

Abstract
The G protein-coupled receptor 52 (GPR52) is an orphan receptor that is selectively expressed in the striatum and regulates various brain functions through activation of cAMP-dependent pathways. GPR52 has been identified as a promising therapeutic target for central nervous system disorders including schizophrenia and substance use disorders. Here, a series of novel GPR52 agonists were designed, synthesized, and evaluated based on compound 4. Several potent and efficacious GPR52 agonists (12c, 23a, 23d, 23e, 23f, and 23h) were identified with nanomolar range potency based on a systematic structure–activity relationship exploration. Further studies of 12c indicate enhanced efficacy, excellent target selectivity, and pharmacokinetic properties including good brain permeability. In vivo proof-of-concept investigations revealed that 12c displayed antipsychotic-like activity by significantly inhibiting amphetamine-induced hyperlocomotor behavior in mice. Collectively, our findings have resulted in an efficacious, brain-penetrant GPR52 agonist as a valuable pharmacological tool for investigating the physiological and therapeutic potential of GPR52 activation.
Graphical Absract
INTRODUCTION
G-protein-coupled receptors (GPCRs) are a superfamily of seven-transmembrane spanning receptor proteins, with over 800 members. GPCRs play critical roles in cellular communication and regulate a wide range of physiological and pathological processes.1 To date, GPCRs have accounted for approximately 30% of all U.S. Food and Drug Administration (FDA)-approved drugs for the therapies of numerous human diseases including cancer, central nervous system (CNS) disorders, metabolic disorders, inflammation diseases, infection, immune diseases, and others.2–5 However, only a fraction of GPCRs are successfully targeted in drug development,6 and roughly one third of the non-sensory GPCRs remain orphan receptors whose endogenous ligands and physiological functions are largely unknown. Despite limited knowledge of these orphan GPCRs, their crucial roles in physiological signaling pathways and putative druggability make them attractive novel therapeutic targets.2,7,8
GPR52 is an understudied brain orphan receptor selectively expressed in the striatum and cortex with the highest level of expression in the nucleus accumbens (NAc).9,10 The selective expression of GPR52 in the NAc and other striatal regions suggests an important function for this receptor in primary striatal neurons and broader corticostriatal circuitry.11 Moreover, GPR52 is colocalized exclusively with dopamine D2 receptors in medium spiny neurons (MSNs) in the striatum and has lesser expression in neurons of the medial prefrontal cortex.10 Notably in transgenic mice, the overexpression of GPR52 significantly decreased methamphetamine-induced locomotion, while GPR52 knockout mice showed an anxiolytic-like phenotype.10 Studies have indicated that GPR52 couples to Gas/olf- G proteins to activate adenylyl cyclase and modulate 5′-cyclic adenosine monophosphate (cAMP) signaling and displays high levels of constitutive activity.12,13 Thus, GPR52 signaling via cAMP could oppose activity of D2 signaling in the striatum while stimulating the D1/N-methyl-d-aspartate (NMDA) function in the frontal cortex.14 Therefore, GPR52 may serve as a promising novel target for psychiatric disorders10 including schizophrenia15 and substance use disorders (SUDs).16
To date, the natural ligand of GPR52 has not been elucidated, but several surrogate ligands have been reported.14,16–18 Most recently, Lin et al. found that the orthosteric ligand binding pocket of GPR52 (PDB codes: 6LI1 and 6LI2) was occupied by its extracellular loop 2 (ECL2).12 They suggested that GPR52 was an unprecedented self-activating receptor and GPR52 ligands could function as allosteric agonists to control GPR52 self-activation.12 Compound 1 (Figure 1) is the first reported potent GPR52 agonist with a half maximal effective concentration (EC50) of about 30 nM, on the basis of their high-throughput screening (HTS) hits.14 Particularly, compound 1 had excellent bioavailability (F = 73%) and brain penetration (brain/plasma AUC ratio = 0.94).14 Additionally, compound 1 significantly inhibited rodent methamphetamine-induced hyperactivity at a dose of 3 mg/kg. However, compound 1 has some drawbacks including high lipophilicity/poor aqueous solubility, which limit its application for further use.18 Further modifications of compound 1 resulted in optimized compound 2 (Figure 1) with slightly improved GPR52 potency (EC50 = 21 nM and Emax = 103%) and water solubility (21 μg/mL at pH 6.8) and similar activity to inhibit stimulant-induced hyperactivity.18 Interestingly, the cocrystal structure of GPR52 and compound 2 (PDB code: 6LI0) was recently solved.12 Compound 2 was found located in a small extracellular pocket of GPR52 surrounded by ECL2, transmembrane helix 1 (TM1), TM2, and TM7. The agonist formed multiple interactions with GPR52 including hydrogen bonds with residues Cys40, Glu191, Ile189, and Asp188 in ECL2, π–π stacking with residue Phe300 of TM7, and hydrophobic contacts with residues Phe117, Thr303, Trp304, and Ile307. The elucidation of this GPR52 ligand-binding pocket is anticipated to facilitate further drug discovery of potent and selective GPR52 modulators. Recently, Tokumaru et al. reported another series of novel GPR52 agonists.17 Among those GPR52 agonists, compound 3 (Figure 1) was an effective agonist with good potency and efficacy (EC50 of 75 nM and Emax of 122%).17 Animal model studies indicated that compound 3 inhibited MK-801-induced hyperactivity and improved memory recognition.15 Likewise, Arena Pharmaceuticals patented and disclosed a series of 1-heteroaryl-indoline-4-carboxamines as GPR52 agonists, among which, compound 4 displayed potent GPR52 agonist activity.16 Here, we choose compound 4 as a starting point for our medicinal chemistry efforts due to its amenability to chemical synthesis, optimal physical–chemical properties, and lack of previously known structure–activity relationship (SAR). Taken together, GPR52 is emerging as a promising neurotherapeutic target of interest for the treatment of schizophrenia and SUDs, and the reported agonists as well as the recently disclosed cocrystal structural analysis serve as new starting points to facilitate further drug discovery of novel GPR52 ligands as pharmacological tools and potential drug candidates for preclinical and clinical development.
Figure 1.
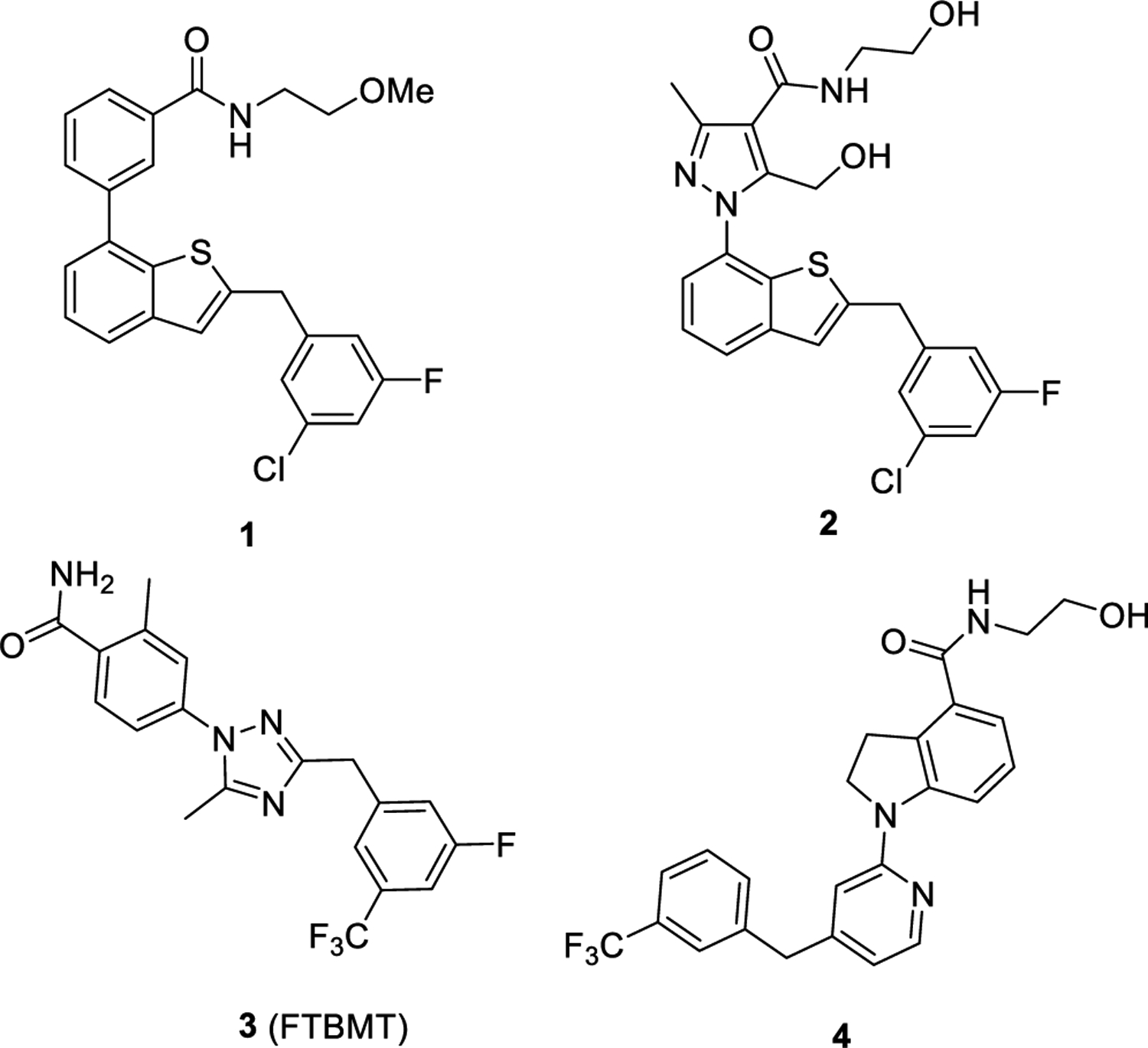
Structures of representative reported GPR52 agonists.
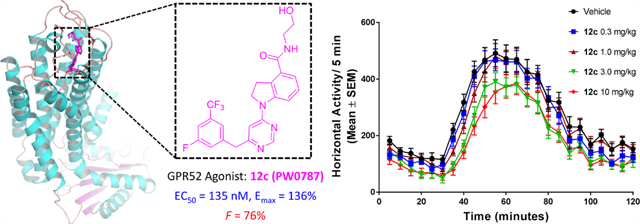
As part of our ongoing research program in understanding the GPR52-associated pharmacology and identifying novel ligands through HTS campaign and structure-based rational drug design, herein, we report our findings in the chemical optimization and pharmacological evaluation of novel GPR52 activators based on compound 416 as the advanced chemical lead to understand the key interactions of agonists with the receptor and to identify improved ligands. Systematic SAR studies around three pharmacological moieties of compound 4 have been explored. A series of novel 1-(pyrimidin-4-yl)indoline-4-carboxamide analogues have been identified as potent and selective GPR52 agonists with nanomolar range potency and varied levels of improved efficacy. This work culminated in discovery of compound 12c (PW0787) with a new pyrimidine core scaffold that is an orally bioavailable, brain-penetrant, potent, and selective GPR52 agonist. This novel ligand dose-dependently suppresses amphetamine-evoked hyperactivity, suggesting a therapeutic potential for neuropsychiatric diseases.
RESULTS AND DISCUSSION
Design.
As described in the Introduction, one of the drawbacks of previously reported GPR52 agonists is the poor aqueous solubility, limiting their use as pharmacological tools or potential drug candidates. The replacement of a CH group with a N atom in aromatic and heteroaromatic rings of the pharmacophore could improve its physicochemical properties for better in vivo pharmacokinetic (PK) properties.19–21 Additionally, compared to the CH group, the N atom has an extra unshared electron pair, which could form hydrogen bonds to potentially improve binding affinity. On this basis, we hypothesized that an extra N atom in the core heteroaromatic ring B of advanced chemical lead 4 could improve its potency as well as physicochemical properties. Therefore, we first altered moiety B (Figure 2, highlight in magenta) by insertion of a nitrogen atom in the pyridine ring of compound 4 as the starting point to systematically pursue SAR studies. Next, we substituted various moieties onto the aromatic ring of moiety C (Figure 2, highlight in cyan). Then, the methylene linker between moieties B and C was also investigated. Finally, we investigated substituents on moiety A (Figure 2, highlight in green) of compound 4 by replacement of aminoethanol with various side chains to understand the critical ligand–receptor interactions.
Figure 2.
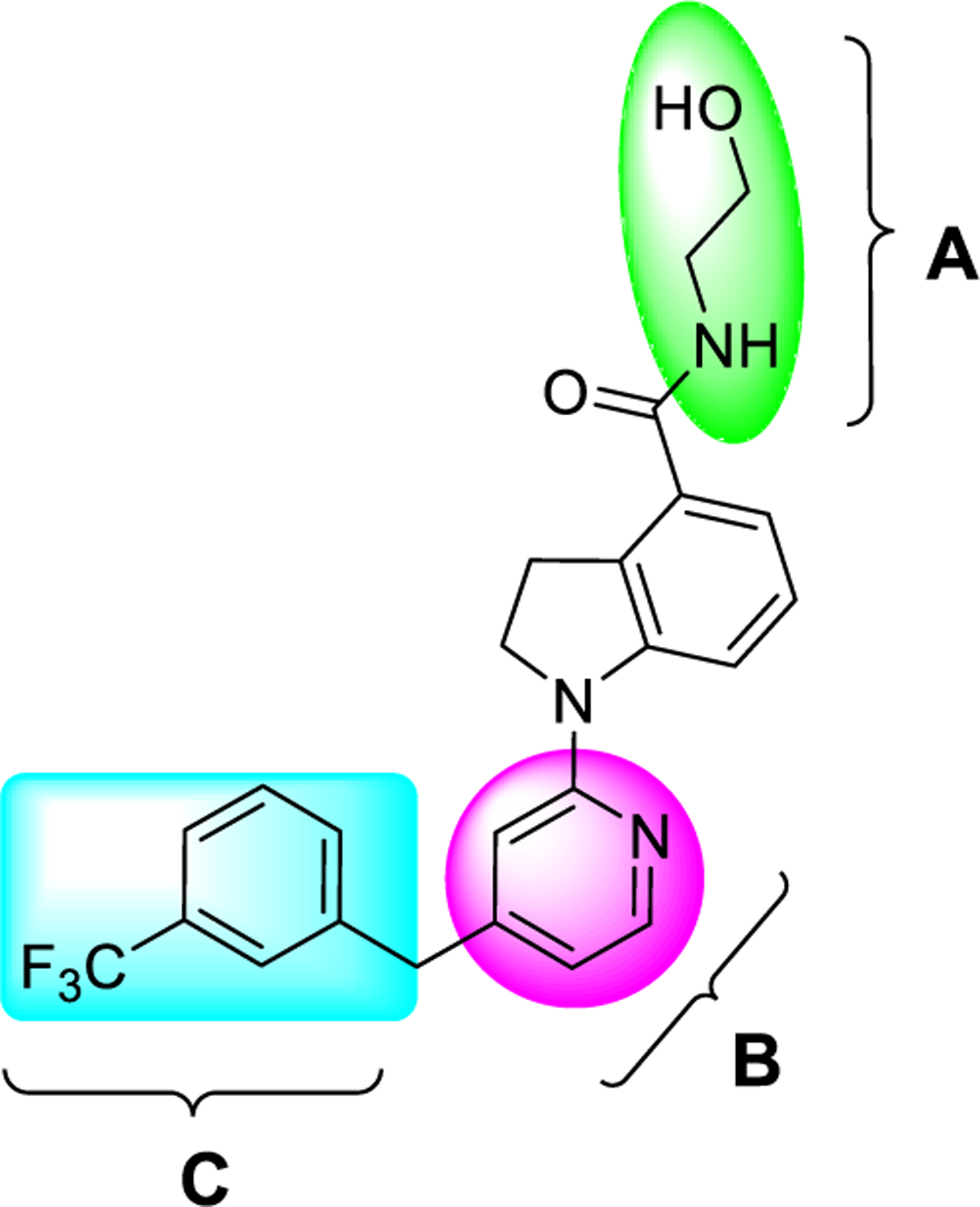
Proposed structural modifications of three substructures (A, B, and C) based on advanced chemical lead 4 for SAR exploration to understand the key interactions of agonists with the receptor GPR52 and to identify newer ligands.
Chemistry.
The synthetic procedures of these newly synthesized GPR52 agonists are depicted in Schemes 1–7. As outlined in Scheme 1, intermediate 6 was prepared by reduction of starting material 5 with triethylsilane and trifluoroacetic acid.22 Intermediates 8a–c were generated by reaction of compound 6 with commercially available compounds 7a–c in the presence of Et3N in yields of 63–67%.23 Intermediates 10a–c were produced via nucleophilic substitution of compounds 8a–c with commercially available 2-(3-fluoro-5-(trifluoromethyl)phenyl)acetonitrile (9a).24 Refluxing of intermediates 10a–c in the HCl/AcOH/H2O mixed solvent system led to key intermediates 11a–c in excellent yields.24 Coupling of intermediates 11a–c with aminoethanol produced compounds 12a–c and resulted in good yields (73–83%).
Scheme 1. Synthetic Routes of Compounds 12a–ca.
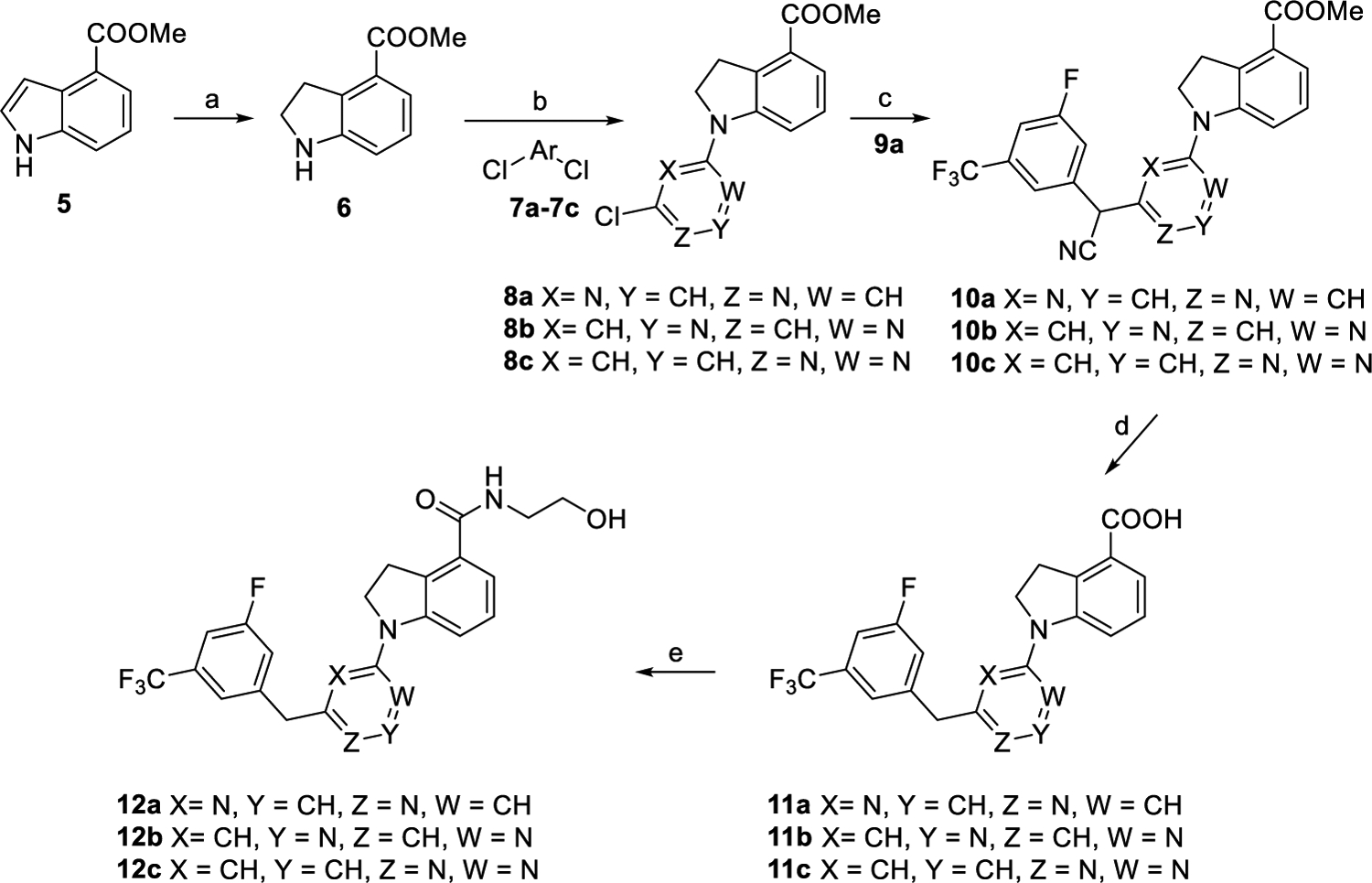
aReagents and conditions: (a) CF3COOH, Et3SiH, CH2Cl2, overnight, and 89%. (b) Et3N, EtOH, rt., overnight, and 63–67%. (c) 2-(3-Fluoro-5-(trifluoromethyl)phenyl)acetonitrile (9a), NaH, DMF, 0 °C to rt., 2 h, and 53–66%. (d) Con. HCl/AcOH/H2O, reflux, overnight, and 91–93%. (e) NH2CH2CH2OH, EDCI, DMAP, DMF, rt., overnight, and 73–83%.
Scheme 7. Synthetic Routes of Compounds 30a–ga.
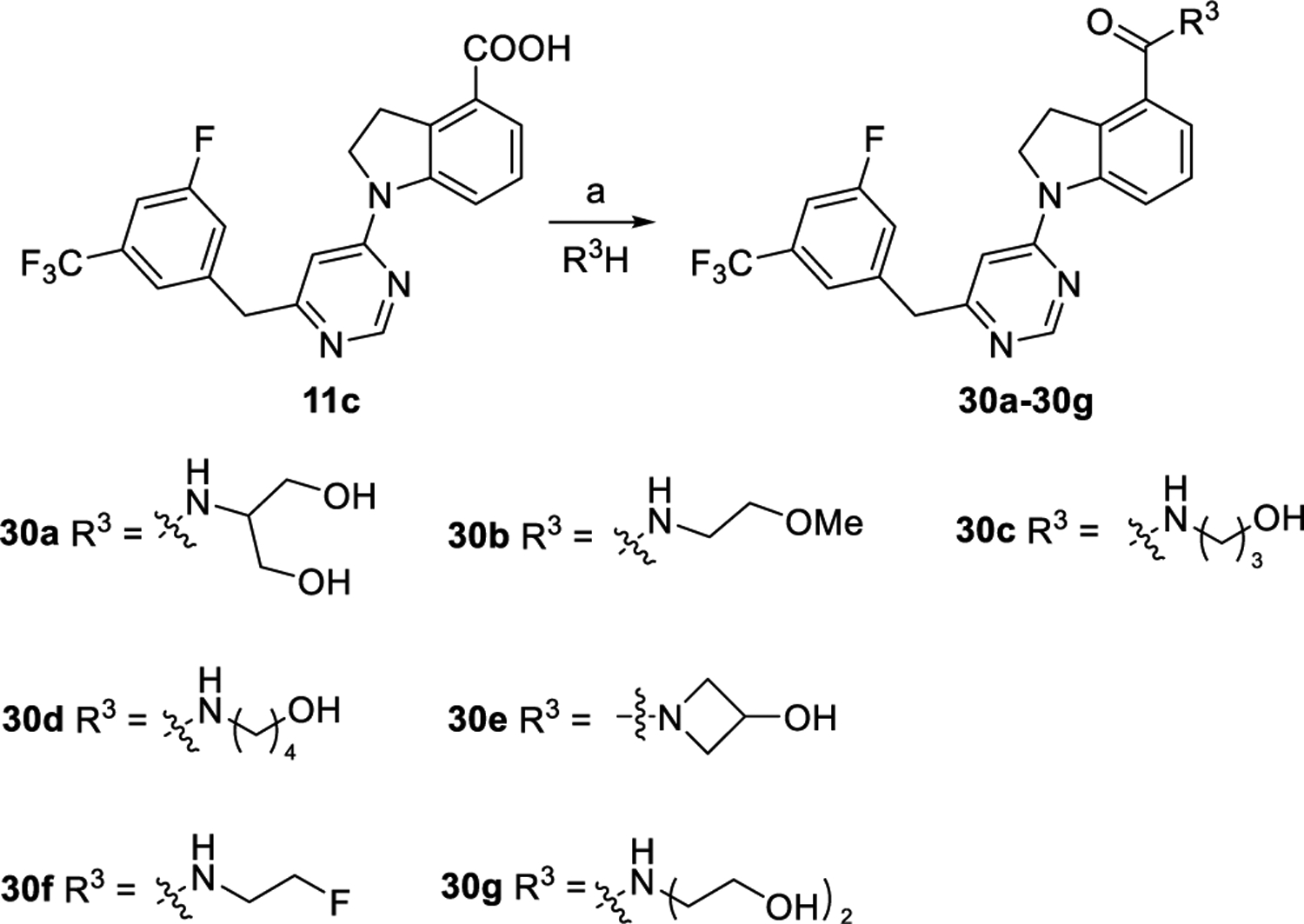
aReagents and conditions: (a) EDCI, DMAP, DMF, rt., overnight, and 37–78%.
As outlined in Scheme 2, intermediate 13 was obtained by reaction of compound 9a with 7a in the presence of NaH. Refluxing of intermediate 13 in the HCl/AcOH/H2O solvent system followed by chlorination with phosphorus oxychloride produced compound 14. Compound 15 was prepared via palladium-catalyzed C–N coupling reaction of 14 with 6 in a yield of 56%. Hydrolysis of intermediate 15 generated key intermediate acid 16. Further, condensation of compound 16 with aminoethanol afforded compound 12d following the same procedure as that of preparing compound 12a.
Scheme 2. Synthetic Routes of Compound 12da.
