

Abstract
Trisomy 21, the presence of a supernumerary chromosome 21, results in a collection of clinical features commonly known as Down syndrome (DS). DS is among the most genetically complex of the conditions that are compatible with human survival post-term, and the most frequent survivable autosomal aneuploidy. Mouse models of DS, involving trisomy of all or part of human chromosome 21 or orthologous mouse genomic regions, are providing valuable insights into the contribution of triplicated genes or groups of genes to the many clinical manifestations in DS. This endeavour is challenging, as there are >200 protein-coding genes on chromosome 21 and they can have direct and indirect effects on homeostasis in cells, tissues, organs and systems. Although this complexity poses formidable challenges to understanding the underlying molecular basis for each of the many clinical features of DS, it also provides opportunities for improving understanding of genetic mechanisms underlying the development and function of many cell types, tissues, organs and systems. Since the first description of trisomy 21, we have learned much about intellectual disability and genetic risk factors for congenital heart disease. The lower occurrence of solid tumours in individuals with DS supports the identification of chromosome 21 genes that protect against cancer when overexpressed. The universal occurrence of the histopathology of Alzheimer disease and the high prevalence of dementia in DS are providing insights into the pathology and treatment of Alzheimer disease. Clinical trials to ameliorate intellectual disability in DS signal a new era in which therapeutic interventions based on knowledge of the molecular pathophysiology of DS can now be explored; these efforts provide reasonable hope for the future.
Down syndrome (DS) is the most common genomic disorder of intellectual disability and is caused by trisomy of Homo sapiens chromosome 21 (HSA21). The eponym of the syndrome is from Down, who described the clinical aspects of the syndrome in 1866 (REF.1). The DS phenotype involves manifestations that affect multiple bodily systems, in particular the musculoskeletal, neurological and cardiovascular systems. Individuals with DS commonly have short stature, muscle hypotonia, atlantoaxial instability, reduced neuronal density, cerebellar hypoplasia, intellectual disability and congenital heart defects (CHDs; particularly atrioventricular septal defects (AVSDs)). Individuals with DS are also more likely to develop certain health conditions, including hypothyroidism, autoimmune diseases, obstructive sleep apnoea, epilepsy, hearing and vision problems, haematological disorders (including leukaemia), recurrent infections, anxiety disorders and early-onset Alzheimer disease (AD) (FIG. 1). Other conditions, such as most solid tumour types, show inverse comorbidity and seem to be less common in individuals with DS than in the general population2.
Fig. 1 |. Symptoms and manifestations in Down syndrome.
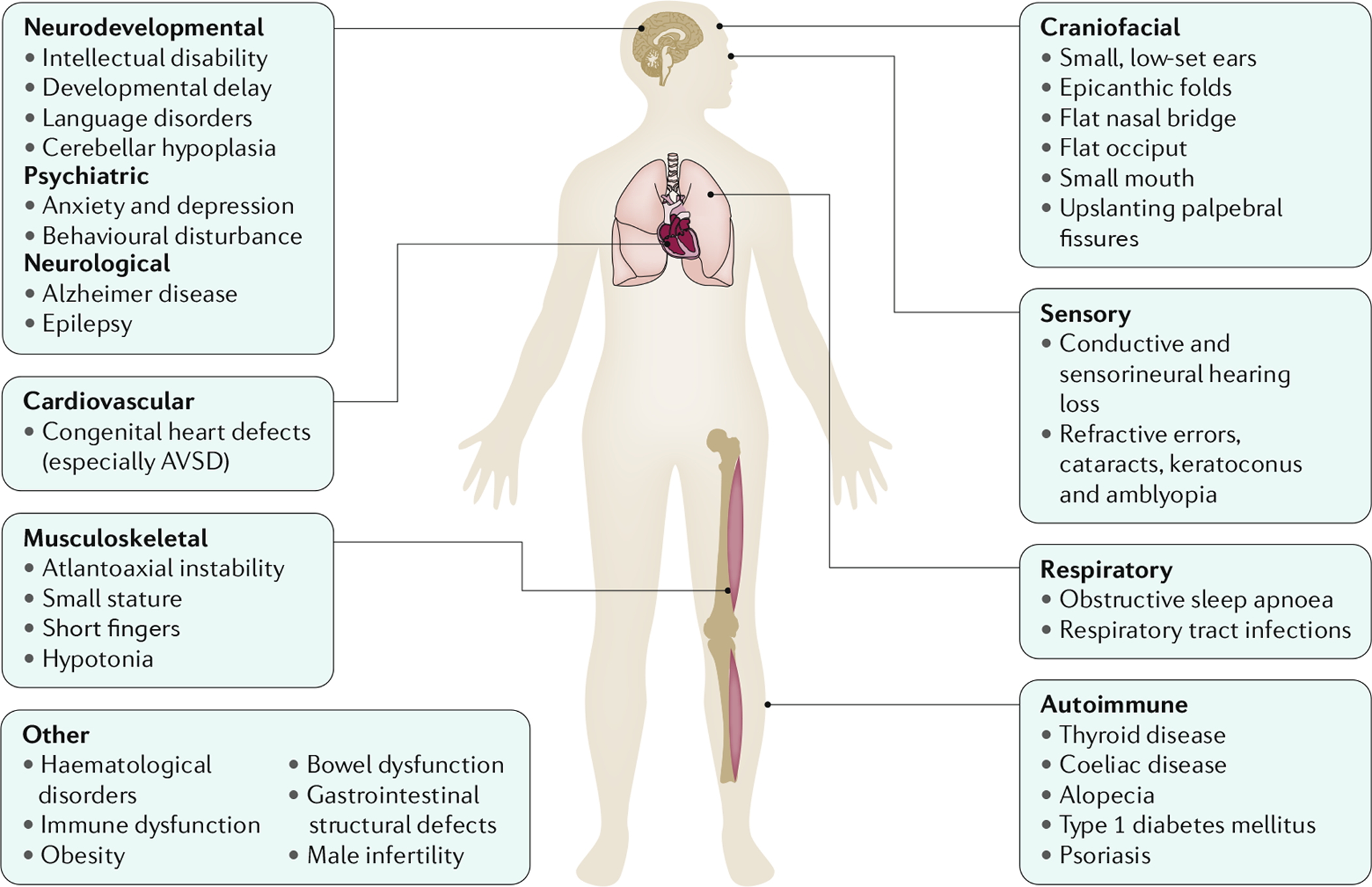
Individuals with trisomy 21 (the presence of a supernumerary chromosome 21; also known as Down syndrome (DS)) present with a distinct collection of symptoms and manifestations that affect multiple body systems, although variation exists between individuals. Individuals with DS are generally of short stature, with short fingers, hypotonia and atlantoaxial instability. Facial characteristics include the presence of epicanthic folds, flat nasal bridge and occiput, small mouth and ears, and up-slanting palpebral fissures. Congenital heart defects are common, particularly atrioventricular septal defect (AVSD). Individuals with DS are also more likely to develop certain health conditions compared with the general population, including hypothyroidism, obstructive sleep apnoea, epilepsy, hearing and vision problems, haematological disorders (including leukaemia), recurrent infections, anxiety disorders, and early-onset Alzheimer disease.
The discovery of a link between a supernumerary chromosome 21 and the DS phenotype was first reported in 1959 (REF.3) and was an important landmark for the development of genetic medicine. Mouse models for the study of DS were first developed in 1990 (REF.4), and the complete nucleotide sequence of the long arm of HSA21 was published in 2000 by a multinational consortium of investigators5. Substantial progress has been made in the ensuing 19 years in understanding the molecular pathophysiology of the different phenotypic manifestations of DS, which is currently considered a disorder of gene expression dysregulation. In addition, widely used screening methods have been introduced for the prenatal detection of DS. The management of the different symptoms and the quality of life of individuals with DS have improved. However, enormous challenges remain, including understanding the precise biological mechanism of each phenotypic component of the syndrome; treatment of the different symptoms, including cognitive dysfunction; and integration of individuals with DS into society in different parts of the world. Furthermore, studies in animal models to unravel the effects of triplication of the >200 protein-coding genes on HSA21, not to mention the triplicated non-coding genes and the downstream and indirect effects of these alterations, are extremely challenging.
In this Primer, we discuss the epidemiology of DS, current understanding of genetics and pathophysiology resulting from the extra chromosome 21, and advances in the diagnosis of trisomy 21. Furthermore, we review the management of the syndrome and the quality of life of individuals with DS, and offer an outlook for the future. Several reviews of DS and trisomy 21 have been published elsewhere and provide additional details about this common and challenging syndrome6–9.
Epidemiology
Prevalence
The lifetime prevalence of DS is increasing substantially as the global population grows. For example, in the USA, the population prevalence of DS increased from ~50,000 in 1950 (3.3 per 10,000 individuals) to ~212,000 in 2013 (6.7 per 10,000 individuals; based on unpublished data10), mostly due to improvements in childhood survival of individuals with DS11 (FIG. 2). The life expectancy of individuals with DS in the USA increased from an estimated mean of 26 years and median of 4 years in 1950 to 53 years and 58 years, respectively, in 2010 (REF.11). As of 2015, estimates of DS population prevalence have been reported for Europe (4.9 per 10,000 individuals)12, Europe excluding former Eastern Bloc countries (6.0 per 10,000 individuals), and former Eastern Bloc countries (3.3 per 10,000 individuals)12. However, a precise global estimate cannot be reliably calculated until more birth registries are created within countries and more data are available on historical and current survival of individuals with DS in different parts of the world.
Fig. 2 |. Prevalence of DS and pregnancy outcomes in the USA.
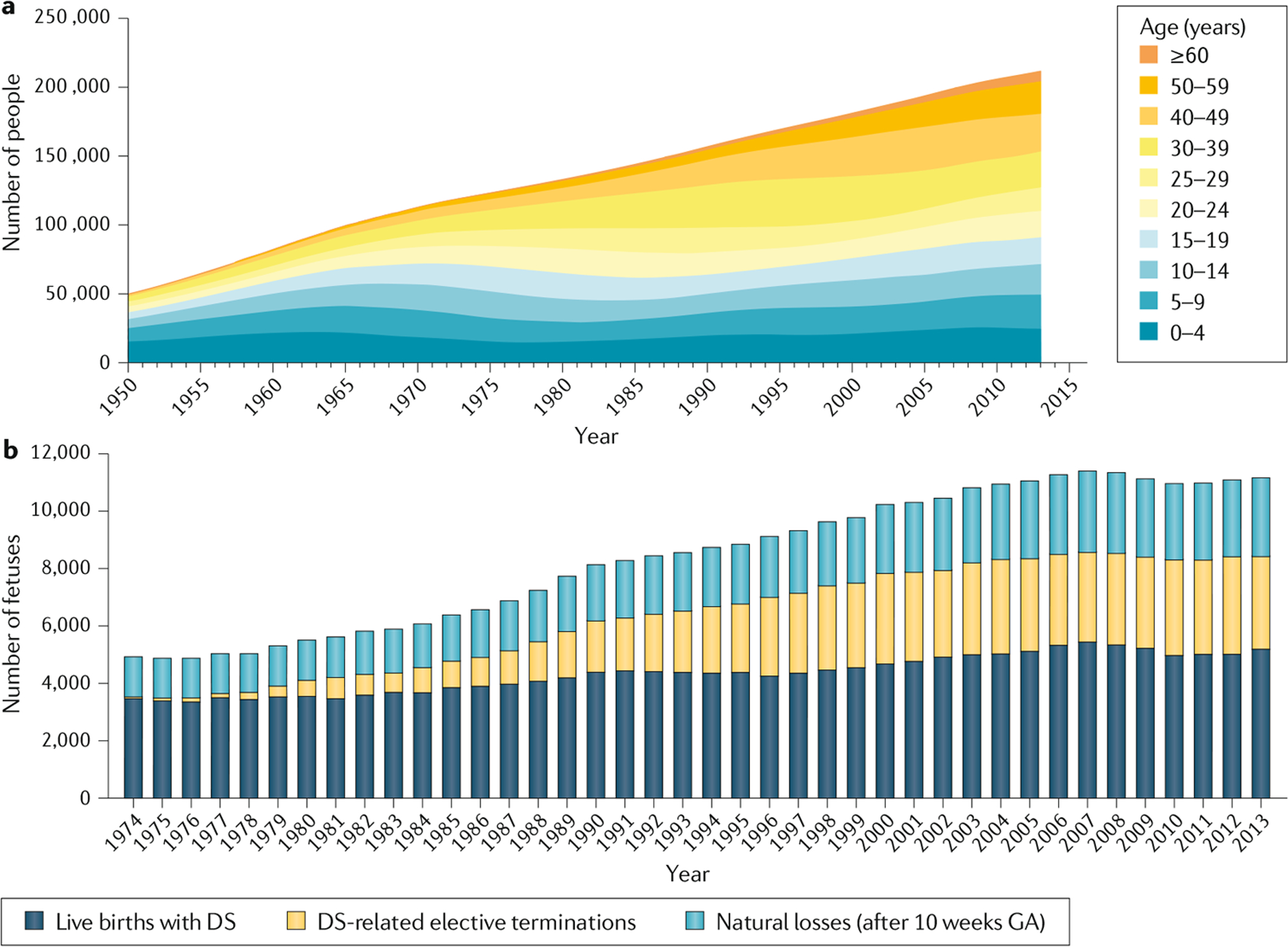
a | Prevalence of Down syndrome (DS) in the USA for the period 1950–2013. This graph combines the prevalence data for 1950–2010 in the USA9 with unpublished prevalence data for 2011–2013 for the same region10. b | Pregnancy outcomes in the USA for the period 1974–2013. This graph combines estimates of live births, natural losses and elective terminations in women carrying a fetus with DS after 10 weeks gestation for the period 1974–2010 in the USA17 with unpublished data for 2011–2013 (REF.10). GA, gestational age. Part a adapted from REF.11, Springer Nature Limited. Part b adapted with permission from REF.17, Wiley-VCH.
DS occurs in all populations, but differences in maternal age at conception between countries and ethnicities influence the number of live births13–16. As of 2013 in the USA, unpublished data suggest that ~1 in every 779 babies born had DS10 (~12.8 per 10,000 live births) (FIG. 2a). The prevalence of DS is influenced by maternal age at conception, which varies among countries, and is estimated to be ~1 in 365 fetuses at 10 weeks gestation17 (FIG. 2b). Some of these pregnancies will result in spontaneous miscarriage, ~32% between 10 weeks gestation and the expected date of birth and ~25% between 16 weeks gestation and the expected date of birth, the risk depending on maternal age, as estimated from reports from England and Wales18. The number of pregnancies that are electively terminated is influenced by the availability and accuracy of screening tests within each country, the number of people choosing prenatal screening and subsequently prenatal testing, and parental decisions once a prenatal diagnosis of DS is made. In 2013, an estimated 3,400 DS-related elective terminations were carried out in the USA, resulting in a 33% reduction in the number of babies with DS that would have been born that year. By comparison, the estimated percentage reduction was 55% in Australia for 2004 (REF.19), and 54% in Europe, 66% in Europe excluding the former Eastern Bloc countries and 32% in former Eastern Bloc countries, for the period 2010–2015 (REF.12). In China, during the period 2003–2011, the termination rate for fetuses with DS led to a 55% reduction in the overall perinatal prevalence20.
Studies from England and Wales21, Slovenia22, Australia19,23 and EUROCAT regions24 have shown that increasing maternal age has counterbalanced the uptake in prenatal screening, resulting in a stable or slightly decreasing prevalence of DS in live births from the 1990s until now. In Europe, DS prevalence in live births has been decreasing slightly since the 1990s, although there are substantial regional differences12. In southern Europe, DS prevalence in live births almost halved in the period 1980–2015 (REF.12). By contrast, in the Netherlands, the live birth prevalence increased slightly in the period 1980–2000 but seems to have been decreasing slightly since 2005 (REF.25). In the USA, the increased prevalence of DS in live births began in the 1980s and levelled off after 2005 (REF.17). Sequencing cell-free placental DNA in maternal plasma, termed noninvasive prenatal screening (NIPS) or noninvasive prenatal testing (NIPT), has been available since 2011 in many countries; however, not enough time has elapsed for the full impact of surveillance programmes on birth rates to be measured. Furthermore, variability in access to prenatal care is also expected to affect the uptake of surveillance programmes.
Risk factors
Advanced maternal age at conception is a major risk factor for trisomy 21, as is true for all human autosomal trisomies26. This risk is associated with non-disjunction of homologous chromosomes or chromatids occurring during the meiotic divisions that occur in the formation of oocytes27. Advanced maternal age has been associated with HSA21 segregation errors in both maternal meiosis I and meiosis II28–32. In addition, specific altered recombination patterns have been observed for these types of maternal errors, only some of which are associated with maternal age33.
Meiotic processes, such as recombination, involve the action of many genes. Thus, there is a clear rationale for examining whether genetic variants predispose to meiotic nondisjunction in humans. A study including a candidate gene analysis and an untargeted genome-wide association study (GWAS) of HSA21 nondisjunction in mothers who gave birth to an infant with trisomy 21 derived from a maternal error was conducted using the mothers as cases and the fathers as controls34. These analyses were stratified by maternal MI or MII errors. Results from this study emphasize the heterogeneity of risk factors based on type of error. For example, variants in candidate genes coding for components of the synaptonemal complex showed an association limited to meiosis I errors (for example, SYCE2) while others were associated with MII errors (for example, SYCP2)34.
Environmental factors also influence the risk of non-disjunction but are difficult to identify due to the inherent problem of defining the exposure, dosage and timing of each factor. Again, studies must define the type of error (parental origin and the type of meiotic or mitotic error) leading to trisomy 21 due to their heterogeneous aetiology. Environmental factors that influence the risk of trisomy 21 include tobacco use, folic acid supplementation, oral contraceptive use and several others (the studies investigating environmental risk factors and their limitations, and possible biomarkers of exposure such as telomere length, have been reviewed elsewhere35).
