

Abstract
Introduction:
Aromatase inhibitors are effective in lowering breast cancer incidence among postmenopausal women, but adverse events represent a barrier to their acceptability and adherence as a preventive treatment. This study aims to assess whether lowering exemestane schedule may retain biological activity while improving tolerability in breast cancer patients.
Methods/design:
We are conducting a, pre-surgical, non-inferiority phase IIb study in postmenopausal women with newly diagnosed estrogen receptor-positive breast cancer. Participants are randomized to receive either exemestane 25 mg/day or 25 mg/three times-week or once a week for 4 to 6 weeks prior to surgery.
The primary endpoint is the percentage change of serum estradiol concentration between baseline and surgery comparing the three arms. Sample size of 180 women was calculated assuming a 6% non-inferiority of the percent change of estradiol in the lower dose arms compared with the 80% decrease predicted in the full dose arm, with 80% power and using a one-sided 5% significance level and a two-sample t-test.
Main secondary outcomes are: safety; change in Ki-67 in cancer and adjacent pre-cancer tissue, circulating sex hormones, adipokines, lipid profile, insulin and glucose changes, in correlation with drug and metabolites concentrations.
Results and discussion:
The present paper is focused on methodology and operational aspects of the study. A total of 180 participants have ben enrolled. The trial is still blinded, and the analyses are ongoing.
Despite the short term duration, results may have relevant implications for clinical management of women at increased risk of developing a ER positive breast cancer.
Keywords: Phase II study, Breast cancer, Preventive medicine, Exemestane, Drug schedule and alternative dose
1. Introduction
The pre-surgical - window of opportunity - trial design has successfully been used to streamline drug development and check tumor sensitivity to a given drug [1]. The treatment effect on the adjacent intraepithelial neoplasia, including ductal or lobular carcinoma in situ (DCIS, LCIS) and atypical hyperplasia can be a reliable readout for the preventive activity generating new insight into new preventive strategies [2,3].
Despite the strong positive findings in breast cancer prevention with tamoxifen, the attitude towards preventive medicine remains ambivalent, and the associated side effects, particularly hot flashes and sexual disturbances, hamper the drug uptake by high-risk women who could benefit from it [4].
Among the strategies to overcome tamoxifen side effects, our group has intensively investigated alternative low doses of tamoxifen in several phase II studies [5–7], and in two phase III studies [8,9] in women at increased risk of breast cancer.
Likewise, aromatase inhibitors (AIs) have proven their capability to lower cancer incidence in postmenopausal high risk women. The Mammary Protocol 3 (MAP.3) trial [10], a randomized, placebo-controlled, double-blind trial with exemestane 25 mg/day for 5 years, at a median follow-up of 35 months, showed a 65% relative reduction in the annual incidence of invasive breast cancer compared with placebo (0.19% vs 0.55%; HR 0.35, 95% CI 0.18–0.70; p = 0.002). Similar results were obtained in the IBIS II Prevention trial where high-risk postmenopausal women were randomly assigned to receive either anastrozole 1 mg daily or placebo for 5 years. After a median follow-up of 5 years, 2% of women in the anastrozole group vs. 4% in the placebo group developed breast cancer (HR 0.47 95% CI 0.32, 0.68; p < 0.0001) [11]. Musculoskeletal adverse events (AEs) were common in the anastrozole group and vasomotor symptoms were present in both groups, but the incidence was higher in women on anastrozole. In the adjuvant setting it has been reported that approximately one in four women adhere sub-optimally to anastrozole therapy during the first year, and that adherence may drop up to 62% at the third year [12]. In the prevention setting, quality of life (QoL) is even more relevant: in the MAP.3 study a worsening of menopausal symptoms determined a discontinuation rate up to 80% irrespective of treatment arm [13].
A significant step forward to promote breast cancer prevention in postmenopausal women is certainly to improve QoL by seeking to determine the minimally active AIs dose and using an intermittent schedule. Previous studies have shown the possibility to use lower or intermittent dose of letrozole, alternative doses as lower as 1 mg three times a week showed similar estradiol reduction compared to standard dose [14]. Colleoni at al showed a similar disease-free survival between continuous versus intermittent letrozole schedules [15]. Exemestane is a steroidal, irreversible AI. This drug is highly specific and inhibits peripheral conversion of androstenedione to estrogen down to 98%. Importantly, the estradiol suppression persists for at least 5 days after administration of a single dose [16]. Despite the shorter half-life, compared to anastrozole and letrozole, exemestane shows a long-lasting inhibitory effect on aromatase, which is attributable to tight binding to the enzyme [17]. This suggests the possibility to reduce the dose and the frequency, while still maintaining a significant estradiol reduction. A study of 10 patients found that women who took a daily dose of 10 mg of exemestane achieved equivalent estradiol suppression as women who took a dose of 25 mg daily [18].
For this purpose we designed a pre-surgical phase IIb, randomized, double-blind, multi-center study for postmenopausal women with histologically confirmed estrogen receptor (ER)-positive breast cancer (Stage 0-II) comparing the exemestane standard daily dose regimen versus two alternative, less frequent dose regimens.
