

Abstract
Motivation
Infectious diseases caused by novel viruses have become a major public health concern. Rapid identification of virus–host interactions can reveal mechanistic insights into infectious diseases and shed light on potential treatments. Current computational prediction methods for novel viruses are based mainly on protein sequences. However, it is not clear to what extent other important features, such as the symptoms caused by the viruses, could contribute to a predictor. Disease phenotypes (i.e. signs and symptoms) are readily accessible from clinical diagnosis and we hypothesize that they may act as a potential proxy and an additional source of information for the underlying molecular interactions between the pathogens and hosts.
Results
We developed DeepViral, a deep learning based method that predicts protein–protein interactions (PPI) between humans and viruses. Motivated by the potential utility of infectious disease phenotypes, we first embedded human proteins and viruses in a shared space using their associated phenotypes and functions, supported by formalized background knowledge from biomedical ontologies. By jointly learning from protein sequences and phenotype features, DeepViral significantly improves over existing sequence-based methods for intra- and inter-species PPI prediction.
Availability and implementation
Code and datasets for reproduction and customization are available at https://github.com/bio-ontology-research-group/DeepViral. Prediction results for 14 virus families are available at https://doi.org/10.5281/zenodo.4429824.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Infectious diseases emerging unexpectedly from novel and reemerging pathogens have been a major enduring public health concern around the globe (Jones et al., 2008). Pathogens disrupt host cell functions (Finlay and Cossart, 1997) and target immune pathways (Dyer et al., 2010) through complex inter-species interactions of proteins (Dyer et al., 2008), RNA (Fajardo et al., 2015) and DNA (Weitzman et al., 2004). The study of pathogen–host interactions (PHI) can therefore provide insights into the molecular mechanisms underlying infectious diseases and guide the discoveries of novel therapeutics or provide a basis for the repurposing of available drugs. For example, a previous study of many PHIs showed that pathogens typically interact with the protein hubs (those with many interaction partners) and bottlenecks (those of central locations to important pathways) in human protein–protein interaction (PPI) networks (Dyer et al., 2008). However, due to cost and time constraints, experimentally validated pairs of interacting pathogen–host proteins are limited in number. Therefore, the computational prediction of PHIs is a useful complementary approach in suggesting candidate interaction partners from the human proteome.
Existing PHI prediction methods for novel viruses typically utilize protein sequence features of the interacting proteins (Alguwaizani et al., 2018 ; Eid et al., 2016; Yang et al., 2020; Zhou et al., 2018). While protein functions have been shown to predict intra-species (e.g. human) PPIs (Guzzi et al., 2012; Jain and Bader, 2010; Pesquita et al., 2009) and such protein specific features exist for some extensively studied pathogens, such as Mycobacterium tuberculosis (Huo et al., 2015) and HIV (Mukhopadhyay et al., 2014), for most pathogens, these features are rare and expensive to obtain. As new virus species continue to be discovered (Woolhouse et al., 2012), a method is needed to rapidly identify candidate interactions from information that can be obtained quickly, such as the signs and symptoms exhibited by the host, which may be utilized as a proxy for the underlying molecular interactions between host and pathogen proteins.
The phenotypes elicited by pathogens, i.e. the signs and symptoms observed in a patient, may provide information about molecular mechanisms (Gkoutos et al., 2018). The information that phenotypes provide about molecular mechanisms is commonly exploited in computational studies of Mendelian disease mechanisms (Oellrich et al., 2016), for example, to suggest candidate genes (Hoehndorf et al., 2011; Meehan et al., 2017) or diagnose patients (Köhler et al., 2009), but the information can also be used to identify drug targets (Hoehndorf et al., 2013a) or gene functions (Hoehndorf et al., 2013b). We hypothesize that the host phenotypes elicited by an infection with a pathogen are, among others, the result of molecular interactions, and that knowledge of the phenotypes exhibited by the host can be used to suggest the protein perturbations from which these phenotypes arise.
One major challenge of the novel PHI prediction problem is the lack of ground truth negative data. A recent method, DeNovo (Eid et al., 2016), adopted a ‘dissimilarity-based negative sampling’: for each virus protein, the negatives are sampled from human proteins that do not have known interactions with other similar virus proteins (above a sequence similarity threshold T). Another method based on protein sequences (Zhou et al., 2018), samples negatives from only the set of host proteins that are less than 80% similar (in terms of sequence similarity) to the host proteins in the positive training data. However, the influence of sequence similarity on function is not uniform and while there is evidence for a number of general evolutionary rules, we are unable to determine cutoffs for any specific protein or function (Ponting, 2001; Whisstock and Lesk, 2003). By construction, these sampling schemes make the human proteins in the negative set different from the positive set; when used not only for training a model but also for evaluating its performance, this sampling scheme has the potential to over-estimate the actual performance for finding novel PHIs. In a more realistic evaluation for a novel virus species, a model would be evaluated on all the host proteins with which it could potentially interact, regardless of sequence similarity.
From these motivations, we developed a machine learning method, DeepViral, to predict potential interactions between viruses and all human proteins for which we can generate the relevant features. Firstly, the features of phenotypes, functions and taxonomic classifications are embedded in a shared space for human proteins and viruses. We then extended a sequence model by incorporating the phenotype features of viruses into the model. We show that the joint model trained on both the sequences and phenotypes can significantly outperform state-of-the-art methods and predict potential PHIs in realistic experimental setups for novel viruses.
2 Materials and methods
DeepViral is a model that predicts potential protein interactions between viruses and human hosts from the protein sequences and feature embeddings of phenotypes, functions and taxonomies. To enable predictions based on such different features we embedded them in a shared representation space. We then combine these feature embeddings with a protein sequence model to predict potential PHIs of novel viruses. The workflow of DeepViral is illustrated in Figure 1.
Fig. 1.