For example, maternal socioeconomic status (SES) is associated only with maternal meiosis II errors36–39. As SES is a proxy for specific exposures, a follow-up study examined maternal occupation as a risk factor and found that some job classification categories were more prevalent in mothers of infants with DS than in mothers of infants with no chromosomal abnormality or major birth defect, and were associated with specific types of meiotic errors40. A preliminary analysis revealed that these occupations involved exposure to solvents in the work environment. Further studies of this kind are needed to examine specific exposures to toxic agents in the work and home environments and their relationship with maternal and paternal HSA21 nondisjunction. This is emphasized by the finding that exposure to endocrine-disrupting chemicals affects meiosis and increases the prevalence of aneuploidy41. For example, exposure to the ubiquitous environmental contaminant bisphenol A (BPA) and other endocrine-disrupting chemicals affects the reproductive system in both sexes (including the ovaries, testes and reproductive tract)41,42.
It is important to point out that not only do different types of meiotic and mitotic errors leading to aneuploidy most likely have different susceptibility to specific environmental exposures but that exposures of multiple generations (for example, grandmother and mother) must also be considered35,43. For example, there is now mounting evidence of transgenerational effects of BPA exposure on sperm and oocytes in experimental models42.
Mechanisms/pathophysiology
Genetics of DS
Partial or complete trisomy 21 (that is, the presence of part of or a complete supernumerary HSA21) is the genomic cause of DS3. A free trisomy 21 is present in 95% of individuals with DS and results from an error in maternal meiosis I (~66%) or meiosis II (~21%); paternal meiosis I (~3%) or meiosis II (5%); or mitosis, after the formation of the zygote (5%)15,44,45. Translocation accounts for trisomy 21 in ~5% of affected individuals, usually t(14;21) or t(21;21)46,47. Mosaicism for trisomy 21 occurs in ~2% of individuals with DS. Partial trisomy 21 is rare48,49 and is associated with a range of symptoms that vary according to the length of the partial triplication of HSA21.
The first published sequence of HSA21 annotated 225 genes on chromosome 21q5. With increased knowledge of the elements of coding and non-coding genes and regulatory motifs, the number of gene structures recognized on HSA21 has increased considerably. The strengths and limitations of genome annotations available in existing public databases have been reviewed elsewhere50. The current version of GENCODE/ENSEMBL (GENCODE release 32) lists 233 protein-coding genes, 423 non-protein-coding genes (69 small, 330 long and 24 miscellaneous non-coding genes) and 188 pseudogenes. Of note, 48% of HSA21 has not been annotated6, the vast majority of which contains repetitive elements (as is the case for all human chromosomes).
Understanding the genetic aetiology of the increased susceptibility of individuals with DS to the multiple manifestations or conditions that are associated with the DS phenotype is an enormous challenge51. Compounding the difficulty of identifying the specific genetic and other effects of trisomy 21, studies of DS in humans are restricted to those conceptuses that survive to term; among pregnancies with a confirmed prenatal diagnosis of DS at 9–14 weeks, ~30–40% subsequently spontaneously miscarry18,52. Extrapolating to all pregnancies, ~80% of trisomy 21 conceptuses are lost during pregnancy53. Thus, it is essential to acknowledge that in studying any DS-associated condition, only the combinations of genetic variants that allow the conceptus to survive to term and to manifest the phenotype of interest are being considered.
Two main hypotheses have been proposed to explain the biological perturbations that underlie the phenotypic manifestations of DS: first, a specific HSA21 gene-dosage effect, which includes both the direct effects of overexpressed HSA21 genes and the downstream consequences of this overexpression; and second, developmental instability, in which nonspecific global disturbance of gene expression from the extra HSA21 results in disruption of overall biological homeostasis54,55. It is likely that the aetiology of DS-associated phenotypes involves both proposed mechanisms. Single-cell transcriptome analyses in trisomy 21 and other trisomies have suggested that the gene dosage effect for trisomic genes with low-to-average expression is mainly due to the higher fraction of trisomic cells expressing these genes56. This results in the expected 1.5-fold higher average expression of trisomic genes in the various tissues of individuals with trisomy 21.
Gene dosage effects.
The simplest effect of HSA21 trisomy is the direct effect of an increased dosage of a single HSA21 gene. For example, an increased dosage of APP, an HSA21 gene that encodes amyloid precursor protein (APP), increases susceptibility to early-onset AD in individuals with DS. Although APP is clearly an ‘effector’ gene, whether only the direct and downstream effects of APP overexpression affect the penetrance and severity of AD, or whether other aspects of trisomy 21 also have a role, remains to be determined. Of note, returning the App copy number to two copies in a mouse model of trisomy 21 (Ts65Dn) alleviates some but not all of the effects of App dosage57.
To define the direct and indirect consequences of increasing gene dosage, many studies have characterized the expression profile of HSA21 genes and the effect of overexpressing these genes on the rest of the genome. Common findings among these studies emerge. First, the expression of most but not all HSA21 genes is increased. It will be important to identify the mechanisms that underlie these discordant expression levels, which may include negative feedback, dosage compensation and epigenetic alterations. Second, in many affected individuals, the expression of some non-HSA21 genes is also altered, which suggests that trisomy 21 leads to perturbation of downstream transcription and signalling networks. A meta-analysis compared the expression profiles of trisomy 21 and euploid samples from various human tissues (such as brain and thymus) or cells (such as lymphoblastoid cell lines (LCLs), blood cells, fibroblasts and induced pluripotent stem cells (iPSCs))58. This analysis found that the trisomy 21:euploid expression ratio was typically close to 3:2 for dysregulated genes and close to 1:1 for unaffected genes. These data are consistent with the hypothesis that upregulation of effector genes (that is, those that enhance or suppress gene expression) on HSA21 leads to similar changes in the expression of associated downstream genes, resulting in a similar altered ratio of 3:2 or 2:3 for these genes. There are exceptions to this pattern, with more extreme ratios that may result from gene–gene interactions or other types of chain effects that amplify the final expression levels. For example, JAKMIP3 is highly upregulated (256.13:1 ratio) in the thymus transcriptome map, whereas BEX5 is highly downregulated (0.07:1 ratio) in the brain transcriptome map.
Finally, as expected, the expression patterns of both HSA21 genes and non-HSA21 genes in trisomy 21 cells differ depending on the tissue being examined. For example, the interferon response is highly upregulated in trisomy 21 fibroblasts and LCLs, most likely due to increased expression of four interferon receptor genes (IFNAR1, IFNAR2, INGR2 and IL10RB) that are located on HSA21 (REF.59). An increased interferon response was also present in iPSCs generated from trisomy 21 fibroblasts but not in neuronal cultures derived from these iPSCs60.
Disruption of homeostasis by genome-wide effects on transcription regulation.
Trisomy 21 may also affect global transcription, either directly if an HSA21 gene functions in transcription regulation or indirectly as a by-product of the additional genetic material61,62. For example, at least three HSA21 genes encode proteins involved in transcription regulation: ADARB1 (one of two proteins involved in adenosine-to-inosine RNA editing), the constitutive splicing factor U2AF1, and DYRK1A, a dual specificity kinase that phosphorylates splicing factors60. Altered ADARB1 transcript levels did not change global adenosine-to-inosine editing levels in trisomy 21 iPSC-derived neuronal cells, whereas trisomy 21-dependent splicing changes were observed in both iPSCs and the neuronal cultures derived from them60.
The possibility that increased genetic material could lead to altered genome-wide gene expression was suggested based on the observation that genes whose expression is similarly altered (that is, upregulated or downregulated) are clustered in regions termed gene expression dysregulation domains (GEDDs) in trisomic iPSCs and fibroblasts61. However, other studies using a range of methods found no evidence for the existence of GEDDs specifically in trisomy 21 cells63,64.
Studies of nuclear genome organization using three-dimensional fluorescence in situ hybridization (3D-FISH) have shown that chromosomes are preferentially localized to discrete regions within the nucleus, termed chromosome territories (reviewed elsewhere65). Homeostasis in trisomic cells can be disrupted by the alteration of chromosome territories. Although studies of chromosome territories in trisomy 21 cells and their effects on cellular processes are just beginning, initial studies have found that an extra HSA21 does not change the overall organization of chromosome territories in the interphase nucleus66 but does alter chromosome compaction and displaces other chromosome territories from their usual nuclear position66.
Trisomy 21 also alters regional and/or global methylation patterns (reviewed elsewhere62), although the methylation changes are evenly distributed on all chromosomes and are not specifically enriched on HSA21. One observed pattern is an overall bias towards hypermethylation in DS cells compared with euploid cells, particularly in brain samples67–69. Furthermore, differential methylation in trisomy 21 cells seems to be localized to discrete regulatory regions of single genes and not to domain-like regions. A meta-analysis identified a small set of genes for which the methylation patterns were different between trisomy 21 and euploid cells in all tissues examined, suggesting that these altered methylation patterns are established early in development and thereby persist in multiple tissue types in DS62.
Two mechanisms have been proposed for the globally altered methylation patterns in trisomy 21. First, some triplicated HSA21 genes might function directly in methylation pathways, including SLC19A1, FTCD, GART, CBS, PRMT2, N6AMT1, MIS18A and DNMT3L. SLC19A1, FTCD, GART and CBS all have a role in the one-carbon metabolism pathway, which is central to DNA methylation. PRMT2 encodes a protein arginine methyltransferase with multiple targets, including histones70. N6AMT1 encodes a methyltransferase responsible for DNA N6-adenosine methylation71. The protein encoded by MIS18A is involved in maintaining the heterochromatic state of centromeres by recruiting the DNA methyltransferases DNMT3A and DNMT3B, thereby repressing the production of non-coding transcripts from centromeric satellite repeats72. DNMT3L also interacts with DNMT3A and DNMT3B and enhances their de novo methylation activity, but lacks DNA methyltransferase activity itself73. DNMT3L is overexpressed in DS neuroprogenitors, which leads to increased expression of APP and PSD95 in differentiating neurons74. These examples illustrate the various mechanisms by which overexpression of these HSA21 genes may lead to trans-epigenetic changes in DS.
The second proposed mechanism is that differential methylation is a result of altered transcription factor occupancy of their binding sites. For example, RUNX1, an HSA21-encoded transcription factor, is overexpressed in trisomy 21 T lymphocytes and differentially methylated sites in trisomy 21 cells are enriched for the RUNX1 binding motif67. Thus, the higher occupancy of RUNX1 binding sites in trisomy 21 cells affects CpG methylation at these sites. Additional support for this hypothesis comes from the observation that several types of sequence motifs, including the binding site for CTCF (an insulator protein that blocks the interaction between enhancers and promoters), are enriched at loci that are differentially methylated in all trisomy 21 tissues examined67,75. Because CTCF binding to its recognition site is sensitive to methylation, the pattern of CTCF occupancy may be particularly affected by DS-associated epigenetic perturbations, which may result in altered 3D conformation of chromatin in the nucleus.
Histone deacetylation and acetylation are other epigenetic mechanisms that are potentially influenced by triplication of some HSA21 genes. For example, DYRK1A, which is overexpressed in trisomy 21, promotes histone deacetylation by phosphorylation and activation of the deacetylase SIRT1 (REF.76). Furthermore, DYRK1A affects chromatin remodelling through its interaction with the transcription repressor REST (also known as NRSF)77, resulting in neuronal gene dysregulation that might contribute to the neural phenotypic changes associated with DS.
Although genomic and transcriptomic data are important for understanding the consequences of trisomy 21 and how they relate to interindividual variation in clinical manifestations, integration with other ‘omics’ datasets is necessary to obtain a complete picture of the effects of trisomy 21. For example, in a proteomic study78 using a method that analysed a large fraction of the proteome79 of fibroblasts from a pair of monozygotic twins discordant for DS (with follow up on samples from 11 unrelated individuals with DS and matched controls), extensive changes were detected in the levels of proteins encoded by HSA21 genes and non-HSA21 genes. In transcriptome data from both of these twins61, steady-state transcript levels were moderately correlated with protein levels. However, the trisomy 21:euploid ratios for gene expression and protein levels were only weakly correlated (Spearman rank correlation ρ = 0.34–0.51), which was mostly due to incongruence of transcript and protein levels for non-HSA21 genes. Thus, substantial post-transcriptional regulation has a role in the differential effect of trisomy 21 on the expression levels of different genes. For example, for HSA21-encoded proteins that are components of heteromeric protein complexes with non-HSA21-encoded proteins, protein degradation seems to buffer against increased transcript levels. Gene enrichment set analyses have shown that protein levels are substantially altered for cell cycle-related functions, cell morphogenesis, lipoprotein metabolism and cellular respiration in mitochondria78.
In another study80 using a proteomics method focused primarily on secreted proteins and those with extracellular domains81, plasma samples from 165 individuals with DS and 98 euploid individuals were compared. Among the proteins that were dysregulated in trisomy 21 samples, many are involved in immune control, the complement cascade and growth factor signalling. However, no clear link exists between the identity or levels of dysregulated proteins and specific clinical manifestations of DS.
Indicators of mitochondrial dysfunction have been observed in trisomy 21 cells (for example, fibroblasts) and organs, including the heart (their properties and consequences have been reviewed elsewhere82–84). The mitochondrial phenotype in DS includes reduced ATP production by oxidative phosphorylation; decreased respiratory capacity; impaired generation of mitochondrial membrane potential; and alterations in mitochondrial structure. These altered functions result in perturbed mitochondrial energy metabolism and oxidative stress, which in turn could increase susceptibility of individuals with DS to a wide range of conditions, including intellectual disability and AD. The molecular basis for mitochondrial dysfunction involves effector HSA21 genes and key regulatory signalling pathways (reviewed elsewhere82). Of 77 HSA21 genes consistently dysregulated identified by a meta-analysis of 45 DS gene expression studies85, NRIP1, SUMO3, DYRK1A, RCAN1, SOD1, APP and CBS are directly or indirectly involved in mitochondrial function and thus represent candidates for the observed DS-associated mitochondrial phenotypes.
Animal models
Animal models of DS, especially mouse models, have been instrumental in advancing DS research. However, a caveat when studying these models is that DS is a human condition that cannot be precisely replicated in other species. Nonetheless, insights into the mechanisms that underlie the developmental and functional consequences of trisomy 21 have been obtained by overexpression of groups of HSA21 genes or orthologues of HSA21 genes from other species. Important questions about the usefulness of animal models for studying trisomy 21 pathophysiology include the question as to what types of studies are informative in mice, whether results in mice are relevant to understanding human outcomes of trisomy 21 and whether mouse models of DS can be used as a translational platform for drug testing.
Mouse models have transformed basic research in DS, beginning with development of the Ts65Dn mouse, which contains a partial trisomy of Mus musculus chromosome 16 (MMU16)4,86. Prior to the introduction of this mouse model, the identification, cloning and expression of individual genes in transgenic mouse models was laborious, and studies using these approaches led to wide-ranging conclusions about individual genes that could ‘cause’ DS. However, overexpression of dozens or hundreds of genes simultaneously, supported by more precisely defined terminal phenotypes, has enabled a more realistic interpretation of studies of development, function and genome-wide perturbations in gene expression.
To understand the challenges of using mouse models, it is necessary to understand the complexity of trisomy 21. Trisomy 21 has profound phenotypic effects, as HSA21 contains >200 protein-coding genes and ~400 additional transcripts of known or presumed importance, which are present in an extra copy in every cell from conception onwards. Conversely, the gene overexpression changes in trisomy 21 are subtle, with an average increase in expression of ~1.5-fold in trisomic cells compared with euploid cells. Expression levels of individual genes vary widely and mRNA levels do not always correlate with the levels of the corresponding protein78. For genes expressed at a low level, this difference cannot be reliably measured with precision in many high-throughput assays. The consequences of this modest overexpression are substantial, as up to 80% of conceptuses with trisomy 21 do not survive to term15.
Relevance of mouse phenotypes to DS.
Several phenotypic features of DS manifest (to varying extents) in all individuals with trisomy 21, including cerebellar hypoplasia, retrusion of the midface skeleton and mandible, Alzheimer-disease-type histopathology at an early age, and intellectual impairment. The risk of CHDs is greatly increased in individuals with DS. Whereas adults with DS show an overall lower risk of developing solid tumour cancers, including breast cancers, when compared to the general population, children with DS have an increased incidence of leukaemia2. All of these features are present in some form in the animal models of DS that have been generated to date. For example, whereas the overt histopathology of AD (plaques and tangles) does not occur in mouse models of trisomy 21, accumulation of tau protein and endosomal changes have been reported in these mice51.
An overall finding from animal models of DS is that the penetrance (occurrence) and expressivity (severity) of a given phenotype is a function of overexpression of multiple genes; DS is not a collection of independent single gene effects. However, many mouse models that are trisomic for different subsets of mouse orthologues of HSA21 genes have been used effectively — especially in combination with individual gene knockouts that allow specific genes to be returned to the normal two copies in a trisomic background — to identify genes with major effects that are important for understanding a number of phenotypes57,87,88. Rather than asking the simplistic question of whether a gene causes DS, different mouse models provide information about whether altered expression of a specific gene or genes is necessary and/or sufficient to contribute to a phenotype. Interpretation is then limited by the ability to quantify the effects of trisomy in an animal model. Saying that a given model has incompletely penetrant CHDs is of little use in understanding the contributions of specific genes to specific outcomes; failure of ventricular or atrial septum development, AVSDs or outflow tract anomalies all occur in different developmental fields involving different cells at different stages of heart development with the contribution of different genes.
Genetic basis of animal models.
