

Abstract
Caffeine has been shown to promote calcium-dependent activation of AMP-activated protein kinase (AMPK) and AMPK-dependent glucose and fatty acid uptake in mammalian skeletal muscle. Though caffeine has been shown to promote autophagy in various mammalian cell lines it is unclear if caffeine-induced autophagy is related to the calcium-dependent activation of AMPK. The purpose of this study was to examine the role of calcium-dependent AMPK activation in regulating caffeine-induced autophagy in mammalian skeletal muscle cells. We discovered that the addition of the AMPK inhibitor Compound C could significantly reduce the expression of the autophagy marker microtubule-associated protein 1 light chain 3b-II (LC3b-II) and autophagic vesicle accumulation in caffeine treated skeletal muscle cells. Additional experiments using pharmacological inhibitors and RNA interference (RNAi) demonstrated that the calcium/calmodulin-activated protein kinases CaMKKβ and CaMKII contributed to the AMPK-dependent expression of LC3b-II and autophagic vesicle accumulation in a caffeine dose-dependent manner. Our results indicate that in skeletal muscle cells caffeine increases autophagy by promoting the calcium-dependent activation of AMPK.
Keywords: Autophagy, Calcium, Calcium/calmodulin-activated protein, kinase II, AMP-activated protein kinase, Caffeine
1. Introduction
The release of endogenous calcium stores from the extensive sarcoplasmic reticular (SR) network represents one of the primary signaling mechanisms in skeletal muscle cells [1]. During periods of exercise and physical activity, the increased frequency and amplitude of intracellular calcium spikes can substantially contribute to cellular protein turnover and removal of dysfunctional or damaged organelles [2]. Eukaryotic cells degrade long-lived proteins, organelles, and bits of cytoplasm via the lysosome-dependent mechanism of macroautophagy, here after referred to as autophagy [3,4]. Autophagy is a highly conserved and tightly regulated process, which involves the formation of double membrane vesicles (autophagosomes) around molecules that have been targeted for degradation [5]. Once the autophagosome has enveloped the targeted molecules it shuttles its cargo to the lysosome where it subsequently fuses with the lysosomal membrane to form an autophagolysosome [5]. Though autophagy is a constitutively active process in skeletal muscle cells it is also highly inducible under specific physiological conditions [6]. Nutrient and energy deprivation, as well as muscle contraction, can promote a calcium-induced increase in autophagosome formation in eukaryotic cells [7–9].
Multiple pathways have been shown to regulate calcium-induced autophagy in mammalian cells [10]. Perhaps the most well characterized pathway is the calcium/calmodulin-dependent protein kinase kinase β (CaMKKβ) activation of AMP-activated protein kinase (AMPK) [11]. Activated AMPK phosphorylates UNC-51-like kinase-1 (ULK1) and forkhead box O3a (FOXO3a), both of which promote autophagosome formation [11]. Previous investigations have found that increasing cytoplasmic calcium levels in several mammalian cell types, including skeletal muscle, can promote an AMPK-independent increase in calcium-induced autophagy [12,13]. In addition, other calcium-induced protein kinases, besides CaMKKβ, have been shown to play a direct role in promoting autophagy [14]. Calcium/calmodulin-dependent protein kinase I (CaMKI) was previously shown to promote autophagy in an AMPK-independent manner in human osteosarcoma cells [14]. However, CaMKI is not highly expressed in mammalian skeletal muscle cells [15]. The most highly expressed CaMK in skeletal muscle cells is CaMKII [15]. Previous investigations have shown that CaMKII can contribute to AMPK activity in mammalian skeletal muscle cells [16,17]. It remains unclear, however, if CaMKII can contribute to AMPK-dependent autophagy induction in skeletal muscle cells.
Caffeine specifically increases the calcium-sensitivity of the luminal domain of the reticular ryanodine receptor (RyR) [18]. Therefore, caffeine significantly reduces the threshold for reticular calcium-induced calcium release, which greatly increases the frequency, but not necessarily the amplitude, of intracellular calcium spikes [18–20]. Previous studies have demonstrated that caffeine can significantly increase autophagy in mammalian liver [21] and cancer cell lines [22]. Caffeine has been shown to promote increased AMPK-dependent glucose uptake and fatty acid utilization by increasing the calcium-dependent activation of AMPK in mammalian skeletal muscle cells [16]. However, it remains unclear if caffeine can promote autophagy in skeletal muscle cells by specifically promoting the calcium-dependent activation of AMPK. The purpose of this study was to examine the role of calcium-induced AMPK activation in regulating caffeine-induced autophagy in mammalian skeletal muscle cells. We hypothesized that caffeine would promote a calcium-dependent increase in AMPK-mediated autophagy in mammalian skeletal muscle cells. Our findings suggest that caffeine promotes autophagy in an AMPK-dependent manner and that both CaMKKβ and CaMKII significantly contribute to AMPK-dependent autophagy in caffeine treated skeletal muscle cells.
2. Materials and methods
2.1. Materials
Fetal bovine serum (FBS), horse serum (HS), penicillin/streptomycin (pen/strep), and Dulbecco’s modified Eagle’s medium (DMEM) were purchase from Life Technologies (Grand Island, NY, USA). Primary antibodies for tubulin, phospho-AMPKα1/2 (p-AMPK, Thr172) and AMPKα1/2 and small interfering RNA (siRNA) for CaMKIIβ, CaMKIIδ and CaMKIIγ were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Primary antibodies for phospho-CaMKII (p-CaMKII, Ser286), and CaMKII were purchased from Cell Signaling Technology (Beverly, MA, USA). Primary antibodies for microtubule-associated protein 1 light chain 3b (LC3b) were purchased from Novus Biologicals (Littleton, CO, USA). All secondary antibodies were purchased from Vector Laboratories (Burlingame, CA, USA). All pharmacological agents were purchased from EMD Millipore (Darmstadt, Germany).
