

Key Points
Anticoagulation by GPRP allows thrombin generation in human whole blood.
Thrombin does not cleave plasma C5 in its native form.
pH-induced C5 conformational change allows thrombin-mediated cleavage of C5.
Abstract
Thrombin activation of C5 connects thrombosis to inflammation. Complement research in whole blood ex vivo necessitates anticoagulation, which potentially interferes with the inflammatory modulation by thrombin. We challenged the concept of thrombin as an activator of native C5 by analyzing complement activation and C5 cleavage in human whole blood anticoagulated with Gly-Pro-Arg-Pro (GPRP), a peptide targeting fibrin polymerization downstream of thrombin, allowing complete endogenous thrombin generation. GPRP dose-dependently inhibited coagulation but allowed for platelet activation in accordance with thrombin generation. Spontaneous and bacterial-induced complement activation by Escherichia coli and Staphylococcus aureus, analyzed at the level of C3 and C5, were similar in blood anticoagulated with GPRP and the thrombin inhibitor lepirudin. In the GPRP model, endogenous thrombin, even at supra-physiologic concentrations, did not cleave native C5, despite efficiently cleaving commercially sourced purified C5 protein, both in buffer and when added to C5-deficient serum. In normal serum, only exogenously added, commercially sourced C5 was cleaved, whereas the native plasma C5 remained intact. Crucially, affinity-purified C5, eluted under mild conditions using an MgCl2 solution, was not cleaved by thrombin. Acidification of plasma to pH ≤ 6.8 by hydrochloric or lactic acid induced a C5 antigenic change, nonreversible by pH neutralization, that permitted cleavage by thrombin. Circular dichroism on purified C5 confirmed the structural change during acidification. Thus, we propose that pH-induced conformational change allows thrombin-mediated cleavage of C5 and that, contrary to previous reports, thrombin does not cleave plasma C5 in its native form, suggesting that thrombin cleavage of C5 may be restricted to certain pathophysiological conditions.
Introduction
The plasma protein cascades comprising the complement, the coagulation, the fibrinolytic, and the contact activation systems have evolved to offer host defense and homeostasis. Complement typically induces inflammation in response to exogenous and endogenous danger (1, 2), whereas coagulation as part of the hemostatic system maintains vascular integrity (3). These systems cross-talk intensively, and the pathophysiological effects are collectively termed thromboinflammation (4). Complement activation can be triggered via three pathways, all converging at C3, leading to its cleavage into C3a and C3b. At high local C3b concentrations, the convertase will shift substrate specificity from C3 to C5 (5). Cleavage of C5 initiates two main effector functions of complement activation: production of proinflammatory C5a and formation of the terminal C5b-9 complement complex (TCC), which can be inserted as the membrane attack complex into lipid membranes or released to the fluid-phase in a soluble form (sC5b-9). The coagulation cascade is triggered through the biologically and pathophysiologically important extrinsic pathway by exposure of tissue factor or by the intrinsic pathway by activation of FXII. The final effect from both pathways is the generation of thrombin and thrombin-mediated cleavage of fibrinogen, which liberates fibrinopeptides A and B (6). Cleaved fibrin monomers are linked via knob-to-hole interaction into fibrin polymers and clot formation.
Activation of complement and coagulation is often triggered simultaneously in systemic inflammatory responses, such as trauma and sepsis, and several nodes for cross-talk between the two entities have been demonstrated (7–9). Cleavage of complement components by serine proteases in the coagulation cascade has been reported in several publications (10–13), in which thrombin-mediated cleavage of C5 is highly acknowledged (14–16). Thrombin was shown to cleave the α-chain of C5 at R947 position efficiently and, subsequently but less efficiently, cleave at the same site as the native C5 convertases, R751 position, releasing of C5a (15). However, the in vivo importance was recently questioned when a strong burst of thrombin and plasmin failed to produce measurable levels of complement activation fragments in baboons in vivo (17).
One common limitation of the numerous in vitro studies on this topic is the use of purified components as the substrate in various buffer systems, which presumes that the purified component is indistinguishable from the native components. Such purified protein models preclude interactions between proteins under physiological conditions where a number of regulatory cascade molecules are present.
The action of thrombin is highly regulated in whole blood by inhibitors targeting thrombin and by the presence of the primary substrate, fibrinogen, in high concentration. Therefore, studying the role of thrombin on complement necessitates the use of whole blood or plasma models anticoagulated downstream the level of thrombin. At the same time, complement should not be influenced by the anticoagulant. The standard whole blood model in complement research has been the lepirudin-based model, which utilizes blockade of thrombin (18). Lepirudin does not interact with complement, which is then free to act mutually with any other biologic system in blood. The main limitation of the model is that thrombin-dependent effects cannot be studied because of the presence of lepirudin.
To circumvent this limitation, we exploited a synthetic peptide comprising four amino acids, Gly-Pro-Arg-Pro (GPRP), corresponding to the sequence of the knob in the α-chain of fibrinogen exposed after fibrinopeptide liberation, as a competitive inhibitor of fibrin polymerization (19, 20). The unique advantage of this novel model is that GPRP exerts its effect downstream of thrombin, leaving this molecule free to react. Thus, we aimed to characterize this novel human whole blood ex vivo model based on the GPRP peptide for anticoagulation and examine its utility to assess the effect of thrombin on complement activation in human whole blood, focusing on the potency for thrombin to cleave C5 under physiological conditions.
Materials and Methods
Reagents
The GPRP peptide Pefabloc FG (Pefa-6003) was obtained from Pentapharm (Basel, Switzerland), and lepirudin (Refludan) from Celgene (Uxbridge, UK). Purified C5 from commercial sources were from Comptech (Tyler, TX) and Quidel Corporation (San Diego, CA), with purified C5b6 also sourced from Comptech. Neutralizing Abs to C2 (mAb 172-62), factor D (mAb 166-32), and C5a (mAb 137-26) were kindly provided by Genentech, CA.
Blood sampling and sample preparation
Human whole blood was collected from healthy volunteers of both genders. Blood was sampled from the antecubital vein via an 18-gauge needle (Nipro, Bridgewater, NJ) connected to a 2-mm silicone tubing (Thermo Fisher, Gothenburg, Sweden) by using gravity flow. No tourniquet was used. Blood was collected in 50-ml Falcon tubes (Sarstedt, Nümbrecht, Germany) or 4.5-ml Nunc polypropylene tubes (Nalgene Nunc, Roskilde, Denmark) in the presence of GPRP (2–8 mg/ml final concentration), lepirudin (50 μg/ml final concentration), or PBS. Whole blood for ROTEM thromboelastometry, hematology, and blood gas analysis was diluted 30% with PBS before analysis. Plasma was prepared from whole blood by centrifugation for 20 min, 3000 × g at 4°C. Blood for fibrinogen function analysis in plasma was collected in BD Vacutainer citrate tubes (BD Biosciences, San Jose, CA) with 0.109 M sodium citrate.
Fibrinogen analysis
The effect of GPRP on fibrin formation was analyzed in a pool of citrated plasma from six donors on a STA-R Evolution (Diagnostica Stago, Asnières, France) with the reagent STA-Liquid Fib (Diagnostica Stago). Plasma was incubated with increasing concentrations of GPRP (1–8 mg/ml) in three separate experiments, and the concentration of fibrin monomers accessible for fibrin formation in the presence of GPRP was related to standard samples with different concentrations of fibrinogen. Plasma used in the analysis was freeze-thawed one time.
ROTEM thromboelastometry
Coagulation kinetics in fresh human whole blood was analyzed using thromboelastometry on a ROTEM instrument (Tem Innovations GmbH, Germany). Human whole blood (300 µl) was added to the preheated plastic cup (37°C) with an automatic pipette in the absence or presence of GPRP (2–8 mg/ml). Thromboelastometry analyses (NATEM) were then performed immediately on the ROTEM instrument, utilizing a slightly modified method by replacing the Star-TEM reagent containing CaCl2 (Tem Innovations GmbH) with PBS (20 µl).
