the EPR spectrum of Tamine shown in Fig. 2a).
Fig. 2.
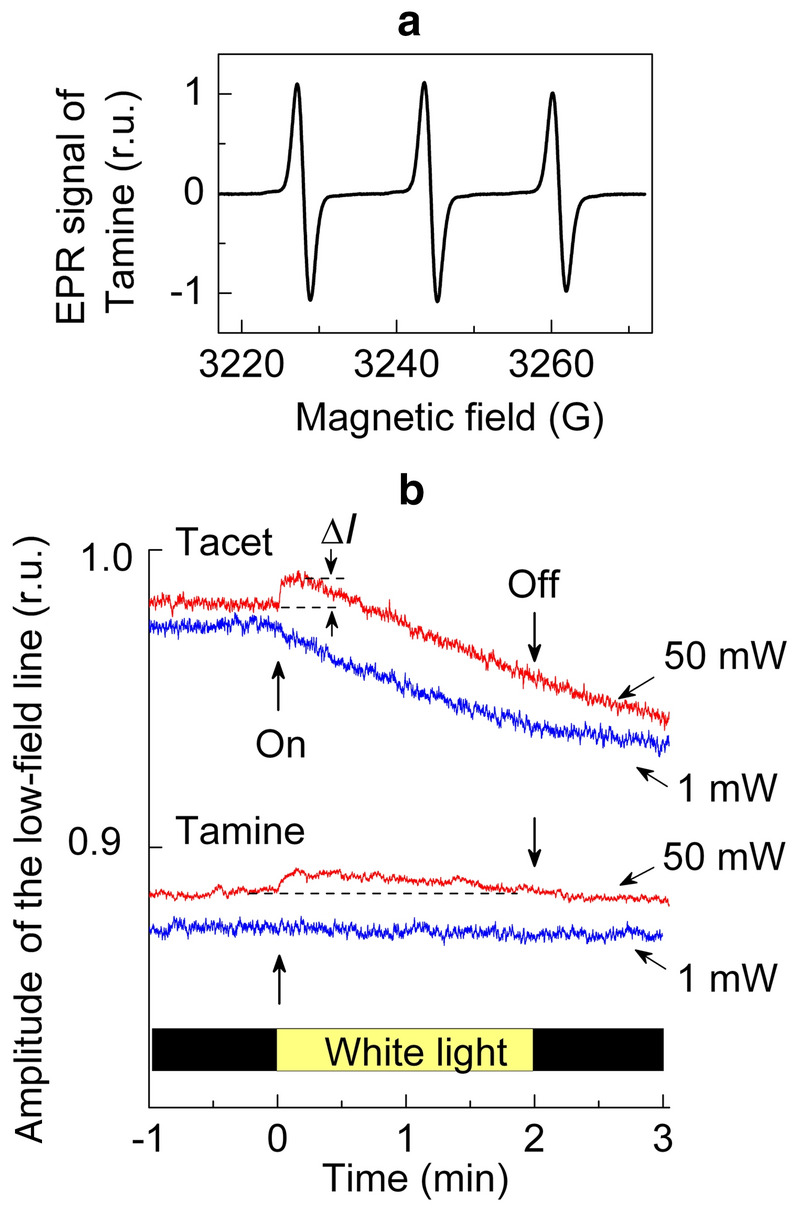
Panel a EPR spectrum of Tamine in the aqueous solution. Panel b the light-induced changes in the amplitude of the low-field lines of the EPR signals of Tacet (top) and Tamine (bottom) recorded at microwave power 1 mW (blue) and 50 mW (red) (color figure online)
The light-induced changes in the amplitude of the EPR signals were monitored by measuring the amplitudes of the low-field components of the EPR spectrum (Fig. 2). This choice is based on the fact that the low-field line is less sensitive to distortions caused by anisotropy of radical rotations. The response of water-soluble spin labels to illumination of PS II can be caused, in general, by different effects, including (i) the redox transients of nitroxide radical and (ii) the influence of paramagnetic 3O2 molecules on relaxation properties of radicals. We can discern these effects by recording the EPR signals at different levels of microwave power. Figure 2b shows the light-induced changes in the amplitude of the low-field lines of the EPR signals of Tacet and Tamine recorded at 1 and 50 mW. As one can see, the light-induced response of the signal amplitude depends on the structure of the spin label and the microwave power level. For Tacet recorded at 1 mW, the amplitude of the EPR signal gradually decreases after switching the light on. The decrease in the signal is caused by Tacet interaction (reduction or oxidation) with PS II complexes. In contrast to Tacet, the EPR signal of Tamine remains at the same level regardless of illumination. This indicates that Tamine is unable to interact with PS II. We assume that such a difference is accounted for by different availabilities of Tacet and Tamine to electron carriers of PS II.
