

Key Points
Fc galactosylation enhances complement activity of WT IgG1, but not hexameric IgG.
Fc galactosylation improves hexamerization potential of IgG1.
Abstract
Human IgG contains one evolutionarily conserved N-linked glycan in its Fc region at position 297. This glycan is crucial for Fc-mediated functions, including its induction of the classical complement cascade. This is induced after target recognition through the IgG–Fab regions, allowing neighboring IgG–Fc tails to associate through Fc:Fc interaction, ultimately leading to hexamer formation. This hexamerization seems crucial for IgG to enable efficient interaction with the globular heads of the first complement component C1q and subsequent complement activation. In this study, we show that galactose incorporated in the IgG1–Fc enhances C1q binding, C4, C3 deposition, and complement-dependent cellular cytotoxicity in human erythrocytes and Raji cells. IgG1–Fc sialylation slightly enhanced binding of C1q, but had little effect on downstream complement activation. Using various mutations that decrease or increase hexamerization capacity of IgG1, we show that IgG1–Fc galactosylation has no intrinsic effect on C1q binding to IgG1, but enhances IgG1 hexamerization potential and, thereby, complement activation. These data suggest that the therapeutic potential of Abs can be amplified without introducing immunogenic mutations, by relatively simple glycoengineering.
Introduction
Antibodies of the IgG isotype are the most abundant Igs in humans and play a crucial role in immunity. After specific recognition of Ags by either one or two Fab regions of the Y-shaped structure, the Fc region can activate the complement system and/or recruit and activate effector cells through IgG–Fc receptors (FcγRs) on myeloid cells.
These effector functions can be modulated by a conserved N-glycan at N297 in the CH2 domain of the Fc region. The N-glycan consists of a biantennary core structure composed of N-actelyglucosamines and mannose residues but can be further extended by a fucose, bisecting N-actelyglucosamine, galactose, and sialic acid residues. The composition of the N-glycans is highly variable and differs depending on the targeted Ag (1), among individuals, and even over time. The exact composition of these sugar residues can, in turn, drastically affect the Abs’ effector functions and, therefore, the effector output in a specific immune response. Whereas sialylation may possibly be affected by enzymes found in plasma (2), the IgG-producing B cells seem mostly responsible for the glycan composition, as Ag-specific IgG glycosylation can have unique characteristics that differ vastly from total IgG (1, 3–5).
Functionally, the best-characterized change in Fc glycosylation is the absence of fucose, which increases the affinity for FcγRIIIa and FcγRIIIb up to 40-fold (6, 7). This can translate into even larger functional effects—up to binary on/off—for Ab-dependent cellular cytotoxicity and phagocytosis through improved avidity (3, 6–8). It was previously reported that afucosylated Abs are formed primarily against Ags found on cellular surfaces, such as membrane proteins of enveloped viruses, and polymorphic variants of proteins on blood cells (alloantigens) (1).
More recent data also suggest the functional importance of Fc galactosylation and sialylation, and these modifications are often increased in recent or active immunization, as seen after COVID-19 infection (1), vaccination (9, 10), or alloimmunization (3, 5). Whereas galactose has a slight positive effect on IgG binding to FcγRs (11, 12), especially in the context of IgG afucosylation (6), sialic acid incorporation seems to have either no or very minor effects on the binding to FcγR (6, 11). In addition, recent data suggest that both Fc galactosylation and sialylation affect binding of IgG to C1q and, thereby, complement-dependent cytotoxicity (CDC). Whereas IgG–Fc galactosylation seems to promote C1q binding and downstream complement activity (6, 13), reports on the effect of IgG–Fc sialylation on this are conflicting, showing either increased or decreased binding of C1q due to sialylation (6, 14). Bisection seems to have no effect on either FcγR- or complement-mediated activities (6).
CDC can be initiated upon binding of many molecules of IgG to a multivalent Ag, which allows multimerization of neighboring IgG–Fc tails. The prototypic structure is a hexamer, which forms an optimal platform for C1q binding (15, 16). This leads to activation of the C1r and C1s complex, starting the complement cascade by depositing C4b and C3b in the process, which ultimately leads to the formation of the membrane attack complex and lysis of target cells. However, the mechanism by which Fc galactosylation or sialylation affects complement activity has not been elucidated.
In this paper, we tested two hypothetical scenarios by which the Fc glycosylation could impact the binding of C1q. These are that either the galactosylation alters Fc:Fc interaction and facilitates the formation of hexamers or it improves the intrinsic affinity/avidity of individual Fc tails to C1q. To study this, we made use of combinations of Fc mutations facilitating IgG hexamerization, glycoengineering, functional complement assays, and size-exclusion multiangle light scattering (SEC-MALS) to follow hexamerization activity in solution.
Materials and Methods
Donor blood
Peripheral blood was obtained from anonymous, healthy volunteers with informed written consent, in accordance with Dutch regulations.
Expression vectors
pcDNA3.1 expression vectors coding for the anti-biotin heavy and L chain were designed previously, according to Bagçi et al. (17) and Kohen et al. (18). A linear DNA fragment coding for the E345R, E430G, and S440Y mutated Fc region (RGY) was ordered from Integrated DNA Technologies. The 5'-NheI and 3'-EcoRI restriction sites were included in the sequence for the subcloning process. The DNA fragment was digested and inserted proximal to the anti-biotin VH sequence in the pcDNA3.1 expression vector, which was used for the transformation of DH5α competent bacteria (Thermo Fisher Scientific). The single Fc mutations E430G, K439E, and S440K were incorporated using the QuikChange II Site-Directed Mutagenesis Kit from Agilent Technologies, according to the manufacturer’s protocol. Corresponding 5' and 3' primers were designed using the QuikChange Primer Design Program (Agilent Technologies) and ordered at Eurogentec. Plasmids were isolated using a NucleoBond Xtra Maxi Kit (Macherey-Nagel) according to the manufacturer’s instructions. For glycoengineering, the pEE6.4 or pEE14.4 expression vector, encoding for β-1,4 galactosyltransferase 1 (B4GALT1) or β-galactoside α-2,6-sialyltransferase 1 (ST6GALT), respectively, was used as described previously (6, 19). To enhance IgG production, vectors encoding for porf-hp21 (p21; InvivoGen), pORF-hp27 v02 (p27; InvivoGen), and adenovirus large–T Ag (pSVLT) were used (20).
