

Abstract
Arthropod-borne rickettsial pathogens cause mild and severe human disease worldwide. The tick-borne pathogen Rickettsia parkeri elicits skin lesions (eschars) and disseminated disease in humans; however, inbred mice are generally resistant to infection. We report that intradermal infection of mice lacking both interferon receptors (Ifnar1-/-;Ifngr1-/-) with as few as 10 R. parkeri elicits eschar formation and disseminated, lethal disease. Similar to human infection, eschars exhibited necrosis and inflammation, with bacteria primarily found in leukocytes. Using this model, we find that the actin-based motility factor Sca2 is required for dissemination from the skin to internal organs, and the outer membrane protein OmpB contributes to eschar formation. Immunizing Ifnar1-/-;Ifngr1-/- mice with sca2 and ompB mutant R. parkeri protects against rechallenge, revealing live-attenuated vaccine candidates. Thus, Ifnar1-/-;Ifngr1-/- mice are a tractable model to investigate rickettsiosis, virulence factors, and immunity. Our results further suggest that discrepancies between mouse and human susceptibility may be due to differences in interferon signaling.
Research organism: Rickettsia parkeri, Mouse, Listeria monocytogenes
eLife digest
Tick bites allow disease-causing microbes, including multiple species of Rickettsia bacteria, to pass from arthropods to humans. Being exposed to Rickettsia parkeri, for example, can cause a scab at the bite site, fever, headache and fatigue.
To date, no vaccine is available against any of the severe diseases caused by Rickettsia species. Modelling human infections in animals could help to understand and combat these illnesses. R. parkeri is a good candidate for such studies, as it can give insight into more severe Rickettsia infections while being comparatively safer to handle. However, laboratory mice are resistant to this species of bacteria, limiting their use as models.
To explore why this is the case, Burke et al. probed whether an immune mechanism known as interferon signalling protects laboratory rodents against R. parkeri. During infection, the immune system releases molecules called interferons that stick to ‘receptors’ at the surface of cells, triggering defense mechanisms that help to fight off an invader.
Burke et al. injected R. parkeri into the skin of mice that had or lacked certain interferon receptors, showing that animals without two specific receptors developed scabs and saw the disease spread through their body. Further investigation showed that two R. parkeri proteins, known as OmpB or Sca2, were essential for the bacteria to cause skin lesions and damage internal organs.
Burke et al. then used R. parkeri that lacked OmpB or Sca2 to test whether these modified, inoffensive microbes could act as ‘vaccines’. And indeed, vulnerable laboratory mice which were first exposed to the mutant bacteria were then able to survive the ‘normal’ version of the microbe.
Together, this work reveals that interferon signalling protects laboratory mice against R. parkeri infections. It also creates an animal model that can be used to study disease and vaccination.
Introduction
Obligate cytosolic bacterial pathogens in the family Rickettsiaceae are a diverse group of arthropod-borne microbes that cause severe human disease worldwide, including spotted fever, scrub typhus, and typhus (Bonell et al., 2017; Fang et al., 2017; Sahni et al., 2019). Human disease caused by the tick-borne spotted fever group (SFG) pathogen Rickettsia parkeri is characterized by a necrotic, dry skin lesion (eschar) at the infection site, as well as generalized rash, headache, fatigue, and fever (Paddock et al., 2008). There is no approved vaccine for R. parkeri or for the more virulent rickettsial pathogens that can cause fatal or latent disease (Osterloh, 2017). Moreover, many critical aspects of disease caused by obligate cytosolic bacterial pathogens, including the mechanisms of virulence and immunity, remain unknown. R. parkeri can be handled under biosafety level 2 (BSL2) conditions, and characterizing mouse models to recapitulate major features of human disease would enhance research efforts into understanding rickettsial pathogens and disease (Osterloh, 2017; Grasperge et al., 2012; Sunyakumthorn et al., 2013).
R. parkeri is genetically similar to the more virulent human pathogens R. rickettsii and R. conorii (Roux and Raoult, 2000; Goddard, 2009), and it can be handled under BSL2 conditions. Moreover, mutants can be generated using transposon mutagenesis (Reed et al., 2014; Lamason et al., 2018), and small rodents including mice have been found as seropositive for R. parkeri in the wild (Moraru et al., 2013a; Moraru et al., 2013b; Krawczak et al., 2016; Barbieri et al., 2019). Thus, a mouse model for R. parkeri that recapitulates key features of human infection would greatly enhance investigations into understanding rickettsial disease. However, inbred mice including C57BL/6 and BALB/c develop no or minor skin lesions upon intradermal (i.d.) infection with R. parkeri (Grasperge et al., 2012). Strategies to develop a mouse model, including infecting Toll-like receptor 4 (TLR4)-deficient mice (Grasperge et al., 2012), delivering high doses of bacteria (Londoño et al., 2019), and delivering R. parkeri via a tick vector (Saito et al., 2019), have been examined. However, a model that recapitulates eschar formation and dissemination via needle inoculation with low doses of R. parkeri has remained elusive. A mouse model for R. parkeri that mimics eschar formation and disseminated disease in C57BL/6 mice, the most widely used genetic background with a large variety of available mutants, would aid investigations into rickettsial virulence mechanisms, the host response to infection, and spotted fever disease.
Toward better understanding the host response to R. parkeri infection, we recently investigated the relationship between R. parkeri and interferons (IFNs), which are ubiquitous signaling molecules of the innate immune system that mobilize the cytosol to an antimicrobial state. Type I IFN (IFN-I) generally restricts viral replication, whereas IFN-γ generally restricts intracellular bacterial pathogens (Raniga and Liang, 2018; Billiau and Matthys, 2009; Meunier and Broz, 2016). We observed that mice lacking either gene encoding the receptors for IFN-I (Ifnar1) or IFN-γ (Ifngr1) are resistant to i.v. infection with R. parkeri, whereas double knockout (DKO) Ifnar1-/-;Ifngr1-/- mice succumb (Burke et al., 2020). This demonstrates that IFNs redundantly protect against systemic R. parkeri. However, the i.v. infection route does not recapitulate eschar formation and i.d. infection may more closely mimic infection by tick bite. Further investigations into whether IFNs redundantly protect against R. parkeri in the skin may improve the mouse model for SFG Rickettsia.
A robust mouse model would facilitate investigations into conserved rickettsial virulence factors, whose role in pathogenesis in vivo remain poorly understood. One virulence mechanism shared by divergent cytosolic bacterial pathogens including Rickettsia, Listeria, Burkholderia, Mycobacterium, and Shigella species is the ability to undergo actin-based motility, which facilitates cell to cell spread (Choe and Welch, 2016; Lamason and Welch, 2017). However, the pathogenic role for many actin-based motility factors in vivo remains unknown. R. parkeri actin-based motility differs from that of other pathogens in that it occurs in two phases. The first phase requires the RickA protein, which elicits actin-based motility by activating the host Arp2/3 complex (Gouin et al., 2004; Jeng et al., 2004); however, RickA is dispensable for cell to cell spread in vitro (Reed et al., 2014). The second phase requires the Sca2 protein (Reed et al., 2014; Kleba et al., 2010), which mimics eukaryotic formins to directly nucleate and elongate actin filaments (Reed et al., 2014; Haglund et al., 2010). Sca2 is required for efficient cell to cell spread, although it is not required for replication in epithelial cells or for avoiding antimicrobial autophagy (Reed et al., 2014; Kleba et al., 2010; Haglund et al., 2010; Engström et al., 2019). sca2 mutant R. rickettsii elicit reduced fever in guinea pigs when compared with wild-type (WT) R. rickettsii (Kleba et al., 2010), yet the explanation for reduced fever and the pathogenic role for Sca2 in vivo remains unclear. Additionally, Sca2 is not essential for dissemination of R. parkeri within ticks (Harris et al., 2018).
A second virulence strategy employed by intracellular pathogens is the ability to avoid autophagy, which for R. parkeri requires the abundant, conserved outer membrane protein B (OmpB) (Engström et al., 2019; Engström et al., 2021). ompB mutant R. parkeri are ubiquitylated and restricted by antimicrobial autophagy in mouse macrophages, and OmpB is important for R. parkeri infection of internal organs in WT mice and for causing lethal disease in IFN receptor-deficient mice after i.v. infection (Burke et al., 2020; Engström et al., 2019). However, the role for R. parkeri OmpB upon i.d. infection remains unknown. An improved mouse model may improve our understanding of how conserved virulence factors including Sca2 and OmpB enhance rickettsial pathogenesis.
Here, we use IFN receptor-deficient mice to examine the effects of i.d. inoculation of R. parkeri, mimicking the natural route of infection. We observe skin lesions that appear similar to human eschars, as well as disseminated lethal disease with as few as 10 bacteria. Using this model, we find that Sca2 promotes dissemination and is required for causing lethality and that OmpB contributes to eschar formation and to lethal disease. We demonstrate that immunization with sca2 or ompB mutant R. parkeri protects IFN receptor-deficient mice against subsequent challenge with WT bacteria, revealing live-attenuated vaccine candidates. Our study establishes a mouse model to investigate numerous aspects of Rickettsia pathogenesis, including eschar formation, virulence factors, and immunity. More broadly, this work also reveals that a potent, redundant IFN response protects mice from eschar-associated rickettsiosis.
Results
Ifnar1-/-;Ifngr1-/- mice are susceptible to eschar-associated rickettsiosis
We sought to develop an i.d. murine infection model to better recapitulate the natural route of tick-borne R. parkeri infection. WT, single mutant Tlr4-/-, Ifnar1-/-, or Ifngr1-/-, and DKO Ifnar1-/-;Ifngr1-/- C57BL/6J mice, as well as outbred WT CD-1 mice, were infected i.d. with 107 WT R. parkeri and monitored over time. No or minor dermal lesions appeared at the site of infection in WT, single mutant Tlr4-/-, Ifnar1-/-, or Ifngr1-/- C57BL/6J mice or CD-1 mice (Figure 1, Figure 1—figure supplement 1a). In contrast, DKO Ifnar1-/-;Ifngr1-/- C57BL/6J mice developed delimited skin lesions measuring >1 cm in diameter that were necrotic, hardened, non-pruritic, and surrounded by an indurated red halo (Figure 1b), similar to human eschars (Figure 1c; Paddock et al., 2008; Paddock et al., 2004; Kaskas, 2014; Cragun et al., 2010; Herrick et al., 2016). In some cases, tails of DKO Ifnar1-/-;Ifngr1-/- or single mutant Ifngr1-/- mutant mice became inflamed after i.d. or i.v. infection (Figure 1—figure supplement 1b). These findings demonstrate that interferons redundantly control disease caused by R. parkeri in the skin and that i.d. infection of DKO Ifnar1-/-;Ifngr1-/- mice recapitulates the hallmark manifestation of human disease caused by R. parkeri.
Figure 1. I.d. infection of Ifnar1-/-;Ifngr1-/- mice with R. parkeri elicits skin lesions that are grossly similar to human eschars.
(a) Representative images of WT, Ifnar1-/-, Ifngr1-/-, and Tlr4-/- single mutant mice, infected intradermally with 107 WT R. parkeri PFU, at 8 d.p.i. WT mice were injected with PBS. White arrows indicate the infection site on the right flank of the mouse. Scale bar, 1 cm. Data are representative of three independent experiments. (b) Representative images of a DKO Ifnar1-/-;Ifngr1-/- mouse after i.d. inoculation with 107 R. parkeri PFU. Data are representative of three independent experiments. The white arrow indicates the injection site on the right flank of the mouse. Scale bar, 1 cm. (c) Gross pathology of a human R. parkeri infection, reproduced from Figure 2a of Paddock et al., 2008, with permission from Oxford Academic. It is not covered by the CC-BY 4.0 license and further reproduction of this panel would require permission from the copyright holder. (d) Representative images of Ifnar1-/-;Ifngr1-/- mice infected intradermally with the indicated amounts of WT R. parkeri at 9 d.p.i. Scale bar, 1 cm. Data are representative of two independent experiments.
© 2008, Oxford Academic Permissions
Gross pathology of a human R. parkeri infection, reproduced from Figure 2a of Paddock et al., 2008, with permission from Oxford Academic. It is not covered by the CC-BY 4.0 license and further reproduction of this panel would need permission from the copyright holder.