2. Methods
2.1. Trial design
This is a multicenter, pre-surgical double-blind non-inferiority phase IIb study in which participants have been randomized to receive either exemestane 25 mg/day or 25 mg/ three times a week or a single dose of 25 mg/week for 4 to 6 weeks. Participants with an histologically-confirmed ER-positive (ER expression ≥10% by immunohistochemistry [IHC]) breast cancer, candidate for breast surgery have been accrued. Complete physical exam and safety lab tests were performed at baseline and at end of treatment. Phone contacts have been made at day 1 of treatment and a week before surgery. Blood samples were collected at baseline and the final visit; tissue biomarkers will be assessed from the diagnostic biopsy and the surgical specimen (Fig. 1).
Fig. 1.
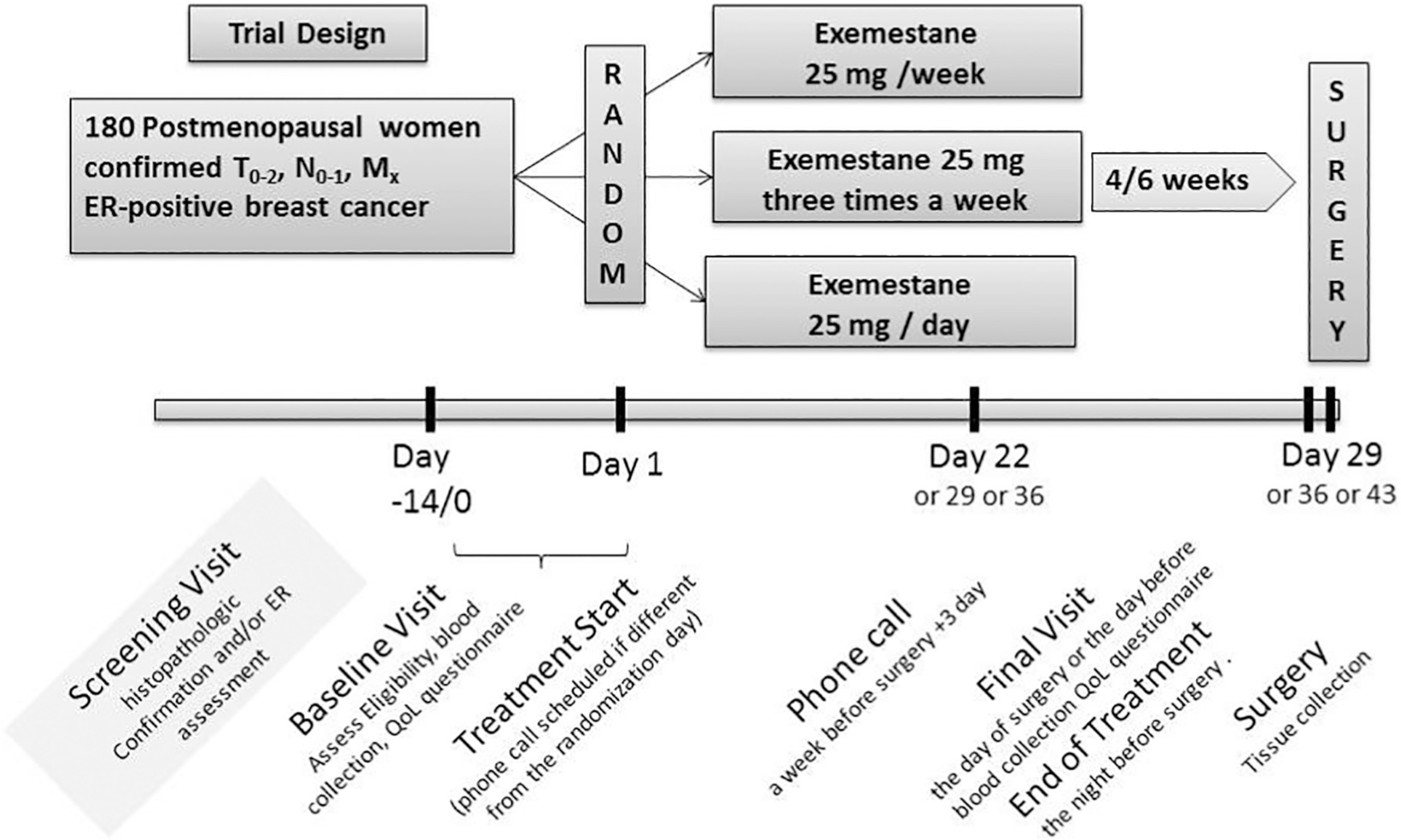
Trial design and procedures schedule.
2.2. Study setting
The study is coordinated by the European Institute of Oncology (IEO) located in Milan, Italy and additional recruiting centers are: Galliera Hospital in Italy, Columbia University Irving Medical Center (CUMC), MD Anderson Cancer Center, and Moffitt Cancer Center in the USA.
The Haukeland Hospital, Bergen Norway is involved in tissue and circulating biomarkers measurements.
2.3. Ethical oversight
The protocol and its amendments has been approved by the National Cancer Institute (NCI) Central Institutional Review Board (IRB) and the local Italian IRBs (registered at Clinical Trials.gov NCT02598557 and IEO 370 EudraCT 2015–005063–16). All participants signed a written informed consent.
2.4. Eligibility criteria
Main inclusion criteria: Postmenopausal patients defined as: women ≥60 years of age, or amenorrhea ≥12 months, or bilateral oophorectomy, or hysterectomized patients with FSH in the menopausal levels as per local institutional guidelines if <60 years old; histologically-confirmed ER-positive (≥ 10%) primary breast cancer stage cT0–2, cN0–1, Mx. Patients with larger tumors who refused neoadjuvant chemotherapy and/or endocrine therapy were also eligible. Blood tests within the normal limits or with no clinical relevance.