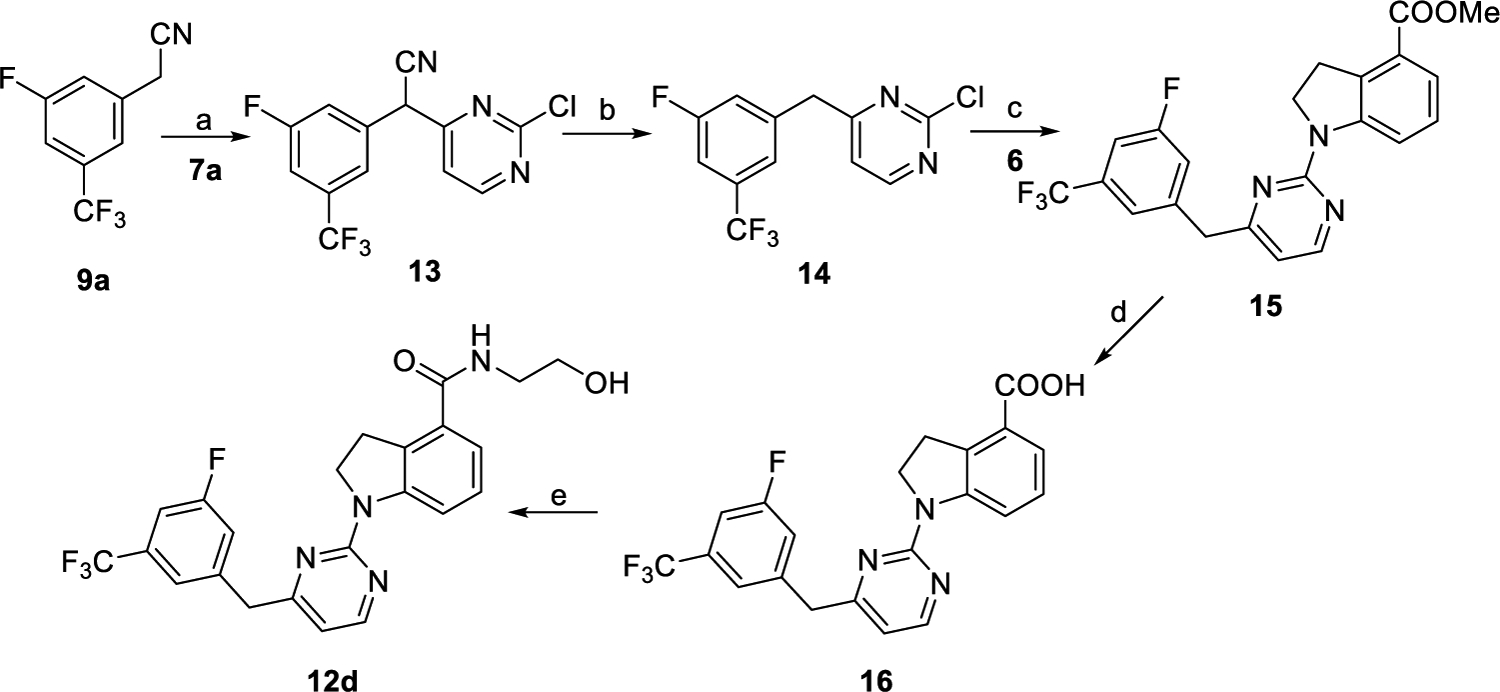
aReagents and conditions: (a) NaH, DMF, 0 °C to rt., 2 h, and 73%. (b) (1) Con. HCl/AcOH/H2O, reflux, and overnight; (2) POCl3, reflux, 3 h, and 51% for two steps. (c) 6, Pd(OAc)2, XantPhos, Cs2CO3, 1,4-dioxane, 100 °C, overnight, and 56%. (d) (1) 2 N NaOH, MeOH, reflux, and 1 h; (2) 2 N HCl, rt., and 95%. (e) NH2CH2CH2OH, EDCI, DMAP, DMF, rt., overnight, and 83%.
As outlined in Scheme 3, with starting material 9a and compounds 7d25 and 7e,25 intermediates 17a and 17b were obtained following the similar synthetic procedure to that of preparing compound 13, respectively. Removing the cyano and the tetrahydro-2H-pyran protection groups of 17a and 17b at the same time led to compounds 18a and 18b in the HCl/AcOH/H2O solvent system. Compounds 18a and 18b were converted into compounds 19a and 19b by a standard chlorination reaction. Key intermediates 20a and 20b were synthesized by C–N coupling reaction of intermediates 19a and 19b with compound 6, respectively, under the palladium-catalyzed conditions. Acids 21a and 21b were prepared by hydrolysis of intermediates 20a and 20b. Compounds 12e and 12f were synthesized following the similar procedure to that of preparing 12a by coupling 21a and 21b with aminoethanol, respectively.
Scheme 3. Synthetic Routes of Compounds 12e and 12fa.
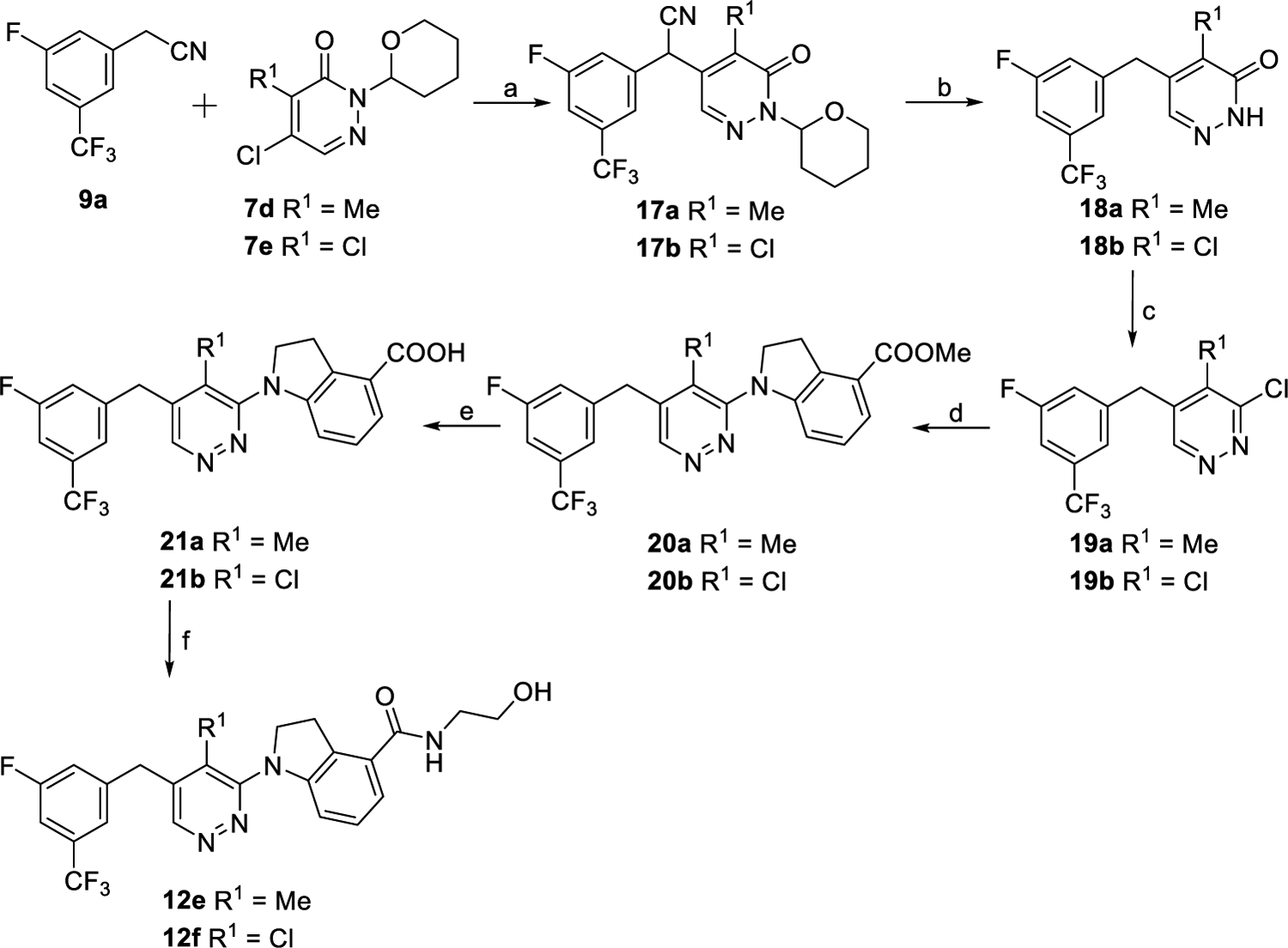
aReagents and conditions: (a) NaH, DMF, 0 °C to rt., 2 h, 36% for 17a, and 63% for 17b. (b) HCl, AcOH, H2O, 120 °C, overnight, 78% for 18a, and 83% for 18b. (c) POCl3, 120 °C, 12 h, 83% for 19a, and 80% for 19b. (d) 6, Pd(OAc)2, XantPhos, Cs2CO3, 1,4-dioxnae, 100 °C, overnight, 64% for 20a, and 60% for 20b. (e) (i) 2 N NaOH, MeOH, reflux, and 1 h; (ii) 2 N HCl, rt., 92% for 21a, and 90% for 21b. (f) NH2CH2CH2OH, EDCI, DMAP, DMF, rt., overnight, 79% for 12e, and 59% for 12f.
Compounds 23a–k were synthesized following the same procedures for preparing 12a from various commercially available substituted 2-phenylacetonitriles (9b–l) (Scheme 4). As outlined in Scheme 5, intermediate 24a was obtained by reaction of compound 9a with compound 7a in the presence of NaH followed by oxidation of the intermediate and then coupling with compound 6. Compounds 24b and 24c were synthesized via reaction of compounds 9a and 9b with compound 8c, respectively, following similar processes to the synthesis of compound 24a. Hydrolysis of esters 24a–c followed by condensation with aminoethanol provided compounds 25a–c in yields of 63–77% over two steps. Treatment of 25a–c with NaBH4 as the reducing agent afforded compounds 25d–f accordingly. Ester intermediates 24a–c were converted into intermediates 26a–c by a reduction reaction and then a hydrolysis reaction. Fluoridation of acid intermediates 26a–c by using diethylaminosulfur trifluoride followed by hydrolysis reactions led to acid compounds 27a–c. Compounds 25g–i were produced following the same procedure as that of preparing 12a from compounds 27a–c.
Scheme 4. Synthetic Routes of Compounds 23a–ka.
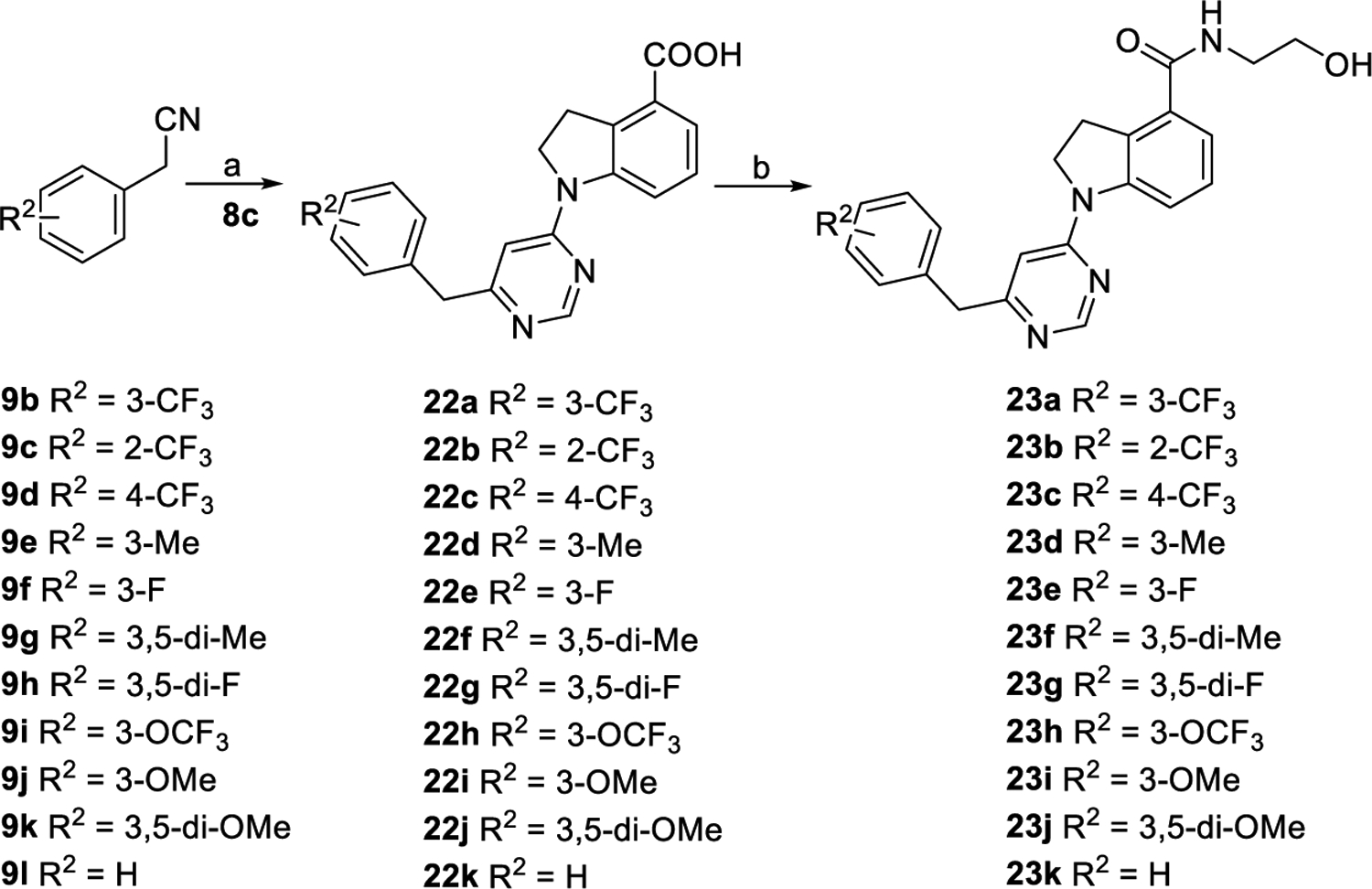
aReagents and conditions: (a) (1) NaH, DMF, 0 °C to rt., and 2 h; (2) Con. HCl/AcOH/H2O, reflux, overnight, and 54–90% for two steps. (b) NH2CH2CH2OH, EDCI, DMAP, DMF, rt., overnight, and 29–83%.
Scheme 5. Synthetic Routes of Compounds 25a–ia.
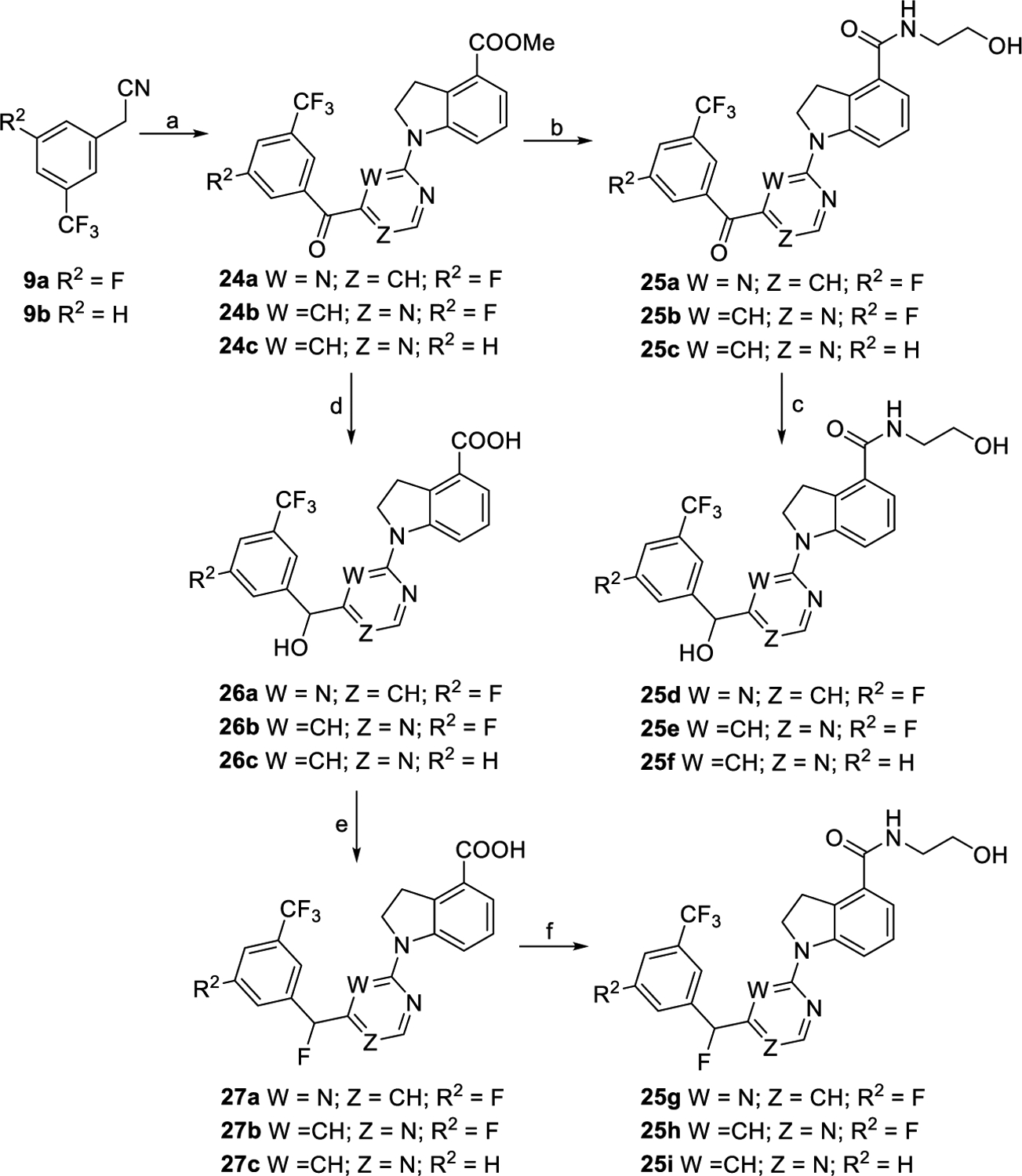
aReagents and conditions: (a) For 24a, (i) 7a, NaH, DMF, 0 °C to rt., and 2 h; (ii) mCPBA, 0 °C to rt., 10 min, and 47% for two steps; (iii) 6, XantPhos, Cs2CO3, Pd(OAc)2, 1,4-dioxane, 100 °C, overnight, and 82%; for 24b and 24c, (i) 8c, NaH, DMF, 0 °C to rt., and 2 h; (ii) mCPBA, 0 °C to rt., 10 min, 49% over two steps for 24b, and 79% over two steps for 24c. (b) (i) Con. HCl/AcOH/H2O, reflux, and overnight; (ii) NH2CH2CH2OH, EDCI, DMAP, DMF, rt., overnight, and 63–77% for two steps. (c) NaBH4, DMF/MeOH, 0 °C to rt., 30 min, and 68–90%. (d) (i) NaBH4, DMF/MeOH, 0 °C to rt., and 30 min; (ii) con. HCl/AcOH/H2O, reflux, overnight, and 78–90%. (e) (i) DAST, CH2Cl2, 0 °C to rt., and 10 min; (ii) THF/H2O, reflux, overnight, and 60–76% for two steps. (f) NH2CH2CH2OH, EDCI, DMAP, DMF, rt., overnight, and 68–86%.
As depicted in Scheme 6, compounds 29a–c were produced via coupling reaction of 8c with corresponding commercially available compounds 28a–c in yields of 63–71%. Subsequently, intermediates 29a–c were converted to corresponding final compounds 25j–l by acidolysis and coupling with aminoethanol, following the similar procedures to that of preparing 25a. Compounds 30a–g were prepared by coupling 11c with corresponding aminoethanol derivatives following the similar procedure to that of preparing 12a, as outlined in Scheme 7.
Scheme 6. Synthetic Routes of Compounds 25j–la.
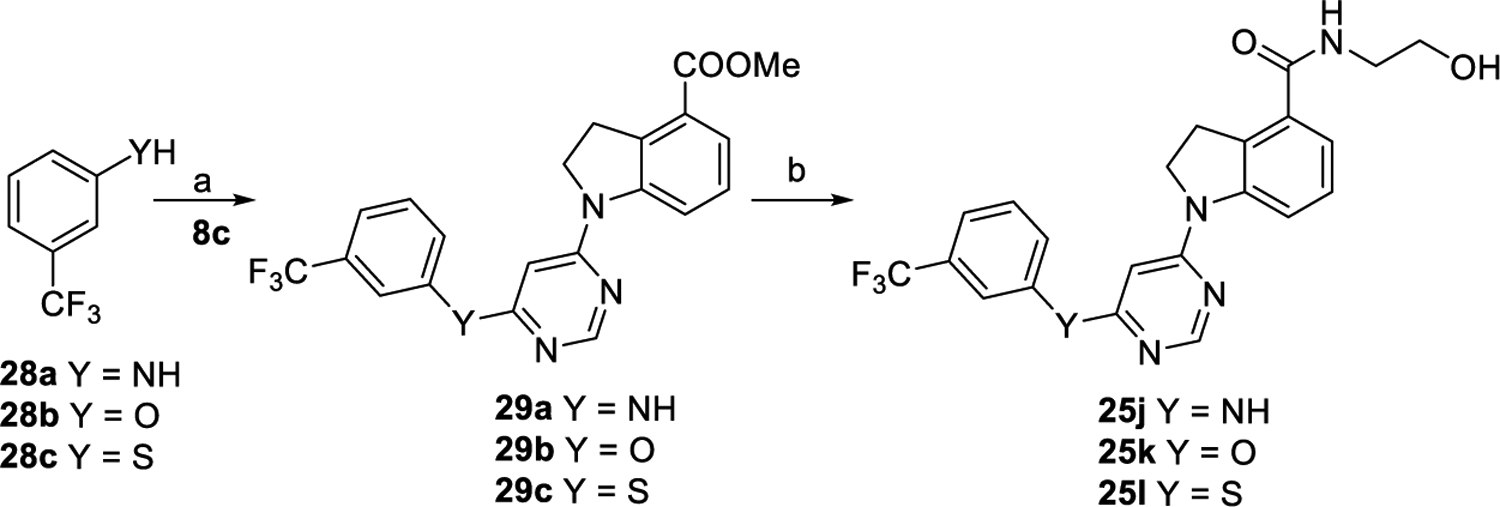
aReagents and conditions: (a) For 29a, Pd(OAc)2, XantPhos, Cs2CO3, 1,4-dioxnae, 100 °C, overnight, and 69%; for 29b and 29c, Cs2CO3, DMF, 120 °C, overnight, 63% for 29b, and 71% for 29c. (b) (i) Con. HCl/AcOH/H2O, reflux, and overnight; (ii) NH2CH2CH2OH, EDCI, DMAP, DMF, rt., overnight, and 43–73% for two steps.
In Vitro Evaluation of GPR52 Activation.
Newly synthesized compounds were evaluated in a twelve-point concentration response for GPR52 agonist activity using the Glosensor cAMP assay in HEK293 cells transiently expressing human GPR52. Transient expression of GPR52 resulted in a dramatic increase in basal cAMP levels (>100-fold over an empty vector, Supporting Information, Figure S1), indicating that GPR52 displays high constitutive activity for the cAMP pathway. As summarized in Tables 1–4, the EC50 (nM) indicates the potency of these novel GPR52 agonists, while the efficacy (Emax) indicates the maximal activity to increase cAMP. Previously reported GPR52 agonist compound 4 was used as the reference compound to highlight SAR comparisons.16 Emax (%) was evaluated with compound 4 as 100% and 0.5% DMSO (vehicle control) as 0%. Compound 4 exhibited very good potency and robust efficacy in the cAMP assay (EC50 of 119 nM and Emax of ~3-fold over basal values, Supporting Information, Figure S1). All synthesized ligands were screened in the assay in a full concentration response (0.1 nM to 30 μM). Some data points, at higher concentrations, were excluded from pharmacological analysis due to limited aqueous solubility (as shown in Supporting Information, Figure S1).
Table 1.
EC50 and Emax of Compounds 12a–f
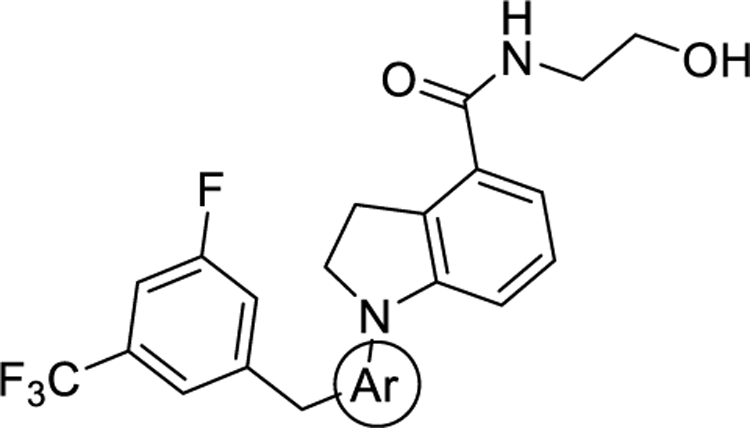
| ||||
|---|---|---|---|---|
| Compound | Ar | ClogPa | EC50 (nM)b | Emax (%)b |
| 4 | 4.11 | 119 ± 18 | 100 ± 5 | |
| 12a |
|
3.64 | 373 ± 35 | 144 ± 7 |
| 12b |
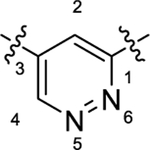
|
3.64 | 158 ± 48 | 158 ± 12 |
| 12c |
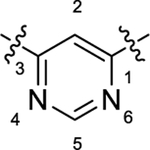
|
3.64 | 135 ± 16 | 136 ± 6 |
| 12d |
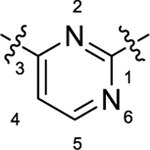
|
3.64 | 186 ± 44 | 138 ± 8 |
| 12e |
|
3.95 | 754 ± 62 | 119 ± 6 |
| 12f |
|
4.30 | 562 ± 80 | 97 ± 7 |
The values are the mean ± SEM of at least three independent experiments. Emax (%) is the efficacy maximum of the compounds in the cAMP assay relative to compound 4 as 100% and 0.5% DMSO as 0%.
Table 4.