When the EPR signals were recorded at microwave power 50 mW, we observed an insignificant initial increase in the amplitudes of the EPR lines of both radicals, Tacet and Tamine. This effect can be explained by the light-induced evolution of O2 dring illumination of PS II. Molecular oxygen is paramagnetic species (3O2, spin S = 1), which can influence the relaxation properties of nitroxide radicals [35–40]. The O2-dependent modulation of the signal amplitude is associated with the action of 3O2 as a paramagnetic “relaxator”. It is important to note that the sign of the effect caused by 3O2 depends on microwave power upon measuring the EPR signals of nitroxide radicals. Comparing the EPR signals of nitroxides recorded at low and high levels of microwave power (P), an experimenter can discriminate between the effects of O2 as the “relaxator” from other factors that can influence the EPR spectra (e.g., the loss of paramagnetism caused by redox transients [29]). At low microwave power (P < 1–5 mW), when the EPR signals of nitroxides are not saturated, increasing with the rise of P, 3O2 molecules reveal themselves as a line-broadening agent. In this case, the interaction of 3O2 with nitroxide radicals leads to broadening the EPR signal, accompanied by a decrease in the signal amplitude. Otherwise, at sufficiently high microwave power (P > 50 mW), molecular oxygen causes an opposite effect: 3O2 induces an increase in the signal amplitude [29]. This is because at high microwave power the rate of thermal relaxation of nitroxide radicals to the ground state is not sufficient to establish thermal equilibrium. In the meantime, paramagnetic 3O2 molecules accelerate the spin–lattice relaxation of nitroxide radicals, thereby releasing the broadening of the EPR signal at high microwave power. Thus, 3O2 attenuates the broadening of the radical signals at high microwave power, leading to an increase in the signal amplitude [24, 29, 36–40]. Comparing the time-courses of the light-induced changes in the EPR signal amplitude at P = 1 mW and P = 50 mW (Fig. 2), we observed the initial increase in the EPR line amplitudes at 50 mW; however, this effect was not observed at 1 mW. This demonstrates that the native PS II samples used in our experiments were photochemically active, producing molecular oxygen upon their illumination. Note that de-oxygenation of samples placed inside the gas-permeable TFE tubes by 10-min blowing with N2 gas eliminated the oxygen effect (data not shown).
Figure 3 shows the light-induced changes in the EPR signals of four different spin labels in suspensions of apo-WOC–PSII. In these cases, electron flow to the primary plastoquinone PQA was supported due to electrons donated to P680+ (via Yz) by NH2OH molecules. All the measurements with the apo-WOC–PSII complexes have been performed on de-oxygenated suspensions. This precluded the potential effects of the interactions of molecular oxygen with spin labels that might interfere with the redox transformations of nitroxide radicals. As noted above, paramagnetic molecules 3O2 may influence the relaxation parameters of nitroxide radicals that could reveal themselves in the time-course of the light-induced changes in the EPR signal. Also, we cannot exclude that superoxide radicals formed upon one-electron reduction of O2 molecules may act as the oxidant which would re-oxidize the > N–OH fragments of spin labels. Comparing the interaction of different spin labels with apo-WOC–PSII complexes, we have found that the rate of the light-induced reduction of EPR signal increases in the following sequence: TEMPOL < Tamine < Tacet < NTI. TEMPOL and Tamine exhibit lower reactivity. Similar to intact PS II complexes, apo-WOC–PSII reduces Tacet more rapidly than Tamine. Note that NTI, which contains the polar group > NO2, revealed much more rapid reduction than Tacet.
Fig. 3.