The workflow of DeepViral. (a) Generation of an embedding: the arrows of human proteins and virus taxa represent their annotations to the ontology classes. The dashed lines between viruses represent their taxonomic relations. The annotations, taxonomic relations and ontologies were fed into DL2Vec to generate feature embeddings of dimension 100 for each human protein and virus taxa. (b) Joint prediction model: latent representations learned from feature embeddings and protein sequences are concatenated into a joint representation, for human protein and virus protein respectively, on which a dot product is performed to predict interactions
2.1 Data sources
Interactions between hosts and pathogens were obtained from the Host Pathogen Interaction Database (HPIDB; version 3) (Ammari et al., 2016). The phenotypes associated with pathogens were collected from the PathoPhenoDB (Kafkas et al., 2019), a database of manually curated and text-mined associations of pathogens, infectious diseases and phenotypes. We downloaded the PathoPhenoDB database version 1.2.1 (http://patho.phenomebrowser.net/).
The phenotypes associated with human genes were collected from the Human Phenotype Ontology (HPO) database (Köhler et al., 2019), and the phenotypes associated with mouse genes and the orthologous gene mappings from mouse genes to human genes originated from the Mouse Genome Informatics (MGI) database (Smith et al., 2018). The Entrez gene IDs in HPO and MGI were mapped to reviewed Uniprot protein IDs using the Uniprot Retrieve/ID mapping tool (https://www.uniprot.org/uploadlists) on March 6, 2020. The Gene Ontology annotations of human proteins (release date 2020-02-22) were downloaded from the Gene Ontology Consortium (Ashburner et al., 2000; The Gene Ontology Consortium, 2017). Human PPI networks were downloaded from String (Szklarczyk et al., 2019) and filtered to only include the interactions with experimental evidence. The human protein sequences were obtained from the Swiss-Prot database (UniProt Consortium, 2019).
To add background knowledge from biomedical ontologies of phenotypes and GO classes, we downloaded the cross-species PhenomeNET ontology (Hoehndorf et al., 2011; Rodríguez-García et al., 2017), from the AberOWL ontology repository (Hoehndorf et al., 2015a) on September 13, 2018. We obtained the NCBI Taxonomy classification (Sayers et al., 2009) as an ontology in OWL format (version 2018-07-27) from EMBL-EBI ontology repository (https://www.ebi.ac.uk/ols/ontologies/ncbitaxon).
The SARS-CoV-2 interactions are from a recently released dataset of 332 PHIs from 27 viral proteins (Gordon et al., 2020). The PHIs of other Coronaviruses are from a recently curated dataset of Coronaviridae–host PPIs (Perrin-Cocon et al., 2020). The protein sequences of the Coronaviruses in our study are retrieved from the Swiss-Prot database (UniProt Consortium, 2019).
2.2 Learning feature embeddings
To generate feature embeddings, we used DL2Vec (Chen et al., 2020), a recent method for learning features for entities (in our case, the human proteins and viruses) from their associations to ontological classes. DL2Vec first converted the ontologies and entity associations into a graph, with the classes and entities as the nodes and the associations and ontology axioms as the edges. Then a number of random walks were performed, starting from the entities over to the ontology graph and thereby generating a corpus of walks in the form of sentences capturing the graph neighborhoods and thereby the ontology axioms. After the construction of such sentences, a Word2vec skipgram model (Mikolov et al., 2013) was used to learn an embedding for each entity by learning from the corpus. Following the recommendations of the authors of DL2Vec, we fixed the number of walks to 100, the walk length to 30, the embedding dimension to 100 and the number of training epochs to 30. The embeddings were trained with the Word2Vec library in Julia (version 1.0.4). The resultant embedding was a vector representation of an entity capturing its co-occurrence relations with other entities within the walks generated by DL2Vec. As an example, the walks starting from a virus node explored its graph neighborhood, i.e. its associated phenotypes and its taxonomic relatives, and as an result, its feature embedding captured this information according to the co-occurrence patterns.
2.3 Supervised prediction models and parameter tuning
The neural network model of DeepViral consists of two components: a phenotype model based on the feature embeddings of viruses and human proteins, and a sequence model based on the amino acid sequences of the human and viral proteins. The maximum input length of protein sequences is set to 1000 amino acids and all shorter sequences are repeated up to the maximum length. The sequence length cut-off of 1000 is chosen to cover the majority of proteins in the databases from which we constructed our dataset, i.e. 88.2% and 83.7% of the human proteins in Swiss-Prot and HPIDB, respectively, and 91.6% of the virus proteins in HPIDB. The input protein sequences are encoded as a one-hot encoding matrix of 22 rows that represents each amino acid type and the original sequence length (before being repeated), and 1000 columns representing each position of the amino acid sequence.
To predict the likelihood of an interaction between a pair of proteins, we trained the network as a binary classifier, to minimize the binary cross-entropy loss defined below,
where N is the total number of predictions, yt and yp is the true label and predicted likelihood of y.
We implemented our model using the Keras library and performed training on Nvidia Tesla V100 GPUs. The phenotype model consists of a fully connected layer with the feature embeddings as input. The sequence model, adapted from DeepGOPlus (Kulmanov and Hoehndorf, 2020), is a convolutional neural network (CNN) with the sequences as input and consists of 1-dimensional convolution, max pooling and fully connected layers. We tuned the following hyperparameters of the model through a grid search: the maximum size of the convolution filters (i.e. 16, 32 and 64), the number of the filters (i.e. 8 and 16), the size of the max pooling layers (i.e. 50 and 200) and the number of neurons in the fully connected layers (i.e. 8, 16 and 32). We then fixed these hyperparameters throughout all the experiments: 16 convolutional layers for each filter of 8, 16,…, 64 in length, a pool size of 200 and 8 neurons for the dense layers. We also used dropouts (Srivastava et al., 2014) for the convolutional and dense layers with a rate of 0.5 and LeakyReLU as the activation function for the dense layer with an alpha set to 0.1.
