

Abstract
Motivation
Trans-acting expression quantitative trait loci (eQTLs) collectively explain a substantial proportion of expression variation, yet are challenging to detect and replicate since their effects are often individually weak. A large proportion of genetic effects on distal genes are mediated through cis-gene expression. Cis-association (between SNP and cis-gene) and gene-gene correlation conditional on SNP genotype could establish trans-association (between SNP and trans-gene). Both cis-association and gene-gene conditional correlation have effects shared across relevant tissues and conditions, and trans-associations mediated by cis-gene expression also have effects shared across relevant conditions.
Results
We proposed a Cross-Condition Mediation analysis method (CCmed) for detecting cis-mediated trans-associations with replicable effects in relevant conditions/studies. CCmed integrates cis-association and gene-gene conditional correlation statistics from multiple tissues/studies. Motivated by the bimodal effect-sharing patterns of eQTLs, we proposed two variations of CCmed, CCmed and CCmed for detecting cross-tissue and tissue-specific trans-associations, respectively. We analyzed data of 13 brain tissues from the Genotype-Tissue Expression (GTEx) project, and identified trios with cis-mediated trans-associations across brain tissues, many of which showed evidence of trans-association in two replication studies. We also identified trans-genes associated with schizophrenia loci in at least two brain tissues.
Availability and implementation
CCmed software is available at http://github.com/kjgleason/CCmed.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
The impact of genetic variation on the human transcriptome is well-established (Dixon et al., 2007; Morley et al., 2004). To date, the majority of known expression quantitative trait loci (eQTLs) affect expression of local genes (cis-eQTLs; SNPs within 1M bp from the gene transcriptional start site) (Battle et al., 2017). It is known that genetic variation may affect distal or inter-chromosomal gene expression as trans-eQTLs. Though trans-eQTLs collectively explain a substantial proportion of expression variation in the genome (Liu et al., 2019), their effects are often individually weak. It is challenging to detect replicable trans-eQTLs with existing eQTL data.
Standard trans-association tests examine the associations of pairs of SNPs and trans-gene expression levels in the genome, adjusting for multiple comparison. The Genotype-Tissue Expression (GTEx) project has conducted both cis- and trans-eQTL analysis across multiple human tissue types (Battle et al., 2017). Bimodal patterns of tissue sharing were observed among both cis- and trans-eQTLs, with some eQTLs having effects shared across many tissues and many others having effects in only specific tissue types. Compared with cis-associations, trans-association effects tend to be more tissue-specific. It is challenging to detect and replicate trans-associations based on standard association tests in single tissue types, given the limited available sample sizes of eQTL data from most tissue/cell types and the ultra large number of potential tests.
Recently, an alternative and complementary strategy to standard trans-association tests has been proposed, to detect trans-associations mediated by cis-gene expression levels (Battle et al., 2014; Pierce et al., 2014; Yang et al., 2017). If an SNP (or a set of SNPs) is associated with the expression levels of a cis-gene (i.e. cis-association) and the cis-gene expression further affects the expression of a distal gene, the trans-association from the SNP(s) to the distal gene is established. Studies have shown that many cis-eQTLs affect distal gene expression (Pierce et al., 2014), and a large proportion of trans-eQTL effects are at least partially mediated by cis-gene expression levels (Yang et al., 2017). That is, trans-associations mediated by cis-gene expression (termed as ‘cis-mediated trans-associations’ hereafter) are abundant in the genome. While the standard trans-association test for total effects between SNP and trans-gene is more powerful in detecting large effects with the available sample sizes of eQTL data, mediation-based association tests can be powerful for detecting small-to-moderate effects when one or more true mediators are measured and accounted for (O’Rourke and MacKinnon, 2015). In studying the trans-association between SNP(s) and distal gene expression, the cis-gene(s) of the SNP(s) is a natural mediator to consider.
Importantly, since both cis-association and gene-gene regulation tend to have effects shared across related cellular contexts, cis-mediated trans-associations often have replicable effects shared across relevant conditions/studies, compared to the condition-specific trans-associations detected via standard tests. Here condition refers to tissue-type, cell composition, micro-environment, study, population and others. Previous literature has reported that many cis ‘hub’ genes (with cis-eQTLs) may affect multiple trans-genes in functionally related tissue types (Yang et al., 2017), supporting the existence and even prevalence of cross-tissue and cross-condition patterns for cis-mediated trans-associations. Most existing trans-association tests were proposed to test for total association effects between SNP and gene-expression, with improved control of false discovery rates (FDR) and enhanced computational efficiency (Ongen et al., 2016; Peterson et al., 2016). Single-tissue mediation methods were also proposed to detect cis-mediated trans-associations in a single study (Yang et al., 2017). With the increasing availability of eQTL data from different studies, it is desirable to develop a method for detecting cross-condition cis-mediated trans-associations by integrating data from multiple studies allowing sample overlap and study heterogeneity.
Motivated by these challenges and needs, we propose a Cross-Condition Mediation (CCmed) method (Fig. 1) to detect the replicable indirect trans-associations mediated by cis-gene expression levels with effects shared across relevant conditions. Considering the often bimodal tissue-sharing pattern of eQTL effects, we developed two variations of CCmed, CCmed and CCmed, for detecting cis-mediated trans-associations across most conditions and those with effects specific to certain conditions, respectively. The proposed method substantially reduces false positive findings as fewer of them may arise by chance in multiple conditions. The CCmed algorithm takes as input two sets of statistics from K eQTL studies: the cis-association statistics and the cis- trans- expression correlation statistics conditional on eQTL genotype. CCmed calculates the posterior probabilities of a trio of cis-SNP(s), cis-gene transcript and trans-gene transcript having non-zero cis-mediated trans-associations in multiple out of K studies, allowing sample overlap. As a proof-of-concept, we first applied CCmed to study gene-level cis-mediated trans-associations with effects shared across 12 out of 13 brain tissues from the GTEx project (V8) (Aguet et al., 2020). Our identified trans-associations from GTEx analysis showed enrichment of trans-associations in two replication studies of different tissue types—whole blood samples from the eQTLGen consortium (Võsa et al., 2018) and dorsolateral prefrontal cortex samples from the CommonMind Consortium (CMC) (Fromer et al., 2016). We also applied CCmed to detect trans-genes for GWAS SNPs from 108 known schizophrenia (SCZ)-associated loci (Ripke et al., 2014), with shared effects in at least two brain tissues. Some identified genes showed strong evidence of SCZ risk associations based on complementary validation analyses.
