

Abstract
With few exceptions, ribosomal protein synthesis begins with methionine (or its derivative N-formyl-methionine) across all domains of life. The role of methionine as the initiating amino acid is dictated by the unique structure of its cognate tRNA known as tRNAfMet. By mis-acylating tRNAfMet, we and others have shown that protein synthesis can be initiated with a variety of canonical and noncanonical amino acids both in vitro and in vivo. Furthermore, because the α-amine of the initiating amino acid is not required for peptide bond formation, translation can be initiated with a variety of structurally disparate carboxylic acids that bear little resemblance to traditional α-amino acids. Herein, we provide a detailed protocol to initiate in vitro protein synthesis with substituted benzoic acid and 1,3-dicarbonyl compounds. These moieties are introduced at the N-terminus of peptides by mis-acylated tRNAfMet, prepared by flexizyme-catalyzed tRNA acylation. In addition, we describe a protocol to initiate in vivo protein synthesis with aromatic noncanonical amino acids (ncAAs). This method relies on an engineered chimeric initiator tRNA that is acylated with ncAAs by an orthogonal aminoacyl-tRNA synthetase. Together, these systems are useful platforms for producing N-terminally modified proteins and for engineering the protein synthesis machinery of Escherichia coli to accept additional nonproteinogenic carboxylic acid monomers.
1. Introduction
Ribosomal protein synthesis begins with the amino acid methionine across all domains of life. This unique ability of methionine to initiate protein synthesis is derived from the structure of its cognate tRNA, which exists in two forms: elongator and initiator methionine tRNA. The elongator tRNA (tRNAMet) exclusively installs methionine at elongating AUG codons; whereas, the initiator tRNA (tRNAfMet) exclusively installs methionine at AUG codons downstream of a Shine-Dalgarno sequence, thereby initiating synthesis of a new polypeptide chain (Fig. 1A and B) (Bhattacharyya & Varshney, 2016; Rodnina, 2018). Despite almost universal conservation in Nature, methionine is not the only amino acid that can initiate protein synthesis. Several decades ago, it was shown that mutants of tRNAfMet, that were mis-acylated with non-methionine amino acids, can initiate protein synthesis in vivo (Chattapadhyay, Pelka, & Schulman, 1990; Varshney & RajBhandary, 1990). More recently, it was shown that mis-acylated tRNAfMet can initiate in vitro translation with any of the 20 canonical amino acids, including both the d- and l-stereoisomers (Goto, Ohta, et al., 2008; Goto, Murakami, & Suga, 2008). Initiation is also not limited to the canonical amino acids. Using native and engineered variants of the methionyl-tRNA synthetase, tRNAfMet can be mis-acylated and initiate translation with numerous structural analogs of methionine in vivo (Fig. 2, 1–7) (Budisa et al., 1995; Ngo, Schuman, & Tirrell, 2013). Moreover, we have developed a chimeric variant of tRNAfMet that is acylated with noncanonical amino acids (ncAAs) by an orthogonal aminoacyl-tRNA synthetase (aaRS, vide infra). Using this system we have initiated translation with a variety of aromatic ncAAs (Fig. 2, 8–13) in Escherichia coli to produce proteins containing these unnatural moieties at their N-terminus (Tharp et al., 2020; Tharp, Vargas-Rodriguez, Schepartz, & Söll, 2021).
Fig. 1.

(A) Genetic encoding of N-formyl-l-methionine and l-methionine in response to initiating and elongating AUG codons. N-formyl-l-methionine is selectively incorporated at the N-terminus by tRNAfMet while l-methionine is selectively incorporated at elongating positions by tRNAMet. (B) The cloverleaf structure of tRNAfMet from E. coli. Highlighted residues are conserved among initiator tRNAs and contribute to the ability of tRNAfMet to initiate translation.
Fig. 2.

Representative amino and non-α-amino acids that have been used to initiate translation in vitro and in vivo.
A unique feature of the initiating (N-terminal) amino acid is that it is the only residue whose α-amine is not involved in peptide bond formation. Therefore, Nα-modified amino acids can be installed into proteins at the initiating position. For example, in prokaryotes translation initiates with the Nα-modified variant of methionine, N-formyl-l-methionine (Fig. 1A) (Bhattacharyya & Varshney, 2016). Similarly, using synthetically mis-acylated tRNAfMet, translation has been initiated with various Nα-acyl and Nα-alkyl amino acids in vitro (Fig. 2, 14–17) (Goto, Ohta, et al., 2008; Tadayoshi et al., 2010). In addition to these Nα-modified amino acids, synthetically mis-acylated initiator tRNAs have been used to initiate translation with structurally disparate non-α-amino acids that bear little resemblance to traditional proteinogenic amino acids. These include, for example, long-chain carboxylic acids, substituted benzoic acids, 1,3-dicarbonyls, peptides, and helical aromatic oligomer-peptide hybrids (Fig. 2, 18–23) (Ad et al., 2019; Goto & Suga, 2009; Lee, Schwarz, Kim, Moore, & Jewett, 2020; Rogers et al., 2018). Taken together, these studies demonstrate exceptional versatility of wild type ribosomes to catalyze peptide bond formation with diverse carboxylic acids at the initiating position.
Some non-α-amino acids that have been introduced into proteins at the initiating position are precursors of polyamide polymers. For example, substituted benzoic acids (e.g., 18) are precursors of aramids; a class of aromatic polyamides with diverse properties. Similarly, long-chain amino acids (e.g., 22) are monomeric precursors of Nylon. Using wild type ribosomes, these molecules are primarily limited to incorporation at the initiating position; however, we hypothesize that mutant ribosomes could be engineered to catalyze peptide bond formation between consecutive polyamide monomers. Such ribosomes would enable DNA-encoded synthesis of sequence-defined chemical polymers with unprecedented properties. Furthermore, we envision that platforms that enable noncanonical initiation can provide a starting point to engineer translational components (ribosomes, aaRSs, elongation factors, etc.) toward this end. In this chapter, we provide detailed methods for initiating in vitro translation with substituted benzoic acid and 1,3-dicarbonyl monomers via flexizyme-catalyzed acylation of tRNAfMet. In addition, we also describe methods for applying orthogonal aaRSs to initiate translation with various aromatic ncAAs in vivo.