3 Results
3.1 Embedding features of viruses and human proteins from phenotypes, functions and taxonomies
We started with the biological hypothesis that phenotypes (i.e. symptoms) elicited by viruses in their hosts can act as a proxy for the underlying molecular mechanisms of the infection, and therefore may provide additional information to the prediction of potential PHIs for novel viruses.
To generate feature embeddings for human proteins and virus taxa, we applied a recent representation learning method DL2Vec (Chen et al., 2020), which learned feature embeddings for entities based on their annotations to ontology classes (see Section 2.2). DL2Vec takes two types of inputs: the associations of the entities with ontology classes (e.g. human proteins and their functions), and the ontologies themselves.
For representing virus taxa through the phenotypes they elicit in their hosts, we used the phenotype associations for viruses from PathoPhenoDB (Kafkas et al., 2019), a database of pathogen to host phenotype (signs and symptoms) associations. To increase the coverage of phenotypes beyond PathoPhenoDB, the taxonomic relations of the viruses were added from the NCBI Taxonomy (Sayers et al., 2009). By adding these taxonomic relations (as annotations of viruses to DL2Vec), we propagated the known phenotypes along the taxonomic hierarchies and learned a generalized embedding for viruses that do not have any phenotype annotations in PathoPhenoDB but have close relatives that do.
Similarly, for representing human proteins, we used the annotations of their associated phenotypes from the Human Phenotype Ontology (HPO) database (Köhler et al., 2019), the phenotypes associated with their mouse orthologs from the Mouse Genome Informatics (MGI) database (Smith et al., 2018), and their protein functions from the Gene Ontology (GO) database (Ashburner et al., 2000; The Gene Ontology Consortium, 2017). We propagated these annotations through the human PPI network, which has been shown to improve prediction for gene-disease associations (Alshahrani and Hoehndorf, 2018).
To provide DL2Vec with structured background knowledge of human and mouse phenotypes as well as protein functions, we used the cross-species phenotype ontology PhenomeNET (Hoehndorf et al., 2011; Rodríguez-García et al., 2017), which is built upon and includes the Gene Ontology (Ashburner et al., 2000; The Gene Ontology Consortium, 2017). These ontologies contain formalized biological background knowledge (Hoehndorf et al., 2015b), which has the potential to significantly improve the performance of these features in machine learning and predictive analyses (Kulmanov et al., 2020; Smaili et al., 2020).
3.2 A joint model for PPI prediction from sequences and phenotypes
DeepViral consists of a phenotype model trained on phenotypes caused by a viral infection and a sequence model trained on protein sequences, as shown in Figure 1b. The two models take a pair of virus and human proteins as input and predicts the probability score of their interaction. The inputs for a human protein are its feature embedding and its sequence, and the features for a viral protein are its sequence and the feature embedding of the virus species to which it belongs. The sequence model projects the protein sequence into a low dimension vector representation, which is concatenated with the vector projected from the embedding by the phenotype model to form a joint representation of the proteins. A dot product was performed over the two vector representations of the pair of proteins to compute their similarity, which was then used as input to a sigmoid activation function to compute their predicted probability of interaction. In an evaluation where the inputs were not symmetric, e.g. only using the feature embeddings of human proteins but not viruses (or vice versa), an additional dense layer was added to project the longer representation to the same dimension as the other so that the dot product could be performed.
Existing prediction methods for inter-species PPI (e.g. virus–human interactions) have rarely been compared with methods designed for intra-species (e.g. human) PPI prediction. To compare with the existing sequence-based methods for both intra- and inter-species PPI prediction, we evaluated DeepViral and RCNN (Chen et al., 2019), a recent method designed for intra-species prediction, on an existing dataset (Eid et al., 2016) that has been used to evaluate a number of PHI prediction methods (Alguwaizani et al., 2018; Yang et al., 2020; Zhou et al., 2018). The respective model performances and implementation details are shown in Supplementary Section S1. DeepViral trained only on sequences achieves comparable performance with other sequence based methods, while the joint model is able to achieve the best performances in most metrics. However, the evaluation dataset suffers from several drawbacks: (i) negative sampling (to create a balanced dataset) was based on sequence dissimilarity; (ii) the training and test sets only cover 39 viral proteins from 26 virus strains and 11 families, which is highly limited relative to the current size and taxonomic diversity of the PHI databases; (iii) there are overlapping virus proteins (i.e. data leakage) at species level between the training and test sets, which makes it unsuitable for the problem of novel PHI prediction.
3.3 Experimental setup, negative sampling and evaluation metrics for novel viruses
Motivated by the need for more representative datasets to evaluate methods for novel PHI prediction, we constructed a larger dataset from the curated virus–host interactions in HPIDB (Ammari et al., 2016), a database of host–pathogen protein–protein interactions. We constructed our positive set by filtering HPIDB to include all virus–host interactions that (i) are provided with an MIscore, a confidence score for molecular interactions (Villaveces et al., 2015); (ii) are associated with an existing virus family in the NCBI taxonomy (Sayers et al., 2009); (iii) are within 1000 amino acids in length (for both human and viral proteins). After filtering, the dataset includes 24 678 positive interactions and 1066 viral proteins from 14 virus families and 292 virus taxa.
To realistically evaluate the prediction performance, we performed a leave-one-family-out (LOFO) cross validation: at each run, one virus family in our positive set was left out for testing, 20% of the remaining families for validation, and the rest 80% for training. The objective of the LOFO cross-validation is to evaluate the model under a scenario in which the novel virus emerges from a novel virus family—in our study, ‘novel’ is defined as the situation in which we have no or very little knowledge about its protein interactions and the molecular functions of the viral proteins.