Fig. 1.

An illustration of the CCmed algorithms for identifying cis-mediated trans-associations across most conditions or in specific conditions. (A) An illustration of the model for mapping gene-level mediation and trans-association in a single tissue type. Note that direct effect from SNPs to trans-gene is allowed. (B) An illustration of the model for identifying trans-genes of a (GWAS) SNP mediated by cis-gene expression in a single tissue type. (C) A flowchart of the CCmedmost and CCmedspec algorithms to establish cross-condition cis-mediated trans-associations in most ( and is close to K) or in a few specific () out of K conditions, respectively
Many (cis-mediated) trans-associations have only moderate-to-weak total association effects in a single condition/study, and would be undetectable via standard trans-association tests after stringent multiple testing adjustment. The GTEx consortium has also conducted a multi-tissue analysis integrating trans-association effects calculated by standard association tests in single tissues, and found limited power improvement in the multi-tissue analysis due to limited effect-sharing among tissues (Battle et al., 2017). A major innovation of our work is that by considering the natural mediator, cis-gene expression, for trans-associations, we focused on cis-mediated trans-association effects and reported that those effects could be shared across relevant conditions/studies and be replicable. The proposed CCmed method can detect effects shared across most conditions as well as effects shared among a few specific conditions. By applying CCmed to data from GTEx brain tissues, we showed that cross-condition cis-mediated trans-association effects are not only prevalent in the genome, but also are replicable across different studies and samples.
2 Materials and methods
We propose a cross-condition mediation method, CCmed, for detecting replicable cis-mediated trans-associations across tissues/conditions. While the CCmed methods can be applied to many conditions (e.g. studies, cellular contexts, tissue types), in this work we applied the methods to multi-tissue data from GTEx (V8) project. For clarity and consistency between descriptions of the methods and applications, we used the term ‘tissue’ in lieu of ‘condition’ in describing the methods. CCmed takes as input K sets of statistics for cis-association and K sets of gene-gene correlation statistics conditional on eQTL genotype from K tissues, and calculated the probability of ‘SNP → cis-gene → trans-gene’ in most or a few tissue types. In mediation analysis, we assume that the confounders were properly modeled and adjusted. See Figure 1 for an illustration of CCmed and CCmed.
2.1 CCmed for mapping robust gene-level cis-mediated trans-associations in most tissues
CCmed detects trios (SNP or SNP-set, cis-gene transcript, trans-gene transcript) showing evidence of cis-mediated trans-association effects across most tissues/studies by quantifying the joint probability of non-zero cis-association and non-zero gene-gene conditional correlation being simultaneously satisfied in at least K1 out of K tissues with K1 being close to K.
Specifically for detecting gene-level trans-association, we consider each trio , where is the genotype matrix of a set of cis-eSNPs for a cis-gene i, Ci denotes the cis-gene i’s expression level and Tj is the expression level of a trans-gene j. Given a user-specified K1, the null hypothesis for each test of a trio is that ‘ in less than K1 out of K tissue types’ and alternative hypothesis is that ‘ in at least K1 out of K tissue types’. We calculated the probability that Ci mediates the effects of on Tj in at least K1 tissue types, , as follows:
| (1) |
| (2) |
| (3) |
where sl is a set of tissue indices and is a subset of with at least K1 tissue types. The set contains all unique combinations of tissue indices that have at least K1 elements. The is a vector of cis-association effects for the set of eQTLs in a single tissue type, and β1 measures the conditional correlation of cis- and trans-gene expression levels in a single tissue type. We estimated and β1 for each tissue type and obtained the test statistics as input. In Supplementary Material, we described in detail the models to calculate the input statistics. Note that here we omitted the subscript for tissue type to make notation simpler.
In (1), the first probability in each product involved in the summation describes the probability of a gene i having at least one cis-eQTL in at least tissues , and the lead cis-eQTL may differ by tissue type; given robust cross-tissue cis-associations in at least tissues , the second probability quantifies the probability of non-zero cis-trans gene expression correlations conditioning on eQTL genotypes in exactly tissues . The derivation from (1) to (2) is based on the assumption that genes with cis-associations are equally or more likely to affect downstream genes compared with other randomly selected genes in the genome. In other words, in tissue/cell types where genes are under- or not expressed, their expression levels are equally or less likely to affect downstream gene expression levels. For highly correlated tissues where there is less heterogeneity in cis-associations, a more stringent lower bound can be considered as presented in (3) by requiring that cis-associations exist in all tissue types to further improve the robustness of mediation detected. For illustration purpose, below we describe our estimation strategy using derivation result (3). Analogous strategy applies equally to the derivation result (2) and we provide both options in our R software package CCmed.
To estimate in all K tissues), we applied the integrative association method Primo (Gleason et al., 2020a). Primo takes as input the M × K matrix of cis-association statistics , considers all possible cross-tissue association patterns and estimates the density function for each pattern, and returns the probabilities of in all K tissue types) for each gene . For each cis-gene i, to estimate the probabilities of correlations for the cis-genes i and its trans-genes in at least K1 tissue types, we apply the Primo algorithm to the matrix of conditional correlation statistics (; and ), and obtain the probabilities . For each trio , the cross-tissue mediation probability can be estimated as . Algorithm 1 provides a summary of the algorithm.For multiple testing adjustment, we can calculate the estimated false discovery rate (FDR) (Storey and Tibshirani, 2003),
where λ is the probability threshold.