Main exclusion criteria were: body mass index (BMI) < 18.5 Kg/m2; previous treatment for breast cancer; uncontrolled intercurrent illness; other co-existing invasive malignancies (with the exclusion of basal cell carcinoma or skin squamous cell carcinoma) diagnosed during the last 2 years before randomization; history of severe osteoporosis; use of systemic hormone replacement therapy in the last 30 days prior to the randomization; use of any chemopreventive agents (selective estrogen receptor modulators or AIs) in the last 3 months; concomitant use of CYP3A4 inducer medication (rifampicin, phenytoin, carbamazepine, phenobarbital, and St. John’s wort).
2.5. Study procedures
Dedicated personnel proposed the trial to the incoming postmenopausal patients with a newly diagnosed ER-positive in situ or invasive breast cancer, or a highly suspicious lesion. For those who agreed, a baseline visit was scheduled. The Italian centers were allowed a screening visit with a dedicated informed consent to determine the histology nature of the lesion. At the baseline visit, medical history, physical exam, concomitant medication, tobacco/alcohol use, and any symptoms have been evaluated together with a self-reported questionnaire (MENQOL) [19] (by Mapi Research Trust, France) and blood collection for safety and circulating biomarkers. Participants who fulfilled the requirements were randomized. Treatment could be started the day of visit or the following days to allow for the appropriate timing from start of treatment to surgery. The participants were advised to take one tablet per day after dinner, approximately at 8 PM, starting from day 1 till the night before surgery. If the start date was different from the randomization day a telephone contact was performed as a reminder for the patient. To monitor compliance, concomitant medications, and toxicity a telephone contact was also scheduled a week before surgery (+3 days).
Given the weekly drug schedule participant has to undergo breast surgery exactly at the 29th or 36th or 43rd day, and the final visit occurred the same day or the day before. Final visit included physical exam, toxicity assessment, concomitant medications, blood collection, compliance/review of pill diary, and self-reported MENQOL questionnaire. The blood withdrawal schedule in this study is crucial to respect the timing since last active pill intake for each arm (range of 12 h, 1.5–2.5 days and 5.5–6.5 days).
If a toxicity “possibly”, “probably” or “definitely” related to the investigational drug was reported, it was monitored by a telephone call at 20–30 days after surgery.
The enrollment was steady throughout the study. Due to its short treatment plan no specific retention strategies were applied, except for a telephone contact a week before surgery.
The three double blind treatment arms were: arm 1, one tablet of exemestane 25 mg per week; arm 2, one tablet of exemestane 25 mg three times a week; arm 3, one tablet of exemestane 25 mg a day.
Tablets were packaged in blister card packaging, each blister card containing 7 tablets. Participants received two wallets of 3 blister cards each (6 blister cards for up to 6 weeks of treatment). Tablets within the blister were numbered from 1 to 7 to clearly mark the order to be taken by the participants. Based on the arm, the blister contained 7 active tablets 1 through 7; or three active tablets corresponding to days 1, 3 and 5 and four placebo tablets at days 2, 4, 6 and 7; or one active tablet at day 1 and 6 placebo in the remaining 6 days (see Fig. 2, as an example). If for any reason they skipped a tablet they were instructed to leave it behind and take the next of the corresponding day.
Fig. 2.
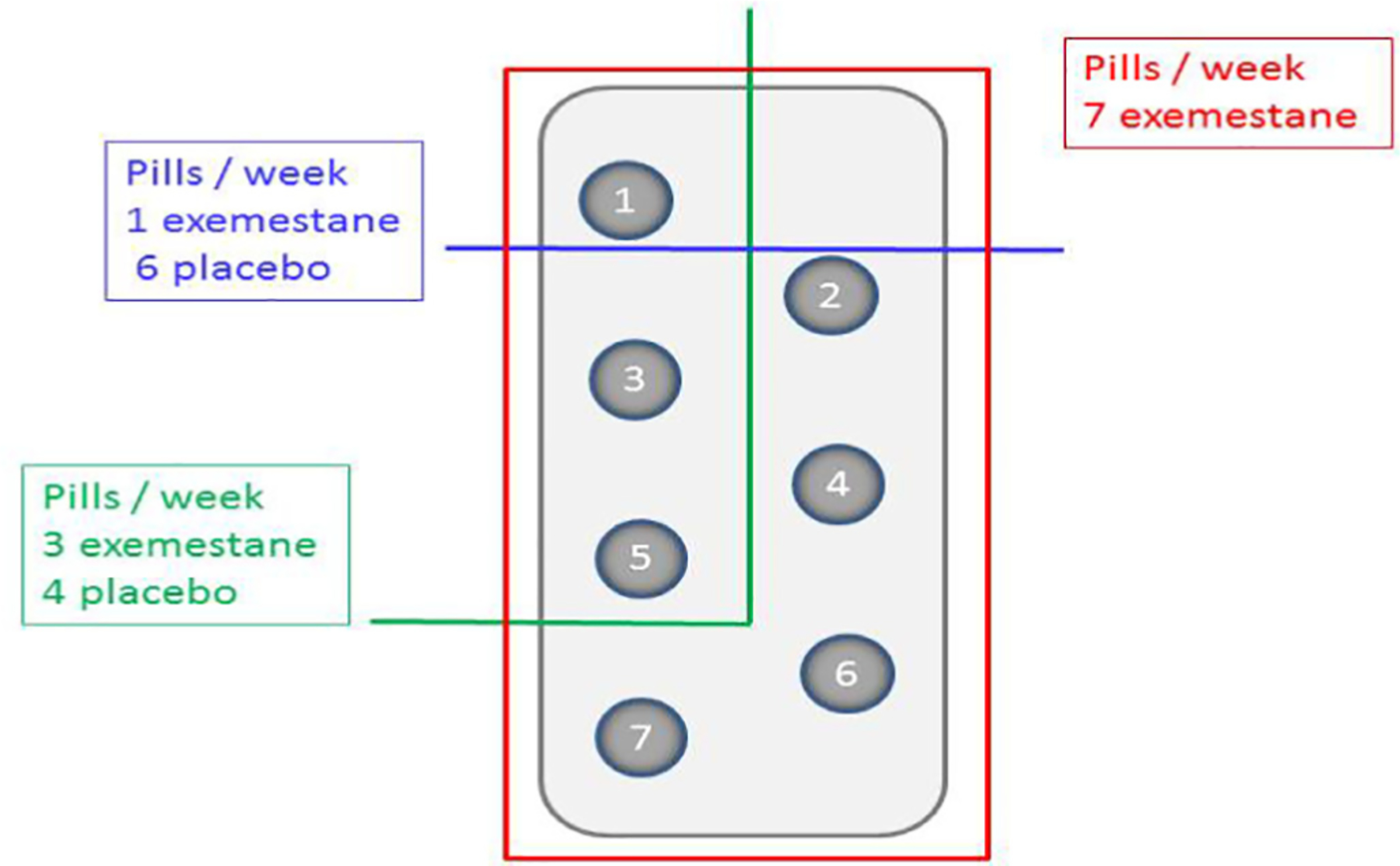
Example of 1 week blister for the three arms: cut 1, one active pill six placebos, cut 2, three active pills and four placebos, cut 3, seven active pills.