Figure 1—figure supplement 1. Ifnar1-/-;Ifngr1-/- mice develop disseminated disease upon intradermal R. parkeri infection.

Our previous observations using the i.v. route revealed dose-dependent lethality in Ifnar1-/-;Ifngr1-/- mice, with 107 R. parkeri eliciting 100% lethality and 105 R. parkeri eliciting no lethality (Burke et al., 2020). R. parkeri are present in tick saliva at a concentration of approximately 104 per 1 μl, and approximately 107 R. parkeri are found in tick salivary glands (Suwanbongkot et al., 2019). However, the number of bacteria delivered from tick infestation likely varies depending on many factors, and we therefore sought to examine the effects of different doses of R. parkeri upon i.d. infection of Ifnar1-/-;Ifngr1-/- mice. We observed skin lesion formation at all infectious doses, from 107 to 10 bacteria (Figure 1d), suggesting that i.d. infection of Ifnar1-/-;Ifngr1-/- mice elicits lesions with doses similar to what is delivered by tick infestation.
We next sought to quantitatively evaluate the effects of i.d. infection by monitoring animal weight, body temperature, the degree of lesion formation, and lethality. Intradermally infected DKO Ifnar1-/-;Ifngr1-/ mice lost significant body weight (Figure 2a, Figure 2—figure supplement 1a) and body temperature (Figure 2b; animals were euthanized when body temperature fell below 90°F / 32.2°C) when compared with WT mice, whereas infected single mutant Tlr4-/-, Ifnar1-/-, or Ifngr1-/- mice did not. To evaluate lesion severity, we scored lesions upon infection with different doses of R. parkeri. Whereas 107 bacteria elicited similar responses as 105, 104, 103, and 102 bacteria (Figure 2c), lesions were less severe when mice were infected with 101 bacteria compared with 107 bacteria. If mice survived, lesions healed over the course of approximately 15–40 days post infection (d.p.i.) at all doses (Figure 2—figure supplement 1b).
Figure 2. I.d. infection of Ifnar1-/-;Ifngr1-/- mice by R. parkeri elicits disseminated, lethal disease.
(a) Weight changes over time in mice infected i.d. with R. parkeri. Data are shown as a percentage change to initial weight. In the left panel, all mice were infected with 107 PFU of R. parkeri: n = 7 (WT), n = 11 (Ifnar1-/-), n = 7 (Ifngr1-/-), n = 9 (DKO Ifnar1-/-;Ifngr1-/-), and n = 4 (Tlr4-/-) individual mice. In the right panel, DKO Ifnar1-/-;Ifngr1-/- mice were infected with the indicated R. parkeri PFU: n = 7 (105), n = 7 (104), n = 8 (103), n = 7 (102), n = 7 (101) individual mice. WT data is the same in both panels. Data for each genotype are combined from two or three independent experiments. (b) Temperature changes over time in mice intradermally infected with 107 R. parkeri PFU. Each line is an individual mouse. Mice were euthanized if their temperature fell below 90°F, as indicated by the dotted line. Data are the combination of three independent experiments with n = 7 (WT) and n = 9 (Ifnar1-/-;Ifngr1-/-) individual mice. (c) Analysis of gross skin pathology after i.d. infection. Ifnar1-/-;Ifngr1-/- mice were infected with the indicated PFU of R. parkeri and monitored over time. WT mice were infected with 107 R. parkeri PFU. Data are the combination of three independent experiments for WT and the 107 dose in Ifnar1-/-Ifngr1-/- mice; data for all other doses are the combination of two independent experiments. n = 9 (107), n = 5 (105), n = 5 (104), n = 8 (103), n = 7 (102), n = 7 (101), and n = 7 (WT) individual mice. (d) Mouse survival after i.d. infection with R. parkeri. In the left panel, all mice were infected with 107 R. parkeri PFU: n = 7 (WT), n = 11 (Ifnar1-/-), n = 7 (Ifngr1-/-), n = 4 Tlr4-/-, and n = 12 (DKO Ifnar1-/-;Ifngr1-/-) individual mice. Data are the combination of three separate experiments for WT, Ifnar1, and DKO Ifnar1-/-;Ifngr1-/- and two separate experiments for Ifngr1-/- and Tlr4-/-. In the right panel, DKO Ifnar1-/-;Ifngr1-/- mice were infected with the indicated amounts of R. parkeri. Data are the combination of two independent experiments: n = 7 (105), n = 7 (104), n = 8 (103), n = 7 (102), and n = 7 (101) individual mice. (e) Bacterial burdens in organs of intradermally infected WT and Ifnar1-/-;Ifngr1-/-mice. Mice were intradermally inoculated with 107 R. parkeri, and spleens and livers were harvested and plated for p.f.u. at 72 h.p.i. Dotted lines indicate the limit of detection. Data are the combination of two independent experiments. n = 4 (WT) and 7 (Ifnar1-/-;Ifngr1-/-) individual mice. Data in (a), (c) are the mean ± SEM. Statistical analyses in (a) used a two-way ANOVA where each group was compared to WT at t = 20 d.p.i. Statistical analyses in (c) used a two-way ANOVA at t = 20 d.p.i. Statistical analyses in (d) used a log-rank (Mantel−Cox) test to compare Ifnar1-/- to DKO Ifnar1-/-;Ifngr1-/- at each dose. Statistical analysis in (e) used a two-tailed Mann–Whitney U test. NS, not significant; **p<<0.01; ***p<0.001; ****p<0.0001.

Figure 2—figure supplement 1. Ifnar1-/- or Ifngr1-/- mice develop limited disease upon intradermal infection, and Ifnar1-/-;Ifngr1-/- develop lesions of dose-dependent severity.

To investigate whether i.d. infection by R. parkeri caused lethal disease, we monitored mouse survival over time. Upon i.d. delivery of 107 R. parkeri, 8 of 12 Ifnar1-/-;Ifngr1-/- mice exhibited lethargy, paralysis, or body temperatures below 90oF, at which point they were euthanized, whereas delivery of the same dose of bacteria to WT and single mutant mice did not elicit lesions and all survived (Figure 2d). Lower doses of R. parkeri also elicited body weight loss (Figure 2a), body temperature loss (Figure 2—figure supplement 1), and lethal disease (Figure 2d) in Ifnar1-/-;Ifngr1-/- mice. Degrees of lethality between different doses in Ifnar1-/-;Ifngr1-/- mice were not significantly different from one another, and the cause of lethality in this model remains unclear. Nevertheless, these findings reveal that i.d. infection can cause lethal disease in Ifnar1-/-;Ifngr1-/- mice with ~10,000-fold lower dose of bacteria than i.v. infection.
It remained unclear whether i.d. infection could also be used to model dissemination from the skin to internal organs. We therefore evaluated bacterial burdens in spleens and livers of WT and Ifnar1-/-;Ifngr1-/- mice at 5 d.p.i. by measuring R. parkeri plaque-forming units (p.f.u.). Bacteria were not recoverable from spleens and livers of intradermally infected WT mice, suggesting that they did not disseminate from the skin to internal organs in high numbers (Figure 2e). In contrast, bacteria were recovered from spleens and livers of intradermally infected Ifnar1-/-;Ifngr1-/- mice at 5 d.p.i. (Figure 2e). This demonstrates that i.d. infection of Ifnar1-/-;Ifngr1-/- mice with R. parkeri causes systemic infection and can be used as a model for dissemination from the skin to internal organs.
Histology and immunohistochemistry reveal necrosis, inflammation, and bacteria within neutrophils and macrophages
To examine whether lesions in Ifnar1-/-;Ifngr1-/- mice had similar inflammation and necrosis as those observed in human eschars at the tissue and cellular level, we performed histologic evaluation to further characterize the tissue alterations of i.d.-infected Ifnar1-/-;Ifngr1-/- mice and used anti-Rickettsia immunohistochemistry to identify the infected cell types in the skin. WT mice infected i.d. with 103 R. parkeri exhibited no discernible inflammation in the skin at 4, 7, or 12 d.p.i. (Figure 3a). In contrast, at 4 d.p.i., Ifnar1-/-;Ifngr1-/- mice infected with 103 R. parkeri exhibited mild inflammation characterized by dermal inflammatory foci composed of neutrophils and macrophages, with high numbers of intralesional rickettsiae (Figure 3b). By 7 d.p.i., the inflammation throughout the skin was severe, with large coalescing foci of predominantly neutrophils and fewer macrophages within areas of fibrosis, associated with abundant rickettsiae. The epidermis had multifocal coagulative necrosis associated with fibrin thrombosis of the underlying dermal capillaries (Figure 3c). The inflammation was still severe and associated with large numbers of intralesional rickettsiae at 12 d.p.i., accompanied by extensive coagulative necrosis of the skin (Figure 3d). Together, this indicates that R. parkeri causes extensive necrosis and inflammatory cell infiltration of the skin of i.d.-infected Ifnar1-/-;Ifngr1-/- mice, with primary staining within neutrophils and macrophages.
Figure 3. Pathology of infected Ifnar1-/-;Ifngr1-/- mice reveals necrosis, inflammation, and rickettsial staining of macrophage and neutrophils.

(a–d) Representative images of the skin from WT and Ifnar1-/-;Ifngr1-/- mice, infected i.d. with 103 R. parkeri. Top row panels are skin stained with hematoxylin and eosin. Bottom row panels are a nearby Z plane, stained with an anti-Rickettsia antibody (brown). (e) Representative image of lungs from Ifnar1-/-;Ifngr1-/- mice, infected i.d. with 103 R. parkeri at 7 d.p.i. (f) Representative image of brains (choroid plexus) from Ifnar1-/-;Ifngr1-/- mice, infected i.d. with 106 R. parkeri at 7 d.p.i. (g, h) Representative images of livers and spleens from WT and Ifnar1-/-;Ifngr1-/- mice, infected i.v. with 107 R. parkeri at 7 d.p.i. The arrow in (g) for WT mice indicates rickettsial staining within a Kupffer cell. The asterisk for Ifnar1-/-;Ifngr1-/- indicates a granuloma. Scale bars are indicated in each panel. Data are representative of two independent replicates.
We also examined internal organ tissues of mice infected i.d. by performing immunohistochemistry of brain and lung at 7 d.p.i., as infection of these organs may result in lethality. In the lungs of Ifnar1-/-;Ifngr1-/- mice, inflammatory foci were scattered throughout the parenchyma, with several cells morphologically compatible with neutrophils containing rickettsiae (Figure 3e). In one tissue section, rickettsiae were also evident in some cells lining small vessels and therefore interpreted as endothelial cells. In the brain, rickettsial immunostaining was not observed after i.d. infection with 103 R. parkeri, but infection with a higher dose (106) resulted in low number of rickettsial bacteria in the leptomeninges and the choroid plexus, in regions moderately infiltrated by neutrophils and macrophages (Figure 3f). These data demonstrate that R. parkeri disseminates from the skin to internal organs, where they are again primarily found in macrophages and neutrophils.
Finally, we used histology and immunohistochemistry after i.v. infection to analyze spleens and livers, as R. parkeri is abundant and easily recoverable from these organs after i.v. infection (Burke et al., 2020; Engström et al., 2019). In WT mice, we observed little to no inflammation or lesions. In contrast, both organs of Ifnar1-/-;Ifngr1-/- mice had fibrinoid vascular wall degeneration, endothelial hypertrophy, fibrin thrombi in medium caliber vessels, and marked inflammation that was composed predominantly of macrophages. In WT mice, rickettsiae were infrequent and were found in macrophages in the red pulp of the spleen and scarcely throughout the liver in Kupffer cells (Figure 3g,h; Bonell et al., 2017). In contrast, in Ifnar1-/-;Ifngr1-/- mice, rickettsiae were abundant in the splenic red pulp, primarily infecting histiocytes/macrophages (Figure 3g). In the liver of Ifnar1-/-;Ifngr1-/- mice, rickettsiae were abundant in macrophages within the granulomas (Figure 3h). We did not observe any immunostaining for Rickettsia in endothelial cells or vessels in the spleen or liver after i.v. infection. Together, these findings reveal that macrophages are the primary cell type affected by R. parkeri in the spleen and liver after i.v. infection.