2.2. Cell culture
C2C12 myoblasts were purchased from ATCC (Manassas, VA, USA). Myoblasts were grown to confluence in normal growth media (DMEM plus 10% FBS, 100 U/ml penicillin, 100 μg/ml streptomycin) at 37 °C in a water-saturated atmosphere of 5% CO2. To promote differentiation of mono-nucleated myoblasts into multi-nucleated myotubes the media was switched to differentiation media (DMEM plus 2% HS, 100 U/ml penicillin, 100 μg/ml streptomycin). Fully differentiated myotubes were normally present upon 5–6 d incubation in differentiation media.
2.3. Pharmacological inhibitor-based experiments
Myoblasts were seeded on 6-well plates in growth media at a density of 1 × 104 cell/well. After 2–3 d in growth media, all cells were switched to differentiation media and incubated for an additional 5–6 d. For caffeine experiments, myotubes either received vehicle (DMSO) or a caffeine dose of 2.5 mM or 10 mM. To assess the overall role of AMPK in regulating caffeine-induced autophagy cells either received vehicle or a 20 μM dose of the specific AMPK-inhibitor Compound C. To determine the role of CaMKII, myotubes either received 10 μM of the specific CaMKII inhibitor KN93 or 10 μM of its negative control KN92. To examine the role of CaMKKβ, myotubes either received vehicle, a 30 μM dose of the CaMKKβ-inhibitor STO. All experiments were conducted for 6 h and all possible experimental scenarios were tested.
2.4. RNA interference (RNAi) experiments
Myoblasts were seeded on 6-well plates in antibiotic free growth media at a density of 1 × 104 cell/well and incubated for 48 h and then switched into antibiotic free differentiation media for an additional 48 h. To inhibit the expression of CaMKII isoforms expressed in skeletal muscle cells, each well (experimental replicate) received 60 nM siRNA for CaMKIIβ, CaMKIIδ, and CaMKIIγ (180 nM total). Control cells received 180 nM control siRNA (scramble) per well. All samples were transfected according to the manufacturers protocol and incubated in antibiotic free differentiation media for an additional 72 h. Triplicate groups of control and CaMKII siRNA transfected cells received either vehicle or 10 mM caffeine for 6 h.
2.5. Western blotting
Cell lysates were collected in lysis buffer (1× TBS, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS, 0.004% sodium azide, 2 mM PMSF, 1 mM sodium orthovanadate, 10 μl protease inhibitor cocktail), immediately snap frozen in liquid nitrogen, and stored at −20°. All samples were vortexed and centrifuged at 15,000g for 15 min at 4 °C and the supernatants were collected. The total protein concentration of each sample was determined using a Bradford assay. An equal volume of supernatant was combined with 2× Laemmli buffer and boiled for 10 min. Twenty micrograms of each denatured sample was submitted to SDS–PAGE using either 7.5% or 12% polyacrylamide gels and subsequently transferred on to a PVDF membrane (EMD Millipore, Darmstadt, Germany). All membranes were blocked for 1 h in 5% bovine serum albumin (BSA) dissolved in Tris-buffered saline plus 0.1% Tween 20 (TBST), incubated with primary antibody (1/1000) over night at 4 °C and subsequently labeled with an appropriate HRP-labeled secondary antibody (1/10,000) for 1 h at room temperature. Once satisfactory images were obtained each membrane was stained with coomassie brilliant blue (R-250) for 5 min, washed in TBST and imaged for total protein assessment. Unless otherwise stated, all blots were normalized to total protein. Blots were developed using standard ECL detection and images were acquired on a FluorChem E System imager (ProteinSimple, Santa Clara, CA, USA). Digital images were analyzed using ImageJ 1.47v (National Institutes of Health, Bethesda, MD, USA).
2.6. Fluorescence microscopy
Myoblasts were seeded on 35 mm dishes that contained a 1.5 mm thick collagen coated coverslip (MatTek Corporation, Ashland, MA, USA) in normal growth media. Once the cells were 75% confluent they were switched into differentiation media for 5–6 d to promote the formation of multi-nucleated myotubes. Triplicate groups of cells were exposed to vehicle (DMSO, control) or a combination of 10 mM caffeine, 20 μM Compound C, 10 μM KN92, 10 μM KN93 and 30 μM STO. All possible experimental scenarios were tested using these treatment groups. All samples were incubated in normal differentiation media containing 2 μl/ml of Cyto-ID Green (fluorescein) Detection Reagent (Enzo Life Sciences, Farmingdale, NY, USA) and 1 μl/ml of Hoechst 33342 nuclear stain for 30 min at 37 °C. All samples were washed in TBS and fixed with 4% paraformaldehyde for 20 min. Samples were analyzed using a Nikon Eclipse TS100-F microscope and NIS Elements Br software (Nikon Instruments, Melville, NY, USA). Autophagic vesicle accumulation was analyzed using the method of Williams et al. [23]. Briefly, images were captured in the FITC channel using a 40× fluorite objective. Because the fluorescein containing Cyto-ID dye selectively labels autophagic vesicles (autophagosomes and auto-phagolysosomes) the mean FITC intensity for a given sample was used as a quantitative measure of autophagic vesicle accumulation. Ten images were randomly captured on each coverslip (replicate sample), which were subsequently used to calculate the mean FITC intensity for each given sample and thus for each experimental treatment (n = 3).
2.7. Statistical analysis
Each experiment was repeated at least three times. Statistical differences between groups were analyzed using a student’s t-test or a one-way ANOVA with subsequent post hoc analysis, as appropriate. A p-value <0.05 was considered significant.