Production of rmAbs
The production of rmAbs was described previously by members from our department (19, 21). In brief, human embryonic kidney (HEK)293F cells (Thermo Fisher Scientific) were cultured in FreeStyle 293 Expression Medium (Thermo Fisher Scientific) using 1-l Erlenmeyer flasks (Corning) at 37°C and 8% CO2 on an orbital shaker with a rotation speed of 125 rpm. Prior to transfection, the cell concentration was measured by CASY cell counter (Roche Innovatis) and adjusted to 1 × 106 cells/ml in fresh medium. A DNA mix was made for the transfection consisting of pcDNA3.1 expression vectors coding for the H and L chain of the anti-biotin Abs and a pSVLT/p21/p27 mix (20). The DNA mix was added to Opti-MEM (Thermo Fisher Scientific) containing 45 mg/ml linear polyethyleneimine HCl MAX (Polysciences) and incubated for 20 min at room temperature (RT). The transfection mixture was added to the cell cultures and incubated at 37°C and 8% CO2 on an orbital shaker with a speed of 125 rpm. Four hours posttransfection, 100 U/ml penicillin and 100 µg/ml streptomycin (Thermo Fisher Scientific) were added. After 6 d, the cells were centrifuged, and the supernatant was collected and filtered with a 0.45-µm syringe filter (Whatman).
Glycoengineering of mAbs
The above-described production of rAbs was adjusted to obtain the desired glycosylation profile, employing glycoengineering. To increase the Fc galactosylation, 1 h prior to the transfection, 5 mM d-galactose (Sigma-Aldrich) was added to the cell culture (19). On top of that, 1% of the pEE6.4 expression vector, encoding for B4GALT1, was added to the DNA mix for transfection. This percentage was calculated based on the total amount of DNA used. To increase Fc sialylation, 5 mM d-galactose and 1% pEE6.4 B4GALT1 were also required, as sialic acid is the terminal sugar residue of the N-glycan structure (Fig. 1B). Furthermore, 2.5% of the pEE14.4 expression vector, encoding for ST6GALT, was also added to the DNA mix for transfection (19).
FIGURE 1.

IgG–Fc glycosylation. (A) Schematic representation of the IgG Y-shaped structure; the Fc-N297 glycan and Fab and Fc regions are indicated. (B) Composition of the N297-glycan biantennary structure with the distinct sugar groups and their respective locations. (C) Ribbon structure of dimeric IgG–Fc regions with N-glycans (1HZH); highlighted areas indicate the N297-glycosylation site (pink) or mutated amino acids involved in Fc interaction presented in this study (E345 [red], E430 [blue], K439 [green], and S440 [orange]).
Purification of mAbs
Abs were purified from the collected supernatant using an AKTA Prime Plus (GE Healthcare) equipped with a 5-ml protein A HiTrap HP column (GE Healthcare). The Abs were eluted from the column with a low pH buffer (0.08 M citric acid per 0.04 M Na2HPO4 [pH 3]). After elution, the pH was neutralized with 1 M Tris-HCL (pH 9), and the obtained fractions were pooled and concentrated using the 2–6-ml Pierce Protein Concentrator PES, 10K MWCO (Thermo Fisher Scientific). The Abs were dialyzed overnight (O/N) to 5 mM sodium acetate buffer (pH 4.5) to avoid aggregation during storage. For the functional assays, the Abs were diluted significantly in the respective assay buffers. After dilution, the Abs were used immediately, unless stated otherwise.
Digestion of IgG for mass spectrometry
Five micrograms of Ab was incubated in 100 µl of 100 mM formic acid for 30 min at RT while shaking, for denaturing the Abs. Then, the Abs were dried for 2 h at 50°C in a centrifugal vacuum concentrator and dissolved in 20 µl of 50 mM ammonium bicarbonate. Twenty microliters of 10 ng/µl sequencing-grade modified trypsin (Promega) was added to digest the Abs O/N at 37°C.
Mass spectrometric glycosylation analysis
The glycopeptides were analyzed by nano-RP-LC-ESI-qTOF-MS on an Ultimate 3000 RSLCnano system (Thermo Fisher Scientific) coupled to a MaXis quadruple–time-of-flight mass spectrometry using a CaptiveSpray and nanoBooster (Bruker Daltonics) (22). A total of 200 nl of sample was injected on a trap column (PepMap 100 C18, 5 µm, 0.3 × 5 mm; Thermo Fisher Scientific) and washed at 25 µl/min with 0.1% trifluoroacetic acid (TFA; Merck) for 1 min before separation on a BEH C18 column (nanoEase M/Z peptide, 1.7-µm particle size, 75 µm × 100 mm; Waters). A binary linear gradient was applied at 600 nl/min with 0.1% TFA (solvent A) and 0.1% TFA in 95% acetonitrile (liquid chromatography–mass spectrometry grade, BioSolve; solvent B): gradient from 3% B to 21.7% B in 0–4.5 min, to 50% B in 4.5–5.5 min, 50% B for 2.5 min and in 1 min to 3% B, and re-equilibration for 2.5 min. Electrospray ionization was performed with an endplate offset of 500 V, capillary voltage of 1200 V, nanoBooster pressure of 0.2 bar using acetonitrile-enriched nitrogen, dry gas flow of 3.0 l/min, and dry temperature of 180°C. Mass spectra were recorded from m/z 550–1800 at 1 Hz with active focus setting, using 5.0-V collision cell voltage, 110-µs transfer time, and 21-µs prepulse storage. The first isotopic peak of triple-protonated glycopeptide signals was manually integrated, and the relative abundance was calculated.
ELISAs
Nunc MaxiSorp flat-bottom 96-well plates (Thermo Fisher Scientific) were coated O/N with 100 µl of 0.5 or 0.25 µg/ml biotinylated BSA (5× biotin/BSA; BioVision) at 4°C. The plates were washed five times with 0.05% PBS–Tween 20. Then, the plates were incubated with 100 µl of anti-biotin Abs for 1 h at RT. A 2-fold dilution series was made, starting with a concentration of 5 µg/ml for the anti-IgG ELISA or 20 µg/ml for the anti-C1q, -C3, and -C4 ELISA. Subsequently, the plates for the anti-IgG ELISA were washed, and 100 µl of 1/1000 Mouse Anti-Human IgG Fc-HRP (SouthernBiotech) or Mouse Anti-Human κ-HRP (SouthernBiotech) was added and incubated for 1 h at RT. The complement ELISAs required an additional step before adding the secondary Ab, and 100 µl of 1:35 pooled human serum in veronal buffer (6) with 0.1% poloxamer 407, 2 mM MgCl2, and 10 mM CaCl2 was added and incubated for 1 h at RT. Consequently, the plates were washed, and 100 µl of either 1/1000 anti–C1q-HRP, anti–C3-HRP, or anti–C4-HRP (23–25) was added and incubated for 1 h at RT. Lastly, after the incubation of the secondary Abs, all ELISA plates were washed and developed with 100 µl of 0.1 mg/ml tetramethylbenzidine solution with 0.11 M NaAc and 0.003% H2O2. The reaction was terminated with 100 µl of 2 M H2SO4, and the absorbance was measured using the BioTek Synergy 2 Multi-Detection Microplate Reader at 450–540 nm.