Ifnar1-/-;Ifngr1-/- mice do not succumb to intradermal infection with Sca2 mutant R. parkeri
We next examined whether Ifnar1-/-;Ifngr1-/- mice could be used to characterize R. parkeri virulence factors. Sca2 is a surface protein that mediates actin-based motility in rickettsial pathogens; however, its contribution to virulence in vivo remains unclear. We examined if i.v. and i.d. infections of WT and Ifnar1-/-;Ifngr1-/- mice could reveal a pathogenic role for R. parkeri Sca2. Upon i.v. infection with 5 × 106 bacteria (Figure 4) or 107 bacteria (Figure 4b), we observed that sca2::Tn mutant R. parkeri caused reduced lethality compared to WT bacteria. Similarly, i.d. infection with sca2::Tn mutant bacteria elicited significantly less lethality (Figure 4c) and weight loss (Figure 4d) as compared to WT bacteria and no severe temperature loss (Figure 4—figure supplement 1a). Although we sought to evaluate infection using a sca2 complement strain of R. parkeri, our attempts to generate such a strain were unsuccessful. As an alternative strategy, we examined whether the transposon insertion itself had an effect on R. parkeri survival in vivo. We evaluated infection of an R. parkeri strain that harbors a transposon insertion in MC1_RS08740 (previously annotated as MC1_05535), which has no known role in virulence (Engström et al., 2019). I.v. infection with MC1_RS08740::Tn R. parkeri caused lethality to a similar degree as WT R. parkeri (Figure 4a), demonstrating that the transposon likely does not significantly impact R. parkeri fitness in vivo. Together, these findings suggest that the actin-based motility factor Sca2 is required for causing lethal disease in Ifnar1-/-;Ifngr1-/- mice.
Figure 4. R. parkeri Sca2 contributes to dissemination from skin to spleens and livers.
(a) Survival of Ifnar1-/-;Ifngr1-/- mice upon i.v. infection with 5 × 106 PFU of R. parkeri. n = 7 (WT), 10 (sca2::Tn), and 7 (MC1_RS08740::Tn R. parkeri) individual mice. Data are the combination of two independent experiments. (b) Survival of Ifnar1-/-;Ifngr1-/- mice upon i.v. infection with 107 R. parkeri. n = 7 (WT) and 10 (sca2::Tn) individual mice. Data are the combination of two independent experiments. (c) Survival of Ifnar1-/-;Ifngr1-/- mice upon i.d. infection with 107 PFU of R. parkeri. n = 6 (WT) and 8 (sca2::Tn) individual mice. Data are the combination of two independent experiments. (d) Weight changes of Ifnar1-/-;Ifngr1-/- mice upon i.d. infection with 107 PFU of R. parkeri. n = 6 (WT) and 8 (sca2::Tn) individual mice. Data are the combination of two independent experiments. (e) Analysis of gross skin pathology after i.d. infection. Ifnar1-/-;Ifngr1-/- mice were infected with 107 PFU of the indicated strains of R. parkeri and monitored over time. n = 9 (WT) and 8 (sca2::Tn) individual mice. Data are the combination of two independent experiments. (f) Representative images of fluorescence in mouse skin after i.d. infection with 106 R. parkeri and delivery of a fluorescent dextran, at 5 d.p.i. Scale bars, 1 cm. The larger black dashed circle represents the area that was measured for fluorescence for each sample, as indicated in (g) (~80,000 pixels). The smaller, black-dashed circle represents of the injection site area that was measured for fluorescence for each sample, as indicated in Figure 4—figure supplement 1 (~7800 pixels). (g) Quantification of fluorescence in mouse skin after i.d. infection. Mice were infected with 107 R. parkeri, and 150 µl fluorescent dextran was intravenously delivered at 5 d.p.i. Skin was harvested 2 hr later, and fluorescence was measured using a fluorescence imager. Data indicate measurements of larger areas of skin, as indicated in (f) by the larger black circle. n = 6 (WT R. parkeri) and n = 6 (sca2::Tn R. parkeri) individual Ifnar1-/-;Ifngr1-/- mice; n = 6 (WT R. parkeri) individual WT mice. For each experiment, the average of uninfected samples was normalized to 100; each sample was divided by the average for uninfected mice and multiplied by 100; the dotted horizontal line indicates 100 arbitrary units, corresponding to uninfected (unin.) mice. Data are the combination of two independent experiments. (h) Quantification of Listeria monocytogenes abundance in organs of WT C57BL/6J mice upon i.v. infection with 104 bacteria, at 72 h.p.i. Data are the combination of two independent experiments. n = 6 (WT), n = 7 (ΔactA) individual mice. (i) Quantification of R. parkeri abundance in spleens and livers of WT C57BL/6J mice upon i.v. infection, at 72 h.p.i. Data are the combination of two independent experiments. n = 10 (WT) and 10 (Sca2::Tn) individual mice. (j) Quantification of R. parkeri abundance in organs upon i.d. infection with 107 PFU of R. parkeri. n = 7 (WT) and 7 (sca2::Tn) individual mice. Data are the combination of two independent experiments. Data for WT R. parkeri in Ifnar1-/-;Ifngr1-/- mice are the same as in Figure 2e. (k) Quantification of R. parkeri abundance in organs upon i.d. infection with 105 PFU of R. parkeri. n = 7 (WT) and 6 (sca2::Tn). Data are the combination of two independent experiments. Solid horizontal bars in (g) indicate means; solid horizontal bars in (h–k) indicate medians; error bars indicate SEM. Statistical analyses for survival in (a–c) used a log-rank (Mantel–Cox) test. Statistical analysis in (d) used a two-way ANOVA at t = 20. Statistical analysis in (e) used a two-way ANOVA from 0 to 10 d.p.i. Statistical analyses in (g) used a two-tailed Student’s T test. Statistical analyses in (h–k) used a two-tailed Mann–Whitney U test. The fold change in (h–k) indicates differences of medians. *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001.

Figure 4—figure supplement 1. Intradermal infection of Ifnar1-/-;Ifngr1-/- mice with sca2::Tn R. parkeri causes less severe temperature loss as compared to WT bacteria.
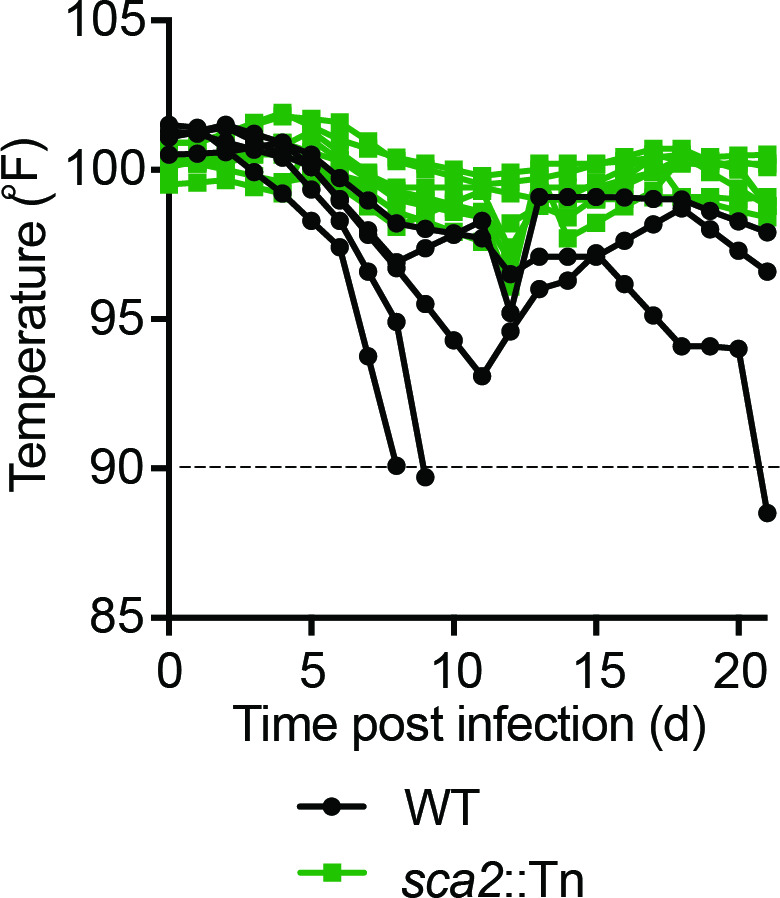
Figure 4—figure supplement 2. WT and sca2::Tn R. parkeri elicit similar amounts of vascular damage in skin upon i.d. infection of Ifnar1-/-;Ifngr1-/- mice.

Figure 4—figure supplement 3. Sca2 does not significantly enhance R. parkeri avoidance of antibacterial innate immune responses in vitro.

Ifnar1-/-;Ifngr1-/- mice exhibit similar skin lesion formation and vascular damage upon i.d. infection with WT and sca2::Tn R. parkeri
We next examined whether Sca2 facilitates R. parkeri dissemination throughout the skin and whether Sca2 is required for lesion formation. Unexpectedly, upon i.d. inoculation, Ifnar1-/-;Ifngr1-/- mice infected with sca2::Tn mutant bacteria developed skin lesions that were of similar severity to lesions caused by WT R. parkeri; however, the lesions elicited by sca2 mutant bacteria appeared significantly earlier than lesions caused by WT bacteria (Figure 4e). Further examinations will be required to better evaluate this observation; however, it may suggest that actin-based motility enables R. parkeri to avoid a rapid onset of inflammation in the skin. To evaluate R. parkeri dissemination within the skin, we used a fluorescence-based assay that measures vascular damage as a proxy for pathogen dissemination (Glasner et al., 2017). Mice were intradermally infected with WT and sca2::Tn R. parkeri. At 5 d.p.i., fluorescent dextran was intravenously delivered, and fluorescence was measured at the infection site (Figure 4f, representative small black circle) and in the surrounding area (Figure 4f, representative large black circle). No significant differences were observed when comparing WT and sca2::Tn R. parkeri infections in Ifnar1-/-Ifngr1-/- mice using an infectious dose of 107 R. parkeri in the larger surrounding area (Figure 4g) or at the site of infection (Figure 4—figure supplement 2a). Similar results were observed upon infection with 106 or 105 bacteria (Figure 4—figure supplement 2b,c). However, significantly more fluorescence was observed in the skin of infected Ifnar1-/-;Ifngr1-/- mice as compared to WT mice (Figure 4g), demonstrating that interferons protect against increased vascular permeability during R. parkeri infection. Fluorescence was also measured in spleens and livers; however, no differences between control or experiment groups was observed, suggesting that this assay as described is most appropriate for the skin. Together, the gross pathological analysis and fluorescence-based assay suggest that Sca2 likely does not significantly enhance R. parkeri spread throughout the skin during i.d. infection of Ifnar1-/-;Ifngr1-/- mice.
R. parkeri Sca2 promotes dissemination from the skin to spleens and livers
Among the factors that mediate actin-based motility, the Listeria monocytogenes actin-based motility factor ActA is one of the best understood. ActA enables L. monocytogenes to spread from cell to cell (Choe and Welch, 2016; Lamason and Welch, 2017), escape antimicrobial autophagy (Cheng et al., 2018; Mitchell et al., 2018; Yoshikawa et al., 2009b; Yoshikawa et al., 2009a), proliferate in mouse organs after i.v. infection (Auerbuch et al., 2001; Le Monnier et al., 2007), and cause lethal disease in mice (Goossens et al., 1992; Brundage et al., 1993). We initially hypothesized that R. parkeri Sca2 plays a similar pathogenic role in vivo to ActA, which we found is required for bacterial survival in spleens and livers upon i.v. delivery (Figure 4h), in agreement with previous experiments (Auerbuch et al., 2001; Le Monnier et al., 2007). However, when we examined bacterial burdens upon i.v. infection of Ifnar1-/-;Ifngr1-/- mice with R. parkeri, similar amounts of WT and sca2::Tn bacteria were recovered in spleens (Figure 4i). We were also surprised to find that significantly more sca2::Tn than WT R. parkeri were recovered in livers (Figure 4i). The explanation for higher sca2::Tn burdens in livers remains unclear. Nevertheless, these data reveal that Sca2 is likely not essential for R. parkeri survival in blood, invasion of host cells, or intracellular survival in spleens and livers.