Physiological and hematological parameters
Physiological parameters, including pH and blood gases, were analyzed on an ABL800 instrument from Radiometer (Radiometer Medical ApS, Brønshøj, Denmark). Hematological parameters were analyzed in whole blood anticoagulated with GPRP (8 mg/ml) and analyzed on a CELL-DYN Sapphire hematology analyzer from Abbott Diagnostics (Abbott Park, IL). Samples were analyzed repeatedly for up to 4 h.
Thrombin and platelet activation
Thrombin and platelets were investigated in whole blood supplemented with GPRP (8 mg/ml), lepirudin (50 µg/ml), or sterile PBS pH 7.4 (Sigma-Aldrich, St Louis, MO) and incubated in 1.8-ml Nunc tubes for up to 20 min at 37°C. A mixture of EDTA (final concentration 10 mM) and 10× citrate-theophylline-adenosine-dipyridamole stock solution (BD Biosciences) was added after incubation to block further activation. Activation of thrombin was measured in plasma by quantification of thrombin anti-thrombin (TAT) complexes by a commercial kit (Siemens Healthcare Diagnostics Products GmbH, Marburg, Germany). Platelet activation was assessed by measuring β-thromboglobulin (βTG) in plasma by a commercial kit (Asserachrom βTG, Diagnostica Stago). Both assays were run according to the instructions from the manufacturer. Samples from time zero and 15 min were also analyzed for prothrombin cleavage by Western blot. Plasma was separated on a 4–15% gradient SDS-PAGE (Bio-Rad, Hercules, CA) under nonreduced conditions. Proteins were transferred onto an immunoblot polyvinylidene fluoride membrane (Bio-Rad) and blotted using rabbit polyclonal IgG anti-prothrombin (Molecular innovations; Novi, MI) followed by an HRP-linked goat anti-rabbit-IgG (Southern Biotech, Birmingham, AL).
Complement activation in whole blood induced by Escherichia coli and Staphylococcus aureus
Heat-inactivated Escherichia coli strain LE392 (ATCC 33572) and Staphylococcus aureus Cowan strain 1 (ATCC 12598) (both from American Type Culture Collection, Manassas, VA), or sterile PBS pH 7.4 (Sigma-Aldrich) as control were incubated in GPRP- and lepirudin-anticoagulated whole blood for 30 min at 37°C. The bacteria were without delay, (i.e., within 2 min) added to the whole blood after blood donation. The bacterial concentration was kept at 105–107/ml for E. coli and 106–108/ml for S. aureus. Following incubation, EDTA was added to a final concentration of 10 mM, and the blood was centrifuged. Complement activation was characterized by measuring plasma activation markers at the level of C3 (C3bc), C5 (C5a), and TCC assembly (sC5b-9). In-house ELISAs were employed to measure C3bc and sC5b-9, as described in detail previously (21). Briefly, these assays are based on capturing monoclonal Abs binding to activation-specific neoepitopes. C5a was measured using a commercial kit (BD Biosciences).
Affinity purification of C5
C5 was affinity-purified from human serum (TCS Biosciences) by an E141A, H164A OmCI column (22). Briefly, serum diluted 1:1 in PBS, 20 mM EDTA was applied to a 5 ml Hi-Trap NHS column (GE Healthcare), with immobilized E141A H164A OmCI protein (20 mg) at a flow rate of 1 ml/min. The column was then washed with five column volumes of PBS, and C5 was eluted using 2 M MgCl2. The eluant was immediately loaded onto an S200 Superdex HiLoad 16/60 gel filtration column (GE Healthcare), which had been pre-equilibrated with PBS, and run at 1 ml/min. Fractions containing the central portion of the protein peak were collected and pooled. C5 protein was aliquoted for storage at −80°C.
Cleavage and detection of purified C5
Thrombin-dependent cleavage of purified C5 was assessed by performing incubations in PBS pH 7.4 at 37°C. Purified C5, bought from Comptech and Quidel, and MgCl2-based affinity-purified C5 was incubated with thrombin (Sigma-Aldrich) in a 5:1 molar ratio (C5, 500 nM and thrombin, 100 nM) from time zero up to 1 h at 37°C. With C5 from Comptech, also incubations for up to 20 h were performed. Lepirudin in 10 times molar excess of thrombin (1 μM) was added to block thrombin activity at the end of incubation. C5 cleaved with cobra venom factor (CVF, Quidel) served as a positive control for C5a. CVF was then mixed with factor B and factor D (both Comptech) in a 1:1:0.1 molar ratio in PBS and 5 mM MgCl2 and incubated for 1 h at 37°C. C5 was then added in a 10 molar excess and incubated furthermore for 20 h at 37°C before loading on the SDS-PAGE. Cleavage of C5 was analyzed by separating C5 fragments on a 4–15% gradient SDS-PAGE under reduced conditions followed either by direct protein staining with Coomassie Brilliant Blue G-250 (Sigma-Aldrich) or transferred onto a polyvinylidene difluoride membrane (Bio-Rad) and blotted using a rabbit pAb anti-C5a (Comptech) followed pAb anti-rabbit-HRP (Southern Biotech, Birmingham, AL). In separate experiments, purified C5b6 (Comptech) in a final concentration of 50 µg/ml was added to tubes containing PBS at pH 7.4, 6.8, and 6.4. Thrombin (400 nM) or 0.145 M NaCl were added, and the tubes were incubated for 60 min at 37°C. Laemmli sample buffer (Bio-Rad) with a reducing agent (2-ME, Bio-Rad) was added at incubation stop, and the proteins were separated on 4–15% gradient SDS-PAGE. Proteins were stained with SYPRO Ruby Protein Gel Stain (Sigma-Aldrich) and detected using ultraviolet transillumination.
Cleavage and detection of C5 in plasma and serum
To assess thrombin-dependent cleavage of C5 in plasma, a pool of GPRP-plasma (three donors) or serum from a C5-deficient individual +/− thrombin (400 nM) +/− lepirudin (7 μM) +/− purified C5 (Comptech, 60 μg/ml) was incubated in 1.8-ml Nunc tubes for 16 h at 37°C. C5a-containing fragments were specifically enriched by immunoprecipitation using mAb anti-C5a 137-26 (23) coupled to Dynabeads M-270 Epoxy (Thermo Fisher) according to the manufacturer’s instructions. The rationale for the immunoprecipitation was to concentrate the samples and increase the signal-to-noise ratio by the Western blot detection by loading more of each sample in each well of the SDS-PAGE. Proteins were eluted using Laemmli sample buffer (Bio-Rad) with a reducing agent (2-ME, Bio-Rad), separated on 4–15% gradient SDS-PAGE and detected by Western blot as described above for purified C5. In separate experiments, GPRP-plasma incubated with blocking Abs targeting C2 (mAb 172-62) and factor D (mAb 166-32), thereby inhibiting classical/lectin and alternative pathway activation prior to C3 convertase formation (24), was acidified to pH 6.4 and pH 6.8 by addition of 5% (v/v) 0.5 and 0.33 M hydrochloric acid, respectively. Plasma was thereafter incubated +/− thrombin (400 nM) and +/− lepirudin (7 μM) for 1 or 24 h. C5a-containing fragments were isolated by immunoprecipitation and detected by Western blot, as described above. In separate experiments, GPRP-plasma, +/− lepirudin (7 μM), was acidified to pH 6.4 and pH 6.8 by addition of 5% (v/v) 0.5 M and 0.33 M hydrochloric acid, respectively, or 0.50 M and 0.33 M lactic acid (Sigma-Aldrich), respectively. Half of the acidified plasma was within 1 min reneutralized with 0.25 M NaOH to pH 7.4. C5a-containing fragments were isolated with Dynabeads and detected with Western blot as described above.
Cleavage of C5 in clotting blood
Human whole blood was sampled in Vacutainer serum tubes (BD Biosciences). Five percent (v/v) hydrochloric acid (0.165 M, 0.330 M, or 0.500 M), lactic acid (0.165 M, 0.330 M, or 0.500 M), or NaCl (0.145 M) was immediately added to the whole blood after sampling. The blood was let to coagulate at 37°C for 60 min and thereafter centrifugated 20 min 3000 × g at 4°C to obtain serum. C5a-containing fragments were specifically enriched by immunoprecipitation and detected by Western blot as described above for the C5 in plasma and serum.