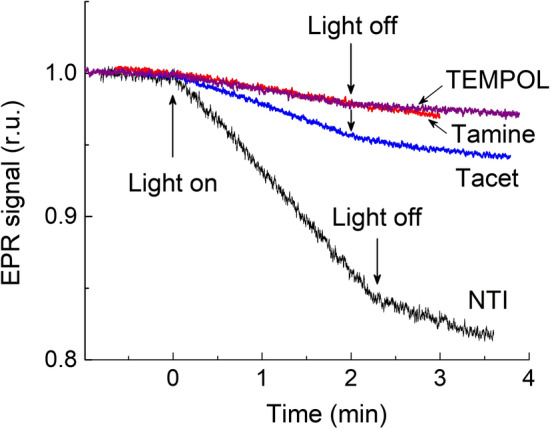
Kinetics of the light-induced changes in the amplitude of the low-field lines of the EPR signals of spin labels TEMPOL, Tamine, Tacet, and NTI in de-oxygenated solutions of apo-WOC–PSII; microwave power 10 mW
Figure 4 depicts a general scheme of spin label interactions with PS II. The light-induced loss of the nitroxide radical (> N–O·) paramagnetism may proceed, in principle, either due the radical > N–O·reduction to > N–OH or as the result of > N–O·oxidation to > N+ = O. Considering the line of arguments pro and contra these points, we can appeal, first of all, to structural data (see Figs. 5 and 6, and explanations in the text below). The architecture of the PS II complex suggests that the light-induced loss of paramagnetism of water-soluble nitroxide radicals occurs predominantly due to electron donation to nitroxides by electron carriers located on the acceptor side of PS II, most likely via PQA–. We can exclude PQB– as a potential donor of electrons for spin labels, because in isolated core complexes of PS II the binding sites of PQB were unoccupied. Concerning a hypothetical possibility of the light-induced oxidation of spin labels, we must note that the high-potential redox cofactors (oxidized centers P680+ and/or oxidized redox species in WOC) might be considered as the potential recipients of electrons from nitroxide radicals. However, we can exclude P680+ centers as direct acceptors of electrons from spin labels, because P680 is deeply buried into the core of the PS II protein complex. In WOC, high-potential redox intermediates are also sequestered from the water-soluble nitroxides. Thus, we conclude that it is the reduction of the nitroxide radicals by PQA– that is responsible predominantly for the light-induced loss of spin label paramagnetism.
Fig. 4.

The potential pathways of the nitroxide radical interactions with PS II
Fig. 5.
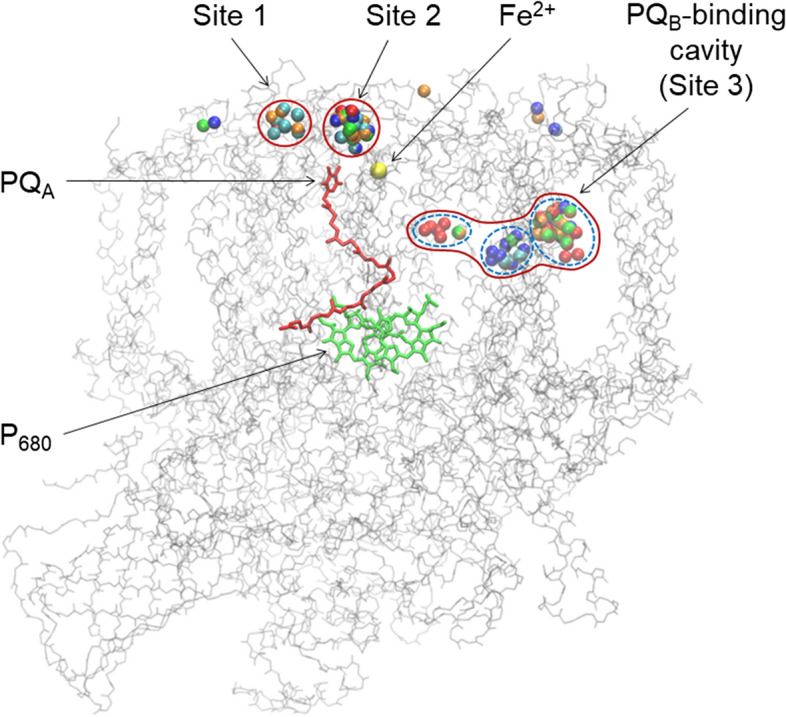
Binding sites of spin labels near the acceptor side of PS II, according to Autodock Vina prediction. Nitroxyl oxygen atoms of the spin labels are indicated by color: TEMPOL-acetate (red), TEMPOL (orange), Tamine (green), NTI (blue), ATI (cyan) (color figure online)
Fig. 6.

Panel a The sites of spin label binding to PS II closest to the redox group of plastoquinone PQA. Distances (in Angstroms) between the oxygen atom of the nitroxide groups of TEMPO-acetate and the O atom of the redox group of PQA are shown. Binding sites numbering is the same as in Fig. 5. Panel b shows the top view of the protein environment of the binding sites 1 and 2 on the surface of PS II. The coloration of the PS II residues is according to their properties: positively charged (blue), negatively charged (red), polar (green), nonpolar (white). Panel c shows the protein environment of the binding site 3. Position of the quinone PQB is depicted in black wireframe (color figure online)
Note that PS II complexes devoid of the Mn4CaO5 cluster can reduce the EPR signals of spin labels during illumination (Fig. 3). In this case, however, the rates of the signal decay were lower than in the intact PS II complexes. We assume that this might be caused by two reasons, (1) the changes in the midpoint redox potential of the redox pair PQA−/PQA in the apo-WOC–PSII, and (2) kinetic effects. The redox potential of the PQA−/PQA pair has been a subject of numerous researches and heated debates for decades [41–50]. Recently, this point was critically analyzed in view of the data from the Rutherford’s laboratory. According to [41, 42, 46], the primary plastoquinone PQA in the apo-WOC–PSII is characterized by higher (about 100–150 mV) redox potential of PQA than in the native PS II (Em(PQA−/PQA) = − 144 ± 2 mV). The bicarbonate ligation to the non-heme iron markedly increased the redox potential Em of PQA in both the native and the apo-WOC–PSII complexes [46]. Shibamoto et al. [43], confirmed the 150-mV redox difference with and without the Mn cluster. Thus, one might expect that the apo-WOC–PSII complexes will be of lower reducing power as compared to intact preparations of PS II, and this factor could slow down the loss of the radical EPR signal in the apo-WOC–PSII system.