2. Initiating translation with substituted benzoic acid and 1,3-dicarbonyl monomers in vitro
In vivo ribosomal translation of chemical polymers is the holy grail of polymer science. However, biosynthesis in living cells is multifaceted and requires each monomer to be cell-permeable (bioavailable), as well as compatible with an aaRS and the ribosomal translation machinery (Chin, 2017). In this way, in vivo experiments can hinder our ability to evaluate the capacity of ribosomal translation to tolerate new chemical moieties. Therefore, in vitro techniques that enable direct interrogation of ribosomal translation are a powerful complement to in vivo biosynthesis efforts. In vitro translation (IVT) systems, such as the commercially available Protein Synthesis Using Purified Recombinant Elements (PURE) system and extract-based systems, are cell-free reconstitutions of all of the necessary components for ribosomal translation (Hammerling, Krüger, & Jewett, 2020; Shimizu et al., 2001). The absence of a cell membrane enables researchers to circumvent issues related to monomer bioavailability and to make user-defined adjustments to reaction conditions (translation factors, template concentration, mis-acylated tRNA concentration, etc.). A complementary technique to IVT is flexizyme-mediated tRNA acylation. Flexizymes are aaRS-like ribozymes designed to charge diverse non-natural amino acids onto a variety of tRNAs (Lee, Bessho, Wei, Szostak, & Suga, 2000; Murakami, Ohta, Ashigai, & Suga, 2006; Murakami, Saito, & Suga, 2003; Niwa, Yamagishi, Murakami, & Suga, 2009; Ramaswamy, Saito, Murakami, Shiba, & Suga, 2004). We and others have shown that flexizymes are capable of mis-acylating tRNA with diverse unnatural monomers such as α-hydroxy, N-alkyl-amino, d-α-amino, α-thio, and α-amino(carbothio) acids, β-amino acids, long chain- and cyclic-amino acids, aramids, and 1,3-dicarbonyls (Ad et al., 2019; Katoh & Suga, 2018, 2020a, 2020b; Katoh, Tajima, & Suga, 2017; Kawakami, Murakami, & Suga, 2008; Lee et al., 2019, 2020; Maini et al., 2019; Ohta, Murakami, Higashimura, & Suga, 2007; Takatsuji et al., 2019). This eliminates the need to engineer an aaRS for each monomer of interest (Chin, 2017). Using flexizyme-mediated tRNA mis-acylation together with IVT, we investigated the tolerance of wild type E. coli ribosomes for diverse aramids and 1,3-dicarbonyls (Fig. 3) (Ad et al., 2019).We found that wild type E.coli ribosomes accepted tRNAfMet mis-acylated with both aramids and 1,3-dicarbonyls. These substrates were elongated to create diverse aramid-peptide and polyketide-peptide hybrid molecules. This work expanded the scope of reactions catalyzed by flexizymes and wild type ribosomes as well as established a starting point for in vivo translation of these novel materials.
Fig. 3.

Structures of benzoic acid, aramid, and malonate monomers used to initiate translation in this work.
2.1. Protocol
2.1.1. Equipment
Analytical balance
Vortex mixer
Centrifuge
Thermocycler
PAGE gel casting apparatus
PAGE gel electrophoresis chamber and power supply
Gel imaging system
Liquid chromatography-mass spectrometer (LC-MS)
2.1.2. Materials
DNA Oligonucleotide Sequences (Integrated DNA Technologies)
| 1 | MetT-F | ATTCCTGCAGTAATACGACTCACTATACGC GGGGTGGAGCAGCCTGGTAGC TCGTCGGGCTCATA |
| 2 | MetT-R* | TmGGTTGCGGGGGCCGGATTTGAACCGACG ACCTTCGGGTTATGAGCCCGA CGAGCTA *mG represents 2′ O-methyl-deoxymethylguanosine |
| 3 | MVFflag-1 | TAATACGACTCACTATAGGGTTAACTTTAA CAAGGAGAAAAACATGGTATTTG ACTACAAGG |
| 4 | MVFflag-2 | CGAAGCTTACTTGTCGTCGTCGTCCTTGTA GTCAAATACCATGTTTTTCTCCT TGTTAAAG |
| 5 | MVFflag-3 | GCGAATTAATACGACTCACTATAGGGTTAA CTTTAACA |
| 6 | MVFflag-4 | AAACCCCTCCGTTTAGAGAGGGGTTATGCT AGTTACTTGTCGTCGTCGTCC TTG |
RNA Oligonucleotide Sequences (Integrated DNA Technologies)
| 1 | Microhelix (MH) RNA | GGCUCUGUUCGCAGAGCCGCCA |
| 2 | aFx | GGUAGGAAAUGUUUCCGCACCCCCGAAAGG GGUAAGGGCGUCAGGU |
| 3 | dFx | GGAUCGAAAGAUUUCCGCAUCCCCGAAAGG GUACAUGGCGUUAGGU |
| 4 | eFx | GGAUCGAAAGAUUUCCGCGGCCCCGAAAGG GGAUUAGCGUUAGGU |
2.1.2.1. Reagents
RNase-free water (Invitrogen, Catalog # 10977–015)
HEPES Buffer: 500 mM HEPES, pH 7.5
Bicine Buffer: 500 mM Bicine, pH 9.0
Magnesium chloride, 1 M
Acrylamide/bis-acrylamide (19:1 crosslinker), 40% (w/v) (Bio-Rad, Catalog # 1610144)
Urea, 72% (w/v)
Sodium Acetate Buffer: 3 M sodium acetate, pH 5.2 (AmericanBio, Catalog # AB13168–01000)
Ammonium persulfate, 10 mgmL−1
Tetramethylethylenediamine (TEMED)
RNA Loading Buffer: 95% (v/v) formamide, 150 mM sodium acetate pH 5.2, 10 mM EDTA, and 200 μg mL−1 bromophenol blue
TBE Buffer: 10.8 gL−1 tris, 5.5 gL−1 boric acid, 10 mM EDTA, pH 8.0
Ethidium bromide
RNase A: 1.5 U μL−1 in 200 mM sodium acetate, pH 5.2 (Millipore Sigma, Catalog # 10109142001)
Trichloroacetic acid, 50% (w/v)
Basic phenol solution (10 mM Tris, 1 mM EDTA, pH 8.0) (Millipore Sigma, Catalog # P4557–100 ML)
Acidic phenol solution (saturated with citric acid, pH 4.5) (Millipore Sigma, Catalog # 6702–400 ML)
Chloroform
Ethanol, 200 proof (AmericanBio, Catalog # AB00515–00500)
RNase-free DNase I (Fisher Scientific, Catalog # EN0521)
l-methionine, 33 mM
l-valine, 33 mM
l-tyrosine, l-phenylalanine, and l-lysine stock, 33 mM each
l-aspartic acid, 7 mM (adjusted to pH 7.0)
Ni-NTA Agarose (Qiagen, Catalog # 30210)
2.1.2.2. Kits and consumables
Hiscribe™T7 Quick High Yield RNA Synthesis Kit (New England Biolabs, Catalog # E2050S)
PURExpress® (ΔtRNA, Δaa) In Vitro Translation (IVT) Kit (New England Biolabs, Catalog # E6840)
RNase-free Micro Bio-spin™ P-30 Columns, Tris Buffer (Bio-Rad, Catalog # 7326250)
2.1.3. Selection of flexizyme and synthesis of flexizyme-compatible monomers
Flexizymes are aaRS-like ribozymes that recognize various tRNAs via base pairing interactions between the 3′-end CCA of the tRNA and the 3′-end GGU of flexizyme (Morimoto, Hayashi, Iwasaki, & Suga, 2011). Originally, flexizyme (Fx) was designed to recognize aromatic amino acids that contain an activated cyanomethyl ester (CME) leaving group (Lee et al., 2000). Later, efforts to create flexizyme variants that tolerate a variety of amino acids yielded dFx and eFx, which recognize their substrates via the aromatic leaving groups 3,5-dinitrobenzyl ester (DNB) and p-chlorobenzyl thioester (CBT) respectively (Murakami et al., 2006). A third flexizyme was later developed to aid in the acylation of poorly soluble monomers (Niwa et al., 2009). This flexizyme, aFx, recognizes its substrates via the hydrophilic leaving group (2-aminoethyl) amidocarboxybenzyl thioester (ABT). The structure of flexizyme leaving groups and the primary sequence of flexizyme variants are provided in Fig. 4.