3. Results
3.1. Caffeine promotes autophagy in skeletal muscle cells in an AMPK-dependent manner
To determine if AMPK significantly contributes to autophagy induction in caffeine treated skeletal muscle cells we first conducted Western blot analysis to examine the dose-dependent expression of the autophagy marker LC3b-II. Our analysis revealed that the addition of 20 μM Compound C could significantly attenuate the expression of LC3b-II in 10 mM caffeine treated cells (Fig. 1A). To verify these results we conducted an autophagy assay in which autophagic vesicles were labeled with fluorescein-containing Cyto-ID dye and analyzed using fluorescence microscopy (Fig. 1B). Our analysis revealed that 10 mM caffeine significantly increased the accumulation of autophagic vesicles, which was completely inhibited by the addition of Compound C.
Fig. 1.
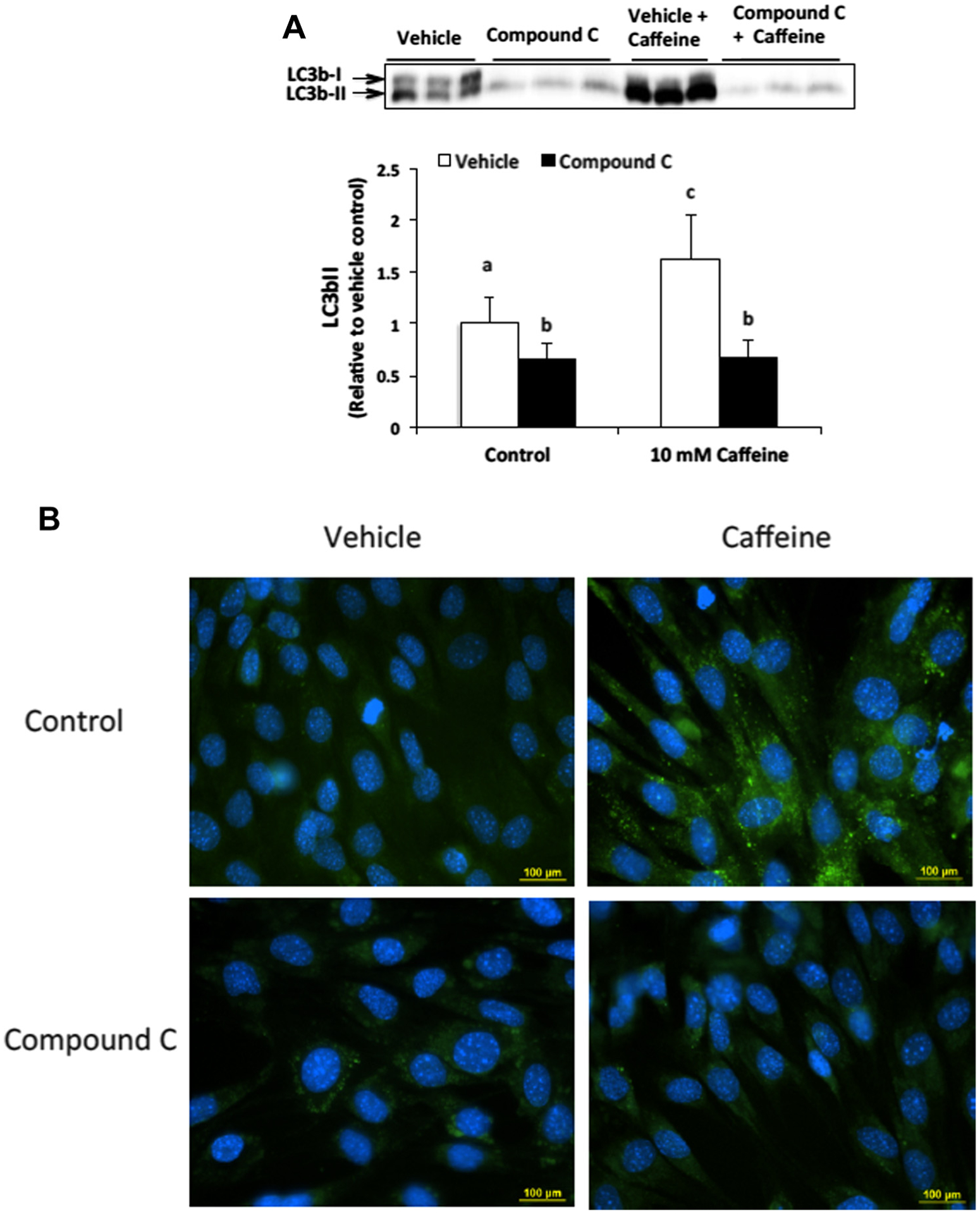
AMPK contributes to autophagy induction in caffeine treated skeletal muscle cells. (A) C2C12 myotubes were treated with either vehicle or Compound C (20 μM) alone or in combination with 10 mM caffeine for 6 h (n = 3). Western blot analysis revealed that caffeine significantly increased LC3b-II expression in an AMPK-dependent manner. Once a satisfactory image was obtained each membrane was stained with coomassie brilliant blue (R-250) and imaged for total protein assessment to allow each sample to be normalized to total protein content of each lane (data not shown). (B) Fluorescence microscopy analysis confirmed that Compound C inhibited autophagic vesicle formation in caffeine treated myotubes (n = 3). a–cIndicate significant difference between treatments without same letters (P < 0.05).