CDC: RBCs
Whole blood (EDTA) from healthy donors was centrifuged at 3000 rpm using a tabletop centrifuge for the isolation of packed RBCs. The cells were washed with PBS and centrifuged two more times. Afterwards, the cells were centrifuged at 3500 rpm, the supernatant was removed, and the cells were incubated for 30 min in the dark with 0.075 mM EZ-Link NHS-PEG12-Biotin (Thermo Fisher Scientific). Generally, a final concentration of 2–5 mM biotin reagent is effective to biotinylate cell surface proteins, according to the manufacturer’s protocol. Initial titration experiments showed that 0.075 mM was optimal with a large experimental window to analyze differences between the anti-biotin Ab variants. After biotinylation, cells were washed two times with PBS and stored in saline–adenine–glucose–mannitol buffer at 4°C before use. One hundred microliters of biotinylated packed RBCs were washed and resuspended in 6 ml of veronal buffer supplemented with 0.05% gelatin. Thirty-five-microliter biotinylated cells were added to a 96-well round-bottom plate with a glass bead in each well and incubated for 90 min at 37°C while shaking, with a 2-fold dilution series of anti-biotin Abs, starting with a concentration of 20 µg/ml, 10% pooled human serum in veronal buffer supplemented with 0.05% gelatin, 1 mM CaCl2, and 0.5 mM MgCl2. Ten microliters of 5% saponin was added to the positive control for setting the maximum level of cell lysis. After incubation, the cells were centrifuged for 2 min at 1800 rpm, and 60 µl of the supernatant was transferred to Nunc MaxiSorp flat-bottom, 96-well plates. The absorbance was measured using the BioTek Synergy 2 Multi-Detection Microplate Reader at 412–690 nm.
CDC: Raji cells
Raji cells (American Type Culture Collection) were cultured at 37°C with 5% CO2 in RPMI 1640 medium (Life Technologies) supplemented with 10% FCS and 100 U/ml penicillin and 100 µg/ml streptomycin (Thermo Fisher Scientific). A total of 18E06 cells were washed and incubated in 0.05 mM EZ-Link NHS-PEG12-Biotin (Thermo Fisher Scientific) in PBS for 30 min at RT. Then, the biotinylated cells were washed with the culture medium and two times with RPMI 1640 and resuspended in 1% PBS/BSA. A total of 5E05 cells were incubated with equal volumes of anti-biotin Abs (0.01–20 µg/ml) and pooled human serum (20%), for 30 min at 37°C while shaking. The cells were washed twice with PBS/BSA and incubated with 40 µl of 1/1000 LIVE/DEAD Fixable Near-IR Dead Cell Stain (Thermo Fisher Scientific) for 30 min on ice. The cells were washed twice with PBS/BSA and measured by flow cytometry. The data were analyzed using FlowJo vX for Windows (BD Biosciences). The Raji cells were gated based on the forward-/side-scatter, and the percentage of dead cells was calculated using the LIVE/DEAD Fixable Near-IR Dead Cell Stain.
SEC-MALS
The Abs were dialyzed to PBS O/N with the Slide-A-Lyzer G2 Dialysis Cassettes, 3.5K MWCO, 3 or 0.5 ml, depending on the volume. Samples were prepared 24 h before measuring and adjusted to the proper concentrations using PBS (0.05, 0.10, 0.20, 0.30, and 1 mg/ml). The samples were run on the Agilent 1260 Infinity II HPLC System equipped with UV/VIS detector SPD-20A (Shimadzu), miniDAWN (Wyatt Technology), Optilab (Wyatt Technology), and Superose 6 Increase 10/300 GL column (GE Healthcare) for size-exclusion chromatography (SEC). Filtered PBS with 0.02% Tween 20 was used as a running buffer with a flow speed of 0.75 ml/min. Further analysis and estimations of the m.w. were calculated via multiangle and dynamic light scattering using the Astra software (version 7) of Wyatt Technology.
Statistics
Statistical analyses were executed within GraphPad Prism 8.02 (263) for Windows 64-bit. Curve fitting was performed using nonlinear regression dose-response curves with log(agonist) versus response–variable slope (four parameters). Bar graphs were analyzed using ordinary one-way ANOVAs with Tukey multicomparison test. The level of significance was set at p ≤ 0.05. Statistical significance is denoted by *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, and ****p ≤ 0.0001.
Results
IgG glycoengineering of variants with different hexamerization potential
To investigate the mechanism by which Fc galactosylation and sialylation (Fig. 1A, 1B) affect the classical complement pathway, we produced glycoengineered variants of monoclonal anti-biotin IgG1 Abs and incorporated amino acid mutations affecting Fc:Fc interaction and hexamerization. These mutations were RGY and E430G, known for increasing Fc:Fc interaction, whereas K439E and S440K suppress Fc:Fc interaction by charge repulsion (15, 16, 26–29) (Fig. 1C). After production (Fig. 2A), the different glycoengineered Abs were subjected to glycoanalysis by liquid chromatography–mass spectrometry (Fig. 2B, 2C, Supplemental Fig. 1). The glycosylation profiles of the unmodified Abs showed nearly 100% fucosylation, 20% galactosylation, <1% sialylation, and 3% bisection (Fig. 2B). By elevated substrate availability and expression of B4GALT1, Fc galactosylation of IgG1 was increased to 80–85%. As observed previously, this was accompanied by a slight increase in sialylation and a decreased bisection (6). Increasing sialylation by overexpression of ST6GALT increased sialylation to ∼30%. Fucosylation was not affected by any of these glycoengineering techniques. The different IgG1 sequence variants showed very similar changes of glycosylation patterns upon glycoengineering, the only exception being RGY, which showed slightly lower levels of sialylation than the other glycoengineered sequence variants (Fig. 2C). For the unmodified Abs, the most abundant glycans were G0F ≫ G1F. For the highly galactosylated Abs, the majority of the glycans had either two or one galactose residues attached with a minor fraction containing one terminal sialic acid (G2F ≫ G1F > G2FS). The glycans of the highly sialylated Abs were the most diverse (G2FS > G2F > G1F > G2FS2 = G0F).
FIGURE 2.