ELISA for detection of a C5a neoepitope in C5
Microtiter plates, 96-well high binding polystyrene plates (Costar, Cambridge, MA), were coated with the C5a neoepitope–specific mAb C17/5 (Thermo Fisher) by overnight incubation (25). C17/5 captures C5 by a neoepitope exposed in C5a and the C5a moiety of conformational changed, but not cleaved, C5 (“C5b-like”) (26). PBS with 0.1% BSA and 0.05% Tween 20 was used for blocking and sample dilution. The samples (purified Comptech C5, GPRP-plasma at pH 7.4, 6.8, and 6.4, C5-deficient serum pH 7.4 +/− purified Comptech C5 incubated for 15 min at 37°C) were diluted in PBS with 0.05% Tween and incubated for 60 min. C5 was detected by eculizumab (Alexion, Cheshire, CT), which had been biotinylated with EZ-Link Sulfo-NHS-Biotin (Thermo Fisher). Eculizumab binds to the C5b part of C5 (27) and can in combination with C17/5 be used to detect “C5b-like”. Streptavidin-HRP (GE Healthcare, Uppsala, Sweden) followed by tetramethylbenzidine substrate solution (R&D Systems, Abingdon, UK) was used for development.
Circular dichroism on purified C5
Purified C5 from Comptech and our affinity-purified C5 were prepared for circular dichroism by buffer exchanging into 20 mM sodium phosphate, 150 mM sodium fluoride, pH 7.2 or 20 mM sodium phosphate, 150 mM sodium fluoride, pH 6.4 using Zeba desalting columns according to the manufacturer’s instructions (Thermo Fisher). The concentration was determined from A280 and samples diluted to 0.5 μM. Far-ultraviolet circular dichroism spectra were recorded on a Chirascan CD spectrophotometer (Applied Photophysics, Leatherhead, UK) equipped with a Peltier controlled cuvette holder set at 20°C. Spectra were recorded in the range 185–250 nm, with a step size of 0.5 nm, a bandwidth of 1 nm, and a cuvette pathlength of 10 mm and in triplicate. Scans were averaged, buffer subtracted, and converted to mean residue ellipticity [θ] according to
where θ is the mean residue ellipticity (deg⋅cm2/dmol), mdeg is the raw circular dichroism signal, MRW is the mean residue weight (g/mol), l is the pathlength of the cuvette (cm), and C is the concentration (g/l).
For the temperature dependence of unfolding, a continuous temperature ramp from 20–90°C, at 2°C intervals, and a rate of 0.23°C/min was applied. The transition midpoint (Tm) temperatures of the spectra were analyzed using a multiwavelength fitting algorithm in Global 3 Analysis Software (Applied Photophysics, Leatherhead, UK).
Statistics
Statistical differences were analyzed using one-way ANOVA, two-tailed, followed by Tukey’s multiple comparison test. GraphPad Prism version 7.0b (GraphPad Software, San Diego, CA) was used for statistical analyses and data presentation.
Ethics statement
The study was designed and performed according to the ethical guidelines from the declaration of Helsinki. Informed written consent was obtained from the blood donors. The study was approved by the ethical committee of the Norwegian Regional Health Authority, ethical permit REK#S-04114, 2010/934.
Results
GPRP-anticoagulated human whole blood by inhibiting fibrin formation
For evaluating GPRP as a competitive fibrin polymerization inhibitor, GPRP was added in increasing concentrations to a pool of citrated plasma, and the concentration of fibrin monomers accessible for fibrin polymerization was quantified (Fig. 1A). Fibrin formation decreased dose-dependently in the presence of GPRP and reached background at 4 mg/ml. The anticoagulant activity of GPRP was further evaluated in whole blood from six individual donors by incubating for up to 8 h in ROTEM thromboelastometry (Fig. 1B). Time until clotting was GPRP dose-dependent; whole blood from all donors treated with 2 mg/ml clotted within the first hour, blood from three out of the six donors coagulated after 4 h with 4 mg/ml, whereas 8 mg/ml prevented clotting in all donors for 8 h. Thus, 8 mg/ml was chosen for all subsequent whole blood experiments.
FIGURE 1.

Effect of GPRP on coagulation in plasma and whole blood. (A) The functional fibrinogen concentration was analyzed in pooled citrate plasma from six donors in three independent experiments (n = 3) on a STA-R Evolution. Plasma was prepared from citrated blood and treated with GPRP (1–8 mg/ml). Calcium was added at the start of the analysis. Data are shown as the concentration (mean ± SD) of fibrin monomers accessible for fibrin formation. (B) Coagulation of whole blood ex vivo from single donors (n = 6) with GPRP in doses of 2, 4, and 8 mg/ml (blue, red, and black lines) and PBS buffer control (purple line) was analyzed for up to 8 h by ROTEM thromboelastometry (NATEM).
Blood physiological and hematological parameters were analyzed in whole blood incubated with GPRP for up to 4 h (Supplemental Tables I and II). All physiological parameters were stable for the first 30 min (Supplemental Table I). pH and pCO2 remained stable throughout the 4-h incubation, HCO3- and base excess decreased gradually, and pO2 increased. The platelet counts rapidly dropped initially but recovered throughout the incubation time (Supplemental Table II).
Thrombin was generated in GPRP-anticoagulated whole blood
Thrombin generation and platelet activation in whole blood anticoagulated with either GPRP, lepirudin, or PBS as a control, were compared by incubation for up to 20 min (Fig. 2). Thrombin was spontaneously generated in GPRP whole blood. Prothrombin was consumed entirely in GPRP-treated whole blood after 15 min, but not in lepirudin-treated whole blood (Fig. 2A). The level of TAT complexes was close to the baseline in GPRP whole blood in the first 5 min postsampling but increased thereafter rapidly to reach a plateau after a 15 min incubation (Fig. 2B). In lepirudin-anticoagulated blood, TAT was at baseline levels for the whole incubation period (Fig. 2B). Whole blood treated with PBS clotted within 10 min in all three donors. The release of platelet βTG followed the same pattern and kinetics as that of TAT (Fig. 2C), consistent with thrombin-dependent activation of protease-activated receptors on platelets (28).
FIGURE 2.

Thrombin activation of GPRP- and lepirudin-anticoagulated blood. Whole blood from three individual donors (n = 3), anticoagulated with GPRP (8 mg/ml), lepirudin (50 μg/ml), or without anticoagulant (PBS) was incubated separately at 37°C for up to 20 min in polypropylene tubes. (A) Consumption of prothrombin, indicated in a representative Western blot, under nonreduced conditions, of prothrombin at 72 kDa in GPRP and lepirudin whole blood. Samples at time zero and 15 min were analyzed. Intact prothrombin and the intermediate prothrombin-1 is indicated on the blot. The unlabeled band, equally stained in all lanes, is not identified. Note, the blot is split between lane 2 and 3 for the sake of presentation. (B) Thrombin generation was evaluated by quantification of thrombin anti-thrombin (TAT) complex formation by ELISA, and (C) platelet activation was assessed by quantification of β-thromboglobulin (βTG). GPRP-blood is shown in red circles, lepirudin in blue circles, and PBS in gray squares (B and C). Data are presented as mean ± SD (A). M, m.w. markers.
Complement activation was similar in GPRP- and lepirudin-anticoagulated whole blood
Complement activation at the level C3 (C3bc), C5 (C5a), and terminal complement complex (sC5b-9) was compared in GPRP- and lepirudin-anticoagulated whole blood, in the presence or absence of E. coli (Fig. 3, left panels) and S. aureus (Fig. 3, right panels). Both bacteria caused dose-dependent complement activation with elevated levels of C3bc (Fig. 3A, 3B), C5a (Fig. 3C, 3D) and sC5b-9 (Fig. 3E, 3F). Complement activation was virtually identical in GPRP and lepirudin whole blood for the analyzed complement activation markers. Thus, complement activation at the level of C3 or C5, including terminal pathway activation, was not affected by the anticoagulants. As a note, the assay detecting the TCC is not specific for the convertase-dependent formation of sC5b-9 but also detects TCC generated from nonconvertase-dependent activation of C5 (Supplemental Fig. 1).