The extent of the triplicated region of HSA21 or the orthologous mouse genomic regions (that is, a segment of a mouse chromosome) is an important consideration in determining the relevance of a mouse model of DS. Trisomy for mouse genomic regions orthologous to HSA21 has the advantage that these regions are subject to endogenous transcription regulation, whereas genetic variation in, for example, promoter or enhancer sequences between mouse and human might be expected to affect transcription. Similarly, any normal human-specific variation in processing signals in DNA and RNA, or amino acid substitutions in proteins, could affect the stoichiometry of interactions between HSA21-encoded proteins and non-HSA21-encoded proteins. Data from GWAS and RNA sequencing suggest that these effects are small, but their potential importance cannot be dismissed. Conversely, although the gene content of HSA21 and the orthologous segments on MMU16, MMU17 and MMU10 are similar, they are not identical (FIG. 3). Of the 233 predicted protein-coding genes on HSA21, only 168 are well conserved in mice. Furthermore, the difference in number and sequence between human and mouse in the non-coding transcripts of presumed function is even greater. The limitations of individual mouse models are emphasized in a comparative study of three of these models, Ts65Dn, Ts1Cje and Ts1Yey89.
Fig. 3 |. Conserved synteny of human chromosome 21 with mouse chromosomes and mouse models of trisomy 21.
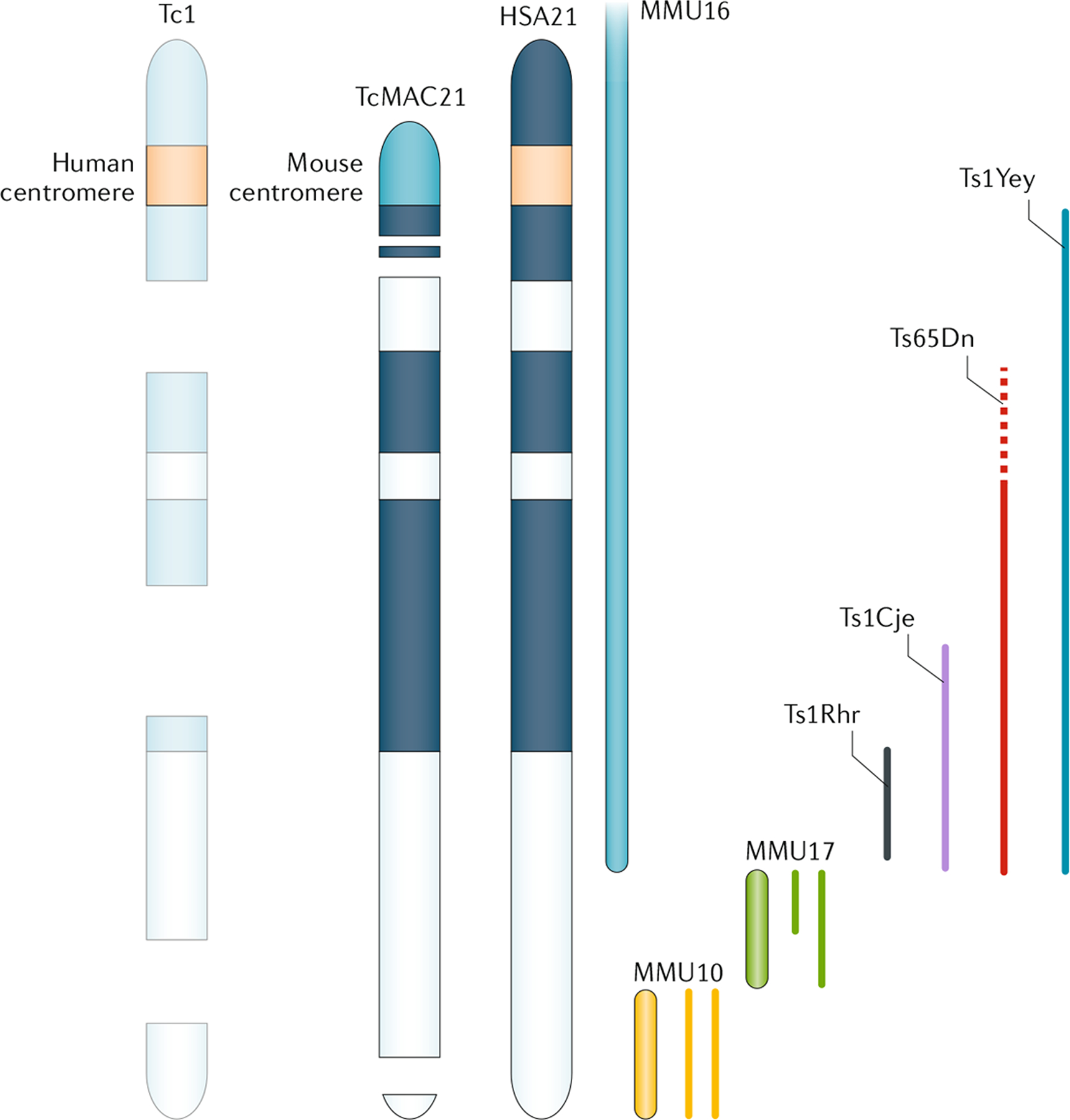
Down syndrome (DS) results from the presence of a supernumerary Homo sapiens chromosome 21 (HSA21). More than 20 mouse models of DS have been created, which are designed to overexpress part of or a complete HSA21, or the orthologous mouse genomic regions7,91,246. Mouse orthologues of HSA21 genes occur on Mus musculus chromosome 16 (MMU16), MMU17 and MMU10. Tc1 mice carry a mutated HSA21 and are mosaic animals, with a mix of trisomic and euploid cells that is unique to each individual, apparently due to suboptimal function of the human centromere in mice. Ts65Dn animals contain a duplication of ~140 genes on MMU16, some of which are not orthologous to HSA21247,248 (dashed line). TcMAC21 animals contain the long arm of HSA21 (HSA21q) as a mouse artificial chromosome (that is, with a mouse centromere to ensure that the chromosome is retained in every cell); however, this artificial chromosome contains deletions that affect ~8% of HSA21q genes. All of these models except Ts65Dn are direct duplications; that is, the genes in each are trisomic but they do not contain an extra chromosome or centromere. Ts1Rhr, Ts1Cje, Ts1Yey and Ts65Dn mouse models are discussed in the text.
These differences have not been emphasized in the past, as only one mouse model (Tc1) that contained a freely segregating HSA21 existed. To date, Ts65Dn is the only reported mouse model of DS that segregates an extra mouse chromosome; all other models are made by direct duplication of a mouse chromosome segment that is orthologous to HSA21 (detailed mapping of the partial mouse trisomies of segments orthologous to HSA21 has been reviewed elsewhere6,7). Genome sequencing in Tc1 mice has revealed substantial deletions, mutations and duplications of HSA21 that compromise the expression of >20% of HSA21 genes in these mice90. However, the biggest drawback of Tc1 is the spontaneous loss of the entire HSA21 in a substantial proportion of cells, resulting in mosaic trisomy (that is, each animal is a unique mosaic of trisomic and euploid cells). As development is driven and regulated through cell–cell communication, many aspects of an individual are liable to vary if neighbouring cells suddenly change ploidy. As the loss of HSA21 is random, the development of every animal is unique. Consequently, these rearrangements and mosaicism necessitate cautious interpretation of studies using Tc1 mice. A new model containing 92% of the protein-coding genes on the long arm of HSA21 as a mouse artificial chromosome avoids the problem of mosaicism and has many of the DS manifestations of trisomy 21 that have been observed in previous mouse models91.
The demonstration that directed interchromosomal recombination could be achieved using the Cre–Lox system paved the way for the creation of new mouse models for dosage imbalance studies92,93. To date, >20 mouse models that are trisomic for different HSA21-homologous portions of MMU16, MMU17 and MMU10 have been created7 (FIG. 3). A set of three mouse strains that together contain a complete duplication of all mouse genomic regions orthologous to HSA21 has been described94. In theory, a three-way cross of these mouse strains should produce ‘full trisomy’ in one out of eight offspring. Although this low frequency is only acceptable for some applications, in practice, the frequency of full trisomy is far lower than the predicted Mendelian frequencies, making models such as Tc1 or the MAC21 mice the most viable options for maximum gene representation.
Whereas 95% of individuals with DS have an extra freely segregating HSA21 and therefore an extra centromere and telomeres, the segmental duplication mouse models of DS do not. In these models, trisomy occurs through direct intrachromosomal duplications so that chromosome (and centromere) number is unchanged. One effect of an extra centromere in mammals is reduced male fertility, which is observed in human males with DS and in mouse models of DS. The exact mechanism of this is not well understood95. Furthermore, trisomic cells seem to proliferate more slowly and/or have a prolonged cell cycle compared with euploid cells96,97. Although the results of many of these studies are contradictory, it is feasible that very small changes in the cell cycle could have a major effect on development. As human development requires 43 population doublings to progress from a single cell to the estimated one trillion cells in a fetus (if all of the cells survived — the actual number is clearly much higher), a very small increase in cell cycle length could result in recognizable phenotypes, such as small stature, short fingers, skeletal retrusion, decreased neuronal density in the brain and hypocellular tissues.
Pathophysiological insights from animal models.
In bio-medical research, mouse models provide an important translational tool to identify and prioritize therapeutic approaches to ameliorate the effects of a condition. Efforts to correlate DS-like phenotypes in mice with trisomy of specific genes (and groups of genes) have led to the development of more than 20 mouse models with partial trisomy7 (FIG. 3). Although the mouse provides a good mammalian genetic model for understanding DS pathogenetic mechanisms, it is important to keep in mind that mice do not develop DS. However, mouse models do illustrate several fundamental principles of gene effects in trisomy and preclinical pharmacological amelioration seems promising, especially for cognitive effects. Trisomic genes that contribute to a deleterious phenotype are targets for drug development to ameliorate features of DS. To illustrate the application of mouse models, examples of the use of mouse models to identify the genetic basis of trisomy effects in brain function, heart development and cancer risk are discussed.
Many preclinical studies emphasize improving cognitive function, possibly the most difficult target for pharmacological intervention. Ts65Dn and many subsequent models exhibit learning and memory deficits and electrophysiological changes consistent with altered synaptic function in the hippocampus86,98,99. Interestingly, Ts65Dn mice showed a significant learning deficit compared with Ts1Cje mice in a standard memory test57. Ts65Dn is trisomic for a larger number of genes than Ts1Cje (FIG. 3), one of which is APP. Returning APP copy number to two by crossing a null allele of APP into the Ts65Dn background substantially improved the performance of these mice, although not to the level of a euploid mouse.
CHDs are present in ~50% of babies born with DS and mouse models display some analogous developmental deficits, albeit at a lower frequency than in human babies100. To identify a gene or genes that cause CHDs when present in three copies, seven (partial or complete) trisomies of the ~23 Mb region of MMU16 orthologous to HSA21 were studied101, which narrowed down the region responsible for causing CHDs (especially septal defects reminiscent of those in DS) to a 4.9 Mb portion of MMU16. However, three smaller trisomies subdividing the 4.9 MB segment did not have heart defects, suggesting that at least two genes must be trisomic to cause CHDs. In a different approach, interactions between trisomy and disomic modifiers of CHD risk were examined. A mouse hemizygous for Creld1 (a gene implicated in familial AVSD) was crossed with Ts65Dn and Ts1Cje mice102. Whereas Creld1+/− mice had normal heart development, the occurrence of septal defects was markedly increased in Creld1+/− Ts65Dn mice but not in Creld1+/− Ts1Cje mice. Of the genes that are trisomic in Ts65Dn but not in Ts1Cje, 14 are expressed in heart development, including junctional adhesion molecule 2 (Jam2), which consistently increases the frequency of heart abnormalities when overexpressed in zebrafish103. Restoration of two copies of Jam2 in Creld1+/− Ts65Dn mice blocked the increased occurrence of CHD in these mice. Thus, trisomy for Jam2 is required for the disomic risk factor gene Creld1 to affect heart development.
Animal models have also been important in understanding cancer risk in individuals with DS. Epidemiological studies provide strong evidence that the incidence of many cancers is reduced in adults with DS104. Importantly, studies in mouse models of DS have confirmed these results, showing that partial trisomy 21 protects against various cancers. For example, familial adenomatous polyposis (FAP) is a congenital condition involving the formation of precancerous adenomatous polyps (predominantly in the colon) at an early age, and is caused by mutations in the tumour suppressor adenomatous polyposis coli (APC). Interestingly, Ts65Dn mice carrying an Apc mutation found in patients with FAP (ApcMin) develop 50% fewer adenomatous polyps than euploid mice87. Ts1Rhr mice, which are trisomic for just 33 of the genes triplicated in Ts65Dn, have a similar reduction in adenomatous polyp formation. This reduction is substantially reversed by restoring just one of these 33 genes, the proto-oncogene Ets2, to two copies. Furthermore, ApcMin mice monosomic for these 33 genes (including Ets2) develop more tumours than euploid ApcMin mice. Thus, assessment in mouse models of trisomy and monosomy has uncovered a protective effect of Ets2 overexpression and a tumorigenesis-permissive effect of reduced Ets2 expression.
Preclinical studies and drug development.
Several clinical trials of pharmaceutical and nutraceutical agents to improve cognition in individuals with DS were initiated on the basis of preclinical observations in mouse models of DS (for example, NCT00748007 for rivastigmine, NCT02484703 and NCT02024789 for RG1662, and NCT01112683 for memantine). Although in-depth discussion of all of these trials is beyond the scope of this Primer, for illustrative purposes we discuss preclinical studies that led to phase I and II trials of RG1662 (also known as basmisanil or RO5186582), an inverse agonist of GABAA α5 receptors. Cognitive tests in Ts65Dn mice showed that these mice have deficits in learning and memory in tasks that depend on hippocampal function86,105,106, prompting electrophysiological studies demonstrating that long-term potentiation (LTP) is reduced in hippocampal slices from Ts65Dn mice98,99,107. The reduced LTP stemmed from excess GABA-mediated inhibition, which could be reversed by treatment with the GABAA inhibitor pentylenetetrazol (PTZ), restoring the inhibitory–excitatory balance and thereby improving performance in cognitive tests, even in young adult mice108.
These studies profoundly changed the understanding of cognitive dysfunction in DS and its treatment, by showing not only that treatment of cognitive deficits in DS is possible but that it is effective in adults, when previously it was widely thought that cognitive improvements could only be made during a small window early in life. Furthermore, this treatment had long-lasting effects, as the improved LTP was measured 3 months after cessation of PTZ treatment108. Thus, treatment for cognitive deficits is not a nebulous goal far in the future but can and should be addressed immediately. An inverse agonist of GABAA α5 receptors restores LTP and improves performance in cognitive tests in Ts65Dn mice109,110. RG1662 was assessed for safety and tolerability in individuals with DS in a successful phase I trial (NCT01436955) that concluded in 2013. However, a phase II trial begun in 2014 was stopped prematurely in 2016 because RG1662 did not show efficacy.
Although the lack of efficacy in this specific trial design was disappointing, the trial represented several advances. First, it demonstrated that the pharmaceutical industry now had the confidence to treat intellectual disability, a first for the DS community. Second, the ability to enlist multiple centres throughout the USA and Europe that were capable of the complex measurements used in the trials, combined with strong support from the DS community in recruiting individuals for the trials, was a clear statement of interest by researchers, clinicians and individuals with DS. Finally, the application of cognitive tests designed specifically for DS was an important part of the trial. Although the official summary of results from this trial have not yet been published, the principles involved in treating individuals with DS have been reported by a number of the investigators involved in the trial111. Overall, the disappointment of the trial result was overlaid with optimism for improved treatment and trial design approaches in the future, and informed further trials such as a recent phase II trial using green tea extract containing epigallocatechin-3-gallate (EGCG) to improve cognitive outcomes112.
Mechanisms of Alzheimer disease
Almost all adults with DS develop AD-like neuropathology by 40 years of age. AD is characterized by a long asymptomatic preclinical stage in which amyloid pathology develops, ~15–20 years before any cognitive impairment is observed113. This preclinical stage is present in autosomal dominant forms of AD114 and in early-onset AD associated with DS115. The evidence is compelling that an increased dosage of APP in trisomy 21 is necessary for increased risk of AD in individuals with DS, although the underlying mechanisms that link APP dosage to neurodegeneration are unknown. However, at least three molecular mechanisms have been proposed to explain the increased risk of AD in individuals with DS (FIG. 4).
Fig. 4 |. Mechanisms of Alzheimer disease in Down syndrome.