Algorithm 1 CCmed for detecting (gene-level) cis-mediated trans-associations with effects across most tissue types
Step 1. Obtain tissue-specific cis-association and conditional gene-gene correlation statistics. In each tissue type k (), calculate the (gene-level) cis-association statistics for each gene i () to its cis-eQTL set , and the gene-gene conditional correlation statistics for each pair of genes i and j ().
Step 2. Estimate the cross-tissue cis-association probabilities. Apply the Primo algorithm to the M × K matrix of cis-association statistics, and estimate the probability of cis-association for each gene in all K tissues, .
Step 3. Estimate the cross-tissue gene-gene conditional correlation probabilities. For each cis-gene i, apply Primo to its matrix of gene-gene conditional correlation statistics for all trans-genes, and estimate for all trans-genes.
Step 4. Estimate the cross-tissue mediation probabilities. For each trio , estimate .
CCmed was proposed to detect the more robust and replicable cross-tissue cis-mediated trans-associations. Here we proposed and illustrated the method in a gene-level analysis for two reasons: first, there might be a limited number of individual SNPs with consistent effects across multiple tissues and the statistical power for detecting SNP-level cross-tissue association is low; second and more importantly, there are trait-relevant gene-level cis-mediated trans-association effects shared across related tissues. It was reported that a large majority of known trait-associated loci are in linkage disequilibrium (LD) with a cis-eQTL in relevant pathogenic tissues (Gamazon et al., 2018). When applying CCmed, the choice of tissues depends on the scientific questions of interest, and we recommend more homogeneous tissues for power concerns. The algorithm can be applied to sets of heterogeneous tissues, with potentially limited power. The choice of K1 balances between the number of discoveries and the replicability of findings. When choosing K1, we recommend first examining effect-sharing patterns across the selected tissues (see Supplementary Fig. S1 for two examples of effect-sharing patterns across 49 tissues and across 13 brain tissues). For sets of homogeneous tissues, a higher K1 (e.g. ) is recommended to detect the more robust trios while still maintaining a good power, and the algorithm is expected to be relatively robust to the choice of K1 to some extent. Whereas, for sets of heterogeneous tissues, lowering K1 may increase the number of discoveries at a cost of potentially reduced reproducibility of the results. In Supplementary materials, we presented simulations to illustrate the impact of varying K1 on the trade-offs between power and replicability of results.
2.2 CCmed for detecting (GWAS) SNP-level cis-mediated trans-associations in specific tissues
Compared with gene-level cis-mediated trans-associations, SNP-level trans-associations are more likely to be present in specific tissues than cross-tissue. Many GWAS SNPs are also eQTLs with cis-association effects on their local gene expression levels (Giambartolomei et al., 2014) and such cis-association effects are likely to be present in some disease/trait-relevant tissue types but not necessarily all or most tissues.
To detect trans-genes associated with a (GWAS) SNP mediated by cis-genes, we propose to integrate the statistics from all K tissues to calculate the probability of mediation in at least K2 tissue types, .
| (4) |
| (5) |
where Ci is the cis-gene i’s expression level of a GWAS SNP, and Gi is the genotype of the GWAS SNP in the cis region of Ci( cis-genes of GWAS SNPs), and Tj is the trans-gene expression; and is a set of tissue indices and is a subset of with K2 distinct tissue types. A GWAS SNP may have multiple cis-genes and may be considered in multiple trios. The parameter is the conditional cis-association effect of the GWAS SNP of interest to a local gene’s expression conditioning on other lead eSNPs (an eSNP is an eQTL SNP, and a lead eSNP is the eSNP with the smallest eQTL P-value in the region), and adjusting other lead eSNPs reduces spurious association to a local gene due to Gi being in LD with other eSNPs. And is the conditional correlation parameter of cis- and trans-gene expression levels conditioning on eQTL and GWAS SNPs. Both cis-association and conditional correlation statistics are calculated separately for each tissue type, and we again omitted the subscript for tissue type for simpler notation. See Supplementary Materials for detailed models in calculating the input statistics. We apply the Primo method to calculate the probabilities in (5) to obtain a lower bound of the probability of trans-gene association of a GWAS SNP via cis-mediation.
The inequality (4) follows from the fact that the probability of mediation in at least K2 tissue types is lower bounded by the maximum probability of mediation in at least any specific set of K2 tissues. The inequality (5) holds under the assumption that cis-genes affected by GWAS SNPs are at least equally likely to affect downstream trans-genes compared to those not affected by GWAS SNPs. The maximum value of the probability products across all possible combinations of K2 tissue types in (5) provides a lower bound estimate for the probability of a GWAS SNP i being associated with a trans-gene j in tissue types, . The probabilities involved can be estimated by applying Primo separately to the matrix of cis-association statistics and to the conditional correlation statistics matrix for each GWAS SNP i.
For CCmed, we suggest . As discussed above, increasing K2 would increase the replicability of findings while fewer trios may have effects shared in more than two tissues. Since it is expected that trait/disease-relevant trans-associations may have effects shared in only specific disease-relevant tissues, further increasing K2 may lose many true cis-mediated trans-associations while the gain in replicability is limited. Moreover, there are a total of unique ’s and the calculation quickly expands as K2 increases if K is not small. We suggest applying CCmed to K disease-relevent tissue types. For some diseases/traits, the disease-relevant tissues or relevant pathogenic tissues are well known, and for other diseases, there are existing methods to identify the disease-relevant tissues (Lu et al., 2016).
Algorithm 2 summarized CCmed for GWAS SNPs.In both CCmed and CCmed, the key innovation of our method is that by integrating data across multiple studies/conditions, our method improves power and precision by aggregating concerted effects from multiple studies. This is based on the rationale that true associations, even if tissue-specific, are more likely to be shared across relevant tissues due to similar cell type compositions, while false positive findings from a single tissue are more likely to be punished when aggregating results from multiple studies (due to the winner’s curse).