FIGURE 3.
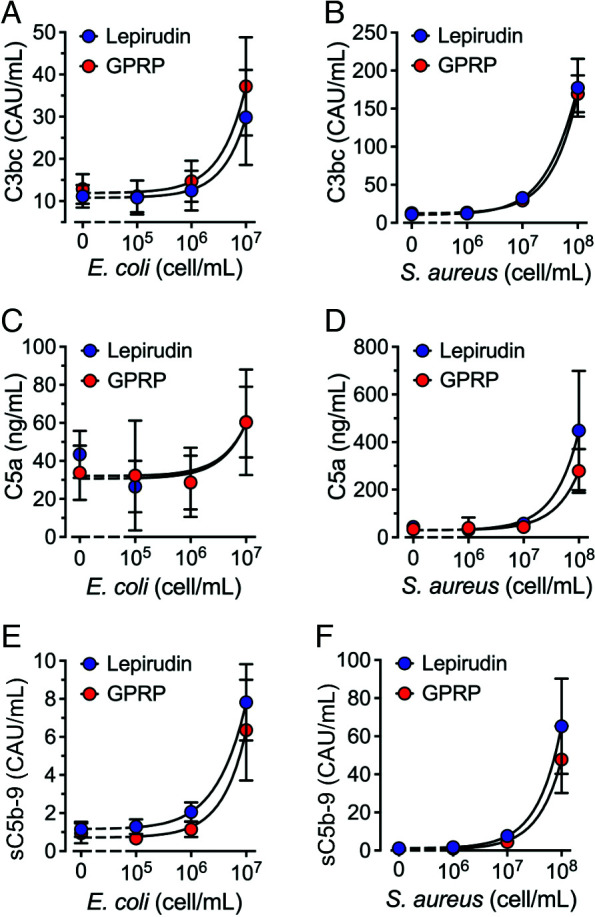
Complement activation in GPRP- and lepirudin-anticoagulated blood. Whole blood was collected in the presence of GPRP (8 mg/ml, red circles) or lepirudin (50 μg/ml, blue circles). Bacteria Escherichia coli (A, C, and E) and Staphylococcus aureus (B, D, and F) were incubated in increasing doses. Following 30 min of incubation at 37°C, complement activation was analyzed by ELISA quantification of C3bc (A and B), C5a (C and D), and sC5b-9 (E and F). Data from six individual blood donors (n = 6) are presented as the mean with a 95% confidence interval.
Thrombin-cleaved purified C5 in buffer and plasma
Because the thrombotic response in whole blood did not influence the degree of complement activation, we aimed to reinvestigate the impact of thrombin on C5 cleavage. Purified thrombin (100 nM) was when incubated with purified C5 (500 nM) in PBS pH 7.4 able to cleave the C5 α-chain in purified C5 from the commercial sources Comptech and Quidel (Fig. 4A, left and middle lanes). However, affinity-purified C5, eluted using mild conditions with 2 M MgCl2 and immediately gel filtered into PBS, was inert to thrombin (Fig. 4A, right lanes). To further evaluate thrombin-mediated C5 degradation, C5 (500 nM) from Comptech was incubated with thrombin (100 nM) for up to 20 h (Fig. 4B). Again, we found that an initial cleavage of the α-chain of C5 occurred rapidly, generating a 35-kDa fragment. This fragment was subsequently split into a 24-kDa fragment and the 11-kDa N-terminal C5a, but this occurred at a much slower rate (Fig. 4B). The initial cleavage was complete already after 7.5 min, whereas the second cleavage was not complete even after 20 h of incubation. This degradation pattern of C5 by thrombin was previously reported by Krisinger et al. and we here refer to the sizes of the 35- (α′), 24- (α′′), and 11-kDa (C5a) C5 fragments reported by them (15). The C5 degradation pattern was also confirmed by incubating C5 with thrombin for 20 h. The 35-kDa fragment and C5a were detected by Western blot using a polyclonal antibody binding the C5a domain of the C5 α-chain and C5a (Fig. 4C). In contrast to the buffer system with purified C5, no cleavage of plasma C5 was seen in GPRP-plasma (Fig. 4D), despite the substantial endogenous thrombin generation in the GPRP model (Fig. 2A, 2B).
FIGURE 4.

Cleavage of purified C5 by thrombin, as detected by protein staining and Western blot. (A) SDS-PAGE on purified C5 (500 nM) from Comptech, Quidel, and our MgCl2 affinity-purified C5 eluted under mild conditions using a 2 M MgCl2 solution, incubated with thrombin (100 nM) for 0, 15, and 60 min at 37°C before the addition of lepirudin (1 μM). At 0 min, lepirudin was added before thrombin. Intact and cleaved α-chain is indicated by α and α′ respectively, β-chain is indicated with β. The gel was stained with Coomassie brilliant blue. (B) Purified C5 (500 nM) from Comptech was incubated with thrombin (100 nM) for different time intervals (7.5 min to 20 h), as indicated on top of a representative SDS-PAGE stained with Coomassie brilliant blue. Reference proteins are shown in separate lanes; thrombin (Thr), lepirudin (Lep), and C5 (C5), and an m.w. reference (M). C5 α-chain (α) and β-chain (β) are indicated in the label on the right-hand side of the figure, so are the primary (α′) and secondary (α′′ cleavage fragments, together with thrombin and C5a. (C) Purified Comptech C5 (500 nM) was incubated for 1 h at 37°C with PBS, thrombin (100 nM), thrombin (100 nM) in the presence of lepirudin (1 μM), or cobra venom factor (CVF). C5a-containing fragments were detected by Western blot using a polyclonal antibody against C5a. (D) GPRP-plasma was incubated with thrombin (400 nM) alone or in the presence of lepirudin (7 μM), purified Comptech C5 (60 μg/ml) alone or in the presence of thrombin, or the combination of purified C5 (60 μg/ml), thrombin (400 nM), and lepirudin (7 μM), for 16 h at 37°C. C5a-containing fragments were immunoprecipitated using a monoclonal antibody against C5a (clone 137-26) and detected by Western blot as described for (B). (E) Identical conditions as described for (C), but incubations were performed in serum from a C5-deficient individual instead of in GPRP-plasma. All samples were run under reducing conditions (A–E). M, m.w. markers with size indicated in kDa.
Furthermore, exogenous addition of thrombin to supra-physiological concentrations (400 nM) did not lead to any cleavage of native C5 (Fig. 4D). However, when plasma was incubated with purified C5 (Comptech) in an equimolar concentration as native plasma C5, cleavage of the C5 α-chain occurred at the primary site, generating the 35-kDa fragment (Fig. 4D). Cleavage occurred to a similar degree both with and without the extra addition of thrombin, indicating that endogenously generated thrombin in GPRP-plasma was sufficient to mediate the cleavage of purified C5 in the amount corresponding to normal C5 plasma levels. As in the purified protein system, this C5 cleavage was inhibited by lepirudin (Fig. 4D). Performing the analogous experiment in serum from a C5-deficient patient confirmed the observation that purified C5 could be cleaved (Fig. 4E). However, because of the depletion of thrombin in serum in contrast to plasma, exogenous addition of thrombin was necessary for the cleavage of C5.
Thrombin-cleaved C5 in hydrochloric acid–acidified plasma
The discrepancy in susceptibility to thrombin-mediated cleavage between C5 from the commercial sources (Comptech and Quidel) and the native C5 in plasma and the C5 affinity-purified and eluted under mild conditions with an MgCl2 solution led us to hypothesize that C5 may be altered under the large-scale C5 purification. Therefore, we challenged the physiological conditions in GPRP-plasma by acidification by adding 5% (v/v) 0.33–0.50 M hydrochloric acid. Using a conformationally selective sandwich ELISA combining Abs binding a neoepitope in C5a (mAb C17/5) and another mAb (eculizumab) binding C5b, respectively, C5 in acidified GPRP-plasma was found to expose the neoepitope in the C5a domain at pH 6.8 and pH 6.4, not present in native C5 at pH 7.4 (Fig. 5A). The differences in neoepitope detection in between pH 6.4 and 6.8 and comparison with pH 7.4 were all statistically significant (p < 0.001). Neoepitope expression was also detected in purified Comptech C5 added to C5-deficient serum.