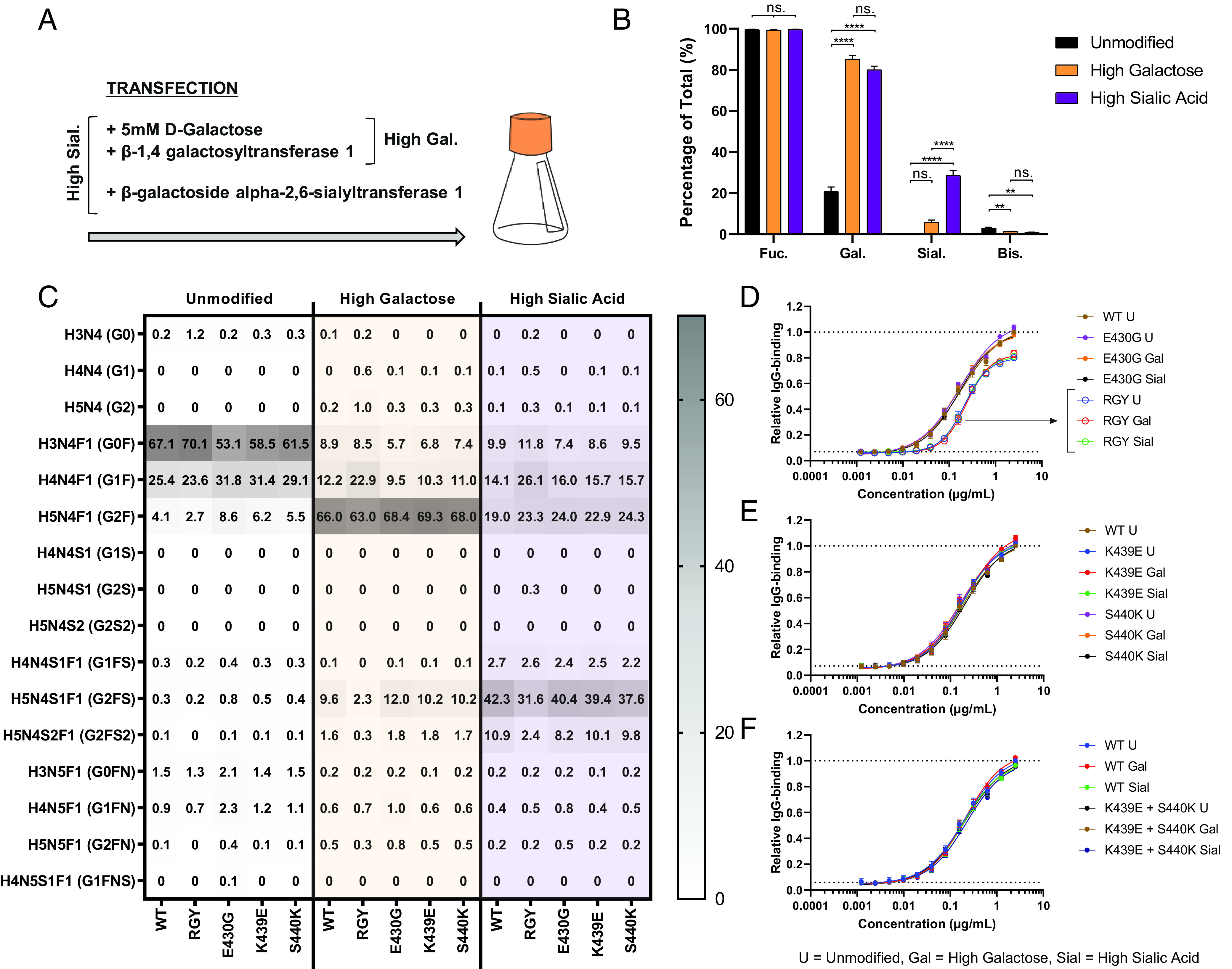
IgG glycoengineering for altered galactosylation and sialylation. (A) Schematic setup for the production process in HEK FreeStyle cells, with the addition of relevant substrates and constructs coding for enzymes prior/during transfection. (B and C) Fc glycosylation profiles of produced anti-biotin mAbs using different glycoengineering techniques to increase Fc galactosylation and sialylation, analyzed by mass spectrometry. The bar graphs represent the mean and SEM of five different mAbs. For the statistical analysis, an ordinary one-way ANOVA with Tukey multicomparison test was performed. (D–F) Relative levels of IgG binding of produced anti-biotin mAbs to 5× biotin/BSA are presented as relative value to the maximum response of the unmodified WT mAb, determined by ELISA (n = 3). Curve fitting was performed using nonlinear regression dose-response curves with log(agonist) versus response–variable slope (four parameters) in GraphPad Prism 8.0.2. No differences in opsonization were observed between glycovariants. **p ≤ 0.01, ****p ≤ 0.0001.
Glycoengineering does not affect Ag binding
We then tested Ag binding of these Ab variants by ELISA. No differences were observed between the wild-type (WT), K439E, S440K, and E430G mutated Abs (Fig. 2D–F). The RGY variants, however, had lower maximum binding capacity, and the EC50 was larger compared with those of all other Abs (Fig. 2D), possibly because of their tendency to form hexamers and thereby affect the binding of the secondary Ab (15). Therefore, the binding capacity was also analyzed with a mouse anti-human κ secondary Ab, which showed identical anti-biotin binding behavior in RGY compared with WT and other mutants (Supplemental Fig. 2). No differences were observed upon glycoengineering, not for the WT or for any other mutated Ab.
IgG–Fc galactosylation primarily enhances C1q binding for WT IgG, not for hexameric IgG
We then tested the potential of these Abs to activate the complement system—that is, C1q binding ((Fig. 3A), C4 deposition ((Fig. 3B), C3 deposition ((Fig. 3C), and RBC lysis ((Fig. 3D). As expected, the unmodified RGY IgG1 was superior in activating the complement system, followed by E430G ((Fig. 3A1, B1, C1, D1). Both showed a higher amount of C1q binding, C4/C3 deposition, and cell lysis than the WT showed. As expected, for both K439E and S440K, less-complement activation was observed than for the WT, whereas the combination of these complementing mutations (15, 27) resulted in similar levels as for the WT. This difference was particularly pronounced in the CDC assay, in which incubation of the individual Abs (K439E or S440K) resulted in no cell lysis ((Fig. 3D), most likely attributable to overall less C1q binding and C4/C3 deposition with these mutants. The combination of both K439E and S440K led to lysis similar to that in WT, likely because of a compensating nature for the electrostatic repulsion effect when these two mutants are mixed (15, 27).
FIGURE 3.

Complement-activating properties of glycoengineered and mutated anti-biotin mAbs shown for (A) all unmodified anti-biotin variants and (B–G) glycoengineered variants. In (A1–A7), relative binding of C1q (n = 4) is shown. (B1–B7) Relative deposition of C4 (n = 4). (C1–C7) Relative deposition of C3 (n = 4). (D1–D7) Complement-mediated lysis of biotinylated RBCs (n = 4). All C1q binding and C4 and C3 deposition were determined by ELISA. Data represent the mean and SEM of four independent experiments; all values were presented as relative value to the maximum response of the unmodified WT mAb. Curve fitting was performed using nonlinear regression dose-response curves with log(agonist) versus response–variable slope (four parameters) in GraphPad Prism 8.0.2. The maximum response and EC50 were calculated, which are presented in Fig. 4. Mutants RGY and E430G are more potent in activating the complement system compared with the WT, whereas E439K and S440K perform worse. Differences in complement activation between glycoengineered variants were only observed for the WT, E439K, and S440K, not for RGY and E430G.