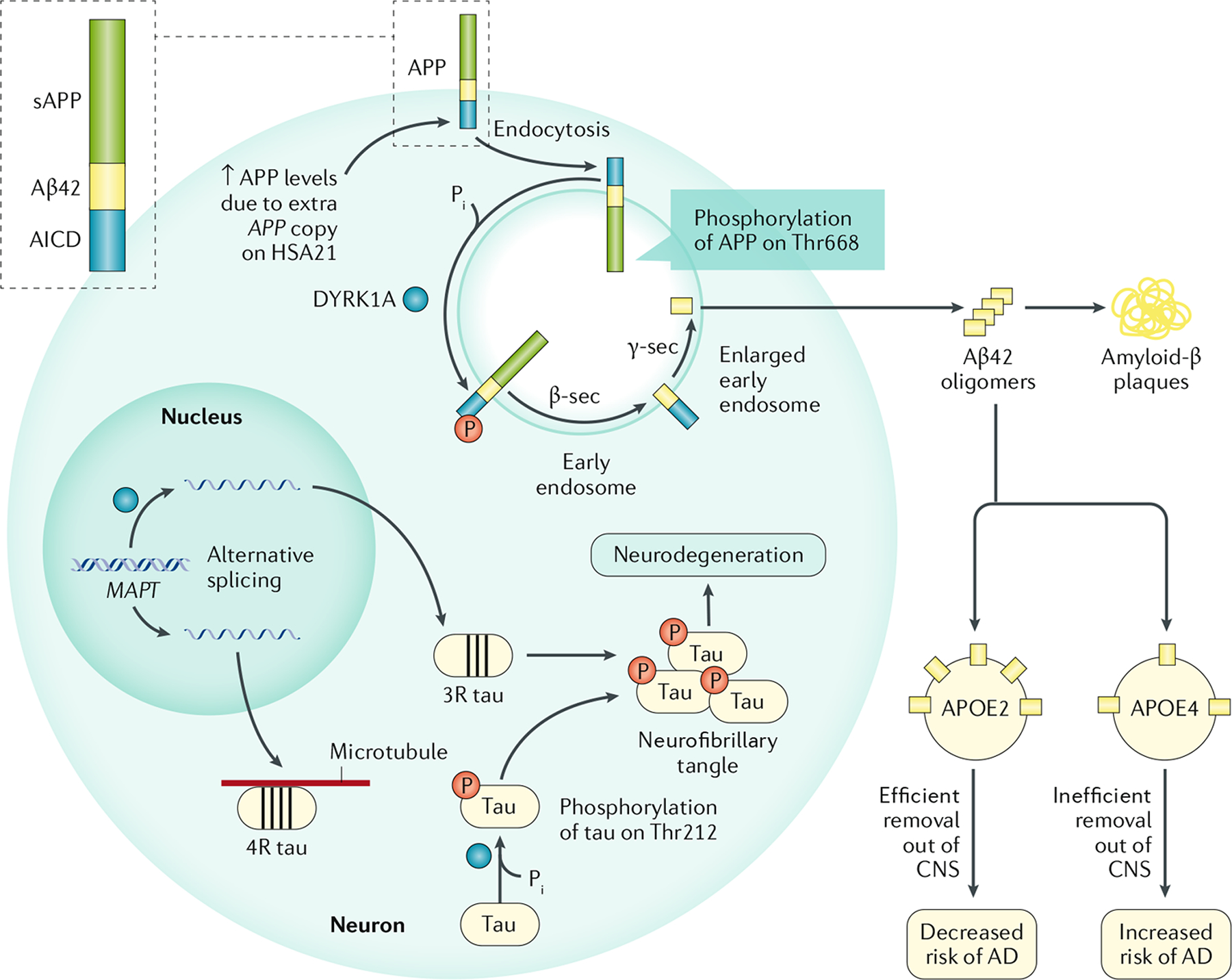
The increased gene dosage of APP (encoding amyloid precursor protein (APP)) and DYRK1A (encoding dual-specificity tyrosine-phosphorylation-regulated kinase 1A (DYRK1A)) on Homo sapiens chromosome 21 (HSA21) in individuals with Down syndrome (DS; trisomy 21) increases their risk of developing Alzheimer disease (AD). APP can undergo non-amyloidogenic processing (not shown) and amyloidogenic processing by β-secretase 1 (β-sec) and γ-secretase (γ-sec) to produce the neurotoxic Aβ42 peptide. The increased APP levels result in higher levels of Aβ42, and phosphorylation of APP at Thr668 by DYRK1A increases the amyloidogenic processing of APP. DYRK1A has other targets in the cell, including splicing factors and the microtubule-binding protein tau. DYRK1A phosphorylation of splicing factors alters the splicing of the MAPT mRNA (encoding tau), resulting in increased levels of tau containing three microtubule-binding domains (three-repeat (3R) tau) and reduced levels of 4R tau. This imbalance leads to neurofibrillary tangle (NFT) formation, possibly due to the lower binding affinity of 3R tau for microtubules. Furthermore, DYRK1A phosphorylation of Thr212 in tau alters the conformation of the protein, reducing the affinity of tau for microtubules and leading to NFT formation. The presence of the ε4 allele of APOE (APOE⋆ε4) alters the processing, deposition and clearance of Aβ249, and is therefore a major risk allele for AD. AICD, APP intracellular domain; APOE, apolipoprotein E; CNS, central nervous system; Pi, inorganic phosphate; sAPP, soluble amyloid precursor protein.
APP and Aβ42.
Increased APP expression resulting from the presence of an additional copy of the APP gene is strongly correlated with early-onset AD116,117. Genomic duplication of the APP locus is the cause of some cases of familial, early-onset AD118,119. In individuals with DS with complete HSA21 trisomy, the extra copy of HSA21-located APP results in increased levels of APP and its cleavage products, including the neurotoxic Aβ42 peptide, in the brain120. However, individuals with partial trisomy of HSA21 who are disomic for APP do not develop early-onset AD and have normal APP expression levels121.
DYRK1A.
A second mechanism involves multiple neuropathological effects of DYRK1A overexpression. HSA21-encoded DYRK1A phosphorylates APP at Thr668, resulting in increases phosphoAPP levels in cells from a mouse model of DS that overexpresses the human DYRK1A gene122. This phosphorylation facilitates the amyloidogenic processing of APP by β-secretase and γ-secretase, leading to increased production of neurotoxic Aβ42 peptide123,124. The levels of phosphoAPP and Aβ42 are increased in the brain of these mice, and phosphoAPP, DYRK1A and Aβ42 levels are increased in the brain of individuals with DS.
DYRK1A also has effects on another molecule implicated in the pathophysiology of AD, the microtubule-stabilizing protein tau. DYRK1A phosphorylates tau at Thr212, a residue that is hyperphosphorylated in individuals with AD125. Abnormal hyperphosphorylation of tau changes its conformation, leading to a reduced affinity for microtubules126 and resulting in microtubule instability, neurofibrillary tangle (NFT) formation and cell death127. Furthermore, DYRK1A also phosphorylates splicing factors and thereby alters the splicing of the mRNA of the tau-encoding gene MAPT. Alternative splicing of MAPT results in the production of tau protein isoforms with three or four microtubule-binding domains (three-repeat (3R) tau and 4R tau, respectively). Equal levels of 3R tau and 4R tau are required for maintaining normal brain function and an imbalance in their levels has been detected in sporadic tauopathies128. DYRK1A overexpression increases the abundance of 3R tau at the expense of 4R tau, which is thought to lead to the formation of NFTs, as 3R tau has lower affinity for microtubules than 4R tau129,130.
Endosomal dysfunction.
An extra copy of APP is sufficient to cause endosomal enlargement and intracellular trafficking defects by an Aβ-independent mechanism131. RAB5 mediates the endocytosis of cell-surface proteins and the homotypic fusion of early endosomes132. Enlargement of RAB5-positive early endosomes, a phenotype consistent with excessive activation of RAB5 (REF.133), is present in neurons in the brain of individuals with sporadic or familial AD134,135 and in individuals with DS who develop AD136. In fact, endosomal size in circulating peripheral blood mononuclear cells might serve as a blood-based biomarker of AD-related endosomal pathology137.
Endosomal function is also affected by other factors in DS. The ε4 allele of APOE (APOE⋆ε4) is the most important genetic risk factor for late-onset AD, as ~80% of all patients with AD have at least one APOE⋆ε4 allele138. APOE⋆ε4 is also thought to increase the risk of dementia in older adults with DS, albeit to a lower extent than for sporadic AD in euploid individuals139. Endosomal enlargement in trisomic neurons is thought to cause axonal trafficking defects that contribute to neuronal degeneration140. Furthermore, APOE regulates the endosomal trafficking of amyloid fibrils141 and early endosomal enlargement is present in the brain of mice carrying APOE⋆ε4 (REF.142). APP gene dose, Aβ42 and the C-terminal fragment of APP, C99 (also known as βCTF), also have important roles in the development of endosome dysfunction. An increased level of C99 is the only APP-related alteration associated with abnormal endosome enlargement and proliferation that is common to all forms of AD143–146.
As APP gene dosage is an important determinant of AD risk in individuals with DS, various therapies to reduce APP levels in individuals with DS might have efficacy in treating AD, including reducing APP expression (using anti-sense approaches) and APP production (using translational inhibitors such as Posiphen), blocking APP cleavage (such as by BACE or DYRK1A inhibition or using γ-secretase modulators) or removing Aβ (by active or passive Aβ immunization).
Diagnosis, screening and prevention
Prenatal screening
In developed countries, laboratory-based prenatal screening for DS is offered as part of routine antenatal care. Screening is a way to identify pregnancies at high risk, thereby limiting diagnostic procedures to minimize the risk of an iatrogenic miscarriage. Since the 1980s, the primary prenatal screening approach has involved a combination of measuring maternal serum biochemical analytes and, more recently, the size of the fetal nuchal translucency (NT; a pouch of fluid behind the neck) in the first trimester147. Initially, levels of α-fetoprotein in maternal serum and amniotic fluid in the second trimester were measured and a level ~70% of that in a normal pregnancy indicated an elevated risk of a DS conceptus. More recently, additional maternal serum analytes have been measured, including β-human chorionic gonado-tropin, unconjugated oestriol, inhibin A and pregnancy-associated plasma protein A. The levels of all of these markers change with gestational age, and some are better at distinguishing between trisomy 21 and euploid fetuses in the first trimester and others in the second trimester. Thus, correct dating of the pregnancy by ultrasonography examination is needed for accurate interpretation of serum analyte test results. For each woman, the risk of DS in the fetus is calculated using a computer algorithm with inputs of raw analyte values, gestational age and demographic information, such as maternal age, geo-ethnic background, smoking status and whether or not she has diabetes. The numerical risk cut-off values used in clinical practice differ in their reference points (that is, the risk of an affected fetus versus a liveborn infant). The chance of having a liveborn infant with DS is lower than the chance of having a fetus with DS in the second trimester because some of the fetuses will spontaneously miscarry in the second trimester18. Professional guidelines recommend that pregnant women with positive screening results are offered post-test counselling and a diagnostic test, such as amniocentesis or chorionic villus sampling followed by genetic analysis.
In the late 1980s and early 1990s, prenatal ultrasonography was incorporated into routine care, and some abnormalities were demonstrated to be associated with DS. Of note, no fetal anatomical findings are diagnostic of DS and, in fact, many neonates with DS have apparently normal prenatal sonograms. First-trimester ultrasonography features that may indicate DS include an increased NT measurement for gestational age and four other first trimester markers: absent nasal bone, increased frontomaxillary angle, tricuspid valve regurgitation and absent or reduced flow in the ductus venosus147. A second trimester anomaly scan has become routine at 18–20 weeks of gestation and includes quantifiable markers, such as a thickened nuchal skin fold and femoral and humeral length measurements. Additional so-called ‘soft’ ultrasonography markers include cystic hygroma, prominent tongue, choroid plexus cysts, mild ventriculomegaly, heart defects, echogenic bowel, duodenal atresia, pyelectasis, bilateral fifth finger clinodactyly and a wide space between the great and second toes148.
Using a combination of either first or second trimester maternal serum analyte quantification and NT measurement, the positive predictive values (PPV) for positive screening results are ~3–5% (refs149,150). Because of these low PPVs, there has been longstanding interest in a more precise NIPS for fetal chromosome aneuploidies. In 2011, sequencing of cell-free DNA in maternal serum became clinically available151. Using massively parallel sequencing, the DNA fragments are counted, mapped to specific areas of the genome, and compared to a reference value; an excess number of fragments that map to HSA21 is suggestive (but not diagnostic) of DS151. Within a very short time, NIPS has become the most clinically implemented example of genomic medicine, with ~10 million tests performed to date152. Currently, in the USA, a pregnant woman at high risk of having a fetus with DS (maternal age >35 years, positive family history, positive serum analyte and/or NT measurements, suggestive ultrasonographic abnormalities) is offered NIPS as a primary screen. In many countries in Europe, NIPS is beginning to be offered as a secondary test following a positive primary screen. At present, only in Belgium and the Netherlands is NIPS offered as a primary screen for all pregnant women, regardless of a priori risk.
A meta-analysis examined the PPV of NIPS in pregnant women at high or low risk of having a fetus with DS153. For DS, the PPVs were 91% and 82% for high-risk and low-risk pregnant women, respectively. Studies that directly compared the predictive performance of analyte and/or NT measurement to that of NIPS in the same pregnant woman have demonstrated a 10–20-fold increase in PPVs using sequencing149,150. Importantly, NIPS is not dependent on gestational age and can be performed at any time between 10 weeks of gestation and delivery. Because NIPS performs better than current standards of care, in some countries with nationalized health systems there is a move towards providing NIPS as a primary screen for DS151.
Because NIPS is noninvasive and has a very good PPV, there have been concerns about what effect this testing will have on the live birth rates of infants with DS. A preliminary study found no difference in the number of live births of babies with DS before and after NIPS became available in eight countries154. However, without access to all prenatal testing results, it is unclear how many more babies with DS would have been born during this time period. More extensive studies are needed to assess the effect of NIPS on the incidence of DS. Although many women choose not to have prenatal testing because it would not influence their reproductive decisions, in one study, women who knew prenatally that their babies had DS had better results on psychological tests than women who discovered at birth that their baby had DS155. A prenatal diagnosis of DS has been reported to have several benefits, including opportunity for parental education, meetings with paediatric subspecialists ahead of delivery, and change of the location of delivery so that qualified paediatric subspecialists are available for care, thereby enabling mother and baby to remain in the same hospital together155. A future benefit of accurate prenatal screening could include the opportunity to initiate treatment during fetal life to improve cognition (discussed later).
Management
Each individual with DS has a distinct set of strengths and challenges that can change throughout his or her life156. Some individuals will require high levels of medical input from birth, whereas others may have few health complications. Similarly, some individuals will require social care and support throughout adulthood, whereas others live independently. Several health problems are more common in individuals with DS than in the general population, including CHDs, obstructive sleep apnoea, thyroid disease, dementia, epilepsy, gastrointestinal disease, hearing and vision problems, intellectual disability and developmental disorders, mental illness, immunological dysfunction, haematological disorders and musculoskeletal issues. Screening for these manifestations should be carried out regularly and there are consensus screening guidelines for children with DS (such as the guidelines of the American Academy of Pediatrics157) but not yet for adults158–160. Services for adults are often more specialized than those for children, and care usually needs to be managed by a number of different medical teams. No consensus currently exists about who should oversee care in adults with DS, but primary care physicians often take on this role100; occasionally, paediatricians may remain involved until early adulthood, and in some countries, such as the UK, intellectual disability psychiatrists are most likely to provide medical support within community teams. Due to the lack of consensus, adults with DS may miss out on regular screening and proactive treatments, with interventions only occurring when difficulties are clinically apparent161.
Illness and disease may present differently in people with DS. There is a risk of diagnostic overshadowing and misattribution of symptoms, which can be compounded by the high prevalence of communication difficulties in individuals with DS. Due to the multisystem involvement in DS, care requires multidisciplinary input from a range of medical, social care and educational teams.
Perinatal management
Pregnant women carrying a fetus with a confirmed DS diagnosis require regular monitoring and support throughout the perinatal period. Pregnant women carrying a fetus with DS are at increased risk of miscarriage, with an estimated rate of spontaneous fetal death after 12 weeks of 30%, which increases with maternal age18. Monitoring recommendations suggest that a detailed ultrasonography examination and fetal echocardiography should be performed at 18–20 weeks gestation, with further ultrasonography examinations at 28–30, 34–36 and 38 weeks to assess for evidence of upper gastrointestinal obstruction, chylothorax, fetal hydrops and intra-uterine growth restriction. If abnormalities are detected, then increased fetal surveillance is recommended. Local paediatric teams should be informed if abnormalities are detected so that they can be involved in planning postnatal care and treatment (for guidance see, for example, the Down’s Syndrome Association and the Down Syndrome Medical Interest Group fact sheet).
Congenital heart defects
CHDs occur in ~50% of individuals with DS, most commonly AVSD (42% of CHDs in individuals with DS), ventricular septal defect (22%) and atrial septal defect (16%)162. Although the frequency of the specific type of CHD depends on age and ethnicity, the primary point is that CHDs have a severe effect on the quality of life of the individual. During pregnancy, a fetal echocardiography examination is recommended. A cardiology examination should take place postnatally, and another echocardiography examination should be performed within the first month after birth. Management is the same as for the general population, including surgical repair163. The mortality rate after surgery in children with DS is equal to or lower than that in the general population164. All individuals with DS should have annual screening throughout life for signs of acquired valve disease and heart failure.
Sleep apnoea
Obstructive sleep apnoea is common in individuals with DS, with an estimated prevalence of 54–90%159,165. Screening for symptoms should be carried out at every health check; these symptoms include loud snoring, heavy breathing, restless nights and daytime sleepiness, as well as neurocognitive symptoms, such as irritability, depression, paranoia, cognitive decline and behavioural problems166. Overnight polysomnography is recommended for all children with DS before 4 years of age, regardless of symptoms157,167. Alternative approaches, such as home oximetry, have been suggested to enable identification of at-risk individuals and to reduce the number of children that require full diagnostic studies168. Management of sleep apnoea includes the use of continuous positive airway pressure (CPAP), mandibular advancement devices and weight loss. Surgical interventions, including tonsillectomy and adenoidectomy, can be considered, although sleep apnoea may persist after surgery165,169.
Thyroid dysfunction
Congenital hypothyroidism is present in ~1% of individuals with DS, and abnormal thyroid function tests have been reported in >50% of neonates with DS170–172. An increased risk of developing thyroid disease remains throughout life, and the risk of developing autoimmune-related thyroid dysfunction increases with age173. As clinical diagnosis can be difficult, it is important to perform regular blood screening. Thyroid-stimulating hormone (TSH) and thyroxine (T4) levels should be obtained postnatally and at 6 months and 12 months of age. Measurements of TSH should then be repeated annually157.
Alzheimer disease
A substantial proportion of individuals with DS develop early-onset AD, which is related to APP overproduction120,174, and dementia is the proximal cause of death in 70% of older adults with DS175. Clinical symptoms appear after 40 years of age, and 77% of individuals with DS aged 60–69 years, and up to 100% of those aged >70 years, will develop cognitive decline176–178 (FIG. 5). The cognitive decline needs to be understood in the context of the existing cognitive phenotype and individual risk, including APOE genotype, as the presence of the APOE⋆ε4 allele seems to increase the risk of earlier cognitive decline due to AD and of mortality179 (FIG. 5b).