We next evaluated the role for Sca2 in R. parkeri dissemination by measuring p.f.u. in spleens and livers following i.d. infection of Ifnar1-/-;Ifngr1-/- mice. After i.d. infection, sca2::Tn mutant bacteria were ~20-fold reduced in their abundance in spleens and ~2-fold reduced in their abundance in livers as compared to WT R. parkeri (Figure 4j). Similar results were seen upon i.d. infection with lower doses of sca2::Tn and WT bacteria (Figure 4k). Together, these results suggest that Sca2 is required for R. parkeri dissemination from the skin to internal organs.
R. parkeri actin-based motility does not contribute to avoiding innate immunity in vitro
Sca2-mediated actin-based motility is required for efficient plaque formation and cell to cell spread by R. parkeri in vitro (Reed et al., 2014; Kleba et al., 2010). However, it remains unclear if Sca2 enables R. parkeri to escape detection or restriction by innate immunity. The actin-based motility factor ActA enables L. monocytogenes to avoid autophagy (Yoshikawa et al., 2009b; Yoshikawa et al., 2009a), and the antimicrobial guanylate binding proteins (GBPs) inhibit Shigella flexneri actin-based motility (Piro et al., 2017). We therefore sought to evaluate whether Sca2-mediated actin-based motility enables R. parkeri to evade innate immunity in vitro. We found that the sca2::Tn mutant grew similarly to WT bacteria in endothelial cells (Figure 4—figure supplement 3a), consistent with previous reports in epithelial cells (Reed et al., 2014; Kleba et al., 2010). We also examined whether Sca2 contributed to R. parkeri survival or growth in bone marrow-derived macrophages (BMDMs), which can restrict other R. parkeri mutants that grow normally in endothelial cells (Engström et al., 2019). However, no significant difference in bacterial survival was observed between WT and sca2::Tn bacteria in BMDMs in the presence or absence of IFN-β (Figure 4—figure supplement 3b). WT and sca2 mutant R. parkeri also elicited similar amounts of host cell death (Figure 4—figure supplement 3c) and IFN-I production (Figure 4—figure supplement 3d). Moreover, we found that the anti-rickettsial factor GBP2 localized to the surface of sca2::Tn mutant R. parkeri at similar frequency as with WT bacteria in the presence or absence of IFN-β (Figure 4—figure supplement 3e,f). Together, these data suggest that Sca2 does not significantly enhance the ability of R. parkeri to evade interferon-stimulated antimicrobial genes or inflammasomes in vitro.
Ifnar1-/-;Ifngr1-/- mice exhibit less severe skin lesions upon infection with a highly attenuated R. parkeri mutant
Because sca2 mutant R. parkeri showed no defect in eschar formation compared to WT, it remained unclear whether skin lesion formation in Ifnar1-/-;Ifngr1-/- mice was influenced by bacterial virulence factors. We therefore investigated i.d. infection with ompB::TnSTOP R. parkeri, which harbors both a transposon and a stop codon in ompB but no other mutations as determined by whole genome sequencing (Engström et al., 2019). ompB mutant R. parkeri are severely attenuated in mice, as evaluated by measuring p.f.u. in organs of WT mice after i.v. infection (Engström et al., 2019) or by measuring lethality in Ifnar1-/-;Ifngr1-/- mice after i.v. infection (Burke et al., 2020). In contrast with WT bacteria, i.d. infection of Ifnar1-/-;Ifngr1-/- mice with ompB::TnSTOP R. parkeri caused no lethality (Figure 5a) or weight loss (Figure 5b). The ompB::TnSTOP mutant R. parkeri also caused significantly less severe skin lesions than WT bacteria (Figure 5c). These findings suggest that Ifnar1-/-;Ifngr1-/- mice can be used as a model to identify bacterial genes important for eschar formation.
Figure 5. ompB mutant R. parkeri elicit no lethality and reduced skin lesion formation in Ifnar1-/-;Ifngr1-/-mice.

(a) Survival of Ifnar1-/-;Ifngr1-/- mice upon i.d. infection with 107 PFU of R. parkeri. n = 6 (WT) and 8 (ompB::TnSTOP) individual mice. Data are the combination of two independent experiments. Data for WT are the same as in Figure 4c. (b) Weight changes of Ifnar1-/-;Ifngr1-/- mice upon i.d. infection with 107 PFU of R. parkeri. n = 6 (WT) and 8 (ompB::TnSTOP) individual mice. Data are the combination of two independent experiments. Data for WT are the same as in Figure 4d. (c) Analysis of gross skin pathology after i.d. infection. Ifnar1-/-;Ifngr1-/- mice were infected with 107 of the indicated strains of R. parkeri and monitored over time. n = 9 (WT) and 8 (ompB::TnSTOP) individual mice. Data are the combination of two independent experiments. Data for WT are the same as in Figure 4e. Error bars indicate SEM. Statistical analyses in (a) used a log-rank (Mantel–Cox) test. Statistical analysis in (b) used a two-way ANOVA from 0 to 20 d.p.i. Statistical analysis in (c) used a two-way ANOVA from 0 to 10 and 20 d.p.i.; **p<0.01; ***p<0.001.
Immunizing Ifnar1-/-;Ifngr1-/- mice with attenuated R. parkeri mutants protects against subsequent rechallenge
There is currently no available vaccine to protect against SFG Rickettsia, which can cause severe and lethal human disease (Osterloh, 2017; Dantas-Torres, 2007), and identifying mouse models that develop protective immunity to R. parkeri would aid investigations into identifying live attenuated vaccine candidates. We therefore examined whether immunization with attenuated R. parkeri mutants would protect against subsequent re-challenge with a lethal dose of WT bacteria. Ifnar1-/-;Ifngr1-/- mice were immunized i.v. with 5 × 106 sca2::Tn or ompB::TnSTOP R. parkeri and 40 d later were re-challenged i.v. with 107 WT R. parkeri, which is approximately 10 times a 50% lethal dose (LD50) (Burke et al., 2020). All mice immunized with sca2 or ompB mutant R. parkeri survived, whereas all naïve mice succumbed to i.v. challenge by 6 d.p.i. (Figure 6a). Upon i.v. rechallenge, mice immunized with ompB and sca2 mutants also did not lose significant weight (Figure 6b) or body temperature (Figure 6c). We next examined whether immunized mice were protected from i.d. rechallenge. Ifnar1-/-;Ifngr1-/- mice were immunized with ompB or sca2 mutant R. parkeri by i.d. infection or with sca2 mutant R. parkeri by i.v infection and rechallenged 40 d later by i.d. infection with 105 WT bacteria. Immunized mice were protected from lethal disease (Figure 6d) and had less severe skin lesions than naïve mice (Figure 6e). These data indicate that attenuated R. parkeri mutants elicit robust protective immune responses, and that Ifnar1-/-;Ifngr1-/- mice may serve as tools to develop live attenuated R. parkeri vaccine candidates.
Figure 6. ompB and sca2 mutant R. parkeri elicit immunity in Ifnar1-/-Ifngr1-/- mice.

(a) Survival of immunized and naïve Ifnar1-/-;Ifngr1-/- mice upon i.v. R. parkeri infection. Immunized mice were first infected with 5 × 106 PFU of sca2::Tn or 107 PFU of ompB:TnSTOP R. parkeri and were re-challenged 40 d later with 107 WT R. parkeri. n = 6 (naïve); n = 5 (sca2::Tn immunized); n = 5 (ompB::TnSTOP immunized) individual mice. Data are the combination of two independent experiments. (b) Weight changes over time in mice infected i.d. with 107 R. parkeri. Data are representative of two independent experiments. n = 3 (naïve); n = 3 (sca2::Tn immunized); n = 3 (ompB::Tnstop immunized) individual mice. Each line represents an individual mouse. (c) Temperature changes over time in mice infected i.d. with 107 R. parkeri. Data are representative from two independent experiments. n = 3 (naïve); n = 3 (sca2::Tn immunized); n = 3 (ompB::TnSTOP immunized) individual mice. Each line represents an individual mouse. (d) Survival of naïve or immunized Ifnar1-/-;Ifngr1-/- mice that were infected i.d. with 105 R. parkeri. (e) Skin lesion pathology of naïve or immunized Ifnar1-/-;Ifngr1-/- mice that were infected i.d. with 105 R. parkeri; n = 7 (naïve); n = 8 (sca2::Tn i.d. immunized); n = 5 (sca2::Tn i.v. immunized); n = 8 (ompB::TnSTOP) individual mice. Statistical analyses in (a, d) used a log-rank (Mantel–Cox) test to compare each group of immunized mice to naïve mice; **p<0.01. Statistical analysis in (e) used a two-way ANOVA from 0 to 10 and 20 d.p.i.; **p<0.01; ***p<0.001.
Discussion
In this study, we find that IFN-I and IFN-γ redundantly protect inbred mice from eschar-associated rickettsiosis and disseminated disease by R. parkeri. Eschar formation is a hallmark clinical feature of human disease caused by R. parkeri (Paddock et al., 2008; Paddock et al., 2004), and thus these findings suggest that the striking difference between human and mouse susceptibilities to R. parkeri may be due to IFN signaling in the skin. Using this mouse model, we uncover a role for R. parkeri Sca2 in dissemination, for OmpB in skin lesion formation, and for both proteins in causing lethal disease. We further demonstrate that attenuated R. parkeri mutants elicit long-lasting immunity, revealing live attenuated vaccine candidates. Obligate cytosolic bacterial pathogens cause a variety of severe human diseases on six continents (Bonell et al., 2017; Abdad et al., 2018), and the animal model described here will facilitate future investigations into rickettsial virulence factors, the host response to infection, and the molecular determinants of human disease.
Our observation that i.d. infection of DKO Ifnar1-/-;Ifngr1-/- mice causes eschar formation highlights the critical importance of interferons in restricting R. parkeri in mice and may reveal a key molecular determinant of human disease. IFN-γ restricts virulent rickettsial species in vitro and in mice, including R. prowazekii (Turco and Winkler, 1983), R. conorii (Feng et al., 1994; Li et al., 1987; Manor and Sarov, 1990), and R. australis (Walker et al., 2001). Moreover, IFN-I has modest anti-rickettsial activity towards R. prowazekii, R. conorii, and R. rickettsii in cell lines in vitro (Turco and Winkler, 1990; Colonne et al., 2011; Hanson, 1991). However, the contribution made by IFN-I in restricting Rickettsia species besides R. parkeri in mice remains unknown (Osterloh, 2017). Importantly, the gross similarities we observe between human eschars (Paddock et al., 2008; Herrick et al., 2016) and skin lesions in Ifnar1-/-;Ifngr1-/- mice may indicate that the IFN response in humans is less well adapted to control R. parkeri than that in mice. Cytokine profiling and mRNA transcript analysis of human rickettsial infections reveals IFN-I and IFN-γ activation (Cillari et al., 1996; Jia et al., 2020; de Sousa et al., 2007); however, it remains unclear whether human interferon-stimulated genes (ISGs) are as protective as mouse ISGs. Future investigations into the ISGs that restrict R. parkeri in mouse versus human cells may improve our understanding of human susceptibility to SFG Rickettsia.
Human eschars resulting from R. parkeri infection are characterized by necrosis of the epidermis, vasculitis of small- to medium-sized dermal vessels, fibrin thrombi, infiltration by macrophages and neutrophils, and infection of macrophage/mononuclear cells (Paddock et al., 2008; Herrick et al., 2016). Eschars elicited by R. parkeri in non-human primates revealed similar histological findings and also bacteria within macrophages and neutrophils (Banajee et al., 2015). Similar histological findings were noted in guinea pigs after intraperitoneal infection with R. parkeri, as well as infection of mononuclear cells (Paddock et al., 2017). In Ifnar1-/-;Ifngr1-/- mice, we observed necrosis, vasculitis, fibrin thrombi, and rickettsial staining that coincided primarily with infiltrating macrophages and neutrophils. We note that R. parkeri primary targeting of macrophages and neutrophils differs from cell types infected by R. rickettsii, which primarily targets endothelial cells (Walker and Ismail, 2008), and therefore infection of Ifnar1-/-;Ifngr1-/- mice with R. parkeri is likely not an appropriate model for lethal Rocky Mountain spotted fever disease. One contrasting finding in eschars of Ifnar1-/-;Ifngr1-/- mice as compared to humans is the number of bacteria observed - we find that the pathogen is numerous throughout the skin of Ifnar1-/-;Ifngr1-/- mice, but their numbers are fewer in reported human eschar biopsies (Paddock et al., 2008; Herrick et al., 2016). This discrepancy may be due to differences in infection kinetics or in interferon-mediated restriction of bacteria. Nevertheless, our pathological findings of infected interferon-receptor deficient mice are similar in many ways to the previous findings in humans, primates, and mice. Our results suggest that interferon receptor-deficient mice may be useful tools for modeling eschar-associated rickettsiosis at the cellular level.