To be compliant, a participant had to have taken ≥80% of the active scheduled pills overall and also specifically in the last 7 days before surgery. Compliance has been measured using subject pill diary and tablets count. The main measure of compliance is the number of tablets taken (i.e. [number of tablets given minus number of tablets returned]) divided by the number of tablets that should have been taken during that period of time. Since the blister may have a mix of placebo and active pills, the position of any missed pill in each blister has been recorded to allow the unblinded statistician to determine the final compliance of active pills.
Each participant received a 42 day diary to facilitate tracking and recording of pill intake. Each participant was told to write Yes/No in the corresponding day of the diary and to register the time of drug intake. The diary had some additional space for participant’s notes (to mark symptoms or other clinical issues). Each pill diary has been collected at the final visit. In cases where the tablets were not returned, the compliance was calculated only from the pill diary and vice versa.
Due to the short treatment time and the evaluation of drug level as secondary endpoint we did not choose to use an electronic devise to monitor the compliance, even if it may improve adherence to the treatment.
2.6. Clinical evaluations and outcomes
Biological samples are serum and whole blood, collected at baseline and final visits and tissue sample collected from the diagnostic biopsy and the surgical specimen. Symptoms/toxicity and quality of life were also collected at baseline and final visits. The study procedures are summarized in Fig. 1.
The primary endpoint, i.e., the percent change in serum unconjugated estradiol concentration will be measured by high performance liquid chromatography tandem mass spectrometry (LC-MS/MS) detection using a solid-phase extraction followed by derivatization. Duplicate standard curves, water blanks, and four assay control pools will be processed with samples to assess accuracy and precision of the assay. The lower limit of quantification is 1.0 pg/mL. The low detection limit of this method will enable us to effectively detect estradiol concentrations at the very lowest levels, a characteristic of older postmenopausal women and in patients treated with AIs. Since lowering estradiol is the most direct action of the drug, this was chosen as primary endpoint.
2.7. Secondary endpoints
Exemestane safety and toxicity recorded at the clinic visit according to the Common Terminology Criteria for Adverse Events v4.0 (CTCAE). QoL with a focus on menopausal symptoms will be evaluated by a self-administered questionnaire (MENQOL) comparing pre- and post-treatment answers.
2.7.1. Circulating biomarkers
Serum drug measurement of exemestane and 17-dihydroxyexemestane, unconjugated and total estrone, androstenedione and testosterone will be measured by LC-MS/MS.
Sex hormone binding globulin (SHBG) serum levels will be measured by a chemiluminescent immunoassay designed for the IDS-iSYS Multi-Discipline Automated System (Immunodiagnostic Systems Limited, United Kingdom). Lower Limit of Quantitation is 0.30 nmol/L.
Insulin concentrations will be measured by a chemiluminescent microparticle immunoassay (CMIA) on the ARCHITECT i System (Abbott Laboratories, Weisbaden, Germany). Glucose will be measured at each local lab. Adipokines: change in leptin and adiponectin serum concentrations will be analyzed and compared among the different treatments arms. These measurements will be performed using an automated platform for immunoassays (ELLA system, ProteinSimple). Lipid profile (total cholesterol, HDL and triglycerides) will be determined locally at baseline and before surgery.