Also, it is highly likely that there may be a kinetic effect, which reveals itself when the WOC is removed (apo-WOC–PSII) and the electron transfer from YZ to P680+ slows down [4, 6, 7]. It is well known that the properties of YZ in the apo-WOC–PSII differ dramatically from those in native PS II [4]. Although the light-induced electron flow through P680 can be maintained due to the presence of artificial electron donors, like NH2OH, the rate at which NH2OH in solution re-reduces YZ must be much slower than in intact PS II complexes. That is why the apo-WOC–PSII complexes revealed slower rates of nitroxide reduction as compared to the native PS II (Table 1).
Table 1.
Rates of the spin label reduction: parameter ΔA characterizes a decrease in the amplitude of the low-field line of the EPR signal with respect to its initial level A0
| Photosystem variant | Spin label | Spin label reduction rate per minute (ΔA/A0) × 102 |
|---|---|---|
| Intact PSII | Tacet | 6.9 ± 1.8 |
| TEMPOL | 1.4 ± 0.5 | |
| Tamine | 0.6 ± 0.4 | |
| Apo-WOC–PSII | NTI | 8.3 ± 2.2 |
| Tacet | 2.9 ± 0.9 | |
| ATI | 2.0 ± 0.6 | |
| TEMPOL | 1.1 ± 0.3 | |
| Tamine | 0.5 ± 0.1 |
Mean values ± SE (n = 4)
Results of the comparative study of the reduction of different nitroxide radicals by PS II are summarized in Table 1. Note that the reduction of different spin labels by apo-WOC–PSII revealed a similar regularity as in intact PS II complexes. We have found that the most significant rate of radical reduction was observed for spin labels NTI and Tacet, which contain the polar groups. Otherwise, electrically neutral spin labels TEMPOL and ATI (pKa = 6.2, ATI is deprotonated at pH 7.5), and positively charged Tamine (pKa = 8.9) showed lower rates of reduction then NTI and Tacet.
Taking together the above results, we assume that the basic reasons for different redox activities of the nitroxide radicals examined in this work are (1) various affinities (docking) of spin labels to PS II and (2) the differences in the reduction potentials of nitroxide radicals. To analyze the role of the first factor, we consider below the potential sites of spin label binding to PS II complex and their positions towards PQA. There are good reasons to believe that the reduced plastoquinone PQA– serves as the immediate electron donor for nitroxide radicals. The redox potential of the PQA–/PQA pair (Em ~ − 100 mV) is appropriate for the reduction of nitroxide radicals, and this process could be realized if the distance between the redox groups of PQA– and spin labels is sufficiently short to provide the electron transfer. In the context of electron transfer from PQA– to water-soluble nitroxides, we have also to mention a potential influence of the non-heme iron (NHI) on the rate of this reaction. The role of the NHI in PS II in general is not well understood [3–5, 48, 49]. Presumably, the NHI may serve as regulator of electron transport on the acceptor side of PS II, which influences the rate of electron transfer from PQA– to PQB [48, 49, 51–53]. We might suggest that the NHI could also act in a similar fashion, accelerating electron transfer from PQA− to nitroxide radicals.
Binding of Spin Labels to Photosystem II
Figure 5 highlights the suitable positions of the spin labels used in this work that can be clustered on the PS II surface in the close vicinity of the redox group of PQA (sites 1 and 2) and the binding sites in the PQB-binding pocket in PS II (site 3)1. The colored balls designate the selected positions of different spin labels determined by the docking method. We can see that different spin labels are clustered approximately in the areas, collectively designated here as sites 1, 2, and 3. The energies of spin label binding to these sites have been estimated as ΔE ≈ 6.0–6.5 kcal/mole. Characteristic distances between the redox moieties of PQA and spin labels positioned in sites 1, 2 and 3 are about 12, 14 and 22 Å, respectively (Fig. 6). According to the semi-empirical Dutton’s ruler [54], these distances fall in the interval that may ensure the electron transfer tunneling between PQA– and the redox centers of spin labels, provided there is an appropriate free energy difference between the donor (PQA–) and acceptor (the nitroxide group).