3.2. CaMKII contributes to increased AMPK phosphorylation in caffeine treated skeletal muscle cells in a dose-dependent manner
We utilized the specific CaMKII inhibitor KN93 and its negative control KN92 to examine the contribution of CaMKII to AMPK phosphorylation in caffeine treated myotubes. Our Western blot analysis revealed that treating myotubes with 2.5 mM caffeine for 6 h promoted a significant increase in AMPK phosphorylation that was independent of CaMKII (Fig. 2A–C). In contrast, treating myotubes with 10 mM caffeine for 6 h promoted a significant increase in AMPK phosphorylation that was partially dependent upon CaMKII activation (Fig. 2A–C). However, the addition of 30 μM STO largely inhibited caffeine induced AMPK phosphorylation, indicating that CaMKKβ was the primary mediator of calcium-induced AMPK phosphorylation in caffeine treated myotubes (Fig. 2A and C). Further analysis revealed that treating myotubes with 10 mM caffeine promoted a significant increase in CaMKII phosphorylation, while treating myotubes with 2.5 mM caffeine had no significant effect on CaMKII phosphorylation (Fig. 2D). To verify the results of our pharmacological inhibitor-based experiments, we conducted experiments using siRNAs for all three isoforms of CaMKII that are expressed in skeletal muscle cells. We were able to consistently reduce the expression of CaMKII isoforms by approximately 75% (Fig. 2E). Western blot analysis revealed a significant reduction in AMPK phosphorylation in 10 mM caffeine-treated myotubes that were transfected with CaMKII siRNA versus myotubes transfected with control siRNA (Fig. 2F).
Fig. 2.
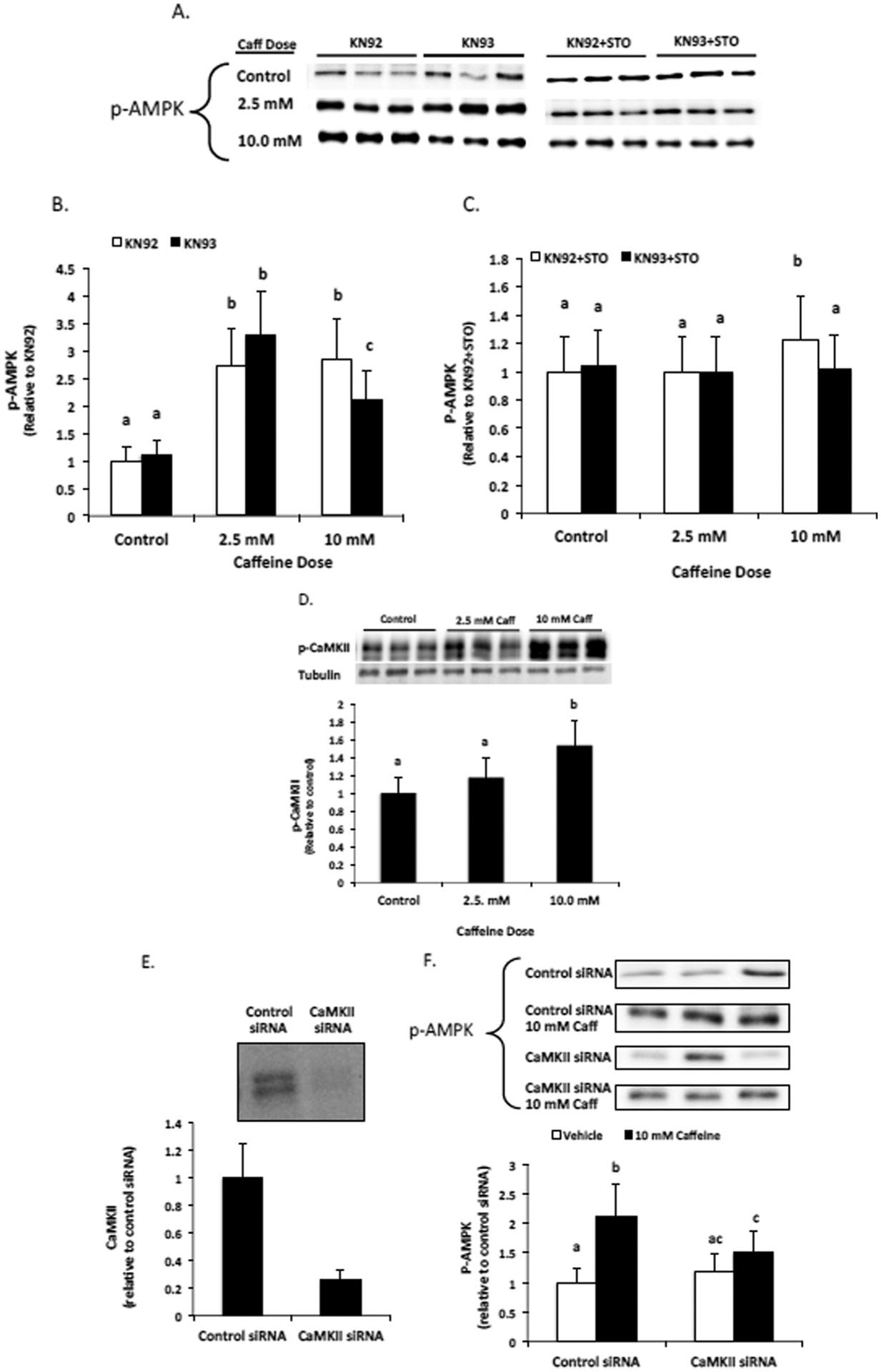
CaMKII contributes to AMPK phosphorylation in caffeine treated skeletal muscle cells. C2C12 myotubes were treated with the CaMKII inhibitor KN93 (10 μM) or its positive control KN92 (10 μM) alone or in combination with CaMKKβ inhibitor STO (30 μM) for 6 h. (A–C) Western blot analysis revealed that CaMKII contributed to AMPK phosphorylation in 10 mM, but not 2.5 mM, caffeine treated myotubes. (D) Further analysis revealed that CaMKII phosphorylation was significantly increased in only the 10 mM caffeine treated cells. (E) The efficiency of siRNA knockdown of CaMKII and (F) the effect of CaMKII knockdown on 10 mM caffeine-induced AMPK phosphorylation was assessed by Western blot. Once a satisfactory image was obtained each membrane was stained with coomassie brilliant blue (R-250) and imaged for total protein assessment to allow each sample to normalized to total protein content of each lane (data not shown). a–cIndicate significant difference between treatments without same letters (P < 0.05).