Instead of using ‘dissimilarity-based negative sampling’ to construct a balanced dataset, we sampled our negatives from all the possible pairwise combinations of human and viral proteins, as long as the pair did not occur in the positive set. Essentially, we treated all ‘unknown’ interactions as negatives. As the dataset was at this point unbalanced with more negatives than positives, we evaluated the model with the area under the receiver operating characteristic (ROC) curve (Fawcett, 2006). A high ROCAUC indicates the ability of the model to rank the true positive interacting proteins higher than proteins for which no such interaction is known. We computed a ROCAUC for each virus family, and also for each viral protein and virus taxon in that family, for which we reported the mean across them, i.e. macro averages. Each model was evaluated 5 times independently to compute the 95% confidence interval of the ROCAUC, which is bounded by mean±1.96×σn, where n is the sample size and σ is the standard deviation. Additionally, the mean ranks of the true positive proteins were provided as a more interpretable metric: for each viral protein, we ranked all of the 16 627 human proteins in Swiss-Prot (with a length limit of 1000) as its potential interaction partner based on the prediction score and obtained the mean ranks of the true positives.
3.4 Phenotypes improve prediction for novel viruses
With the newly constructed dataset, we further evaluated the existing methods as well as the variants of DeepViral, under the scenario in which a novel virus (from a novel family) emerges and no previous knowledge (except about its protein sequences and the phenotypes elicited in its hosts) is known.
We compared DeepViral with two existing state-of-the-art methods based on protein sequences: Doc2Vec + RF (Yang et al., 2020), a recent method predicting for virus–human interactions; and RCNN (Chen et al., 2019), a recent deep learning based method for intra-species (e.g. human) PPI prediction. To adapt Doc2Vec + RF on our dataset, we used the pretrained Doc2Vec model provided by the authors and the same parameters for the random forest model for training. Similarly, for RCNN, we used the pre-trained embeddings for amino acids and the same model parameters for training. Since the stop criterion for Doc2Vec + RF was to have at most 2 samples at each leaf node, we did not use validation data and trained it with the entirety of the training data, while a validation set was used for both RCNN and DeepViral as described in the experimental setup.
For each model, the summary statistics of the predictive performance are shown in Table 1. For models using only sequence features, DeepViral and Doc2Vec + RF perform on a similar level across the metrics. As the current state-of-the-art method for intra-species PPI prediction, RCNN consistently yields the lowest performances. Adding human or virus embeddings individually shows a slight improvement in most metrics, compared to the sequence-only models, while the joint model with both embeddings achieved the best performances overall. The distributions of the ranks of true positives (Fig. 2) are in general correspondence with the summary statistics, with the joint model having lowest ranks overall.
Table 1.
Comparison with the state-of-the-art methods on our dataset to evaluate the performances for novel viruses
| Method | Family-wise ROCAUC | Taxon-wise ROCAUC | Protein-wise ROCAUC | Mean rank |
|---|---|---|---|---|
| RCNN (Chen et al., 2019) | 0.726 [0.717–0.734] | 0.759 [0.750–0.768] | 0.737 [0.731–0.743] | 4669 |
| Doc2Vec + RF (Yang et al., 2020) | 0.764 [0.763–0.765] | 0.768 [0.766–0.770] | 0.751 [0.751–0.752] | 3740 |
| DeepViral (seq) | 0.770 [0.763–0.777] | 0.768 [0.759–0.777] | 0.749 [0.742–0.756] | 4064 |
| DeepViral (seq + human embedding) | 0.778 [0.766–0.790] | 0.789 [0.776–0.801] | 0.757 [0.742–0.771] | 4245 |
| DeepViral (seq + viral embedding) | 0.788 [0.776–0.801] | 0.782 [0.773–0.790] | 0.757 [0.746–0.767] | 3496 |
| DeepViral (joint) | 0.813 [0.808–0.817] | 0.829 [0.822–0.836] | 0.800 [0.797–0.804] | 3156 |
Note: The brackets after DeepViral indicate the features used for the model: seq—protein sequences, joint—both sequences and embeddings of human proteins and viruses. The square brackets behind ROCAUC scores indicate the 95% confidence interval. The bold numbers indicate the best performing method for the respective metrics.
Fig. 2.
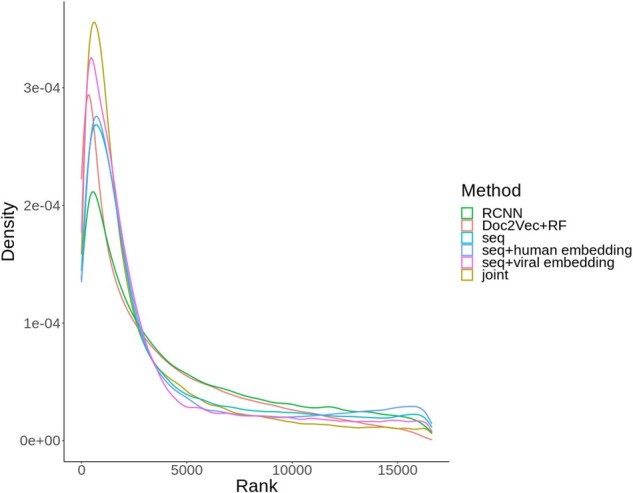
Density plot of the predicted ranks of true positives for each PHI prediction method. The last four methods correspond to the variants of DeepViral
4 Discussion
4.1 Species-level optimization of DeepViral for novel viruses
The continued emergence of novel viruses is an issue of increasing relevance to global public health (Woolhouse et al., 2012) and economic stability (Chakraborty and Maity, 2020). Accurate prediction of potential PHIs for novel viruses with rapidly obtainable features, such as sequences and phenotypes, would be important for understanding infectious disease mechanisms and the repurposing of existing drugs. The LOFO cross-validation excludes the taxonomic relatives from the same family of the test virus, simulating a challenging scenario where the virus is from an entirely novel family. While this provides a stringent evaluation scheme for DeepViral, it likely leads to an underestimate of performance when applied to real world PHI data as most emerging viruses arise from existing virus families (Woolhouse et al., 2012). To investigate whether the inclusion of data from viruses in the same family can improve DeepViral’s ability to predict interactions for viral species, we additionally designed and implemented a leave-one-species-out (LOSO) training and evaluation method. Due to the large number of species, we only applied this method to three viral species from three different RNA virus families, as well as the novel coronavirus SARS-CoV-2 based on a recently released dataset (Gordon et al., 2020).