Fig. 5 |. Alzheimer disease prevalence and cognitive decline in Down syndrome.
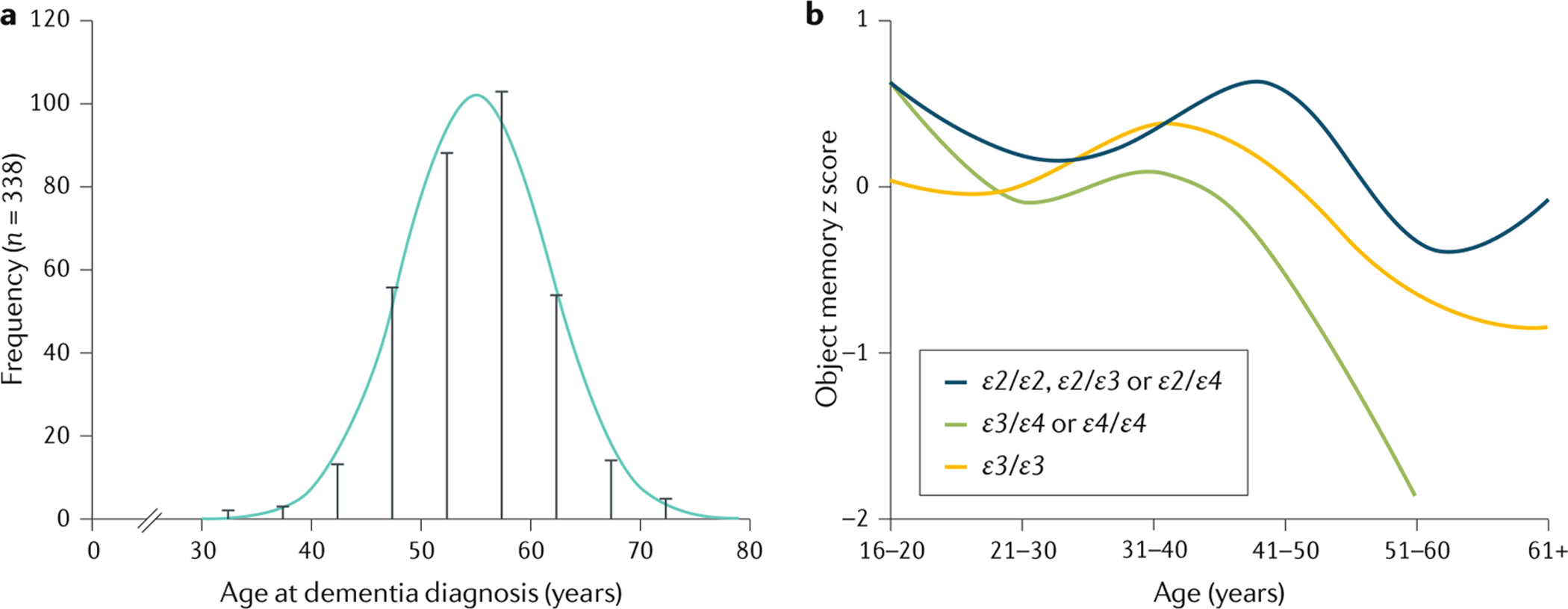
a | Distribution of age at dementia diagnosis in individuals with Down syndrome (DS). The risk of developing Alzheimer disease in individuals with DS is closely related to age. The mean age of dementia diagnosis is 55 years, although some individuals already show cognitive decline starting from 40 years of age, whereas others are not diagnosed until after 60 years of age. b | Cognitive decline in individuals with DS, measured by performance (z score) in a memory test. Cross-sectional data from the London Down Syndrome Consortium showing the distribution of test scores on an object memory task by age and apolipoprotein E (APOE) genotype in individuals with DS. The task is a measure of short-term and delayed memory, adapted for individuals with DS. Participants are instructed to name and recall seven objects with two immediate recall trials, and one delayed recall trial after 5 minutes250. The data are split by APOE genotype to compare the effect of the ε4 allele of APOE (APOE⋆ε4; which increases the risk of late-onset sporadic Alzheimer disease) with that of the APOE⋆ε2 allele (which decreases AD risk) and APOE⋆ε3 allele (which has a neutral effect on AD risk) on cognitive performance. Cognitive performance in individuals with an APOE⋆ε3/APOE⋆ε4 or APOE⋆ε4/APOE⋆ε4 genotype declines from 40 years of age, noticeably earlier than the average age of dementia diagnosis and earlier than in individuals with an APOE⋆ε2/APOE⋆ε2, APOE⋆ε2/APOE⋆ε3, APOE⋆ε2/APOE⋆ε4 or APOE⋆ε3/APOE⋆ε3 genotype. Part a is adapted from REF.251, CC-BY-4.0 (https://creativecommons.org/licenses/by/4.0/). Part b adapted with permission from REF.115, Elsevier.
Deficits in memory and attention occur early in DS, as in individuals with sporadic AD180, but often go unnoticed until behavioural changes appear181. Some individuals with DS and dementia may present with seizures and, as the disease progresses, the development of neurological symptoms, such as myoclonus and seizures, is commonly seen182.
A baseline assessment of cognitive and adaptive functioning at 30 years of age is recommended in all individuals with DS, to aid future monitoring and diagnosis183. Management should focus on early detection and supportive measures. Positive benefit may be obtained from the use of acetylcholinesterase inhibitors184,185, although some individuals may develop a reduced heart rate and other adverse effects. Individuals with DS and dementia should have access to adequate support, as their needs will increase as the disease progresses. Efforts should be made to keep individuals within their familiar homes, and transfer to other providers should be based on individual circumstances and needs186.
Epilepsy
Epilepsy is present in 8% of children with DS, with a bimodal age of onset (one peak before 3 years of age and the other after 30 years of age160). Infant onset, most frequently infantile spasms, has been associated with a severe form of epilepsy termed West syndrome, which includes psychomotor regression and hypsarrhythmia (an abnormal pattern of activity between convulsions) on electroencephalography, and occurs in ~6–32% of seizures reported in infants187. Later onset of epilepsy is linked to the development of AD136. The management of epilepsy depends on the aetiology, but usually involves anticonvulsant medication, which is generally effective in reducing seizure activity156,178,188.
Hearing and vision
Conductive hearing loss is common in individuals with DS, with a high prevalence of otitis media with effusion189,190. Baseline hearing tests should be performed postnatally using, for example, brainstem auditory acoustic response, and then every 6 months until school age and at least annually thereafter157. Early recognition and treatment can reduce the risk of later hearing loss191. Sensorineural loss becomes increasingly common in adulthood. Treatment with hearing aids and cochlear implantation has been successful. The use of speech therapy, communication aids and sign language, including the language programme Makaton, can also be beneficial160.
Because there is increased risk of refractive errors, cataracts (congenital and developmental), keratoconus and amblyopia in individuals with DS, ophthalmological examination should be performed at birth and regularly throughout life, ideally every 1–2 years157. Treatment and corrective aids should be given promptly157. Cataract surgery is routinely performed, usually with good outcomes192,193.
Atlantoaxial instability
Atlantoaxial subluxation (misalignment of the first and second cervical vertebrae) develops in ~1–2% of children with DS194. If the condition is present, parents should be advised that participation in sports increases the risk of spinal cord injury in the child. Symptoms such as neck pain, weakness, spasticity, gait difficulties and hyperreflexia should be evaluated with cervical spine radiography157.
Mental health
Individuals with DS have increased prevalence of behavioural and mental health problems compared with the general population195, in particular, depressive and anxiety disorders196,197. A small proportion of adolescents and young adults with DS undergo acute regression (also known as Down syndrome disintegrative disorder), which involves a loss of skills and independence compared with their previous levels of functioning. At present, the cause of this decline is unknown, although it often seems to occur after exposure to emotional stressors198. To date, there is no definitive treatment for this presentation.
Diagnosis of mental health issues in individuals with DS can be complicated by the presence of intellectual disability, communication difficulties and atypical symptoms199. The effect of psychological stressors should not be underestimated, including transitions in accommodation, school or care arrangements, and bereavement166,200. Treatment for mental illness should be based on standard clinical guidelines, and individuals with DS show positive responses to behavioural interventions, psychotropic medication and psychological therapy.
Neurodevelopment
DS is the most common genetic cause of intellectual disability, with the majority of individuals with DS classified as having mild–moderate disability. Their cognitive profile demonstrates strengths in visual learning, but weaknesses in expressive language, verbal working memory, and episodic memory201. However, there is a wide range in cognitive function, with variations in IQ, language, attention, memory and functional abilities.
Individuals with DS often have autism spectrum disorder (~10–15%) and attention-deficit–hyperactivity disorder (ADHD; ~6%) and appropriate screens for these should be performed160,202. Clinical presentations may differ from those of the general population and assessments may require input from specialists. Standard treatments for ADHD are recommended, although there can be a less marked response to stimulant medication in individuals with intellectual disability than in those without intellectual disability203.
Prenatal therapy to improve neurocognition.
Increasing attention has been paid to using prenatal diagnosis of DS as an opportunity to provide antenatal therapy to improve neurocognition204,205. Impairment of neurogenesis in individuals with DS begins prenatally206–208 and is one of several factors209 that result in abnormalities in fetal brain growth and shape that are recognizable in third trimester prenatal ultrasound and MR images210. Thus, treatment initiated in fetal or early neonatal life is hypothesized to have maximal effect211 (the ethical issues posed by treatment of fetuses with DS are discussed elsewhere212).
To date, most experiments involving fetal therapy have been performed using mouse models of DS. Antenatal treatments that have resulted in postnatal improvement in learning and neurobehaviour in Ts65Dn mice include dosing with the vasoactive intestinal pep-tides NAPVSIPQ (NAP) and SALLRSIPA (SAL)213, maternal dietary choline supplementation214, and inhibition of DYRK1A. For example, prenatal treatment with the DYRK1A inhibitor EGCG, which crosses both the placental and blood–brain barriers in mice, improved some aspects of the cranial vault morphological defects in Ts65Dn mice215. Furthermore, oral administration of the novel DYRK1A inhibitor ALGERNON (altered generation of neurons) to pregnant Ts1Cje dams from embryonic day 10 (E10) to E15 improved neurogenesis, corticogenesis and behavioural performance of their pups, possibly by enhancing the proliferation of neural stem cells216.
Other potential therapeutic targets have been identified from studies of mouse models. For example, mitochondrial dysfunction in fibroblasts from human fetuses with DS is reversed by activation of the PGC1α pathway using metformin217. The mRNA and protein levels of PGC1α are markedly reduced in fibroblasts from fetuses with DS, and metformin treatment increased oxygen consumption and cellular ATP concentration, improved respiratory activity, increased mitochondrial membrane potential and reversed mitochondrial ultrastructural abnormalities in these cells217. Metformin already has FDA regulatory approval for the treatment of maternal gestational diabetes, so its use in clinical trials in pregnant women carrying fetuses with DS would potentially face fewer regulatory hurdles. Studies in mouse models of DS have also shown that prenatal treatment with the selective serotonin reuptake inhibitor fluoxetine restores hippocampal neurogenesis and connectivity, granule cell number and dendritic patterns, and hippocampus-dependent memory functions218. A clinical trial of high-dose fluoxetine in pregnant women (without psychiatric disorders) is ongoing at the University of Texas (USA)211, although no information about the trial is publicly available. However, fluoxetine use in the first trimester may have adverse effects, such as increasing the risk of congenital heart disease and other malformations219.
Apigenin, a natural flavone found in citrus fruits and leafy green vegetables, also shows promise in reducing oxidative stress and improving total oxidative capacity in human cells from fetuses with trisomy 21, as well as improving some behaviours in neonatal and adult Ts1Cje mice treated prenatally204. The recognition of the possibilities for improving fetal brain development, and testing of an increasing number of compounds for eventual use in clinical trials, make this a very exciting time in DS research, although recommendations for supplementation cannot be made owing to a paucity of evidence.
Immune and haematological systems
Individuals with DS are more prone to infections, especially of the respiratory tract220–222. Infections should be recognized and treated promptly157. There is no contraindication to immunizations, and the standard childhood schedule should be followed.
Individuals with DS also show a predisposition to autoimmune diseases, particularly coeliac disease160,223. Testing the levels of immunoglobulin A antibodies to tissue transglutaminase 2 is recommended if symptoms of coeliac disease are present157, but screening of asymptomatic individuals has not proved cost-effective224. Type 1 diabetes mellitus, alopecia areata and Addison disease have also been reported in individuals with DS at higher rates than in the general population.
Children with DS are at a markedly increased risk of developing acute leukaemia, compared with children without DS. Transient myeloproliferative disorder (TMD), also referred to as transient leukaemia of DS, is observed in 5–30% of individuals with DS before 3 months of age, and a blood count and film should be performed within 3 days of birth to enable the identification and monitoring of this disorder225. Although TMD usually resolves without treatment in the first few months of life, it seems to increase the risk of developing leukaemia by 5 years of age, and it is estimated that 20–30% of individuals with TMD will go on to develop myeloid leukaemia of DS157,226,227.
Quality of life
Research into the quality of life of individuals with DS is limited, although it is recognized that stigma and cultural norms can be barriers to the active participation of individuals with DS in the community. If adequately supported during their life, many individuals with DS can live fairly independently. The perspectives of family members of individuals with DS have been somewhat studied228. Support groups and DS networks can provide valuable input, and individuals with DS and their families should be aware of local and national organizations.
Children with DS are now regularly educated within mainstream schools, often with additional one-to-one or specific Special Educational Needs support229. Some children with DS can gain additional benefits in terms of language and literacy skills when educated in inclusion classrooms rather than in substantially separate classrooms, whereas others may require a specialist school230.
Many individuals with DS are employed and report fairly high levels of satisfaction, and many have become successful professionals, including artists, speakers and actors. However, there is a disparity between the number of individuals with DS who wish to work and those who have secured employment. They may access voluntary work and this seems to be more common than in the general adult population. Cultural assumptions, social care cuts and inadequate adjustments in work can make it difficult for individuals with DS to access employment231,232.
Quality of life can be affected by physical health, and proactive screening programmes and early intervention can reduce the negative effect of prolonged hospitalizations and ill health. Recognition and correction of hearing and vision problems can reduce the negative effect on communication and development233. Obesity, cervical spine abnormalities and muscular difficulties may contribute to the exclusion of individuals with DS from physical activity, and in the USA, children with DS have lower levels of physical activity than their peers234. However, the majority would be able to participate in exercise programmes if available, which can have a positive effect on health and wellbeing235.
Owing to early cognitive decline, many adults with DS have increased support needs as they age. Adequate proactive support, with regular reassessment of needs and services set up to anticipate functional impairments, is crucial for their quality of life.
Outlook
Although the lives of individuals with DS have been much improved by the evolution of medical care and the benefits of societal changes, there is no treatment for DS at present that is based on the current understanding of the molecular pathophysiology of DS. Therefore, to design effective therapies, further research is needed to improve understanding of the biology of each symptom and feature of the syndrome. The most important topics for further research are discussed below.
Understanding the contribution of genetic variation to the different and variable phenotypic characteristics and manifestations of DS is an important goal that could be achieved by genome sequencing of thousands of individuals with DS, and linking the genetic variation with the detailed phenotypic characterization of each individual6. This effort is based on the reasonable hypothesis that genomic differences are major determinants of the extensive phenotypic variation. Furthermore, genes and other functional genomic elements that in three copies contribute to the phenotype of DS need to be identified. There is supportive evidence that genes causing disease states with haploinsufficiency, in which one functional copy is not sufficient, may also be important when present in three copies, as is the case with DS. For example, DYRK1A is haploinsufficient (probability of loss-of-function intolerant (pLI) score of 1), and the loss of one copy of DYRK1A causes intellectual disability236. Several studies have also shown that an extra copy of DYRK1A is involved in the biological dysfunction that results from trisomy 21 (REF.237). However, the pLI score may not be an absolute discriminator of genes involved in the pathophysiology of DS. For example, APP has been strongly linked to AD in individuals with DS120 but has a pLI score of 0.06, which is compatible with a haplosufficient gene. Furthermore, HMGN1, a gene that is linked to the transcriptional dysregulation in trisomy 21(REF.238), has a pLI score of 0.22. In addition to the causative contribution of specific genes on HSA21, the phenotypic contribution of the extra-chromosomal material, regardless of its gene content, needs to be investigated.
Experimental models, such as the mouse and other organisms7, need to be used to study the brain and other organs in the different stages of development. Advances in single-cell technologies239 that enable study of the genome, transcriptome, histone modifications, chromatin contacts and protein levels in individual cells will certainly provide insights into the cellular mechanisms of the altered developmental pathways in DS and the effect of chromatin conformation on the dysregulation of gene expression.
Experimental efforts to modify the dysregulated expression of genes on HSA21 and other chromosomes may result in novel therapeutic approaches that should be explored further. For example, experimental silencing of large chromosomal regions by introducing the X chromosome-inactivation effector XIST on one copy of HSA21, or silencing one of the three alleles of HMGN1, DYRK1A or APP, may rescue some phenotypic characteristics of DS.
Early-onset AD in individuals with DS is genetically analogous to some forms of autosomal dominant AD, and the amyloid pathway is strongly implicated in AD pathology due to APP triplication. Therefore, amyloid-targeting drugs should be trialled in individuals with DS, as they are at very high risk of early-onset AD, preferably before any symptoms develop in order to prevent or delay the onset of dementia115. PET imaging of amyloid or tau deposits in the brain240,241 or measures of Aβ peptide ratios in cerebrospinal fluid242 have been shown to be feasible biomarkers for patient stratification, whereas plasma levels of the neurodegeneration markers neurofilament light chain and neuronal pentraxin 2 show promise as biomarkers of treatment response243–245.