EC50 and Emax of Compounds 30a–g
| ||||
|---|---|---|---|---|
| Compound | R3 | ClogPa | EC50 (nM)b | Emax (%)b |
| 4 | 4.11 | 119 ± 18 | 100 ±5 | |
| 30a |
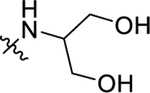
|
3.00 | 346 ± 19 | 115 ± 15 |
| 30b |
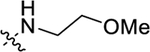
|
4.30 | 489 ± 86 | 119 ± 7 |
| 30c |
|
4.03 | 275 ± 13 | 120 ± 15 |
| 30d |
|
4.42 | 399 ± 51 | 120 ± 18 |
| 30e |
|
3.73 | 760 ± 176 | 111 ± 10 |
| 30f |
|
4.62 | 431 ± 7 | 129 ± 17 |
| 30g |
|
3.35 | 673 ± 135 | 119 ± 15 |
The values are the mean ± SEM of at least three independent experiments. Emax (%) is the efficacy maximum of the compounds in the cAMP assay relative to compound 4 as 100% and 0.5% DMSO as 0%.
As summarized in Table 1, to begin a systematic SAR study of the scaffold of compound 4,16 we first investigated the core ring B (Figure 2, highlight in magenta) by insertion of an additional N atom in the pyridine ring of compound 4. Replacement of the pyridine ring by a pyrimidine ring with two N atoms at positions 2 and 4, leading to compound 12a, resulted in ~3-fold loss of potency with an EC50 value of 373 nM relative to compound 4. However, compared to compound 4, the efficacy of compound 12a increased with an Emax of 144%. In addition, 12a has a more favorable ClogP relative to that of 4 (3.64 vs 4.11), suggesting that 12a may have a better PK profile. In the further SAR studies, the pyridine ring of compound 4 was replaced with a pyridazine ring to obtain compound 12b (EC50 = 158 nM and Emax = 158%), which exhibited comparable potency to that of 4, with improved efficacy. Next, modification of the pyridine ring of compound 4 to a pyrimidine ring with the two N atoms fixed at positions 4 and 6 led to compound 12c (Table 1 and Figure 3A). Compound 12c showed equal potency as that of lead 4 (135 nM vs 119 nM) but interestingly resulted in a superior efficacy (Emax of 136%), which was the best compound among this series. However, when the two N atoms were moved to positions 2 and 6 to obtain 12d, the potency decreased slightly (EC50 = 186 nM) compared to 12c. To explore the effect of adding a substituent on potency, we introduced a methyl or a chlorine at the 2-position on compound 12b, leading to compounds 12e and 12f, respectively. However, both compounds showed substantial loss of potency (>4-fold decreased EC50), indicating that adding a substituent on core moiety B was not favorable.
Figure 3.
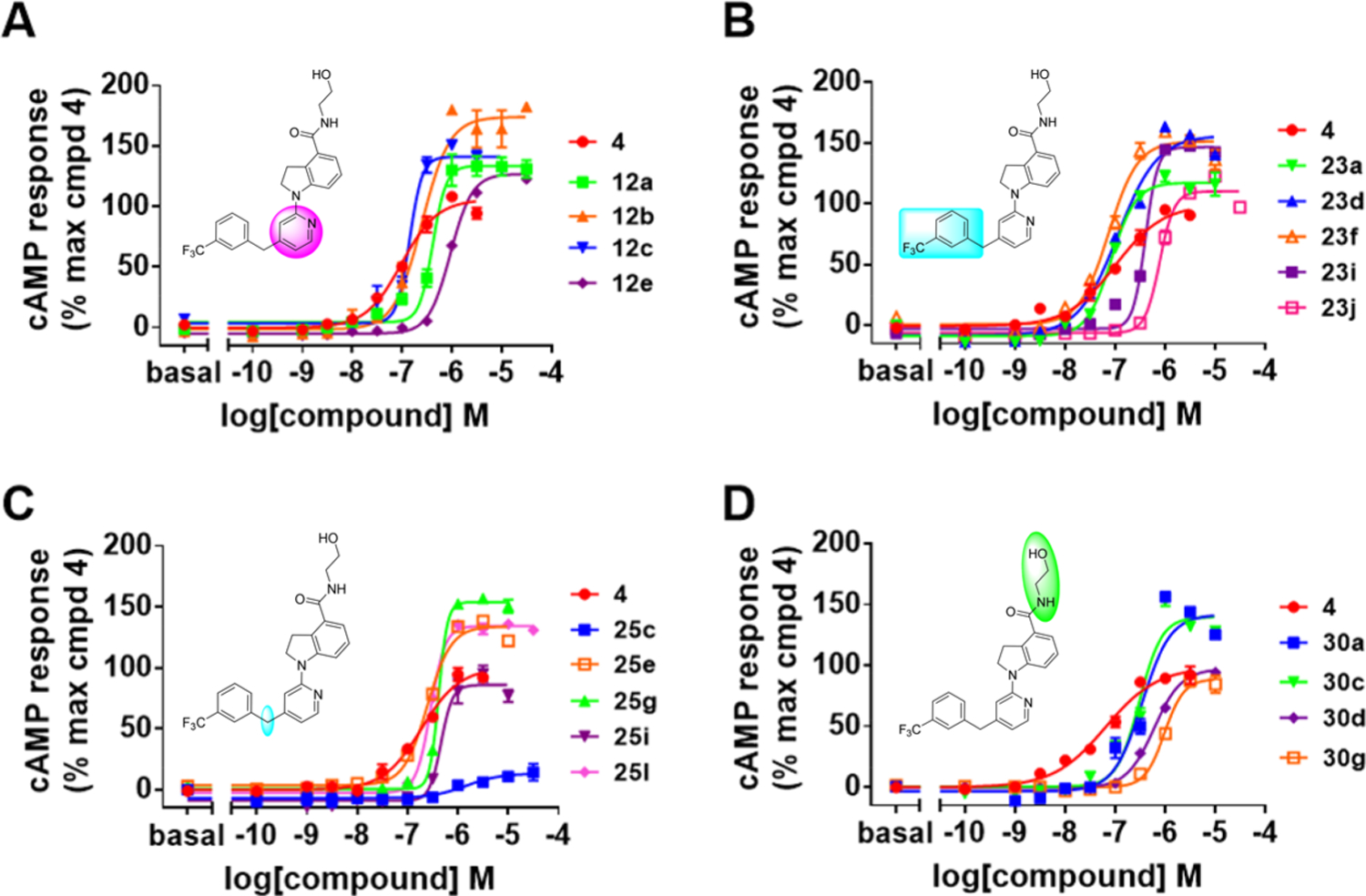
Concentration responses of select GPR52 agonists on cAMP signaling in HEK293 cells. HEK293 cells expressing human GPR52 and the cAMP Glosensor reporter were subjected to twelve-point concentration response (0.3 nM–30 μM) testing. (A) Modifications around ring B (magenta): comparison with compounds 4, 12a, 12b, 12c, and 12e. (B) Modifications around ring C (cyan): comparison with compounds 4, 23a, 23d, 23f, 23i, 23j. (C) Modifications around carbon linker (cyan): comparison with compounds 4, 25c, 25e, 25g, 25i, and 25l. (D) Modifications around head group A (green): comparison with compounds 4, 30a, 30c, 30d, and 30g. All results are presented as the mean ± SEM from triplicate testing in a representative experiment, with similar results observed in 3–17 experiments.
Having identified an optimal heterocycle in moiety B that maintains potency while increasing efficacy, we next focused on probing the role of the substituents on the benzene ring of moiety C (Figure 2, highlight in cyan), with results summarized in Table 2. To evaluate the importance of the F substituent, we removed the F from compound 12c, leading to compound 23a, resulting in a slight increase in potency (EC50 of 101 nM) and minor decrease in efficacy relative to compound 12c (127% vs 136%) (Table 2). Compounds 23b and 23c were then designed and synthesized to explore whether the CF3 position had an influence on agonist potency. CF3 at the ortho-position (23b) resulted in a dramatic loss of potency (>7-fold loss) compared with CF3 at the meta-position (23a). Remarkably, when the CF3 moiety was moved to the para-position (23c), a similar potency and efficacy was observed to that of 23a (109 nM vs 101 nM for EC50 and 136% vs 127% for Emax (Table 2 and Figure 3B)). From these results, we can conclude that the substituent position on the benzene ring of moiety C has an important role in compound potency and their preferred order is the meta > para > ortho position. Further, we probed the electronic properties of substituents on the benzene ring of moiety C. Replacement of meta-CF3 of 23a with a methyl group led to compound 23d, which displayed the best potency among this series with an EC50 of 90 nM while maintaining excellent efficacy with an Emax of 144% (Table 2 and Figure 3B). Similarly, compound 23e was obtained by substitution of meta-CF3 of 23a with a F atom, which displayed no change in potency, an EC50 of 106 nM, and maintained excellent efficacy, an Emax of 148%, (Table 2). Next, we probed whether a second additional electron-withdrawing moiety in the 5-position would further improve potency and efficacy. Disubstituted variants of 23d and 23e, leading to compounds 23f and 23g, respectively, displayed identical pharmacologic profiles as their monosubstituted counter parts (Table 2 and Figure 3B). These data suggest that the addition of an extra electron-withdrawing substituent at the 5-position of the benzene ring C has limited influence on the potency and efficacy. However, substitution of meta-CF3 of 23a with an electron-donating group such as OCF3 (23h), OMe (23i), and 3,5-di-OMe (23j) dramatically decreased the potency as well as the efficacy (Table 2 and Figure 3B). Moreover, compound 23k was synthesized without any substituent on the benzene ring C, resulting in ~3-fold loss of potency relative to 23a, suggesting that an electron-withdrawing substituent (e.g., 23a, 23c, and 23e) on the benzene ring of moiety C is favorable for the potency. Taken together, these findings suggest that electron-withdrawing or small alkyl substituents at the 3-position are favorable for improved potency, with an additional small substituent at the 5-position also well-tolerated, while compounds without any substituent on the benzene ring or with electron-donating groups are not tolerated.
Table 2.
EC50 and Emax of Compounds 23a–k
| ||||
|---|---|---|---|---|
| compound | R2 | ClogPa | EC50 (nM)b | Emax (%)b |
| 4 | 4.11 | 119 ± 18 | 100 ± 5 | |
| 23a | 3-CF3 | 3.50 | 101 ± 31 | 127 ± 10 |
| 23b | 2-CF3 | 3.50 | 711 ± 160 | 115 ± 6 |
| 23c | 4-CF3 | 3.50 | 109 ± 19 | 136 ± 7 |
| 23d | 3-Me | 2.79 | 90 ± 19 | 144 ± 9 |
| 23e | 3-F | 2.62 | 106 ± 35 | 148 ± 14 |
| 23f | 3,5-di-Me | 3.10 | 97 ± 20 | 140 ± 11 |
| 23g | 3,5-di-F | 2.76 | 115 ± 19 | 142 ± 10 |
| 23h | 3-OCF3 | 3.38 | 131 ± 24 | 113 ± 11 |
| 23i | 3-OMe | 2.49 | 351 ± 6 | 122 ± 8 |
| 23j | 3,5-di-OMe | 2.50 | 850 ± 35 | 113 ± 2 |
| 23k | H | 2.48 | 292 ± 17 | 134 ± 4 |
The values are the mean ± SEM of at least three independent experiments. Emax (%) is the efficacy maximum of the compounds in the cAMP assay relative to compound 4 as 100% and 0.5% DMSO as 0%.
To probe the impact of the flexibility and steric availability around the linker between moieties B and C, compounds 25a–l were designed, synthesized, and evaluated, with the results summarized in Table 3. We first replaced the methylene linker with a carbonyl moiety to yield compounds 25a–c, which were found to dramatically decrease the potency and efficacy, likely due to limited flexibility. Reduction of the carbonyl moiety of compounds 25a–c produced compounds 25d–f, and this restored a certain degree of the potency and efficacy but still at least 3-fold less potency than compound 23a. Fluorination of compounds 25d–f yielded compounds 25g–i, which failed to improve the potency or efficacy (Table 3 and Figure 3C). Replacement of the methylene linker of compound 23a with either an NH, O, or S led to corresponding compounds 25j–l; however, none of these three compounds showed favorable potency and efficacy (Table 3 and Figure 3C). Taken together, these results suggest that GPR52 ligands of this scaffold need to have a flexible and sterically limited linker to achieve good potency. These SAR results are consistent with the recently disclosed cocrystal structure of compound 2 and GPR52, suggesting that the GPR52 binding region for these molecules is a narrow sterically limited pocket.12
Table 3.
EC50 and Emax of Compounds 25a–l
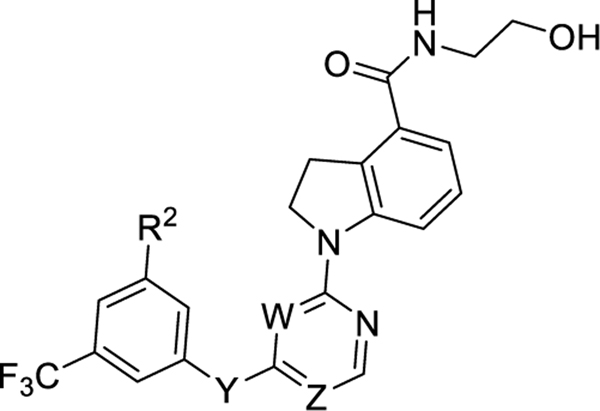
|
|||||||
|---|---|---|---|---|---|---|---|
| compound | R2 | Y | W | Z | ClogPa | EC50 (nM)b | Emax (%)b |
| 4 | 4.11 | 119 ± 18 | 100 ± 5 | ||||
| 25a | F | CO | N | CH | 3.28 | 1589 ± 166 | 96 ± 7 |
| 25b | F | CO | CH | N | 3.28 | 1693 ± 437 | 22 ± 3 |
| 25c | H | CO | CH | N | 3.14 | 1008 ± 237 | 22 ± 4 |
| 25d | F | CHOH | N | CH | 3.13 | 399 ± 68 | 130 ± 9 |
| 25e | F | CHOH | CH | N | 3.13 | 338 ± 26 | 134 ± 28 |
| 25f | H | CHOH | CH | N | 2.99 | 560 ± 19 | 89 ± 4 |
| 25g | F | CHF | N | CH | 4.10 | 329 ± 113 | 144 ± 9 |
| 25h | F | CHF | CH | N | 4.10 | 330 ± 38 | 85 ± 5 |
| 25i | H | CHF | CH | N | 3.97 | 557 ± 273 | 83 ± 5 |
| 25j | H | NH | CH | N | 3.66 | 1105 ± 24 | 81 ± 2 |
| 25k | H | O | CH | N | 3.70 | 2404 ± 292 | 99 ± 4 |
| 25l | H | s | CH | N | 4.06 | 371 ± 47 | 123 ± 6 |
The values are the mean ± SEM of at least three independent experiments. Emax (%) is the efficacy maximum of the compounds in the cAMP assay relative to compound 4 as 100% and 0.5% DMSO as 0%.
Finally, to detect the impact of an amino alcohol side chain in moiety A, compounds with various lengths and flexibilities were designed through different strategies, including introducing extra hydroxyl groups (30a and 30g), modification of the terminal hydroxyl group (30b and 30f), and changing the length of the alkyl linker between amino and hydroxyl groups (30c–e) (Figure 3D). Unfortunately, these seven compounds substantially lost GPR52 potency compared to compound 12c (Table 4), indicating that moiety A is critical and unlikely amenable for further optimization.
Molecular Docking Study of Compound 12c with GPR52.
To understand the potential binding mode and the receptor–ligand interactions that are important for agonist activity at GPR52, molecular docking using Schrödinger Drug Discovery Suite was employed. Interestingly, the cocrystal structure of GPR52 bound to compound 2 was recently solved (PDB code: 6LI0), affording an unprecedented opportunity to pursue computational docking studies with an orphan GPCR.12 Compound 12c was selected as the example of our optimized compounds to further explore the binding interactions with GPR52. As shown in Figure 4, the docking results indicated that compound 12c has energetically favorable interactions with the narrow pocket surrounded by ECL2, TM1, TM2, and TM7 and forms a similar binding pose to compound 2 with GPR52 in the solved cocrystal structure.12 Notably, there are three critical hydrogen bonding interactions between compound 12c and the residues of the ECL2 and TM1 of the receptor: the 12c terminal hydroxyl group forms hydrogen bonds with residues Glu191 and Ile189 of ECL2, and the 12c carbonyl group of the amide forms a hydrogen bond with residue Cys40 from TM1. This binding model may explain the significant activity loss when replacing the amino alcohol side chain into other side chains. The pyrimidine ring (core ring B) forms a π–π stacking interaction with the residue Phe300 of TM7. Moreover, the benzene ring (moiety C) of compound 12c pointed into a narrow hydrophobic pocket and formed hydrophobic interactions with residues Phe117, Cys94, Tyr185, Ile47, Ile121, Ile307, and Trp304. This may explain why modifications with only small substitutions on moiety C are tolerated, and why the hydrophobic groups (e.g., F and CF3) are more favorable for the binding to drive potency. This docking also indicates that the binding pocket near ECL2 is a hot spot of interaction for both previously identified GPR52 agonists as well as 12c and likely other indoline-carboxamide-based ligands.
Figure 4.
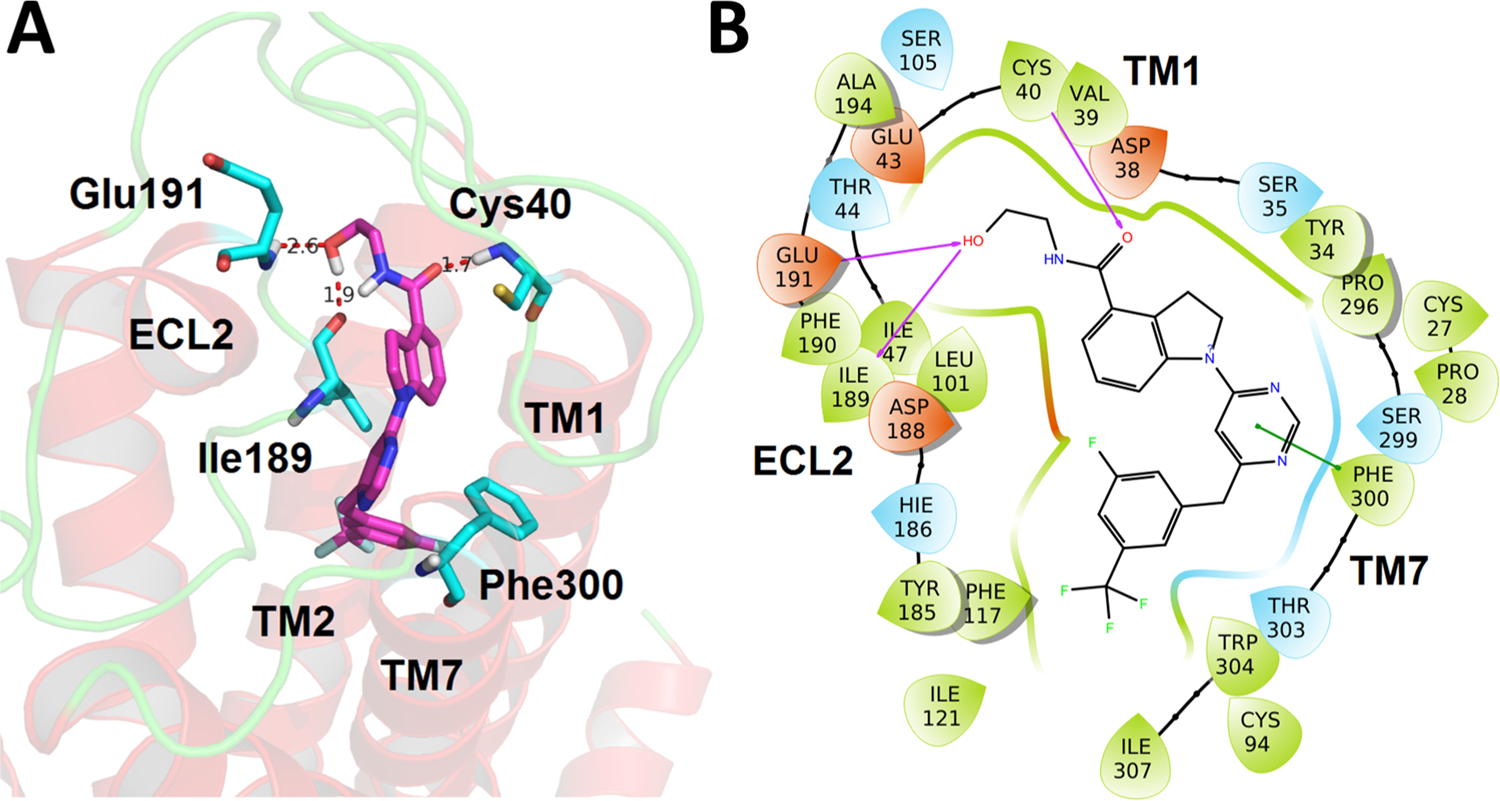
Putative binding mode and molecular docking of compound 12c with GPR52 (PDB code: 6LI0). (A) Docking of compound 12c (magenta) into the binding pocket of GPR52. Important residues are drawn as sticks. Hydrogen bonds are shown as dashed red lines. (B) Docking of compound 12c into the binding pocket of GPR52 in 2D view. Hydrogen bonds are shown as magenta lines, and π–π interaction is shown as a green line.
In Vitro Assessment of Off-Target Effects of Compound 12c.
Considering the potential drug-like properties of chemical scaffolds and overall in vitro activities of GPR52 activation, compound 12c was selected as a representative chemical probe among the identified active analogs to pursue further pharmacological evaluations in a proof-of-concept study. To evaluate if the cAMP signaling activity of optimized compound 12c is indeed due to GPR52 agonist activation, 12c was further tested in control studies using HEK293 cells lacking GPR52. Results determined that 12c did not increase cAMP in the cells lacking GPR52 (Supporting Information, Figure S2). Therefore, compound 12c regulates cAMP increases through a direct interaction with GPR52 in this cellular system. To determine the selectivity of 12c against other GPCRs, a broad-panel counter screening was tested by the National Institute of Mental Health Psychoactive Drug Screening Program (NIMH-PDSP).26 This testing determined that compound 12c at a concentration of 10 μM displayed no significant binding affinity (Ki) at over 30 brain receptors or channels, including important GPCRs that are current targets of antipsychotic medications (e.g., 5-HT2A and D2 receptors), as shown in Table 5. In addition, compound 12c at 10 μM did not show any human ether-a-go-go-related gene (hERG) potassium channel binding activity (Table 5), indicating that compound 12c should not have undesirable hERG inhibition known to cause cardiotoxicity.27
Table 5.
Broad-Panel Counter Screening of 12c against Other GPCRs and Transportersa
| GPCRs | % inhibition (10 μM)b | Ki (μM)b | GPCRs, transporters, ion channels | % inhibition (10 μM) | Ki (μM)b |
|---|---|---|---|---|---|
| 5-HT1A | 8.68 | ND | D1 | 12.29 | ND |
| 5-HT1B | 6.3 | ND | D2 | 2.51 | ND |
| 5-HT1D | 29.64 | ND | D3 | 10.64 | ND |
| 5-HT1E | 7.71 | ND | D4 | 29.91 | ND |
| 5-HT2A | 10.62 | ND | D5 | −7.17 | ND |
| 5-HT2B | 28.69 | ND | DAT | 24.83 | ND |
| 5-HT2C | 52.62 | >10 | GABAA | 5.98 | ND |
| 5-HT3 | 26.82 | ND | H1 | 6.43 | ND |
| 5-HTSA | 13.95 | ND | H2 | 22.36 | ND |
| 5-HT6 | 7.98 | ND | KOR | 1.48 | ND |
| 5-HT7A | 5.16 | ND | M1 | 2.86 | ND |
| Alpha1A | 13.56 | ND | M2 | −1.87 | ND |
| Alpha1B | −5.42 | ND | M3 | 45.38 | ND |
| Alpha2A | 4.45 | ND | M4 | 29.26 | ND |
| Beta1 | 17.2 | ND | M5 | 0.82 | ND |
| Beta2 | −14.07 | ND | MOR | 16.24 | ND |
| hERG | 11.04 | ND |
The broad-panel counter screening of 12c against a panel of GPCRs and transporters was generously provided by the NIMH Psychoactive Drug Screening Program.
The values are the mean percent inhibition of binding from at least three independent experiments, SEM < 20%. “ND” means that a Ki binding affinity was not detected.
In Vivo PK Profile of Compound 12c.
Based on its potency and efficacy as well as potential drug-like properties, compound 12c was selected as the representative compound for further PK evaluations. Compound 12c was evaluated in rats after a single dose of 20 mg/kg by oral (PO) or 10 mg/kg by intravenous (IV) administration, and the results are summarized in Table 6. Compound 12c has excellent plasma exposure after PO (AUC0-inf = 13,749 ng·h/mL) and IV dosing (AUC0-inf = 9030 ng·h/mL), as well as high maximum serum concentration following PO (Cmax = 3407 ng/mL) and IV administration (Cmax = 6726 ng/mL). Additionally, compound 12c displayed good volume of plasma distribution (Vss = 1.5 L/kg) and acceptable plasma clearance (CL = 1.1 L/h/kg) after 10 mg/kg IV. Excellent oral bioavailability (F) with the value of 76% was observed. Compound 12c was evaluated in rats for brain permeability and concentration after a single dose of 10 mg/kg by IV administration, and the concentrations of 12c in the brain and plasma were measured at 0.25 and 1 h after administration. As shown in Table 7, compound 12c exhibits a good concentration in the brain at 0.25 h with the value of 1807 ng/g and also exhibits an acceptable brain permeability with a brain/plasma ratio of 0.28. The high brain concentration and brain permeability of 12c persisted for at least 1 h, with a brain concentration over 1000 ng/g and a brain/plasma ratio of 0.39 after 1 h injection. Based upon these collective findings, compound 12c was selected for further in vivo evaluation.
Table 6.
In Vivo Pharmacokinetic Parameters of Compound 12c in Ratsa
| route | AUC0-∞ (ng·h/mL) | t1/2 (h) | Cmax (ng/mL) | CL (L/h/kg) | Vss (L/kg) | F (%) |
|---|---|---|---|---|---|---|
| PO | 13,749 ± 2710 | 2.5 ± 0.2 | 3407 ± 179 | l.5 ± 0.3 | 5.5 ± 1.16 | 76 ± 15 |
| IV | 9030 ± 1018 | 1.0 ± 0.1 | 6726 ± 727 | 1.1 ± 0.1 | 1.5 ± 0.2 |
AUC0-∞, area under the curve (t = 0 to 24 h); t1/2, terminal half-life; Cmax, maximum concentration of the drug in plasma; CL, plasma clearance; Vss, volume of distribution at steady state; F, absolute oral bioavailability. Experiments were studied in biological triplicates, and data values are shown as the mean ± SEM.
Table 7.