3.3. CaMKII contributes to increased LC3b-II expression in caffeine treated skeletal muscle cells
To determine if CaMKII contributed to AMPK-dependent autophagy induction we conducted Western blotting to analyze the expression of autophagy marker protein LC3b-II in samples from both pharmacological inhibitor and RNAi experiments. Our results indicate that caffeine increases LC3b-II expression in a dose-dependent manner and that CaMKII activation is required for maximal LC3b-II expression in 10 mM, but not 2.5 mM, caffeine treated myotubes (Fig. 3A, B and D). To determine if caffeine-induced autophagy was dependent upon CaMKKβ activation of AMPK, we treated myotubes with the CaMKKβ inhibitor STO. Using Western blot analysis we found that adding 30 μM STO completely inhibited the increased expression of LC3b-II in 2.5 mM and 10 mM caffeine treated myotubes (Fig. 3A and C).
Fig. 3.
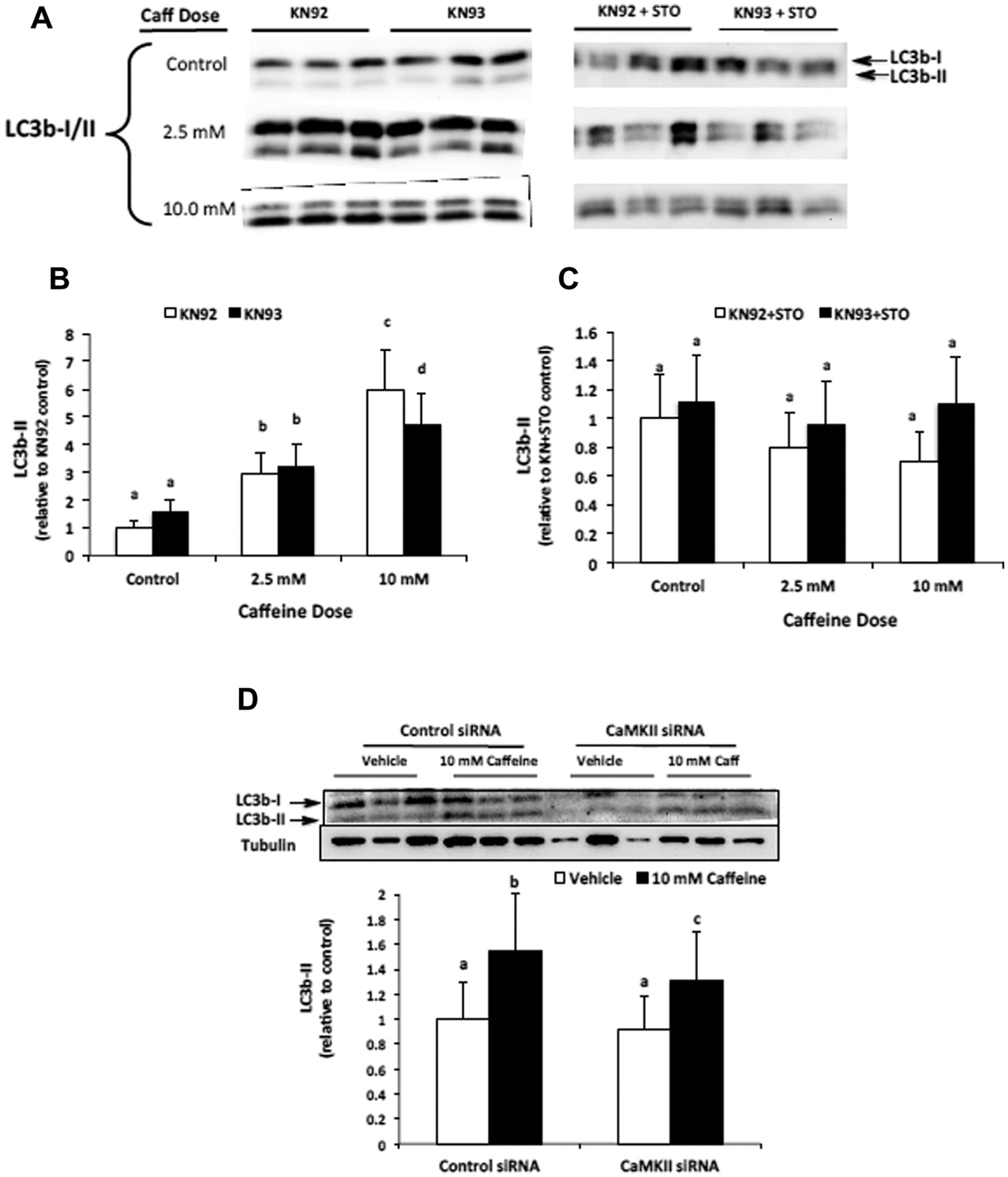
CaMKII contributes to AMPK-mediated LC3b-II expression in caffeine treated myotubes. C2C12 myotubes were treated with the CaMKII inhibitor KN93 (10 μM) or its positive control KN92 (10 μM) alone or in combination with CaMKKβ inhibitor STO (30 μM) for 6 h. (A and B) Western blot analysis revealed that caffeine increased LC3b-II expression in a dose-dependent manner and that CaMKII contributed to increased LC3b-II expression in 10 mM, but not 2.5 mM, caffeine treated myotubes (n = 6). (A and C) The addition of CaMKKβ inhibitor STO (30 μM) completely attenuated the caffeine-induced increase in LC3b-II expression (n = 6). (D) Western blot analysis revealed a significant reduction in LC3b-II expression in 10 mM caffeine-treated myotubes transfected with CaMKII siRNA vs. control siRNA (n = 3). Once a satisfactory image was obtained each membrane was stained with coomassie brilliant blue (R-250) and imaged for total protein assessment to allow each sample to be normalized to total protein content of each lane (data not shown). a–dIndicate significant difference between treatments without same letters (P < 0.05).