Population-based, longitudinal, large-scale cohort studies following individuals with DS would be helpful to the further understanding of the co-occurring medical conditions, family attitudes, educational achievement, individual experiences, and societal successes that now exist for individuals with DS. Increasingly sophisticated analytical methods combined with improved genetic models offer hope for more translation of promising therapies.
Acknowledgements
The authors thank the members of the London Down Syndrome (LonDownS) Consortium, G. de Graaf of the Dutch Down Syndrome Foundation and F. Buckley of Down Syndrome Education International for their review of the epidemiology section of this article. Work in the authors’ laboratories and clinics was supported by grants from the SNF, EU, ERC, Jerome Lejeune, and ChildCare Foundations to S.E.A.; a Wellcome Trust Strategic Award (grant number 098330/Z/12/Z) conferred upon the LonDownS Consortium, an MRC project grant (LonDownsPREVENT MR/S011277/1), and grants from the EU Joint Programme - Neuro-degenerative Disease Research (MR/R024901/1, as part of the HEROES consortium), Network of Centres of Excellence in Neurodegeneration (COEN) (MR/S005145/1), Lumind Foundation and Jerome Lejeune Foundation to A.S.; the Jerome Lejeune Foundation USA, Anna and John Sie Foundation, US National Institutes of Health (NIH; HD42053-10, UL1TR001064, ZIA HG200399-04) to D.W.B.; NIH and Lumind Foundation to S.L.S.; HD038384-20, HD098540 and the Lumind Foundation to R.H.R.; and the Alzheimer’s Society and BRC to S.P. The authors thank all past and present members of their laboratories, their collaborators and the patients and their families for their inspiration and support.
Competing interests
S.E.A. is the co-founder and CEO of MediGenome, a clinical and laboratory diagnostic company. B.G.S. occasionally consults on the topic of Down syndrome through Gerson Lehrman Group. B.G.S. receives remuneration from Down syndrome non-profit organizations for speaking engagements and associated travel expenses. B.G.S. receives annual royalties from Woodbine House, Inc., for the publication of his book, Fasten Your Seatbelt: A Crash Course on Down Syndrome for Brothers and Sisters. Within the past 2 years, B.G.S. has received research funding from F. Hoffmann-La Roche, Inc., and LuMind IDSC Down Syndrome Foundation to conduct clinical trials for people with Down syndrome. B.G.S. is occasionally asked to serve as an expert witness in legal cases where Down syndrome is discussed. B.G.S. serves in a non-paid capacity on the Honorary Board of Directors for the Massachusetts Down Syndrome Congress and the Professional Advisory Committee for the National Center for Prenatal and Postnatal Down Syndrome Resources. B.G.S. has a sister with Down syndrome. M.S.R. is a consultant to AC Immune SA. A.S. has consulted for Roche Pharmaceuticals, ONO Pharma, Aelis Farma and AC Immune, and he serves on the Scientific Advisory Board of ProMIS Neurosciences. A.S. and S.E.P. provide clinical services within the UK National Health Service to individuals with Down syndrome. The remaining authors declare no competing interests.
References
- 1.Down JLH Observations on an ethnic classification of idiots. Lond. Hosp. Rep 3, 259–262 (1866). [Google Scholar]
- 2.Hasle H, Clemmensen IH & Mikkelsen M Risks of leukaemia and solid tumours in individuals with Down’s syndrome. Lancet 355, 165–169 (2000). [DOI] [PubMed] [Google Scholar]
- 3.LeJeune J, Gautier M & Turpin R Study of somatic chromosomes from 9 mongoloid children [French]. C. R. Hebd. Seances. Acad. Sci 248, 1721–1722 (1959). [PubMed] [Google Scholar]; This study is the first description of the chromosomal abnormality in DS.
- 4.Davisson MT, Schmidt C & Akeson EC Segmental trisomy of murine chromosome 16: a new model system for studying Down syndrome. Prog. Clin. Biol. Res 360, 263–280 (1990). [PubMed] [Google Scholar]
- 5.Hattori M et al. The DNA sequence of human chromosome 21. Nature 405, 311–319 (2000). [DOI] [PubMed] [Google Scholar]; A landmark paper on the sequencing of the long arm of HSA21.
- 6.Antonarakis SE Down syndrome and the complexity of genome dosage imbalance. Nat. Rev. Genet 18, 147–163 (2017). [DOI] [PubMed] [Google Scholar]
- 7.Herault Y et al. Rodent models in Down syndrome research: impact and future opportunities. Dis. Model. Mech 10, 1165–1186 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen XQ & Mobley WC Exploring the pathogenesis of Alzheimer disease in basal forebrain cholinergic neurons: converging insights from alternative hypotheses. Front. Neurosci 13, 446 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reeves RH et al. Paving the way for therapy: the Second International Conference of the Trisomy 21 Research Society. Mol. Syndromol 9, 279–286 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.de Graaf G, Buckley F & Skotko B People living with Down syndrome in the USA: births and population. Down Syndrome Education International https://dsuri.net/us-population-factsheet (2019). [Google Scholar]
- 11.de Graaf G, Buckley F & Skotko BG Estimation of the number of people with Down syndrome in the United States. Genet. Med 19, 439–447 (2017). [DOI] [PubMed] [Google Scholar]
- 12.de Graaf G, Buckley F & Skotko BG Birth and population prevalence of Down syndrome in European countries. (Poster presented at the World Down Syndrome Congress 2018). [Google Scholar]
- 13.Bray I, Wright DE, Davies C & Hook EB Joint estimation of Down syndrome risk and ascertainment rates: a meta-analysis of nine published data sets. Prenat. Diagn 18, 9–20 (1998). [PubMed] [Google Scholar]
- 14.Hecht CA & Hook EB Rates of Down syndrome at livebirth by one-year maternal age intervals in studies with apparent close to complete ascertainment in populations of European origin: a proposed revised rate schedule for use in genetic and prenatal screening. Am. J. Med. Genet 62, 376–385 (1996). [DOI] [PubMed] [Google Scholar]
- 15.Hassold T & Hunt P To err (meiotically) is human: the genesis of human aneuploidy. Nat. Rev. Genet 2, 280–291 (2001). [DOI] [PubMed] [Google Scholar]
- 16.Morris JK, Wald NJ, Mutton DE & Alberman E Comparison of models of maternal age-specific risk for Down syndrome live births. Prenat. Diagn 23, 252–258 (2003). [DOI] [PubMed] [Google Scholar]
- 17.de Graaf G, Buckley F & Skotko BG Estimates of the live births, natural losses, and elective terminations with Down syndrome in the United States. Am. J. Med. Genet. A 167, 756–767 (2015). [DOI] [PubMed] [Google Scholar]
- 18.Savva GM, Morris JK, Mutton DE & Alberman E Maternal age-specific fetal loss rates in Down syndrome pregnancies. Prenat. Diagn 26, 499–504 (2006). [DOI] [PubMed] [Google Scholar]
- 19.Bittles AH, Bower C, Hussain R & Glasson EJ The four ages of Down syndrome. Eur. J. Public Health 17, 221–225 (2007). [DOI] [PubMed] [Google Scholar]
- 20.Deng C et al. Recent trends in the birth prevalence of Down syndrome in China: impact of prenatal diagnosis and subsequent terminations. Prenat. Diagn 35, 311–318 (2015). [DOI] [PubMed] [Google Scholar]
- 21.Morris JK & Alberman E Trends in Down’s syndrome live births and antenatal diagnoses in England and Wales from 1989 to 2008: analysis of data from the National Down Syndrome cytogenetic register. BMJ 339, b3794 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tul N, Verdenik I, Srsen TP & Antolic ZN P31.05: incidence of Down syndrome in Slovenia in the last 15 years. Ultrasound Obstet. Gynecol 30, 569–570 (2007). [Google Scholar]
- 23.Collins VR, Muggli EE, Riley M, Palma S & Halliday JL Is Down syndrome a disappearing birth defect? J. Pediatr 152, 20–24. e1 (2008). [DOI] [PubMed] [Google Scholar]
- 24.Dolk H et al. Trends and geographic inequalities in the prevalence of Down syndrome in Europe, 1980–1999. Rev. Epidemiol Sante Publique 53, 2S87–2S95 (2005). [PubMed] [Google Scholar]
- 25.de Graaf G et al. Estimates of live birth prevalence of children with Down syndrome in the period 1991–2015 in the Netherlands. J. Intellect. Disabil. Res 61, 461–470 (2017). [DOI] [PubMed] [Google Scholar]
- 26.Nagaoka SI, Hassold TJ & Hunt PA Human aneuploidy: mechanisms and new insights into an age-old problem. Nat. Rev. Genet 13, 493–504 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gruhn JR et al. Chromosome errors in human eggs shape natural fertility over reproductive life span. Science 365, 1466–1469 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Antonarakis SE et al. The meiotic stage of nondisjunction in trisomy 21: determination by using DNA polymorphisms. Am. J. Hum. Genet 50, 544–550 (1992). [PMC free article] [PubMed] [Google Scholar]
- 29.Allen EG et al. Maternal age and risk for trisomy 21 assessed by the origin of chromosome nondisjunction: a report from the Atlanta and National Down Syndrome Projects. Hum. Genet 125, 41–52 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yoon PW et al. Advanced maternal age and the risk of Down syndrome characterized by the meiotic stage of chromosomal error: a population-based study. Am. J. Hum. Genet 58, 628–633 (1996). [PMC free article] [PubMed] [Google Scholar]
- 31.Freeman SB et al. The National Down Syndrome Project: design and implementation. Public Health Rep. 122, 62–72 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ghosh S, Feingold E & Dey SK Etiology of Down syndrome: evidence for consistent association among altered meiotic recombination, nondisjunction, and maternal age across populations. Am. J. Med. Genet. A 149, 1415–1420 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Oliver TR et al. New insights into human nondisjunction of chromosome 21 in oocytes. PLoS Genet. 4, e1000033 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chernus JM et al. A candidate gene analysis and GWAS for genes associated with maternal nondisjunction of chromosome 21. PLoS Genet. 15, e1008414 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Coppede F Risk factors for Down syndrome. Arch. Toxicol 90, 2917–2929 (2016). [DOI] [PubMed] [Google Scholar]
- 36.Hunter JE et al. The association of low socioeconomic status and the risk of having a child with Down syndrome: a report from the National Down Syndrome Project. Genet. Med 15, 698–705 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Torfs CP & Christianson RE Socioeconomic effects on the risk of having a recognized pregnancy with Down syndrome. Birth Defects Res. A Clin. Mol. Teratol 67, 522–528 (2003). [DOI] [PubMed] [Google Scholar]
- 38.Christianson RE, Sherman SL & Torfs CP Maternal meiosis II nondisjunction in trisomy 21 is associated with maternal low socioeconomic status. Genet. Med 6, 487–494 (2004). [DOI] [PubMed] [Google Scholar]
- 39.Ghosh S, Ghosh P & Dey SK Altered incidence of meiotic errors and Down syndrome birth under extreme low socioeconomic exposure in the Sundarban area of India. J. Community Genet 5, 119–124 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Keen C et al. The association between maternal occupation and Down syndrome: a report from the national Down syndrome project. Int. J. Hyg. Environ. Health 223, 207–213 (2020). [DOI] [PubMed] [Google Scholar]
- 41.Sartain CV & Hunt PA An old culprit but a new story: bisphenol A and “NextGen” bisphenols. Fertil. Steril 106, 820–826 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Horan TS et al. Replacement bisphenols adversely affect mouse gametogenesis with consequences for subsequent generations. Curr. Biol 28, 2948–2954. e3 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Grandjean P et al. Timescales of developmental toxicity impacting on research and needs for intervention. Basic Clin. Pharmacol. Toxicol 125, 70–80 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Antonarakis SE, Avramopoulos D, Blouin JL, Talbot CC Jr. & Schinzel AA Mitotic errors in somatic cells cause trisomy 21 in about 4.5% of cases and are not associated with advanced maternal age. Nat. Genet 3, 146–150 (1993). [DOI] [PubMed] [Google Scholar]
- 45.Antonarakis SE Parental origin of the extra chromosome in trisomy 21 as indicated by analysis of DNA polymorphisms. Down Syndrome Collaborative Group. N. Engl. J. Med 324, 872–876 (1991). [DOI] [PubMed] [Google Scholar]; This paper reports the use of DNA polymorphic markers to determine the parental origin of the supernumerary HSA21 in DS.
- 46.Antonarakis SE 10 years of genomics, chromosome 21, and Down syndrome. Genomics 51, 1–16 (1998). [DOI] [PubMed] [Google Scholar]
- 47.Morris JK, Alberman E, Mutton D & Jacobs P Cytogenetic and epidemiological findings in Down syndrome: England and Wales 1989–2009. Am. J. Med. Genet. A 158, 1151–1157 (2012). [DOI] [PubMed] [Google Scholar]
- 48.Lyle R et al. Genotype-phenotype correlations in Down syndrome identified by array CGH in 30 cases of partial trisomy and partial monosomy chromosome 21. Eur. J. Hum. Genet 17, 454–466 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Barlow GM et al. Down syndrome congenital heart disease: a narrowed region and a candidate gene. Genet. Med 3, 91–101 (2001). [DOI] [PubMed] [Google Scholar]
- 50.Gupta M, Dhanasekaran AR & Gardiner KJ Mouse models of Down syndrome: gene content and consequences. Mamm. Genome 27, 538–555 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Epstein CJ et al. Protocols to establish genotype-phenotype correlations in Down syndrome. Am. J. Hum. Genet 49, 207–235 (1991). [PMC free article] [PubMed] [Google Scholar]
- 52.Bray IC & Wright DE Estimating the spontaneous loss of Down syndrome fetuses between the times of chorionic villus sampling, amniocentesis and livebirth. Prenat. Diagn 18, 1045–1054 (1998). [PubMed] [Google Scholar]
- 53.Hassold TJ & Jacobs PA Trisomy in man. Ann.Rev. Genet 18, 69–97 (1984). [DOI] [PubMed] [Google Scholar]
- 54.Shapiro BL Down syndrome – a disruption of homeostasis. Am. J. Med. Genet 14, 241–269 (1983). [DOI] [PubMed] [Google Scholar]
- 55.Pritchard M & Kola I The ‘gene dosage effect’ hypothesis versus the ‘amplified developmental instability’ hypothesis in Down syndrome. J. Neural Transm 57, 293–303 (1999). [PubMed] [Google Scholar]
- 56.Stamoulis G et al. Single cell transcriptome in aneuploidies reveals mechanisms of gene dosage imbalance. Nat. Commun 10, 4495 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Salehi A et al. Increased App expression in a mouse model of Down’s syndrome disrupts NGF transport and causes cholinergic neuron degeneration. Neuron 51, 29–42 (2006). [DOI] [PubMed] [Google Scholar]; A study that provided support for the hypothesis that neuronal degeneration in DS is due to increased APP expression.
- 58.Pelleri MC et al. Integrated quantitative transcriptome maps of human trisomy 21 tissues and cells. Front. Genet 9, 125 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sullivan KD et al. Trisomy 21 consistently activates the interferon response. eLife 5, e16220 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gonzales PK et al. Transcriptome analysis of genetically matched human induced pluripotent stem cells disomic or trisomic for chromosome 21. PLoS One 13, e0194581 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Letourneau A et al. Domains of genome-wide gene expression dysregulation in Down’s syndrome. Nature 508, 345–350 (2014). [DOI] [PubMed] [Google Scholar]
- 62.Do C, Xing Z, Yu YE & Tycko B Trans-acting epigenetic effects of chromosomal aneuploidies: lessons from Down syndrome and mouse models. Epigenomics 9, 189–207 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Do LH, Mobley WC & Singhal N Questioned validity of gene expression dysregulated domains in Down’s syndrome. F1000Res. 4, 269 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ahlfors H et al. Gene expression dysregulation domains are not a specific feature of Down syndrome. Nat. Commun 10, 2489 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Umlauf D & Mourad R The 3D genome: from fundamental principles to disease and cancer. Semin. Cell Dev. Biol 90, 128–137 (2019). [DOI] [PubMed] [Google Scholar]
- 66.Kemeny S et al. Spatial organization of chromosome territories in the interphase nucleus of trisomy 21 cells. Chromosoma 127, 247–259 (2018). [DOI] [PubMed] [Google Scholar]
- 67.Mendioroz M et al. Trans effects of chromosome aneuploidies on DNA methylation patterns in human Down syndrome and mouse models. Genome Biol. 16, 263 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.El Hajj N et al. Epigenetic dysregulation in the developing Down syndrome cortex. Epigenetics 11, 563–578 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Horvath S et al. Accelerated epigenetic aging in Down syndrome. Aging Cell 14, 491–495 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Scott HS et al. Identification and characterization of two putative human arginine methyltransferases (HRMT1L1 and HRMT1L2). Genomics 48, 330–340 (1998). [DOI] [PubMed] [Google Scholar]
- 71.Xiao CL et al. N6-methyladenine DNA modification in the human genome. Mol. Cell 71, 306–318. e7 (2018). [DOI] [PubMed] [Google Scholar]
- 72.Kim IS et al. Roles of Mis18α in epigenetic regulation of centromeric chromatin and CENP-A loading. Mol. Cell 46, 260–273 (2012). [DOI] [PubMed] [Google Scholar]
- 73.Veland N et al. DNMT3L facilitates DNA methylation partly by maintaining DNMT3A stability in mouse embryonic stem cells. Nucleic Acids Res. 47, 152–167 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lu J et al. Global hypermethylation in fetal cortex of Down syndrome due to DNMT3L overexpression. Hum. Mol. Genet 25, 1714–1727 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sailani MR et al. DNA-methylation patterns in trisomy 21 using cells from monozygotic twins. PLoS One 10, e0135555 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Guo X, Williams JG, Schug TT & Li X DYRK1A and DYRK3 promote cell survival through phosphorylation and activation of SIRT1. J. Biol. Chem 285, 13223–13232 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lepagnol-Bestel AM et al. DYRK1A interacts with the REST/NRSF-SWI/SNF chromatin remodelling complex to deregulate gene clusters involved in the neuronal phenotypic traits of Down syndrome. Hum. Mol. Genet 18, 1405–1414 (2009). [DOI] [PubMed] [Google Scholar]
- 78.Liu Y et al. Systematic proteome and proteostasis profiling in human trisomy 21 fibroblast cells. Nat. Commun 8, 1212 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; First quantitative study of the trisomy 21 proteome, using fibroblasts from individuals with DS.