We then compared the effect of glycoengineering on C1q binding, C4 and C3 deposition, and CDC ((Fig. 3A2–7, B2–7, C2–7, D2–7). Elevated galactosylation of WT IgG1 enhanced their complement activity ((Fig. 3A2, B2, C2, D2), and elevated sialylation only showed a slight additional effect on C1q binding. Similar effects were seen for the K439E and S440K Abs, except the overall C1q binding and C3 deposition were reduced compared with those in WT IgG1; importantly, they showed no CDC activity, and the additional improvement of C1q binding for the sialylated Abs was more noticeable ((Fig. 3A3, A4, B3, B4, C3, C4, D3, D4). Enhanced galactosylation of the combination of K439E + S440K also elevated complement activity to a similar degree as WT, including CDC. The most striking result, however, was the observation that neither galactosylation nor sialylation of Abs with improved Fc:Fc interaction and hexamerization potential, RGY and E430G, had any effect on complement activity at any stage ((Fig. 3A6, A7, B6, B7, C6, C7, D6, D7).
The maximum responses (Fig. 4A–D) and EC50 values (Fig. 4E–H) were extracted from these dose-response curves. In terms of increasing activities, the mutants ranked in three categories, K439E and S440K < WT and K439E + S440K < RGY and E430G. In general, both the highly galactosylated and sialylated Abs showed significantly higher maximum responses and lower EC50s compared with those of the unmodified Abs, except for the RGY and E430G variants. However, no significant differences were found between the highly galactosylated and sialylated Abs.
FIGURE 4.

The maximum response and EC50 of complement-activating properties of glycoengineered and mutated anti-biotin mAbs extracted from (Fig. 3. (A–D) Maximum response. (E–H) EC50 values. Data represent the mean and SEM of four independent experiments; the maximum response is presented as relative value to the maximum response of the unmodified WT mAb. For the statistical analysis, an ordinary one-way ANOVA with Tukey multicomparison test was performed. Nonsignificant comparisons were not shown. Higher levels of Fc galactosylation and Fc sialylation significantly induce the maximum response and reduce the EC50 of complement activation by WT, E439K, and S440K mAbs. These differences were not observed for RGY and E430G mAbs. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001.
Next, we investigated the potential of these anti-biotin Abs to activate the complement system on biotinylated Raji cells, as these cells are commonly used as substrates for human nucleated cells and are more resistant to CDC than RBCs are. The CDC was measured by flow cytometry, using the gating strategy as visualized in (Fig. 5A. Despite the difference in cell type, the Raji cells behaved very similarly to the RBCs, showing the same tendencies. In terms of complement-dependent cell lysis, the mutants ranked in three categories, K439E and S440K < WT and K439E + S440K < RGY and E430G (Fig. 5B). Enhanced Fc galactosylation boosted complement activity of WT IgG1 and K439E + S440K, but not those with enhanced hexamerization potential (E430G and RGY) or isolated K439E or S440K (Fig. 5C–J).
FIGURE 5.

Complement-mediated lysis of biotinylated Raji cells. (A) Flow cytometric gating strategy; cells were gated based on the forward-/side-scatter, and the percentage of dead cells was calculated using the LIVE/DEAD Fixable Near-IR Dead Cell Stain. (B) Complement-mediated lysis with unmodified anti-biotin variants. (C–H) Complement-mediated lysis using glycoengineered anti-biotin variants. Data represent the mean and SEM of three independent experiments; curve fitting was performed using nonlinear regression dose-response curves with log(agonist) versus response–variable slope (four parameters) in GraphPad Prism 8.0.2. (I) The maximum response and (J) EC50 of complement-mediated lysis of glycoengineered and mutated anti-biotin mAbs extracted from (Fig. 5C–H. For the statistical analysis, an ordinary one-way ANOVA with Tukey multicomparison test was performed. Nonsignificant comparisons were not shown. *p ≤ 0.05, **p ≤ 0.01, ****p ≤ 0.0001.
We therefore hypothesized that these glycan adducts enhance Fc:Fc interaction potential and, thereby, Ab oligomerization and complement activity.
Fc galactosylation affects hexamerization potential of Abs
RGY IgG1 Abs form hexamers spontaneously in solution in a concentration-dependent manner (15, 16). We therefore tested the oligomerization properties of RGY Abs at limiting concentrations with our glycoengineered Abs using SEC. Approximately 90% of the RGY variant was present as hexamer in solution at high concentrations (Fig. 6A). As expected, a higher abundancy of monomeric IgG was found at lower concentrations. We noted that the retention time of the peaks was also dependent on the concentration, likely because (relatively fast) association and dissociation take place during the high performance size exclusion chromatography runs, and peaks reflect the effective average complex size at intermediate concentrations. Estimation of the molecular mass of these peaks by multiangle laser light scattering confirmed their molecular identities (estimation lacking at the lowest concentrations because of the low intensity of the UV signals; (Fig. 6B–E). Enhanced galactosylation, but not sialylation, of these variants shifted the relative balance of the RGY at lower concentrations in the direction of higher-m.w. complexes, consistent with formation of hexamers (Fig. 6B–E). Next, we quantified the oligomerization based on their intensities and found that only galactose significantly enhanced IgG1 hexamerization (Fig. 6F). Together, these data strongly suggest that IgG1 Abs with high levels of Fc galactosylation have higher potential to activate the complement cascade because of their improved hexamerization capabilities and, therefore, its interaction with the hexameric C1q.
FIGURE 6.

Hexamerization of RGY mAbs is facilitated by galactosylation. (A) The oligomerization of the RGY Abs was visualized by SEC-MALS and was observed in a concentration-dependent manner. Approximately 90% of the Ab was present as hexamer in solution at a concentration of 1 mg/ml. However, the lower the concentration the higher the abundancy of monomeric IgG. Absorbance was measured at 280 nm (OD), and normalization was performed using the sum of all values in the data set (area under the curve [AUC]), shown on the left y-axis. Molecular mass was measured using multiangle light scattering (MALS) and shown on the right y-axis. (B–E) Representative SEC curves of glycoengineered RGY mAbs at different concentrations. Normalization was performed based on the AUC. (F) Fc galactosylation significantly induces oligomerization of RGY mAbs at all concentrations. Data represent the mean and SEM of three to five independent samples. For the statistical analysis, an ordinary one-way ANOVA with Tukey multicomparison test was performed. *p ≤ 0.05, ****p ≤ 0.0001.