3.4. CaMKKβ is the primary mediator of AMPK-dependent autophagic vesicle accumulation in caffeine treated skeletal muscle cells
To further analyze the role of CaMKKβ and CaMKII in regulating AMPK-dependent autophagy in caffeine treated skeletal muscle cells we conducted an autophagy assay using fluorescence microscopy. Skeletal muscle cells treated 10 μM KN92 + 10 mM caffeine for 6 h displayed a significant increase in autophagic vesicle accumulation (Fig. 4.) Though the addition of 10 μM KN93 was able to reduce the mean FITC intensity of 10 mM caffeine treated cells by approximately 30% the difference was not statistically significantly (P = 0.06). However, treating cells with either KN92 or KN93 + 30 μM STO completely inhibited the 10 mM caffeine-induced increase in autophagic vesicle accumulation in skeletal muscle cells (Fig. 4).
Fig. 4.
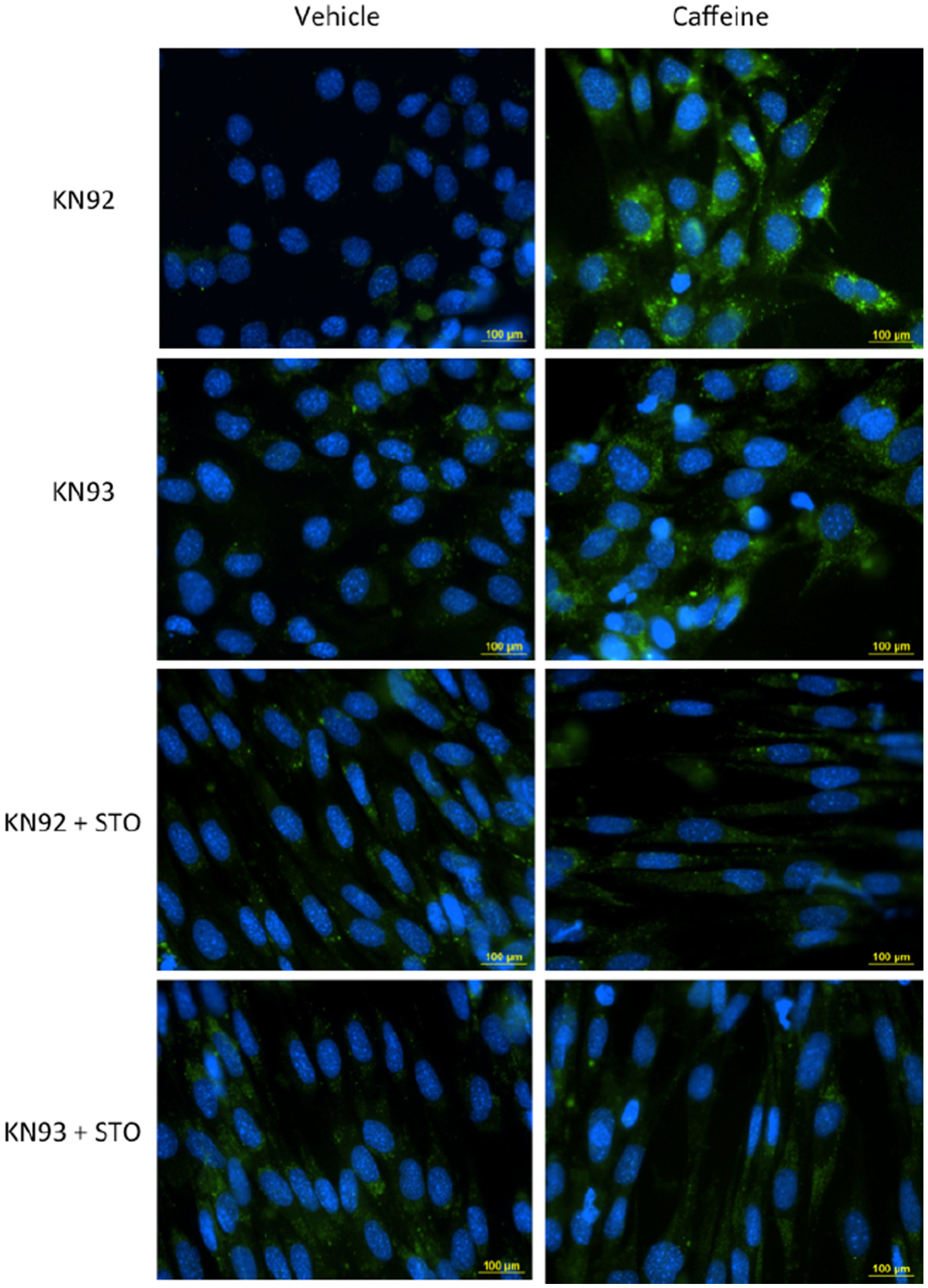
CaMKKβ is the primary mediator of AMPK-dependent autophagic vesicle accumulation in caffeine treated skeletal muscle cells. C2C12 myotubes were treated with the CaMKII inhibitor KN93 (10 μM) or its positive control KN92 (10 μM) alone or in combination with CaMKKβ inhibitor STO (30 μM) and incubated in media containing vehicle (DMSO) or 10 mM caffeine for 6 h. For the last 30 min of incubation the cells received 1 μM of Hoechst 33342 and 2 μl/ml of Cyto-ID Green (fluorescein) dye to label autophagic vesicles (autophagosomes and autolysosomes). Fluorescence microscopy analysis was conducted to examine the accumulation autophagic vesicles in each experimental treatment (n = 3). Ten individual images were taken in random locations on each coverslip (sample). The mean FITC fluorescence intensity was calculate for each sample and was used to for the quantitative assessment of autophagic vesicle accumulation between experimental treatments. Cells treated with KN92 and KN93 + 10 mM caffeine displayed a significant increase in autophagic vesicle accumulation (FITC intensity) compared to other experimental treatments (P < 0.05). The addition of STO completely inhibited the caffeine-induced increase in autophagic vesicle accumulation (P > 0.05).
