

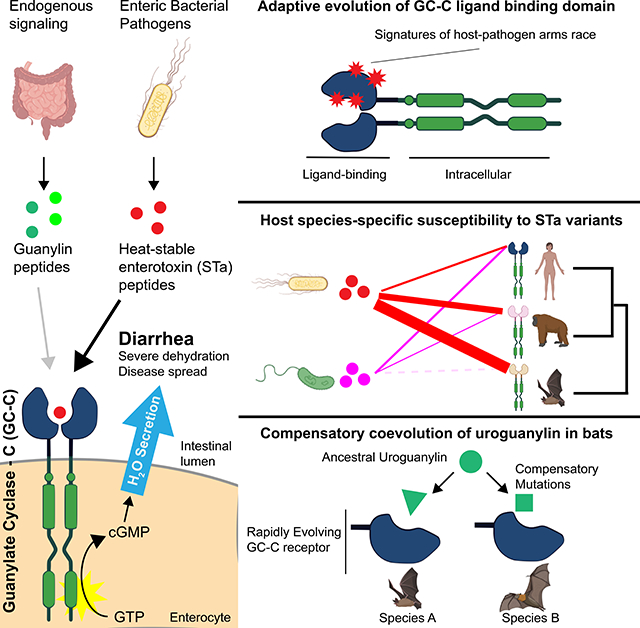
SUMMARY
The pathogenesis of infectious diarrheal diseases is attributed to enterotoxins that cause dehydration by disrupting intestinal water absorption. We investigated patterns of genetic variation in mammalian Guanylate Cyclase-C (GC-C), an intestinal receptor targeted by bacterially-encoded heat-stable enterotoxins (STa), to determine how host species adapt in response to diarrheal infections. Our phylogenetic and functional analysis of GC-C supports longstanding evolutionary conflict with diarrheal bacteria in primates and bats, with highly variable susceptibility to STa across species. In bats, we further show that GC-C diversification has sparked compensatory mutations in the endogenous uroguanylin ligand, suggesting an unusual scenario of pathogen-driven evolution of an entire signaling axis. Together these findings suggest that conflicts with diarrheal pathogens have had far-reaching impacts on the evolution of mammalian gut physiology.
eTOC blurb
Studies of evolutionary arms-races have primarily focused on interactions between host immune factors and infectious microbes. Carey et al. describe an unusual genetic conflict between diarrheal bacterial pathogens and a core intestinal signaling axis critical for maintaining proper gut physiology, broadening conceptions of the reach of pathogen-driven evolution.
Graphical Abstract
INTRODUCTION
Symptoms of diarrhea enhance the ability of enteric pathogens to multiply and facilitates transmission to new hosts (Viswanathan et al., 2009). These pathogens directly promote the symptoms of diarrhea by secreting enterotoxin proteins that disrupt the normal physiological state of the intestine. Enterotoxins often act by manipulating host ion transport proteins in intestinal enterocytes to induce water secretion into the lumen of the gut, resulting in watery diarrhea. Enterotoxins primarily benefit the pathogen by increasing its dissemination to new hosts, but can also increase the availability of nutrients in the intestine enabling enhanced proliferation of these microbes (Nelson et al., 2009; Rivera-Chávez and Mekalanos, 2019). Dehydration resulting from the watery diarrhea caused by host-enterotoxin interactions represents a significant cause of mortality and morbidity in humans, accounting for over one million deaths annually (Troeger et al., 2018). Despite this widespread health impact, it is unknown to what extent mammals have adapted to counteract threats posed by infectious diarrheal microbes.
The receptor Guanylate Cyclase-C (GC-C) is a master regulator of intestinal water balance and a frequent target of enterotoxins encoded by bacterial pathogens. Located on the membranes of enterocytes lining the small intestine and colon, GC-C signaling fine-tunes the luminal water content of the intestine (Basu et al., 2010). GC-C is activated by the peptide hormones guanylin and uroguanylin, which bind to its extracellular domain to activate cGMP production by the intracellular catalytic domain (Figure 1A). Elevated enterocyte cGMP levels promote an osmotic shift that proceeds via two main mechanisms. First, cGMP production leads to the activation of the cystic fibrosis transmembrane conductance regulator (CFTR), which pumps chloride into the intestinal lumen. Second, elevated cGMP levels promote the accumulation of sodium in the lumen through inhibition of the sodium importer NHE3. Increased lumenal ion concentrations create an osmotic gradient that leads to secretion of water from enterocytes. Secretion of GC-C-activating guanylin hormones is controlled by feeding and circadian rhythms to promote normal digestive function and is stringently regulated to prevent excessive loss of water (Forte, 2004).
