

Abstract
Summary
Unsupervised machine learning provides tools for researchers to uncover latent patterns in large-scale data, based on calculated distances between observations. Methods to visualize high-dimensional data based on these distances can elucidate subtypes and interactions within multi-dimensional and high-throughput data. However, researchers can select from a vast number of distance metrics and visualizations, each with their own strengths and weaknesses. The Mercator R package facilitates selection of a biologically meaningful distance from 10 metrics, together appropriate for binary, categorical and continuous data, and visualization with 5 standard and high-dimensional graphics tools. Mercator provides a user-friendly pipeline for informaticians or biologists to perform unsupervised analyses, from exploratory pattern recognition to production of publication-quality graphics.
Availabilityand implementation
Mercator is freely available at the Comprehensive R Archive Network (https://cran.r-project.org/web/packages/Mercator/index.html).
1 Introduction
Visualization of patterns in data through unsupervised machine learning (ML) is an important tool of scientific analysis, helping researchers understand underlying relationships as they search for biological meaning. As large-scale, high-throughput experiments increase, new visualization methods must be developed to keep pace with both technical demands and research potential. This problem is especially acute in biomedical research with the advent of ‘omics’ technologies that can measure tens of thousands of features across thousands of samples.
Here, we present Mercator, an R-package providing a flexible and user-friendly pipeline for unsupervised ML and visualization of any high-throughput data. Mercator makes it easy to quickly cluster and visualize any distance matrix using standard and high-dimensional graphical methods. These visualizations use consistent color schemes, enabling users to compare and contrast different metrics or different clustering algorithms. Critically, since cluster labels from different algorithms are arbitrary, Mercator can synchronize them making it easier to compare unsupervised analyses on large high-dimensional datasets. Mercator implements additional tools for binary matrices, including tools for removal of duplicate features, outlier detection, feature reduction and estimates of the number of clusters. In our internal tests, Mercator has successfully processed binary data containing 70 000 samples, 2700 features and at least 130 clusters. Package outputs from unsupervised analysis are inter-operable with other R tools for downstream analyses.
2 Implementation
Mercator streamlines rigorous and reproducible unsupervised ML on a matrix of binary or continuous high-throughput data in a user-friendly pipeline. For binary data, initial filtering is performed using Thresher (Wang et al., 2018) an R package implementing outlier detection, principal components analysis and von Mises Fisher mixture models. By identifying significant features, Thresher performs feature reduction through the identification and removal of non-informative features and the unbiased calculation of the number of groups (K) for downstream use.
Many unsupervised ML analyses rely on calculated similarities and differences between observed samples in a large feature space, based on a chosen distance metric. There is not one distance metric that is seen as superior to all others in all contexts for all data types. For continuous data, Mercator relies on existing distance metrics in R. For binary data, Mercator supports 10 metrics, representing major subgroups defined by Choi and colleagues (Choi et al., 2010): Jaccard, Sokal & Michener, Hamming, Russell-Rao, Pearson, Goodman & Kruskal, Manhattan, Canberra, Binary and Euclidean. These 10 representative metrics capture appropriate solutions in common and uncommon use for asymmetric and symmetric binary, categorical and continuous data. To cluster, Mercator uses Partitioning Around Medoids (PAM), an efficient k-medoids algorithm compatible with multiple methods of distance calculation (Kaufman and Rousseeuw, 1990). However, Mercator allows researchers to use any desired clustering algorithm and link import its results for use with its visualization tools. Mercator also provides a simple interface to computation and display of the silhouette width (Kaufman and Rousseeuw, 1990) for any distance metric or clustering algorithm. Combined with the other visualization methods, the ease-of-use of the silhouette width plots can help select appropriate distance metrics or clustering algorithms that provide a good fit to the data.
Finally, Mercator facilitates five graphical methods, including both standard techniques (e.g. hierarchical clustering) and large-scale multi-dimensional visualizations [multi-dimensional scaling (MDS), t-distributed Stochastic Neighbor Embedding (t-SNE) (van der Maaten and Hinton, 2008) and iGraph (Csardi and Nepusz, 2006)]. Flexible graphics parameters consistently aid the creation of descriptive, showcase-quality visualizations. Mercator supports users who mix and match distance metrics and visualization techniques to gain a richer understanding of patterns in their data. These functionalities are elaborated in the package vignettes.