Algorithm 2 CCmed for detecting SNP-level cis-mediated trans-associations for (GWAS) SNPs with effects shared in at least K2 tissues
Step 1. Obtain tissue-specific cis-association and conditional gene-gene correlation statistics. In each tissue type k (), calculate the cis-association statistics for each GWAS SNP adjusting for other eQTLs. Note that if a GWAS SNP is in cis with multiple genes, the pairs are separately considered and estimated. Calculate the gene-gene conditional correlation statistics .
Step 2. Estimate the probabilities of GWAS SNPs being also eQTLs. We apply the Primo algorithm to the matrix of cis-association statistics for GWAS SNPs conditioning on other eQTLs and estimate the probability of GWAS SNP i being an eQTL in at least tissue(s) , for each possible tissue set with K2 tissue types, .
Step 3. Estimate the gene-gene conditional correlation probabilities. We apply the Primo algorithm to the matrix of gene-gene conditional correlation statistics for each GWAS SNP and estimate the probability of non-zero gene-gene conditional correlation in at least tissue(s) , for each possible .
Step 4. Estimate the probabilities of trans-association via cis-mediation in at least K2 tissue types. For each trio of , we estimate the probability of trans-association of a (GWAS) SNP i to a trans-gene j via cis-mediation by (5).
CCmed and CCmed differ in the following major aspects: First, they were motivated to detect effects that are shared across most tissues (higher replicability), and those that are specific to only certain tissues, respectively. Second, in terms of calculation of statistics, the cis-association statistic of CCmed calculated the gene-level (SNP-set level) statistics based on an F-test for a set of cis-eQTLs. In constrast, CCmed was motivated and used to detect SNP-level association effects that do not have strong tissue-sharing. Third and importantly, CCmed will likely yield more replicable findings across conditions, and the results of CCmed is replicable in only relevant conditions.
3 Results
3.1 Simulations to evaluate CCmed in detecting robust cis-mediated trans-associations across conditions
In this section, we evaluated the performance of CCmed and CCmed through simulation studies. We simulated data where trans-eQTLs’ total effects (including both direct and indirect effects) are moderate in each tissue type, and where indirect effects mediated by cis-gene expression are shared across tissue-types. The CCmed algorithms can borrow information across tissue types to improve power and detect cis-mediated trans-associations, while controlling FDRs. We also compared with standard association tests for trans-associations in each single tissue type and showed the advantages of the proposed algorithms. In each of the algorithm evaluations, we simulated trios (SNP(s), cis-gene expression, trans-gene expression) for subjects in K = 10 tissues.
3.2 The performance of CCmed in identifying robust gene-level trans-associations
We simulated data on 500 sets of cis-eQTLs for 500 cis-gene expression levels from 350 subjects. For each set of cis-eQTLs, we simulated the genotypes of 10 correlated eSNPs with pairwise correlation of 0.3. Based on the genotypes, in each tissue type (K = 10), we randomly selected 1 SNP as the causal eSNP and generated 1 cis- and 500 trans-gene expression levels. The causal eSNPs varied across tissues. There were a total of 250 000 trios of (cis-eQTL set, cis-gene, trans-gene) from 10 correlated tissue types. Expression data was simulated such that cis-gene expression levels were associated with the cis-eQTL set in a varying proportion of the tissues (including none), and that non-zero conditional cis-trans gene expression correlations were present in a subset of the cis-trans gene pairs in a varying proportion of the tissues (including none). Among those trios with non-zero cis-mediated trans-associations, 50% of them also had non-zero direct effects from SNPs on the trans-gene expression levels. Effect sizes were simulated to mimic the observed total effects of trans-associations in the GTEx study. See Supplemental Materials for additional simulation details. In the simulation studies, we are interested in detecting the trios with (cis-mediated) trans-associations in at least 9 out of 10 tissue types.
Table 1A presented the power as well as the true and estimated FDRs to detect gene-level trans-associations mediated by cis-gene in at least out of K = 10 tissue types at a threshold of , 0.8 and 0.9, respectively, based on the calibration result (2). As a comparison, we also obtained the P-values of F-statistics based on standard trans-association tests for total gene-level trans-association effects. For each of the thresholds used in the CCmed approach, we applied the corresponding estimated FDR to the 250 000 by 10 matrix of P-values of standard association tests for total effects and obtained the trios with significant total gene-level trans-associations in at least 9 tissues. The power of the standard association test was low as expected due to the weak trans-association effects, the stringent cross-tissue association criteria (i.e. 9 out of 10) and multiple testing burden. In addition, there is a slight inflation in the FDRs. This is because controlling the FDR for individual association tests (there are of them) does not guarantee the control of the FDR for testing trans-associations in at least 9 out of 10 correlated tissues types (there are 250 000 of them). In contrast, CCmed accounted for tissue-tissue correlations, and it greatly improved the power by borrowing information across tissues and detecting robust cis-mediated trans-associations while well controlling the FDRs.
Table 1.