Investigating the IFN response in the skin may also lead to a better understanding of other arthropod-borne diseases. One example may be scrub typhus, caused by Orientia tsutsugamushi (Rajapakse et al., 2017), a prevalent but poorly understood tropical disease endemic to Southeast Asia (Bonell et al., 2017; Kelly et al., 2015; Richards and Jiang, 2020). O. tsutsugamushi elicits eschar formation in humans, but inbred mice do not recapitulate eschar formation during O. tsutsugamushi infection (Sunyakumthorn et al., 2013), similar to R. parkeri. A second example may be Borrelia burgdorferi, a tick-borne pathogen that causes a skin rash at the site of tick bite as a hallmark feature of Lyme disease (Bratton et al., 2008), the most prevalent tick-borne disease in the United States (Bratton et al., 2008; Schwartz et al., 2008). Existing mouse models also do not recapitulate skin rash formation following B. burgdorferi infection (Barthold et al., 1990; Wang et al., 2001). Lastly, other rickettsial pathogens including R. conorii, R. typhi, and R. akari cause no or limited disease in WT C57BL/6 mice (Osterloh, 2017; Anderson and Osterman, 1980; Eisemann et al., 1984; Osterloh et al., 2016) and it remains unclear if interferons redundantly protect mice from these pathogens. Further investigations into how IFNs protect the skin from arthropod-borne pathogens may reveal critical aspects of the innate immune response to zoonotic disease.
Our study further highlights the utility of mouse models that mimic natural routes of infection. Our observation that i.d. infection can cause lethal disease with as few as 10 bacteria, ~ 10,000 fewer bacteria than i.v. infection (Burke et al., 2020), suggests that R. parkeri may be highly adapted to reside in the skin. However, this model could be further improved by investigating the role for tick vector components in pathogenesis. Saliva from ticks, mosquitos, and sand flies enhances pathogenesis of arthropod-borne bacterial, viral, and parasitic pathogens (Pingen et al., 2017; Lestinova et al., 2017; Šimo et al., 2017), and non-human primates inoculated with R. parkeri exhibit altered inflammatory responses when administered after tick-bite (Banajee et al., 2015). This may suggest a potential role for tick vector components such as tick saliva in R. parkeri pathogenesis. Developing improved murine infection models that mimic the natural route of infection, including with tick saliva or the tick vector, is critical to better understand the virulence and transmission of tick-borne pathogens.
Many Rickettsia species, as well as many facultative cytosolic pathogens including L. monocytogenes, undergo actin-based motility to spread from cell to cell. For L. monocytogenes, the actin-based motility factor ActA enables the pathogen to survive in vivo, as actA mutant bacteria are over 1,000-fold attenuated by measuring lethality (Goossens et al., 1992; Brundage et al., 1993) and by enumerating bacteria in spleens and livers of mice after i.v. infection (Auerbuch et al., 2001; Le Monnier et al., 2007). However, the pathogenic role for actin-based motility in the Rickettsiae has remained unclear. We find that Sca2 is not required for intracellular survival in organs upon i.v. infection of Ifnar1-/-;Ifngr1-/- mice, but rather, is required for dissemination from skin to internal organs and lethality upon i.d. infection. Consistent with an important role for Sca2 in pathogenesis, a previous study reported that i.v. infection of guinea pigs with sca2 mutant R. rickettsii did not elicit fever (Kleba et al., 2010). Our results suggest that Sca2-mediated actin-based motility by Rickettsia may facilitate dissemination in host reservoirs, although we cannot rule out other roles for Sca2 that do not involve actin assembly. R. prowazekii and R. typhi, which cause severe human disease, encode a fragmented sca2 gene (Ngwamidiba et al., 2005), and undergo no or dramatically reduced frequency of actin-based motility, respectively (Teysseire et al., 1992; Heinzen et al., 1993). Although it remains unclear why some Rickettsia species lost the ability to undergo actin-based motility, Sca2 is dispensable for R. parkeri dissemination in the tick vector (Harris et al., 2018), suggesting that actin-based motility may play a specific role in dissemination within mammalian hosts.
We find that sca2 or ompB mutant R. parkeri elicit a robust protective immune response in Ifnar1-/-;Ifngr1-/- mice. These findings complement previous observations that sca2 mutant R. rickettsii elicits antibody responses in guinea pigs (Kleba et al., 2010), and expands upon these findings by demonstrating protection from rechallenge and revealing additional vaccine candidates. There are currently limited vaccine candidates that protect against rickettsial disease (Osterloh, 2017). Identifying new vaccine candidates may reveal avenues to protect against tick-borne infections and aerosolized Rickettsia, which are extremely virulent and potential bioterrorism agents (Walker, 2009), as well as against Brill-Zinsser disease, caused by latent R. prowazekii (Osterloh, 2017). Future studies exploring whether attenuated R. parkeri mutants provide immunity against other Rickettsia species are warranted to better define the mechanisms of protection. These findings on immunity may also help develop R. parkeri as an antigen delivery platform. R. parkeri resides directly in the host cytosol for days and could potentially be engineered to secrete foreign antigens for presentation by major histocompatibility complex I. In summary, the mouse model described here will facilitate future investigations into numerous aspects of R. parkeri infection, including actin-based motility and immunity, and may serve as model for other arthropod-borne pathogens.
Materials and methods
Key resources table.
| Reagent type (species) or resource | Designation | Source or reference | Identifiers | Additional information |
|---|---|---|---|---|
| Biological sample (Mus musculus) | WT C57Bl/6J | Jackson labs | Stock #: 000664 | |
| Biological sample (Mus musculus) | DKO Ifnar1-/-;Ifngr1-/- | Jackson labs | Stock #: 029098 | |
| Biological sample (Mus musculus) | B6.129S2-Ifnar1tm1Agt/Mmjax; aka Ifnar1-/- | Jackson labs | Stock #: 32045-JAX | RRID:MMRRC_032045-JAX |
| Biological sample (Mus musculus) | B6.129S7-Ifngr1tm1Agt/J; akaIfngr1-/- | Jackson labs | Stock #: 003288 | |
| Antibody | I7205 anti-Rickettsia (Rabbit polyclonal) | Dr. Ted Hackstadt | IHC (1:1000);IF (1:1000) | |
| Biological sample (Rickettsia parkeri) | WT R. parkeri strain Portsmouth | Originally from Dr. Chris Paddock (CDC) | ||
| Biological sample (Rickettsia parkeri) | Sca2::Tn R. parkeri | Welch lab (UC Berkeley)PMID:24361066 | ||
| Biological sample (Rickettsia parkeri) | OmpB::TnSTOPR. parkeri | Welch lab (UC Berkeley)PMID:31611642 |
Bacterial preparations
R. parkeri strain Portsmouth was originally obtained from Dr. Christopher Paddock (Centers for Disease Control and Prevention). To amplify R. parkeri, confluent monolayers of female African green monkey kidney epithelial Vero cells (obtained from UC Berkeley Cell Culture Facility, tested for mycoplasma contamination, and authenticated by mass spectrometry) were infected with 5 × 106 R. parkeri per T175 flask. Vero cells were grown in DMEM (Gibco 11965–092) containing 4.5 g l–1 glucose and 2% fetal bovine serum (FBS; GemCell). Infected cells were scraped and collected at five or 6 d.p.i. when ~90% of cells were rounded due to infection. Scraped cells were then centrifuged at 12,000 g for 20 min at 4°C. Pelleted cells were resuspended in K-36 buffer (0.05 M KH2PO4, 0.05 M K2HPO4, 100 mM KCl, 15 mM NaCl, pH 7) and dounced for ~40 strokes at 4°C. The suspension was then centrifuged at 200 g for 5 min at 4°C to pellet host cell debris. Supernatant containing R. parkeri was overlaid on a 30% MD-76R (Merry X-Ray) gradient solution in ultracentrifuge tubes (Beckman/Coulter Cat 344058). Gradients were centrifuged at 18,000 r.p.m. in an SW-28 ultracentrifuge swinging bucket rotor (Beckman/Coulter) for 20 min at 4°C. These ‘30% prep’ bacterial pellets were resuspended in brain heart infusion (BHI) media (BD, 237500), aliquoted, and stored at −80°C. Titers were determined by plaque assays by serially diluting the R. parkeri in six-well plates containing confluent Vero cells. Plates were spun for 5 min at 300 g in an Eppendorf 5810 R centrifuge and at 24 hr post infection (h.p.i.); the media from each well was aspirated, and the wells were overlaid with 4 ml/well DMEM with 5% FBS and 0.7% agarose (Invitrogen, 16500–500). At 6 d.p.i., an overlay of 0.7% agarose in DMEM containing 2.5 % neutral red (Sigma, N6264) was added. Plaques were then counted 24 hr later. For infections with ompB mutant bacteria, the ompBSTOP::Tn mutant was used, which contains a transposon and an upstream stop codon in ompB, as previously described (Engström et al., 2019).
Deriving bone marrow macrophages
For obtaining bone marrow, male or female mice were euthanized, and femurs, tibias, and fibulas were excised. Bones were sterilized with 70% ethanol and washed with BMDM media (20% FBS (HyClone), 0.1% β-mercaptoethanol, 1% sodium pyruvate, 10% conditioned supernatant from 3T3 fibroblasts, in DMEM (Gibco) with 4.5 g l–1 glucose and 100 μg/ml streptomycin and 100 U/ml penicillin), and ground with a mortar and pestle. Bone homogenate was passed through a 70 μm nylon cell strainer (Thermo Fisher Scientific, 08-771-2) for particulate removal. Filtrates were then centrifuged at 290 g in an Eppendorf 5810 R centrifuge for 8 min, supernatant was aspirated, and the pellet was resuspended in BMDM media. Cells were plated in 30 ml BMDM media in non-TC-treated 15 cm petri dishes at a ratio of 10 dishes per two femurs/tibias and incubated at 37°C. An additional 30 ml of BMDM media was added 3 d later. At 7 d the media was aspirated, 15 ml cold PBS (Gibco, 10010–023) was added, and cells were incubated at 4°C with for 10 min. BMDMs were scraped from the plate, collected in a 50 ml conical tube, and centrifuged at 290 g for 5 min. PBS was aspirated, and cells were resuspended in BMDM media with 30% FBS and 10% DMSO at 107 cells/ml. One milliliter of aliquots was stored at –80°C for 24 hr in Styrofoam boxes and then moved to long-term storage in liquid nitrogen.
Infections in vitro
HMEC-1 cells (obtained from the UC Berkeley Cell Culture Facility and authenticated by short-tandem-repeat analysis) were passaged two to three times weekly and grown at 37°C with 5% CO2 in DMEM containing 10 mM L-glutamine (Sigma, M8537), supplemented with 10% heat-inactivated FBS (HyClone), 1 μg/ml hydrocortisone (Spectrum Chemical, CO137), 10 ng/ml epidermal growth factor (Thermo Fisher Scientific, CB40001; Corning cat. no. 354001), and 1.18 mg/ml sodium bicarbonate. HMEC media was prepared every 1–2 months and aliquoted and stored at 4°C. To prepare HMEC-1 cells for infection, cells were treated with 0.25% trypsin-EDTA (Thermo Fisher Scientific); the number of cells was counted using a hemocytometer (Bright-Line), and 3 × 104 cells were plated into 24-well plates 48 hr prior to infection.
To plate macrophages for infection, BMDM aliquots were thawed on ice, diluted into 9 ml of DMEM, centrifuged at 290 g for 5 min in an Eppendorf 5810 R centrifuge, and the pellet was resuspended in 10 ml BMDM media without antibiotics. 5 × 105 cells were plated into 24-well plates. Approximately 16 hr later, ‘30 % prep’ R. parkeri were thawed on ice and diluted into fresh BMDM media to either 106 p.f.u./ml or 2 × 105 p.f.u./ml. Media was then aspirated from the BMDMs and replaced with 500 µl media containing R. parkeri, and plates were spun at 300 g for 5 min in an Eppendorf 5810 R centrifuge. Infected cells were incubated in a humidified CEDCO 1600 incubator set to 33°C and 5% CO2. Recombinant mouse IFN-β (PBL, 12405–1) was added directly to infected cells after infection.