Discussion
IgG–Fc glycosylation plays an important role in the modulation of its effector functions. The composition of the single and apparently evolutionarily conserved biantennary N-linked glycan at position 297 is highly variable. Afucosylated IgG are specifically evoked in alloimmune responses and against enveloped viruses, elevating binding affinity to FcγRIIIa and FcγRIIIb by ∼20-fold and thereby increasing effector functions (1, 3, 6, 8, 30, 31). Galactosylation is transiently increased during pregnancy (3, 32) and decreased with advancing age (33). Fc galactosylation of Ag-specific IgG is often increased with recent or active immunization, as seen after COVID-19 infection (1), vaccination (9, 10), or alloimmunization (3, 5). Low levels of IgG–Fc galactose in total IgG are also associated with development of autoimmune diseases (34–40). However, a mechanistic explanation for this association is lacking (41). In this study, we extend previous observations showing that elevated Fc galactosylation enhances C1q binding and complement activity of IgG1 (6, 13, 14) and provide new mechanistic insights into how that occurs. It has been suggested that Fc glycans are associated with the structural stability of Fc region of mAbs and are required for the open CH2 conformation. Structurally, the mannose (Man) α1–6 arm extends toward the CH2–CH3 elbow region, and the Man α1–3 arm extends into the inner space of the CH2 domain in the Fc region (42). Multiple interactions between the glycan components of the Man α1–6 arm and protein structure of an Ab affect the conformational stability of the CH2 domain (43–47). These studies lend support for at least three hypothetical scenarios on how IgG galactosylation and/or sialylation affects complement activity through C1q binding. The first is that it alters the flexibility of the CH2 domains and thereby directly affects binding to C1q, as the critical residues for C1q binding reside in this region (31). The second possibility is that the glycan adducts are in direct contact with the globular head of C1q, and therefore, the binding and activity of C1q is altered. Third, the altered flexibility or orientation of the CH2–CH3 region is altered, enhancing Fc:Fc interaction and, therefore, hexamerization that require optimal CH2–CH3 configuration (15, 48), which is reminiscent of a similarly suggested difference in flexibility between IgG1 and IgG3 Fc (49).
Our results seem to eliminate the first two possibilities and lend support for the last scenario, as elevated galactosylation only improved complement activation for WT IgG1 and variants with weaker hexamerization potential, but not for variants with strongly elevated capacity to form hexamers, even in solution. Those variants reached their maximum capacity to form hexamers and inherently show elevated binding to C1q. In line with this, the tendency of the RGY variant to form spontaneous hexamers in solution, which is known to be decreased at lower concentrations, is increased with elevated galactosylation. However, these concentrations are challenging to translate to the in vitro or in vivo situation, as the effective concentration of the Ab on the cellular surface, when bound to an Ag, is probably much higher on a small surface area. Therefore, higher concentrations were required in solution in which Fc:Fc interaction is the only factor affecting the equilibrium between monomer and multimers, improving the probability to come into vicinity and intermolecular interaction.
These results are in agreement with recently published data from Wei et al. (50), who showed that Fc galactosylation leads to conformational changes in the CH2 domain, stabilizing hexameric IgG. The effect of Fc sialylation on complement activity is less clear. Quast et al. (14) found that Fc sialylation impaired CDC, whereas Dekkers et al. (6) and Wada et al. (43) found no, or possibly slightly increased, C1q binding by sialylated IgG. In this study, a slight (nonsignificant) positive effect of sialylation on C1q binding was observed only for the WT and K439E and S440K mutant Abs, but not on downstream complement deposition or CDC. If Fc sialylation had a direct effect on C1q binding, we would have expected to see differences in all IgG variants, including E430G and RGY. Furthermore, sialylation had no hexamerization effect in addition to what galactosylation had on RGY IgG1 in solution.
In conclusion, IgG–Fc galactosylation elevates the Fc:Fc interaction potential of IgG, resulting in enhanced hexamerization capabilities and, therefore, C1q binding. Subsequently, this results in complement activation, ultimately leading to increased CDC. These data suggest that the therapeutic potential of Abs can be amplified without introducing immunogenic mutations, by relatively simple glycoengineering.
Supplementary Material
Acknowledgments
We thank Maximilian Brinkhaus, Nienke Oskam, and Steven W. de Taeye for sharing knowledge about the complement assays used in this manuscript and Jan Voorberg for helpful discussions and comments on the manuscript.
This article is featured in Top Reads, p.1495
This work was supported by Stichting Sanquin Bloedvoorziening (PPO 17-44).
T.L.J.v.O., T.R., C.E.v.S, M.W., and G.V. designed and supervised experimental work; T.L.J.v.O., J.N., N.I.L.D., and G.v.M. performed experiments and collected data. G.V. conceived the study. T.L.J.v.O. and G.V. wrote the manuscript, which was edited by all authors. All authors analyzed and interpreted data and approved the manuscript.
The online version of this article contains supplemental material.
- B4GALT1
- β-1,4 galactosyltransferase 1
- CDC
- complement-dependent cytotoxicity
- Man
- mannose
- O/N
- overnight
- RGY
- E345R, E430G, and S440Y mutated Fc region
- RT
- room temperature
- SEC
- size-exclusion chromatography
- SEC-MALS
- size-exclusion multiangle light scattering
- ST6GALT
- β-galactoside α-2,6-sialyltransferase 1
- TFA
- trifluoroacetic acid
- WT
- wild-type
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Larsen M. D., de Graaf E. L., Sonneveld M. E., Plomp H. R., Nouta J., Hoepel W., Chen H.-J., Linty F., Visser R., Brinkhaus M., et al. 2020. Afucosylated IgG characterizes enveloped viral responses and correlates with COVID-19 severity. Science 371: eabc8378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jones M. B., Oswald D. M., Joshi S., Whiteheart S. W., Orlando R., Cobb B. A.. 2016. B-cell-independent sialylation of IgG. Proc. Natl. Acad. Sci. USA 113: 7207–7212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kapur R., Kustiawan I., Vestrheim A., Koeleman C. A. M., Visser R., Einarsdottir H. K., Porcelijn L., Jackson D., Kumpel B., Deelder A. M., et al. 2014. A prominent lack of IgG1-Fc fucosylation of platelet alloantibodies in pregnancy. Blood 123: 471–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kapur R., Della Valle L., Verhagen O. J. H. M., Hipgrave Ederveen A., Ligthart P., de Haas M., Kumpel B., Wuhrer M., van der Schoot C. E., Vidarsson G.. 2015. Prophylactic anti-D preparations display variable decreases in Fc-fucosylation of anti-D. Transfusion 55: 553–562. [DOI] [PubMed] [Google Scholar]