FIGURE 5.

C5a Ag exposure and C5 cleavage in hydrochloric acid–acidified GPRP-plasma and serum. (A) Conformationally selective ELISA for detecting a C5a neoepitope of C5 in GPRP-plasma and C5-deficient serum including reconstitution with purified C5 (60 μg/ml) from Comptech. The plasma pH was adjusted with hydrochloric acid to 6.4, 6.8, or kept at 7.4, and incubated for 15 min at 37°C. Exposure of the C5a neoepitope in C5 was detected by ELISA, combining capturing and detecting Abs specific for the C5a neoepitope and C5b, respectively. Data are shown as the optical density mean values ± SD (n = 4), *p < 0.001. (B) GPRP-plasma was pH-adjusted to 6.4 and 6.8 with hydrochloric acid or kept at 7.4, and incubated with 0.9% NaCl, lepirudin (Lep) and/or thrombin (Thr, 400 nM) for 1 h at 37°C. C5a-containing fragments were specifically enriched by immunoprecipitation using a mAb against C5a (clone 137-26) and detected by Western blot, under reduced conditions, using a polyclonal Ab against C5a. Intact and cleaved C5 α-chain are indicated by α and α′, respectively, on the representative Western blot from two replicates. M, m.w. markers with size indicated in kDa.
The antigenic similarity between the commercially purified C5 and the C5 modified by GPRP-plasma-acidification led us to hypothesize that C5 in acidified plasma could have become sensitive for thrombin-mediated cleavage. Therefore, incubations of GPRP-plasma were carried out for 1 and 24 h at physiologic pH 7.4 and acidic conditions at pH 6.8 and 6.4 (Fig. 5B). C5 cleavage occurred under the acidic but not under physiologic conditions. There was no difference between the 1-h and the 24-h incubations. In contrast to the cleavage of the purified C5 from the commercial sources, no C5a band was observed, even at incubations for up to 24 h.
C5 was weakly susceptible to thrombin-mediated cleavage in lactic acid–acidified plasma
To evaluate whether C5 cleavage in plasma was a general phenomenon to acidification or unique to hydrochloric acid, plasma was acidified with the more physiologically relevant lactic acid. Lactic acid or hydrochloric acid were added to GPRP-plasma until the pH reached 6.8 (Fig. 6). In separate vials, the pH of lactic acid– and hydrochloric acid–acidified plasma was neutralized to pH 7.4 with NaOH. The C5a neoepitope Ag was similarly expressed in both hydrochloric acid– and lactic acid–acidified plasma (Fig. 6A, 6B), and the expression was not reversed by neutralization to pH 7.4 with NaOH. The neoepitope Ag was expressed in Comptech C5 (Fig. 6A, 6B) also at neutral pH. Cleavage of C5 was evaluated in GPRP-plasma acidified with hydrochloric acid and lactic acid to pH 6.8 and in the corresponding reneutralized samples (Fig. 6C). Cleavage occurred in hydrochloric acid–acidified plasma, irrespective of whether the plasma had been neutralized or not (Fig. 6C). In the corresponding lactic acid–acidified sample, the 35-kDa band was not detected in two out of three blots. In the third blot, the band was weakly detectable. The presence of lepirudin blocked all cleavages.
FIGURE 6.

C5a Ag exposure and C5 cleavage in hydrochloric acid– and lactic acid–acidified GPRP-plasma. (A and B) Conformationally selective ELISA for the detection of C5a neoepitope of C5 in normal GPRP-plasma (pH 7.4) and plasma acidified to pH 6.8 with hydrochloric acid (HCl) (A) or lactic acid (B), with or without neutralization to pH 7.4 with NaOH 1 min after acidification. Exposure of the C5a neoepitope in C5 was detected by ELISA, combining capturing and detecting Abs specific for the C5a neoepitope and C5b, respectively. Purified C5 (Comptech) at 60 μg/ml was included as a control. Data are shown as the optical density mean values ± SD (n = 3), *p < 0.001. (C) GPRP-plasma with and without lepirudin was pH-adjusted to 6.8 with hydrochloric acid (HCl) or lactic acid, with or without neutralization to pH 7.4 with NaOH 1 min after acidification. All samples, including GPRP-plasma, were kept at 7.4, and purified C5 with thrombin (400 nM) +/− lepirudin were incubated for 1 h at 37°C. C5a-containing fragments were specifically enriched by immunoprecipitation using a mAb against C5a (clone 137-26) and detected by Western blot, under reduced conditions, using a polyclonal Ab against C5a. Samples, as indicated on top of the membrane, are GPRP-plasma pH 7.4 (lanes 1 and 2), GPRP-plasma acidified to pH 6.8 with either hydrochloric acid (HCl) (lanes 3–6) or with lactic acid (lanes 7–10). Samples in lanes 4, 6, 8, and 10 are neutralized to pH 7.4 with NaOH after acidification. It is indicated which samples contain lepirudin. Intact and cleaved C5 α-chain are indicated by α and α′, respectively, on the representative Western blot from three replicates. M, m.w. markers with size indicated in kDa.
C5 was cleaved in acidified clotting blood
To further understand the prerequisites for C5 cleavage, human whole blood was collected without anticoagulant and let to clot in glass tubes under neutral and acidic conditions. After 60 min at 37°C, a stable clot was formed in all tubes. C5 α-chain cleavage and formation of the 35-kDa fragment occurred in all tubes containing either hydrochloric or lactic acid (Fig. 7A) but, consistent with the GPRP-treated plasma, no cleavage was seen under neutral conditions.
FIGURE 7.

Cleavage of C5 in clotting blood and thrombin-mediated cleavage of C5b in the C5b6 complex. (A) Human whole blood was collected in additive-free glass serum tubes. The blood was immediately acidified with 5% (v/v) lactic acid (0.165, 0.330, 0.500 M) or hydrochloric acid (HCl) (0.165, 0.330, 0.500 M) or added physiologic NaCl for volume control. The blood was let to clot for 60 min at 37°C and then centrifuged to serum. C5a-containing fragments were specifically enriched by immunoprecipitation using a mAb against C5a (clone 137-26) and detected by Western blot, under reduced conditions, using a polyclonal Ab against C5a. Intact and cleaved C5 α-chain are indicated by α and α′, respectively, on the representative Western blot from two replicates. (B) SDS-PAGE on purified C5b6 (50 μg/ml) from Comptech incubated with and without thrombin (400 nM) in PBS at pH 7.4, 6.8, and 6.4 for 60 min at 37°C. The samples were run on an SDS-PAGE under reduced conditions and stained with SYPRO Ruby Protein Gel Stain. Intact and cleaved α-chain is indicated by α and α′, respectively, β-chain is indicated with β, and C6 with C6. M, m.w. markers with size indicated in kDa.
Thrombin-cleaved C5b in the C5b6 complex
Cleavage of C5b in a C5b6 complex was investigated by incubating purified C5b6 with and without thrombin at neutral and acidic pH. Cleavage of the α-chain occurred both at neutral (pH 7.4) and acidic (pH 6.8 and pH 6.4) pH in the tubes containing thrombin (Fig. 7B). The size of the smaller fragment, derived from cleavage of the α-chain, corresponded to the 35-kDa a′ fragment without the 9-kDa C5a.