For measuring p.f.u., supernatants from infected BMDMs were aspirated, and each well was washed twice with 500 µl sterile milli-Q-grade water. After adding 1 ml of sterile milli-Q water to each well, macrophages were lysed by repeated pipetting. Serial dilutions of lysates were added to confluent Vero cells in 12-well plates. Plates were spun at 300 g using an Eppendorf 5810 R centrifuge for 5 min at room temperature and incubated at 33°C overnight. At ~16 h.p.i., media was aspirated and replaced with 2 ml/well of DMEM containing 0.7% agarose and 5% FBS (GemCell). At ~6 d.p.i., 1 ml of DMEM containing 0.7% agarose, 1% FBS (GemCell), 200 µg/ml amphotericin B (Invitrogen, 15290–018), and 2.5 % neutral red (Sigma) was added to each well. Plaques were then counted after 24 hr.
Microscopy, LDH, and IFN-I experiments were performed as described (Burke et al., 2020).
Animal experiments
Animal research was conducted under a protocol approved by the University of California, Berkeley Institutional Animal Care and Use Committee (IACUC) in compliance with the Animal Welfare Act and other federal statutes relating to animals and experiments using animals (Welch lab animal use protocol AUP-2016-02-8426). The University of California, Berkeley IACUC is fully accredited by the Association for the Assessment and Accreditation of Laboratory Animal Care International and adheres to the principles of the Guide for the Care and use of Laboratory Animals (National Research Council, 2011). Mouse infections were performed in a biosafety level two facility. All animals were maintained at the University of California, Berkeley campus, and all infections were performed in accordance with the approved protocols. Mice were between 8 and 20 weeks old at the time of initial infection. Mice were selected for experiments based on their availability, regardless of sex. The sex of mice used for survival after i.d. infection and raw data for mouse experiments is provided in the Source Data for each figure. A statistical analysis was not performed to predetermine sample size prior to initial experiments. Initial sample sizes were based on availability of mice and the capacity to process or measure samples within a given time. After the first experiment, a Power Analysis was conducted to determine subsequent group sizes. All mice were of the C57BL/6J background, except for outbred CD-1 mice. All mice were healthy at the time of infection and were housed in microisolator cages and provided chow, water, and bedding. No mice were administered antibiotics or maintained on water with antibiotics. Experimental groups were littermates of the same sex that were randomly assigned to experimental groups. For experiments with DKO mice deficient in Ifnar1 and Ifngr1, mice were immediately euthanized if they exhibited severe degree of infection, as defined by a core body temperature dropping below 90°F or lethargy that prevented normal movement.
Mouse genotyping
Single mutant Tlr4-/- (Hoshino et al., 1999, Ifnar1-/- Müller et al., 1994, Ifngr1-/- Huang et al., 1993), DKO Ifnar1-/-;Ifngr1-/-, and WT C57BL/6J mice were previously described and originally obtained from Jackson Laboratories. CD-1 mice were obtained from Charles River. For genotyping, ear clips were boiled for 15 min in 60 µl of 25 mM NaOH, quenched with 10 µl Tris–HCl pH 5.5, and 2 µl of lysate was used for PCR using SapphireAMP (Takara, RR350) and gene-specific primers. Primers used were: Ifnar1 forward (F): CAACATACTACAACGACCAAGTGTG; Ifnar1 WT reverse (R): AACAAACCCCCAAACCCCAG; Ifnar1-/- R: ATCTGGACGAAGAGCATCAGG; Ifngr1 (F): CTCGTGCTTTACGGTATCGC; Ifngr1 (R): TCGCTTTCCAGCTGATGTACT; WT Tlr4 (F): CACCTGATACTTAATGCTGGCTGTAAAAAG; WT Tlr4 (R): GGTTTAGGCCCCAGAGTTTTGTTCTTCTCA; Tlr4-/- (F): TGTTGCCCTTCAGTCACAGAGACTCTG; and Tlr4-/- (R): TGTTGGGTCGTTTGTTCGGATCCGTCG.
Mouse infections
For mouse infections, R. parkeri was prepared by diluting 30%-prep bacteria into cold sterile PBS on ice. Bacterial suspensions were kept on ice during injections. For i.d. infections, mice were anaesthetized with 2.5% isoflurane via inhalation. The right flank of each mouse was shaved with a hair trimmer (Braintree CLP-41590), wiped with 70% ethanol, and 50 µl of bacterial suspension in PBS was injected intradermally using a 30.5-gauge needle. Mice were monitored for ~3 min until they were fully awake. No adverse effects were recorded from anesthesia. For i.v. infections, mice were exposed to a heat lamp while in their cages for approximately 5 min and then each mouse was moved to a mouse restrainer (Braintree, TB-150 STD). The tail was sterilized with 70% ethanol, and 200 µl of bacterial suspension in sterile PBS was injected using 30.5-gauge needles into the lateral tail vein. Body temperatures were monitored using a rodent rectal thermometer (BrainTree Scientific, RET-3).
For fluorescent dextran experiments, mice were intravenously injected with 150 µl of 10 kDa dextran conjugated with Alexa Fluor 680 (D34680; Thermo Fisher Scientific) at a concentration of 1 mg/ml in sterile PBS (Glasner et al., 2017). As a negative control, mice with no R. parkeri infection were injected with fluorescent dextran. As an additional negative control, uninfected mice were injected intravenously with PBS instead of fluorescent dextran. At 2 hr post-injection, mice were euthanized with CO2 and cervical dislocation, doused with 70% ethanol, and skin surrounding the injection site (approximately 2 cm in each direction) was removed. Connective tissue between the skin and peritoneum was removed, and skin was placed hair-side-up on a 15 cm Petri dish. Skin was imaged with an LI-COR Odyssey CLx (LI-COR Biosciences), and fluorescence was quantified using ImageStudioLite v5.2.5. The skin from mice with no injected fluorescent dextran was used as the background measurement. Skin from mice injected with fluorescent dextran but no R. parkeri was normalized to an arbitrary number (100), and R. parkeri-infected samples were normalized to this value (R. parkeri-infected/uninfected × 100). The number of pixels at the injection site area was maintained across experiments (7,800 for small area and 80,000 for the large area).
All mice in this study were monitored daily for clinical signs of disease throughout the course of infection, such as hunched posture, lethargy, scruffed fur, paralysis, facial edema, and lesions on the skin of the flank and tail. If any such manifestations were observed, mice were monitored for changes in body weight and temperature. If a mouse displayed severe signs of infection, as defined by a reduction in body temperature below 90°F or an inability to move normally, the animal was immediately and humanely euthanized using CO2 followed by cervical dislocation, according to IACUC-approved procedures. Pictures of skin and tail lesions were obtained with permission from the Animal Care and Use Committee Chair and the Office of Laboratory and Animal Care. Pictures were captured with an Apple iPhone 8, software v13.3.1.
For harvesting spleens and livers, mice were euthanized at the indicated pre-determined times and doused with ethanol. Mouse organs were extracted and deposited into 50 ml conical tubes containing 4 ml sterile cold PBS for the spleen and 8 ml PBS for the liver. Organs were kept on ice and were homogenized for ~10 s using an immersion homogenizer (Fisher, Polytron PT 2500E) at ~22,000 r.p.m. Organ homogenates were spun at 290 g for 5 min to pellet the cell debris (Eppendorf 5810 R centrifuge). Twenty microliters of organ homogenates was then serial diluted into 12-well plates containing confluent Vero cells. The plates were then spun at 260 g for 5 min at room temperature (Eppendorf 5810 R centrifuge) and incubated at 33°C. To reduce the possibility of contamination, organ homogenates were plated in duplicate and the second replicate was treated with 50 µg/ml carbenicillin (Sigma) and 200 µg/ml amphotericin B (Gibco). The next day, at approximately 16 h.p.i., the cells were gently washed by replacing the existing media with 1 ml DMEM containing 2% FBS (GemCell). The media were then aspirated and replaced with 2 ml/well of DMEM containing 0.7% agarose, 5% FBS, and 200 µg/ml amphotericin B. When plaques were visible at 6 d.p.i., 1 ml of DMEM containing 0.7% agarose, 1% FBS, and 2.5% neutral red (Sigma) was added to each well, and plaques were counted at 24 h.p.i.
Histology
For histology, spleens and livers were harvested from infected mice and immediately stored in 10% neutral buffered formalin (Sigma HT501128). Histology was performed by HistoWiz Inc (http://histowiz.com) using a standard operating procedure and fully automated workflow. Samples were processed, embedded in paraffin, and sectioned at 4 μm thickness. BOND Polymer Refine Detection (Leica Biosystems) was used according to manufacturer’s protocol. After staining with hematoxylin and eosin, sections were dehydrated and film coverslipped using a TissueTek-Prisma and Coverslipper (Sakura). Whole slide scanning (40×) was performed on an Aperio AT2 (Leica Biosystems). Histological consultation was blindly performed by a pathologist. Immunohistochemistry was performed with a rabbit anti-Rickettsia I7205 antibody (1:500 dilution; gift from Ted Hackstadt). Shareable links for all histology and immunohistochemistry data are available upon request to the authors.
Statistical analysis
Statistical parameters and significance are reported in the figure legends. For comparing two sets of data, a two-tailed Student’s T test was performed. For comparing two sets of in vivo p.f.u. data, Mann–Whitney U tests were used. For comparing two survival curves, log-rank (Mantel–Cox) tests were used. For comparing curves of two samples (mouse health, weight, and temperature), two-way ANOVAs were used. For two-way ANOVAs, if a mouse was euthanized prior to the statistical end point, the final value that was recorded for the mouse was repeated until the statistical endpoint. For two-way ANOVAs, if a measurement was not recorded for a timepoint, the difference between values at adjacent time points was used. Data were determined to be statistically significant when p<0.05. Asterisks denote statistical significance as: *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001, compared to indicated controls. Error bars indicate standard deviation (SD) for in vitro experiments and standard error of the mean (SEM) for in vivo experiments. All other graphical representations are described in the figure legends. Statistical analyses were performed using GraphPad PRISM v7.0.
Acknowledgements
We thank Neil Fisher for editing this manuscript. PE was supported by postdoctoral fellowships from the Sweden-America Foundation. MDW was supported by NIH/NIAID grants R01AI109044 and R21AI138550. DRG, DAE, and EH were partially supported by NIH/NIAID grant R01 AI24493 (EH).
Funding Statement
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Contributor Information
Thomas P Burke, Email: tburke@berkeley.edu.
Matthew D Welch, Email: welch@berkeley.edu.
Kim Orth, HHMI/University of Texas Southwestern Medical Center, United States.
Dominique Soldati-Favre, University of Geneva, Switzerland.
Funding Information
This paper was supported by the following grants:
National Institute of Allergy and Infectious Diseases R01AI109044 to Matthew D Welch.
National Institute of Allergy and Infectious Diseases R21AI138550 to Matthew D Welch.
National Institute of Allergy and Infectious Diseases R01AI124493 to Eva Harris.
Additional information
Competing interests
None.
none.
Author contributions
Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Supervision, Validation, Visualization, Writing – original draft, Writing – review and editing.
Resources, Writing – review and editing.
Investigation, Writing – review and editing.
Formal analysis, Investigation, Writing – review and editing.
Investigation, Resources.
Investigation, Resources.
Resources, Supervision.
Conceptualization, Funding acquisition, Project administration, Supervision, Writing – review and editing.
Ethics
Animal research was conducted under a protocol approved by the University of California, Berkeley Institutional Animal Care and Use Committee (IACUC) in compliance with the Animal Welfare Act and other federal statutes relating to animals and experiments using animals (Welch lab animal use protocol AUP-2016-02-8426). The University of California, Berkeley IACUC is fully accredited by the Association for the Assessment and Accreditation of Laboratory Animal Care International and adheres to the principles of the Guide for the Care and use of Laboratory Animals. Mouse infections were performed in a biosafety level 2 facility. All animals were maintained at the University of California, Berkeley campus, and all infections were performed in accordance with the approved protocols. Mice were immediately euthanized if they exhibited severe degree of infection, as defined by a core body temperature dropping below 90 F or lethargy that prevented normal movement.