Brain Permeability of Compound 12c in Ratsa
| route | time (h) | brain conc. (ng/g) | plasma conc. (ng/g) | brain/plasma ratio |
|---|---|---|---|---|
| IV | 0.25 | 1807 ± 198 | 6324 ± 271 | 0.28 ± 0.02 |
| 1 | 1006 ± 43 | 2589 ± 241 | 0.39 ± 0.02 |
Concentrations of compound 12c in the brain and plasma were determined at 0.25 and 1 h after a single dose of 10 mg/kg by IV. Experiments were studied in biological triplicates, and data values are shown as the mean ± SEM.
Efficacy Evaluation of Compound 12c In Vivo.
Compound 12c exhibited potent GPR52 activity in vitro and a good PK profile to support further in vivo assessment. Amphetamine-induced hyperlocomotion in rodents is a well-characterized task with high translational validity for predicting preclinical antipsychotic-like activity.28,29 Given that GPR52 agonists inhibit hyperactivity induced by psychostimulants,10,30 we employed this assay to investigate the efficacy of compound 12c in vivo. Mice were injected intraperitoneally (IP) with a vehicle or compound 12c (0.3, 1, 3, or 10 mg/kg in vehicle; Figure 5A); automated activity monitoring began immediately and continued for 30 min. Mice were then injected with amphetamine (AMPH; 3 mg/kg IP), and activity was monitored for the next 90 min (Figure 5A). A one-way ANOVA indicated that compound 12c suppressed horizontal activity (F4,54 = 3.287, P < 0.05); Dunnett’s a priori comparisons indicated that 3 mg/kg (P < 0.05) and 10 mg/kg (P < 0.05) suppressed horizontal activity (Figure 5B). Additionally, compound 12c suppressed AMPH-induced horizontal activity (F4,54 = 4.736, P < 0.05; Figure 5C) at both 3 mg/kg (P < 0.05) and 10 mg/kg doses (P < 0.05). Taken together, compound 12c is an orally bioavailable and brain-penetrant GPR52 agonist, which dose-dependently inhibits amphetamine-evoked hyperactivity, suggesting therapeutic potential for neuropsychiatric diseases.
Figure 5.
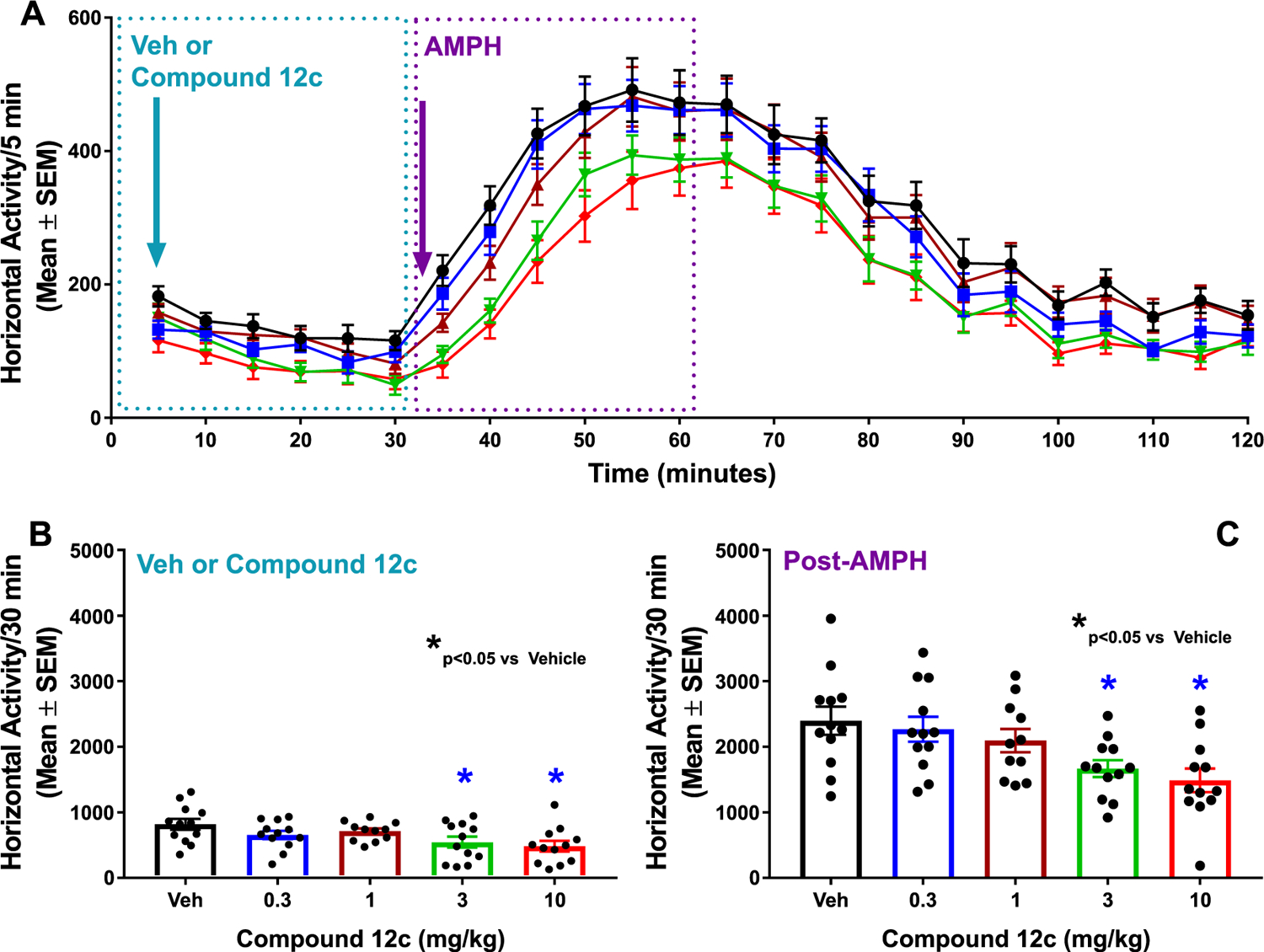
Compound 12c shows antipsychotic-like activity in mice. (A) Time course of horizontal activity (counts/5 min; mean ± SEM) following injection of the vehicle (black circles) or compound 12c (0.3 (blue squares), 1 (burgundy triangles), 3 (green inverted triangles), or 10 mg/kg IP (red diamonds) is shown during automated activity monitoring for 30 min (blue dotted box)). Mice were then injected with amphetamine (AMPH; 3 mg/kg IP), and activity was monitored for the next 90 min. Horizontal activity (counts/5 min; mean ± SEM) during the first 30 min following AMPH injection is illustrated (purple dotted box). (B) Bar graph showing horizontal activity/30 min (mean ± SEM) following injection of the vehicle (Veh) or compound 12c. (C) Bar graph showing horizontal activity (mean ± SEM) in the first 30 min period after AMPH injection. Filled circles indicate horizontal activity for individual animals. n = 12 mice/dose except for the 1 mg/kg AMPH (n = 11 mice/dose). *P < 0.05 vs Veh.
CONCLUSIONS
To understand the key ligand–receptor interactions and identify potent and efficacious GPR52 agonists, we have conducted a comprehensive structural optimization campaign based on previously reported advanced chemical lead 4 through a systematic SAR study by introduction of various amino alcohol side chains and modifications of numerous substituents on the core B ring and C ring. Among them, several compounds 12c (PW0787), 23a (PW0860), 23d (PW0878), 23e (PW0885), 23f (PW0888), and 23h (PW0890) were identified as novel GPR52 agonists that elevated cAMP signaling and were more potent than reference compound 4. Compound 12c has a favorable ClogP and a good in vivo PK profile. Therefore, compound 12c was selected for further in vivo efficacy determination. Molecular docking studies of compound 12c and GPR52 suggest a binding mode with three critical hydrogen bond pairs, π–π stacking with ring B, and hydrophobic interactions with ring C. This binding model offers theoretical studies for further SAR studies of compound 12c. Compound 12c was also counter screened and displayed no off-target affinities at other important brain GPCRs and ion channels. In addition, compound 12c has excellent oral plasma exposure, maximum serum concentration, bioavailability, and brain concentration. In vivo studies showed that compound 12c significantly inhibited amphetamine-induced hyperactivity in mice. Therefore, newly discovered GPR52 agonist 12c provides a useful pharmacological tool for studying GPR52 functions and for evaluating GPR52 agonist therapeutic potential for the treatment of brain diseases such as neuropsychiatric disorders.
EXPERIMENTAL SECTION
General.
All commercially available starting materials and solvents were reagent grade and used without further purification. Reactions were performed under a nitrogen atmosphere in dry glassware with magnetic stirring. Preparative column chromatography was performed using silica gel 60, particle size 0.063–0.200 mm (70–230 mesh, flash). Analytical TLC was carried out by employing silica gel 60 F254 plates (Merck, Darmstadt). Visualization of the developed chromatograms was performed with detection by UV (254 nm). NMR spectra were recorded on a Bruker-600 (1H, 300 MHz; 13C, 75 MHz) spectrometer. 1H and 13C NMR spectra were recorded with TMS as an internal reference. Chemical shifts downfield from TMS were expressed in ppm, and J values were given in Hz. High-resolution mass spectra (HRMS) were obtained from a Thermo Fisher LTQ Orbitrap Elite mass spectrometer. Parameters include the following: nano ESI spray voltage was 1.8 kV, capillary temperature was 275 °C, and the resolution was 60,000; ionization was achieved by positive mode. Purity of final compounds was determined by analytical HPLC, which was carried out on a Shimadzu HPLC system (model: CBM-20A LC-20AD SPD-20A UV/vis). HPLC analysis conditions were a Waters μBondapak C18 (300 mm × 3.9 mm), a flow rate 0.5 mL/min, UV detection at 270 and 254 nm, and linear gradient from 10% acetonitrile in water (0.1% TFA) to 100% acetonitrile (0.1% TFA) in 20 min followed by 30 min of the last-named solvent. Compounds 4 and 6 were resynthesized in-house following reported synthetic procedures.16,22 All biologically evaluated compounds are >95% pure.
Methyl 1-(2-Chloropyrimidin-4-yl)indoline-4-carboxylate (8a).
To a solution of 6 (354 mg, 2 mmol) and 2,4-dichloropyrimidine (7a) (300 mg, 2 mmol) in EtOH (5 mL) was added DIPEA (387 mg, 3 mmol) at rt., and the mixture solution was stirred at rt. overnight. After the reaction was completed (detected by TLC), the white solid was filtered and washed with water and cold EtOH. The cake was collected and dried to afford the product as white solid 8a (387 mg, 67%). 1H NMR (300 MHz, DMSO-d6) δ 8.53 (d, J = 8.1 Hz, 1H), 8.34 (d, J = 6.0 Hz, 1H), 7.57 (d, J = 7.8 Hz, 1H), 7.40 (t, J = 8.0 Hz, 1H), 6.87 (d, J = 6.1 Hz, 1H), 4.08 (t, J = 8.6 Hz, 2H), 3.85 (s, 3H), 3.50 (s, 2H).
Methyl 1-(5-Chloropyridazin-3-yl)indoline-4-carboxylate (8b).
Compound 8b (364 mg, 63%) was synthesized by a procedure similar to that used to prepare compound 8a as a light yellow solid. 1H NMR (300 MHz, DMSO-d6) δ 8.89 (s, 1H), 8.68 (d, J = 8.2 Hz, 1H), 7.55–7.47 (m, 2H), 7.37 (s, 1H), 4.11 (t, J = 8.7 Hz, 2H), 3.86 (s, 3H), 3.53 (t, J = 8.7 Hz, 2H).
Methyl 1-(6-Chloropyrimidin-4-yl)indoline-4-carboxylate (8c).
Compound 8c (370 mg, 64%) was synthesized by a procedure similar to that used to prepare compound 8a as a white solid. 1H NMR (300 MHz, DMSO-d6) δ 8.69 (dd, J = 8.2, 1.1 Hz, 1H), 8.61 (d, J = 0.8 Hz, 1H), 7.56 (dd, J = 7.9, 1.0 Hz, 1H), 7.37 (t, J = 8.0 Hz, 1H), 6.97 (d, J = 0.9 Hz, 1H), 4.07 (t, J = 8.5 Hz, 2H), 3.85 (s, 3H), 3.50 (t, J = 8.5 Hz, 2H).
Methyl 1-(2-(Cyano(3-fluoro-5-(trifluoromethyl)phenyl)-methyl)pyrimidin-4-yl)indoline-4-carboxylate (10a).
2-(3-Fluoro-5-(trifluoromethyl)phenyl)acetonitrile (9a) (336 mg, 1.6 mmol) was dissolved in 5 mL of DMF, and the mixture solution was cooled to 0 °C with an ice bath. NaH (141 mg, 3.5 mmol) was added to the solution at 0 °C, and the mixture solution was stirred at 0 °C for 30 min. Then, 8a (480 mg, 1.6 mmol) was added to the solution at 0 °C, and the mixture solution was stirred at rt. for 2 h. After the reaction was completed (detected by TLC), the reaction was quenched with NH4Cl(sat. aq.). The solution was worked up by the addition of water and then extracted with EtOAc (20 mL × 3). The combined EtOAc extracts were washed with brine, dried over Na2SO4, filtered, and condensed by rotary evaporation to yield a yellow oil. The residue was purified by silica gel chromatography (gradient: 1 to 5% MeOH in CH2Cl2) and provided product 10a (300 mg, 66%) as a yellow solid. 1H NMR (300 MHz, chloroform-d) δ 8.67 (d, J = 8.3 Hz, 1H), 8.40 (d, J = 6.1 Hz, 1H), 7.71 (dd, J = 8.0, 1.0 Hz, 2H), 7.54 (dt, J = 8.8, 2.0 Hz, 1H), 7.36 (t, J = 8.0 Hz, 2H), 6.53 (d, J = 6.1 Hz, 1H), 5.40 (s, 1H), 4.06 (dd, J = 9.3, 7.8 Hz, 2H), 3.94 (s, 3H), 3.66 (t, J = 8.5 Hz, 2H).
Methyl 1-(5-(Cyano(3-fluoro-5-(trifluoromethyl)phenyl)-methyl)pyridazin-3-yl)indoline-4-carboxylate (10b).
Compound 10b (120 mg, 53%) was synthesized by a procedure similar to that used to prepare compound 10a as a yellow solid. 1H NMR (300 MHz, chloroform-d) δ 8.74–8.62 (m, 2H), 7.66 (dd, J = 7.9, 1.0 Hz, 1H), 7.48 (s, 1H), 7.43 (d, J = 7.8 Hz, 1H), 7.33 (ddd, J = 8.1, 4.9, 3.0 Hz, 2H), 7.01 (dd, J = 1.9, 0.8 Hz, 1H), 5.25 (s, 1H), 4.14 (t, J = 8.6 Hz, 2H), 3.94 (s, 3H), 3.69 (t, J = 8.5 Hz, 2H).
Methyl 1-(6-(Cyano(3-fluoro-5-(trifluoromethyl)phenyl)-methyl)pyrimidin-4-yl)indoline-4-carboxylate (10c).
Compound 10c (130 mg, 57%) was synthesized by a procedure similar to that used to prepare compound 10a as a yellow solid. 1H NMR (300 MHz, chloroform-d) δ 8.83–8.70 (m, 2H), 7.71 (dd, J = 7.9, 1.0 Hz, 1H), 7.60 (s, 1H), 7.48 (d, J = 8.7 Hz, 1H), 7.36 (t, J = 8.1 Hz, 2H), 6.80 (s, 1H), 5.22 (s, 1H), 4.19–4.07 (m, 2H), 3.95 (s, 3H), 3.69 (t, J = 8.5 Hz, 2H).
1-(2-(3-Fluoro-5-(trifluoromethyl)benzyl)pyrimidin-4-yl)-indoline-4-carboxylic acid (11a).
To a solution of 10a (300 mg, 0.7 mmol) in con. HCl (4 mL), H2O (1 mL) and AcOH (1 mL) were added, and the mixture solution was stirred at reflux overnight. The reaction was cooled to room temperature, and a yellow solid precipitated from the solution. The yellow solid was filtered and washed with water, and the cake was collected and dried to afford the product as light yellow solid 11a (256 mg, 93%). 1H NMR (300 MHz, DMSO-d6) δ 13.12 (s, 1H), 8.62–8.51 (m, 1H), 7.89 (d, J = 8.3 Hz, 1H), 7.83–7.55 (m, 4H), 7.10–6.93 (m, 2H), 4.51 (s, 2H), 4.16 (t, J = 8.4 Hz, 2H), 3.52 (t, J = 8.3 Hz, 4H).
1-(5-(3-Fluoro-5-(trifluoromethyl)benzyl)pyridazin-3-yl)-indoline-4-carboxylic acid (11b).
Compound 11b (101 mg, 92%) was synthesized by a procedure similar to that used to prepare compound 11a as a white solid. 1H NMR (300 MHz, DMSO-d6) δ 8.99 (d, J = 1.4 Hz, 1H), 8.48 (d, J = 8.1 Hz, 1H), 7.74–7.69 (m, 2H), 7.69–7.63 (m, 1H), 7.57 (dd, J = 10.7, 7.6 Hz, 2H), 7.35 (d, J = 8.0 Hz, 1H), 4.23 (s, 2H), 4.17 (t, J = 8.5 Hz, 2H), 3.54 (s, 2H).
1-(6-(3-Fluoro-5-(trifluoromethyl)benzyl)pyrimidin-4-yl)-indoline-4-carboxylic Acid (11c).
Compound 11c (100 mg, 91%) was synthesized by a procedure similar to that used to prepare compound 11a as a white solid. 1H NMR (300 MHz, DMSO-d6) δ 8.92 (s, 1H), 8.71 (d, J = 8.2 Hz, 1H), 7.73 (s, 1H), 7.71–7.65 (m, 2H), 7.62 (dd, J = 8.8, 1.9 Hz, 1H), 7.43 (t, J = 8.0 Hz, 1H), 7.21 (s, 1H), 4.28 (s, 2H), 4.21 (t, J = 8.3 Hz, 2H), 3.59 (t, J = 8.3 Hz, 2H).
1-(2-(3-Fuoro-5-(trifluoromethyl)benzyl)pyrimidin-4-yl)-N-(2-hydroxyethyl)indoline-4-carboxamide (12a).
Compound 11a (42 mg, 0.1 mmol) and 2-aminoethan-1-ol (13 mg, 0.2 mmol) were dissolved in 2 mL of DMF, and the mixture solution was cooled to 0 °C with an ice bath. HOBt (14 mg, 0.1 mmol), EDCI (39 mg, 0.2 mmol), and DMAP (24 mg, 0.2 mmol) were added to the solution at 0 °C. Then, the ice bath was removed, and the mixture solution was stirred at room temperature overnight. After the reaction was completed (detected by TLC), the reaction was worked up by the addition of water and then extracted with EtOAc (20 mL × 3). The combined EtOAc extracts were washed with brine, dried over Na2SO4, filtered, and condensed by rotary evaporation to yield a yellow oil. This material was further purified by preparative TLC plates using CH2Cl2/MeOH = 50:1 as the eluent to yield 12a as a white solid (33 mg, 73%). 1H NMR (300 MHz, chloroform-d and MeOD) δ 8.26 (dd, J = 6.3, 2.6 Hz, 1H), 8.14 (dd, J = 7.9, 2.4 Hz, 1H), 7.45 (s, 1H), 7.29–7.04 (m, 5H), 6.39 (dd, J = 6.5, 2.5 Hz, 1H), 4.20 (s, 2H), 3.93 (t, J = 8.6 Hz, 2H), 3.71 (t, J = 5.0 Hz, 2H), 3.48 (qd, J = 8.7, 7.3, 2.6 Hz, 4H). 13C NMR (75 MHz, chloroform-d and MeOD) δ 168.9, 167.3, 159.0, 155.9, 144.1, 142.0, 132.4, 130.9, 127.5, 122.4–122.1 (m), 120.3, 120.0, 110.8 (d, J = 24.5 Hz), 103.1, 61.2, 49.2, 45.0, 42.3, 27.5. HRMS (ESI): (M + H)+ calcd for C23H21F4N4O2, 461.1595; found, 461.1591.
1-(5-(3-Fluoro-5-(trifluoromethyl)benzyl)pyridazin-3-yl)-N-(2-hydroxyethyl)indoline-4-carboxamide (12b).
Compound 12b (35 mg, 76%) was synthesized by a procedure similar to that used to prepare compound 12a as a white solid. 1H NMR (300 MHz, methanol-d4) δ 8.58 (s, 1H), 8.45 (dd, J = 7.5, 1.7 Hz, 1H), 7.53 (s, 1H), 7.39 (tt, J = 6.3, 2.0 Hz, 2H), 7.29–7.17 (m, 2H), 7.11 (d, J = 1.6 Hz, 1H), 4.14 (s, 2H), 4.03 (t, J = 8.6 Hz, 2H), 3.74 (t, J = 5.8 Hz, 2H), 3.51 (t, J = 5.8 Hz, 2H), 3.45 (t, J = 8.6 Hz, 2H). 13C NMR (75 MHz, MeOD) δ 169.6, 164.4, 161.1, 144.6, 142.7 (d, J = 7.7 Hz), 131.7, 131.6, 127.1, 121.6–121.4 (m), 119.7, 119.4, 116.7, 114.1, 111.1 (d, J = 4.1 Hz), 111.0, 110.7 (d, J = 4.0 Hz), 60.23, 48.81, 41.92, 37.30, 27.06. HRMS (ESI): (M + H)+ calcd for C23H21F4N4O2, 461.1595; found, 461.1591.
1-(6-(3-Fluoro-5-(trifluoromethyl)benzyl)pyrimidin-4-yl)-N-(2-hydroxyethyl)indoline-4-carboxamide (12c).
Compound 12c (38 mg, 83%) was synthesized by a procedure similar to that used to prepare compound 12a as a white solid. 1H NMR (300 MHz, chloroform-d) δ 8.70 (s, 1H), 8.57 (d, J = 8.1 Hz, 1H), 7.37 (s, 1H), 7.30–7.16 (m, 3H), 7.10 (d, J = 7.7 Hz, 1H), 6.71 (t, J = 5.6 Hz, 1H), 6.33 (s, 1H), 4.04 (s, 2H), 3.91 (t, J = 8.6 Hz, 2H), 3.83 (t, J = 4.9 Hz, 2H), 3.60 (d, J = 5.1 Hz, 2H), 3.49 (d, J = 8.4 Hz, 3H). 13C NMR (75 MHz, DMSO) δ 167.5, 166.7, 163.9, 160.7, 159.5, 157.9, 144.3, 143.8, 143.7, 132.6, 132.5, 131.2 (qd, J = 32.1, 8.3 Hz), 127.6, 122.5 (p, J = 3.6 Hz), 123.8 (qd, J = 270.8, 3.0 Hz), 121.1, 120.9, 120.6, 117.9, 111.4 (q, J = 4.0 Hz), 111.1 (q, J = 3.7 Hz), 105.0, 60.2, 48.9, 42.7, 42.4, 27.7. HRMS (ESI): (M + H)+ calcd for C23H21F4N4O2, 461.1595; found, 461.1591.
2-(2-Chloropyrimidin-4-yl)-2-(3-fluoro-5-(trifluoromethyl)-phenyl)acetonitrile (13).
2-(3-Fluoro-5-(trifluoromethyl)phenyl)-acetonitrile (9a) (406 mg, 2 mmol) was dissolved in 5 mL of DMF, and the mixture solution was cooled to 0 °C with an ice bath. NaH (190 mg, 4.4 mmol) was added to the solution at 0 °C, and the mixture solution was stirred at 0 °C for 30 min. Then, 2,4-dichloropyrimidine (7a) (450 mg, 3 mmol) was added to the solution at 0 °C, and the mixture solution was stirred at rt. for 2 h. After the reaction was completed (detected by TLC), the reaction mixture was worked up by the addition of water and then extracted with EtOAc (20 mL × 3). The combined EtOAc extracts were washed with brine, dried over Na2SO4, filtered, and condensed by rotary evaporation to yield a yellow oil. The residue was purified by silica gel chromatography (gradient: 10 to 20% EtOAc in hexane) to provide product 13 (460 mg, 73%) as a yellow oil. 1H NMR (300 MHz, chloroform-d) δ 8.76 (d, J = 5.0 Hz, 1H), 7.57 (s, 1H), 7.52 (d, J = 5.0 Hz, 1H), 7.43 (ddd, J = 14.3, 7.2, 1.8 Hz, 2H), 5.30 (s, 1H).
2-Chloro-4-(3-fluoro-5-(trifluoromethyl)benzyl)pyrimidine (14).
To a solution of 13 (460 mg, 1.5 mmol) in con. HCl (4 mL), H2O (1 mL) and AcOH (1 mL) were added, and the mixture solution was stirred at reflux overnight. The reaction was cooled to room temperature, and a yellow solid precipitated from the solution. The yellow solid was filtered and washed with water, and the cake was collected and dried to afford the product as a light yellow solid. The yellow solid was dissolved in 2 mL of POCl3, and the mixture solution was stirred at reflux overnight. The reaction was cooled to room temperature. After the reaction was completed (detected by TLC), the reaction was poured into ice water, and the pH of the mixture solution was adjusted to pH = 8 with Na2CO3(sat. aq.) solution. The solution was extracted with EtOAc (20 mL × 3). The organic phase was washed with brine, dried over Na2SO4, and then concentrated under reduced pressure. The residue was purified by silica gel chromatography (gradient: 10 to 20% EtOAc in hexane) to provide product 14 as a brown colorless oil (222 mg, 51% for two steps). 1H NMR (300 MHz, chloroform-d) δ 8.57 (d, J = 5.0 Hz, 1H), 7.36 (s, 1H), 7.30–7.26 (m, 1H), 7.22 (dd, J = 8.8, 1.9 Hz, 1H), 7.09 (d, J = 5.0 Hz, 1H), 4.17 (s, 2H).
Methyl 1-(4-(3-Fluoro-5-(trifluoromethyl)benzyl)pyrimidin-2-yl)indoline-4-carboxylate (15).