Simulation results evaluating the performance of CCmed
| (A) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Method | Association in at least out of K = 10 tissue types |
||||||||
| estP |
estP |
estP |
|||||||
| Power | true | estFDR | Power | true | estFDR | Power | true | estFDR | |
| (%) | FDR(%) | (%) | (%) | FDR(%) | (%) | (%) | FDR(%) | (%) | |
| CCmed | 68.0 | 3.2 | 7.7 | 60.2 | 2.2 | 4.0 | 54.7 | 2.2 | 3.1 |
| 7.7% estFDR | 4.0% estFDR | 3.1% estFDR | |||||||
| Standard association test | 31.6 | 9.4 | – | 25.9 | 4.8 | – | 23.9 | 3.6 | – |
| (B) | |||||||||
| Method | Association in at least out of K = 10 tissue types | ||||||||
| estP | estP | estP | |||||||
| Power | true | estFDR | Power | true | estFDR | Power | true | estFDR | |
| (%) | FDR(%) | (%) | (%) | FDR(%) | (%) | (%) | FDR(%) | (%) | |
| CCmed | 83.7 | 2.4 | 2.7 | 80.0 | 0.8 | 0.7 | 76.7 | 0.4 | 0.4 |
| Sobel’s test | 82.4 | 1.3 | 2.9 | 78.8 | 0.2 | 0.9 | 76.6 | 0.0 | 0.5 |
| 2.7% estFDR | 0.7% estFDR | 0.4% estFDR | |||||||
| Standard association test | 59.4 | 0.8 | – | 53.6 | 0.2 | – | 51.1 | 0.1 | – |
| (C) | |||||||||
| Method | Association in at least out of K = 10 tissue types | ||||||||
| estP | estP | estP | |||||||
| Power | true | estFDR | Power | true | estFDR | Power | true | estFDR | |
| (%) | FDR(%) | (%) | (%) | FDR(%) | (%) | (%) | FDR(%) | (%) | |
| CCmed | 64.2 | 3.5 | 7.6 | 55.5 | 1.2 | 2.7 | 49.8 | 0.5 | 1.2 |
| Sobel’s test | 58.1 | 1.5 | 9.9 | 46.8 | 0.4 | 3.7 | 39.8 | 0.2 | 1.8 |
| 7.6% estFDR | 2.7% estFDR | 1.2% estFDR | |||||||
| Standard association test | 34.2 | 2.8 | – | 25.2 | 0.9 | – | 20.6 | 0.4 | – |
Note: estP estimated probability; estFDR estimated FDR.(A) Simulation to compare CCmed with standard association tests for trans-association in detecting associations in at least out of K = 10 tissue types. (B) Simulation to compare CCmed with standard total association tests and Sobel’s test in detecting trans-associations in at least out of K = 10 tissue types when the sample size is moderate. (C) Simulation to compare CCmed with standard association tests and Sobel’s test in detecting trans-associations in at least out of 10 tissue types when the sample size is small.
3.3 The performance of CCmed in identifying cis-mediated trans-genes for each (GWAS) SNP in selected tissue-types
In this setting, we simulated cis-gene expression levels being affected by 3 correlated eQTLs with correlation 0.3. We focused on one of them as the (GWAS) SNP of interest and generated the trans-gene expression levels being affected by the SNP in selected tissue types. See Supplemental Materials for additional simulation details. Table 1B and 1C presented the power as well as the true and estimated FDRs to detect trans-associations in at least tissues in moderate sample size (350 subjects) and in small sample size (100 subjects), respectively. Here we also include a cross-tissue analysis based on a commonly used mediation test, Sobel’s test (Sobel, 1982), for comparison. Note that the Sobel’s test is not applicable to a gene-level test. For each tissue type, Sobel’s statistic is calculated based on the product of the cis-association α and the conditional correlation between cis- and trans- gene expression levels β, and is approximately normally distributed when the sample size is large. To make a comparison with CCmed, we applied the Primo method to Sobel’s t-statistics to quantify the probabilities of mediation in at least tissues. When the sample size was 350 as in Table 1B, with stronger effect sizes on average and smaller than those in the previous simulation, the standard test had reasonable power and FDR control this time. And with CCmed, the power can still be improved by nearly 50% in this simulation setup with the same FDR control. Sobel’s test performed similar to CCmed with slightly lower power with a sample size of 350. However, when the sample size was decreased to 100 as in Table 1C, CCmed enjoyed improved power over Sobel’s test by 10% to 25%. Previous literature has reported that the Sobel’s test (taking the product of associations) was less powerful than the joint significance test requiring both cis-association and conditional correlation to be non-zero in a single study especially when sample size was limited (Fritz and MacKinnon, 2007). Our simulation results echoed this conclusion and generalized it to cross-conditions settings.
3.4 Mapping gene-level cis-mediated trans-associations with effects in most of the 13 GTEx brain tissue types
We applied CCmed to data from the 13 brain tissue types of the GTEx project (V8) to identify gene-level cis-mediated trans-associations across most brain tissues. There are 5347 autosomal genes with a posterior probability of gene-level cis-association in all 13 brain tissues. For each of these 5347 cis-genes, we further assessed the conditional correlation to each of the trans-gene expression levels in each brain tissue type, and subsequently calculated the cross-tissue probability of cis-mediated trans-association across 12 out of 13 tissues for those cis-genes. At a threshold of , we identified a total of 9323 trios (cis-eSNP set, cis-gene expression, trans-gene expression) showing evidence of gene-level cis-mediated trans-associations in at least 12 out of 13 brain tissue types (with an estimated FDR at 90% posterior probability). These trios included 617 unique cis- and 2511 unique trans-genes. Those cis-genes were not enriched for known transcription factors.
To replicate the trans-associations identified by CCmed, we used data from the eQTLGen Consortium (Võsa et al., 2018) and the CommonMind Consortium (CMC) (Fromer et al., 2016) as two replication studies (see Supplemental Materials for detailed data descriptions). The eQTLGen Consortium, which focused on 10 317 trait-associated SNPs, performed a meta-analysis of trans-eQTL association statistics based on whole blood samples of 31 684 individuals from 37 datasets. To replicate our gene-level trans-association findings with eQTLGen results, for each (Ci, Tj) cis-trans pair in a CCmed mediation trio, we obtained the eQTLGen trans-association P-values between expression of Tj and each SNP in cis with Ci. As a comparison, we also obtained the eQTLGen trans-association P-values for randomly selected cis-trans gene pairs. In the QQ-plots of Figure 2A, the eQTLGen reported trans-association P-values for cis-SNPs and trans-genes identified by CCmed (cyan points) showed a much stronger enrichment of association than the randomly selected GWAS SNPs in the eQTLGen data (black points). This enrichment was present despite the discovery and replication analyses using different tissue types, brain versus blood, respectively, suggesting that CCmed identifies cis-gene-mediated trans-associations that are robust across tissue types and studies. Note that since the eQTLGen consortium (with a very large sample size) focused on only trait-associated SNPs (enriched for cis-/trans- eQTLs), the randomly selected cis-trans gene pairs (black points) from eQTLGen were also enriched for trans-associations and deviated from the 45 degree line. We varied the K1 in the above analysis and re-examined the replication rates. We found that gives the highest replication rates at different P-value thresholds. As expected, lowering K1 would lead to more discoveries but reduce the replicability of results.