Additional files
Data availability
All data sets generated or analyzed during this study are included in the manuscript and supporting files.
References
- Abdad MY, Abou Abdallah R, Fournier PE, Stenos J, Vasoo S. A concise review of the epidemiology and diagnostics of rickettsioses: Rickettsia and Orientia SPP. Journal of Clinical Microbiology. 2018;56:e01728-17. doi: 10.1128/JCM.01728-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson GW, Osterman J. Host defenses in experimental rickettsialpox: Genetics of natural resistance to infection. Infect. Immun. 1980;28:132–136. doi: 10.1128/iai.28.1.132-136.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auerbuch V, Lenz LL, Portnoy DA. Development of a competitive index assay to evaluate the virulence of Listeria monocytogenes actA mutants during primary and secondary infection of mice. Infect. Immun. 2001;69:5953–5957. doi: 10.1128/IAI.69.9.5953-5957.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banajee KH, Embers ME, Langohr IM, Doyle LA, Hasenkampf NR, Macaluso KR. Correction: Amblyomma maculatum feeding augments Rickettsia parkeri infection in a rhesus macaque model: a pilot study. PLOS ONE. 2015;10:e135175. doi: 10.1371/journal.pone.0137598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbieri ARM, Szabó MPJ, Costa FB, Martins TF, Soares HS, Pascoli G, Torga K, Saraiva DG, Ramos VN, Osava C, Gerardi M, Dias RA, Moraes EA, Jr, Ferreira F, Castro MB, Labruna MB. Species richness and seasonal dynamics of ticks with notes on rickettsial infection in a Natural Park of the Cerrado biome in Brazil. Ticks and Tick-Borne Diseases. 2019;10:442–453. doi: 10.1016/j.ttbdis.2018.12.010. [DOI] [PubMed] [Google Scholar]
- Barthold SW, Beck DS, Hansen GM, Terwilliger GA, Moody KD. Lyme borreliosis in selected strains and ages of laboratory mice. J. Infect. Dis. 1990;162:133–138. doi: 10.1093/infdis/162.1.133. [DOI] [PubMed] [Google Scholar]
- Billiau A, Matthys P. Interferon-γ: A historical perspective. Cytokine and Growth Factor Reviews. 2009;20:97–113. doi: 10.1016/j.cytogfr.2009.02.004. [DOI] [PubMed] [Google Scholar]
- Bonell A, Lubell Y, Newton PN, Crump JA, Paris DH. Estimating the burden of scrub typhus: A systematic review. PLOS Neglected Tropical Diseases. 2017;11:e0005838. doi: 10.1371/journal.pntd.0005838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bratton RL, Whiteside JW, Hovan MJ, Engle RL, Edwards FD. Diagnosis and treatment of Lyme disease. Mayo Clinic Proceedings. 2008;83:566–571. doi: 10.4065/83.5.566. [DOI] [PubMed] [Google Scholar]
- Brundage RA, Smith GA, Camilli A, Theriot JA, Portnoy DA. Expression and phosphorylation of the Listeria monocytogenes ActA protein in mammalian cells. PNAS. 1993;90:11890–11894. doi: 10.1073/pnas.90.24.11890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke TP, Engström P, Chavez RA, Fonbuena JA, Vance RE, Welch MD. Inflammasome-mediated antagonism of type I interferon enhances Rickettsia pathogenesis. Nature Microbiology. 2020;5:688–696. doi: 10.1038/s41564-020-0673-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng MI, Chen C, Engström P, Portnoy DA, Mitchell G. Actin-based motility allows Listeria monocytogenes to avoid autophagy in the macrophage cytosol. Cellular Microbiology. 2018;20:e12854. doi: 10.1111/cmi.12854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choe JE, Welch MD. Actin-based motility of bacterial pathogens: Mechanistic diversity and its impact on virulence. Pathogens and Disease. 2016;74:ftw099. doi: 10.1093/femspd/ftw099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cillari E, Milano S, D’Agostino P, Arcoleo F, Stassi G, Galluzzo A, Richiusa P, Giordano C, Quartararo P, Colletti P, Gambino G, Mocciaro C, Spinelli A, Vitale G, Mansueto S. Depression of CD4 T cell subsets and alteration in cytokine profile in Boutonneuse fever. The Journal of Infectious Diseases. 1996;174:1051–1057. doi: 10.1093/infdis/174.5.1051. [DOI] [PubMed] [Google Scholar]
- Colonne PM, Eremeeva ME, Sahni SK. Beta interferon-mediated activation of signal transducer and activator of transcription protein 1 interferes with Rickettsia conorii replication in human endothelial cells. Infect. Immun. 2011;79:3733–3743. doi: 10.1128/IAI.05008-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cragun WC, Bartlett BL, Ellis MW, Hoover AZ, Tyring SK, Mendoza N, Vento TJ, Nicholson WL, Eremeeva ME, Olano JP, Rapini RP, Paddock CD. The expanding spectrum of eschar-associated rickettsioses in the United States. Archives of Dermatology. 2010;146:641–648. doi: 10.1001/archdermatol.2010.48. [DOI] [PubMed] [Google Scholar]
- Dantas-Torres F. Rocky Mountain spotted fever. The Lancet Infectious Diseases. 2007;7:724–732. doi: 10.1016/S1473-3099(07)70261-X. [DOI] [PubMed] [Google Scholar]
- de Sousa R, Ismail N, Nobrega SD, França A, Amaro M, Anes M, Poças J, Coelho R, Torgal J, Bacellar F, Walker DH. Intralesional expression of mRNA of interferon- gamma , tumor necrosis factor- alpha , interleukin-10, nitric oxide synthase, indoleamine-2,3-dioxygenase, and RANTES is a major immune effector in Mediterranean spotted fever rickettsiosis. The Journal of Infectious Diseases. 2007;196:770–781. doi: 10.1086/519739. [DOI] [PubMed] [Google Scholar]
- Eisemann CS, Nypaver MJ, Osterman JV. Susceptibility of inbred mice to Rickettsiae of the spotted fever group. Infection and Immunity. 1984;43:143–148. doi: 10.1128/iai.43.1.143-148.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engström P, Burke TP, Mitchell G, Ingabire N, Mark KG, Golovkine G, Iavarone AT, Rape M, Cox JS, Welch MD. Evasion of autophagy mediated by Rickettsia surface protein OmpB is critical for virulence. Nature Microbiology. 2019;4:2538–2551. doi: 10.1038/s41564-019-0583-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engström P, Burke TP, Tran CJ, Iavarone AT, Welch MD. Lysine methylation shields an intracellular pathogen from ubiquitylation and autophagy. Science Advances. 2021;7:eabg2517. doi: 10.1126/sciadv.abg2517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang R, Blanton LS, Walker DH. Rickettsiae as emerging infectious agents. Clinics in Laboratory Medicine. 2017;37:383–400. doi: 10.1016/j.cll.2017.01.009. [DOI] [PubMed] [Google Scholar]
- Feng HM, Popov VL, Walker DH. Depletion of gamma interferon and tumor necrosis factor alpha in mice with Rickettsia conorii-infected endothelium: Impairment of rickettsicidal nitric oxide production resulting in fatal, overwhelming rickettsial disease. Infect. Immun. 1994;62:1952–1960. doi: 10.1128/iai.62.5.1952-1960.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glasner DR, Ratnasiri K, Puerta-Guardo H, Espinosa DA, Beatty PR, Harris E. Dengue virus NS1 cytokine-independent vascular leak is dependent on endothelial glycocalyx components. PLOS Pathogens. 2017;13:e1006673. doi: 10.1371/journal.ppat.1006673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goddard J. Historical and recent evidence for close relationships among Rickettsia parkeri, R. conorii, R. africae, and R. sibirica: Implications for rickettsial taxonomy. J. Vector Ecol. 2009;34:238–242. doi: 10.1111/j.1948-7134.2009.00032.x. [DOI] [PubMed] [Google Scholar]
- Goossens PL, Milon G, Bevan M. Induction of protective CD8+ T lymphocytes by an attenuated Listeria monocytogenes actA mutant. Int. Immunol. 1992;4:1413–1418. doi: 10.1093/intimm/4.12.1413. [DOI] [PubMed] [Google Scholar]
- Gouin E, Egile C, Dehoux P, Villiers V, Adams J, Gertler F, Li R, Cossart P. The RickA protein of Rickettsia conorii activates the Arp2/3 complex. Nature. 2004;427:457–461. doi: 10.1038/nature02318. [DOI] [PubMed] [Google Scholar]
- Grasperge BJ, Reif KE, Morgan TD, Sunyakumthorn P, Bynog J, Paddock CD, Macaluso KR. Susceptibility of inbred mice to Rickettsia parkeri. Infection and Immunity. 2012;80:1846–1852. doi: 10.1128/IAI.00109-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haglund CM, Choe JE, Skau CT, Kovar DR, Welch MD. Rickettsia Sca2 is a bacterial formin-like mediator of actin-based motility. Nat. Cell Biol. 2010;12:1057–1063. doi: 10.1038/ncb2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson B. Comparative susceptibility to mouse interferons of Rickettsia tsutsugamushi strains with different virulence in mice and of Rickettsia rickettsii. Infection and Immunity. 1991;59:4134–4141. doi: 10.1128/iai.59.11.4134-4141.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris EK, Jirakanwisal K, Verhoeve VI, Fongsaran C, Suwanbongkot C, Welch MD, Macaluso KR. Role of Sca2 and RickA in the Dissemination of Rickettsia parkeri in Amblyomma maculatum. Infection and Immunity. 2018;86:e00123-18. doi: 10.1128/IAI.00123-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinzen RA, Hayes SF, Peacock MG, Hackstadt T. Directional actin polymerization associated with spotted fever group Rickettsia infection of Vero cells. Infect. Immun. 1993;61:1926–1935. doi: 10.1128/iai.61.5.1926-1935.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrick KL, Pena SA, Yaglom HD, Layton BJ, Moors A, Loftis AD, Condit ME, Singleton J, Kato CY, Denison AM, Ng D, Mertins JW, Paddock CD. Rickettsia parkeri Rickettsiosis, Arizona, USA. Emerging Infectious Diseases. 2016;22:780–785. doi: 10.3201/eid2205.151824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshino K, Takeuchi O, Kawai T, Sanjo H, Ogawa T, Takeda Y, Takeda K, Akira S. Cutting edge: Toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide: evidence for TLR4 as the Lps gene product. Journal of Immunology. 1999;162:3749–3752. [PubMed] [Google Scholar]
- Huang S, Hendriks W, Althage A, Hemmi S, Bluethmann H, Kamijo R, Vilcek J, Zinkernagel RM, Aguet M. Immune response in mice that lack the interferon-gamma receptor. Science. 1993;259:1742–1745. doi: 10.1126/science.8456301. [DOI] [PubMed] [Google Scholar]
- Jeng RL, Goley ED, D’Alessio JA, Chaga OY, Svitkina TM, Borisy GG, Heinzen RA, Welch MD. A Rickettsia WASP-like protein activates the Arp2/3 complex and mediates actin-based motility. Cellular Microbiology. 2004;6:761–769. doi: 10.1111/j.1462-5822.2004.00402.x. [DOI] [PubMed] [Google Scholar]
- Jia N, Liu HB, Zheng YC, Shi WQ, Wei R, Chu YL, Ning NZ, Jiang BG, Jiang RR, Li T, Huo QB, Bian C, Liu X, Sun Y, Li LF, Wang Q, Wei W, Wang YW, Jongejan F, Jiang JF, Song JL, Wang H, Cao WC. Cutaneous Immunoprofiles of Three Spotted Fever Group Rickettsia Cases. Infection and Immunity. 2020;88:e0686-19. doi: 10.1128/IAI.00686-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaskas NM. Rickettsia parkeri: Eschar diagnosis. Journal of the American Academy of Dermatology. 2014;71:E87–E89. doi: 10.1016/j.jaad.2014.03.024. [DOI] [PubMed] [Google Scholar]
- Kelly DJ, Foley DH, Richards AL. A spatiotemporal database to track human scrub typhus using the Vectormap application. PLOS Neglected Tropical Diseases. 2015;9:e0004161. doi: 10.1371/journal.pntd.0004161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleba B, Clark TR, Lutter E, Ellison DW, Hackstadt T. Disruption of the Rickettsia rickettsii Sca2 autotransporter inhibits actin-based motility. Infect. Immun. 2010;78:2240–2247. doi: 10.1128/IAI.00100-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krawczak FS, Binder LC, Oliveira CS, Costa FB, Moraes-Filho J, Martins TF, Sponchiado J, Melo GL, Gregori F, Polo G, Oliveira SV, Labruna MB. Ecology of a tick-borne spotted fever in southern Brazil. Experimental & Applied Acarology. 2016;70:219–229. doi: 10.1007/s10493-016-0070-1. [DOI] [PubMed] [Google Scholar]