2.7.2. Pharmacogenetics
Exemestane and its major metabolite 17-dihydroexemestane are metabolized by UGT enzyme. UGT genetic variations play a role in altered 17-dihydroexemestane glucuronidation and overall exemestane metabolism. We have data from a pre-surgical study, consistent with these observations, showing a significant association of the UGT2B17 deletion with increased serum concentration of 17-dihydroexemestane [20]. To assess UGT genotype DNA will be extracted from whole blood EDTA treated samples (Qiagen, Italy). We will use Taqman copy number variation assay (Life Technologies, Monza, Italy) for the UGT2B17 genotyping. The expected minor allele frequency (UGT2B17 deletion) in the Caucasian population is 0.26.
2.7.3. Tissue biomarkers
Expression of ER, PgR, Her2 and Ki67 pre-treatment (biopsy) and post-treatment (surgical specimen) has been centralized to minimize the variability among different centers. Ki-67 is assessed by IHC according to recent international recommendations [21]. using the Mib-1 monoclonal antibody (1:50 dilution; Dako, Denmark), using an automated immunostainer (Dako) as previously reported [22]. Also ER, PgR (PharmDX) and Her2 (herceptest) expression will be determined by IHC.
Furthermore, on the surgical specimen it is planned to compare Ki-67 labeling index in adjacent intraepithelial neoplasia among the three arms.
Exemestane, and 17-dihydroxyexemestane concentration will be also analyzed on frozen tissue samples both in tumor and normal breast at time of surgery. Estradiol and estrone concentration will also be measured based on tissue sample availability.
2.8. Toxicity and dose modification
Participants have been asked to maintain the full dose throughout the treatment period. Due to the short time of treatment no dose modification has been applied. Toxicity has been evaluated using the NCI terminology criteria (CTCAE version 4.0.3, published 06/14/2010). For grade 1 or 2 toxicity the participant remains on treatment irrespective of attribution to the study drug. In case of grade 3 toxicity unrelated or unlikely related to study treatment, participant may remain on treatment as per physician judgment. Women who experience probable, possible or definite related to study treatment grade 3 or any grade 4 adverse events will be removed from the study. Overall as shown in Fig. 3 only two adverse events, possibly drug related and only one serious adverse event non related determined treatment interruption.
Fig. 3.
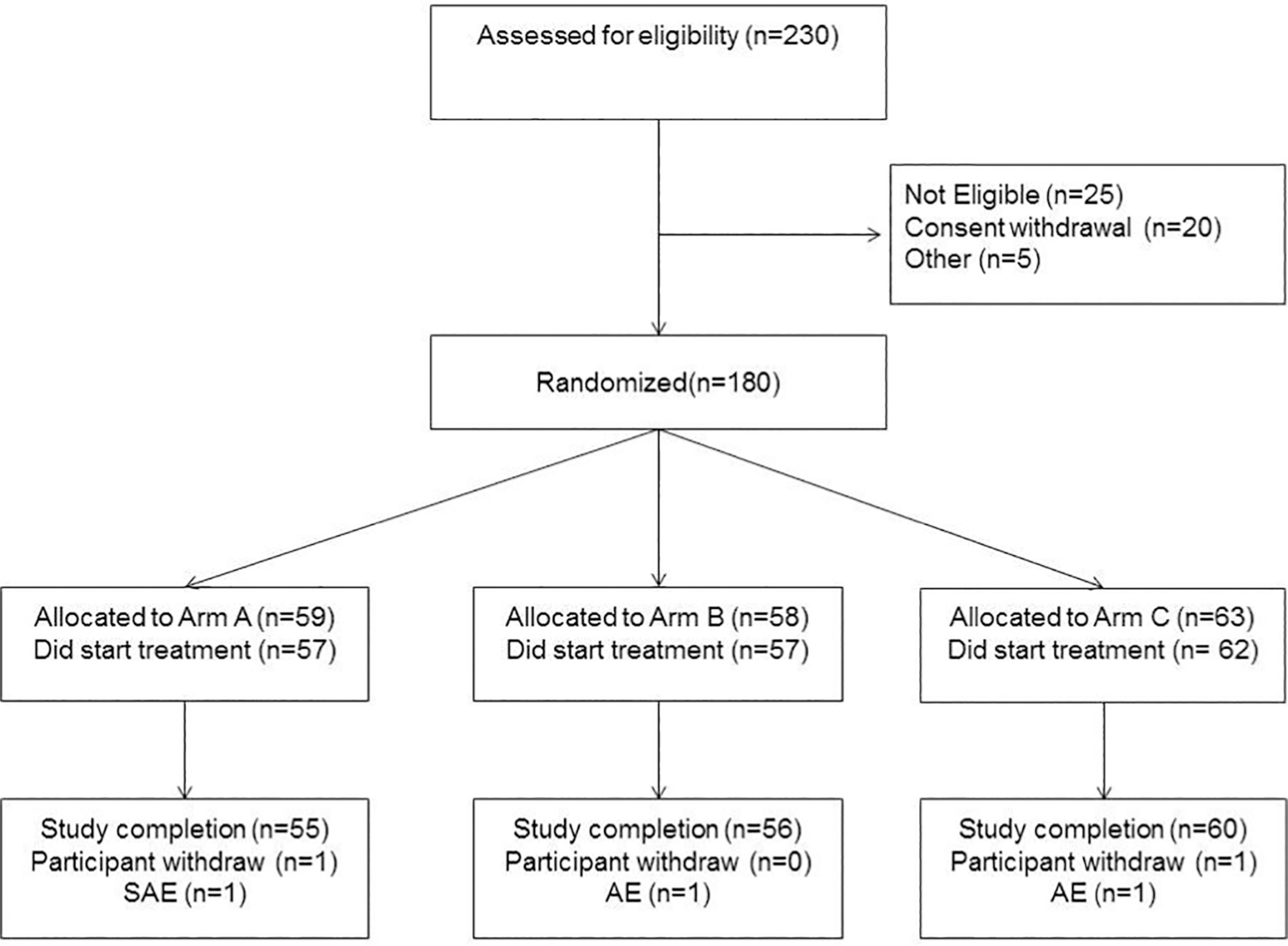
Consort statement.