Fig. 4.
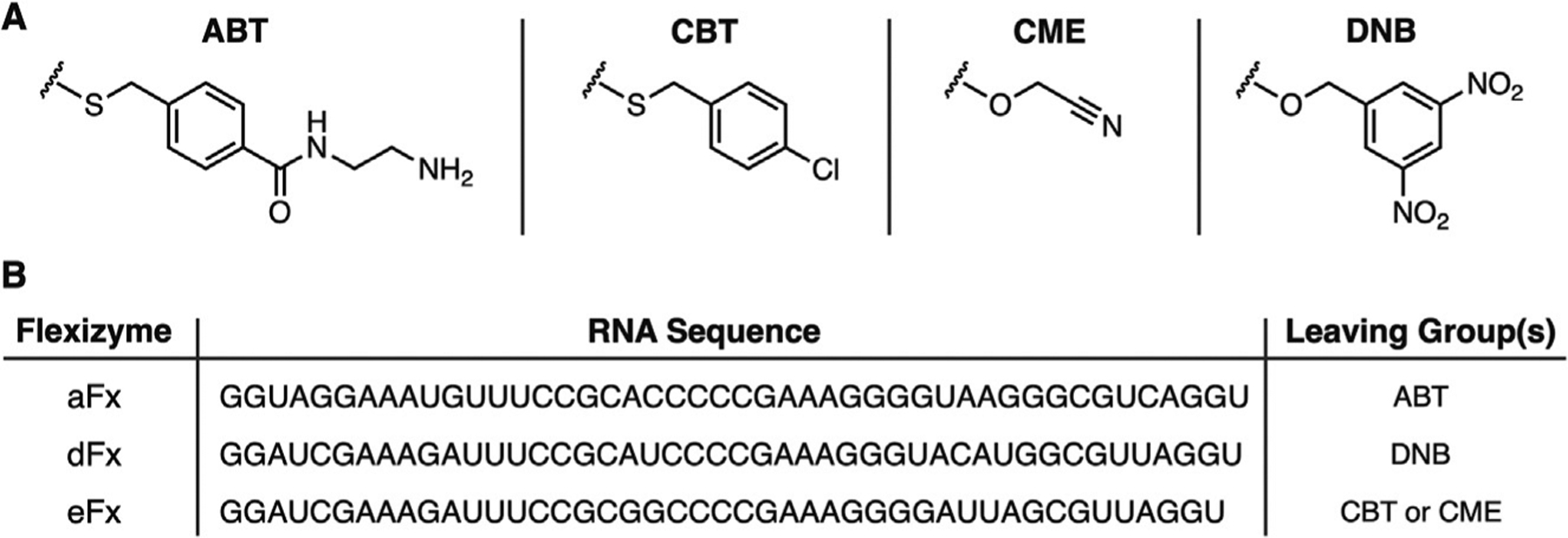
(A) Structures of Leaving groups for flexizyme-compatible substrates. (B) Summary of flexizyme sequences and leaving groups used to identify substrates.
Flexizymes are commonly synthesized by in vitro transcription; however, in our studies, we exclusively use flexizymes synthesized (via solid-phase techniques) by Integrated DNA Technologies. Standard “desalting” purification removes small truncation products and organic contaminants such that there is no noticeable impact on flexizyme activity. The synthesis of flexizyme compatible substrates is unique to each monomer of interest. Within the work presented here we synthesized greater than 20 flexizyme compatible substrates. Thus, a detailed description of the synthetic methodology is beyond the scope of this protocol. However, our lab and others have previously reported the synthesis of dozens of flexizyme compatible monomers. (Ad et al., 2019; Katoh & Suga, 2018, 2020a, 2020b; Katoh et al., 2017; Kawakami et al., 2008; Lee et al., 2020; Lee et al., 2019; Maini et al., 2019; Ohta et al., 2007; Takatsuji et al., 2019).
Identification of the optimal flexizyme variant and leaving group for charging tRNA with a given monomer is somewhat empirical. Generally, eFx and the leaving group CME may be used with monomers that contain aromatic side chains or backbones. However, monomers devoid of an aromatic moiety require the leaving groups ABT, DNB, or CBT along with the corresponding flexizymes aFx, dFx, and eFx.
2.1.4. Optimizing conditions for flexizyme-catalyzed acylation
Microhelix (MH) RNA, a 22-nucleotide surrogate for the acceptor stem of full-length tRNA, is often used to optimize conditions for flexizyme-mediated tRNA acylation. The small size of MH RNA makes it accessible via solid-phase synthesis and enables characterization of acylation via acid-urea PAGE gel-shift analysis. In addition to gel-shift analysis we adapted a highly sensitive RNase A/LC-MS assay to directly observe the 2′ and 3′ acyl-adenosine of the acylated MH RNA (McMurry & Chang, 2017). A summary of MH RNA acylation optimization is provided in Fig. 5.
Fig. 5.

Overview of microhelix acylation and analysis.
2.1.4.1. Flexizyme-catalyzed acylation of microhelix (MH) RNA
Mix 1 μL of appropriate flexizyme (250 μM stock in RNase-free water), 1 μL of MH RNA (250 μM stock in RNase-free water), and 1 μL of buffer (500 mM HEPES pH 7.5 or 500 mM Bicine pH 9.0)
Incubate sample at 95°C for 2 min
Allow sample to cool at room temperature for 5 min
Add 6 μL of magnesium chloride (1 M stock in RNase-free water).