- Lamason RL, Welch MD. Actin-based motility and cell-to-cell spread of bacterial pathogens. Current Opinion in Microbiology. 2017;35:48–57. doi: 10.1016/j.mib.2016.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamason RL, Kafai NM, Welch MD. A streamlined method for transposon mutagenesis of Rickettsia parkeri yields numerous mutations that impact infection. PLOS ONE. 2018;13:e0197012. doi: 10.1371/journal.pone.0197012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Monnier A, Autret N, Join-Lambert OF, Jaubert F, Charbit A, Berche P, Kayal S. ActA is required for crossing of the fetoplacental barrier by Listeria monocytogenes. Infection and Immunity. 2007;75:950–957. doi: 10.1128/IAI.01570-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lestinova T, Rohousova I, Sima M, de Oliveira CI, Volf P. Insights into the sand fly saliva: Blood-feeding and immune interactions between sand flies, hosts, and leishmania. PLOS Neglected Tropical Diseases. 2017;11:e0005600. doi: 10.1371/journal.pntd.0005600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Jerrells TR, Spitalny GL, Walker DH. Gamma interferon as a crucial host defense against Rickettsia conorii in vivo. Infect. Immun. 1987;55:1252–1255. doi: 10.1128/iai.55.5.1252-1255.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Londoño AF, Mendell NL, Walker DH, Bouyer DH. A biosafety level-2 dose-dependent lethal mouse model of spotted fever rickettsiosis: Rickettsia parkeri Atlantic Rainforest strain. PLOS Neglected Tropical Diseases. 2019;13:e0007054. doi: 10.1371/journal.pntd.0007054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manor E, Sarov I. Inhibition of Rickettsia conorii growth by recombinant tumor necrosis factor alpha: Enhancement of inhibition by gamma interferon. Infect. Immun. 1990;58:1886–1890. doi: 10.1128/iai.58.6.1886-1890.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meunier E, Broz P. Interferon-inducible GTPases in cell autonomous and innate immunity. Cell. Microbiol. 2016;18:168–180. doi: 10.1111/cmi.12546. [DOI] [PubMed] [Google Scholar]
- Mitchell G, Cheng MI, Chen C, Nguyen BN, Whiteley AT, Kianian S, Cox JS, Green DR, McDonald KL, Portnoy DA. Listeria monocytogenes triggers noncanonical autophagy upon phagocytosis, but avoids subsequent growth-restricting xenophagy. PNAS. 2018;115:E210–E217. doi: 10.1073/pnas.1716055115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moraru GM, Goddard J, Murphy A, Link D, Belant JL, Varela-Stokes A. Evidence of antibodies to spotted fever group rickettsiae in small mammals and quail from Mississippi. Vector Borne and Zoonotic Diseases. 2013a;13:1–5. doi: 10.1089/vbz.2012.1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moraru GM, Goddard J, Paddock CD, Varela-Stokes A. Experimental infection of cotton rats and bobwhite quail with Rickettsia parkeri. Parasites and Vectors. 2013b;6:1–5. doi: 10.1186/1756-3305-6-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller U, Steinhoff U, Reis LF, Hemmi S, Pavlovic J, Zinkernagel RM, Aguet M. Functional role of type I and type II interferons in antiviral defense. Science. 1994;264:1918–1921. doi: 10.1126/science.8009221. [DOI] [PubMed] [Google Scholar]
- National Research Council Guide for the Care and Use of Laboratory Animals. The Biochemical Journal. 2011;10:12910. doi: 10.17226/12910. [DOI] [Google Scholar]
- Ngwamidiba M, Blanc G, Ogata H, Raoult D, Fournier PE. Phylogenetic study of Rickettsia species using sequences of the autotransporter protein-encoding gene sca2. Annals of the New York Academy of Sciences. 2005;1063:94–99. doi: 10.1196/annals.1355.015. [DOI] [PubMed] [Google Scholar]
- Osterloh A, Papp S, Moderzynski K, Kuehl S, Richardt U, Fleischer B. Persisting Rickettsia typhi causes fatal central nervous system inflammation. Infection and Immunity. 2016;84:1615–1632. doi: 10.1128/IAI.00034-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osterloh A. Immune response against rickettsiae: Lessons from murine infection models. Medical Microbiology and Immunology. 2017;206:403–417. doi: 10.1007/s00430-017-0514-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paddock CD, Sumner JW, Comer JA, Zaki SR, Goldsmith CS, Goddard J, McLellan SLF, Tamminga CL, Ohl CA. Rickettsia parkeri: a newly recognized cause of spotted fever rickettsiosis in the United States. Clinical Infectious Diseases. 2004;38:805–811. doi: 10.1086/381894. [DOI] [PubMed] [Google Scholar]
- Paddock CD, Finley RW, Wright CS, Robinson HN, Schrodt BJ, Lane CC, Ekenna O, Blass MA, Tamminga CL, Ohl CA, McLellan SLF, Goddard J, Holman RC, Openshaw JJ, Sumner JW, Zaki SR, Eremeeva ME. Rickettsia parkeri rickettsiosis and its clinical distinction from Rocky Mountain spotted fever. Clinical Infectious Diseases. 2008;47:1188–1196. doi: 10.1086/592254. [DOI] [PubMed] [Google Scholar]
- Paddock CD, Allerdice MEJ, Karpathy SE, Nicholson WL, Levin ML, Smith TC, Becker T, Delph RJ, Knight RN, Ritter JM, Sanders JH, Goddard J. Unique strain of Rickettsia parkeri associated with the hard tick Dermacentor parumapertus Neumann in the western United States. Applied and Environmental Microbiology. 2017;83:e03463-16. doi: 10.1128/AEM.03463-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pingen M, Schmid MA, Harris E, McKimmie CS. Mosquito biting modulates skin response to virus infection. Trends in Parasitology. 2017;33:645–657. doi: 10.1016/j.pt.2017.04.003. [DOI] [PubMed] [Google Scholar]
- Piro AS, Hernandez D, Luoma S, Feeley EM, Finethy R, Yirga A, Frickel EM, Lesser CF, Coers J. Detection of cytosolic Shigella flexneri via a C-terminal triple-arginine motif of GBP1 inhibits actin-based motility. MBio. 2017;8:e01979-17. doi: 10.1128/mBio.01979-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajapakse S, Weeratunga P, Sivayoganathan S, Fernando SD. Clinical manifestations of scrub typhus. Transactions of the Royal Society of Tropical Medicine and Hygiene. 2017;111:43–54. doi: 10.1093/trstmh/trx017. [DOI] [PubMed] [Google Scholar]
- Raniga K, Liang C. Interferons: Reprogramming the metabolic network against viral infection. Viruses. 2018;10:1–21. doi: 10.3390/v10010036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed SCO, Lamason RL, Risca V, Abernathy E, Welch MD. Rickettsia actin-based motility occurs in distinct phases mediated by different actin nucleators. Curr. Biol. 2014;24:98–103. doi: 10.1016/j.cub.2013.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards AL, Jiang J. Scrub Typhus: Historic perspective and current status of the worldwide presence of Orientia species. Tropical Medicine and Infectious Disease. 2020;5:49. doi: 10.3390/tropicalmed5020049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux V, Raoult D. Phylogenetic analysis of members of the genus Rickettsia using the gene encoding the outer-membrane protein rOmpB (ompB. International Journal of Systematic and Evolutionary Microbiology. 2000;50 Pt 4:1449–1455. doi: 10.1099/00207713-50-4-1449. [DOI] [PubMed] [Google Scholar]
- Sahni A, Fang R, Sahni SK, Walker DH. Pathogenesis of rickettsial diseases: Pathogenic and immune mechanisms of an endotheliotropic infection. Annual Review of Pathology. 2019;14:127–152. doi: 10.1146/annurev-pathmechdis-012418-012800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito TB, Bechelli J, Smalley C, Karim S, Walker DH. Vector tick transmission model of spotted fever rickettsiosis. Am. J. Pathol. 2019;189:115–123. doi: 10.1016/j.ajpath.2018.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz AM, Hinckley AF, Mead PS, Hook SA, Kugeler KJ. Surveillance for Lyme disease - United States, 2008-2015. Morbidity and Mortality Weekly Report. 2008;66:9. doi: 10.15585/mmwr.ss6622a1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Šimo L, Kazimirova M, Richardson J, Bonnet SI. The essential role of tick salivary glands and saliva in tick feeding and pathogen transmission. Frontiers in Cellular and Infection Microbiology. 2017;7:281. doi: 10.3389/fcimb.2017.00281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunyakumthorn P, Paris DH, Chan T-C, Jones M, Luce-Fedrow A, Chattopadhyay S, Jiang J, Anantatat T, Turner GDH, Day NPJ, Richards AL. An intradermal inoculation model of scrub typhus in Swiss CD-1 mice demonstrates more rapid dissemination of virulent strains of Orientia tsutsugamushi. PLOS ONE. 2013;8:e54570. doi: 10.1371/journal.pone.0054570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suwanbongkot C, Langohr IM, Harris EK, Dittmar W, Christofferson RC, Macaluso KR. Spotted fever group Rickettsia infection and transmission dynamics in Amblyomma maculatum. Infection and Immunity. 2019;87:e00804-18. doi: 10.1128/IAI.00804-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teysseire N, Chiche-Portiche C, Raoult D. Intracellular movements of Rickettsia conorii and R. typhi based on actin polymerization. Res. Microbiol. 1992;143:821–829. doi: 10.1016/0923-2508(92)90069-z. [DOI] [PubMed] [Google Scholar]
- Turco J, Winkler HH. Cloned mouse interferon-λ inhibits the growth of Rickettsia prowazekii in cultured mouse fibroblasts. J. Exp. Med. 1983;158:2159–2164. doi: 10.1084/jem.158.6.2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turco J, Winkler HH. Interferon-alpha/beta and Rickettsia prowazekii: induction and sensitivity. Annals of the New York Academy of Sciences. 1990;590:168–186. doi: 10.1111/j.1749-6632.1990.tb42219.x. [DOI] [PubMed] [Google Scholar]
- Walker DH, Olano JP, Feng HM. Critical role of cytotoxic T lymphocytes in immune clearance of rickettsial infection. Infection and Immunity. 2001;69:1841–1846. doi: 10.1128/IAI.69.3.1841-1846.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker DH, Ismail N. Emerging and re-emerging rickettsioses: endothelial cell infection and early disease events. Nature Reviews Microbiology. 2008;6:375–386. doi: 10.1038/nrmicro1866. [DOI] [PubMed] [Google Scholar]
- Walker DH. The realities of biodefense vaccines against Rickettsia. Vaccine. 2009;27:D52–D55. doi: 10.1016/j.vaccine.2009.07.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Ojaimi C, Iyer R, Saksenberg V, McClain SA, Wormser GP, Schwartz I. Impact of genotypic variation of Borrelia burgdorferi sensu stricto on kinetics of dissemination and severity of disease in C3H/HeJ mice. Infection and Immunity. 2001;69:4303–4312. doi: 10.1128/IAI.69.7.4303-4312.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshikawa Y, Ogawa M, Hain T, Chakraborty T, Sasakawa C. Listeria monocytogenes ActA is a key player in evading autophagic recognition. Autophagy. 2009a;5:1220–1221. doi: 10.4161/auto.5.8.10177. [DOI] [PubMed] [Google Scholar]
- Yoshikawa Y, Ogawa M, Hain T, Yoshida M, Fukumatsu M, Kim M, Mimuro H, Nakagawa I, Yanagawa T, Ishii T, Kakizuka A, Sztul E, Chakraborty T, Sasakawa C. Listeria monocytogenes ActA-mediated escape from autophagic recognition. Nature Cell Biology. 2009b;11:1233–1240. doi: 10.1038/ncb1967. [DOI] [PubMed] [Google Scholar]