2.9. Statistical consideration
Statistical analysis will be performed according to intention to treat principle and then a subgroup analysis will be restricted to women who are “compliant”, such as participants who have taken ≥80% of the active scheduled pills, and notably, due to the different arm schedule, ≥ 80% of the active pills has to be reached also in the last 7 days before surgery. The primary objective of this study is to assess if the reduction in unconjugated estradiol with the lower doses is comparable with the standard dose. Thus, the primary endpoint is the percentage change of serum estradiol concentration from baseline and we will compare the median change and percentage changes among arms. We choose the percentage change as the primary endpoint because it will improve generalization of results, given the wide variability among different subjects, and because we predict that a large percentage of participants will have the final values of estradiol below the detectable level.
Differences by arms of percentage change, absolute change and final values will be tested considering t-test and ANCOVA models. Unconjugated estradiol at baseline will be included as explanatory variable together with other possible confounders such as BMI, age and time since last dose. Normal distribution of residuals from full model will be checked and, if needed, a transformation will be considered. We will carry out the two comparisons with the standard dose considering alfa = 0.025.
Given the expected reduction of at least 80% change with exemestane at 25 mg daily [23] which is considered as reference group (control arm) against 25 mg 3 times a week, or 25 mg weekly, we assume a non-inferiority difference of 6% in percentage changes of estradiol after treatment from baseline, using a one-sided, two-sample t-test. A total sample size of 162 participants (54 participants per arm), enables us to achieve 80% power with a margin of equivalence of −6% in the mean percentage changes from baseline at the lower dose compared to the standard dose. The true difference between the means percentage changes is assumed to be 0.0%. The significance level of the test is 0.025 to take into account multiple comparisons (we will compare the changes at the two low doses with the standard dose) [24]. The data are drawn from populations with common standard deviations of 11% [25].
Assuming a 10% drop-out rate while on study, we need to randomize 180 participants (60 participants per arm) to achieve the total sample size of 162 evaluable participants (54 participants per arm).
Eligibility has been verified at each participant site, and confirmed by the PI’s site, electronic case report forms (CRFs) has been filled into a web database. Once eligibility has been verified and confirmed also by the Consortium Lead Organization (CLO) staff, the randomization number was generated by the database and assigned to the participant. In order to minimize the imbalance among arms, participants have been stratified according to center and BMI (<25 kg/m2 versus ≥25 kg/m2). BMI was chosen as possible variable for estrogen concentration, hence the exclusion of underweight women (BMI <18.5 kg/m2). For similar reason each center it was required to have at least 50% of women with BMI ≥ 25 kg/m2. A fixed block randomization has carried out with a block size of 6 patients.
Overall a full distributions and median values of primary and secondary endpoints will be presented at baseline, after treatment and also absolute changes and percentage changes (with interquartile ranges) of all continuous variable, by arms. ANCOVA models will evaluate the associations of post values (after treatment) and changes from baseline by study arms adjusting for baseline values and possible confounders (such as age and BMI). Normal distribution of residuals from full models will be checked and, if needed, a transformation will be considered. For binary variables, when we need to assess differences in frequencies, Chi-squares tests and Odds ratios (ORs) will be calculated. Multivariate logistic models will be considered to assess differences between arms, adjusting for possible confounders. We will report nominal P-values and we will highlight associations that meet a false discovery rate adjusted P less than or equal to 0.05 by the Benjamini and Hochberg method [26].
3. Preliminary results
Between February 2017 to August 2019 a total of 180 participants have been randomized: 59, 58, and 63 are the number of participant in each of the three treatment groups (Fig. 3 shows the Consort Statement). The study protocol required a strong coordination among the investigators to respect the day of surgery to keep the proper timelapse from the last active pill based on treatment arm. Surgery was preferably scheduled on day 29. If, for any reasons, that day was not possible to perform the intervention, the surgery had to be moved to the following weeks (on day 36 or 43).
Overall four subjects did not start the treatment, an additional five dropped out of the study, two for personal reasons, two for adverse events and one for a serious adverse event not related to study treatment. This trial was conducted in compliance with the protocol, Good Clinical Practice (GCP), and the applicable regulatory requirements. The study is still blinded; primary and secondary endpoints are under analysis.
4. Conclusions
Cancer incidence, morbidity, and mortality have a dramatic impact on public health worldwide; and the increasing number of patients and treatment costs will reach an unaffordable level in the near future [27]. For these reasons the development and promotion of more effective cancer prevention programs are needed.
Awareness and education on cancer prevention should be implemented at all levels, from health care professionals to the general population. Risk perception, information and counselling are crucial elements to be discussed between physician and at-risk individual, considering that personal preferences and values have to be strongly supported in the field of prevention, [28].
The individual risk level may lead to the different breast cancer risk reduction options, including life-style changes, preventive medicine, and prophylactic surgery, in case of hereditary cancer syndromes.