LOSO is different from LOFO with respect to the training and validation datasets: for each test species, one species from the same family is chosen as the validation set and the rest of the family are all included in the training set. To ensure there is no taxonomic leakage, i.e. identical virus protein sequences among the training, validation and testing datasets, we excluded virus taxa for which proteins have 100% sequence identity.
The comparison between the LOFO and LOSO evaluation is shown in Table 2 and the taxonomic information of the viruses is shown in Supplementary Section S2. When including data from taxonomic relatives (those of the same virus family) in the training and validation sets, the predictive performance of DeepViral improved in all four test cases. The improvements for different viruses exhibited large variability (see Table 2). For example, the Influenza A virus had the largest increase in performance among the four viral species. A similar difference between the virus families can also be observed from the LOFO experiments, as shown in Figure 3. Both the sequence and joint models show similar family-wise variability, with some occasional differences, e.g. Retroviridae performs better than Herpesviridae in the joint model but not in the sequence-based model. The taxon-wise variabilities in both LOFO and LOSO suggest that the features used to predict PHIs may have different generalization and prediction powers across different virus taxa, or PHIs may be characterized to different degrees of completeness. In the future, explainable models (Ribeiro et al., 2016; Lundberg and Lee, 2017) may provide more interpretable insight into this variability.
Table 2.
Improvements of DeepViral’s predictive performance for four virus species, between leave-one-family-out (LOFO) and leave-one-species-out (LOSO) evaluation
| Test virus | Taxon ID | Taxon-wise ROCAUC | Protein-wise ROCAUC | Mean rank | |||
|---|---|---|---|---|---|---|---|
| LOFO | LOSO | LOFO | LOSO | LOFO | LOSO | ||
| SARS-CoV-2 | 2697049 | 0.710 [0.680–0.740] | 0.729 [0.708–0.750] | 0.750 [0.716–0.784] | 0.776 [0.756–0.796] | 4683 | 4344 |
| Zika virus | 2043570 | 0.729 [0.699–0.760] | 0.771 [0.752–0.790] | 0.731 [0.716–0.746] | 0.748 [0.731–0.765] | 4516 | 4413 |
| HPV 18 | 333761 | 0.747 [0.716–0.779] | 0.801 [0.771–0.831] | 0.820 [0.795–0.846] | 0.890 [0.872–0.907] | 4157 | 3240 |
| Influenza A | 644788 | 0.804 [0.787–0.821] | 0.933 [0.907–0.958] | 0.804 [0.788–0.821] | 0.935 [0.914–0.956] | 3306 | 1112 |
Note: Taxon identifiers are based on the NCBI Taxonomy Database (Sayers et al., 2009). Each experiment was repeated five times to compute the 95% confidence interval. The bold numbers indicate the better performing method between LOFO and LOSO.
Fig. 3.
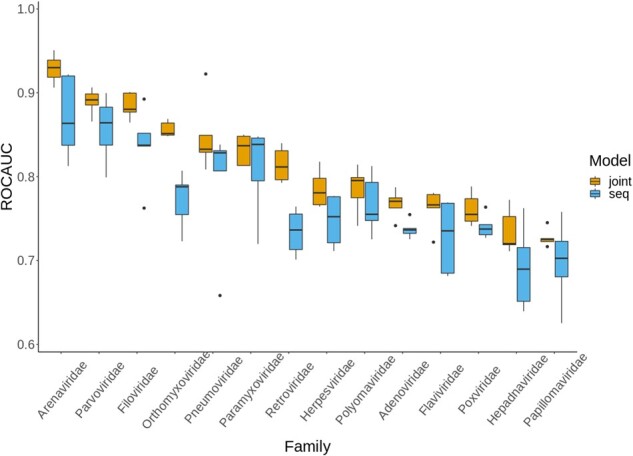
ROCAUC for each of the 14 virus families from the joint model and the sequence model, respectively, ordered by the ROCAUC of the joint model
A contemporary example of a novel virus is the coronavirus SARS-CoV-2, which by the end of 2020 reached more than 83.4 million cases of infections and 1.8 million fatalities globally (Dong et al., 2020) in a timespan of 13 months. In the short time since its emergence, many experimental studies of PHIs between SARS-CoV-2 and human proteins have been published at a historical speed, which enabled biologists to speculate on the infection mechanisms and suggest drug candidates for repurposing (Gordon et al., 2020).
The Coronaviridae M protein constitutes an integral part of the SARS-CoV-2 viral envelope, involved in morphogenesis and assembly via its composite interactions with other structural proteins (Mousavizadeh and Ghasemi, 2020). DeepViral has predicted an interaction between the M protein and the TANK-binding kinase TBK1 (UniProt: Q13158, within top 0.1% of all human proteins). TBK1 plays an important role in the activation of many genes involved in the innate immune response (Fitzgerald et al., 2003; Ran et al., 2016). The interaction between the SARS-CoV-2 M protein and TBK1 was recently validated through affinity capture experiments (Zheng et al., 2020) and proximity-dependent biotinylation methods (Samavarchi-Tehrani et al., 2020). TBK1 has previously been associated with phenotypes related to respiratory distress and respiratory failure through its complex role in amyotrophic lateral sclerosis (Oakes et al., 2017), matching the respiratory phenotypes associated with COVID-19 infections. While the predictions made by DeepViral do not yet allow for a complete understanding of underlying causality, the interaction identified by DeepViral demonstrates how sequence and phenotype information is combined for predicting interactions.