Fig. 2.
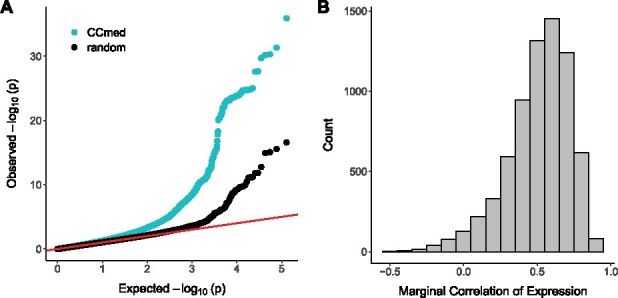
Replication results in two other studies for cis-mediated trans-associations identified from GTEx brain tissues by CCmedmost. (A) QQ-plot of of eQTLGen trans-association P-values comparing trans-associations identified by CCmed (cyan points) to randomly selected trans-associations of trait-associated variants (black points) in the eQTLGen study. (B) Marginal gene-gene correlations in the Dorsolateral Prefrontal Cortex samples from CMC for gene pairs identified by CCmed
Next, we examined the cis-trans gene-gene correlations for gene pairs identified by CCmed using data from the CMC (Fromer et al., 2016). CMC has generated DNA and RNA sequencing data from postmortem brain samples from donors with schizophrenia and bipolar disorder, and from subjects with no neuropsychiatric disorders. For the cis-trans gene pairs identified by CCmed using the GTEx data, Figure 2B showed their marginal expression correlations in the CMC data. As shown by the histogram, most cis-trans pairs in mediation trios identified by CCmed had moderately to strongly correlated expression levels in the dorsolateral prefontal cortex samples of CMC. Since 80% (FDR) of the genes in the CMC data were reported to have at least one cis-eQTL (Fromer et al., 2016), the presence of cis-association and correlation of cis-trans gene expression suggested gene-level trans-association or mediation effects being present in the dorsolateral prefrontal cortex tissue. That is, we observed suggestive evidence of replication in the CMC data for our cis-mediated trans-associations findings from GTEx brain tissue data identified by CCmed.
3.5 Detecting trans-genes associated with 108 known schizophrenia loci in GTEx brain tissues and examining their SCZ risk-associations
To detect trans-genes with expression levels being associated with SCZ GWAS SNPs, we applied CCmed to the 108 SCZ susceptibility loci reported by the PGC consortium (Ripke et al., 2014). Of the 128 reported GWAS SNPs in these loci, 103 were genotyped in the GTEx data, 21 were captured by an SNP in strong LD, 1 SNP was not captured and 3 non-autosomal SNPs were excluded. For each of the 124 SCZ-associated SNPs we analyzed, there were multiple cis-genes in the cis-region. A total of 1643 unique cis-genes were considered for those 124 GWAS SNPs, with all cis-genes being expressed in all brain tissues and having at least 1 brain eQTL reported by GTEx.
For each (GWAS SNP, cis-gene) pair, we estimated the probability of each GWAS SNP being also an eQTL in at least 2 tissue types, conditioning on other cis-eQTLs. There were 40 (GWAS SNP, cis-gene) pairs showing evidence of cis-association in at least 2 brain tissue types (with cross-2-tissue cis-association posterior probability ). Note that here we used a more liberal significance threshold than what was used in gene-level analysis, and this was because the expected power for SNP-level cis-mediated trans-associations was relatively lower. For each of the 40 (GWAS SNP, cis-gene) pairs, we further conducted conditional correlation analysis with each of its trans-genes in each tissue type (see Supplementary Materials for the regression model). We identified 1492 (GWAS SNP, cis-gene, trans-gene) trios with cis-mediated trans-association effects in at least two brain tissue types (FDR ), corresponding to 1418 unique trans-genes.
Those identified suspected trans-gene for SCZ GWAS SNPs may or may not be associated with SCZ risk, due to the potential pleiotropy of SCZ loci. To further examine and validate the SCZ risk-associations for the suspected trans-genes, we conducted a series of analyses on those suspected genes. Those validation analyses are based on a common rationale (as shown in Fig. 3A). Considering a suspected trans-gene identified via CCmed, if the trans-gene is truly associated with the complex trait of interest (here, schizophrenia), then the local eQTLs of the trans-gene would also have associations to the complex trait; otherwise, they would not. Therefore, by examining the trait-association statistics from existing GWAS for SNPs in cis with the suspected trans-genes, one could further identify and validate the trans-genes truly associated with the complex trait (SCZ risk) versus those ones that may be only co-expressed with the SCZ-associated cis-genes. Note that suspected trans-genes not having any local eQTLs cannot be checked nor validated by those analyses.
Fig. 3.