In breast cancer preventive medicine we have obtained remarkable results in terms of feasibility and drug acceptability by lowering tamoxifen dose in healthy at-risk women and in patients with intraepithelial neoplasia [8,9]. In our recent publications we have shown that there is no difference in serious adverse events using 5 mg per day of tamoxifen compared to placebo [9]. The low toxicity profile achieved with low-dose tamoxifen showed the same compliance compared to placebo whereas the adherence to treatment is lower with 20md/day standard dose [29].
The present study aims to investigate the biological activity of two alternative exemestane schedules compared to the standard daily dose. Secondary endpoints, particularly the Ki67 evaluation, may provide an important sign of evidence of clinical efficacy. Whereas, due to the short time of treatment, the evaluation of toxicity/QoL will only assess short-term effects. Drug efficacy will be also evaluated according to BMI and other participants baseline characteristics.
If a significant estradiol reduction will be confirmed with these doses/schedules, further trials to investigate directly the clinical activity and QoL should be developed. The final goal is to achieve new strategies for cancer prevention and treatment. The target population can be postmenopausal women either healthy at increased risk or with a previous breast intraepithelial neoplasia diagnosis. A lighter drug schedule and better QoL may improve the acceptability and the adherence to the treatment. We aim to pursue further phase II and III trials to validate a low-dose/intermittent exemestane, ideally versus standard dose or eventually low dose tamoxifen based on our previous study [9].
Acknowledgements
Mark Sherman MD, former Medical Monitor of the study.
Jemos Costantino PharmD IEO pharmacist.
Local study coordinators.
Funding
NCI Contract HHSN26120120034I.
Partially supported by: Italian Ministry of Health with “Ricerca Corrente” and 5xmille funds.
Abbreviations:
- AE
Adverse Event
- AI
Aromatase Inhibitor
- BMI
Body Mass Index
- DCIS
Ductal Carcinoma in Situ
- IHC
Immunohistochemistry
- LCIS
Lobular Carcinoma in Situ
- QoL
Quality of Life
Footnotes
Declaration of Competing Interest
The authors declare that they have no competing interests.
References
- [1].Yerushalmi R, Woods R, Ravdin PM, Hayes MM, Gelmon KA, Ki67 in breast cancer: prognostic and predictive potential, Lancet Oncol. 11 (2010) 174–183. [DOI] [PubMed] [Google Scholar]
- [2].Decensi A, Puntoni M, Pruneri G, Guerrieri-Gonzaga A, Lazzeroni M, Serrano D, et al. , Lapatinib activity in premalignant lesions and HER-2-positive cancer of the breast in a randomized, placebo-controlled presurgical trial, Cancer Prev. Res. (Phila.) 4 (2011) 1181–1189. [DOI] [PubMed] [Google Scholar]
- [3].Decensi A, Puntoni M, Guerrieri-Gonzaga A, Cazzaniga M, Serrano D, Lazzeroni M, et al. , Effect of metformin on breast ductal carcinoma in situ proliferation in a randomized presurgical trial, Cancer Prev. Res. (Phila.) 8 (2015) 888–894. [DOI] [PubMed] [Google Scholar]
- [4].Waters EA, Cronin KA, Graubard BI, Han PK, Freedman AN, Prevalence of tamoxifen use for breast cancer chemoprevention among U.S. women, Cancer Epidemiol. Biomark. Prev. 19 (2010) 443–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Decensi A, Robertson C, Guerrieri-Gonzaga A, Serrano D, Cazzaniga M, Mora S, et al. , Randomized double-blind 2 × 2 trial of low-dose tamoxifen and fenretinide for breast cancer prevention in high-risk premenopausal women, J. Clin. Oncol. 27 (2009) 3749–3756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Bonanni B, Serrano D, Gandini S, Guerrieri-Gonzaga A, Johansson H, Macis D, et al. , Randomized biomarker trial of anastrozole or low-dose tamoxifen or their combination in subjects with breast intraepithelial neoplasia, Clin. Cancer Res. 15 (2009) 7053–7060. [DOI] [PubMed] [Google Scholar]
- [7].Decensi A, Gandini S, Serrano D, Cazzaniga M, Pizzamiglio M, Maffini F, et al. , Randomized dose-ranging trial of tamoxifen at low doses in hormone replacement therapy users, J. Clin. Oncol. 25 (2007) 4201–4209. [DOI] [PubMed] [Google Scholar]
- [8].Decensi A, Bonanni B, Maisonneuve P, Serrano D, Omodei U, Varricchio C, et al. , A phase-III prevention trial of low-dose tamoxifen in postmenopausal hormone replacement therapy users: the HOT study, Ann. Oncol. 24 (2013) 2753–2760. [DOI] [PubMed] [Google Scholar]
- [9].Decensi A, Puntoni M, Guerrieri-Gonzaga A, Caviglia S, Avino F, Cortesi L, et al. , Randomized placebo controlled trial of low-dose tamoxifen to prevent local and contralateral recurrence in breast intraepithelial neoplasia, J. Clin. Oncol. 37 (2019) 1629–1637. JCO1801779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Goss PE, Ingle JN, es-Martinez JE, Cheung AM, Chlebowski RT, Wactawski-Wende J, et al. , Exemestane for breast-cancer prevention in postmenopausal women, N. Engl. J. Med. 364 (2011) 2381–2391. [DOI] [PubMed] [Google Scholar]