4.2 Using phenotypes to reveal molecular mechanisms of viral infections
DeepViral is, to our knowledge, the first machine learning method that uses clinical phenotypes as a feature to predict PHIs between viruses and human hosts. The use of phenotypes has resulted in a significant improvement (P < 0.05; see confidence intervals in Table 1) over methods that rely on sequences alone. Our model avoids the bottleneck of identifying the molecular functions of pathogen proteins by instead introducing a novel and—in the context of infectious diseases—rarely explored type of feature, the phenotypes elicited by pathogens in their hosts, as a ‘proxy’ for the molecular mechanisms, which in turn eventually produce the observed clinical phenotypes.
One challenge in using phenotypes associated with viral infections or proteins is that they have been derived under different contexts. While phenotypes associated with viral infections are the result of the immune-mediated response and observed in a clinical context (Kafkas et al., 2019), the phenotypes of human proteins are usually associated with a loss or depletion of protein function (Köhler et al., 2019; Smith et al., 2018). However, the phenotypes associated with viral infections obtained from PathoPhenoDB focus on hallmark phenotypes of viral infections that can be used to discriminate between infections of different viruses and thereby de-emphasize the phenotypes resulting from general immune response (Kafkas et al., 2019). Furthermore, the application of neural networks with supervised training can account for differences between observed phenotypes and may even exploit patterns in these differences that are not explicit in the phenotypic representations (Kulmanov et al., 2020; Kulmanov and Hoehndorf, 2020b).
Utilizing phenotypic features observed in humans and mice may have the crucial advantage that we can identify PHIs that may contribute to particular signs and symptoms of infection (Durrant et al., 2011). For example, our model consistently ranks the RNA helicase protein DDX3X (UniProt: O00571) within the top 0.37% of all human proteins as a potential interaction partner of the non-structural protein 4A (UniProt: A0A024B7W1-PRO_0000443029) of Zika virus (NCBITaxon : 2043570). Infections with Zika virus may result in abnormal embryogenesis and, in particular, microcephaly (Wang et al., 2017). Phenotypes associated with DDX3X in the mouse ortholog include abnormal embryogenesis, microcephaly and abnormal neural tube closure (Chen et al., 2016). While DDX3X has previously been linked to the infectivity of the Zika virus (Doñate-Macián et al., 2018) and can result in intellectual disability (Blok et al., 2015), our model further suggests a role of DDX3X in the development of the embryogenesis phenotypes from Zika virus infections.
4.3 Evaluating predictions for novel viruses
While we have demonstrated a quantitative improvement over existing methods on a previously published dataset (Eid et al. 2016; see Supplementary Section S1), we argue that the performance of PHI prediction methods may be over-estimated on datasets where negatives are obtained using a ‘dissimilarity-based negative sampling’ strategy; when only human proteins that are sufficiently different from known interaction partners of viruses are considered for an evaluation, the prediction task is likely to become too simple to reflect performance in a realistic scenario. To address this challenge, we establish an evaluation strategy in which all host proteins are considered as potential interaction partners for novel viruses. Using this evaluation, the predictive performance is considerably lower than using a dissimilarity-based sampling strategy (see Table 1). Another possible explanation for the decrease in performance is that our negative set likely includes some positive interactions that are (falsely) considered as negatives due to absent knowledge of the interaction; this can potentially result in an underestimation of the actual predictive performance.
We use the mean ranks to evaluate model predictions when challenged with a novel virus from a novel family (LOFO), or with known interactions from its taxonomic relatives (LOSO). However, even the best performing model, i.e. DeepViral jointly trained with phenotypes and sequences, has only been able to rank the known true positive proteins up to a mean rank of 3156 out of all 16 627 human proteins in the LOFO evaluation. While the mean rank is sensitive to predictions at a low rank (see Fig. 2), future work is required to further improve PHI prediction methods, especially in regards to the feature selection and engineering, and evaluation methodologies.
4.4 Limitations and future work
DeepViral has several limitations that can be addressed by future work. One is the scarcity of training data for inter-species PPIs. This challenge may be addressed by transfer learning on the much larger intra-species PPI data available for humans and other model organisms. We also did not utilize other types of PHIs outside virus–human interactions in our current study, such as those of other hosts, e.g. plants and fishes, and other types of pathogens, e.g. bacteria and fungi; both may provide further insights in PHIs and the mechanisms underlying viral infections. In particular, in zoonotic diseases, information from PHIs in animals (if available) may be used to identify or suggest interactions that occur in human hosts (Dimonaco et al., 2020; Li et al., 2020). Furthermore, predicting tissue-specific PHIs would also provide additional insights as proteins of both human hosts (Fagerberg et al., 2014) and viruses (Jarosinski et al., 2012) often have tissue-specific expressions and functions.
Supplementary Material
Acknowledgements
The authors thank Maxat Kulmanov and Mona Alshahrani for their advice on earlier versions of this work. They also thank Jeffery Law for making public the mappings of the SARS-CoV-2 proteins. They acknowledge the use of computational resources from the KAUST Supercomputing Core Laboratory.
Funding
This work was supported by funding from King Abdullah University of Science and Technology (KAUST) Office of Sponsored Research (OSR) under Award No. URF/1/3790-01-01.
Conflict of Interest: none declared.
Contributor Information
Wang Liu-Wei, Computer, Electrical and Mathematical Sciences and Engineering Division, King Abdullah University of Science and Technology, Thuwal 23955, Saudi Arabia.
Şenay Kafkas, Computer, Electrical and Mathematical Sciences and Engineering Division, King Abdullah University of Science and Technology, Thuwal 23955, Saudi Arabia; Computational Bioscience Research Center, King Abdullah University of Science and Technology, Thuwal 23955, Saudi Arabia.