Add 1 μL of appropriate monomer (50 mM stock in dimethyl sulfoxide)
-
Pipette mix and incubate at 4°C for desired amount of time.
Note: Typically, incubation for 48h is sufficient to observe acylation.
2.1.4.2. Acid-urea PAGE gel-shift analysis
Prepare unpolymerized acid-urea PAGE gel mixture as follows: 5 mL of 40% (w/v) acrylamide/bis-acrylamide (19:1), 5 mL of 72% (w/v) urea, and 167 μL of 3 M sodium acetate (pH 5.2).
To initiate polymerization, add 100 μL of 10 mg μL−1 ammonium persulfate (freshly prepared) and 8 μL of TEMED. The final gel composition is 20% acrylamide/bis-acrylamide, 36% (w/v) urea, 50 mM sodium acetate (pH 5.2), 0.1% (w/v) ammonium persulfate, and 0.08% (v/v) TEMED
Quickly mix and transfer acid-urea PAGE gel mixture to an appropriate gel casting apparatus. Reserve a small amount of unpolymerized gel mixture to monitor the polymerization status of the PAGE gel
Allow the gel to polymerize until the reserved portion has solidified—typically approximately 30 min
Add 1 μL of acylation reaction mixture to 1 μL of denaturing RNA loading buffer (95% (v/v) formamide, 150 mM sodium acetate (pH 5.2), 10 mM EDTA, and 200 μg μL−1 bromophenol blue).
Run the denatured sample on an acid-urea PAGE gel with 50 mM sodium acetate (pH 5.2) as the running buffer at 120V until the dye has run to the bottom of the gel—approximately 3–4h. Note: It is advisable to run acylation reaction samples against MH RNA and flexizyme controls. These controls can help detect minor changes in sample migration
Stain the gel with ethidium bromide (1 μg mL−1 in TBE Buffer) for 2 min. Note: MH RNA may exhibit poor staining with other detection reagents. In our hands, ethidium bromide has provided the strongest signal for these samples
Destain the gel with TBE Buffer for 1 min
Image with an appropriate gel imaging system and quantify UV densitometry using ImageJ
2.1.4.3. RNase A/LC-MS analysis
Add 1 μL of acylation reaction mixture to 1.1 μL of RNase A (1.5 U μL−1 in 200 mM sodium acetate, pH 5.2) and incubate at room temperature for 5 min
Precipitate RNase A by adding 0.25 μL of 50% (w/v) trichloroacetic acid and incubating the mixture at room temperature for 5 min
Remove insoluble material by centrifugation (20,000 × g) for 2 min at 4°C
Dilute 1 μL of the sample with water to 20 μL and freeze by incubation at −80°C for 5 min
Before analysis, thaw the sample and remove insoluble material by centrifugation at (20,000 × g) for 10 min at 4°C
Analyze the sample via LC-MS to detect the exact mass of the 2′ and 3′ acyl-adenosine
2.1.5. Preparation of mis-acylated tRNAfMet
2.1.5.1. In vitro transcription and purification of tRNAfMet
Prepare DNA template for transcribing E. coli tRNAfMet using polymerase chain reaction (PCR) by annealing and extending the oligonucleotides MetT-F and MetT-R. The oligonucelotides are mixed 1:1 at 10 μM. MetT-R contains a 2′-O-methyl-deoxymethylguanosine at the penultimate nucleotide to prevent run-off transcription
Extract the extended template 1× with 1 volume of a 1:1 (v/v) basic phenol (pH 8.0)/chloroform mixture
Precipitate the extracted template with 3 volumes of 95% (v/v) ethanol
Pellet the precipitated template by centrifugation (20,000 × g) for 30 min at 4°C
Remove the supernatant and allow the pellet to dry at room temperature for 15 min
Suspend the purified template in RNase-free water and quantify by measuring the absorbance at 260 nm
Set up in vitro transcription of tRNAfMet using HiScribe™ T7 Quick High Yield RNA Synthesis Kit from NEB using the manufacturer’s protocol for short transcripts at a total reaction scale of 200 μL (6.7 mM ATP, GTP, UTP, and CTP and 50 ng μL−1 DNA template).
Incubate in vitro transcription reaction at 37°C for 6h
Treat reaction with 10U of RNase-free DNAse at 37°C for 2h
Add sodium acetate buffer, pH 5.2 to a final concentration of 200 mM
Extract the reaction 1× with one volume of a 1:1 (v/v) acidic phenol (pH 4.5)/chloroform mixture and 2× with one volume of chloroform
Precipitate the extracted product with 3 volumes of 95% (v/v) ethanol. If necessary, incubate at −80°C for 1h or −20°C overnight to encourage precipitation
Pellet the precipitated tRNAfMet by centrifugation (20,000 × g) for 30 min at 4°C
Remove the supernatant and wash the pellet 2× with ice cold 70% (v/v) ethanol. Centrifuge (20,000 × g) for 5 min at 4°C
Allow the pellet to dry at room temperature for 15 min
Suspend the product in RNase-free water and remove excess nucleotides using RNase-free Micro Bio-Spin® P-30 columns, Tris Buffer from Bio-Rad using the manufacturer’s protocol
Quantify the recovery of tRNAfMet by measuring the absorbance at 260 nm
Store purified tRNAfMet at −80°C for optimal stability. For short-term (<1 month), −20°C can be used for storage
2.1.5.2. Flexizyme-catalyzed Mis-acylation of tRNAfMet
Mix 10 μL of appropriate flexizyme (250 μM stock in RNase-free water), 10 μL of tRNAfMet (250 μM stock in RNase-free water), and 10 μL of buffer (500 mM HEPES pH 7.5 or 500 mM Bicine pH 9.0).
Incubate sample at 95°C for 2 min
Allow sample to cool at room temperature for 5 min
Add 60 μL of magnesium chloride (1 M stock in RNase-free water).
Add 10 μL of appropriate monomer (50 mM stock in dimethyl sulfoxide)
Pipette mix and incubate at 4°C for amount of time determined by MH RNA optimization experiments
If desired, monitor the reaction by removing a 1 μL aliquot of the tRNAfMet acylation reaction mixture for analysis via the RNase A digestion assay described above
Add sodium acetate buffer (pH 5.2) to a final concentration of 300 mM and precipitate the mis-acylated tRNAfMet by adding ethanol to a final concentration of 70% (v/v).