- [11].Cuzick J, Sestak I, Forbes JF, Dowsett M, Knox J, Cawthorn S, et al. , Anastrozole for prevention of breast cancer in high-risk postmenopausal women (IBIS-II): an international, double-blind, randomised placebo-controlled trial, Lancet 383 (2014) 1041–1048. [DOI] [PubMed] [Google Scholar]
- [12].Partridge AH, LaFountain A, Mayer E, Taylor BS, Winer E, snis-Alibozek A, Adherence to initial adjuvant anastrozole therapy among women with early-stage breast cancer, J. Clin. Oncol. 26 (2008) 556–562. [DOI] [PubMed] [Google Scholar]
- [13].Meggetto O, Maunsell E, Chlebowski R, Goss P, Tu D, Richardson H, Factors associated with early discontinuation of study treatment in the mammary prevention. 3 Breast cancer chemoprevention trial, J. Clin. Oncol. 35 (2017) 629–635. [DOI] [PubMed] [Google Scholar]
- [14].Lòpez AM, Pruthi S, Boughey JC, Perloff M, Hsu CH, Lang JE, et al. , Double-blind, randomized trial of alternative letrozole dosing regimens in postmenopausal women with increased breast cancer risk, Cancer Prev. Res. (Phila.) 9 (2016) 142–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Colleoni M, Luo W, Karlsson P, Chirgwin J, Aebi S, Jerusalem G, et al. , Extended adjuvant intermittent letrozole versus continuous letrozole in postmenopausal women with breast cancer (SOLE): a multicentre, open-label, randomised, phase 3 trial, Lancet Oncol. 19 (2018) 127–138. [DOI] [PubMed] [Google Scholar]
- [16].Evans TR, Di SE, Ornati G, Lassus M, Benedetti MS, Pianezzola E, et al. , Phase I and endocrine study of exemestane (FCE 24304), a new aromatase inhibitor, in postmenopausal women, Cancer Res. 52 (1992) 5933–5939. [PubMed] [Google Scholar]
- [17].Lonning P, Pfister C, Martoni A, Zamagni C, Pharmacokinetics of third-generation aromatase inhibitors, Semin. Oncol. 30 (2003) 23–32. [DOI] [PubMed] [Google Scholar]
- [18].Johannessen DC, Engan T, Di SE, Zurlo MG, Paolini J, Ornati G, et al. , Endocrine and clinical effects of exemestane (PNU 155971), a novel steroidal aromatase inhibitor, in postmenopausal breast cancer patients: a phase I study, Clin. Cancer Res. 3 (1997) 1101–1108. [PubMed] [Google Scholar]
- [19].Hilditch JR, Lewis J, Peter A, van Maris B, Ross A, Franssen E, et al. , A menopause-specific quality of life questionnaire: development and psychometric properties, Maturitas 24 (1996) 161–175. [DOI] [PubMed] [Google Scholar]
- [20].Johansson H, Aristarco V, Gandini S, Gjerde J, Macis D, Guerrieri-Gonzaga A, et al. , Prognostic impact of genetic variants of CYP19A1 and UGT2B17 in a randomized trial for endocrine-responsive postmenopausal breast cancer, Pharmacogenom. J 20 (2019) 19–26. [DOI] [PubMed] [Google Scholar]
- [21].Dowsett M, Nielsen TO, A’Hern R, Bartlett J, Coombes RC, Cuzick J, et al. , Assessment of Ki67 in breast cancer: recommendations from the international Ki67 in breast cancer working group, J. Natl. Cancer Inst. 103 (2011) 1656–1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Viale G, Giobbie-Hurder A, Regan MM, Coates AS, Mastropasqua MG, Dell’Orto P, et al. , Prognostic and predictive value of centrally reviewed Ki-67 labeling index in postmenopausal women with endocrine-responsive breast cancer: results from Breast International Group Trial 1–98 comparing adjuvant tamoxifen with letrozole, J. Clin. Oncol. 26 (2008) 5569–5575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Geisler J, King N, Anker G, Ornati G, Di SE, Lonning PE, et al. , In vivo inhibition of aromatization by exemestane, a novel irreversible aromatase inhibitor, in postmenopausal breast cancer patients, Clin. Cancer Res. 4 (1998) 2089–2093. [PubMed] [Google Scholar]
- [24].Chow SC, Shao J, Wang H, Sample Size Calculations in Clinical Research, Marcel Dekker, New York, 2003. [Google Scholar]
- [25].Hus JC, Multiple Comparisons: Theory and Methods, London, 1996. [Google Scholar]
- [26].Benjamini Y, Hochberg Y, Controlling the false discovery rates: a practical and powerful approach to multiple testing, J. R. Stat. Soc. B 35 (1995) 289–300. [Google Scholar]
- [27].Bray F, Jemal A, Torre LA, Forman D, Vineis P, Long-term realism and cost-effectiveness: primary prevention in combatting cancer and associated inequalities worldwide, J. Natl. Cancer Inst. 107 (2015), djv273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Miller FG, Colloca L, The placebo phenomenon and medical ethics: rethinking the relationship between informed consent and risk-benefit assessment, Theor. Med. Bioeth. 32 (2011) 229–243. [DOI] [PubMed] [Google Scholar]
- [29].Smith SG, Sestak I, Howell A, Forbes J, Cuzick J, Participant-reported symptoms and their effect on long-term adherence in the international breast cancer intervention study I (IBIS I), J. Clin. Oncol. 35 (2017) 2666–2673. [DOI] [PMC free article] [PubMed] [Google Scholar]