Binding sites 1 and 2 have positively charged amino acids (Arg28T and Arg251D in the site 1; Arg28T and Lys227A in the site 2); site 2 also contains negatively charged Glu25T. The rest of the site 2 surface is comprised of uncharged polar and nonpolar residues, including: Ser471C, Asn250D, Ile29T, Leu467C and Pro470C in site 1, Ser262D, Thr30T, Tyr224A, Pro26T and lipid molecules in site 2. Site 3 is comprised entirely of uncharged and mostly nonpolar residues, and lipids [3, 4]. The abundance of uncharged residues allows to suggest that the spin label binding to all three sites is governed by hydrophobic interactions. Positive charge of sites 1 and 2 pose obstacles for positively charged spin labels to bind these sites.
Chemical “Reactivity” of Spin Labels
Comparing the reaction activities of spin labels investigated in this work, we have found that NTI and Tacet, which contain polar groups, reveal relatively high rates of the nitroxide radical reduction by PS II. There are good reasons to believe that electron density in the radical fragment of NTI is reduced as compared to other spin labels. In NTI molecule, the NO2 group contains the positively charged N atom (Fig. 7a), which can attract electron density from the paramagnetic > N–O· fragment, thereby increasing its electrophilicity. A decrease in electron density on the N atom of the > N–O· group reveals itself as a decrease in the hyperfine splitting constant aiso of the NTI molecule. In the bottom panel of Fig. 7, we compare the EPR spectra of NTI and ATI molecules. Note that the NTI and deprotonated ATI molecules show different values of the splitting parameter aiso. Deprotonated (electrically neutral) ATI molecules reveal higher value of aiso than protonated (positively charged) ATI(H+) molecules. The protonation of ATI causes a marked decrease in aiso (Fig. 7, for more details, see [55, 56]). This observation supports the notion that the positive charge in the moiety of the spin label molecule reduces electron density in the radical fragment > N−O·. Interestingly, NTI and protonated ATI molecules reveal close values of the EPR spectrum parameter aiso. These data suggest that changes in the structure of spin labels causes the redistribution of the electron and spin densities over the spin label molecule. A decrease in the electron density of the radical fragment > N−O· might enhance the electrophilicity of this group, thereby increasing the redox potential of the > N−O·/ > N−OH redox pair (for references, see [27]). This can explain an enhanced capacity of NTI as the recipient of an electron donated by PS II.
Fig. 7.
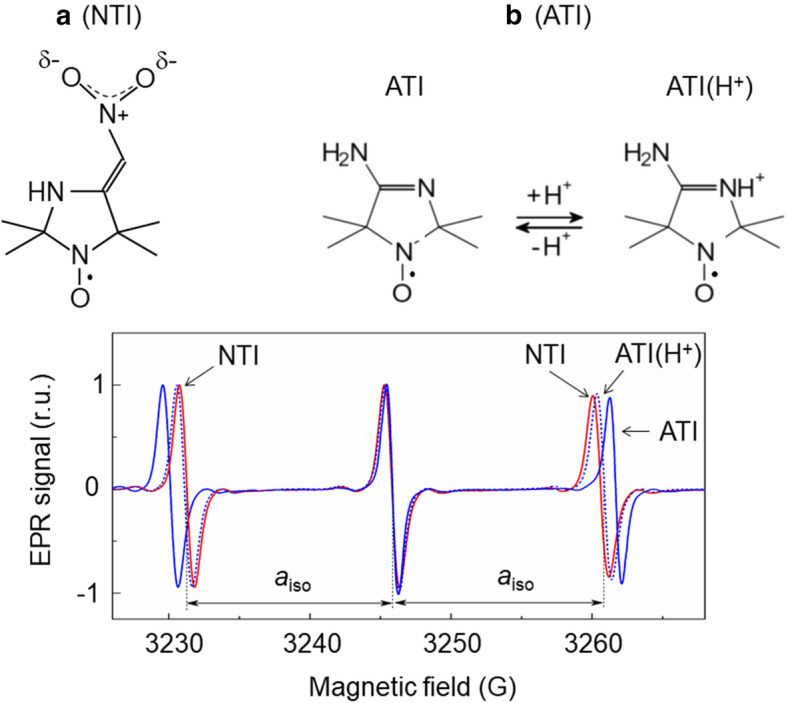
EPR spectra of spin labels NTI and ATI in water. EPR spectra of deprotonated and protonated forms of ATI were obtained for ATI solutions at pH 5 and pH 8
Conclusion
In this work, we have found that the redox activity of water-soluble nitroxide radicals depends on their chemical structure. Nitroxide radicals NTI (2,2,5,5-tetramethyl-4-nitromethylene-3-imidazolidine-N-oxyl) and Tacet (4-hydroxy-2,2,6,6-tetramethylpiperidine-1-oxyl-acetate) with the polar groups appear to be most efficient acceptors of electrons donated by PS II as compared to neutral or positively charged spin labels (TEMPOL and Tamine). We assume that enhanced reactivities of NTI and Tacet are determined by their relatively high redox potentials and their affinities to the binding sites on the surface (and/or in the PQB-binding pocket) of the PS II protein complex located at distances ~ 12–22 Å from the redox group of the primary plastoquinone PQA.