To encourage precipitation, incubate sample at −80°C for 1h
Pellet the mis-acylated tRNAfMet by centrifugation (20,000 × g) for 30 min at 4°C
Wash the pellet 1× with 500 μL of ice cold 70% (v/v) ethanol. Centrifuge (20,000 × g) for 5 min at 4°C
Allow the pellet to dry at room temperature for 5 min
If mis-acylated tRNAfMet will be used immediately, suspend the pellet in 1 mM sodium acetate (pH 5.2). Otherwise, store the dry pellet at −80°C for later use
2.1.6. In vitro translation reactions
The commercially available Protein Synthesis Using Purified Recombinant Elements (PURE) system is an in vitro assembly of translation factors, tRNAs, RNAP polymerase, and ribosomes that have been individually purified and supplemented with amino acids, nucleotides, and co-factors (Hammerling et al., 2020; Shimizu et al., 2001). Supplementing this mixture with mis-acylated tRNA enables direct interrogation of ribosomal translation. A schematic overview of in vitro translation (IVT) with the PURE system and subsequent analysis of translation products via LC-MS is provided in Fig. 6.
Fig. 6.

Overview of reprogrammed ribosomal translation via the PURE system and subsequent LC-MS analysis of the translation products.
2.1.6.1. Synthesis of MVFDYKDDDDK (MVF-FLAG) template DNA
Prepare DNA template for expression of MVFDYKDDDDK by annealing and extending the oligonucleotides MVFflag-1 and MVFflag-2. For extension, the oligonucleotides are mixed 1:1 at 10 μM
Extract the extended template 1× with 1 volume of a 1:1 (v/v) basic phenol (pH8.0)/chloroform mixture
Precipitate the extracted template with 3 volumes of 95% (v/v) ethanol
Pellet the precipitated template by centrifugation (20,000 × g) for 30 min at 4°C
Remove the supernatant and allow the pellet to dry at room temperature for 15 min
Suspend the purified template in RNase-free water and quantify by measuring the absorbance at 260 nm
Amplify the extension product of MVFflag-1 and MVFflag-2 by PCR using primers MVFflag-3 and MVFflag-4
Extract the extended template 1× with 1 volume of a 1:1 (v/v) basic phenol (pH 8.0)/chloroform mixture
Precipitate the extracted template with 3 volumes of 95% (v/v) ethanol
Pellet the precipitated template by centrifugation (20,000 × g) for 30 min at 4°C
Wash the pellet 2× with 500 μL 70% (v/v) ethanol. Centrifuge (20,000 × g) for 5 min at 4°C
Remove the supernatant and allow the pellet to dry at room temperature for 15 min
Suspend the purified template in RNase-free water and quantify by measuring the absorbance at 260 nm
2.1.6.2. In vitro translation with Mis-acylated tRNAfMet
The model FLAG-tagged peptide MVFDYKDDDDK (MVF-FLAG) was expressed using the PureExpress® (ΔtRNA, Δaa) kit by NEB with some modifications
- To initiate ribosomal translation with unnatural monomers, combine the following:
- 5μL of solution A (ΔtRNA, Δaa)
- 0.25 μL of a valine stock solution at 33 mM
- 0.25 μL of a stock solution containing 33 mM tyrosine, 33 mM phenylalanine, and 33 mM lysine
- 1μL of a stock solution containing 7 mM aspartic acid, pH 7.0
- 2.5 μL of tRNA solution
- 7.5 μL of solution B
- 500–1000ng of MVF-FLAG DNA template
- 1.25–2.5 nmols of mis-acylated tRNAfMet freshly prepared in 1 mM sodium acetate, pH 5.2
- RNase-free water to bring the total reaction volume to 25 μL
To perform a control expression of the MVF-FLAG template, omit the mis-acylated tRNAfMet and add 0.25 μL of a methionine stock solution at 33 mM
To demonstrate that the translation product is template-dependent, omit the MVF-FLAG DNA template
Incubate reactions for 6h at 37°C
Quench reactions by placing them on ice and adding 25 μL of dilution buffer (10 mM magnesium acetate and 100 mM sodium chloride).
To remove most proteins and nucleic acids, add 5 μL of Ni-NTA slurry and incubate on ice for 1h
To remove the Ni-NTA resin, centrifuge the sample (20,000 × g) for 10 min at 4°C
Freeze the supernatant at −80°C for 5 min
Remove insoluble material by centrifugation (20,000 × g) for 10 min at 4°C
Analyze the supernatant via LC-MS to detect the exact mass of the translation product
3. Initiating translation with noncanonical amino acids in vivo
In vitro, mis-acylated tRNAfMet can be prepared using various chemical acylation strategies, or by flexizyme-catalyzed acylation. These complementary methods are highly adaptable and can be used to produce tRNAfMet acylated with virtually any desired acid. However, these methods are not compatible with protein synthesis in vivo. In living cells, tRNAs are acylated by the aaRSs, an ancient class of enzymes that have evolved high efficiency and fidelity toward recognition of a single amino acid substrate. Genetic code expansion (GCE) is a technique that uses orthogonal aaRS and tRNA pairs to co-translationally install ncAAs into proteins, typically in response to the amber (UAG) nonsense codon (Fig. 7A) (Chin, 2017; Young & Schultz, 2018). One aaRS•tRNA pair that is frequently used for this purpose is the tyrosyl-tRNA synthetase (MjTyrRS) and tRNATyr pair from Methanocaldococcus jannaschii. Over the past two decades, the MjTyrRS has been engineered to recognize dozens of aromatic ncAAs to facilitate their co-translational incorporation into protein in E. coli (Dumas, Lercher, Spicer, & Davis, 2015). The MjTyrRS recognizes its cognate tyrosine tRNA using a small set of identity elements including the discriminator base (A73) and the first base pair (C1–G72) (Fechter, Rudinger-Thirion, Tukalo, & Giegé, 2001; Sakamoto & Hayashi, 2019). By transplanting these nucleotides into E. coli tRNAfMet we developed a chimeric tRNA (itRNATy2) that is a substrate for MjTyrRS-catalyzed aminoacylation with ncAAs (Fig. 7B). Because this chimeric tRNA was designed using tRNAfMet as a scaffold, once acylated by MjTyrRS, itRNATy2 can initiate translation with ncAAs at UAG codons downstream of a Shine-Dalgarno sequence (Fig. 7C).
Fig. 7.