An examination of SCZ risk-associations for suspected trans-genes for SCZ GWAS SNPs identified from GTEx by CCmed. (A) A conceptual illustration of the validation analyses to examine the risk-associations of the suspected trans-genes for GWAS SNPs. In the discovery analysis by CCmed, we identified the suspected trans-genes for GWAS SNPs. If a trans-gene is trait-associated, we expect the SNPs in cis with the trans-gene to be trait-associated. (B) Among the 1418 trans-genes for 124 SCZ-SNPs detected from GTEx via CCmed, 1158 trans-genes have at least 1 cis-eQTL. The histogram (truncated at P = 0.05) of SCZ GWAS summary statistics for the 124 619 eQTLs of those trans-genes showed enrichment of SCZ risk-associations. (C) An example of a suspected trans-gene with SCZ risk association suggested by both TWAS and TWMR analysis. The median eQTL -values in the GTEx brain tissues (y-axis) are plotted against GWAS -values (x-axis) for cis-SNPs of the gene PRR12. Points are colored by LD correlation (r2) to the lead GWAS SNP in the region. SNPs with stronger eQTL associations also have stronger GWAS associations, implying a non-zero effect from the gene PRR12 on SCZ risk
By examining the single-SNP SCZ GWAS summary statistics from PGC for local eQTLs of suspected trans-genes, we observed that the local eQTLs for suspected trans-genes identified by CCmed were highly enriched for associations with schizophrenia-risk. Specifically, for each suspected trans-gene identified from GTEx via CCmed, we checked the SCZ risk-association P-values from GWAS for the local eQTLs of the suspected trans-gene. There are 1158 out of 1418 suspected trans-genes having at least one GTEx reported local eQTL (Aguet et al., 2020) that could also be mapped to the PGC GWAS summary statistics. We checked the SCZ risk-associations of the local SNPs of those genes and found that 589 genes (50.9%) had at least one GTEx reported eQTL with suggestive SCZ GWAS risk-association (P-value < 0.05). Figure 3B shows the histogram of PGC GWAS P-values for the local eQTLs of the 1158 suspected trans-genes. Next, we obtained the transcriptome-wide association analysis (TWAS) P-values for SCZ risk-associations for all genes from a recent analysis by Barbeira et al. (2019), and transcriptome-wide Mendelian Randomization (TWMR) P-values for SCZ risk-associations from a recent analysis by Gleason et al. (2021). The TWAS analysis performed with S-MultiXcan used predicted transcriptome data from a multitissue eQTL reference panel of 44 GTEx (V6p) tissue types (including brain and non-brain tissue types) while the TWMR analysis performed with MR-MtRobin used eQTL summary statistics from the 13 brain tissues in GTEx V8. Both analyses used PGC SCZ GWAS summary statistics. 1342 CCmed trans-genes were tested in at least one of these analyses. Of these, 403 genes (30.0%) had suggestive evidence of SCZ risk-associations (P < 0.05) by at least one of the two analyses. In other words, we identified suspected trans-genes associated with SCZ GWAS SNPs via CCmed using data from normal GTEx brain tissues (without using disease information), and those suspected trans-genes have a much higher than random gene-level SCZ risk-associations based on two other analyses integrating SCZ GWAS data. The scatterplot in Figure 3C shows an example of a gene implicated by both S-MultiXcan and MR-MtRobin (PRR12, with a S-MultiXcan and a MR-MtRobin ). By plotting median eQTL -values from GTEx brain tissues against the GWAS -values from PGC for the cis-SNPs for PRR12, it shows a clear dependence between the two sets of summary statistics (SNPs with stronger eQTL effects also showing consistent GWAS associations), implying a non-zero gene-level effect of PRR12 on SCZ risk.
In identifying the mechanisms through which GWAS SNPs affect complex traits, most existing research examined the effects of GWAS SNPs on cis-genes. Here we introduced CCmed to identify trans-associations of GWAS-SNPs mediated by cis-gene expression and conducted a series of validation analyses showing that the detected trans-genes are also enriched with SCZ risk-associations.
4 Discussion
In this work, we focused on studying cross-condition trans-associations mediated by cis-gene expression levels, which have a direct mechanistic interpretation. A major innovation of ours is that we showed that cis-mediated trans-associations often have effects shared across relevant tissues/conditions and were replicable in different studies. Motivated by the cross-tissue and tissue-specific bimodal effect-sharing patterns of eQTLs, we proposed two variations of the algorithm, CCmed and CCmed, for detecting gene-level cis-mediated trans-associations with effects shared across most conditions and SNP-level effects present in only a few functionally related conditions, respectively. By applying CCmed to data from the 13 brain tissues of the GTEx project, we identified cis-trans gene pairs with gene-level trans-association effects in most brain tissues. Our findings are replicable with many cis-trans gene pairs showing evidence of trans-association effects in two different tissue types from two other replication studies. As a proof-of-concept for CCmed, we applied the method to 108 schizophrenia susceptibility loci and identified the trans-genes for SCZ GWAS SNPs with cis-mediated trans-association effects in at least 2 out of 13 GTEx brain tissues. We further showed that the identified trans-genes are enriched with SCZ risk associations by two other methods, TWAS and TWMR.
Trans-acting genetic effects on distal gene expression are ubiquitous in the genome. Using standard trans-association tests for pairs of SNPs and distal gene expression levels adjusting for multiple comparison, the detection and replication of trans-association effects generally require very large sample sizes. Due to limited tissue-sharing of effects and the tissue-specific sample sizes, trans-associations detected by standard tests are found to be hard to replicate. It is also possible that many false positive findings arise by chance in single-tissue analyses. Our work describes a complementary approach to the standard single-study association tests, and CCmed borrows information across studies/tissues/conditions and detects replicable associations mediated by cis-gene expression levels with effects shared across relevant conditions. By analyzing multitissue eQTL data from GTEx and replicating our findings, we showed that many cis-mediated trans-associations are robust and replicable in different studies.
There are some caveats of the current work that could be potentially improved in future research. First, we analyzed 13 GTEx brain tissue types as a multi-condition mediation analysis. It should be noted that the brain tissues from GTEx have only modest sample size, and a future multi-condition mediation analysis could benefit from the use of eQTL data from other studies, tissue types and cellular conditions. Second, findings from both CCmed and CCmed should be interpreted as association rather than causation. Third, there are still quite a lot of trans-associations not mediated through cis-gene expression levels. Some might be mediated through other omics traits such as cis-methylation levels, cis- alternative splicing events, etc. The identification and integration of other and multiple mediators are of great interest and will be explored in future works.