Jun Chen, Computer, Electrical and Mathematical Sciences and Engineering Division, King Abdullah University of Science and Technology, Thuwal 23955, Saudi Arabia.
Nicholas J. Dimonaco, Institute of Biological, Environmental and Rural Sciences, Aberystwyth University, Wales SY23 3BQ, UK
Jesper Tegnér, Computer, Electrical and Mathematical Sciences and Engineering Division, King Abdullah University of Science and Technology, Thuwal 23955, Saudi Arabia; Biological and Environmental Science and Engineering Division, King Abdullah University of Science and Technology, Thuwal 23955, Saudi Arabia.
Robert Hoehndorf, Computer, Electrical and Mathematical Sciences and Engineering Division, King Abdullah University of Science and Technology, Thuwal 23955, Saudi Arabia; Computational Bioscience Research Center, King Abdullah University of Science and Technology, Thuwal 23955, Saudi Arabia.
References
- Alguwaizani S. et al. (2018) Predicting interactions between virus and host proteins using repeat patterns and composition of amino acids. J. Healthcare Eng., 2018, 1391265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alshahrani M., Hoehndorf R. (2018) Semantic disease gene embeddings (SmuDGE): phenotype-based disease gene prioritization without phenotypes. Bioinformatics, 34, i901–i907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ammari M.G. et al. (2016) HPIDB 2.0: a curated database for host–pathogen interactions. Database, 2016, baw103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M. et al. (2000) Gene ontology: tool for the unification of biology. Nat. Genet., 25, 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blok L.S. et al. (2015) Mutations in DDX3X are a common cause of unexplained intellectual disability with gender-specific effects on Wnt signaling. Am. J. Hum. Genet., 97, 343–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty I., Maity P. (2020) COVID-19 outbreak: migration, effects on society, global environment and prevention. Sci. Total Environ., 728, 138882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C.-Y. et al. (2016) Targeted inactivation of murine DDX3X: essential roles of DDX3 in placentation and embryogenesis. Hum. Mol. Genet., 25, 2905–2922. [DOI] [PubMed] [Google Scholar]
- Chen J. et al. (2020) Predicting candidate genes from phenotypes, functions and anatomical site of expression. Bioinformatics, 2020, btaa879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M. et al. (2019) Multifaceted protein–protein interaction prediction based on siamese residual RCNN. Bioinformatics, 35, i305–i314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- UniProt Consortium. (2019) UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res., 47, D506–D515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimonaco N.J. et al. (2020) Computational analysis of SARS-CoV-2 and SARS-like coronavirus diversity in human, bat and pangolin populations. Viruses, 13, 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doñate-Macián P. et al. (2018) The TRPV4 channel links calcium influx to DDX3X activity and viral infectivity. Nat. Commun., 9, 2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong E. et al. (2020) An interactive web-based dashboard to track COVID-19 in real time. Lancet Infect. Dis., 20, 533–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durrant C. et al. (2011) Collaborative Cross mice and their power to map host susceptibility to Aspergillus fumigatus infection. Genome Res., 21, 1239–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyer M.D. et al. (2008) The landscape of human proteins interacting with viruses and other pathogens. PLoS Pathogens, 4, e32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyer M.D. et al. (2010) The human-bacterial pathogen protein interaction networks of Bacillus anthracis, Francisella tularensis, and Yersinia pestis. PLoS One, 5, e12089-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eid F.-E. et al. (2016) DeNovo: virus-host sequence-based protein–protein interaction prediction. Bioinformatics, 32, 1144–1150. [DOI] [PubMed] [Google Scholar]
- Fagerberg L. et al. (2014) Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol. Cell. Proteomics, 13, 397–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fajardo T. Jr. et al. (2015) Disruption of specific RNA–RNA interactions in a double-stranded RNA virus inhibits genome packaging and virus infectivity. PLoS Pathogens, 11, e1005321–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fawcett T. (2006) An introduction to ROC analysis. Pattern Recogn. Lett., 27, 861–874. [Google Scholar]
- Finlay B.B., Cossart P. (1997) Exploitation of mammalian host cell functions by bacterial pathogens. Science, 276, 718–725. [DOI] [PubMed] [Google Scholar]
- Fitzgerald K.A. et al. (2003) IKKε and TBK1 are essential components of the IRF3 signaling pathway. Nat. Immunol., 4, 491–496. [DOI] [PubMed] [Google Scholar]
- Gkoutos G.V. et al. (2018) The anatomy of phenotype ontologies: principles, properties and applications. Brief. Bioinf., 19, 1008–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon D.E. et al. (2020) A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature, 583, 459–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzzi P.H. et al. (2012) Semantic similarity analysis of protein data: assessment with biological features and issues. Brief. Bioinf., 13, 569–585. [DOI] [PubMed] [Google Scholar]
- Hoehndorf R. et al. (2011) PhenomeNET: a whole-phenome approach to disease gene discovery. Nucleic Acids Res., 39, e119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoehndorf R. et al. (2013a) Mouse model phenotypes provide information about human drug targets. Bioinformatics, 30, 719–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoehndorf R. et al. (2013b) Systematic analysis of experimental phenotype data reveals gene functions. PLoS ONE, 8, e60847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoehndorf R. et al. (2015a) Aber-OWL: a framework for ontology-based data access in biology. BMC Bioinformatics, 16, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoehndorf R. et al. (2015b) The role of ontologies in biological and biomedical research: a functional perspective. Brief. Bioinf., 16, 1069–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huo T. et al. (2015) Prediction of host – pathogen protein interactions between Mycobacterium tuberculosis and Homo sapiens using sequence motifs. BMC Bioinformatics, 16, 100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain S., Bader G.D. (2010) An improved method for scoring protein–protein interactions using semantic similarity within the gene ontology. BMC Bioinformatics, 11, 562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarosinski K.W. et al. (2012) Fluorescently tagged pUL47 of Marek’s disease virus reveals differential tissue expression of the tegument protein in vivo. J. Virol., 86, 2428–2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones K.E. et al. (2008) Global trends in emerging infectious diseases. Nature, 451, 990–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kafkas Ş. et al. (2019) PathoPhenoDB, linking human pathogens to their phenotypes in support of infectious disease research. Sci. Data, 6, 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köhler S. et al. (2009) Clinical diagnostics in human genetics with semantic similarity searches in ontologies. Am. J. Hum. Genet., 85, 457–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köhler S. et al. (2019) Expansion of the Human Phenotype Ontology (HPO) knowledge base and resources. Nucleic Acids Res., 47, D1018–D1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulmanov,M., and Hoehndorf,R. (2020. a) DeepGOPlus: improved protein function prediction from sequence. Bioinformatics, 36, 422–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulmanov M. et al. (2020) Semantic similarity and machine learning with ontologies. Brief. Bioinf., in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulmanov M., Hoehndorf R. (2020. b) DeepPheno: predicting single gene loss-of-function phenotypes using an ontology-aware hierarchical classifier. PLoS Comput. Biol., 16, e1008453–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X. et al. (2020) Emergence of SARS-CoV-2 through recombination and strong purifying selection. Sci. Adv., 6, eabb9153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundberg S.M., Lee S.-I. (2017) A unified approach to interpreting model predictions. In Proceedings of the 31st International Conference on Neural Information Processing Systems, NIPS’17. Curran Associates Inc., Red Hook, NY, USA, pp. 4768–4777.