- 79.Gillet LC et al. Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: a new concept for consistent and accurate proteome analysis. Mol. Cell Proteom 11, O111.016717 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sullivan KD et al. Trisomy 21 causes changes in the circulating proteome indicative of chronic autoinflammation. Sci. Rep 7, 14818 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gold L et al. Aptamer-based multiplexed proteomic technology for biomarker discovery. PLoS One 5, e15004 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Izzo A et al. Mitochondrial dysfunction in Down syndrome: molecular mechanisms and therapeutic targets. Mol. Med 24, 2 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zamponi E & Helguera PR The shape of mitochondrial dysfunction in Down syndrome. Dev. Neurobiol 79, 613–621 (2019). [DOI] [PubMed] [Google Scholar]
- 84.Valenti D et al. Mitochondria as pharmacological targets in Down syndrome. Free. Radic. Biol. Med 114, 69–83 (2018). [DOI] [PubMed] [Google Scholar]
- 85.Vilardell M et al. Meta-analysis of heterogeneous Down syndrome data reveals consistent genome-wide dosage effects related to neurological processes. BMC Genomics 12, 229 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Reeves R et al. A mouse model for Down syndrome exhibits learning and behaviour deficits. Nat. Genet 11, 177–183 (1995). [DOI] [PubMed] [Google Scholar]; This paper describes the first complex model of trisomy 21, Ts65Dn, and lays out characterizations for comparing the effects of aneuploidy in mice and humans.
- 87.Sussan T, Yang A, Li F, Ostrowski M & Reeves RH Trisomy protects against ApcMin-mediated tumors in mouse models of Down syndrome. Nature 451, 73–75 (2008). [DOI] [PubMed] [Google Scholar]
- 88.Garcia-Cerro S, Rueda N, Vidal V, Lantigua S & Martinez-Cue C Normalizing the gene dosage of Dyrk1A in a mouse model of Down syndrome rescues several Alzheimer’s disease phenotypes. Neurobiol. Dis 106, 76–88 (2017). [DOI] [PubMed] [Google Scholar]
- 89.Aziz NM et al. Lifespan analysis of brain development, gene expression and behavioral phenotypes in the Ts1Cje, Ts65Dn and Dp(16)1/Yey mouse models of Down syndrome. Disease Models Mech 11, dmm031013 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gribble SM et al. Massively parallel sequencing reveals the complex structure of an irradiated human chromosome on a mouse background in the Tc1 model of Down syndrome. PLoS One 8, e60482 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kazuki Y et al. A non-mosaic humanized mouse model of Down syndrome, trisomy of a nearly complete long arm of human chromosome 21 in mouse chromosome background. Preprint at bioRxivhttps://www.biorxiv.org/content/10.1101/862433v1 (2019).
- 92.Ramirez-Solis R, Liu P & Bradley A Chromosome engineering in mice. Nature 378, 720–724 (1995). [DOI] [PubMed] [Google Scholar]
- 93.Olson LE, Richtsmeier JT, Leszl J & Reeves RH A chromosome 21 critical region does not cause specific Down syndrome phenotypes. Science 306, 687–690 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Yu T et al. A mouse model of Down syndrome trisomic for all human chromosome 21 syntenic regions. Hum. Mol. Genet 19, 2780–2791 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper reports a mouse model that is trisomic for all mouse chromosomal regions syntenic with HSA21.
- 95.Stefanidis K et al. Causes of infertility in men with Down syndrome. Andrologia 43, 353–357 (2011). [DOI] [PubMed] [Google Scholar]
- 96.Williams BR et al. Aneuploidy affects proliferation and spontaneous immortalization in mammalian cells. Science 322, 703–709 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Contestabile A et al. Cell cycle alteration and decreased cell proliferation in the hippocampal dentate gyrus and in the neocortical germinal matrix of fetuses with Down syndrome and in Ts65Dn mice. Hippocampus 17, 665–678 (2007). [DOI] [PubMed] [Google Scholar]
- 98.Kleschevnikov AM et al. Hippocampal long-term potentiation suppressed by increased inhibition in the Ts65Dn mouse, a genetic model of Down syndrome. J. Neurosci 24, 8153–8160 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Siarey RJ, Stoll J, Rapoport SI & Galdzicki Z Altered long-term potentiation in the young and old Ts65Dn mouse, a model for Down syndrome. Neuropharmacology 36, 1549–1554 (1997). [DOI] [PubMed] [Google Scholar]
- 100.Ferencz C et al. Congenital cardiovascular malformations associated with chromosome abnormalities: an epidemiologic study. J. Pediatr 114, 79–86 (1989). [DOI] [PubMed] [Google Scholar]
- 101.Lana-Elola E et al. Genetic dissection of Down syndrome-associated congenital heart defects using a new mouse mapping panel. eLife 5, e11614 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Li H et al. Penetrance of congenital heart disease in a mouse model of Down syndrome depends on a trisomic potentiator of a disomic modifier. Genetics 203, 763–770 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Edie S et al. Survey of human chromosome 21 gene expression effects on early development in Danio rerio. G3 8, 2215–2223 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Yang Q, Rasmussen SA & Friedman JM Mortality associated with Down’s syndrome in the USA from 1983 to 1997: a population-based study. Lancet 359, 1019–1025 (2002). [DOI] [PubMed] [Google Scholar]
- 105.Escorihuela RM et al. A behavioral assessment of Ts65Dn mice: a putative Down syndrome model. Neurosci. Lett 199, 143–146 (1995). [DOI] [PubMed] [Google Scholar]
- 106.Holtzman DM et al. Developmental abnormalities and age-related neurodegeneration in a mouse model of Down syndrome. Proc. Natl Acad. Sci. USA 93, 13333–13338 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Hanson JE, Blank M, Valenzuela RA, Garner CC & Madison DV The functional nature of synaptic circuitry is altered in area CA3 of the hippocampus in a mouse model of Down’s syndrome. J. Physiol 579, 53–67 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study establishes the imbalance between excitatory and inhibitory inputs in the hippocampus, which has been the focus of efforts to antagonize signalling through GABA-A receptors, including a trial to improve cognitive ability in individuals with DS (NCT01436955).
- 108.Fernandez F et al. Pharmacotherapy for cognitive impairment in a mouse model of Down syndrome. Nat. Neurosci 10, 411–413 (2007). [DOI] [PubMed] [Google Scholar]; One of the first mouse studies that raised the possibility of pharmacotherapy for the cognitive impairment in DS.
- 109.Braudeau J et al. Specific targeting of the GABA-A receptor α5 subtype by a selective inverse agonist restores cognitive deficits in Down syndrome mice. J Psychopharmacol (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Duchon A et al. Long-lasting correction of in vivo LTP and cognitive deficits of mice modelling Down syndrome with an α5-selective GABAA inverse agonist. Br. J. Pharmacol 10.1111/bph.14903 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Hart SJ et al. Pharmacological interventions to improve cognition and adaptive functioning in Down syndrome: strides to date. Am. J. Med. Genet. A 173, 3029–3041 (2017). [DOI] [PubMed] [Google Scholar]
- 112.de la Torre R et al. Safety and efficacy of cognitive training plus epigallocatechin-3-gallate in young adults with Down’s syndrome (TESDAD): a double-blind, randomised, placebo-controlled, phase 2 trial. Lancet Neurol. 15, 801–810 (2016). [DOI] [PubMed] [Google Scholar]; A successful randomized trial to improve cognitive functioning in young adults with DS.
- 113.Jack CR Jr. et al. A/T/N: An unbiased descriptive classification scheme for Alzheimer disease biomarkers. Neurology 87, 539–547 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Esparza TJ et al. Amyloid-β oligomerization in Alzheimer dementia versus high-pathology controls. Ann. Neurol 73, 104–119 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Strydom A et al. Alzheimer’s disease in Down syndrome: an overlooked population for prevention trials. Alzheimers Dement. 4, 703–713 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Thonberg H et al. Mutation screening of patients with Alzheimer disease identifies APP locus duplication in a Swedish patient. BMC Res. Notes 4, 476 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Wallon D et al. The French series of autosomal dominant early onset Alzheimer’s disease cases: mutation spectrum and cerebrospinal fluid biomarkers. J. Alzheimers Dis 30, 847–856 (2012). [DOI] [PubMed] [Google Scholar]
- 118.Rovelet-Lecrux A et al. APP locus duplication causes autosomal dominant early-onset Alzheimer disease with cerebral amyloid angiopathy. Nat. Genet 38, 24–26 (2006). [DOI] [PubMed] [Google Scholar]; This study provides strong evidence for a link between the triplication of APP in trisomy 21 and early-onset AD in individuals with DS.
- 119.Sleegers K et al. APP duplication is sufficient to cause early onset Alzheimer’s dementia with cerebral amyloid angiopathy. Brain 129, 2977–2983 (2006). [DOI] [PubMed] [Google Scholar]
- 120.Wiseman FK et al. A genetic cause of Alzheimer disease: mechanistic insights from Down syndrome. Nat. Rev. Neurosci 16, 564–574 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Doran E et al. Down syndrome, partial trisomy 21, and absence of Alzheimer’s disease: the role of APP. J. Alzheimers Dis 56, 459–470 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Ryoo SR et al. Dual-specificity tyrosine(Y)-phosphorylation regulated kinase 1A-mediated phosphorylation of amyloid precursor protein: evidence for a functional link between Down syndrome and Alzheimer’s disease. J. Neurochem 104, 1333–1344 (2008). [DOI] [PubMed] [Google Scholar]
- 123.Wegiel J et al. Intraneuronal Aβ immunoreactivity is not a predictor of brain amyloidosis-β or neurofibrillary degeneration. Acta Neuropathol 113, 389–402 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Vingtdeux V et al. Phosphorylation of amyloid precursor carboxy-terminal fragments enhances their processing by a gamma-secretase-dependent mechanism. Neurobiol. Dis 20, 625–637 (2005). [DOI] [PubMed] [Google Scholar]
- 125.Woods YL et al. The kinase DYRK phosphorylates protein-synthesis initiation factor eIF2Bε at Ser539 and the microtubule-associated protein tau at Thr212: potential role for DYRK as a glycogen synthase kinase 3-priming kinase. Biochem. J 355, 609–615 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Lindwall G & Cole RD Phosphorylation affects the ability of tau protein to promote microtubule assembly. J. Biol. Chem 259, 5301–5305 (1984). [PubMed] [Google Scholar]
- 127.LaPointe NE et al. The amino terminus of tau inhibits kinesin-dependent axonal transport: implications for filament toxicity. J. Neurosci. Res 87, 440–451 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Goedert M & Jakes R Mutations causing neurodegenerative tauopathies. Biochim. Biophys. Acta 1739, 240–250 (2005). [DOI] [PubMed] [Google Scholar]
- 129.Liu F et al. Overexpression of Dyrk1A contributes to neurofibrillary degeneration in Down syndrome. FASEB J. 22, 3224–3233 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Yin X et al. Dyrk1A overexpression leads to increase of 3R-tau expression and cognitive deficits in Ts65Dn Down syndrome mice. Sci. Rep 7, 619 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Chen XQ, Sawa M & Mobley WC Dysregulation of neurotrophin signaling in the pathogenesis of Alzheimer disease and of Alzheimer disease in Down syndrome. Free Radic. Biol. Med 114, 52–61 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Stenmark H Rab GTPases as coordinators of vesicle traffic. Nat. Rev. Mol. Cell Biol 10, 513–525 (2009). [DOI] [PubMed] [Google Scholar]
- 133.Roberts RL et al. Endosome fusion in living cells overexpressing GFP-rab5. J. Cell Sci 112, 3667–3675 (1999). [DOI] [PubMed] [Google Scholar]
- 134.Cataldo A et al. Endocytic disturbances distinguish among subtypes of Alzheimer’s disease and related disorders. Ann. Neurol 50, 661–665 (2001). [PubMed] [Google Scholar]
- 135.Cataldo AM, Barnett JL, Pieroni C & Nixon RA Increased neuronal endocytosis and protease delivery to early endosomes in sporadic Alzheimer’s disease: neuropathologic evidence for a mechanism of increased β-amyloidogenesis. J. Neurosci 17, 6142–6151 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Cataldo AM et al. Endocytic pathway abnormalities precede amyloid beta deposition in sporadic Alzheimer’s disease and Down syndrome: differential effects of APOE genotype and presenilin mutations. Am. J. Pathol 157, 277–286 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Corlier F et al. Modifications of the endosomal compartment in peripheral blood mononuclear cells and fibroblasts from Alzheimer’s disease patients. Transl. Psychiatry 5, e595 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Saunders AM et al. Association of apolipoprotein E allele epsilon 4 with late-onset familial and sporadic Alzheimer’s disease. Neurology 43, 1467–1472 (1993). [DOI] [PubMed] [Google Scholar]
- 139.Rohn TT, McCarty KL, Love JE & Head E Is apolipoprotein E4 an important risk factor for dementia in persons with Down syndrome? J. Parkinsons Dis. Alzheimers Dis 1, 7 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Cataldo AM et al. Down syndrome fibroblast model of Alzheimer-related endosome pathology: accelerated endocytosis promotes late endocytic defects. Am. J. Pathol 173, 370–384 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Cossec JC et al. Trisomy for synaptojanin1 in Down syndrome is functionally linked to the enlargement of early endosomes. Hum. Mol. Genet 21, 3156–3172 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Nuriel T et al. The endosomal-lysosomal pathway is dysregulated by APOE4 expression in vivo. Front. Neurosci 11, 702 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Xu W et al. Amyloid precursor protein-mediated endocytic pathway disruption induces axonal dysfunction and neurodegeneration. J. Clin. Invest 126, 1815–1833 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Jiang Y et al. Alzheimer’s-related endosome dysfunction in Down syndrome is Aβ-independent but requires APP and is reversed by BACE-1 inhibition. Proc. Natl Acad. Sci. USA 107, 1630–1635 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Jiang Y et al. Lysosomal dysfunction in Down syndrome is APP-dependent and mediated by APP-βCTF (C99). J. Neurosci 39, 5255–5268 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Kwart D et al. A large panel of isogenic APP and PSEN1 mutant human iPSC neurons reveals shared endosomal abnormalities mediated by APP β-CTFs, not Aβ. Neuron 104, 256–270. e1–e5 (2019). [DOI] [PubMed] [Google Scholar]
- 147.Cuckle H & Maymon R Development of prenatal screening – a historical overview. Semin. Perinatol 40, 12–22 (2016). [DOI] [PubMed] [Google Scholar]
- 148.Bianchi DW, Crombleholme TM, D’Alton ME & Malone FD Fetology: Diagnosis and Management of the Fetal Patient Ch. 131 (McGraw-Hill Medical, 2010). [Google Scholar]
- 149.Bianchi DW, Rava RP & Sehnert AJ DNA sequencing versus standard prenatal aneuploidy screening. N. Engl. J. Med 371, 577–578 (2014). [DOI] [PubMed] [Google Scholar]
- 150.Norton ME & Wapner RJ Cell-free DNA analysis for noninvasive examination of trisomy. N. Engl. J. Med 373, 2581–2582 (2015). [DOI] [PubMed] [Google Scholar]
- 151.Bianchi DW & Chiu RWK Sequencing of circulating cell-free DNA during pregnancy. N. Engl. J. Med 379, 464–473 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Liu S et al. Genomic analyses from non-invasive prenatal testing reveal genetic associations, patterns of viral infections, and Chinese population history. Cell 175, 347–359. e14 (2018). [DOI] [PubMed] [Google Scholar]
- 153.Taylor-Phillips S et al. Accuracy of non-invasive prenatal testing using cell-free DNA for detection of Down, Edwards and Patau syndromes: a systematic review and meta-analysis. BMJ Open. 6, e010002 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Hill M et al. Has noninvasive prenatal testing impacted termination of pregnancy and live birth rates of infants with Down syndrome? Prenat. Diagn 37, 1281–1290 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Ralston SJ, Wertz D, Chelmow D, Craigo SD & Bianchi DW Pregnancy outcomes after prenatal diagnosis of aneuploidy. Obstet. Gynecol 97, 729–733 (2001). [DOI] [PubMed] [Google Scholar]
- 156.Alexander M et al. Morbidity and medication in a large population of individuals with Down syndrome compared to the general population. Dev. Med. Child. Neurol 58, 246–254 (2016). [DOI] [PubMed] [Google Scholar]
- 157.Bull MJ & Committee on Genetics,. Health supervision for children with Down syndrome. Pediatrics 128, 393–406 (2011). [DOI] [PubMed] [Google Scholar]
- 158.Jensen KM & Bulova PD Managing the care of adults with Down’s syndrome. BMJ 349, g5596 (2014). [DOI] [PubMed] [Google Scholar]
- 159.Capone G et al. Co-occurring medical conditions in adults with Down syndrome: a systematic review toward the development of health care guidelines. Am. J. Med. Genet. A 176, 116–133 (2018). [DOI] [PubMed] [Google Scholar]
- 160.Roizen NJ & Patterson D Down’s syndrome. Lancet 361, 1281–1289 (2003). [DOI] [PubMed] [Google Scholar]
- 161.Glasson EJ, Dye DE & Bittles AH The triple challenges associated with age-related comorbidities in Down syndrome. J. Intellect. Disabil. Res 58, 393–398 (2014). [DOI] [PubMed] [Google Scholar]
- 162.Bergstrom S et al. Trends in congenital heart defects in infants with Down syndrome. Pediatrics 138, e20160123 (2016). [DOI] [PubMed] [Google Scholar]
- 163.Morales-Demori R Congenital heart disease and cardiac procedural outcomes in patients with trisomy 21 and Turner syndrome. Congenit. Heart Dis 12, 820–827 (2017). [DOI] [PubMed] [Google Scholar]
- 164.Russell MW, Chung WK, Kaltman JR & Miller TA Advances in the understanding of the genetic determinants of congenital heart disease and their impact on clinical outcomes. J. Am. Heart Assoc 7, e006906 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Churchill SS, Kieckhefer GM, Landis CA & Ward TM Sleep measurement and monitoring in children with Down syndrome: a review of the literature, 1960–2010. Sleep. Med. Rev 16, 477–488 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Smith DS Health care management of adults with Down syndrome. Am. Fam. Physician 64, 1031–1039 (2001). [PubMed] [Google Scholar]
- 167.Esbensen AJ, Hoffman EK, Stansberry E & Shaffer R Convergent validity of actigraphy with polysomnography and parent reports when measuring sleep in children with Down syndrome. J. Intellect. Disabil. Res 62, 281–291 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Hill CM et al. Home oximetry to screen for obstructive sleep apnoea in Down syndrome. Arch. Dis. Child 103, 962–967 (2018). [DOI] [PubMed] [Google Scholar]
- 169.Venekamp RP et al. Tonsillectomy or adenotonsillectomy versus non-surgical management for obstructive sleep-disordered breathing in children. Cochrane Database Syst. Rev 10, CD011165 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Purdy IB, Singh N, Brown WL, Vangala S & Devaskar UP Revisiting early hypothyroidism screening in infants with Down syndrome. J. Perinatol 34, 936–940 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Iughetti L, Lucaccioni L, Fugetto F, Mason A & Predieri B Thyroid function in Down syndrome. Expert Rev. Endocrinol. Metab 10, 525–532 (2015). [DOI] [PubMed] [Google Scholar]
- 172.Sarici D et al. Thyroid functions of neonates with Down syndrome. Ital. J. Pediatr 38, 44 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Hardy O et al. Hypothyroidism in Down syndrome: screening guidelines and testing methodology. Am. J. Med. Genet. A 124, 436–437 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Bittles AH & Glasson EJ Clinical, social, and ethical implications of changing life expectancy in Down syndrome. Dev. Med. Child. Neurol 46, 282–286 (2004). [DOI] [PubMed] [Google Scholar]
- 175.Hithersay R et al. Association of dementia with mortality among adults with Down syndrome older than 35 years. JAMA Neurol. 76, 152–160 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper identifies AD as the most prominent cause of mortality in adults with DS.