Supplementary Material
Acknowledgements
The authors thank the GTEx, eQTLGen, CMC and PGC consortia.
Conflict of Interest: none declared.
Funding
This work was supported by the National Institutes of Health [2R01GM108711 to L.S.C and F.Y., SUB-U24 CA2109993 to L.S.C. and F31CA239557 to K.J.G.].
Data availability
The data that support the findings of this study are openly available in the Genotype-Tissue Expression Portal at https://gtexportal.org/home/datasets and CommonMind Consortium Knowledge Portal at https://www.synapse.org/#!Synapse:syn2759792. Summary statistics were downloaded from eQTLGen, https://www.eqtlgen.org/, and PGC websites, https://www.med.unc.edu/pgc/.
Contributor Information
Fan Yang, Department of Biostatistics and Informatics, Colorado School of Public Health, University of Colorado Anschutz Medical Campus, Aurora, CO 80045, USA.
Kevin J. Gleason, Department of Public Health Sciences, University of Chicago, Chicago, IL 60637, USA
Jiebiao Wang, Department of Biostatistics, University of Pittsburgh, Pittsburgh, PA 15261, USA.
Jubao Duan, Unit of Functional Genomics in Psychiatry, Center for Psychiatric Genetics, NorthShore University HealthSystem, Evanston, IL 60201, USA; Department of Psychiatry and Behavioral Neuroscience, Chicago, IL 60637, USA.
Xin He, Department of Human Genetics, University of Chicago, Chicago, IL 60637, USA.
Brandon L. Pierce, Department of Public Health Sciences, University of Chicago, Chicago, IL 60637, USA Department of Human Genetics, University of Chicago, Chicago, IL 60637, USA.
Lin S. Chen, Department of Public Health Sciences, University of Chicago, Chicago, IL 60637, USA.
References
- Aguet F. et al. (2020) The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science, 369, 1318–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbeira A.N. et al. (2019) Integrating predicted transcriptome from multiple tissues improves association detection. PLoS Genet., 15, e1007889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battle A. et al. (2014) Characterizing the genetic basis of transcriptome diversity through RNA-sequencing of 922 individuals. Genome Res., 24, 14–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battle A. et al. ; eQTL Manuscript Working Group. (2017) Genetic effects on gene expression across human tissues. Nature, 550, 204–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon A.L. et al. (2007) A genome-wide association study of global gene expression. Nat. Genet., 39, 1202–1207. [DOI] [PubMed] [Google Scholar]
- Fritz M.S., MacKinnon D.P. (2007) Required sample size to detect the mediated effect. Psychol. Sci., 18, 233–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fromer M. et al. (2016) Gene expression elucidates functional impact of polygenic risk for schizophrenia. Nat. Neurosci., 19, 1442–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamazon E.R. et al. ; GTEx Consortium. (2018) Using an atlas of gene regulation across 44 human tissues to inform complex disease- and trait-associated variation. Nat. Genet., 50, 956–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giambartolomei C. et al. (2014) Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet., 10, e1004383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gleason K.J. et al. (2020a) Primo: integration of multiple GWAS and omics QTL summary statistics for elucidation of molecular mechanisms of trait-associated SNPs and detection of pleiotropy in complex traits. Genome Biol., 21, 236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gleason K.J. et al. (2021) A robust two-sample transcriptome-wide Mendelian Randomization method integrating GWAS with multi-tissue eQTL summary statistics. Genetic Epidemiology, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X. et al. (2019) Trans effects on gene expression can drive omnigenic inheritance. Cell, 177, 1022–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Q. et al. (2016) Integrative tissue-specific functional annotations in the human genome provide novel insights on many complex traits and improve signal prioritization in genome wide association studies. PLoS Genet., 12, e1005947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morley M. et al. (2004) Genetic analysis of genome-wide variation in human gene expression. Nature, 430, 743–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ongen H. et al. (2016) Fast and efficient QTL mapper for thousands of molecular phenotypes. Bioinformatics, 32, 1479–1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Rourke H.P., MacKinnon D.P. (2015) When the test of mediation is more powerful than the test of the total effect. Behav. Res. Methods, 47, 424–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson C.B. et al. (2016) TreeQTL: hierarchical error control for eQTL findings. Bioinformatics, 32, 2556–2558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce B.L. et al. (2014) Mediation analysis demonstrates that trans-eQTLs are often explained by cis-mediation: a genome-wide analysis among 1,800 South Asians. PLoS Genet., 10, e1004818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ripke S. et al. (2014) Biological insights from 108 schizophrenia-associated genetic loci. Nature, 511, 421–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobel M.E. (1982) Asymptotic confidence intervals for indirect effects in structural equation models. Sociol. Methodol., 13, 290–312. [Google Scholar]
- Storey J.D., Tibshirani R. (2003) Statistical significance for genomewide studies. Proc. Natl. Acad. Sci. USA, 100, 9440–9445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Võsa U. et al. (2018) Unraveling the polygenic architecture of complex traits using blood eQTL meta-analysis. bioRxiv, doi: 10.1101/447367. [Google Scholar]
- Yang F., GTEx Consortium. et al. (2017) Identifying cis-mediators for trans-eQTLs across many human tissues using genomic mediation analysis. Genome Res., 27, 1859–1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are openly available in the Genotype-Tissue Expression Portal at https://gtexportal.org/home/datasets and CommonMind Consortium Knowledge Portal at https://www.synapse.org/#!Synapse:syn2759792. Summary statistics were downloaded from eQTLGen, https://www.eqtlgen.org/, and PGC websites, https://www.med.unc.edu/pgc/.