- 5.Sonneveld M. E., Natunen S., Sainio S., Koeleman C. A. M., Holst S., Dekkers G., Koelewijn J., Partanen J., van der Schoot C. E., Wuhrer M., Vidarsson G.. 2016. Glycosylation pattern of anti-platelet IgG is stable during pregnancy and predicts clinical outcome in alloimmune thrombocytopenia. Br. J. Haematol. 174: 310–320. [DOI] [PubMed] [Google Scholar]
- 6.Dekkers G., Treffers L., Plomp R., Bentlage A. E. H., de Boer M., Koeleman C. A. M., Lissenberg-Thunnissen S. N., Visser R., Brouwer M., Mok J. Y., et al. 2017. Decoding the human immunoglobulin G-glycan repertoire reveals a spectrum of Fc-receptor- and complement-mediated-effector activities. Front. Immunol. 8: 877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shields R. L., Lai J., Keck R., O’Connell L. Y., Hong K., Meng Y. G., Weikert S. H. A., Presta L. G.. 2002. Lack of fucose on human IgG1 N-linked oligosaccharide improves binding to human Fcgamma RIII and antibody-dependent cellular toxicity. J. Biol. Chem. 277: 26733–26740. [DOI] [PubMed] [Google Scholar]
- 8.Temming A. R., de Taeye S. W., de Graaf E. L., de Neef L. A., Dekkers G., Bruggeman C. W., Koers J., Ligthart P., Nagelkerke S. Q., Zimring J. C., et al. 2019. Functional attributes of antibodies, effector cells, and target cells affecting NK cell-mediated antibody-dependent cellular cytotoxicity. J. Immunol. 203: 3126–3135. [DOI] [PubMed] [Google Scholar]
- 9.Selman M. H. J., de Jong S. E., Soonawala D., Kroon F. P., Adegnika A. A., Deelder A. M., Hokke C. H., Yazdanbakhsh M., Wuhrer M.. 2012. Changes in antigen-specific IgG1 Fc N-glycosylation upon influenza and tetanus vaccination. Mol. Cell. Proteomics 11: M111.014563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang T. T., Maamary J., Tan G. S., Bournazos S., Davis C. W., Krammer F., Schlesinger S. J., Palese P., Ahmed R., Ravetch J. V.. 2015. Anti-HA glycoforms drive B cell affinity selection and determine influenza vaccine efficacy. Cell 162: 160–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thomann M., Schlothauer T., Dashivets T., Malik S., Avenal C., Bulau P., Rüger P., Reusch D.. 2015. In vitro glycoengineering of IgG1 and its effect on Fc receptor binding and ADCC activity. PLoS One 10: e0134949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lippold S., Nicolardi S., Domínguez-Vega E., Heidenreich A.-K., Vidarsson G., Reusch D., Haberger M., Wuhrer M., Falck D.. 2019. Glycoform-resolved FcɣRIIIa affinity chromatography-mass spectrometry. MAbs 11: 1191–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Peschke B., Keller C. W., Weber P., Quast I., Lünemann J. D.. 2017. Fc-galactosylation of human immunoglobulin gamma isotypes improves C1q binding and enhances complement-dependent cytotoxicity. Front. Immunol. 8: 646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Quast I., Keller C. W., Maurer M. A., Giddens J. P., Tackenberg B., Wang L. X., Münz C., Nimmerjahn F., Dalakas M. C., Lünemann J. D.. 2015. Sialylation of IgG Fc domain impairs complement-dependent cytotoxicity. J. Clin. Invest. 125: 4160–4170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Diebolder C. A., Beurskens F. J., De Jong R. N., Koning R. I., Strumane K., Lindorfer M. A., Voorhorst M., Ugurlar D., Rosati S., Heck A. J. R., et al. 2014. Complement is activated by IgG hexamers assembled at the cell surface. Science 343: 1260–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang G., de Jong R. N., van den Bremer E. T. J., Beurskens F. J., Labrijn A. F., Ugurlar D., Gros P., Schuurman J., Parren P. W. H. I., Heck A. J. R.. 2016. Molecular basis of assembly and activation of complement component C1 in complex with immunoglobulin G1 and antigen. Mol. Cell 63: 135–145. [DOI] [PubMed] [Google Scholar]
- 17.Bagçi H., Kohen F., Kusçuoglu U., Bayer E. A., Wilchek M.. 1993. Monoclonal anti-biotin antibodies simulate avidin in the recognition of biotin. FEBS Lett. 322: 47–50. [DOI] [PubMed] [Google Scholar]
- 18.Kohen F., Bagci H., Barnard G., Bayer E. A., Gayer B., Schindler D. G., Ainbinder E., Wilchek M.. 1997. Preparation and properties of anti-biotin antibodies. Methods Enzymol. 279: 451–463. [DOI] [PubMed] [Google Scholar]
- 19.Dekkers G., Plomp R., Koeleman C. A. M., Visser R., von Horsten H. H., Sandig V., Rispens T., Wuhrer M., Vidarsson G.. 2016. Multi-level glyco-engineering techniques to generate IgG with defined Fc-glycans. Sci. Rep. 6: 36964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vink T., Oudshoorn-Dickmann M., Roza M., Reitsma J. J., de Jong R. N.. 2014. A simple, robust and highly efficient transient expression system for producing antibodies. Methods 65: 5–10. [DOI] [PubMed] [Google Scholar]
- 21.Brinkhaus M., Douwes R. G. J., Bentlage A. E. H., Temming A. R., de Taeye S. W., Tammes Buirs M., Gerritsen J., Mok J. Y., Brasser G., Ligthart P. C., et al. 2020. Glycine 236 in the lower hinge region of human IgG1 differentiates FcγR from complement effector function. J. Immunol. 205: 3456–3467. [DOI] [PubMed] [Google Scholar]
- 22.Falck D., Jansen B. C., de Haan N., Wuhrer M.. 2017. High-throughput analysis of IgG Fc glycopeptides by LC-MS. Methods Mol. Biol. 1503: 31–47. [DOI] [PubMed] [Google Scholar]
- 23.McGrath F. D. G., Brouwer M. C., Arlaud G. J., Daha M. R., Hack C. E., Roos A.. 2006. Evidence that complement protein C1q interacts with C-reactive protein through its globular head region. J. Immunol. 176: 2950–2957. [DOI] [PubMed] [Google Scholar]
- 24.Hack C. E., Paardekooper J., Smeenk R. J., Abbink J., Eerenberg A. J., Nuijens J. H.. 1988. Disruption of the internal thioester bond in the third component of complement (C3) results in the exposure of neodeterminants also present on activation products of C3. An analysis with monoclonal antibodies. J. Immunol. 141: 1602–1609. [PubMed] [Google Scholar]
- 25.Leito J. T. D., Ligtenberg A. J. M., van Houdt M., van den Berg T. K., Wouters D.. 2011. The bacteria binding glycoprotein salivary agglutinin (SAG/gp340) activates complement via the lectin pathway. Mol. Immunol. 49: 185–190. [DOI] [PubMed] [Google Scholar]