A mixture of 14 (145 mg, 0.5 mmol), Pd(OAc)2 (6 mg, 0.025 mmol), XantPhos (29 mg, 0.05 mmol), Cs2CO3 (325 mg, 1 mmol), and 6 (89 mg, 0.5 mmol) in 1,4-dioxane (3 mL) was subjected to three rounds of vacuum evacuation followed by introduction of nitrogen. The reaction mixture was then stirred at 100 °C overnight. The reaction was cooled to room temperature and poured in water and then extracted with EtOAc (10 mL × 3). The organic phase was washed with brine, dried over Na2SO4, and then concentrated under reduced pressure. The residue was purified by silica gel chromatography (gradient: 10 to 20% EtOAc in hexane) to provide product 15 (120 mg, 56%) as a yellow solid. 1H NMR (300 MHz, chloroform-d) δ 8.52–8.42 (m, 2H), 7.60 (dd, J = 7.8, 1.1 Hz, 1H), 7.45 (s, 1H), 7.26 (dd, J = 8.4, 3.5 Hz, 3H), 6.62 (d, J = 5.0 Hz, 1H), 4.28 (dd, J = 9.3, 8.2 Hz, 2H), 4.10 (s, 2H), 3.93 (s, 3H), 3.60–3.53 (m, 2H).
1-(4-(3-Fluoro-5-(trifluoromethyl)benzyl)pyrimidin-2-yl)-indoline-4-carboxylic Acid (16).
To a solution of 14 (120 mg, 0.28 mmol) in MeOH (4 mL) was added a 2 N solution of NaOH (1 mL), and the mixture solution was stirred at reflux for 1 h. The pH of the mixture solution was adjusted to pH = 1 with 2 N HCl solution, and then, a yellow solid precipitated from the solution. The yellow solid was filtered and washed with water, and the cake was collected and dried to afford the product as light yellow solid 16 (110 mg, 95%). 1H NMR (300 MHz, DMSO-d6) δ 8.51 (d, J = 5.0 Hz, 1H), 8.23 (d, J = 8.1 Hz, 1H), 7.67 (s, 1H), 7.59 (dt, J = 9.5, 2.0 Hz, 2H), 7.44 (dd, J = 7.8, 1.1 Hz, 1H), 7.13 (t, J = 8.0 Hz, 1H), 6.91 (d, J = 5.0 Hz, 1H), 4.23 (s, 2H), 4.14 (t, J = 8.7 Hz, 2H), 3.43 (t, J = 8.6 Hz, 2H).
1-(4-(3-Fluoro-5-(trifluoromethyl)benzyl)pyrimidin-2-yl)-N-(2-hydroxyethyl)indoline-4-carboxamide (12d).
Compound 12d (92 mg, 83%) was synthesized by a procedure similar to that used to prepare compound 12a as a white solid. 1H NMR (300 MHz, chloroform-d) δ 8.37 (d, J = 5.0 Hz, 1H), 8.30 (dd, J = 7.4, 1.5 Hz, 1H), 7.43 (s, 1H), 7.24 (dd, J = 8.9, 1.4 Hz, 2H), 7.16–7.05 (m, 2H), 6.77 (t, J = 5.6 Hz, 1H), 6.57 (d, J = 5.0 Hz, 1H), 4.17 (t, J = 8.7 Hz, 2H), 4.05 (s, 2H), 3.81 (t, J = 4.9 Hz, 2H), 3.58 (d, J = 5.1 Hz, 2H), 3.40 (q, J = 9.6, 8.7 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ 169.2, 167.6, 164.1, 160.8, 159.0, 157.9, 144.6, 141.7 (d, J = 7.7 Hz), 132.6, 130.8, 127.5, 122.1 (dd, J = 7.1, 3.5 Hz), 120.1, 119.8, 119.2, 117.6, 111.3 (q, J = 3.9 Hz), 111.0 (q, J = 3.8 Hz), 110.6, 62.1, 49.0, 43.5, 42.6, 27.3. HRMS (ESI): (M + H)+ calcd for C23H21F4N4O2, 461.1595; found, 461.1591.
2-(3-Fluoro-5-(trifluoromethyl)phenyl)-2-(5-methyl-6-oxo-1-(tetrahydro-2H-pyran-2-yl)-1,6-dihydropyridazin-4-yl)-acetonitrile (17a).
Compound 17a (150 mg, 36%) was synthesized by a procedure similar to that used to prepare compound 10a as a yellow oil. 1H NMR (300 MHz, chloroform-d) δ 7.75 (d, J = 8.8 Hz, 1H), 7.46–7.37 (m, 2H), 7.27 (dt, J = 8.5, 2.3 Hz, 1H), 6.06 (dt, J = 10.6, 1.9 Hz, 1H), 5.30 (d, J = 6.2 Hz, 1H), 4.17–4.09 (m, 1H), 3.75 (ddd, J = 11.7, 9.0, 2.7 Hz, 1H), 2.18 (dd, J = 10.7, 4.2 Hz, 1H), 1.85–1.58 (m, 4H).
2-(5-Chloro-6-oxo-1-(tetrahydro-2H-pyran-2-yl)-1,6-dihydropyridazin-4-yl)-2-(3-fluoro-5-(trifluoromethyl)phenyl)-acetonitrile (17b).
Compound 17b (520 mg, 63%) was synthesized by a procedure similar to that used to prepare compound 10a as a yellow oil. 1H NMR (300 MHz, chloroform-d) δ 7.90 (d, J = 6.1 Hz, 1H), 7.51 (s, 1H), 7.43 (d, J = 8.0 Hz, 1H), 7.39–7.33 (m, 1H), 6.07 (dt, J = 10.6, 2.7 Hz, 1H), 5.59 (d, J = 6.9 Hz, 1H), 4.13 (td, J = 6.9, 3.2 Hz, 1H), 3.77 (ddd, J = 11.5, 8.7, 3.0 Hz, 1H), 2.24–2.06 (m, 2H), 1.87–1.62 (m, 4H).
5-(3-Fluoro-5-(trifluoromethyl)benzyl)-4-methylpyridazin-3(2H)-one (18a).
To a solution of 17a (830 mg, 2 mmol) in con. HCl (8 mL), H2O (2 mL) and AcOH (2 mL) were added, and the mixture solution was stirred at reflux overnight. After the reaction was completed (detected by TLC), the reaction was poured into ice water. The pH of the mixture solution was adjusted to pH = 8 with Na2CO3(sat. aq.) solution. Then, the solution was extracted with EtOAc (20 mL × 3). The organic phase was washed with brine, dried over Na2SO4, and then concentrated under reduced pressure. The residue purification by silica gel chromatography (gradient: 30 to 50% EtOAc in hexane) provided product 18a (480 mg, 78%) as a white solid. 1H NMR (300 MHz, chloroform-d) δ 11.67 (s, 1H), 7.58 (s, 1H), 7.25 (d, J = 7.0 Hz, 2H), 7.07–6.99 (m, 1H), 3.97 (s, 2H), 2.23 (s, 3H).
4-Chloro-5-(3-fluoro-5-(trifluoromethyl)benzyl)pyridazin-3(2H)-one (18b).
Compound 18b (410 mg, 83%) was synthesized by a procedure similar to that used to prepare compound 18a as a yellow oil. 1H NMR (300 MHz, chloroform-d) δ 7.65 (s, 1H), 7.38–7.23 (m, 3H), 7.17–7.10 (m, 1H), 4.12 (s, 2H).
3-Chloro-5-(3-fluoro-5-(trifluoromethyl)benzyl)-4-methylpyridazine (19a).
A solution of 18a (400 mg, 1.3 mmol) in POCl3 (3 mL) was heated to reflux for 3 h. After the reaction was completed (detected by TLC), the reaction was cooled to room temperature and poured in ice water. The pH of the mixture solution was adjusted to pH = 8 with 2 N NaOH solution and then extracted with EtOAc (30 mL × 3). The organic phase was washed with brine, dried over Na2SO4, and then concentrated under reduced pressure. The residue purification by silica gel chromatography (gradient: 10 to 20% EtOAc in hexane) provided product 19a (350 mg, 83%) as a brown solid. 1H NMR (300 MHz, chloroform-d) δ 8.83 (s, 1H), 7.29 (d, J = 9.7 Hz, 2H), 7.22 (s, 1H), 6.99 (d, J = 8.8 Hz, 1H), 4.12 (s, 2H), 2.39 (s, 3H).
3,4-Dichloro-5-(3-fluoro-5-(trifluoromethyl)benzyl)-pyridazine (19b).
Compound 19b (350 mg, 80%) was synthesized by a procedure similar to that used to prepare compound 19a as a brown solid. 1H NMR (300 MHz, chloroform-d) δ 8.87 (s, 1H), 7.35–7.29 (m, 2H), 7.16–7.06 (m, 1H), 4.23 (s, 2H).
Methyl 1-(5-(3-Fluoro-5-(trifluoromethyl)benzyl)-4-methylpyridazin-3-yl)indoline-4-carboxylate (20a).
Compound 20a (142 mg, 64%) was synthesized by a procedure similar to that used to prepare compound 15 as a brown solid. 1H NMR (300 MHz, chloroform-d) δ 8.76 (s, 1H), 7.50 (dd, J = 7.9, 0.9 Hz, 1H), 7.27 (d, J = 7.9 Hz, 4H), 7.10 (q, J = 9.6, 8.7 Hz, 2H), 6.46 (d, J = 7.8 Hz, 1H), 4.27 (t, J = 8.2 Hz, 2H), 4.13 (s, 2H), 3.94 (s, 3H), 3.57 (t, J = 8.2 Hz, 2H), 2.18 (s, 3H).
Methyl 1-(4-Chloro-5-(3-fluoro-5-(trifluoromethyl)benzyl)-pyridazin-3-yl)indoline-4-carboxylate (20b).
Compound 20b (280 mg, 60%) was synthesized by a procedure similar to that used to prepare compound 15 as a white solid. 1H NMR (300 MHz, chloroform-d) δ 8.71 (s, 1H), 7.59 (dd, J = 7.9, 1.0 Hz, 1H), 7.37–7.29 (m, 2H), 7.25–7.12 (m, 2H), 7.00 (dd, J = 8.0, 0.9 Hz, 1H), 4.34 (t, J = 8.3 Hz, 2H), 4.23 (s, 2H), 3.94 (s, 3H), 3.60 (t, J = 8.3 Hz, 2H).
1-(5-(3-Fluoro-5-(trifluoromethyl)benzyl)-4-methylpyridazin-3-yl)indoline-4-carboxylic Acid (21a).
Compound 21a (126 mg, 92%) was synthesized by a procedure similar to that used to prepare compound 16 as a yellow solid. 1H NMR (300 MHz, DMSO-d6) δ 8.91 (d, J = 1.8 Hz, 1H), 7.66–7.47 (m, 3H), 7.38 (d, J = 7.9 Hz, 1H), 7.14 (t, J = 7.9 Hz, 1H), 6.64 (d, J = 7.9 Hz, 1H), 4.29 (s, 2H), 4.12 (t, J = 8.3 Hz, 2H), 3.45 (t, J = 8.0 Hz, 2H), 2.20 (d, J = 1.9 Hz, 3H).
1-(4-Chloro-5-(3-fluoro-5-(trifluoromethyl)benzyl)-pyridazin-3-yl)indoline-4-carboxylic Acid (21b).
Compound 21a (243 mg, 90%) was synthesized by a procedure similar to that used to prepare compound 16 as a yellow solid. 1H NMR (300 MHz, DMSO-d6) δ 9.00 (s, 1H), 7.65–7.51 (m, 3H), 7.44 (dd, J = 7.8, 1.0 Hz, 1H), 7.18 (t, J = 7.9 Hz, 1H), 6.95 (dd, J = 8.0, 1.0 Hz, 1H), 4.34 (s, 2H), 4.21 (t, J = 8.3 Hz, 4H), 3.46 (t, J = 8.2 Hz, 2H).
1-(5-(3-Fluoro-5-(trifluoromethyl)benzyl)-4-methylpyridazin-3-yl)-N-(2-hydroxyethyl)indoline-4-carboxamide (12e).
Compound 12e (107 mg, 79%) was synthesized by a procedure similar to that used to prepare compound 12a as a white solid. 1H NMR (300 MHz, chloroform-d) δ 8.71 (s, 1H), 7.29 (d, J = 9.7 Hz, 1H), 7.24 (s, 1H), 7.07 (d, J = 9.0 Hz, 1H), 7.02–6.84 (m, 3H), 6.29 (dd, J = 5.4, 3.4 Hz, 1H), 4.25–4.08 (m, 4H), 3.86 (t, J = 4.8 Hz, 2H), 3.63 (q, J = 5.1 Hz, 2H), 3.43 (t, J = 8.1 Hz, 3H), 2.15 (s, 3H). 13C NMR (75 MHz, CDCl3) δ 168.9, 158.9, 149.1, 147.8, 140.9 (d, J = 7.3 Hz), 138.7, 132.0, 131.8, 131.2, 126.8, 121.0 (t, J = 3.3 Hz), 119.3, 119. 0, 118.4, 111.9, 62.1, 53.6, 42.8, 36.0, 28.8, 14.5. HRMS (ESI): (M + H)+ calcd for C24H23F4N4O2, 475.1752; found, 475.1748.
1-(4-Chloro-5-(3-fluoro-5-(trifluoromethyl)benzyl)-pyridazin-3-yl)-N-(2-hydroxyethyl)indoline-4-carboxamide (12f).
Compound 12f (29 mg, 59%) was synthesized by a procedure similar to that used to prepare compound 12a as a white solid. 1H NMR (300 MHz, chloroform-d) δ 8.66 (s, 1H), 7.36–7.24 (m, 2H), 7.14 (d, J = 9.0 Hz, 1H), 7.11–7.02 (m, 2H), 6.89 (dd, J = 6.4, 3.6 Hz, 2H), 4.34–4.15 (m, 4H), 3.81 (t, J = 4.9 Hz, 2H), 3.60 (t, J = 5.1 Hz, 2H), 3.47 (t, J = 8.2 Hz, 2H). 13C NMR (75 MHz, CDCl3) δ 168.8, 164.4, 161.0, 156.8, 148.2, 146.2, 139.9 (d, J = 7.4 Hz), 138.7, 132.1, 131.6, 130.2, 126.8, 121.5–121.4 (m), 119.6, 119.4, 119.3, 115.3, 112.2 (d, J = 3.6 Hz), 111.9 (d, J = 3.9 Hz), 62.1, 53.6, 42.7, 36.1, 29.1. HRMS (ESI): (M + H)+ calcd for C23H20ClF4N4O2, 495.1205; found, 495.1201.
1-(6-(3-(Trifluoromethyl)benzyl)pyrimidin-4-yl)indoline-4-carboxylic Acid (22a).
2-(3-(Trifluoromethyl)phenyl)acetonitrile (9b) (370 mg, 2 mmol) was dissolved in 5 mL of DMF, and the mixture solution was cooled to 0 °C with an ice bath. NaH (176 mg, 4.4 mmol) was added to the solution at 0 °C, and the mixture solution was stirred at 0 °C for 30 min. Then, 8c (580 mg, 2 mmol) was added to the solution at 0 °C, and the mixture solution was stirred at rt. for 2 h. After the reaction was completed (detected by TLC), the reaction mixture was worked up by the addition of water and then extracted with EtOAc (20 mL × 3). The combined EtOAc extracts were washed with brine, dried over Na2SO4, filtered, and condensed by rotary evaporation to yield a yellow solid. 1H NMR (300 MHz, DMSO-d6) δ 8.75 (s, 1H), 8.67 (d, J = 8.0 Hz, 1H), 7.93–7.85 (m, 2H), 7.77 (d, J = 7.9 Hz, 1H), 7.71 (d, J = 7.7 Hz, 1H), 7.54 (dd, J = 7.9, 1.0 Hz, 1H), 7.35 (t, J = 7.9 Hz, 1H), 7.07 (s, 1H), 6.00 (s, 1H), 4.08 (dd, J = 8.8, 2.0 Hz, 2H), 3.85 (s, 3H), 3.51 (t, J = 8.6 Hz, 2H).
To a solution of the yellow solid (100 mg, 0.23 mmol) in con. HCl (2 mL), H2O (0.5 mL) and AcOH (0.5 mL) were added, and the mixture solution was stirred at reflux overnight. The reaction was cooled to room temperature, and a yellow solid precipitated from the solution. The yellow solid was filtered and washed with water, and the cake was collected and dried to afford the product as light yellow solid 22a (82 mg, 90% for two steps). 1H NMR (300 MHz, DMSO-d6) δ 8.86 (s, 1H), 8.70 (d, J = 8.1 Hz, 1H), 7.81 (s, 1H), 7.73 (d, J = 7.5 Hz, 1H), 7.68–7.53 (m, 3H), 7.40 (t, J = 8.0 Hz, 1H), 7.13 (s, 1H), 4.26–4.11 (m, 4H), 3.57 (t, J = 8.4 Hz, 2H).
1-(6-(2-(Trifluoromethyl)benzyl)pyrimidin-4-yl)indoline-4-carboxylic Acid (22b).
Compound 22b (65 mg, 54% for two steps) was synthesized by a procedure similar to that used to prepare compound 22a as a white solid. 1H NMR (300 MHz, DMSO-d6) δ 8.86 (s, 1H), 8.70 (d, J = 8.1 Hz, 1H), 7.79 (d, J = 7.7 Hz, 1H), 7.64 (dd, J = 10.4, 7.6 Hz, 2H), 7.55 (d, J = 7.4 Hz, 1H), 7.46 (d, J = 7.8 Hz, 1H), 7.40 (d, J = 8.1 Hz, 1H), 6.77 (s, 1H), 4.32 (s, 2H), 4.07 (t, J = 8.4 Hz, 2H), 3.53 (t, J = 8.4 Hz, 2H).
1-(6-(4-(Trifluoromethyl)benzyl)pyrimidin-4-yl)indoline-4-carboxylic Acid (22c).
Compound 22c (68 mg, 57% for two steps) was synthesized by a procedure similar to that used to prepare compound 22a as a white solid. 1H NMR (300 MHz, DMSO-d6) δ 8.82 (s, 1H), 8.69–8.66 (m, 1H), 7.71 (d, J = 8.1 Hz, 2H), 7.64–7.59 (m, 3H), 7.40 (d, J = 8.2 Hz, 1H), 7.08 (s, 1H), 4.19–4.12 (m, 4H), 3.54 (d, J = 8.7 Hz, 2H).
1-(6-(3-Methylbenzyl)pyrimidin-4-yl)indoline-4-carboxylic Acid (22d).
Compound 22d (86 mg, 83% for two steps) was synthesized by a procedure similar to that used to prepare compound 22a as a white solid. 1H NMR (300 MHz, DMSO-d6) δ 8.86 (s, 1H), 8.70 (d, J = 8.1 Hz, 1H), 7.81 (s, 1H), 7.73 (d, J = 7.5 Hz, 1H), 7.68–7.53 (m, 3H), 7.40 (t, J = 8.0 Hz, 1H), 7.13 (s, 1H), 4.26–4.11 (m, 4H), 3.57 (t, J = 8.4 Hz, 2H), 2.49 (s, 3H).
1-(6-(3-Fluorobenzyl)pyrimidin-4-yl)indoline-4-carboxylic Acid (22e).
Compound 22e (71 mg, 68% for two steps) was synthesized by a procedure similar to that used to prepare compound 22a as a white solid. 1H NMR (300 MHz, DMSO-d6) δ 8.91 (s, 1H), 8.71 (d, J = 8.5 Hz, 1H), 7.67 (d, J = 7.8 Hz, 1H), 7.47–7.38 (m, 2H), 7.31–7.23 (m, 2H), 7.16–7.06 (m, 2H), 4.24–4.13 (m, 4H), 3.58 (t, J = 8.4 Hz, 2H).
1-(6-(3,5-Dimethylbenzyl)pyrimidin-4-yl)indoline-4-carboxylic Acid (22f).
Compound 22f (88 mg, 81% for two steps) was synthesized by a procedure similar to that used to prepare compound 22a as a white solid. 1H NMR (300 MHz, DMSO-d6) δ 8.95 (s, 1H), 8.74 (d, J = 5.7 Hz, 1H), 7.71 (d, J = 7.7 Hz, 1H), 7.58 (s, 1H), 7.43 (d, J = 8.3 Hz, 1H), 7.17 (s, 1H), 7.02 (s, 2H), 4.24 (d, J = 8.1 Hz, 2H), 4.08 (s, 2H), 3.60 (d, J = 8.4 Hz, 2H), 2.25 (s, 6H).
1-(6-(3,5-Difluorobenzyl)pyrimidin-4-yl)indoline-4-carboxylic Acid (22g).
Compound 22g (79 mg, 72% for two steps) was synthesized by a procedure similar to that used to prepare compound 22a as a white solid. 1H NMR (300 MHz, DMSO-d6) δ 8.93 (s, 1H), 8.72 (d, J = 8.2 Hz, 1H), 7.69 (d, J = 7.6 Hz, 1H), 7.43 (t, J = 8.0 Hz, 1H), 7.29–7.11 (m, 4H), 4.20 (d, J = 8.3 Hz, 4H), 3.58 (t, J = 8.3 Hz, 2H).
1-(6-(3-(Trifluoromethoxy)benzyl)pyrimidin-4-yl)indoline-4-carboxylic Acid (22h).
Compound 22h (87 mg, 70% for two steps) was synthesized by a procedure similar to that used to prepare compound 22a as a white solid. 1H NMR (300 MHz, DMSO-d6) δ 8.95 (s, 1H), 8.71 (d, J = 8.1 Hz, 1H), 7.69 (d, J = 7.7 Hz, 1H), 7.50 (d, J = 4.7 Hz, 3H), 7.43 (t, J = 8.0 Hz, 1H), 7.33–7.27 (m, 1H), 7.20 (s, 1H), 4.25–4.16 (m, 4H), 3.58 (t, J = 8.1 Hz, 2H).
1-(6-(3-Methoxybenzyl)pyrimidin-4-yl)indoline-4-arboxylic Acid (22i).
Compound 22i (90 mg, 83% for two steps) was synthesized by a procedure similar to that used to prepare compound 22a as a white solid. 1H NMR (300 MHz, DMSO-d6) δ 8.89 (s, 1H), 8.70 (d, J = 8.0 Hz, 1H), 7.66 (dt, J = 7.8, 1.2 Hz, 2H), 7.45–7.38 (m, 2H), 7.13 (t, J = 7.8 Hz, 1H), 7.08 (d, J = 7.1 Hz, 1H), 6.79 (d, J = 2.0 Hz, 1H), 4.13–4.04 (m, 4H), 3.75 (s, 3H), 3.58 (d, J = 8.2 Hz, 2H).
1-(6-(3,5-Dimethoxybenzyl)pyrimidin-4-yl)indoline-4-carboxylic Acid (22j).
Compound 22j (94 mg, 80% for two steps) was synthesized by a procedure similar to that used to prepare compound 22a as a white solid. 1H NMR (300 MHz, DMSO-d6) δ 13.12 (s, 1H), 8.94 (s, 1H), 8.71 (d, J = 8.3 Hz, 1H), 7.69 (d, J = 7.7 Hz, 1H), 7.43 (t, J = 8.1 Hz, 1H), 7.15 (d, J = 9.7 Hz, 1H), 6.44 (d, J = 19.6 Hz, 2H), 6.24 (d, J = 11.6 Hz, 1H), 4.20 (t, J = 8.4 Hz, 2H), 4.02 (s, 2H), 3.71 (d, J = 14.4 Hz, 6H), 3.58 (t, J = 8.2 Hz, 2H).
1-(6-Benzylpyrimidin-4-yl)indoline-4-carboxylic Acid (22k).
Compound 22k (76 mg, 77% for two steps) was synthesized by a procedure similar to that used to prepare compound 22a as a white solid. 1H NMR (300 MHz, DMSO-d6) δ 13.19 (s, 1H), 8.96 (s, 1H), 8.71 (d, J = 8.2 Hz, 1H), 7.70 (dd, J = 7.9, 1.0 Hz, 1H), 7.48–7.42 (m, 2H), 7.41–7.27 (m, 3H), 7.17 (s, 1H), 4.25–4.16 (m, 4H), 3.57 (t, J = 8.2 Hz, 2H).
N-(2-Hydroxyethyl)-1-(6-(3-(trifluoromethyl)benzyl)-pyrimidin-4-yl)indoline-4-carboxamide (23a).
Compound 23a (52 mg, 59%) was synthesized by a procedure similar to that used to prepare compound 12a as a white solid. 1H NMR (300 MHz, chloroform-d and MeOD) δ 8.62 (s, 1H), 8.48 (d, J = 7.6 Hz, 1H), 7.52–7.37 (m, 4H), 7.21–7.09 (m, 2H), 6.32 (s, 1H), 4.01 (s, 2H), 3.87 (t, J = 8.6 Hz, 2H), 3.70 (t, J = 5.2 Hz, 2H), 3.53–3.37 (m, 4H). 13C NMR (75 MHz, CDCl3 and MeOD) δ 168.8, 166.7, 159.4, 157.5, 144.2, 138.6, 132.5, 132.4, 131.1 (d, J = 5.2 Hz), 131.0, 129.2, 127.7, 125.9–125.6 (m), 123.7 (d, J = 3.9 Hz), 120.3, 118.6, 103.9, 61.1, 48.7, 43.5, 42.2, 27.4. HRMS (ESI): (M + H)+ calcd for C23H22F3N4O2, 443.1689; found, 443.1685.
N-(2-Hydroxyethyl)-1-(6-(2-(trifluoromethyl)benzyl)-pyrimidin-4-yl)indoline-4-carboxamide (23b).
Compound 23b (36 mg, 61%) was synthesized by a procedure similar to that used to prepare compound 12a as a white solid. 1H NMR (300 MHz, chloroform-d and MeOD) δ 8.65 (s, 1H), 8.48 (d, J = 7.8 Hz, 1H), 7.66 (d, J = 7.8 Hz, 1H), 7.49 (t, J = 7.6 Hz, 1H), 7.34 (d, J = 8.1 Hz, 2H), 7.21 (ddd, J = 28.3, 13.0, 6.9 Hz, 3H), 6.19 (s, 1H), 4.18 (s, 2H), 3.83 (t, J = 8.5 Hz, 2H), 3.70 (t, J = 5.3 Hz, 2H), 3.53–3.39 (m, 4H). 13C NMR (75 MHz, CDCl3 and MeOD) δ 168.9, 166.9, 159.4, 157.3, 144.3, 135.7, 132.4, 132.1, 131.0, 127.7, 127.1, 126.2 (d, J = 5.6 Hz), 120.2, 118.5, 104.0, 61.1, 48.6, 42.3, 40.1, 27.4. HRMS (ESI): (M + H)+ calcd for C23H22F3N4O2, 443.1689; found, 443.1685.
N-(2-Hydroxyethyl)-1-(6-(4-(trifluoromethyl)benzyl)-pyrimidin-4-yl)indoline-4-carboxamide (23c).