3 Application
Cytogenetic data from 3387 patients with chronic lymphocytic leukemia (CLL) obtained from the Mitelman Database of Chromosome Alterations and Gene Fusions in Cancer (Mitelman et al., 2021) was processed to a binary matrix using CytoGPS (Abrams et al., 2019). Data were filtered with Thresher and reduced from 2748 raw features to 845 significant features. Principal components analysis suggested 16 clusters, which were recovered based on Jaccard distance. Visualization with t-SNE highlighted clearly defined clusters and closely located groups. MDS plots expand this understanding by finding important components that distinguish clusters; higher dimensional MDS plots are required to elucidate the entire structure. After down-sampling the dataset to underemphasize the strongest signals while preserving the weaker ones, the iGraph visualization provides a better understanding of interactions between the cytogenetics of CLL. MDS visualizations were employed to expand this understanding by separating distinguishing components. Visualization with iGraph elucidates interactions between key cytogenetic abnormalities (Fig. 1). Thus, Mercator provides a streamlined approach to improve understanding of the cytogenetics of CLL.
Fig. 1.
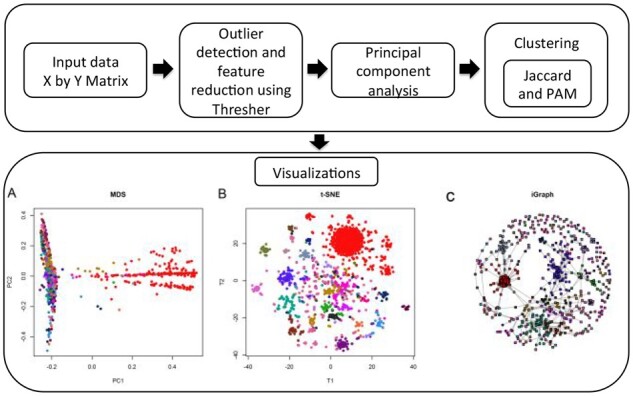
Workflow for unsupervised Mercator pipeline analysis of binary cytogenetic data on 3387 patients with CLL. After data processing by outlier detection, feature reduction and principal components analysis, 16 clusters were recovered with the Jaccard distance and PAM. The first coordinate of MDS (A) shows the dominant signal (trisomy 12, a key cytogenetic abnormality in CLL) in samples colored red. t-SNE (B) shows not only that signal, but clearly reveals other clusters based on patterns of cytogenetic abnormalities. iGraph (C) illustrates the connections between clustered entities
4 Conclusion
The Mercator R package supports a user-friendly pipeline for unsupervised visualization of large-scale, multi-dimensional data using 10 distance methods for binary, continuous and categorical data and 5 visualization techniques. By providing a toolbox to facilitate distance metric selection and high-quality figure generation, Mercator aids researchers (both informaticians and biologists) to achieve richer understanding the underlying patterns present in their data.
Acknowledgements
The authors would like to acknowledge support from the Summer Internship Program at the Ohio State University Department of Biomedical Informatics.
Funding
This work was supported by the National Library of Medicine (NLM) [T15 LM011270], the National Cancer Institute (NCI) [R03 CA235101] and by Pelotonia Intramural Research Funds from the James Cancer Center, Columbus, OH.
Conflict of Interest: none declared.
Data availability
All source data is available upon request. Mercator is freely available at the Comprehensive R Archive Network (https://cran.r-project.org/web/packages/Mercator/index.html).
Contributor Information
Zachary B. Abrams, Department of Biomedical Informatics, The Ohio State University, Columbus, OH 43210, USA
Caitlin E. Coombes, Department of Biomedical Informatics, The Ohio State University, Columbus, OH 43210, USA College of Medicine, The Ohio State University, Columbus, OH 43210, USA.
Suli Li, Department of Operations Research and Information Engineering, College of Engineering, Cornell, New York, NY 10044, USA.
Kevin R. Coombes, Department of Biomedical Informatics, The Ohio State University, Columbus, OH 43210, USA.
References
- Abrams Z.B. et al. (2019) CytoGPS: a web-enabled karyotype analysis tool for cytogenetics. Bioinformatics, 35, 5365–5366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi S.-S. et al. (2010) A survey of binary similarity and distance measures. J. Syst. Cybern. Inf., 8, 43–48. [Google Scholar]
- Csardi G., Nepusz T. (2006) The igraph software package for complex network research. InterJournal, 1695, 1–9. [Google Scholar]
- Kaufman P.J., Rousseeuw L. (1990) Finding Groups in Data: An Introduction to Cluster Analysis. John Wiley & Sons, Hoboken, NJ. [Google Scholar]
- Mitelman, F. (2021) Database of Chromosome Aberrations and Gene Fusions in Cancer. In Mitelman, F. et al., (eds), https://mitelmandatabase.isb-cgc.org.
- van der Maaten L., Hinton G. (2008) Visualizing data using t-SNE. J. Mach. Learn. Res., 9, 2579–2605. [Google Scholar]
- Wang M. et al. (2018) Thresher: determining the number of clusters while removing outliers. BMC Bioinformatics, 19, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All source data is available upon request. Mercator is freely available at the Comprehensive R Archive Network (https://cran.r-project.org/web/packages/Mercator/index.html).