- Meehan T.F. et al. (2017) Disease model discovery from 3,328 gene knockouts by The International Mouse Phenotyping Consortium. Nat. Genet., 49, 1231–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikolov T. et al. (2013) Distributed representations of words and phrases and their compositionality. In Burges C.J.C. et al. (eds.) Advances in Neural Information Processing Systems, Vol. 26. Curran Associates, Inc., Red Hook, NY, pp. 3111–3119. [Google Scholar]
- Mousavizadeh L., Ghasemi S. (2020) Genotype and phenotype of COVID-19: their roles in pathogenesis. J. Microbiol. Immunol. Infect. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay A. et al. (2014) Incorporating the type and direction information in predicting novel regulatory interactions between HIV-1 and human proteins using a biclustering approach. BMC Bioinformatics, 15, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakes J.A. et al. (2017) TBK1: a new player in ALS linking autophagy and neuroinflammation. Mol. Brain, 10, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oellrich A. et al. (2016) The digital revolution in phenotyping. Brief. Bioinf., 17, 819–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrin-Cocon L. et al. (2020) The current landscape of coronavirus-host protein–protein interactions. J. Transl. Med., 18, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pesquita C. et al. (2009) Semantic similarity in biomedical ontologies. PLoS Comput. Biol., 5, e1000443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponting C.P. (2001) Issues in predicting protein function from sequence. Brief. Bioinf., 2, 19–29. [DOI] [PubMed] [Google Scholar]
- Ran Y. et al. (2016) Autoubiquitination of TRIM26 links TBK1 to NEMO in RLR-mediated innate antiviral immune response. J. Mol. Cell Biol., 8, 31–43. [DOI] [PubMed] [Google Scholar]
- Ribeiro M.T. et al. (2016) “Why should I trust you?”: Explaining the predictions of any classifier. In Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, San Francisco, CA, USA, August 13–17, 2016, pp. 1135–1144.
- Rodríguez-García M.Á. et al. (2017) Integrating phenotype ontologies with phenomeNET. J. Biomed. Semant., 8, 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samavarchi-Tehrani P. et al. (2020) A SARS-CoV-2 – host proximity interactome. 10.1101/2020.09.03.282103.. [DOI]
- Sayers E.W. et al. (2009) Database resources of the national center for biotechnology information. Nucleic Acids Res., 37, D5–D15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smaili F.Z. et al. (2020) Formal axioms in biomedical ontologies improve analysis and interpretation of associated data. Bioinformatics, 36, 2229–2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith C.L. et al. (2018) Mouse genome database (MGD)-2018: Knowledgebase for the laboratory mouse. Nucleic Acids Res., 46, D836–D842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava N. et al. (2014) Dropout: a simple way to prevent neural networks from overfitting. J. Mach. Learn. Res., 15, 1929–1958. [Google Scholar]
- Szklarczyk D. et al. (2019) STRING v11: protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res., 47, D607–D613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Gene Ontology Consortium. (2017) Expansion of the gene ontology knowledgebase and resources. Nucleic Acids Res., 45, D331–D338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villaveces J.M. et al. (2015) Merging and scoring molecular interactions utilising existing community standards: tools, use-cases and a case study. Database, 2015, bau131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang A. et al. (2017) Zika virus genome biology and molecular pathogenesis. Emerg. Microbes Infect., 6, e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weitzman M.D. et al. (2004) Interactions of viruses with the cellular DNA repair machinery. DNA Repair, 3, 1165–1173. [DOI] [PubMed] [Google Scholar]
- Whisstock J.C., Lesk A.M. (2003) Prediction of protein function from protein sequence and structure. Q. Rev. Biophys., 36, 307–340. [DOI] [PubMed] [Google Scholar]
- Woolhouse M. et al. (2012) Human viruses: discovery and emergence. Philos. Trans. R. Soc. B Biol. Sci., 367, 2864–2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X. et al. (2020) Prediction of human–virus protein–protein interactions through a sequence embedding-based machine learning method. Comput. Struct. Biotechnol. J., 18, 153–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y. et al. (2020) Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) membrane (M) protein inhibits type I and III interferon production by targeting RIG-I/MDA-5 signaling. Signal Transduct. Targeted Ther., 5, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X. et al. (2018) A generalized approach to predicting protein–protein interactions between virus and host. BMC Genomics, 19, 568. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.