Compound 23c (31 mg, 53%) was synthesized by a procedure similar to that used to prepare compound 12a as a white solid. 1H NMR (300 MHz, chloroform-d and MeOD) δ 8.65 (s, 1H), 8.52 (d, J = 7.8 Hz, 1H), 7.55 (d, J = 7.9 Hz, 2H), 7.38 (d, J = 7.9 Hz, 2H), 7.27–7.12 (m, 3H), 6.35 (s, 1H), 4.03 (s, 2H), 3.91 (t, J = 8.6 Hz, 2H), 3.72 (t, J = 5.2 Hz, 2H), 3.56–3.41 (m, 4H). 13C NMR (75 MHz, CDCl3 and MeOD) δ 168.9, 166.8, 159.4, 157.5, 144.3, 141.8, 132.4, 131.0, 129.4, 127.7, 125.6 (q, J = 3.7 Hz), 120.3, 118.6, 104.0, 61.2, 48.7, 43.6, 42.3, 27.5. HRMS (ESI): (M + H)+ calcd for C23H22F3N4O2, 443.1689; found, 443.1685.
N-(2-Hydroxyethyl)-1-(6-(3-methylbenzyl)pyrimidin-4-yl)-indoline-4-carboxamide (23d).
Compound 23d (43 mg, 83%) was synthesized by a procedure similar to that used to prepare compound 12a as a white solid. 1H NMR (300 MHz, chloroform-d and MeOD) δ 8.63 (s, 1H), 8.49 (d, J = 7.9 Hz, 1H), 7.34–7.09 (m, 4H), 7.03 (d, J = 7.7 Hz, 3H), 6.33 (s, 1H), 3.99–3.82 (m, 4H), 3.71 (t, J = 5.1 Hz, 2H), 3.56–3.34 (m, 5H), 2.30 (s, 3H). 13C NMR (75 MHz, CDCl3 and MeOD) δ 168.9, 168.1, 159.4, 157.3, 144.4, 138.3, 137.5, 132.4, 131.0, 129.8, 128.6, 127.7, 127.5, 126.1, 120.2, 118.5, 103.9, 61.2, 49.1, 43.8, 42.2, 27.4, 21.2. HRMS (ESI): (M + H)+ calcd for C23H25N4O2, 389.1972; found, 389.1968.
1-(6-(3-Fluorobenzyl)pyrimidin-4-yl)-N-(2-hydroxyethyl)-indoline-4-carboxamide (23e).
Compound 23e (38 mg, 72%) was synthesized by a procedure similar to that used to prepare compound 12a as a white solid. 1H NMR (300 MHz, chloroform-d and MeOD) δ 8.68–8.57 (m, 1H), 8.51 (d, J = 7.8 Hz, 1H), 7.32–7.11 (m, 4H), 7.02 (d, J = 7.7 Hz, 1H), 6.98–6.86 (m, 2H), 6.34 (s, 1H), 4.02–3.86 (m, 4H), 3.71 (t, J = 5.2 Hz, 2H), 3.55–3.40 (m, 4H). 13C NMR (75 MHz, CDCl3 and MeOD) δ 168.9, 168.9, 167.1, 164.5, 161.2, 159.5, 157.4, 144.3, 140.0 (d, J = 7.4 Hz), 132.4, 131.0 (2C), 130.2 (d, J = 8.3 Hz), 127.7, 124.8 (d, J = 2.9 Hz), 120.3, 118.6, 115.9 (d, J = 21.4 Hz), 113.73 (d, J = 21.0 Hz), 103.9, 61.1, 48.7, 43.5, 43.4, 42.2, 27.5. HRMS (ESI): (M + H)+ calcd for C22H22FN4O2, 393.1721; found, 393.1716.
1-(6-(3,5-Dimethylbenzyl)pyrimidin-4-yl)-N-(2-hydroxyethyl)indoline-4-carboxamide (23f).
Compound 23f (42 mg, 78%) was synthesized by a procedure similar to that used to prepare compound 12a as a white solid. 1H NMR (300 MHz, chloroform-d and MeOD) δ 8.59 (s, 1H), 8.46 (d, J = 7.9 Hz, 1H), 7.40–7.05 (m, 3H), 6.82 (s, 3H), 6.32 (s, 1H), 3.87 (d, J = 5.0 Hz, 4H), 3.73–3.56 (m, 4H), 3.46 (d, J = 6.6 Hz, 2H), 2.23 (s, 6H). 13C NMR (75 MHz, CDCl3 and MeOD) δ 169.0, 168.1, 159.4, 157.2, 144.3, 138.2, 137.4, 132.3, 131.0, 128.4, 127.6, 126.8, 120.2, 118.4, 103.9, 61.0, 48.7, 43.7, 42.3, 27.4, 21.1 (2C). HRMS (ESI): (M + H)+ calcd for C24H27N4O2, 403.2129; found, 403.2125.
1-(6-(3,5-Dimethylbenzyl)pyrimidin-4-yl)-N-(2-hydroxyethyl)indoline-4-carboxamide (23g).
Compound 23g (37 mg, 67%) was synthesized by a procedure similar to that used to prepare compound 12a as a white solid. 1H NMR (300 MHz, chloroform-d) δ 8.81–8.40 (m, 2H), 7.63–7.05 (m, 3H), 6.96–6.55 (m, 3H), 6.43 (s, 1H), 3.97 (d, J = 9.2 Hz, 4H), 3.89–3.66 (m, 4H), 3.54 (s, 2H). 13C NMR (75 MHz, CDCl3) δ 173.0, 172.9, 170.1, 168.6 (d, J = 13.3 Hz), 165.4, 163.4, 161.4, 148.1, 145.4, 136.3, 135.1, 131.7, 124.4, 122.6, 116.0, 115.7, 108.0, 106.6, 106.2, 105.9, 65.0, 52.7, 47.2, 46.2, 46.1, 31.4. HRMS (ESI): calcd for C22H21F2N4O2, 411.1627; found, 411.1623.
N-(2-Hydroxyethyl)-1-(6-(3-(trifluoromethoxy)benzyl)-pyrimidin-4-yl)indoline-4-carboxamide (23h).
Compound 23h (41 mg, 67%) was synthesized by a procedure similar to that used to prepare compound 12a as a white solid. 1H NMR (300 MHz, methanol-d4) δ 8.57 (d, J = 3.4 Hz, 2H), 7.43 (t, J = 7.9 Hz, 1H), 7.33 (d, J = 7.8 Hz, 1H), 7.29–7.22 (m, 3H), 7.20–7.13 (m, 1H), 6.65 (s, 1H), 4.05 (s, 2H), 3.94 (t, J = 8.6 Hz, 2H), 3.74 (t, J = 5.8 Hz, 2H), 3.51 (t, J = 5.8 Hz, 2H), 3.41 (t, J = 8.5 Hz, 2H). 13C NMR (75 MHz, MeOD) δ 169.4, 166.6, 159.6, 156.9, 149.3, 144.0, 140.7, 132.1, 131.6, 129.9, 127.6, 127.1, 121.4, 120.6, 118.8, 118.4, 104.0, 60.2, 48.4, 42.5, 41.9, 26.9. HRMS (ESI): (M + H)+ calcd for C23H22F3N4O3, 459.1639; found, 459.1636.
N-(2-Hydroxyethyl)-1-(6-(3-methoxybenzyl)pyrimidin-4-yl)-indoline-4-carboxamide (23i).
Compound 23i (26 mg, 48%) was synthesized by a procedure similar to that used to prepare compound 12a as a white solid. 1H NMR (300 MHz, chloroform-d) δ 8.72 (s, 1H), 8.59 (d, J = 8.2 Hz, 1H), 7.28–7.19 (m, 2H), 7.05 (d, J = 7.7 Hz, 1H), 6.95–6.79 (m, 3H), 6.51 (t, J = 5.4 Hz, 1H), 6.36 (s, 1H), 4.01 (s, 2H), 3.94 (t, J = 8.6 Hz, 2H), 3.87 (t, J = 4.9 Hz, 2H), 3.82 (s, 3H), 3.63 (q, J = 5.3 Hz, 2H), 3.54 (t, J = 8.5 Hz, 2H), 2.91 (s, 1H). 13C NMR (75 MHz, CDCl3) δ 168.6, 168.0, 159.9, 159.4, 157.5, 144.7, 139.4, 132.6, 130.7, 129.7, 127.8, 121.6, 119.6, 118.6, 115.1, 112.1, 103.8, 62.4, 55.2, 48.7, 44.3, 42.7, 27.7. HRMS (ESI): (M + H)+ calcd for C23H25N4O3, 405.1921; found, 405.1917.
1-(6-(3,5-Dimethoxybenzyl)pyrimidin-4-yl)-N-(2-hydroxyethyl)indoline-4-carboxamide (23j).
Compound 23j (17 mg, 29%) was synthesized by a procedure similar to that used to prepare compound 12a as a white solid. 1H NMR (300 MHz, chloroform-d) δ 8.67 (d, J = 1.0 Hz, 1H), 8.55 (d, J = 8.1 Hz, 1H), 7.18 (t, J = 7.9 Hz, 1H), 6.98 (dd, J = 7.8, 1.0 Hz, 1H), 6.56 (t, J = 5.6 Hz, 1H), 6.48 (d, J = 2.3 Hz, 2H), 6.39 (t, J = 2.3 Hz, 1H), 6.32 (d, J = 1.1 Hz, 1H), 3.94 (s, 2H), 3.91 (d, J = 8.5 Hz, 2H), 3.87–3.83 (m, 2H), 3.80 (s, 6H), 3.62 (t, J = 5.1 Hz, 2H), 3.50 (t, J = 8.6 Hz, 2H). 13C NMR (75 MHz, CDCl3) δ 168.5, 167.6, 161.0, 159.3, 157.4, 144.6, 140.0, 132.5, 130.7, 127.7, 119.7, 118.5, 107.4, 103.8, 98.6, 62.2, 55.4 (2C), 48.7, 44.5, 42.7, 27.6. HRMS (ESI): (M + H)+ calcd for C24H27N4O4, 435.2027; found, 435.2024.
1-(6-Benzylpyrimidin-4-yl)-N-(2-hydroxyethyl)indoline-4-carboxamide (23k).
Compound 23k (39 mg, 78%) was synthesized by a procedure similar to that used to prepare compound 12a as a white solid. 1H NMR (300 MHz, chloroform-d and MeOD) δ 8.60 (s, 1H), 8.47 (d, J = 7.9 Hz, 1H), 7.33–7.07 (m, 8H), 6.27 (s, 1H), 3.96 (s, 2H), 3.82 (t, J = 8.5 Hz, 2H), 3.71 (t, J = 5.1 Hz, 2H), 3.49 (q, J = 5.3 Hz, 2H), 3.44–3.30 (m, 3H). 13C NMR (75 MHz, CDCl3) δ 168.9, 167.9, 166.6, 159.3, 157.2, 144.3, 137.6, 132.3, 131.0, 129.1, 128.7, 127.6, 126.8, 120.2, 118.5, 103.9, 61.2, 48.6, 43.8, 42.3, 27.4. HRMS (ESI): (M + H)+ calcd for C22H23N4O2, 375.1816; found, 375.1812.
Methyl 1-(4-(3-Fluoro-5-(trifluoromethyl)benzoyl)-pyrimidin-2-yl)indoline-4-carboxylate (24a).
Compound 9a (406 mg, 2 mmol) was dissolved in 5 mL of DMF, and the mixture solution was cooled to 0 °C with an ice bath. NaH (190 mg, 4.4 mmol) was added to the solution at 0 °C, and the mixture solution was stirred at 0 °C for 30 min. Then, 2,4-dichloropyrimidine (450 mg, 3 mmol) was added to the solution at 0 °C. After the mixture solution was stirred at rt. for 2 h, the mixture was cooled to 0 °C with an ice bath. Followed by adding mCPBA (516 mg, 3 mmol), the mixture solution was stirred at 0 °C for 30 min. After the reaction was completed (detected by TLC), the reaction was worked up by the addition of water and EtOAc extraction. The combined EtOAc extracts were washed with brine, dried over Na2SO4, filtered, and condensed by rotary evaporation to yield a yellow oil. The residue purification by silica gel chromatography (gradient: 10 to 20% EtOAc in hexane) provided product (2-chloropyrimidin-4-yl)(3-fluoro-5-(trifluoromethyl)phenyl)methanone (280 mg, 47%) as a yellow solid. 1H NMR (300 MHz, chloroform-d) δ 8.98 (d, J = 4.9 Hz, 1H), 8.30 (s, 1H), 8.14 (dt, J = 8.9, 1.9 Hz, 1H), 7.96 (d, J = 4.9 Hz, 1H), 7.63 (dt, J = 8.1, 2.0 Hz, 1H).
A mixture of (2-chloropyrimidin-4-yl)(3-fluoro-5-(trifluoromethyl)phenyl)methanone (91 mg, 0.3 mmol), Pd(OAc)2 (3 mg, 0.015 mmol), XantPhos (17 mg, 0.03 mmol), Cs2CO3 (196 mg, 0.6 mmol), and 6 (53 mg, 0.3 mmol) in 1,4-dioxane (3 mL) was subjected to three rounds of vacuum evacuation followed by introduction of nitrogen. The reaction mixture was then stirred at 100 °C overnight. The reaction mixture was cooled to room temperature and poured into water and then extracted with EtOAc (10 mL × 3). The organic phase was washed with brine, dried over Na2SO4, and then concentrated under reduced pressure. The residue purification by silica gel chromatography (gradient: 10 to 20% EtOAc in hexane) provided product 24a (110 mg, 82%) as a yellow solid. 1H NMR (300 MHz, chloroform-d) δ 8.84 (d, J = 4.8 Hz, 1H), 8.43 (s, 2H), 8.15 (d, J = 8.8 Hz, 1H), 7.64 (dd, J = 7.9, 1.2 Hz, 2H), 7.39 (d, J = 4.8 Hz, 1H), 7.23 (t, J = 8.0 Hz, 1H), 4.32 (t, J = 8.7 Hz, 2H), 3.94 (s, 3H), 3.62 (t, J = 8.7 Hz, 2H).
Methyl 1-(6-(3-Fluoro-5-(trifluoromethyl)benzoyl)-pyrimidin-4-yl)indoline-4-carboxylate (24b).
2-(3-Fluoro-5-(trifluoromethyl)phenyl)acetonitrile (9a) (406 mg, 2 mmol) was dissolved in 5 mL of DMF, and the mixture solution was cooled to 0 °C with an ice bath. NaH (190 mg, 4.4 mmol) was added to the solution at 0 °C, and the mixture solution was stirred at 0 °C for 30 min. Then, compound 8c (580 mg, 2 mmol) was added to the solution at 0 °C. After the mixture solution was stirred at rt. for 2 h, the mixture was cooled to 0 °C with an ice bath. Followed by adding mCPBA (516 mg, 3 mmol), the mixture solution was stirred at 0 °C for 10 min. After the reaction was completed (detected by TLC), the reaction was worked up by the addition of water and then extracted with EtOAc (20 mL × 3). The combined EtOAc extracts were washed with brine, dried over Na2SO4, filtered, and condensed by rotary evaporation to yield a yellow oil. The residue purification by silica gel chromatography (gradient: 20 to 50% EtOAc in hexane) provided product 24b (440 mg, 49%) as a yellow solid. 1H NMR (300 MHz, chloroform-d) δ 8.97 (d, J = 1.1 Hz, 1H), 8.82 (d, J = 8.1 Hz, 1H), 8.31 (s, 1H), 8.17 (d, J = 9.0 Hz, 1H), 7.73 (dd, J = 7.9, 1.0 Hz, 1H), 7.60 (d, J = 8.0 Hz, 1H), 7.39 (t, J = 8.0 Hz, 1H), 7.31 (s, 1H), 4.21 (t, J = 8.6 Hz, 2H), 3.96 (s, 3H), 3.72 (t, J = 8.6 Hz, 2H).
Methyl 1-(6-(3-(Trifluoromethyl)benzoyl)pyrimidin-4-yl)-indoline-4-carboxylate (24c).
Compound 24c (162 mg, 79%) was synthesized by a procedure similar to that used to prepare compound 24b as a white solid. 1H NMR (300 MHz, DMSO-d6) δ 8.75 (d, J = 0.9 Hz, 1H), 8.67 (d, J = 8.1 Hz, 1H), 7.94–7.84 (m, 2H), 7.77 (d, J = 7.9 Hz, 1H), 7.71 (d, J = 7.7 Hz, 1H), 7.54 (dd, J = 7.9, 1.0 Hz, 1H), 7.35 (t, J = 8.0 Hz, 1H), 7.08 (s, 1H), 6.01 (s, 1H), 4.10 (t, J = 9.8 Hz, 2H), 3.85 (s, 3H), 3.51 (t, J = 8.5 Hz, 2H).
1-(4-(3-Fluoro-5-(trifluoromethyl)benzoyl)pyrimidin-2-yl)-N-(2-hydroxyethyl)indoline-4-carboxamide (25a).
To a solution of 24a (45 mg, 0.1 mmol) in con. HCl (4 mL), H2O (1 mL) and AcOH (1 mL) were added, and the mixture solution was stirred at reflux overnight. The reaction mixture was cooled to room temperature, and a yellow solid precipitated from the solution. The yellow solid was filtered and washed with water, and the cake was collected and dried to afford the product as a light yellow solid. The yellow solid and 2-aminoethan-1-ol (13 mg, 0.2 mmol) were dissolved in 2 mL of DMF, and the mixture solution was cooled to 0 °C with an ice bath. HOBt (14 mg, 0.1 mmol), EDCI (39 mg, 0.2 mmol), and DMAP (24 mg, 0.2 mmol) were added to the solution at 0 °C. Then, the ice bath was removed, and the mixture solution was stirred at room temperature overnight. After the reaction was completed (detected by TLC), the reaction was worked up by the addition of water and then extracted with EtOAc (20 mL × 3). The combined EtOAc extracts were washed with brine, dried over Na2SO4, filtered, and condensed by rotary evaporation to yield a yellow oil. This material was further purified by preparative TLC plates using CH2Cl2/MeOH = 50:1 as the eluent to yield 25a as a yellow solid (36 mg, 77% for two steps). 1H NMR (300 MHz, DMSO-d6) δ 8.94 (d, J = 4.9 Hz, 1H), 8.33 (s, 1H), 8.17 (dd, J = 26.9, 8.6 Hz, 3H), 7.44 (d, J = 4.9 Hz, 1H), 7.21 (d, J = 7.6 Hz, 1H), 7.10 (s, 1H), 4.71 (t, J = 5.6 Hz, 1H), 4.19 (t, J = 8.6 Hz, 2H), 3.51 (q, J = 6.1 Hz, 2H), 3.39 (t, J = 8.7 Hz, 2H), 3.30 (s, 2H). 13C NMR (75 MHz, DMSO) δ 159.9, 143.8, 132.7, 127.4, 122.0, 120.9, 116.7, 110.5, 60.2, 49.4, 42.4, 42.3, 40.8, 27.3. HRMS (ESI): (M + H)+ calcd for C23H19F4N4O3, 475.1388; found, 475.1385.
1-(6-(3-Fluoro-5-(trifluoromethyl)benzoyl)pyrimidin-4-yl)-N-(2-hydroxyethyl)indoline-4-carboxamide (25b).
Compound 25b (35 mg, 75%) was synthesized by a procedure similar to that used to prepare compound 25a as a yellow solid. 1H NMR (300 MHz, chloroform-d and MeOD) δ 8.86 (d, J = 1.1 Hz, 1H), 8.63 (d, J = 7.7 Hz, 1H), 8.21 (s, 1H), 8.07 (dt, J = 8.8, 2.0 Hz, 1H), 7.55 (dt, J = 8.0, 2.0 Hz, 1H), 7.28–7.23 (m, 2H), 7.22–7.20 (m, 1H), 4.10 (t, J = 8.5 Hz, 2H), 3.74 (t, J = 5.1 Hz, 2H), 3.54 (dd, J = 9.3, 7.1 Hz, 4H). 13C NMR (75 MHz, CDCl3 and MeOD) δ 190.3, 168.8, 166.6, 163.7, 160.4, 159.5 (d, J = 59.6 Hz), 157.3, 143.9, 138.0, 132.7, 131.2, 127.8, 123.9–123.3 (m), 121.3 (d, J = 23.2 Hz), 120.9, 119.0, 117.34 (dd, J = 24.4, 3.2 Hz), 105.1, 61.2, 49.0, 42.3, 27.5. HRMS (ESI): (M + H)+ calcd for C23H19F4N4O3, 475.1388; found, 475.1386.
N-(2-Hydroxyethyl)-1-(6-(3-(trifluoromethyl)benzoyl)-pyrimidin-4-yl)indoline-4-carboxamide (25c).
Compound 25c (360 mg, 63%) was synthesized by a procedure similar to that used to prepare compound 25a as a yellow solid. 1H NMR (300 MHz, DMSO-d6) δ 8.92 (d, J = 1.1 Hz, 1H), 8.63 (dd, J = 6.6, 2.6 Hz, 1H), 8.38–8.25 (m, 3H), 8.13–8.05 (m, 1H), 7.88–7.77 (m, 1H), 7.39–7.26 (m, 3H), 4.73 (t, J = 5.6 Hz, 1H), 4.17 (t, J = 8.5 Hz, 2H), 3.59–3.41 (m, 4H), 3.34 (m, 2H). 13C NMR (75 MHz, DMSO) δ 192.0, 167.4, 160.0, 159.9 (d, J = 8.6 Hz), 157.5, 143.9, 136.4, 135.0, 132.9 (d, J = 14.2 Hz), 132.7, 130.2, 129.4, 127.7, 127.3 (d, J = 3.6 Hz), 127.2, 121.8, 118.3, 105.5, 60.2, 49.0, 42.4, 27.7. HRMS (ESI): (M + H)+ calcd for C23H20F3N4O3, 457.1482; found, 457.1480.
1-(4-((3-Fluoro-5-(trifluoromethyl)phenyl)(hydroxy)-methyl)pyrimidin-2-yl)-N-(2-hydroxyethyl)indoline-4-carboxamide (25d).
To a solution of 25a (10 mg, 0.02 mmol) in MeOH (1 mL) was added NaBH4 (1 mg, 0.02 mmol) at 0 °C, and the mixture solution was stirred at rt. for 1 h. After the reaction was completed (detected by TLC), the reaction mixture was worked up by the addition of water and EtOAc extraction. The combined EtOAc extracts were washed with brine, dried over Na2SO4, filtered, and condensed by rotary evaporation to yield a colorless oil. This material was further purified by preparative TLC plates using CH2Cl2/MeOH = 50:1 as the eluent to yield 25d as a white solid (9 mg, 93%). 1H NMR (300 MHz, chloroform-d) δ 8.50–8.35 (m, 2H), 7.57 (s, 1H), 7.37 (d, J = 9.1 Hz, 1H), 7.24 (dd, J = 15.9, 7.8 Hz, 2H), 7.12 (d, J = 7.7 Hz, 1H), 6.66 (t, J = 4.5 Hz, 2H), 5.67 (s, 1H), 4.94 (s, 1H), 4.24 (t, J = 8.8 Hz, 2H), 3.85 (t, J = 4.9 Hz, 2H), 3.63 (q, J = 5.2 Hz, 2H), 3.44 (t, J = 8.6 Hz, 2H). 13C NMR (75 MHz, CDCl3) δ 169.1, 168.7, 164.2, 160.9, 158.4, 158.1, 145.8 (d, J = 6.9 Hz), 144.4, 132.9 (d, J = 8.1 Hz), 132.5 (d, J = 8.0 Hz), 130.9, 127.7, 125.0 (d, J = 3.0 Hz), 121.3 (d, J = 3.0 Hz), 119.5–119.24 (m), 117.7, 117.3 (d, J = 22.2 Hz), 112.5 (ddd, J = 24.2, 7.1, 3.4 Hz), 108.0, 73.8, 62.3, 49.2, 42.6, 27.2. HRMS (ESI): (M + H)+ calcd for C23H21F4N4O3, 477.1544; found, 477.1540.
1-(6-((3-Fluoro-5-(trifluoromethyl)phenyl)(hydroxy)-methyl)pyrimidin-4-yl)-N-(2-hydroxyethyl)indoline-4-carboxamide (25e).
Compound 25e (32 mg, 78%) was synthesized by a procedure similar to that used to prepare compound 25d as a white solid. 1H NMR (300 MHz, chloroform-d and MeOD) δ 8.66–8.27 (m, 2H), 7.51 (s, 1H), 7.34 (d, J = 8.5 Hz, 2H), 7.26–7.01 (m, 3H), 6.68 (s, 1H), 5.60 (s, 1H), 3.90 (t, J = 8.6 Hz, 2H), 3.70 (s, 2H), 3.46–3.27 (m, 4H). 13C NMR (75 MHz, CDCl3 and MeOD) δ 168.7 (d, J = 36.5 Hz), 164.0, 159.4, 156.8, 146.2, 144.1, 132.4, 131.0, 127.6, 120.5, 119.2, 118.7, 117.2 (d, J = 22.5 Hz), 112.4–111.7 (m), 101.0, 73.9, 61.1, 42.3, 42.2, 27.3. HRMS (ESI): (M + H)+ calcd for C23H21F4N4O3, 477.1544; found, 477.1540.
1-(6-(Hydroxy(3-(trifluoromethyl)phenyl)methyl)pyrimidin-4-yl)-N-(2-hydroxyethyl)indoline-4-carboxamide (25f).
Compound 25f (41 mg, 90%) was synthesized by a procedure similar to that used to prepare compound 25d as a white solid. 1H NMR (300 MHz, DMSO-d6) δ 8.64 (s, 1H), 8.54 (d, J = 7.0 Hz, 1H), 8.25 (t, J = 5.8 Hz, 1H), 7.89–7.72 (m, 2H), 7.69–7.52 (m, 2H), 7.28 (d, J = 6.6 Hz, 2H), 7.09 (s, 1H), 6.45 (d, J = 4.5 Hz, 1H), 5.72 (d, J = 4.5 Hz, 1H), 4.71 (t, J = 5.6 Hz, 1H), 4.11 (q, J = 7.9 Hz, 2H), 3.60–3.33 (m, 6H). 13C NMR (75 MHz, DMSO) δ 170.9, 167.5, 159.7, 157.4, 145.2, 144.4, 132.6, 131.5, 129.6, 129.2 (d, J = 31.4 Hz), 127.6, 126.5, 124.5 (t, J = 3.9 Hz), 123.7 (t, J = 3.9 Hz), 121.1, 117.9, 101.3, 74.5, 60.3, 49.0, 42.4, 27.7. HRMS (ESI): (M + H)+ calcd for C23H22F3N4O3, 459.1639; found, 459.1635.