(A) Overview of genetic code expansion. Orthogonal aminoacyl-tRNA synthetase and tRNA pairs are used to introduce ncAAs into proteins during normal ribosomal protein synthesis. Typically, orthogonal suppressor tRNAs are used to introduce ncAAs in response to the UAG nonsense codon. (B) The cloverleaf structure of itRNATy2. Positions highlighted in magenta differ from the sequence of tRNAfMet. (C) Plasmid maps of the two-plasmid system used to introduce ncAAs at the N-terminus of superfolder green fluorescent protein (sfGFP). sfGFP with an M1 → UAG mutation (sfGFP-1 am) is co-expressed along with itRNATy2 and a polyspecific MjTyrRS mutant (pCNFRS) that recognizes para-substituted phenylalanine derivatives.
Using itRNATy2, together with engineered MjTyrRS variants, we have demonstrated efficient ncAA initiation of several genes. These include the common reporter genes chloramphenicol acetyltransferase, superfolder green fluorescent protein, and the bacterial luciferase gene. In addition, we have found that this system is compatible with several commonly employed laboratory strains of E. coli. However, while itRNATy2 can be used to initiate translation with ncAAs in wild type strains, the expression yield is quite low. This is likely due to competition with endogenous tRNAfMet for binding to ribosomal P-sites (Kapoor, Das, & Varshney, 2011). To overcome this limitation, we developed a strain of E. coli in which three of the four genomic copies of tRNAfMet were deleted from the genome (strain DH10BΔmetZWV). Studies have shown that ΔmetZWV strains show improved initiation with mutant initiator tRNAs (Kapoor et al., 2011; Samhita, Virumäe, Remme, & Varshney, 2013). Using strain DH10BΔmetZWV initiation with ncAAs is at least as efficient as elongation with the same ncAA.
3.1. Protocol
3.1.1. Equipment
Stationary and shaking incubators
Water bath
Cell density meter
Analytical balance
2 × 1L shaker flasks
Petri plates (100 mm × 20 mm)
Culture tubes (14 mL; 17 mm × 100 mm)
Polypropylene gravity flow columns (e.g., Bio-Rad Catalog # 7311550)
Centrifuge
Autoclave
Mass spectrometer
3.1.2. Materials
Plasmid pBAD sfGFP[1UAG] itRNATy2 (Tharp et al., 2020) (Plasmids are available from the corresponding authors upon request)
Plasmid pMW pCNFRS (Tharp et al., 2020)
2 × YT media
LB agar
Ampicillin
Spectinomycin
para-acetyl-l-phenylalanine (8, pAcF, Chem-Impex, Catalog # 24756) or desired ncAA
Glycerol solution, 10% v/v (autoclaved)
50× salts solution (autoclaved) (Hammill, Miyake-Stoner, Hazen, Jackson, & Mehl, 2007)
MgSO4, 1 M (filtered)
l-aspartate solution, 5% w/v (pH adjusted to 7.5 and filtered)
l-leucine solution, 30 mM (autoclaved)
25× amino acid solution (filtered) (Tharp et al., 2020)
5000× Trace metal solution (filtered) (Hammill et al., 2007)
Autoclaved MilliQ water
Isopropyl β-d-1 thiogalactopyranoside, 1 M (filtered)
l-arabinose, 20% w/v (filtered)
10× BugBuster® protein extraction reagent (MilliporeSigma, Catalog # 70921)
Benzonase® nuclease (MilliporeSigma, Catalog # E1014–25KU)
Lysozyme
Talon® metal affinity resin (Takara Bio, Catalog # 635501)
IMAC Buffer A: 50 mM Tris pH 8.0, 500 mM NaCl, 10 mM Imidazole
IMAC Buffer B: 50 mM Tris pH 8.0, 300 mM NaCl, 250 mM Imidazole
3.1.3. Expression and purification of sfGFP-1pAcF
3.1.3.1. Transformation
Prepare LB agar plates containing 100 μg mL−1 ampicillin and 50 μg mL−1 spectinomycin
Co-transform DH10BΔmetZWV by adding 2 μL of the plasmid pBAD sfGFP[1UAG] itRNATy2 (~100 ng μL−1) and 3 μL of plasmid pMW pCNFRS (~20 ng μL−1) to a 100 μL aliquot of chemically competent cells in a 1.5 mL Eppendorf™ tube. Gently flick the tube to mix and incubate the cell/plasmid mixture on ice for 10 min
Transfer the tube containing the cell/plasmid mixture to a 42°C water bath and incubate for 45s. Place the tube back on ice and incubate an additional 2 min
Transfer the cell/plasmid mixture to a culture tube containing 900 μL of 2 × YT (without antibiotics) and incubate in a shaking incubator at 37°C for 45 min
Pipette 100 μL of the culture onto the surface of a dry LB agar plate (containing ampicillin and spectinomycin). Using a cell spreader or glass beads, evenly spread the solution across the entire surface of the agar plate until the liquid is absorbed. Incubate the plate upside down, overnight at 37°C
3.1.3.2. Protein expression
The day before protein expression, use a sterile toothpick or disposable pipette tip to pick a single colony of co-transformed DH10BΔmetZWV. Grow the cells overnight at 37°C in 10 mL 2 × YT supplemented with 100 μg mL−1 ampicillin and 50 μg mL−1 spectinomycin
Prepare 200 mL of chemically defined media (CDM) by mixing the following components in an autoclaved 1 L shaker flask: 20 mL glycerol (10%, v/v), 4 mL 50× salts, 400 μL MgSO4 (1 M), 10 mL l-aspartate (5% w/v), 4 mL l-leucine (30 mM), 8 mL amino acid solution (25×), 40 μL trace metals (5000×), and autoclaved MilliQ water to 200 mL (~153 mL). Supplement the prepared media with ampicillin and spectinomycin to 100 and 50 μg mL−1, respectively. Note: Protein expression can be carried out entirely in 2 × YT or other rich media, however, this will result in higher background (protein expressed in the absence of an ncAA) and, depending on which MjTyrRS variant is used, may produce significant amounts of protein containing canonical amino acids
Add the 10 mL overnight culture of co-transformed DH10BmetZWV to the prepared CDM and incubate in a shaking incubator at 37°C until the OD600 reaches 0.3–0.6. Note: DH10BΔmetZWV grow considerably slower in CDM than in rich media. Under these conditions the culture will require ~4–5h to reach OD600=0.3
Prepare a fresh solution (100 mM) of pAcF by dissolving 0.041g of pAcF in 2 mL of 0.3 M NaOH
Once the culture reaches an OD600 of 0.3–0.6 induce protein expression by adding to the culture flask 20 μL IPTG (1 M, 0.1 mM final concentration) and 2 mL of l-arabinose (20%, 0.2% final concentration). Divide the culture by pouring half of the volume (~100 mL) into a second shaker flask. To one flask add 2 mL pAcF solution (100 mM in 0.3 M NaOH, 2 mM final concentration). To the second flask add 2 mL of 0.3M NaOH. The second culture will serve as a negative (no ncAA) control to gauge the degree of canonical amino acid incorporation