C5 undergoes a conformational change upon acidification
Lastly, to evaluate conformational or structural changes in C5, circular dichroism was performed on purified C5 at neutral and acidic pH. At pH 7.2, the initial far-ultraviolet circular dichroism spectra were not identical, suggesting differences in secondary structure content and/or conformation between our affinity-purified and the commercial Comptech C5 (Fig. 8A). This was most apparent in the C5 exchanged into pH 6.4, where a significant amount of protein was lost, consistent with the reduced intensity of the circular dichroism signal. To further understand these differences, we monitored the spectra as a function of temperature (Fig. 8B). Each curve showed a change in secondary structure with temperature as the protein unfolded, with all samples retaining residual structure at 90°C, although all melting profiles were distinct. To probe these structural differences further, we applied a global fitting procedure, monitoring the change in circular dichroism signal at each wavelength as a function of temperature (Fig. 8C). The least number of transitions that accurately described the data are shown in (Fig. 8D. Interestingly, we saw increased thermal stability of our affinity-purified C5 (melting temperature or Tm of 67.2°C) over the commercial source Comptech C5 (Tm of 61.5°C). The acidification of C5 to pH 6.4 was markedly destabilizing and resulted in two unfolding events, one at 40.3°C and a second at 62.4°C. These transitions can be explained by an equilibrium of partially unfolded C5 (Tm1) and folded C5 (Tm2). Importantly, this second unfolding is similar to that observed with Comptech C5, potentially indicating that this transition can be induced by acidification. Overall, these data suggest structural differences between the samples and that a loss of stability can be induced by lowering pH.
FIGURE 8.

Circular dichroism spectra of C5 at pH 7.2 and pH 6.4. (A) Far-ultraviolet circular dichroism spectra of our affinity-purified C5 at pH 7.2 (black) and pH 6.4 (red), and Comptech C5 pH 7.2 (blue). Highlighted at 217 nm are the minima associated with β-sheet content. (B) Circular dichroism spectra as a function of temperature. (C) Unfolding curves and average fit from all wavelengths. (D) Transition midpoints derived from the fitted data.
Discussion
The four amino acid peptide GPRP has a similar sequence to knob A in thrombin-cleaved fibrin monomers. Therefore, it can compete with this domain to prevent the knob-to-hole interaction between fibrin monomers and prevent fibrin polymerization. We used this peptide as an anticoagulant in human whole blood and characterized this model to investigate complement activation in the presence of active thrombin. This is a significant improvement of the lepirudin-based whole blood model (18), which is regarded as the most physiologically relevant model for whole blood incubations until now. However, its main limitation is the irreversible inhibition of thrombin.
Thrombin was instantly generated in whole blood anticoagulated with GPRP, and despite the thrombin response, anticoagulation was achieved for the entire 8-h incubation. The basal, cellular, and physiologic parameters were similar to what has been shown for the lepirudin-based whole blood model (18) except for an initial decrease of platelet number, which later normalized partly. This phenomenon might arise through early platelet activation, with elevated plasma βTG and aggregation, with subsequent dissolvent of unstable platelet aggregates, in the absence of a stable fibrin clot.
Thrombin-dependent complement activation has been regarded as a key axis for cross-talk between complement and coagulation (14). However, to our surprise, complement activation, as detected by the activation of C3 and C5, was similar in GPRP- and lepirudin-anticoagulated blood, both in the presence and absence of bacteria. This implies that activation, at the level of C3 and terminal pathway activation, occurred independently of the substantial thrombin generation in GPRP whole blood, which questions thrombin as a contributor to complement activation in human whole blood under the present conditions.
The ability of thrombin to cleave C5 has been thoroughly studied in buffer milieu by others (10, 15, 16) and was here re-evaluated by including C5 from different preparations. We also extended this by including C5 cleavage in the plasma milieu by employing the GPRP model. We found that thrombin could cleave purified C5 from the two commercial sources, thus confirming the previous results observed in purified systems (15). Commercial purified C5 was cleaved both in buffer milieu and when added to serum and plasma. Further on, we found that plasma C5 could be cleaved if plasma was acidified with hydrochloric and lactic acid when both samples were adjusted to pH ≤ 6.8. Neutralization of plasma back to pH 7.4 did not affect the cleavage pattern if plasma once had been acidified to pH 6.8.
Native C5 in plasma was resistant to thrombin, as was C5 purified by a mild MgCl2-based affinity chromatography protocol using an affinity-attenuated, double mutant form of OmCI (22). This observation was intriguing because both of the commercial C5 preparations were readily cleaved by thrombin. Unfortunately, none of Comptech or Quidel protocols for the purification of C5 were available to us. Still, Comptech referred to the procedure by Tack et al. from 1976 (29) with a minor modification described in Rawal et al. (30). This procedure is a traditional protocol including initial polyethylene glycol precipitation of plasma followed by solubilization and repeated chromatography columns (31–33), sometimes including pH shifts (34). These protocols exert stress on purified C5 protein and, as shown here, make C5 sensitive to thrombin. However, affinity chromatography with the E141A, H164A OmCI mutant, followed by gentle elution with 2 M MgCl2 solution and immediate gel filtration into PBS, kept C5 resistant to thrombin. Other recent protocols have employed affinity chromatography to purify C5, and protein elution has been carried out at mild acidic conditions (pH 5.5) (35). We could, using circular dichroism, confirm that C5 purified using our one-step affinity purification protocol was structurally or conformationally different from Comptech C5. At neutral pH, differences in the far-ultraviolet spectra were apparent for the two samples, alongside a corresponding decrease in thermal stability for the commercial material. Importantly, acidification of the affinity-purified material resulted in a loss of thermal stability with a new transition occurring at a similar temperature as measured for the Comptech material, suggesting that instability may be induced by mild changes in pH. In light of our data, it is desirable to retain C5 at neutral pH at every purification step.
Thrombin-mediated C5 cleavage was, as previously shown by Krisinger et al. (15), efficient in the α-chain at the initial R947 position but inefficient at the second R751 position. Krisinger et al. elegantly supported this by showing that the amino acid sequence surrounding R947 aligned with the consensus sequence for thrombin cleavage, Pro (P2) - Arg (P1) - Ser/Ala/Gly/Thr (P1′) - not acidic amino acid (P2′) (36), in addition, middle-sized hydrophobic residue at P4 and P4′, a sequence in C5 which was evolutionary conserved among vertebrates (15). The sequence surrounding R751 did not align with the consensus sequence for thrombin cleavage (15), and is not present the homologous C3 protein (37). Cleavage at R751 releases the C5a anaphylatoxin, whereas C5 after cleavage at only R947 remains as one unit because of internal disulfide bonds in the α-chain (16). Cleavage at R751 was only observed in purified C5 in buffer and not in C5 in the acidic plasma. The discrimination in C5 cleavage by thrombin in buffer and plasma could possibly be explained by inhibition of thrombin by plasma anti-thrombin or saturation by its natural substrate, fibrinogen. However, this could not explain why the cleavage of purified C5 in normal plasma and plasma C5 under acidic conditions occurred and why affinity-purified C5, eluted with MgCl2 under mild conditions, remained intact in the presence of thrombin.
Instead, we propose that a conformational change in the C5 molecule could explain the cleavage upon purification and acidification of plasma and whole blood. This conformational change would make the cleavage site at R947 accessible for thrombin. Indeed, we observed that commercially purified C5 and C5-acidified plasma exposed a C5a neoepitope, indicating a C5 conformational change. Because the neoepitope was also expressed in the lactic acid–acidified plasma, but this C5 molecule was very weakly cleaved by thrombin, there was no absolute correlation between C5a neoepitope exposure and susceptibility to thrombin-mediated cleavage (i.e., the conformational change induced by lactic acid in plasma was not enough to allow substantial thrombin-mediated cleavage) in contrast to the conformational change caused by hydrochloric acid. Incubation of human whole blood without GPRP, or other anticoagulants, induced clotting, which under acidic but not in neutral conditions was accompanied by C5 cleavage. In contrast to the experiment in GPRP-plasma, C5 cleavage was equally efficient in blood as blood acidified with lactic acid and hydrochloric acid. In accordance with Krisinger et al. (15), we found C5b in the purified C5b6 complex to be cleaved by thrombin. Cleavage occurred both in neutral and acidic pH, indicating that the cleavage site is accessible also in C5b (38).
Several studies have reported complement activation in acidosis in vitro (39–41) and in vivo (42). Acidic conditions have altered the activity of the alternative pathway convertase, promoting activation and limiting its regulation for convertase-mediated cleavage of C5 at R751 for the generation of C5b and C5a (43). It is well known that the C5 molecule can undergo antigenic and functional changes under nonphysiological conditions. We, and others, have shown that C5 in acidified serum is antigenically different from serum C5 at physiological pH (26, 44). In serum at pH 6.4 and 6.8, a neoepitope in C5a is also exposed in the parental C5 protein before the C5a is cleaved (26). By designing an ELISA combining Abs binding a neoepitope in C5a (mAb C17/5) and the C5b (eculizumab), respectively, we could discriminate between native C5 and an intact but antigenically changed C5 and showed that the neoepitope in the C5a moiety was expressed in purified C5 preparations, and in C5 in acidified plasma, still, if the plasma was immediately neutralized.