1-(4-((3-Fluoro-5-(trifluoromethyl)phenyl)(hydroxy)-methyl)pyrimidin-2-yl)indoline-4-carboxylic Acid (26a).
To a solution of 24a (427 mg, 1 mmol) in DMF (5 mL) and MeOH (5 mL) was added NaBH4 (38 mg, 1 mmol) at 0 °C, and the mixture solution was stirred at rt. for 30 min. After the reaction was completed (detected by TLC), the reaction mixture was worked up by the addition of water and then extracted with EtOAc (20 mL × 3). The combined EtOAc extracts were washed with brine, dried over Na2SO4, filtered, and condensed by rotary evaporation to yield a colorless oil. The colorless oil was dissolved in con. HCl (4 mL), H2O (1 mL), and AcOH (1 mL), and the mixture solution was stirred at reflux overnight. The reaction was cooled to room temperature, and a yellow solid precipitated from the solution. The yellow solid was filtered and washed with water, and the cake was collected and dried to afford the product as a light yellow solid (210 mg, 78%). 1H NMR (300 MHz, chloroform-d) δ 8.63 (s, 1H), 8.51 (d, J = 4.8 Hz, 1H), 7.74 (dd, J = 7.9, 1.0 Hz, 1H), 7.57 (s, 1H), 7.36 (t, J = 10.4 Hz, 3H), 6.67 (d, J = 5.1 Hz, 1H), 5.73 (s, 1H), 4.39 (t, J = 8.3 Hz, 2H), 3.66 (t, J = 8.6 Hz, 2H).
1-(6-((3-Fluoro-5-(trifluoromethyl)phenyl)(hydroxy)-methyl)pyrimidin-4-yl)indoline-4-carboxylic Acid (26b).
Compound 26b (390 mg, 90%) was synthesized by a procedure similar to that used to prepare compound 26a as a yellow solid. 1H NMR (300 MHz, DMSO-d6) δ 8.86 (s, 1H), 8.71 (d, J = 7.8 Hz, 1H), 7.84 (s, 1H), 7.77 (d, J = 9.5 Hz, 1H), 7.64 (d, J = 7.8 Hz, 2H), 7.45–7.40 (m, 1H), 7.32 (d, J = 2.9 Hz, 1H), 6.00 (s, 1H), 4.24 (dt, J = 9.0, 3.9 Hz, 2H), 3.61–3.50 (m, 3H).
1-(6-(Hydroxy(3-(trifluoromethyl)phenyl)methyl)pyrimidin-4-yl)indoline-4-carboxylic Acid (26c).
Compound 26c (117 mg, 68%) was synthesized by a procedure similar to that used to prepare compound 26a as a yellow solid. 1H NMR (300 MHz, DMSO-d6) δ 8.86 (d, J = 3.4 Hz, 1H), 8.71 (d, J = 7.8 Hz, 1H), 7.96 (s, 1H), 7.87 (d, J = 7.6 Hz, 1H), 7.73–7.58 (m, 3H), 7.42 (t, J = 8.0 Hz, 1H), 7.31 (d, J = 2.3 Hz, 1H), 5.99 (s, 1H), 4.32–4.14 (m, 2H), 3.58 (t, J = 8.2 Hz, 2H).
1-(4-(Fluoro(3-fluoro-5-(trifluoromethyl)phenyl)methyl)-pyrimidin-2-yl)indoline-4-carboxylic Acid (27a).
To a solution of 26a (210 mg, 0.6 mmol) in DCM (5 mL) was added DAST (322 mg, 1.8 mmol) at 0 °C, and the mixture solution was stirred at 0 °C for 10 min. After the reaction was completed (detected by TLC), the reaction mixture was worked up by the addition of water and then extracted with EtOAc (20 mL × 3). The combined EtOAc extracts were washed with brine, dried over Na2SO4, filtered, and condensed by rotary evaporation to yield a yellow oil. The residue was dissolved in THF (2 mL) and H2O (2 mL), and the mixture solution was stirred at reflux overnight. The reaction was cooled to room temperature, and a yellow solid precipitated from the solution. The yellow solid was filtered and washed with water, and the cake was collected and dried to afford the product as light yellow solid 27a (140 mg, 54% for two steps). 1H NMR (300 MHz, chloroform-d) δ 8.63 (d, J = 5.0 Hz, 1H), 8.49 (d, J = 8.2 Hz, 1H), 7.73–7.64 (m, 2H), 7.46 (d, J = 8.9 Hz, 1H), 7.40–7.29 (m, 2H), 7.04 (dd, J = 5.1, 1.7 Hz, 1H), 6.41 (d, J = 46.4 Hz, 1H), 4.28 (dd, J = 8.3, 3.1 Hz, 2H), 3.61 (t, J = 8.8 Hz, 2H).
1-(6-(Fluoro(3-fluoro-5-(trifluoromethyl)phenyl)methyl)-pyrimidin-4-yl)indoline-4-carboxylic Acid (27b).
Compound 27b (260 mg, 60%) was synthesized by a procedure similar to that used to prepare compound 27a as a yellow solid. 1H NMR (300 MHz, DMSO-d6) δ 13.01 (s, 1H), 8.71 (d, J = 7.6 Hz, 2H), 7.80–7.71 (m, 3H), 7.56 (ddd, J = 7.8, 3.3, 1.2 Hz, 1H), 7.35 (dd, J = 9.1, 6.9 Hz, 1H), 7.11 (s, 1H), 6.75 (d, J = 45.7 Hz, 1H), 4.26–4.13 (m, 2H), 3.55 (t, J = 8.4 Hz, 2H).
1-(6-(Fluoro(3-(trifluoromethyl)phenyl)methyl)pyrimidin-4-yl)indoline-4-carboxylic Acid (27c).
Compound 27c (110 mg, 76%) was synthesized by a procedure similar to that used to prepare compound 27a as a yellow solid. 1H NMR (300 MHz, DMSO-d6) δ 8.71 (s, 1H), 7.90 (s, 1H), 7.83–7.74 (m, 1H), 7.69 (d, J = 7.7 Hz, 1H), 7.56 (dd, J = 7.3, 3.1 Hz, 1H), 7.38 (t, J = 8.0 Hz, 1H), 7.10 (s, 1H), 6.72 (d, J = 45.7 Hz, 1H), 4.20 (dd, J = 9.4, 4.7 Hz, 2H), 3.55 (t, J = 8.6 Hz, 2H).
1-(4-(Fluoro(3-fluoro-5-(trifluoromethyl)phenyl)methyl)-pyrimidin-2-yl)-N-(2-hydroxyethyl)indoline-4-carboxamide (25g).
Compound 25g (140 mg, 86%) was synthesized by a procedure similar to that used to prepare compound 12a as a light yellow solid. 1H NMR (300 MHz, chloroform-d and MeOD) δ 8.54 (d, J = 5.0 Hz, 1H), 8.27 (d, J = 7.7 Hz, 1H), 7.63 (s, 1H), 7.41 (d, J = 8.9 Hz, 1H), 7.31 (d, J = 8.4 Hz, 1H), 7.21–7.09 (m, 2H), 7.07–6.90 (m, 2H), 6.35 (d, J = 46.3 Hz, 1H), 4.17 (tt, J = 9.1, 3.5 Hz, 2H), 3.75 (t, J = 5.1 Hz, 2H), 3.54 (q, J = 5.3 Hz, 2H), 3.41 (t, J = 8.7 Hz, 2H). 13C NMR (75 MHz, CDCl3 and MeOD) δ 169.2, 169.1, 166.6 (d, J = 28.5 Hz), 164.0, 160.7, 159.0, 158.4 (d, J = 3.6 Hz), 144.3, 141.5 (dd, J = 21.8, 7.5 Hz), 132.7, 132.5, 131.0, 131.0, 127.5, 119.13–118.56 (m), 116.8 (dd, J = 22.9, 7.7 Hz), 113.2 (d, J = 23.1 Hz), 106.5 (d, J = 6.7 Hz), 91.95 (d, J = 179.2 Hz), 61.5, 49.1, 42.4, 27.2. HRMS (ESI): (M + H)+ calcd for C23H20F5N4O2, 479.1501; found, 479.1498.
1-(6-(Fluoro(3-fluoro-5-(trifluoromethyl)phenyl)methyl)-pyrimidin-4-yl)-N-(2-hydroxyethyl)indoline-4-carboxamide (25h).
Compound 25h (162 mg, 68%) was synthesized by a procedure similar to that used to prepare compound 12a as a white solid. 1H NMR (300 MHz, chloroform-d) δ 8.74 (s, 1H), 8.66 (d, J = 8.1 Hz, 1H), 7.60 (s, 1H), 7.44 (d, J = 8.9 Hz, 1H), 7.37–7.31 (m, 1H), 7.28 (t, J = 4.0 Hz, 1H), 7.19 (d, J = 7.6 Hz, 1H), 6.86 (s, 1H), 6.61 (t, J = 5.8 Hz, 1H), 6.39 (d, J = 46.4 Hz, 1H), 4.11 (t, J = 8.6 Hz, 2H), 3.87 (q, J = 4.8 Hz, 2H), 3.72–3.53 (m, 4H), 2.73 (t, J = 5.0 Hz, 1H). 13C NMR (75 MHz, CDCl3) δ 168.6, 164.9 (d, J = 26.3 Hz), 164.1, 160.7, 159.6, 157.7 (d, J = 3.4 Hz), 144.4, 141.4, 132.8, 130.9, 127.9, 124.8, 121.2, 120.2, 119.1, 117.3 (d, J = 15.5 Hz), 113.4 (d, J = 23.9 Hz), 100.1 (d, J = 8.2 Hz), 92.07 (d, J = 179.4 Hz), 77.4, 62.3, 49.0, 42.6, 27.7. 19F NMR (282 MHz, chloroform-d) δ −62.8, −109.5 (t, J = 8.7 Hz), −183.9 (d, J = 46.4 Hz). HRMS (ESI): (M + H)+ calcd for C23H20F5N4O2, 479.1501; found, 479.1498.
1-(6-(Fluoro(3-(trifluoromethyl)phenyl)methyl)pyrimidin-4-yl)-N-(2-hydroxyethyl)indoline-4-carboxamide (25i).
Compound 25i (240 mg, 72%) was synthesized by a procedure similar to that used to prepare compound 12a as a white solid. 1H NMR (300 MHz, chloroform-d and MeOD) δ 8.59 (dd, J = 15.6, 5.0 Hz, 2H), 7.76–7.42 (m, 4H), 7.18 (dt, J = 12.7, 7.0 Hz, 3H), 6.80 (d, J = 4.2 Hz, 1H), 6.35 (dd, J = 46.3, 4.4 Hz, 1H), 4.00 (q, J = 7.3 Hz, 2H), 3.74 (d, J = 5.6 Hz, 2H), 3.49 (dt, J = 20.7, 6.7 Hz, 4H). 13C NMR (75 MHz, CDCl3) δ 168.8, 165.3 (d, J = 26.5 Hz), 159.5, 157.4, 144.1, 138.4 (d, J = 20.5 Hz), 132.5, 131.2, 131.1, 130.8, 130.3 (d, J = 6.5 Hz), 129.2, 127.7, 125.9, 123.5 (dt, J = 6.1, 2.8 Hz) 123.5, 120.5, 118.9, 100.2 (d, J = 8.1 Hz), 92.7 (d, J = 177.7 Hz), 61.3, 48.9, 42.4, 27.5. 19F NMR (282 MHz, CDCl3) δ −62.8, −181.1. HRMS (ESI): (M + H)+ calcd for C23H21F4N4O2, 461.1595; found, 461.1590.
Methyl 1-(6-((3-(Trifluoromethyl)phenyl)amino)pyrimidin-4-yl)indoline-4-carboxylate (29a).
A mixture of 28a (48 mg, 0.3 mmol), Pd(OAc)2 (3 mg, 0.015 mmol), XantPhos (17 mg, 0.03 mmol), Cs2CO3 (196 mg, 0.6 mmol), and 8c (87 mg, 0.3 mmol) in 1,4-dioxane (3 mL) was subjected to three rounds of vacuum evacuation followed by introduction of nitrogen. The reaction mixture was then stirred at 100 °C overnight. The reaction mixture was cooled to room temperature and poured into water and then extracted with EtOAc (10 mL × 3). The organic phase was washed with brine, dried over Na2SO4, and then concentrated under reduced pressure. The residue was purified by silica gel chromatography (gradient: 10 to 20% EtOAc in hexane) to provide product 29a (86 mg, 69%) as a yellow solid. 1H NMR (300 MHz, DMSO-d6) δ 9.72 (s, 1H), 8.65 (dd, J = 8.2, 1.1 Hz, 1H), 8.49 (s, 1H), 8.24 (s, 1H), 7.86 (d, J = 8.0 Hz, 1H), 7.55–7.46 (m, 2H), 7.36–7.27 (m, 2H), 6.11 (s, 1H), 4.03 (t, J = 8.7 Hz, 2H), 3.86 (s, 3H), 3.52 (t, J = 8.7 Hz, 2H).
Methyl 1-(6-(3-(Trifluoromethyl)phenoxy)pyrimidin-4-yl)-indoline-4-carboxylate (29b).
To a solution of 28b (49 mg, 0.3 mmol) in DMF (2 mL) were added cesium carbonate (195 mg, 0.6 mmol) and 8c (87 mg, 0.3 mmol), and the reaction was heated to 120 °C overnight. At this point, the reaction mixture was cooled to rt., poured into water (15 mL), and extracted with ethyl acetate (3 × 10 mL). The combined organic extracts were washed with water (10 mL) and brine (10 mL), dried over anhydrous sodium sulfate, filtered, and concentrated in vacuo. The residue was purified by chromatography on silica gel (eluent: 1% MeOH in CH2Cl2) and yielded product 29b (79 mg, 63%) as a white solid. 1H NMR (300 MHz, DMSO-d6) δ 8.69 (t, J = 8.2 Hz, 1H), 8.47 (s, 1H), 7.73–7.50 (m, 5H), 7.36 (t, J = 8.2 Hz, 1H), 6.47 (d, J = 5.4 Hz, 1H), 4.10 (t, J = 8.5 Hz, 2H), 3.86 (s, 3H), 3.54 (t, J = 8.6 Hz, 2H).
Methyl 1-(6-((3-(Trifluoromethyl)phenyl)thio)pyrimidin-4-yl)indoline-4-carboxylate (29c).
Compound 29c (92 mg, 71%) was synthesized by a procedure similar to that used to prepare compound 29b as a white solid. 1H NMR (300 MHz, DMSO-d6) δ 8.63–8.50 (m, 2H), 8.01–7.85 (m, 3H), 7.75 (t, J = 7.8 Hz, 1H), 7.52 (dd, J = 7.9, 1.1 Hz, 1H), 7.32 (t, J = 8.1 Hz, 1H), 6.52 (d, J = 1.1 Hz, 1H), 3.91 (t, J = 8.6 Hz, 2H), 3.84 (s, 3H), 3.45 (t, J = 8.6 Hz, 2H).
N-(2-Hydroxyethyl)-1-(6-((3-(trifluoromethyl)phenyl)-amino)pyrimidin-4-yl)indoline-4-carboxamide (25j).
To a solution of 29a (41 mg, 0.1 mmol) in con. HCl (4 mL), H2O (1 mL) and AcOH (1 mL) were added, and the mixture solution was stirred at reflux overnight. The reaction was cooled to room temperature, and a yellow solid precipitated from the solution. The yellow solid was filtered and washed with water, and the cake was collected and dried to afford the product as a light yellow solid. The yellow solid and 2-aminoethan-1-ol (13 mg, 0.2 mmol) were dissolved in 2 mL of DMF, and the mixture solution was cooled to 0 °C with an ice bath. HOBt (14 mg, 0.1 mmol), EDCI (39 mg, 0.2 mmol), and DMAP (24 mg, 0.2 mmol) were added to the solution at 0 °C. Then, the ice bath was removed, and the mixture solution was stirred at rt. overnight. After the reaction was completed (detected by TLC), the reaction mixture was worked up by the addition of water and then extracted with EtOAc (20 mL × 3). The combined EtOAc extracts were washed with brine, dried over Na2SO4, filtered, and condensed by rotary evaporation to yield a yellow oil. This material was further purified by preparative TLC plates using CH2Cl2/MeOH = 50:1 as the eluent to yield 25j as a white solid (43 mg, 73% for two steps). 1H NMR (300 MHz, chloroform-d and MeOD) δ 8.43–8.25 (m, 2H), 7.66 (s, 1H), 7.55 (d, J = 8.2 Hz, 1H), 7.40 (t, J = 7.9 Hz, 1H), 7.32–7.23 (m, 2H), 7.19–7.03 (m, 2H), 5.89 (s, 1H), 3.80 (t, J = 8.5 Hz, 2H), 3.70 (t, J = 5.2 Hz, 2H), 3.49 (t, J = 5.3 Hz, 2H), 3.34 (t, J = 8.5 Hz, 2H). 13C NMR (75 MHz, CDCl3 and MeOD) δ 169.2, 166.6, 160.5, 159.7, 157.4, 153.0, 144.8, 139.9, 132.0, 131.6, 131.2, 131.0, 129.7, 127.6, 124.1, 119.6, 117.8, 117.5, 85.9, 61.1, 42.3, 27.4. HRMS (ESI): (M + H)+ calcd for C22H21F3N5O2, 444.1642; found, 444.1639.
N-(2-Hydroxyethyl)-1-(6-(3-(trifluoromethyl)phenoxy)-pyrimidin-4-yl)indoline-4-carboxamide (25k).
Compound 25k (48 mg, 43%) was synthesized by a procedure similar to that used to prepare compound 25j as a white solid. 1H NMR (300 MHz, chloroform-d and MeOD) δ 8.50 (d, J = 7.8 Hz, 1H), 8.39 (d, J = 3.5 Hz, 1H), 7.59–7.08 (m, 7H), 6.05 (d, J = 3.5 Hz, 1H), 3.93 (t, J = 8.8 Hz, 2H), 3.69 (q, J = 4.5 Hz, 2H), 3.48 (s, 4H). 13C NMR (75 MHz, CDCl3 and MeOD) δ 169.3, 169.0, 161.2, 157.4, 152.9, 144.4, 132.3, 131.1, 130.3, 127.7, 124.9, 122.1, 120.3, 118.4, 89.3, 61.1, 42.3, 27.5. HRMS (ESI): (M + H)+ calcd for C22H20F3N4O3, 445.1482; found, 445.1480.
N-(2-Hydroxyethyl)-1-(6-((3-(trifluoromethyl)phenyl)thio)-pyrimidin-4-yl)indoline-4-carboxamide (25l).
Compound 25l (64 mg, 56%) was synthesized by a procedure similar to that used to prepare compound 25j as a white solid. 1H NMR (300 MHz, chloroform-d and MeOD) δ 8.47 (d, J = 3.9 Hz, 1H), 8.42–8.28 (m, 1H), 7.92–7.52 (m, 4H), 7.31–7.07 (m, 3H), 6.03 (d, J = 3.8 Hz, 1H), 3.71 (d, J = 5.7 Hz, 4H), 3.49 (t, J = 5.0 Hz, 2H), 3.40 (d, J = 9.4 Hz, 2H). 13C NMR (75 MHz, CDCl3 and MeOD) δ 169.0, 168.8, 158.5, 157.1, 144.1, 138.6, 132.4, 132.3, 131.9, 131.1, 130.2, 130.2, 127.6, 126.5, 120.4, 118.4, 100.4, 61.2, 48.6, 42.3, 42.2, 27.4. HRMS (ESI): (M + H)+ calcd for C22H20F3N4O2S, 461.1254; found, 461.1250.
N-(1,3-Dihydroxypropan-2-yl)-1-(6-(3-fluoro-5-(trifluoromethyl)benzyl)pyrimidin-4-yl)indoline-4-carboxamide (30a).
Compound 30a (28 mg, 57%) was synthesized by a procedure similar to that used to prepare compound 12a as a white solid. 1H NMR (300 MHz, chloroform-d and MeOD) δ 8.68 (s, 1H), 8.55 (d, J = 7.5 Hz, 1H), 7.33 (s, 1H), 7.28–7.11 (m, 4H), 6.40 (s, 1H), 4.10–3.90 (m, 5H), 3.82 (dd, J = 11.3, 4.3 Hz, 2H), 3.70 (dd, J = 11.3, 5.1 Hz, 2H), 3.49 (t, J = 8.5 Hz, 2H). 13C NMR (75 MHz, CDCl3 and MeOD) δ 168.5, 166.6, 166.0, 159.5, 157.7, 144.3, 141.6, 132.3, 130.9, 127.8, 121.8–121.5 (m), 120.5, 119.7, 119.4, 118.8, 111.5 (d, J = 3.9 Hz), 111.13 (d, J = 3.8 Hz), 104.0, 61.6, 52.6, 48.8, 43.4, 27.5. HRMS (ESI): (M + H)+ calcd for C24H23F4N4O3, 491.1701; found, 491.1697.
1-(6-(3-Fluoro-5-(trifluoromethyl)benzyl)pyrimidin-4-yl)-N-(2-methoxyethyl)indoline-4-carboxamide (30b).
Compound 30b (30 mg, 65%) was synthesized by a procedure similar to that used to prepare compound 12a as a white solid. 1H NMR (300 MHz, chloroform-d) δ 8.78 (s, 1H), 8.63 (d, J = 8.1 Hz, 1H), 7.38 (s, 1H), 7.31–7.21 (m, 3H), 7.17 (d, J = 7.7 Hz, 1H), 6.47 (d, J = 6.2 Hz, 1H), 6.43 (s, 1H), 4.08 (s, 2H), 4.02 (t, J = 8.6 Hz, 2H), 3.61 (ddd, J = 16.4, 10.7, 6.9 Hz, 6H), 3.42 (s, 3H). 13C NMR (75 MHz, CDCl3) δ 167.6, 166.2, 164.2, 160.9, 159.5, 158.0, 144.5, 141.8 (d, J = 7.6 Hz), 132.9, 132.5, 132.5, 131.2, 127.8, 125.0, 121.7 (t, J = 3.6 Hz), 120.0, 119.6 (d, J = 21.4 Hz), 118.6, 111.3 (dd, J = 24.6, 3.9 Hz), 103.9, 77.4, 77.0, 76.6, 71.2, 48.8, 43.8, 39.5, 27.7. HRMS (ESI): (M + H)+ calcd for C24H23F4N4O2, 475.1752; found, 475.1749.
1-(6-(3-Fluoro-5-(trifluoromethyl)benzyl)pyrimidin-4-yl)-N-(3-hydroxypropyl)indoline-4-carboxamide (30c).
Compound 30c (26 mg, 56%) was synthesized by a procedure similar to that used to prepare compound 12a as a white solid. 1H NMR (300 MHz, chloroform-d) δ 8.77 (s, 1H), 8.63 (d, J = 8.1 Hz, 1H), 7.38 (s, 1H), 7.29–7.20 (m, 3H), 7.14 (d, J = 7.7 Hz, 1H), 6.64 (t, J = 6.1 Hz, 1H), 6.43 (s, 1H), 4.07 (s, 2H), 4.00 (t, J = 8.6 Hz, 2H), 3.76 (t, J = 5.5 Hz, 2H), 3.69–3.50 (m, 4H), 3.23 (s, 1H), 1.82 (p, J = 5.7 Hz, 2H). 13C NMR (75 MHz, CDCl3) δ 168.7, 166.2, 164.2, 160.9, 159.5, 158.0, 144.6, 141.8 (d, J = 7.5 Hz), 132.6, 131.0, 127.9, 121.8–121.6 (m), 119.8, 119.5, 118.8, 111.7–111.0 (m), 103.9, 59.9, 48.8, 43.8, 37.0, 32.1, 27.7. HRMS (ESI): (M + H)+ calcd for C24H23F4N4O2, 475.1752; found, 475.1749.
1-(6-(3-Fluoro-5-(trifluoromethyl)benzyl)pyrimidin-4-yl)-N-(4-hydroxybutyl)indoline-4-carboxamide (30d).
Compound 30d (30 mg, 61%) was synthesized by a procedure similar to that used to prepare compound 12a as a white solid. 1H NMR (300 MHz, chloroform-d and MeOD) δ 8.66 (s, 1H), 8.52 (d, J = 8.1 Hz, 1H), 7.33 (s, 1H), 7.27–7.06 (m, 5H), 6.39 (s, 1H), 4.02 (s, 2H), 3.94 (t, J = 8.5 Hz, 2H), 3.61 (t, J = 5.8 Hz, 2H), 3.47 (t, J = 8.5 Hz, 2H), 3.38 (t, J = 6.2 Hz, 2H), 1.63 (dq, J = 12.9, 6.7 Hz, 4H). 13C NMR (75 MHz, CDCl3 and MeOD) δ 168.5, 165.9, 164.1, 160.8, 159.5, 157.7, 144.2, 141.6 (d, J = 7.7 Hz), 132.3, 131.5, 127.7, 121.6, 120.3, 119.7, 119.4, 118.5, 111.4 (d, J = 3.8 Hz), 111.1 (d, J = 3.7 Hz), 104.0, 61.7, 48.8, 43.3, 39.6, 29.6, 27.4, 25.9. HRMS (ESI): (M + H)+ calcd for C25H25F4N4O2, 489.1908; found, 489.1904.
(1-(6-(3-Fluoro-5-(trifluoromethyl)benzyl)pyrimidin-4-yl)-indolin-4-yl)(3-hydroxyazetidin-1-yl)methanone (30e).
Compound 30e (17 mg, 37%) was synthesized by a procedure similar to that used to prepare compound 12a as a white solid. 1H NMR (300 MHz, chloroform-d) δ 8.76 (d, J = 1.0 Hz, 1H), 8.56 (d, J = 8.2 Hz, 1H), 7.38 (s, 1H), 7.24 (d, J = 8.4 Hz, 3H), 7.00 (dd, J = 7.7, 1.0 Hz, 1H), 6.45–6.39 (m, 1H), 4.79–4.66 (m, 1H), 4.41 (s, 2H), 4.11–3.94 (m, 6H), 3.52–3.39 (m, 2H), 3.06 (s, 1H). 13C NMR (75 MHz, CDCl3) δ 169.9, 166.2, 164.2, 160.9, 159.5, 158.0, 144.5, 141.7 (d, J = 7.6 Hz), 132.0, 129.5, 127.5, 121.8–121.6 (m), 120.9, 119.6 (d, J = 21.7 Hz), 118.3, 111.3 (d, J = 28.0 Hz), 103.9, 62.2, 61.7, 58.2, 48.7, 43.7, 27.2. HRMS (ESI): (M + H)+ calcd for C24H21F4N4O2, 473.1595; found, 473.1593.