- 26.de Jong R. N., Beurskens F. J., Verploegen S., Strumane K., van Kampen M. D., Voorhorst M., Horstman W., Engelberts P. J., Oostindie S. C., Wang G., et al. 2016. A novel platform for the potentiation of therapeutic antibodies based on antigen-dependent formation of IgG hexamers at the cell surface. PLoS Biol. 14: e1002344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Oostindie S. C., van der Horst H. J., Lindorfer M. A., Cook E. M., Tupitza J. C., Zent C. S., Burack R., VanDerMeid K. R., Strumane K., Chamuleau M. E. D., et al. 2019. CD20 and CD37 antibodies synergize to activate complement by Fc-mediated clustering. Haematologica 104: 1841–1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Strasser J., de Jong R. N., Beurskens F. J., Schuurman J., Parren P. W. H. I., Hinterdorfer P., Preiner J.. 2020. Weak fragment crystallizable (Fc) domain interactions drive the dynamic assembly of IgG oligomers upon antigen recognition. ACS Nano 14: 2739–2750. [DOI] [PubMed] [Google Scholar]
- 29.Strasser J., de Jong R. N., Beurskens F. J., Wang G., Heck A. J. R., Schuurman J., Parren P. W. H. I., Hinterdorfer P., Preiner J.. 2019. Unraveling the macromolecular pathways of IgG oligomerization and complement activation on antigenic surfaces. Nano Lett. 19: 4787–4796. [DOI] [PubMed] [Google Scholar]
- 30.Wuhrer M., Porcelijn L., Kapur R., Koeleman C. A. M., Deelder A., de Haas M., Vidarsson G.. 2009. Regulated glycosylation patterns of IgG during alloimmune responses against human platelet antigens. J. Proteome Res. 8: 450–456. [DOI] [PubMed] [Google Scholar]
- 31.Vidarsson G., Dekkers G., Rispens T.. 2014. IgG subclasses and allotypes: from structure to effector functions. Front. Immunol. 5: 520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bondt A., Rombouts Y., Selman M. H. J., Hensbergen P. J., Reiding K. R., Hazes J. M. W., Dolhain R. J. E. M., Wuhrer M.. 2014. Immunoglobulin G (IgG) Fab glycosylation analysis using a new mass spectrometric high-throughput profiling method reveals pregnancy-associated changes. Mol. Cell. Proteomics 13: 3029–3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Baković M. P., Selman M. H. J., Hoffmann M., Rudan I., Campbell H., Deelder A. M., Lauc G., Wuhrer M.. 2013. High-throughput IgG Fc N-glycosylation profiling by mass spectrometry of glycopeptides. J. Proteome Res. 12: 821–831. [DOI] [PubMed] [Google Scholar]
- 34.Parekh R. B., Dwek R. A., Sutton B. J., Fernandes D. L., Leung A., Stanworth D., Rademacher T. W., Mizuochi T., Taniguchi T., Matsuta K., et al. 1985. Association of rheumatoid arthritis and primary osteoarthritis with changes in the glycosylation pattern of total serum IgG. Nature 316: 452–457. [DOI] [PubMed] [Google Scholar]
- 35.Ercan A., Cui J., Chatterton D. E. W., Deane K. D., Hazen M. M., Brintnell W., Donnell C. I. O., Derber L. A., Weinblatt M. E., Shadick N. A., et al. 2010. Aberrant IgG galactosylation precedes disease onset, correlates with disease activity, and is prevalent in autoantibodies in rheumatoid arthritis. Arthritis Rheum. 62: 2239–2248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Watson M., Rudd P. M., Bland M., Dwek R. A., Axford J. S.. 1999. Sugar printing rheumatic diseases: a potential method for disease differentiation using immunoglobulin G oligosaccharides. Arthritis Rheum. 42: 1682–1690. [DOI] [PubMed] [Google Scholar]
- 37.Kri J., Gudelj I., Teruel M., Keser T., Pezer M., Barrios C., Pavic T., Trbojevic I., Menni C., Wang Y., et al. 2015. Association of systemic lupus erythematosus with decreased immunosuppressive potential of the IgG glycome. Arthritis Rheumatol. 67: 2978–2989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wuhrer M., Selman M. H. J., McDonnell L. A., Kümpfel T., Derfuss T., Khademi M., Olsson T., Hohlfeld R., Meinl E., Krumbholz M.. 2015. Pro-inflammatory pattern of IgG1 Fc glycosylation in multiple sclerosis cerebrospinal fluid. J. Neuroinflammation 12: 235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Selman M. H. J., Niks E. H., Titulaer M. J., Verschuuren J. J. G. M., Wuhrer M., Deelder M.. 2011. IgG Fc N-glycosylation changes in Lambert-Eaton myasthenic syndrome and myasthenia gravis research articles. J. Proteome Res. 10: 143–152. [DOI] [PubMed] [Google Scholar]
- 40.Trbojevi I., Nimmo E. R., Kalla R., Drummond H., Dunlop M. G.. 2015. Inflammatory bowel disease associates with proinflammatory potential of the immunoglobulin G glycome. Inflamm. Bowel Dis. 21: 1237–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dekkers G., Rispens T., Vidarsson G.. 2018. Novel concepts of altered immunoglobulin G galactosylation in autoimmune diseases. Front. Immunol. 9: 553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Krapp S., Mimura Y., Jefferis R., Huber R., Sondermann P.. 2003. Structural analysis of human IgG-Fc glycoforms reveals a correlation between glycosylation and structural integrity. J. Mol. Biol. 325: 979–989. [DOI] [PubMed] [Google Scholar]
- 43.Wada R., Matsui M., Kawasaki N.. 2019. Influence of N-glycosylation on effector functions and thermal stability of glycoengineered IgG1 monoclonal antibody with homogeneous glycoforms. MAbs 11: 350–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Aoyama M., Hashii N., Tsukimura W., Osumi K., Harazono A., Tada M., Kiyoshi M., Matsuda A., Ishii-Watabe A.. 2019. Effects of terminal galactose residues in mannose α1-6 arm of Fc-glycan on the effector functions of therapeutic monoclonal antibodies. MAbs 11: 826–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mimura Y., Church S., Ghirlando R., Ashton P. R., Dong S., Goodall M.. 2001. The influence of glycosylation on the thermal stability and effector function expression of human IgG1-Fc : properties of a series of truncated glycoforms. Mol. Immunol. 37: 697–706. [DOI] [PubMed] [Google Scholar]
- 46.Alsenaidy M. A., Okbazghi S. Z., Kim J. H., Joshi S. B., Middaugh C. R., Tolbert T. J., Volkin D. B.. 2014. Physical stability comparisons of IgG1-Fc variants : effects of N-glycosylation site occupancy and Asp/Gln residues at site Asn 297. J. Pharm. Sci. 103: 1613–1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Harbison A. M., Brosnan L. P., Fenlon K., Fadda E.. 2019. Sequence-to-structure dependence of isolated IgG Fc complex biantennary N -glycans : a molecular dynamics study. Glycobiology 29: 94–103. [DOI] [PubMed] [Google Scholar]
- 48.Ugurlar D., Howes S. C., de Kreuk B.-J., Koning R. I., de Jong R. N., Beurskens F. J., Schuurman J., Koster A. J., Sharp T. H., Parren P. W. H. I., Gros P.. 2018. Structures of C1-IgG1 provide insights into how danger pattern recognition activates complement. Science 359: 794–797. [DOI] [PubMed] [Google Scholar]
- 49.Rispens T., Davies A. M., Ooijevaar-de Heer P., Absalah S., Bende O., Sutton B. J., Vidarsson G., Aalberse R. C.. 2014. Dynamics of inter-heavy chain interactions in human immunoglobulin G (IgG) subclasses studied by kinetic Fab arm exchange. J. Biol. Chem. 289: 6098–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wei B., Gao X., Cadang L., Izadi S., Liu P., Zhang H. M., Hecht E., Shim J., Magill G., Pabon J. R., et al. 2021. Fc galactosylation follows consecutive reaction kinetics and enhances immunoglobulin G hexamerization for complement activation. MAbs 13: 1893427. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.