Using this novel ex vivo model, our data set the coagulation-complement cross-talk into a new context, limiting the impact of general coagulation-driven complement activation and focusing more on a critical C5 conformational change for thrombin-mediated cleavage. Our data are in line with a recent study by Keshari et al. where complement activation was studied in two coagulopathic models in baboons in vivo (17). Despite a massive burst in thrombin, any measurable levels of complement activation markers could not be detected. Whether the situation would have been different under in vivo acidosis remains to be determined. pH-dependent cross-talk between coagulation and complement was discussed by Kenawy et al. in 2015 (45). They highlight the simultaneous activation of the contact and complement systems under acidic pH and the potential of activated contact-specific and common coagulation proteases for cleaving complement components.
Our observations in this study exclude thrombin from cleaving C5 under physiological conditions ex vivo. However, we cannot exclude that pathophysiological conditions such as in ischemic tissue, in the tumor microenvironment, or an abscess, all associated with low pH and production of reactive oxygen species that are also are known to change C5 conformation (46, 47), can turn C5 into a thrombin substrate. Although we could not detect any C5a in acidified GPRP-plasma, and C5a was very slowly generated from C5 cleavage by thrombin in buffer milieu, we cannot exclude a “C5a effect” from the conformational changed C5. Thus, our findings may have pathophysiological and clinical consequences, implying terminal complement pathway activation under certain pathophysiological conditions, provided that such a condition first changes C5 conformation to expose the cleavage motif for thrombin. The same would apply to C5, as well as C3, in relation to other plasma proteases; cleavage can occur as long as the cleavage motif is exposed, which remains to be proven experimentally in a physiologic or pathophysiological relevant context. Factor IXa, FXa, FXIa, and plasmin have all been shown to cleavage C5, as well as C3 in purified systems (12). Apparently, the requirement for a specific cleavage motif is less strict in a purified system because thrombin, in the purified system, can cleave C5 at R751, and cleave C3.
In summary, we have characterized a novel human whole blood model using the four amino acid GPRP peptide as an anticoagulant to study thromboinflammation under close to physiological conditions. Our data show an immediate thrombin generation in GPRP-anticoagulated whole blood, but, despite this, no cleavage of C5 or enhanced complement activation was observed. However, the cleavage of C5 was induced by thrombin when C5 was changed by purification or acidification, supported by the structural changes shown by circular dichroism. We conclude that thrombin is not an activator of native C5 in plasma, but it cannot be excluded that cleavage may occur in vivo in pathophysiological settings. Our data also support that the lepirudin-based model is suitable and reliable for studies on complement activation, for which it has been used since it was developed (18). Combining data from studies in the lepirudin and the GPRP ex vivo models gives an even deeper mechanistic understanding of complement cross-talk in thromboinflammation.
Supplementary Material
This article is featured in Top Reads, p.1495
P.H.N. designed and performed experiments and wrote the paper; C.J., Q.H.Q., A.M., O.D., H.F., A.L., G.B., C.S., and L.M.H.-K. designed and performed experiments; S.E.P., J.v.d.E., and O.-L.B. designed and supervised the project; M.H.-L. provided critical discussions and edited the manuscript; T.E.M. designed and supervised the project and wrote the paper. All authors approved the final version of the manuscript.
This work was supported by the Norwegian Research Council (Project 274332), the Swedish Research Council (Project 2018-04087), the Norwegian Council on Cardiovascular Disease, the Crafoord Foundation, the Odd Fellows Foundation, and the Simon Fougner Hartmann Family Fund.
The online version of this article contains supplemental material.
- βTG
- β-thromboglobulin
- CVF
- cobra venom factor
- GPRP
- Gly-Pro-Arg-Pro
- TAT
- thrombin anti-thrombin complex
- TCC
- terminal C5b-9 complement complex
Disclosures
T.E.M. is an advisory board member of Ra Pharma, now part of UCB. The other authors have no financial conflicts of interest.
References
- 1.Ricklin D., Hajishengallis G., Yang K., Lambris J. D.. 2010. Complement: a key system for immune surveillance and homeostasis. Nat. Immunol. 11: 785–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hajishengallis G., Reis E. S., Mastellos D. C., Ricklin D., Lambris J. D.. 2017. Novel mechanisms and functions of complement. Nat. Immunol. 18: 1288–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tanaka K. A., Key N. S., Levy J. H.. 2009. Blood coagulation: hemostasis and thrombin regulation. Anesth. Analg. 108: 1433–1446. [DOI] [PubMed] [Google Scholar]
- 4.Ekdahl K. N., Teramura Y., Hamad O. A., Asif S., Duehrkop C., Fromell K., Gustafson E., Hong J., Kozarcanin H., Magnusson P. U., et al. 2016. Dangerous liaisons: complement, coagulation, and kallikrein/kinin cross-talk act as a linchpin in the events leading to thromboinflammation. Immunol. Rev. 274: 245–269. [DOI] [PubMed] [Google Scholar]
- 5.Berends E. T., Gorham R. D. Jr., Ruyken M., Soppe J. A., Orhan H., Aerts P. C., de Haas C. J., Gros P., Rooijakkers S. H.. 2015. Molecular insights into the surface-specific arrangement of complement C5 convertase enzymes. BMC Biol. 13: 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hantgan R., Fowler W., Erickson H., Hermans J.. 1980. Fibrin assembly: a comparison of electron microscopic and light scattering results. Thromb. Haemost. 44: 119–124. [PubMed] [Google Scholar]
- 7.Markiewski M. M., Nilsson B., Ekdahl K. N., Mollnes T. E., Lambris J. D.. 2007. Complement and coagulation: strangers or partners in crime? Trends Immunol. 28: 184–192. [DOI] [PubMed] [Google Scholar]
- 8.Lupu F., Keshari R. S., Lambris J. D., Coggeshall K. M.. 2014. Crosstalk between the coagulation and complement systems in sepsis. Thromb. Res. 133(Suppl 1): S28–S31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hamad O. A., Bäck J., Nilsson P. H., Nilsson B., Ekdahl K. N.. 2012. Platelets, complement, and contact activation: partners in inflammation and thrombosis. In Current Topics in Innate Immunity II. Lambris J., Hajishengallis G., eds. Springer, New York, p. 185–205. [DOI] [PubMed] [Google Scholar]
- 10.Amara U., Rittirsch D., Flierl M., Bruckner U., Klos A., Gebhard F., Lambris J. D., Huber-Lang M.. 2008. Interaction between the coagulation and complement system. Adv. Exp. Med. Biol. 632: 71–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Foley J. H., Walton B. L., Aleman M. M., O’Byrne A. M., Lei V., Harrasser M., Foley K. A., Wolberg A. S., Conway E. M.. 2016. Complement Activation in Arterial and Venous Thrombosis is Mediated by Plasmin. EBioMedicine 5: 175–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Amara U., Flierl M. A., Rittirsch D., Klos A., Chen H., Acker B., Brückner U. B., Nilsson B., Gebhard F., Lambris J. D., Huber-Lang M.. 2010. Molecular intercommunication between the complement and coagulation systems. J. Immunol. 185: 5628–5636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wiggins R. C., Giclas P. C., Henson P. M.. 1981. Chemotactic activity generated from the fifth component of complement by plasma kallikrein of the rabbit. J. Exp. Med. 153: 1391–1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huber-Lang M., Sarma J. V., Zetoune F. S., Rittirsch D., Neff T. A., McGuire S. R., Lambris J. D., Warner R. L., Flierl M. A., Hoesel L. M., et al. 2006. Generation of C5a in the absence of C3: a new complement activation pathway. Nat. Med. 12: 682–687. [DOI] [PubMed] [Google Scholar]