1-(6-(3-Fluoro-5-(trifluoromethyl)benzyl)pyrimidin-4-yl)-N-(2-fluoroethyl)indoline-4-carboxamide (30f).
Compound 30f (36 mg, 78%) was synthesized by a procedure similar to that used to prepare compound 12a as a white solid. 1H NMR (300 MHz, chloroform-d) δ 8.83–8.73 (m, 1H), 8.66 (d, J = 8.1 Hz, 1H), 7.39 (s, 1H), 7.33–7.15 (m, 4H), 6.45 (d, J = 9.4 Hz, 2H), 4.71 (t, J = 4.7 Hz, 1H), 4.55 (t, J = 4.7 Hz, 1H), 4.14–3.95 (m, 4H), 3.83 (q, J = 5.1 Hz, 1H), 3.74 (q, J = 5.1 Hz, 1H), 3.58 (t, J = 8.5 Hz, 2H). 13C NMR (75 MHz, CDCl3) δ 167.8, 166.3, 164.2, 160.9, 159.5, 158.0, 144.7, 141.8 (d, J = 7.6 Hz), 132.6, 130.8, 128.0, 121.7 (t, J = 3.5 Hz), 119.9, 119.8, 119.5, 118.9, 111.5 (d, J = 3.7 Hz), 111.2 (d, J = 3.8 Hz), 103.9, 82.8 (d, J = 166.7 Hz), 48.8, 43.8, 40.2 (d, J = 19.5 Hz), 27.7. 19F NMR (282 MHz, chloroform-d) δ −62.7, −110.6 (t, J = 8.8 Hz), −224.2 (tt, J = 47.4, 28.3 Hz). HRMS (ESI): (M + H)+ calcd for C23H20F5N4O, 473.1595; found, 473.1593.
1-(6-(3-Fluoro-5-(trifluoromethyl)benzyl)pyrimidin-4-yl)-N,N-bis(2-hydroxyethyl)indoline-4-carboxamide (30g).
Compound 30g (27 mg, 53%) was synthesized by a procedure similar to that used to prepare compound 12a as a white solid. 1H NMR (300 MHz, chloroform-d) δ 8.71 (s, 1H), 8.45 (d, J = 8.2 Hz, 1H), 7.37 (s, 1H), 7.29–7.17 (m, 3H), 6.97 (d, J = 7.6 Hz, 1H), 6.40 (s, 1H), 4.10–3.57 (m, 12H), 3.44 (t, J = 5.0 Hz, 2H), 3.26 (t, J = 8.5 Hz, 2H). 13C NMR (75 MHz, CDCl3) δ 172.3, 166.2, 164.2, 160.9, 159.5, 158.0, 144.0, 141.7 (d, J = 7.5 Hz), 133.0, 129.7, 127.8, 122.0–121.5 (m), 120.2, 119.8, 119.5, 117.0, 111.3 (d, J = 24.4 Hz), 103.8, 61.1, 60.1, 52.9, 49.1, 48.5, 43.6, 26.1. HRMS (ESI): (M + H)+ calcd for C25H25F4N4O3, 505.1857; found, 505.1853.
Molecular Docking Protocol.
The molecular docking study was performed using Schrödinger Small-Molecule Drug Discovery Suite (Schrödinger, LLC, New York, NY, 2020). The cocrystal structures of GPR52 and compound 2 (PDB code: 6LI0) were downloaded from the RCS PDB bank. The cocrystal structure was preprocessed and minimized with Schrödinger Protein Preparation Wizard using default settings. The grid center was chosen on the centroid of an existing ligand, and the size of the grid box was set to 30 Å on each side. The 3D structure of ligand 12c was created using Schrödinger Maestro, and a low energy conformation was calculated using LigPrep. Docking was employed with Glide using the SP precision. Docked poses were incorporated into Schrödinger Maestro for visualization and analysis of binding site interactions.
In Vitro Pharmacology.
Cell Culture and Plasmids.
Wildtype (WT) human embryonic kidney 293 (HEK293) cells were a generous gift from Dr. Asuka Inoue, Tohoku University.31 The wildtype HEK293 cells were cultured in DMEM (Gibco, Carlsbad, CA) with 10% FBS (Omega Scientific, Tarzana, CA) and 10,000 U/mL penicillin–streptomycin (Gibco, Carlsbad, CA) in a humidified incubator at 37 °C in 5% CO2. A GPR52 TANGO construct was purchased from Addgene (Watertown, MA). A stop codon was placed in the frame immediately after the receptor coding sequence to generate a human GPR52 WT coding construct. Point mutation of GPR52 WT from GPR52 TANGO was made by polymerase chain reaction (PCR) using the QuikChange II Site-Directed Mutagenesis Kit (Agilent Technologies, Santa Clara, CA) according to the manufacturer’s protocol. Mutagenesis and sequencing primers were obtained from ThermoFisher (Pittsburgh, PA). The primers used to make the GPR52 WT point mutant were 5′–gctaacagctgttccatctaagataccggtggacgcacc–3′ (sense) and 5′–cgattgtcgacaaggtagattctatggccacctgcgtgg–3′ (antisense). Parental DNA in the reaction mixture was digested using restriction endonuclease DpnI (from diplococcus pneumoniae) at 37 °C for one hour. A digestion mixture (2 μL) was transformed into XL1-Blue supercompetent cells by heat shock at 42 °C for 45 s. The reaction was incubated in Super Optimal broth with a catabolite repression medium (Sigma-Aldrich) and then plated onto Luria–Bertani (LB) agar plates containing 100 μg/mL ampicillin. A single colony was selected for sequencing, and the point mutation was verified prior to use (Molecular Genomics Core, University of Texas Medical Branch, Galveston, TX).
Transfections.
For the cAMP Glosensor assays, 6.5 × 105 wildtype HEK293 cells were plated in 6-well plates (Corning, Oneonta, NY) 24 h before transfection to achieve 70–80% confluency the following day. Each well of wildtype HEK293 cells was transiently transfected with 0.10 μg of wildtype human GPR52 plasmid cDNA and 1 μg of the 22F Glosensor plasmid (Promega, Madison, WI) using 10 μL of Lipofectamine 2000 (Invitrogen, Waltham, MA) in 1 mL of OptiMEM (Gibco, Carlsbad, CA) and 1 mL of growth media.
cAMP Glosensor Assay.
Eighteen hours after transfection, wildtype HEK293 cells with GPR52 and Glosensor constructs (see Transfections above), cells were seeded at 60,000 cells/well in growth media into poly-l-lysine (Trevigen, Gaithersburg, MD) coated 96-well white clear-bottom cell culture plates (Greiner Bio-One, Monroe, NC). Four hours later, complete media was aspirated and replaced with 90 μL of a 1% Glosensor reagent (Promega, Madison, WI) in 1× Hanks’ balanced salt solution (HBSS) and 20 mM HEPES. Cells were serum-starved for 2 h at room temperature in the dark. Test compounds were weighed and diluted to 2 mM in DMSO. Serial dilutions of each test compound were prepared at 10× final concentration and transferred to a 96-well source plate. Cells were incubated with 10 μL of 10× drug solutions for 15 min. LCPS were recorded on a Microbeta2 Microplate counter (PerkinElmer, Waltham, MA). Data from at least three independent experiments (n = 3 to 21) conducted in technical triplicate are presented as percentage of compound 4 response. All in vitro pharmacological data were analyzed using GraphPad Prism 7.05 software (La Jolla, CA). Data from cAMP ligand dose response assays are presented as the half maximum (EC50, nM) and maximum effect (Emax) (means ± SEM) as computed by GraphPad using a four-parameter nonlinear regression curve-fitting algorithm. All Emax ligand responses were scaled relative to 3 μM compound 4 in cells treated for each assay (3 μM compound 4 response set to 100%).
In Vivo PK and Brain Permeability Studies.
Male Sprague Dawley rats (n = 3 per treatment group; Beijing Vital River Laboratory, Animal Technology Co., Ltd., Beijing, China) weighing 200–250 g at the beginning of the experiment were housed three per cage in a pathogen-free, temperature-controlled (20–26 °C), and humidity-controlled (40–70%) environment with a 12 h light–dark cycle and ad libitum access to food and filtered water. Rats were randomly assigned to treatment groups. A vehicle [10% dimethyl sulfoxide (DMSO) and 90% 2-hydroxypropyl-β-cyclodextrin (HP-β-CD); Cyclodextrin Technologies Development, Inc., High Springs, FL, USA] or compound 12c dissolved in the vehicle was administered to rats IV at 10 mg/kg or PO at 20 mg/kg. The rat was restrained manually at the designated time points (0.08, 0.25, 0.5, 1.0, 2.0, 4.0, 8.0, and 24 h post-dosing for IV; 0.25, 0.5, 1.0, 2.0, 4.0, 8.0, and 24 h post-dosing for PO). Blood (yielding ~50 μL of plasma) samples (100 μL) are taken from the animal’s jugular vein and placed into micro K2EDTA tubes. Blood samples were placed on ice and centrifuged at 8000 rpm for 5 min at 4 °C to generate plasma samples within 0.5 h of collection. Brain samples were collected at 0.25 and 1 h post-dosing. All samples were stored at −20 °C. The concentration of 12c in each sample was analyzed by Sundia MediTech Co., Ltd. The study and the related standard operating procedures were reviewed and approved by the Sundia Institutional Animal Care and Use Committee. The Sundia animal facility is approved with yearly inspection by the Shanghai Laboratory Animal Management Committee. All treatment assignments were blinded to investigators who performed PK assays and end point statistical analyses.
The PK parameters of compound 12c were calculated according to a non-compartmental model using WinNonlin (Pharsight Corporation, ver 5.3, Mountain View, CA, USA). The peak concentration (Cmax) was directly obtained by visual inspection of the plasma concentration–time profile. The elimination rate constant (λ) was obtained by the least-squares fitted terminal log-linear portion of the slope of the plasma concentration–time profile. The elimination half-life (t1/2) was evaluated according to 0.693/λ. The area under the plasma concentration–time curve from 0 to time t (AUC0–t) was evaluated using the linear trapezoidal rule and further extrapolated to infinity (AUC0–∞) according to the following equation: AUC0–∞ = AUC0–t + Clast/λ. The pharmacokinetic parameters were presented as mean ± SEM.
In Vivo Pharmacology of Compound 12c.
Animals.
Naïve male C57/BL6 mice (n = 60; Jackson Laboratory, Bar Harbor, ME) weighing between 24 and 31 g (~60 days of age) at the beginning of the experiment were housed four/cage in a temperature- (21–23 °C) and humidity-controlled (40–50%) environment on a 12 h light/dark cycle (lights on at 0600). Mice were provided ad libitum access to chow and water for 14–20 days prior to experiments, which were carried out in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals (2011) and with the approval of the Institutional Animal Care and Use Committee at the University of Texas Medical Branch.
Drugs.
Compound 12c was dissolved in 0.9% saline containing 20% HP-β-CD with a final pH of the solution adjusted to 7.4. Dextroamphetamine (Sigma-Aldrich, St Louis, MO, USA) was prepared in 0.9% saline. Compounds were then administered IP at 2 mL/1 kg body weight.
Locomotor Activity Assessment.
A modified open field activity system (San Diego Instruments, San Diego, California, U.S.A.) was employed to quantify activity under low-light conditions.32–34 Clear Plexiglas chambers (40 × 40 × 40 cm) were surrounded by a 4 × 4 photobeam matrix positioned ~1 cm above the chamber floor. Horizontal activity was quantified as the sum of photobeam breaks that occurred within the inner central 16 × 16 cm area and in the surrounding outer 12 × 12 cm perimeter of the activity monitor.
A between-subjects design was employed to study the efficacy of compound 12c to impact horizontal activity. Mice were handled three times/week for two weeks prior to the study and were habituated to the motor activity monitors 1 h/day for three days. Mice were transported in their home cages and allowed to acclimate to the testing room for 1 h prior to evaluation. On the day of the test, mice were weighed and randomized into five treatment groups for compound 12c administration (n = 12/dose). One animal was excluded as an outlier (i.e., horizontal activity two standard deviations from the mean) with n = 11 for 1 mg/kg compound 12c). Following IP administration of the vehicle or compound 12c (0.3, 1, 3, and 10 mg/kg), automated activity monitoring began immediately and continued for 30 min. Mice were then injected with AMPH (3 mg/kg IP), and activity was monitored for the next 90 min. All testings occurred between 0800 and 1700 h.
Statistical Analyses.
Locomotor activities are presented as mean horizontal activity (mean ± SEM). A one-way ANOVA for a between-subjects design was employed to analyze horizontal activity. A priori comparisons were defined prior to the start of experimentation and conducted by Dunnett’s procedure,35 with an experiment-wise error rate set at α = 0.05. Investigators who performed compound administration were blinded to treatment assignments and endpoint statistical analyses.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by grants T32 DA 007287 and U18 DA052543 from the National Institutes of Health, the PhRMA foundation, and the Center for Addiction Research at the University of Texas Medical Branch (UTMB). J.Z. is also partially supported by the John D. Stobo, M. D., Distinguished Chair Endowment Fund. The authors would like to acknowledge Robert Fox for in vivo assay assistance and the preparation of amphetamine solutions for in vivo injections. The authors would also like to acknowledge Dr. Asuka Inoue, Tohoku University, for the generous gift of the HEK cell line, Dr. Lawrence C. Sowers at the UTMB Department of Pharmacology and Toxicology as well as Dr. Tianzhi Wang at the NMR core facility of UTMB for the NMR spectroscopy assistance, and Dr. Xuemei Luo at the UTMB Mass Spectrometry Core with the funding support from the University of Texas System Proteomics Network for the HRMS analysis.
ABBREVIATIONS USED
- GPCRs
G-protein-coupled receptors
- FDA
U.S. Food and Drug Administration
- CNS
central nervous system
- NAc
nucleus accumbens
- MSNs
medium spiny neurons
- cAMP
cyclic adenosine monophosphate
- NMDA
N-methyl-d-aspartate
- SUD
substance use disorder
- ECL2
extracellular loop 2
- EC50
half maximal effective concentration
- HTS
high-throughput screening
- TM
transmembrane helix
- SAR
structure–activity relationship
- PK
pharmacokinetic
- DMF
N,N-dimethylformamide
- EDCI
N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride
- DMAP
4-dimethylaminopyridine
- XantPhos
4,5-bis(diphenylphosphino)-9,9-dimethylxanthene
- mCPBA
meta-chloroperoxybenzoic acid
- DAST
diethylaminosulfur trifluoride
- DMSO
dimethyl sulfoxide
- NIMH
National Institute of Mental Health
- PDSP
psychoactive drug screening program
- hERG
human ether-a-go-go-related gene
- PO
oral
- IV
intravenous
- AMPH
amphetamine
- WT
wildtype
- HEK293
human embryonic kidney 293
- PCR
polymerase chain reaction
- HBSS
Hanks’ balanced salt solution
- IP
intraperitoneal
Footnotes
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.jmedchem.0c01498
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.0c01498.
Figures S1 and S2, HPLC analysis spectra of representative compounds, and 1H and 13C NMR spectra of all new compounds (PDF)
Molecular formula strings and data (CSV)
Docking compound 12c with GPR52 (PDB)
The authors declare no competing financial interest.
Contributor Information
Pingyuan Wang, Chemical Biology Program, Department of Pharmacology and Toxicology, University of Texas Medical Branch, Galveston, Texas 77555, United States.
Daniel E. Felsing, Center for Addiction Research, University of Texas Medical Branch, Galveston, Texas 77555, United States.
Haiying Chen, Chemical Biology Program, Department of Pharmacology and Toxicology, University of Texas Medical Branch, Galveston, Texas 77555, United States.
Sonja J. Stutz, Center for Addiction Research, University of Texas Medical Branch, Galveston, Texas 77555, United States
Ryan E. Murphy, Center for Addiction Research, University of Texas Medical Branch, Galveston, Texas 77555, United States
Kathryn A. Cunningham, Chemical Biology Program, Department of Pharmacology and Toxicology and Center for Addiction Research, University of Texas Medical Branch, Galveston, Texas 77555, United States;.
John A. Allen, Chemical Biology Program, Department of Pharmacology and Toxicology and Center for Addiction Research, University of Texas Medical Branch, Galveston, Texas 77555, United States;.
Jia Zhou, Chemical Biology Program, Department of Pharmacology and Toxicology and Center for Addiction Research, University of Texas Medical Branch, Galveston, Texas 77555, United States;.
REFERENCES
- (1).Allen JA; Roth BL Strategies to discover unexpected targets for drugs active at G protein-coupled receptors. Annu. Rev. Pharmacol. Toxicol 2011, 51, 117–144. [DOI] [PubMed] [Google Scholar]
- (2).Wold EA; Chen J; Cunningham KA; Zhou J Allosteric modulation of class A GPCRs: targets, agents, and emerging concepts. J. Med. Chem 2019, 62, 88–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Zhou J; Wild C GPCR drug discovery: emerging targets, novel approaches and future trends. Curr. Top. Med. Chem 2019, 19, 1363–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Overington JP; Al-Lazikani B; Hopkins AL How many drug targets are there? Nat. Rev. Drug Discovery 2006, 5, 993–996. [DOI] [PubMed] [Google Scholar]
- (5).Lagerström MC; Schiöth HB Structural diversity of G protein-coupled receptors and significance for drug discovery. Nat. Rev. Drug Discovery 2008, 7, 339–357. [DOI] [PubMed] [Google Scholar]
- (6).Hauser AS; Attwood MM; Rask-Andersen M; Schiöth HB; Gloriam DE Trends in GPCR drug discovery: new agents, targets and indications. Nat. Rev. Drug Discovery 2017, 16, 829–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Roth BL; Irwin JJ; Shoichet BK Discovery of new GPCR ligands to illuminate new biology. Nat. Chem. Biol 2017, 13, 1143–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Ye N; Li B; Mao Q; Wold EA; Tian S; Allen JA; Zhou J Orphan receptor GPR88 as an emerging neurotherapeutic target. ACS Chem. Neurosci 2019, 10, 190–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Sawzdargo M; Nguyen T; Lee DK; Lynch KR; Cheng R; Heng HHQ; George SR; O’Dowd BF Identification and cloning of three novel human G protein-coupled receptor genes GPR52, ΨGPR53 and GPR55: GPR55 is extensively expressed in human brain. Mol. Brain Res 1999, 64, 193–198. [DOI] [PubMed] [Google Scholar]
- (10).Komatsu H; Maruyama M; Yao S; Shinohara T; Sakuma K; Imaichi S; Chikatsu T; Kuniyeda K; Siu FK; Peng LS; Zhuo K; Mun LS; Han TM; Matsumoto Y; Hashimoto T; Miyajima N; Itoh Y; Ogi K; Habata Y; Mori M Anatomical transcriptome of G protein-coupled receptors leads to the identification of a novel therapeutic candidate GPR52 for psychiatric disorders. PLoS One 2014, 9, No. e90134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Yang RY; Quan J; Sodaei R; Aguet F; Segrè AV; Allen JA; Lanz TA; Reinhart V; Crawford M; Hasson S; Ardlie KG; Guigö R; Xi HS A systematic survey of human tissue-specific gene expression and splicing reveals new opportunities for therapeutic target identification and evaluation. bioRxiv 2018, 311563. [Google Scholar]
- (12).Lin X; Li M; Wang N; Wu Y; Luo Z; Guo S; Han GW; Li S; Yue Y; Wei X; Xie X; Chen Y; Zhao S; Wu J; Lei M; Xu F Structural basis of ligand recognition and self-activation of orphan GPR52. Nature 2020, 579, 152–157. [DOI] [PubMed] [Google Scholar]
- (13).Martin AL; Steurer MA; Aronstam RS Constitutive activity among orphan class-A G protein coupled receptors. PLoS One 2015, 10, No. e0138463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Setoh M; Ishii N; Kono M; Miyanohana Y; Shiraishi E; Harasawa T; Ota H; Odani T; Kanzaki N; Aoyama K; Hamada T; Kori M Discovery of the first potent and orally available agonist of the orphan G-protein-coupled receptor 52. J. Med. Chem 2014, 57, 5226–5237. [DOI] [PubMed] [Google Scholar]
- (15).Nishiyama K; Suzuki H; Harasawa T; Suzuki N; Kurimoto E; Kawai T; Maruyama M; Komatsu H; Sakuma K; Shimizu Y; Shimojo M FTBMT, a novel and selective GPR52 agonist, demonstrates antipsychotic-like and procognitive effects in rodents, revealing a potential therapeutic agent for schizophrenia. J. Pharmacol. Exp. Ther 2017, 363, 253–264. [DOI] [PubMed] [Google Scholar]
- (16).Xiong YGAJ; Korman H; Lehmann J; Ren AS; Semple G 1-Heteroaryl-indoline-4-carboxamines as modulators of GPR52 usedul for the treatment or prevention of disorders related thereto. WO2016176571 A1, November3, 2016. [Google Scholar]
- (17).Tokumaru K; Ito Y; Nomura I; Nakahata T; Shimizu Y; Kurimoto E; Aoyama K; Aso K Design, synthesis, and pharmacological evaluation of 4-azolyl-benzamide derivatives as novel GPR52 agonists. Bioorg. Med. Chem 2017, 25, 3098–3115. [DOI] [PubMed] [Google Scholar]
- (18).Nakahata T; Tokumaru K; Ito Y; Ishii N; Setoh M; Shimizu Y; Harasawa T; Aoyama K; Hamada T; Kori M; Aso K Design and synthesis of 1-(1-benzothiophen-7-yl)-1H-pyrazole, a novel series of G protein-coupled receptor 52 (GPR52) agonists. Bioorg. Med. Chem 2018, 26, 1598–1608. [DOI] [PubMed] [Google Scholar]
- (19).Meanwell NA Synopsis of some recent tactical application of bioisosteres in drug design. J. Med. Chem 2011, 54, 2529–2591. [DOI] [PubMed] [Google Scholar]
- (20).Meanwell NA The influence of bioisosteres in drug design: tactical applications to address developability problems. Tactics Contemp. Drug Des 2015, 9, 283–382. [Google Scholar]
- (21).Pennington LD; Moustakas DT The necessary nitrogen atom: a versatile high-impact design element for multiparameter optimization. J. Med. Chem 2017, 60, 3552–3579. [DOI] [PubMed] [Google Scholar]
- (22).Cesta DJ; Somersan Karakaya SS; Saito K; Javidnia PE; Roberts J; Ling Y; Gold BS; Lopez Quezada L; Zhang D; Abramyan TM; Kireev DB; Aubé J; Scarry SM Synthesis and antibacterial evaluation of cephalosporin isosteres. Asian J. Org. Chem 2019, 8, 1053–1057. [Google Scholar]
- (23).Sengmany S; Lebre J; Le Gall E; Léonel E Selective monoamination of dichlorodiazines. Tetrahedron 2015, 71, 4859–4867. [Google Scholar]
- (24).Loksha YM Novel synthetic route for 5-substituted 6-arylmethylluracils from 2,4,6-trichloropyrimidines. J. Heterocycl. Chem 2009, 46, 1296–1301. [Google Scholar]
- (25).Gray DL; Allen JA; Mente S; O’Connor RE; DeMarco GJ; Efremov I; Tierney P; Volfson D; Davoren J; Guilmette E; Salafia M; Kozak R; Ehlers MD Impaired β-arrestin recruitment and reduced desensitization by non-catechol agonists of the D1 dopamine receptor. Nat. Commun 2018, 9, 674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Besnard J; Ruda GF; Setola V; Abecassis K; Rodriguiz RM; Huang X-P; Norval S; Sassano MF; Shin AI; Webster LA; Simeons FRC; Stojanovski L; Prat A; Seidah NG; Constam DB; Bickerton GR; Read KD; Wetsel WC; Gilbert IH; Roth BL; Hopkins AL Automated design of ligands to polypharmacological profiles. Nature 2012, 492, 215–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Kalyaanamoorthy S; Barakat KH Development of safe drugs: the hERG challenge. Med. Res. Rev 2018, 38, 525–555. [DOI] [PubMed] [Google Scholar]
- (28).Jones CA; Watson DJG; Fone KCF Animal models of schizophrenia. Br. J. Pharmacol 2011, 164, 1162–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Dichter GS; Damiano CA; Allen JA Reward circuitry dysfunction in psychiatric and neurodevelopmental disorders and genetic syndromes: animal models and clinical findings. J. Neurodev. Disord 2012, 4, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Komatsu H Novel therapeutic GPCRs for psychiatric disorders. Int. J. Mol. Sci 2015, 16, 14109–14121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Grundmann M; Merten N; Malfacini D; Inoue A; Preis P; Simon K; Rüttiger N; Ziegler N; Benkel T; Schmitt NK; Ishida S; Müller I; Reher R; Kawakami K; Inoue A; Rick U; Kühl T; Imhof D; Aoki J; König GM; Hoffmann C; Gomeza J; Wess J; Kostenis E Lack of beta-arrestin signaling in the absence of active G proteins. Nat. Commun 2018, 9, 341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Simmler LD; Anacker AMJ; Levin MH; Vaswani NM; Gresch PJ; Nackenoff AG; Anastasio NC; Stutz SJ; Cunningham KA; Wang J; Zhang B; Henry LK; Stewart A; Veenstra-VanderWeele J; Blakely RD Blockade of the 5-HT transporter contributes to the behavioural, neuronal and molecular effects of cocaine. Br. J. Pharmacol 2017, 174, 2716–2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Cortez I; Bulavin DV; Wu P; McGrath EL; Cunningham KA; Wakamiya M; Papaconstantinou J; Dineley KT Aged dominant negative p38α MAPK mice are resistant to age-dependent decline in adult-neurogenesis and context discrimination fear conditioning. Behav. Brain Res 2017, 322, 212–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Cunningham KA; Anastasio NC; Fox RG; Stutz SJ; Bubar MJ; Swinford SE; Watson CS; Gilbertson SR; Rice KC; Rosenzweig-Lipson S; Moeller FG Synergism between a serotonin 5-HT2A receptor (5-HT2AR) antagonist and 5-HT2CR agonist suggests new pharmacotherapeutics for cocaine addiction. ACS Chem. Neurosci 2013, 4, 110–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Keppel G; Wickens TD Design and analysis: a researcher’s handbook; Pearson Prentice Hall: 2004. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.