4. Discussion
Calcium-activated protein kinases play an important role in regulating calcium-mediated pathways in skeletal muscle cells. In the present study we found that CaMKII contributed to AMPK phosphorylation in myotubes treated with the highest caffeine dose. Our data regarding the contribution of CaMKII to increased AMPK phosphorylation in 10 mM, but not 2.5 mM, caffeine treated myotubes is similar the study of Raney et al. [16], which found that CaMKII directly contributed to AMPK activity in electrically stimulated, but not 3 mM caffeine treated, rat skeletal muscle. Given that our highest caffeine dose was more than three times that of Raney et al. [16], it is possible that the CaMKII-dependent increase in AMPK activity that was observed in the present study was similar to that observed in electrically stimulated skeletal muscle. Though CaMKII contributed significantly to AMPK phosphorylation at the highest caffeine dose, CaMKKβ contributed more substantially to AMPK phosphorylation at both caffeine doses (Fig. 1). A previous investigation found that as the frequency of calcium spikes increased the level of CaMKII activity increased significantly, as well [24]. We observed a 54% increase in CaMKII phosphorylation in 10 mM caffeine treated myotubes, which likely explains why CaMKII significantly contributed to increased AMPK phosphorylation in 10 mM caffeine, but not 2.5 mM, treated myotubes.
While caffeine has been shown to promote autophagy in numerous mammalian cell lines, to the best of our knowledge the current work represents the first published report of caffeine promoting autophagy in skeletal muscle cells. We found that the addition of the AMPK inhibitor Compound C prevented the significant increase in LC3b-II expression and autophagic vesicle accumulation in 10 mM caffeine treated myotubes (Fig. 1). Caffeine-induced autophagy was largely dependent upon the calcium-induced activation of AMPK (Figs. 3 and 4). Our data suggests that CaMKKβ is the primary calcium-activated regulator of AMPK-dependent autophagy in caffeine treated skeletal muscle cells as LC3b-II expression and autophagic vesicle accumulation were completely inhibited in cells treated with the CaMKKβ inhibitor STO (Figs. 3 and 4). The addition of STO similarly inhibited LC3b-II and autophagic vesicle accumulation in the presence of either KN92 or KN93. These data strongly suggests that even though CaMKII was able to promote a small but significant increase in AMPK phosphorylation when CaMKKβ activity was inhibited (Fig. 2), CaMKII alone was not able to significantly increase AMPK-dependent autophagy in caffeine treated skeletal muscle cells. However, CaMKII did significantly contribute to increased LC3b-II expression in 10 mM caffeine treated muscle cells (Fig. 3). Also, the addition of the CaMKII inhibitor KN93 was able to reduce the 10 mM caffeine-induce increase in autophagic vesicle accumulation (mean FITC intensity) by approximately 30% (Fig. 4), though again the difference was statistically significant. These data indicate that CaMKII not only plays an role in promoting the maximal calcium-induced activation AMPK, it also plays an important role in promoting the maximal level of AMPK-dependent autophagy in caffeine treated skeletal muscle cells. To the best of our knowledge the current study represents the first published report of CaMKII directly contributing to AMPK-dependent autophagy in any mammalian cell line.
Our conclusions are seemingly in contrast with a previous investigation by Saiki et al. [22], which found that 10 mM caffeine significantly increased autophagy in human neuroblasts (SH-SY5Y) and HeLa cells by inhibiting the PI3K/Akt/mTOR pathway. AMPK is a well-known inhibitor of the PI3K/Akt/mTOR pathway [25]. However, Saiki et al. [22] did not specifically address the possible involvement of AMPK in caffeine-dependent autophagy. Also, in the current study we did not investigate the specific AMPK-dependent mechanism that was responsible for caffeine-induced autophagy. Additional investigations are needed to determine the contribution of AMPK-dependent phosphorylation of Foxo3a and ULK1 and the regulation of the PI3K/Akt/mTOR pathway in promoting caffeine-induced autophagy.
In summary, our results strongly suggest that caffeine promotes autophagy in skeletal muscle cells in an AMPK-dependent manner. In addition, we found that both CaMKKβ and CaMKII significantly contributed to AMPK-dependent autophagy in caffeine treated skeletal muscle cells, though CaMKKβ was the primary mediator AMPK-dependent autophagy.
Acknowledgments
The authors thank Melissa Hughes, Atelia Harper and Elvis Foley for their assistance in completing this project.
Grant support
Financial support was provided Magellan Scholars Fellowships that were awarded by the University of South Carolina Office of Undergraduate Research to T.S. Mathew, R.K. Ferris and R.M. Downs. In addition, this work was partially supported by a RISE grant awarded to B.L. Baumgarner by the Office of the Vice President for Research at the University of South Carolina. Additional support was provided by a Magellan Mentors Award that was awarded by the University of South Carolina Upstate Office of Sponsored Awards and Research Support to B.L. Baumgarner.
Footnotes
Conflict of interest
The authors declare no conflict of interest.