We dedicate this article to our dear and long-term collaborators, Klaus Moebius and Kev Salikhov, on the occasion of their glorious anniversaries.
Acknowledgements
We thank professors E.K. Ruuge and I.A. Grigor’ev, and Dr. I.A. Kirilyuk for the gifts of spin labels Tacet, ATI and NTI.
Author’s Contributions
BVT performed EPR measurements and EPR data processing; GEM analyzed the data using the docking procedure; MDA isolated and modified PS II complexes; AYuS participated in planning the work, discussion of the results and writing the manuscript; ANT participated in planning the work, supervision of EPR measurements, data processing and writing the manuscript.
Funding
This work was partly supported by the Russian Science Foundation (Grant 21–74-20047). The authors acknowledge partial support from Lomonosov Moscow State University Program of Development.
Declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Footnotes
Positions of remote binding sites are not shown.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Maio N, Lafont BAP, Sil D, Li Y, Bollinger JM, Jr, Krebs C, Pierson TC, Linehan WM, Rouault TA. Science. 2021 doi: 10.1126/science.abi5224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zielonka J, Joseph J, Sikora A, Hardy M, Ouari O, Vasquez-Vivar J, Cheng G, Lopez M, Kalyanaraman B. Chem. Rev. 2017;117:9973. doi: 10.1021/acs.chemrev.7b00042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Umena Y, Kawakami K, Shen JR, Kamiya N. Nature. 2011;473:55. doi: 10.1038/nature09913. [DOI] [PubMed] [Google Scholar]
- 4.Guskov A, Kern J, Gabdulkhakov A, Brose M, Zouni A, Saenger W. Nat. Struct. Molec. Biol. 2009;16:334. doi: 10.1038/nsmb.1559. [DOI] [PubMed] [Google Scholar]
- 5.Renger G. Biochim. Biophys. Acta Bioenerg. 2004;1655:195. doi: 10.1016/j.bbabio.2003.07.007. [DOI] [PubMed] [Google Scholar]
- 6.Styring S, Sjöholm J, Mamedov F. Biochim. Biophys. Acta Bioenerg. 2012;1817:76. doi: 10.1016/j.bbabio.2011.03.016. [DOI] [PubMed] [Google Scholar]
- 7.Lubitz W, Chrysina M, Cox N. Photosynth. Res. 2019;142:105. doi: 10.1007/s11120-019-00648-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Semenov A, Cherepanov D, Mamedov M. Photosynth. Res. 2008;98:121. doi: 10.1007/s11120-008-9377-z. [DOI] [PubMed] [Google Scholar]
- 9.Mamedov MDMD, Kurashov VNVN, Cherepanov DADA, Semenov AYAY. Front. Biosci. 2010;15:1007. doi: 10.2741/3658. [DOI] [PubMed] [Google Scholar]
- 10.Rao KK, Hall DO, Vlachopoulos N, Grätzel M, Evans MCW, Seibert M. J. Photochem. Photobiol. B Biol. 1990;5:379. doi: 10.1016/1011-1344(90)85052-X. [DOI] [Google Scholar]
- 11.Fang X, Sokol KP, Heidary N, Kandiel TA, Zhang JZ, Reisner E. Nano. Lett. 2019;19:1844. doi: 10.1021/acs.nanolett.8b04935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang JZ, Reisner E. Nat. Rev. Chem. 2020;4:6. doi: 10.1038/s41570-019-0149-4. [DOI] [Google Scholar]
- 13.Kato M, Cardona T, Rutherford AW, Reisner E. J. Am. Chem. Soc. 2012;134:8332. doi: 10.1021/ja301488d. [DOI] [PubMed] [Google Scholar]
- 14.Ikeda T, Senda M, Shiraishi T, Takahashi M, Asada K. Chem. Lett. 1989;18:913. doi: 10.1246/cl.1989.913. [DOI] [Google Scholar]
- 15.Shevela D, Messinger J. Biochim. Biophys. Acta Bioenerg. 2012;1817:1208. doi: 10.1016/j.bbabio.2012.03.038. [DOI] [PubMed] [Google Scholar]