- 15.Krisinger M. J., Goebeler V., Lu Z., Meixner S. C., Myles T., Pryzdial E. L., Conway E. M.. 2012. Thrombin generates previously unidentified C5 products that support the terminal complement activation pathway. Blood 120: 1717–1725. [DOI] [PubMed] [Google Scholar]
- 16.Wetsel R. A., Kolb W. P.. 1983. Expression of C5a-like biological activities by the fifth component of human complement (C5) upon limited digestion with noncomplement enzymes without release of polypeptide fragments. J. Exp. Med. 157: 2029–2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Keshari R. S., Silasi R., Lupu C., Taylor F. B. Jr., Lupu F.. 2017. In vivo-generated thrombin and plasmin do not activate the complement system in baboons. Blood 130: 2678–2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mollnes T. E., Brekke O. L., Fung M., Fure H., Christiansen D., Bergseth G., Videm V., Lappegård K. T., Köhl J., Lambris J. D.. 2002. Essential role of the C5a receptor in E coli-induced oxidative burst and phagocytosis revealed by a novel lepirudin-based human whole blood model of inflammation. Blood 100: 1869–1877. [PubMed] [Google Scholar]
- 19.Laudano A. P., Doolittle R. F.. 1980. Studies on synthetic peptides that bind to fibrinogen and prevent fibrin polymerization. Structural requirements, number of binding sites, and species differences. Biochemistry 19: 1013–1019. [DOI] [PubMed] [Google Scholar]
- 20.Chernysh I. N., Nagaswami C., Purohit P. K., Weisel J. W.. 2012. Fibrin clots are equilibrium polymers that can be remodeled without proteolytic digestion. Sci. Rep. 2: 879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bergseth G., Ludviksen J. K., Kirschfink M., Giclas P. C., Nilsson B., Mollnes T. E.. 2013. An international serum standard for application in assays to detect human complement activation products. Mol. Immunol. 56: 232–239. [DOI] [PubMed] [Google Scholar]
- 22.Macpherson A., Liu X., Dedi N., Kennedy J., Carrington B., Durrant O., Heywood S., van den Elsen J., Lawson A. D. G.. 2018. The rational design of affinity-attenuated OmCI for the purification of complement C5. J. Biol. Chem. 293: 14112–14121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fung M., Lu M., Fure H., Sun W., Sun C., Shi N. Y., Dou Y., Su J., Swanson X., Mollnes T. E.. 2003. Pre-neutralization of C5a-mediated effects by the monoclonal antibody 137-26 reacting with the C5a moiety of native C5 without preventing C5 cleavage. Clin. Exp. Immunol. 133: 160–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Harboe M., Ulvund G., Vien L., Fung M., Mollnes T. E.. 2004. The quantitative role of alternative pathway amplification in classical pathway induced terminal complement activation. Clin. Exp. Immunol. 138: 439–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Oppermann M., Schulze M., Götze O.. 1991. A sensitive enzyme immunoassay for the quantitation of human C5a/C5a(desArg) anaphylatoxin using a monoclonal antibody with specificity for a neoepitope. Complement Inflamm. 8: 13–24. [DOI] [PubMed] [Google Scholar]
- 26.Nilsson P. H., Thomas A. M., Bergseth G., Gustavsen A., Volokhina E. B., van den Heuvel L. P., Barratt-Due A., Mollnes T. E.. 2017. Eculizumab-C5 complexes express a C5a neoepitope in vivo: Consequences for interpretation of patient complement analyses. Mol. Immunol. 89: 111–114. [DOI] [PubMed] [Google Scholar]
- 27.Jore M. M., Johnson S., Sheppard D., Barber N. M., Li Y. I., Nunn M. A., Elmlund H., Lea S. M.. 2016. Structural basis for therapeutic inhibition of complement C5. Nat. Struct. Mol. Biol. 23: 378–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Covic L., Gresser A. L., Kuliopulos A.. 2000. Biphasic kinetics of activation and signaling for PAR1 and PAR4 thrombin receptors in platelets. Biochemistry 39: 5458–5467. [DOI] [PubMed] [Google Scholar]
- 29.Tack B. D., Prahl J. W.. 1976. Third component of human complement: purification from plasma and physicochemical characterization. Biochemistry 15: 4513–4521. [DOI] [PubMed] [Google Scholar]
- 30.Rawal N., Pangburn M. K.. 1998. C5 convertase of the alternative pathway of complement. Kinetic analysis of the free and surface-bound forms of the enzyme. J. Biol. Chem. 273: 16828–16835. [DOI] [PubMed] [Google Scholar]
- 31.Sottrup-Jensen L., Andersen G. R.. 2014. Purification of human complement protein C5. Methods Mol. Biol. 1100: 93–102. [DOI] [PubMed] [Google Scholar]
- 32.Hammer C. H., Wirtz G. H., Renfer L., Gresham H. D., Tack B. F.. 1981. Large scale isolation of functionally active components of the human complement system. J. Biol. Chem. 256: 3995–4006. [PubMed] [Google Scholar]
- 33.Tack B. F., Morris S. C., Prahl J. W.. 1979. Fifth component of human complement: purification from plasma and polypeptide chain structure. Biochemistry 18: 1490–1497. [DOI] [PubMed] [Google Scholar]
- 34.DiScipio R. G., Sweeney S. P.. 1994. The fractionation of human plasma proteins. II. The purification of human complement proteins C3, C3u, and C5 by application of affinity chromatography. Protein Expr. Purif. 5: 170–177. [DOI] [PubMed] [Google Scholar]
- 35.Zelek W. M., Stott M., Walters D., Harris C. L., Morgan B. P.. 2018. Characterizing a pH-switch anti-C5 antibody as a tool for human and mouse complement C5 purification and cross-species inhibition of classical and reactive lysis. Immunology 155: 396–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gallwitz M., Enoksson M., Thorpe M., Hellman L.. 2012. The extended cleavage specificity of human thrombin. PLoS One 7: e31756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.de Bruijn M. H., Fey G. H.. 1985. Human complement component C3: cDNA coding sequence and derived primary structure. Proc. Natl. Acad. Sci. USA 82: 708–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Aleshin A. E., DiScipio R. G., Stec B., Liddington R. C.. 2012. Crystal structure of C5b-6 suggests structural basis for priming assembly of the membrane attack complex. J. Biol. Chem. 287: 19642–19652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Emeis M., Sonntag J., Willam C., Strauss E., Walka M. M., Obladen M.. 1998. Acidosis activates complement system in vitro. Mediators Inflamm. 7: 417–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sonntag J., Emeis M., Strauss E., Obladen M.. 1998. In vitro activation of complement and contact system by lactic acidosis. Mediators Inflamm. 7: 49–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hecke F., Hoehn T., Strauss E., Obladen M., Sonntag J.. 2001. In-vitro activation of complement system by lactic acidosis in newborn and adults. Mediators Inflamm. 10: 27–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sonntag J., Wagner M. H., Strauss E., Obladen M.. 1998. Complement and contact activation in term neonates after fetal acidosis. Arch. Dis. Child. Fetal Neonatal Ed. 78: F125–F128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fishelson Z., Horstmann R. D., Müller-Eberhard H. J.. 1987. Regulation of the alternative pathway of complement by pH. J. Immunol. 138: 3392–3395. [PubMed] [Google Scholar]
- 44.Hänsch G., Hammer C., Jiji R., Rother U., Shin M.. 1983. Lysis of paroxysmal nocturnal hemoglobinuria erythrocytes by acid-activated serum. Immunobiology 164: 118–126. [DOI] [PubMed] [Google Scholar]
- 45.Kenawy H. I., Boral I., Bevington A.. 2015. Complement-Coagulation Cross-Talk: A Potential Mediator of the Physiological Activation of Complement by Low pH. Front. Immunol. 6: 215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vogt W.1996. Complement activation by myeloperoxidase products released from stimulated human polymorphonuclear leukocytes. Immunobiology 195: 334–346. [DOI] [PubMed] [Google Scholar]
- 47.Vogt W., Damerau B., von Zabern I., Nolte R., Brunahl D.. 1989. Non-enzymic activation of the fifth component of human complement, by oxygen radicals. Some properties of the activation product, C5b-like C5. Mol. Immunol. 26: 1133–1142. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.