Return both flasks to the shaking incubator and incubate overnight (~18h) at 37°C
Pellet the cells by centrifugation (3500 × g, 20 min) and discard the supernatant. Store cell pellets at −80°C until purification
3.1.3.3. Protein purification
Prepare 1× Lysis Buffer by combining the following components: 18 mL IMAC Buffer A, 2 mL BugBuster® protein extraction reagent, 20 mg lysozyme (1 mg mL−1 final concentration), and 500 units Benzonase® nuclease
Transfer 1 mL of Talon® metal affinity resin to a polypropylene gravity flow column. Wash the resin with 5 column volumes of MilliQ water followed by 10 column volumes of IMAC Buffer A
Thaw cell pellets at room temperature and then resuspend each pellet in 10 mL 1× Lysis Buffer and incubate at room temperature for 30 min with gentle agitation
Pellet insoluble material by centrifugation (10,000 × g, 45 min) and transfer the soluble lysate to the column containing equilibrated Talon® metal affinity resin. Allow the lysate to slowly flow through the column
Wash the resin with 20 column volumes of IMAC Buffer A and then elute bound proteins with ~5 mL of IMAC Buffer B collecting ~0.5 mL fractions
Analyze the fractions by gel electrophoresis and combine fractions containing purified sfGFP-1pAcF. Recommended: Further analysis of the protein by mass spectroscopy is recommended to confirm ncAA incorporation
4. Summary
In this chapter we provided a detailed protocol to initiate in vitro protein synthesis with substituted benzoic acids and 1,3-dicarbonyls. The method described here relies on flexizyme-catalyzed acylation to generate tRNAfMet that is mis-acylated with the desired acid. The mis-acylated tRNAfMet is then used in cell-free protein synthesis reactions to initiate mRNA translation and introduce the benzoic acid or 1,3-dicarbonyl at the N-terminus of peptides. In addition to those described here, the E. coli ribosome has been shown to accept many diverse non-α-amino acids at the initiating position. More recently, some of these acids, including substituted benzoic acids, have been shown to be compatible with ribosomal elongation (Katoh & Suga, 2020a, 2020b; Lee et al., 2020). Thus, the present represents an exciting time in the history of synthetic biology, as we are on the cusp of engineering ribosomes that can catalyze bond formation between consecutive non-α-amino acid monomers. Such technology will open the door to the eventual ribosomal synthesis of DNA-encoded chemical polymers with precisely defined sequences.
Herein we also described a protocol to initiate in vivo protein synthesis with ncAAs. This method relies on an engineered chimeric initiator tRNA (itRNATy2) that is acylated by the MjTyrRS—an archaeal aaRS that is orthogonal in E. coli and has been engineered to recognize numerous aromatic ncAAs (Dumas et al., 2015). When co-expressed along with MjTyRS, in media containing a desired ncAA, itRNATy2 can initiate protein synthesis at UAG codons to produce proteins containing the ncAA at their N-terminus. Although not described here, this system is compatible with the pyrrolysyl-tRNA synthetase (PylRS)•tRNAPyl pair and can be used along with mutually orthogonal PylRS•tRNAPyl pairs to co-translationally install two or three distinct ncAA monomers into a single protein in E. coli (Tharp et al., 2020, 2021). Although MjTyrRS is currently limited to α-amino and α-hydroxy acid substrates, using itRNATy2 with further engineered MjTyrRS variants could enable genetic encoding of more disparate non- α-amino acids at the N-terminus of proteins in vivo.
Acknowledgments
This work was funded by the Center for Genetically Encoded Materials, an NSF Center for Chemical Innovation (NSF CHE-2002182 to A.S.). During the writing of this review, J.M.T was supported by NIGMS (R35GM122560 to D.S.).
References
- Ad O, Hoffman KS, Cairns AG, Featherston AL, Miller SJ, Söll D, et al. (2019). Translation of diverse aramid- and 1,3-dicarbonyl-peptides by wild type ribosomes in vitro. ACS Central Science, 5, 1289–1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharyya S, & Varshney U (2016). Evolution of initiator tRNAs and selection of methionine as the initiating amino acid. RNA Biology, 13, 810–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budisa N, Steipe B, Demange P, Eckerskorn C, Kellermann J, & Huber R (1995). High-level biosynthetic substitution of methionine in proteins by its analogs 2-aminohexanoic acid, selenomethionine, telluromethionine and ethionine in Escherichia coli. European Journal of Biochemistry, 230, 788–796. [DOI] [PubMed] [Google Scholar]
- Chattapadhyay R, Pelka H, & Schulman LH (1990). Initiation of in vivo protein synthesis with non-methionine amino acids. Biochemistry, 29, 4263–4268. [DOI] [PubMed] [Google Scholar]
- Chin JW (2017). Expanding and reprogramming the genetic code. Nature, 550, 53–60. [DOI] [PubMed] [Google Scholar]
- Dumas A, Lercher L, Spicer CD, & Davis BG (2015). Designing logical codon reassignment—Expanding the chemistry in biology. Chemical Science, 6, 50–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fechter P, Rudinger-Thirion J, Tukalo M, & Giegé R (2001). Major tyrosine identity determinants in Methanococcus jannaschii and Saccharomyces cerevisiae tRNATyr are conserved but expressed differently. European Journal of Biochemistry, 268, 761–767. [DOI] [PubMed] [Google Scholar]
- Goto Y, Murakami H, & Suga H (2008). Initiating translation with d-amino acids. RNA, 14, 1390–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto Y, Ohta A, Sako Y, Yamagishi Y, Murakami H, & Suga H (2008). Reprogramming the translation initiation for the synthesis of physiologically stable cyclic peptides. ACS Chemical Biology, 3, 120–129. [DOI] [PubMed] [Google Scholar]
- Goto Y, & Suga H (2009). Translation initiation with initiator tRNA charged with exotic peptides. Journal of the American Chemical Society, 131, 5040–5041. [DOI] [PubMed] [Google Scholar]
- Hammerling MJ, Krüger A, & Jewett MC (2020). Strategies for in vitro engineering of the translation machinery. Nucleic Acids Research, 48, 1068–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammill JT, Miyake-Stoner S, Hazen JL, Jackson JC, & Mehl RA (2007). Preparation of site-specifically labeled fluorinated proteins for 19F-NMR structural characterization. Nature Protocols, 2, 2601–2607. [DOI] [PubMed] [Google Scholar]