- 16.Kato M, Zhang JZ, Paul N, Reisner E. Chem. Soc. Rev. 2014;43:6485. doi: 10.1039/C4CS00031E. [DOI] [PubMed] [Google Scholar]
- 17.Longatte G, Fu HY, Buriez O, Labbé E, Wollman FA, Amatore C, Rappaport F, Guille-Collignon M, Lemaître F. Biophys. Chem. 2015;205:1. doi: 10.1016/j.bpc.2015.05.003. [DOI] [PubMed] [Google Scholar]
- 18.Ravi SK, Tan SC. Energy Environ. Sci. 2015;8:2551. doi: 10.1039/C5EE01361E. [DOI] [Google Scholar]
- 19.Brinkert K, Le Formal F, Li X, Durrant J, Rutherford AW, Fantuzzi A. Biochim. Biophys. Acta Bioenerg. 2016;1857:1497. doi: 10.1016/j.bbabio.2016.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.N. Plumeré, and M. M. Nowaczyk, Adv. Biochem. Eng. Biotechnol. (Springer Science and Business Media Deutschland GmbH, 2016), pp. 111–136 [DOI] [PubMed]
- 21.Li Z, Wang W, Ding C, Wang Z, Liao S, Li C. Energy Environ. Sci. 2017;10:765. doi: 10.1039/C6EE03401B. [DOI] [Google Scholar]
- 22.Saboe PO, Conte E, Farell M, Bazan GC, Kumar M. Energy Environ. Sci. 2017;10:14. doi: 10.1039/C6EE02801B. [DOI] [Google Scholar]
- 23.Fu H-Y, Picot D, Choquet Y, Longatte G, Sayegh A, Delacotte J, Guille-Collignon M, Lemaître F, Rappaport F, Wollman F-A. Nat. Commun. 2017;8:15274. doi: 10.1038/ncomms15274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Larom S, Kallmann D, Saper G, Pinhassi R, Rothschild A, Dotan H, Ankonina G, Schuster G, Adir N. Photosynth. Res. 2015;126:161. doi: 10.1007/s11120-015-0075-3. [DOI] [PubMed] [Google Scholar]
- 25.R. I. Pinhassi, D. Kallmann, G. Saper, S. Larom, A. Linkov, A. Boulouis, M. A. Schöttler, R. Bock, A. Rothschild, N. Adir, and G. Schuster, PLoS One 10, (2015) [DOI] [PMC free article] [PubMed]
- 26.A. N. Tikhonov, and W. K. Subczynski, in Biomed. EPR, Part A Free Radicals, Met. Med. Physiol. Biol. Magn. Reson., (ed.) S. R. Eaton, G. R. Eaton, and L. J. Berliner (Springer, Boston, MA, 2005), pp. 147–194
- 27.Kocherginsky N, Swartz HM. Nitroxide spin labels: reactions in biology and chemistry. 1. Boca Raton: CRC Press; 1995. [Google Scholar]
- 28.Tikhonov AN. Cell Biochem. Biophys. 2017;75:421. doi: 10.1007/s12013-017-0797-2. [DOI] [PubMed] [Google Scholar]
- 29.Ptushenko VV, Ikryannikova LN. Appl. Magn. Reson. 2006;30:329. doi: 10.1007/BF03166204. [DOI] [Google Scholar]
- 30.Haag E, Irrgamg K-D, Boekema EJ, Renger G. Eur. J. Biochem. 1990;189:47. doi: 10.1111/j.1432-1033.1990.tb15458.x. [DOI] [PubMed] [Google Scholar]
- 31.Gopta OA, Tyunyatkina AA, Kurashov VN, Semenov AY, Mamedov MD. Eur. Biophys. J. 2008;37:1045. doi: 10.1007/s00249-007-0231-6. [DOI] [PubMed] [Google Scholar]
- 32.Kuvykin IV, Ptushenko VV, Vershubskii AV, Tikhonov AN. Biochim. Biophys. Acta Bioenerg. 2011;1807:336. doi: 10.1016/j.bbabio.2010.12.012. [DOI] [PubMed] [Google Scholar]
- 33.Trott O, Olson AJ. J. Comput. Chem. 2010;31:455. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Humphrey W, Dalke A, Schulten K. J. Mol. Graph. 1996;14:33. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 35.Backer JM, Budker VG, Eremenko SI, Molin YN. Biochim. Biophys. Acta Bioenerg. 1977;460:152. doi: 10.1016/0005-2728(77)90161-X. [DOI] [PubMed] [Google Scholar]
- 36.J. S. Hyde and W. K. Subczynski, in Biol. Magn. Reson., edited by L. J. Berliner and J. Reuben (Plenum Press, New York, NY, 1989), pp. 399–425.