- 176.Strydom A et al. Dementia in older adults with intellectual disabilities—epidemiology, presentation, and diagnosis. J. Policy Pract. Intellect. Disabil 7, 96–110 (2010). [Google Scholar]
- 177.Ballard C, Mobley W, Hardy J, Williams G & Corbett A Dementia in Down’s syndrome. Lancet Neurol. 15, 622–636 (2016). [DOI] [PubMed] [Google Scholar]
- 178.Bayen E, Possin KL, Chen Y, Cleret de Langavant L & Yaffe K Prevalence of aging, dementia, and multimorbidity in older adults with Down syndrome. JAMA Neurol. 75, 1399–1406 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Lott IT & Head E Dementia in Down syndrome: unique insights for Alzheimer disease research. Nat. Rev. Neurol 15, 135–147 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Startin CM et al. Cognitive markers of preclinical and prodromal Alzheimer’s disease in Down syndrome. Alzheimers Dement. 15, 245–257 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Firth NC et al. Aging related cognitive changes associated with Alzheimer’s disease in Down syndrome. Ann. Clin. Transl. Neurol 5, 741–751 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.De Simone R, Puig XS, Gelisse P, Crespel A & Genton P Senile myoclonic epilepsy: delineation of a common condition associated with Alzheimer’s disease in Down syndrome. Seizure 19, 383–389 (2010). [DOI] [PubMed] [Google Scholar]
- 183.Royal College of Psychiatrists & The British Psychological Society. Dementia and People with Intellectual Disabilities: Guidance on the assessment, diagnosis, interventions and support of people with intellectual disabilities who develop dementia. https://www.dsrf.org/media/REP77%20final%20proof%20(3).pdf (The British Psychological Society, 2015). [Google Scholar]
- 184.Eady N et al. Impact of cholinesterase inhibitors or memantine on survival in adults with Down syndrome and dementia: clinical cohort study. Br. J. Psychiatry 212, 155–160 (2018). [DOI] [PubMed] [Google Scholar]
- 185.Livingstone N, Hanratty J, McShane R & Macdonald G Pharmacological interventions for cognitive decline in people with Down syndrome. Cochrane Database Syst. Rev 10, CD011546 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Dodd K et al. Consensus statement of the international summit on intellectual disability and Dementia related to post-diagnostic support. Aging Mental Health 22, 1406–1415 (2018). [DOI] [PubMed] [Google Scholar]
- 187.Sanmaneechai O et al. Treatment outcomes of West syndrome in infants with Down syndrome. Pediatric Neurol. 48, 42–47 (2013). [DOI] [PubMed] [Google Scholar]
- 188.Gholipour T, Mitchell S, Sarkis RA & Chemali Z The clinical and neurobehavioral course of Down syndrome and dementia with or without new-onset epilepsy. Epilepsy Behav. 68, 11–16 (2017). [DOI] [PubMed] [Google Scholar]
- 189.Austeng ME et al. Otitis media with effusion in children with in Down syndrome. Int. J. Pediatr Otorhinolaryngol 77, 1329–1332 (2013). [DOI] [PubMed] [Google Scholar]
- 190.Fisher PG Congenital hearing loss in Down syndrome. J. Pediatr 166, 1–3 (2015). [DOI] [PubMed] [Google Scholar]
- 191.Shott SR, Joseph A & Heithaus D Hearing loss in children with Down syndrome. Int. J. Pediatr. Otorhinolaryngol 61, 199–205 (2001). [DOI] [PubMed] [Google Scholar]
- 192.Krinsky-McHale SJ et al. Vision deficits in adults with Down syndrome. J. Appl. Res. Intellect. Disabil 27, 247–263 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Li EY, Chan TC, Lam NM & Jhanji V Cataract surgery outcomes in adult patients with Down’s syndrome. Br. J. Ophthalmol 98, 1273–1276 (2014). [DOI] [PubMed] [Google Scholar]
- 194.Brockmeyer D Down syndrome and craniovertebral instability. Topic review and treatment recommendations. Pediatr. Neurosurg 31, 71–77 (1999). [DOI] [PubMed] [Google Scholar]
- 195.Capone G, Goyal P, Ares W & Lannigan E Neurobehavioral disorders in children, adolescents, and young adults with Down syndrome. Am. J. Med. Genet. C 142, 158–172 (2006). [DOI] [PubMed] [Google Scholar]
- 196.Mantry D et al. The prevalence and incidence of mental ill-health in adults with Down syndrome. J. Intellect. Disabil. Res 52, 141–155 (2008). [DOI] [PubMed] [Google Scholar]
- 197.Tassé MJ et al. Psychiatric conditions prevalent among adults with Down syndrome. J. Policy Pract. Intellect. Disabil 13, 173–180 (2016). [Google Scholar]
- 198.Mircher C et al. Acute regression in young people with Down syndrome. Brain Sci 7, 57 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Spendelow JS Assessment of mental health problems in people with Down syndrome: key considerations. Br. J. Learn. Disabil 39, 306–313 (2011). [Google Scholar]
- 200.Nevill RE & Benson BA Risk factors for challenging behaviour and psychopathology in adults with Down syndrome. J.Intellect. Disabil. Res 62, 941–951 (2018). [DOI] [PubMed] [Google Scholar]
- 201.Liogier d’Ardhuy X et al. Assessment of cognitive scales to examine memory, executive function and language in individuals with Down syndrome: implications of a 6-month observational study. Front. Behav. Neurosci 9, 300 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Dykens EM Psychiatric and behavioral disorders in persons with Down syndrome. Ment. Retard. Dev. Disabil. Res. Rev 13, 272–278 (2007). [DOI] [PubMed] [Google Scholar]
- 203.Aman MG, Buican B & Arnold LE Methylphenidate treatment in children with borderline IQ and mental retardation: analysis of three aggregated studies. J. Child. Adolesc. Psychopharmacol 13, 29–40 (2003). [DOI] [PubMed] [Google Scholar]
- 204.Guedj F, Bianchi DW & Delabar JM Prenatal treatment of Down syndrome: a reality? Curr. Opin. Obstet. Gynecol 26, 92–103 (2014). [DOI] [PubMed] [Google Scholar]
- 205.Bianchi DW From prenatal genomic diagnosis to fetal personalized medicine: progress and challenges. Nat. Med 18, 1041–1051 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Stagni F, Giacomini A, Emili M, Guidi S & Bartesaghi R Neurogenesis impairment: an early developmental defect in Down syndrome. Free. Radic. Biol. Med 114, 15–32 (2018). [DOI] [PubMed] [Google Scholar]
- 207.Guidi S et al. Neurogenesis impairment and increased cell death reduce total neuron number in the hippocampal region of fetuses with Down syndrome. Brain Pathol. 18, 180–197 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Guidi S, Ciani E, Bonasoni P, Santini D & Bartesaghi R Widespread proliferation impairment and hypocellularity in the cerebellum of fetuses with Down syndrome. Brain Pathol. 21, 361–373 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Dierssen M & de Lagran MM DYRK1A (dual-specificity tyrosine-phosphorylated and -regulated kinase 1A): a gene with dosage effect during development and neurogenesis. ScientificWorldJournal 6, 1911–1922 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Tarui T et al. Quantitative MRI analyses of regional brain growth in living fetuses with Down syndrome. Cereb. Cortex 10.1093/cercor/bhz094 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Stagni F, Giacomini A, Guidi S, Ciani E & Bartesaghi R Timing of therapies for Down syndrome: the sooner, the better. Front. Behav. Neurosci 9, 265 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.de Wert G, Dondorp W & Bianchi DW Fetal therapy for Down syndrome: an ethical exploration. Prenat. Diagn 37, 222–228 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Incerti M et al. Prenatal treatment prevents learning deficit in Down syndrome model. PLoS One 7, e50724 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Kelley CM et al. Effects of maternal choline supplementation on the septohippocampal cholinergic system in the Ts65Dn mouse model of Down syndrome. Curr. Alzheimer Res 13, 84–96 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.McElyea SD et al. Influence of prenatal EGCG treatment and Dyrk1a dosage reduction on craniofacial features associated with Down syndrome. Hum. Mol. Genet 25, 4856–4869 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Nakano-Kobayashi A et al. Prenatal neurogenesis induction therapy normalizes brain structure and function in Down syndrome mice. Proc. Natl Acad. Sci. USA 114, 10268–10273 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Izzo A et al. Metformin restores the mitochondrial network and reverses mitochondrial dysfunction in Down syndrome cells. Hum. Mol. Genet 26, 1056–1069 (2017). [DOI] [PubMed] [Google Scholar]
- 218.Guidi S et al. Prenatal pharmacotherapy rescues brain development in a Down’s syndrome mouse model. Brain 137, 380–401 (2014). [DOI] [PubMed] [Google Scholar]
- 219.Gao SY et al. Fluoxetine and congenital malformations: a systematic review and meta-analysis of cohort studies. Br. J. Clin. Pharmacol 83, 2134–2147 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Chan M et al. The burden of respiratory syncytial virus (RSV) associated acute lower respiratory infections in children with Down syndrome: a systematic review and meta-analysis. J. Glob. Health 7, 020413 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Grut V, Söderström L & Naumburg E National cohort study showed that infants with Down’s syndrome faced a high risk of hospitalisation for the respiratory syncytial virus. Acta Paediatr. 106, 1519–1524 (2017). [DOI] [PubMed] [Google Scholar]
- 222.Bloemers BLP et al. Increased risk of respiratory tract infections in children with Down syndrome: the consequence of an altered immune system. Microbes Infect. 12, 799–808 (2010). [DOI] [PubMed] [Google Scholar]
- 223.Giménez-Barcons M et al. Autoimmune predisposition in Down syndrome may result from a partial central tolerance failure due to insufficient intrathymic expression of AIRE and peripheral antigens. J. Immunol 193, 3872–3879 (2014). [DOI] [PubMed] [Google Scholar]
- 224.Swigonski NL, Kuhlenschmidt HL, Bull MJ, Corkins MR & Downs SM Screening for celiac disease in asymptomatic children with Down syndrome: cost-effectiveness of preventing lymphoma. Pediatrics 118, 594–602 (2006). [DOI] [PubMed] [Google Scholar]
- 225.Tunstall O et al. Guidelines for the investigation and management of transient leukaemia of Down syndrome. Br. J. Haematol 182, 200–211 (2018). [DOI] [PubMed] [Google Scholar]
- 226.Mateos MK, Barbaric D, Byatt S-A, Sutton R & Marshall GM Down syndrome and leukemia: insights into leukemogenesis and translational targets. Transl. Pediatr 4, 76–92 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Gamis AS & Smith FO Transient myeloproliferative disorder in children with Down syndrome: clarity to this enigmatic disorder. Br. J. Haematol 159, 277–287 (2012). [DOI] [PubMed] [Google Scholar]
- 228.Skotko BG, Levine SP, Macklin EA & Goldstein RD Family perspectives about Down syndrome. Am. J. Med. Genet. A 170, 930–941 (2016). [DOI] [PubMed] [Google Scholar]
- 229.Van Herwegen J, Ashworth M & Palikara O Parental views on special educational needs provision: cross-syndrome comparisons in Williams syndrome, Down syndrome, and autism spectrum disorders. Res. Dev. Disabil 80, 102–111 (2018). [DOI] [PubMed] [Google Scholar]
- 230.Buckley S, Bird G, Sacks B & Archer T A comparison of mainstream and special education for teenagers with Down syndrome: implications for parents and teachers. Syndrome Res. Pract 9, 54–67 (2006). [DOI] [PubMed] [Google Scholar]
- 231.Kumin L & Schoenbrodt L Employment in adults with Down syndrome in the United States: results from a national survey. Appl. Res. Intellect. Disabil 29, 330–345 (2015). [DOI] [PubMed] [Google Scholar]
- 232.Mihaila I et al. Leisure activity and caregiver involvement in middle-aged and older adults with Down syndrome. Intellect. Dev. Disabil 55, 97–109 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Xanthopoulos MS et al. Caregiver-reported quality of life in youth with Down syndrome. J. Pediatr 189, 98–104. e1 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Diaz KM Physical inactivity among parents of children with and without Down syndrome: the National Health Interview Survey. J. Intellect. Disabil. Res 64, 38–44 (2020). [DOI] [PubMed] [Google Scholar]
- 235.Hardee JP & Fetters L The effect of exercise intervention on daily life activities and social participation in individuals with Down syndrome: a systematic review. Res. Dev. Disabil 62, 81–103 (2017). [DOI] [PubMed] [Google Scholar]
- 236.O’Roak BJ et al. Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science 338, 1619–1622 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Becker W, Soppa U & Tejedor FJ DYRK1A: a potential drug target for multiple Down syndrome neuropathologies. CNS Neurol. Disord. Drug Targets 13, 26–33 (2014). [DOI] [PubMed] [Google Scholar]
- 238.Mowery CT et al. Trisomy of a Down syndrome critical region globally amplifies transcription via HMGN1 overexpression. Cell Rep. 25, 1898–1911. e5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Tanay A & Regev A Scaling single-cell genomics from phenomenology to mechanism. Nature 541, 331–338 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Annus T et al. The Down syndrome brain in the presence and absence of fibrillar β-amyloidosis. Neurobiol. Aging 53, 11–19 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; Describes the age of onset of amyloid deposition in the brain of individuals with DS using PET imaging.
- 241.Rafii MS Tau PET imaging for staging of Alzheimer’s disease in Down syndrome. Dev. Neurobiol 79, 711–715 (2019). [DOI] [PubMed] [Google Scholar]
- 242.Fortea J et al. Plasma and CSF biomarkers for the diagnosis of Alzheimer’s disease in adults with Down syndrome: a cross-sectional study. Lancet Neurol. 17, 860–869 (2018). [DOI] [PubMed] [Google Scholar]; Explores age-related changes in fluid biomarkers associated with AD in individuals with DS to show similarities with sporadic AD.
- 243.Strydom A et al. Neurofilament light as a blood biomarker for neurodegeneration in Down syndrome. Alzheimer’s Res. Ther 10, 39 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Xiao MF et al. NPTX2 and cognitive dysfunction in Alzheimer’s disease. eLife 6, e23798 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Rafii MS et al. Plasma neurofilament light and Alzheimer’s disease biomarkers in Down syndrome: results from the Down syndrome biomarker initiative (DSBI). J. Alzheimers Dis 70, 131–138 (2019). [DOI] [PubMed] [Google Scholar]
- 246.Das I & Reeves RH The use of mouse models to understand and improve cognitive deficits in Down syndrome. Dis. Model.Mech 4, 596–606 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Duchon A et al. Identification of the translocation breakpoints in the Ts65Dn and Ts1Cje mouse lines: relevance for modeling Down syndrome. Mamm. Genome 22, 674–684 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Reinholdt L et al. Molecular characterization of the translocation breakpoints in the Down syndrome mouse model Ts65Dn. Mamm. Genome 22, 685–691 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Kanekiyo T, Xu H & Bu G ApoE and Aβ in Alzheimer’s disease: accidental encounters or partners? Neuron 81, 740–754 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Startin CM et al. The LonDownS adult cognitive assessment to study cognitive abilities and decline in Down syndrome. Wellcome Open Res. 1, 11 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Sinai A et al. Predictors of age of diagnosis and survival of Alzheimer’s disease in Down syndrome. J. Alzheimers Dis 61, 717–728 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]