- Kapoor S, Das G, & Varshney U (2011). Crucial contribution of the multiple copies of the initiator tRNA genes in the fidelity of tRNAfMet selection on the ribosomal P-site in Escherichia coli. Nucleic Acids Research, 39, 202–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh T, & Suga H (2018). Ribosomal incorporation of consecutive β-amino acids. Journal of the American Chemical Society, 140, 12159–12167. [DOI] [PubMed] [Google Scholar]
- Katoh T, & Suga H (2020a). Ribosomal elongation of aminobenzoic acid derivatives. Journal of the American Chemical Society, 142, 16518–16522. [DOI] [PubMed] [Google Scholar]
- Katoh T, & Suga H (2020b). Ribosomal elongation of cyclic ɣ-amino acids using a reprogrammed genetic code. Journal of the American Chemical Society, 142, 4965–4969. [DOI] [PubMed] [Google Scholar]
- Katoh T, Tajima K, & Suga H (2017). Consecutive elongation of D-amino acids in translation. Cell Chemical Biology, 24, 46–54. [DOI] [PubMed] [Google Scholar]
- Kawakami T, Murakami H, & Suga H (2008). Messenger RNA-programmed incorporation of multiple N-methyl-amino acids into linear and cyclic peptides. Chemistry & Biology, 15, 32–42. [DOI] [PubMed] [Google Scholar]
- Lee N, Bessho Y, Wei K, Szostak JW, & Suga H (2000). Ribozyme-catalyzed tRNA aminoacylation. Nature Structural Biology, 7, 28–33. [DOI] [PubMed] [Google Scholar]
- Lee J, Schwarz KJ, Kim DS, Moore JS, & Jewett MC (2020). Ribosome-mediated polymerization of long chain carbon and cyclic amino acids into peptides in vitro. Nature Communications, 11, 4304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Schwieter KE, Watkins AM, Kim DS, Yu H, Schwarz KJ, et al. (2019). Expanding the limits of the second genetic code with ribozymes. Nature Communications, 10, 5097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maini R, Kimura H, Takatsuji R, Katoh T, Goto Y, & Suga H (2019). Ribosomal formation of thioamide bonds in polypeptide synthesis. Journal of the American Chemical Society, 141, 20004–20008. [DOI] [PubMed] [Google Scholar]
- McMurry JL, & Chang MCY (2017). Fluorothreonyl-tRNA deacylase prevents mistranslation in the organofluorine producer Streptomyces cattleya. Proceedings of the National Academy of Sciences of the United States of America, 114, 11920–11925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morimoto J, Hayashi Y, Iwasaki K, & Suga H (2011). Flexizymes: Their evolutionary history and the origin of catalytic function. Accounts of Chemical Research, 44, 1359–1368. [DOI] [PubMed] [Google Scholar]
- Murakami H, Ohta A, Ashigai H, & Suga H (2006). A highly flexible tRNA acylation method for non-natural polypeptide synthesis. Nature Methods, 3, 357–359. [DOI] [PubMed] [Google Scholar]
- Murakami H, Saito H, & Suga H (2003). A versatile tRNA aminoacylation catalyst based on RNA. Chemistry & Biology, 10, 655–662. [DOI] [PubMed] [Google Scholar]
- Ngo JT, Schuman EM, & Tirrell DA (2013). Mutant methionyl-tRNA synthetase from bacteria enables site-selective N-terminal labeling of proteins expressed in mammalian cells. Proceedings of the National Academy of Sciences of the United States of America, 110, 4992–4997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niwa N, Yamagishi Y, Murakami H, & Suga H (2009). A flexizyme that selectively charges amino acids activated by a water-friendly leaving group. Bioorganic & Medicinal Chemistry Letters, 19, 3892–3894. [DOI] [PubMed] [Google Scholar]
- Ohta A, Murakami H, Higashimura E, & Suga H (2007). Synthesis of polyester by means of genetic code reprogramming. Chemistry & Biology, 14, 1315–1322. [DOI] [PubMed] [Google Scholar]
- Ramaswamy K, Saito H, Murakami H, Shiba K, & Suga H (2004). Designer ribozymes: Programming the tRNA specificity into flexizyme. Journal of the American Chemical Society, 126, 11454–11455. [DOI] [PubMed] [Google Scholar]
- Rodnina MV (2018). Translation in prokaryotes. Cold Spring Harbor Perspectives in Biology, 10, a032664. 10.1101/cshperspect.a032664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers JM, Kwon S, Dawson SJ, Mandal PK, Suga H, & Huc I (2018). Ribosomal synthesis and folding of peptide-helical aromatic foldamer hybrids. Nature Chemistry, 10, 405–412. [DOI] [PubMed] [Google Scholar]
- Sakamoto K, & Hayashi A (2019). Synthetic tyrosine tRNA molecules with noncanonical secondary structures. International Journal of Molecular Sciences, 20, 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samhita L, Virumäe K, Remme J, & Varshney U (2013). Initiation with elongator tRNAs. Journal of Bacteriology, 195, 4202–4209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu Y, Inoue A, Tomari Y, Suzuki T, Yokogawa T, Nishikawa K, et al. (2001). Cell-free translation reconstituted with purified components. Nature Biotechnology, 19, 751–755. [DOI] [PubMed] [Google Scholar]
- Tadayoshi E, Kaori S, Takuya C, Yasunori T, Issei I, & Takahiro H (2010). Incorporation of non-natural amino acids with two labeling groups into the N-terminus of proteins. Bulletin of the Chemical Society of Japan, 83, 176–181. [Google Scholar]
- Takatsuji R, Shinbara K, Katoh T, Goto Y, Passioura T, Yajima R, et al. (2019). Ribosomal synthesis of backbone-cyclic peptides compatible with in vitro display. Journal of the American Chemical Society, 141, 2279–2287. [DOI] [PubMed] [Google Scholar]
- Tharp JM, Ad O, Amikura K, Ward FR, Garcia EM, Cate JHD, et al. (2020). Initiation of protein synthesis with non-canonical amino acids in vivo. Angewandte Chemie International Edition, 59, 3122–3126. [DOI] [PubMed] [Google Scholar]
- Tharp JM, Vargas-Rodriguez O, Schepartz A, & Söll D (2021). Genetic encoding of three distinct noncanonical amino acids using reprogrammed initiator and nonsense codons. ACS Chemical Biology, 16, 766–774. 10.1021/acschembio.1c00120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varshney U, & RajBhandary UL (1990). Initiation of protein synthesis from a termination codon. Proceedings of the National Academy of Sciences of the United States of America, 87, 1586–1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young DD, & Schultz PG (2018). Playing with the molecules of life. ACS Chemical Biology, 13, 854–870. [DOI] [PMC free article] [PubMed] [Google Scholar]