- 37.Molin YN, Salikhov KM, Zamaraev KI. Spin exchange Principles and Applications in Chemistry and Biology. Berlin: (Springer-Verlag; 1980. [Google Scholar]
- 38.Walczak T, Sarna T, Swartz HM, Strzalka K. Arch. Biochem. Biophys. 1990;281:312. doi: 10.1016/0003-9861(90)90449-9. [DOI] [PubMed] [Google Scholar]
- 39.Ligeza A, Tikhonov AN, Subczynski WK. Biochim. Biophys. Acta Bioenerg. 1997;1319:133. doi: 10.1016/S0005-2728(96)00122-3. [DOI] [Google Scholar]
- 40.Tikhonov AN, Subczynski WK. Cell Biochem. Biophys. 2019;77:47. doi: 10.1007/s12013-018-0861-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Krieger A, Rutherford AW, Johnson GN. Biochim. Biophys. Acta Bioenerg. 1995;1229:193. doi: 10.1016/0005-2728(95)00002-Z. [DOI] [Google Scholar]
- 42.Johnson GN, Rutherford AW, Krieger A. Biochim. Biophys. Acta Bioenerg. 1995;1229:202. doi: 10.1016/0005-2728(95)00003-2. [DOI] [Google Scholar]
- 43.Shibamoto T, Kato Y, Sugiura M, Watanabe T. Biochemistry. 2009;48:10682. doi: 10.1021/bi901691j. [DOI] [PubMed] [Google Scholar]
- 44.Kato Y, Nagao R, Noguchi T. Proc. Natl. Acad. Sci. U. S. A. 2016;113:620. doi: 10.1073/pnas.1520211113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kato Y, Ohira A, Nagao R, Noguchi T. Biochim. Biophys. Acta Bioenerg. 2019;1860:148082. doi: 10.1016/j.bbabio.2019.148082. [DOI] [PubMed] [Google Scholar]
- 46.Brinkert K, De Causmaecker S, Krieger-Liszkay A, Fantuzzi A, Rutherford AW. Proc. Natl. Acad. Sci. U. S. A. 2016;113:12144. doi: 10.1073/pnas.1608862113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Messant M, Timm S, Fantuzzi A, Weckwerth W, Bauwe H, Rutherford AW, Krieger-Liszkaya A. Plant Physiol. 2018;177:1277. doi: 10.1104/pp.18.00341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cardona T, Sedoud A, Cox N, Rutherford AW. Biochim. Biophys. Acta Bioenerg. 2012;1817:26. doi: 10.1016/j.bbabio.2011.07.012. [DOI] [PubMed] [Google Scholar]
- 49.Müh F, Glöckner C, Hellmich J, Zouni A. Biochim. Biophys. Acta Bioenerg. 2012;1817:44. doi: 10.1016/j.bbabio.2011.05.021. [DOI] [PubMed] [Google Scholar]
- 50.Saito K, Rutherford AW, Ishikita H. (2013) Mechanism of proton-coupled quinone reduction in Photosystem II. Proc. Natl. Acad. Sci. U.S.A. 2013;110:954. doi: 10.1073/pnas.1212957110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wraight CA. FEBS Lett. 1978;93:283. doi: 10.1016/0014-5793(78)81122-3. [DOI] [PubMed] [Google Scholar]
- 52.Diner BA, Petrouleas V, Wendoloski JJ. Physiol. Plant. 1991;81:423. doi: 10.1111/j.1399-3054.1991.tb08753.x. [DOI] [Google Scholar]
- 53.Petrouleas V, Crofts AR. In: Photosystem II: the light-driven water: plastoquinone oxidoreductase. Wydrzynski T, Satoh K, editors. Dordrecht: Springer; 2005. pp. 177–206. [Google Scholar]
- 54.Page CC, Moser CC, Chen X, Dutton PL. Nature. 1999;402:47. doi: 10.1038/46972. [DOI] [PubMed] [Google Scholar]
- 55.Ikryannikova LN, Ustynyuk LY, Tikhonov AN. J. Phys. Chem. A. 2004;108:4759. doi: 10.1021/jp037943d. [DOI] [Google Scholar]
- 56.Tikhonov AN, Agafonov RV, Grigor’e Kirilyuk Ptushenko Trubitsin IAIAVVBV. Biochim. Biophys. Acta Bioenerg. 2008;1777:285. doi: 10.1016/j.bbabio.2007.12.002. [DOI] [PubMed] [Google Scholar]