References
- [1].Cheng H, Lederer WJ, Calcium sparks, Physiol. Rev 88 (2008) 1491–1545. [DOI] [PubMed] [Google Scholar]
- [2].Grumati P, Bonaldo P, Autophagy in skeletal muscle homeostasis and in muscular dystrophies, Cells 1 (2012) 325–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Cecconi F, Levine B, The role of autophagy in mammalian development: cell makeover rather than cell death, Dev. Cell 3 (2008) 344–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Reggiori F, Klionsky DJ, Autophagy in the eukaryotic cell, Eukaryot. Cell 1 (2002) 11–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Wang C-W, Klionsky DJ, The molecular mechanism of autophagy, Mol. Med 9 (2003) 65–76. [PMC free article] [PubMed] [Google Scholar]
- [6].Bonaldo P, Sandri M, Cellular and molecular mechanisms of muscle atrophy, Dis. Model Mech 6 (2013) 25–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Brady NR, Hamacher-Brady A, Yuan H, Gottlieb RA, The autophagic response to nutrient deprivation in the hl-1 cardiac myocyte is modulated by Bcl-2 and sarco/endoplasmic reticulum calcium stores, FEBS J. 274 (2007) 3184–3197. [DOI] [PubMed] [Google Scholar]
- [8].Harr MW, Distelhorst CW, Apoptosis and autophagy: decoding calcium signals that mediate life or death, Cold Spring Harbor Perspect. Biol 2 (2010) a005579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Xi H, Barredo JC, Merchan JR, Lampidis TJ, Endoplasmic reticulum stress induced by 2-deoxyglucose but not glucose starvation activates AMPK through CaMKKβ leading to autophagy, Biochem. Pharmacol 85 (2013) 1463–1477. [DOI] [PubMed] [Google Scholar]
- [10].Decuypere J-P, Bultynck G, Parys JB, A dual role for Ca2+ in autophagy regulation, Cell Calcium 50 (2011) 242–250. [DOI] [PubMed] [Google Scholar]
- [11].Høyer-Hansen M, Bastholm L, Szyniarowski P, et al. , Control of macroautophagy by calcium, calmodulin-dependent kinase kinase-beta, and Bcl-2, Mol. Cell 25 (2007) 193–205. [DOI] [PubMed] [Google Scholar]
- [12].Grotemeier A, Alersa S, Pfistererb SG, Paascha F, Daubrawaa M, Dieterlea A, Violletc B, Wesselborga S, Proikas-Cezanneb T, Storka B, AMPK-independent induction of autophagy by cytosolic Ca2+ increase, Cell. Signal 6 (2010) 914–925. [DOI] [PubMed] [Google Scholar]
- [13].Madaro L, Marrocco V, Carnio S, Sandri M, Bouché M, Intracellular signaling in ER stress-induced autophagy in skeletal muscle cells, FASEB J. 27 (2013) 1990–2000. [DOI] [PubMed] [Google Scholar]
- [14].Pfisterer SG, Mauthe M, Codogno P, Proikas-Cezanne T, Ca2+/calmodulin-dependent kinase (CaMK) signaling via CaMKI and AMPK-activated protein kinase contributes to the regulation of WIPI-1 at the onset of autophagy, Mol. Pharmacol 80 (2011) 1066–1075. [DOI] [PubMed] [Google Scholar]
- [15].Rose AJ, Kiens B, Richter EA, Ca2+-calmodulin-dependent protein kinase expression and signalling in skeletal muscle during exercise, J. Physiol 574 (2006) 889–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Raney MA, Turcotte LP, Evidence for the involvement of CaMKII and AMPK in Ca2+-dependent signaling pathways regulating FA uptake and oxidation in contracting rodent muscle, J. Appl. Physiol 104 (2008) 1366–1373. [DOI] [PubMed] [Google Scholar]
- [17].Gutierrez-Martin Y, Martin-Romero FJ, Henao F, Store-operated calcium entry in differentiated C2C12 skeletal muscle cells, Biochim. Biophys. Acta 1711 (2005) 33–40. [DOI] [PubMed] [Google Scholar]
- [18].Lamb GD, Cellini MA, Stephenson DG, Different Ca2+releasing action of caffeine and depolarisation in skeletal muscle fibres of the rat, J. Physiol 531 (2001) 715–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Kong H, Jones PP, Koop A, Zhang L, Duff HJ, Chen SRW, Caffeine induces Ca2+ release by reducing the threshold for luminal Ca2+ activation of the ryanodine receptor, Biochem. J 414 (2008) 441–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Ding S, Contrevas JR, Abramov AY, Qi Z, Duchen MR, Mild stress of caffeine increased mtDNA content in skeletal muscle cells: the interplay between Ca2+ transients and nitric oxide, J. Muscle Res. Cell Motil 33 (2012) 327–337. [DOI] [PubMed] [Google Scholar]
- [21].Sinha RA, Farah BL, Singh BK, Siddique MM, Li Y, Wu Y, Ilkayeva OR, Gooding J, Ching J, Zhou J, Martinez L, Xie S, Bay B-H, Summers SA, Newgard CB, Yen PM, Caffeine stimulates hepatic lipid metabolism by the autophagy–lysosomal pathway in mice, Hepatology 59 (2014) 1366–1380. [DOI] [PubMed] [Google Scholar]
- [22].Saiki S, Sasazawa Y, Imamichi Y, Kawajiri S, Fujimaki T, Tanida I, Kobayashi H, Sato F, Sato S, Ishikawa K-I, Imoto M, Hattori N, Caffeine induces apoptosis by enhancement of autophagy via PI3K/Akt/mTOR/p70S6K inhibition, Autophagy 7 (2011) 176–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Williams RT, Yu AL, Diccianni MB, Theodorakis EA, Batova A, Renal cancer-selective Englerin A induces multiple mechanisms of cell death and autophagy, J. Exp. Clin. Cancer Res 32 (2013) 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Li L, Stefan MI, Le Nove’re N, Calcium input frequency, duration and amplitude differentially modulate the relative activation of calcineurin and CaMKII, PLoS One 7 (2012) e43810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Hahn-Windgassen A, Nogueira V, Chen CC, Skeen JE, Sonenberg N, Hay N, Akt activates the mammalian target of rapamycin by regulating cellular ATP level and AMPK activity, J. Biol. Chem 280 (2005) 32081–32089. [DOI] [PubMed] [Google Scholar]