Figure 1: Rapid evolution of the heat-stable enterotoxin receptor in primates and bats.

(A) Amino acid sequences of STa peptides encoded by four human pathogens are shown in comparison to human guanylin and uroguanylin. Disulfide bonds are indicated with connecting lines (top). Schematic of GC-C signaling in intestinal enterocytes. Activation of cGMP synthesis via ligand binding to GC-C results in chloride secretion through CFTR and inhibition of sodium transport via NHE3. Resulting shifts in ion balance facilitate water secretion into the intestinal lumen. Prolonged activation by STa causes excess water secretion and diarrhea (bottom). (B) Evolutionary analysis of GC-C in primates and bats. Phylogenetic trees indicate patterns of rapid evolution in primates (left) and bats (right). Branches with dN/dS >1 are highlighted in blue (primates) or magenta (bats) Codons with evidence of positive selection are indicated on the GC-C primary structure with blue (primates) or magenta (bats) triangles (PAML posterior probability >95%).
GC-C signaling is hijacked by several bacterial pathogens that secrete heat-stable enterotoxins (STa). These toxin peptides mimic sequence and structural features of guanylins and bind with high affinity to GC-C, causing prolonged cGMP production that results in secretory diarrhea (Weiglmeier et al., 2010). Despite their short sequence length, there is considerable diversity in the sequence of STa between bacterial strains. Enterotoxigenic E. coli (ETEC) are among the most common agent of infectious diarrhea in humans, with most strains expressing a combination of STa variants termed STp and STh (Fleckenstein et al., 2010). Pathogens from other genera are also known to produce STa peptides with unique amino acid sequences, including Vibrio (V-ST) and Yersinia (Y-ST) species (Takao et al., 1985a, 1985b). STa producing pathogens have been shown to cause disease in humans and livestock but are also isolated from wild animals. Despite the considerable diversity in the sequence of STa, it is unknown whether these changes represent adaptations to different host GC-C receptors.
Given the central role of STa-GC-C interactions in the pathogenesis of many diarrheal infections, we hypothesized that GC-C might adapt resistance to STa variants as part of an ongoing evolutionary conflict with diarrheal bacteria. Traditional host-pathogen evolutionary conflicts are often characterized as ‘arms-race’ scenarios between coevolving hosts and obligate parasites. Enteric bacterial pathogens like ETEC, however, are able to inhabit environmental niches and can transmit virulence genes, including STa, horizontally (Gonzales-Siles and Sjöling, 2016). These lifestyle characteristics potentially change the dynamics of host-pathogen conflicts, where hosts might face pressure to adapt to effector variants shared among pathogenic strains, rather than any particular microbe. Previous work has detailed conflicts between the iron-sequestering protein transferrin and bacterial iron scavenging factors, demonstrating how bacterial infections can shape host evolution (Barber and Elde, 2014). However, whether bacterial pathogens can similarly drive adaptations in genes like GC-C that mediate organ-level physiology has not been explored.
In this report, we show that the GC-C ligand-binding domain bears signatures of adaptive evolution in primates and bats, consistent with selective pressures imposed by interactions with STa. Cellular and physiological assays further revealed species-specific patterns of STa susceptibility, with some bat species encoding GC-C receptors that are highly resistant to specific toxin variants. Remarkably, we find that the endogenous uroguanylin peptide is highly divergent among bats, which supports a unique model of pathogen-driven compensatory coevolution within this signaling axis. These findings highlight the evolutionary impact of diarrheal disease on host species and show how pathogens can alter the evolution of core physiological processes.
RESULTS
Rapid evolution of the heat-stable enterotoxin receptor in primates and bats
To determine whether GC-C evolved under selective pressure to modulate interactions with enterotoxins, we first collected the sequences of GC-C orthologs from 19 primate genomes for phylogenetic analysis (Table S1). Using statistical models that compare ratios of nonsynonymous to synonymous mutations (dN/dS) across species (Murrell et al., 2015; Yang, 2007), we find strong evidence of recurrent positive selection in primate GC-C (Table S2). Similar patterns of abundant nonsynonymous substitutions are widely reported for genes encoding dedicated immune functions that are targeted by pathogen factors (Daugherty and Malik, 2012). This suggests that GC-C may similarly be subject to recurrent selection based on temporary advantages conferred by mitigating pathogen interactions. Unlike dedicated immune proteins, however, GC-C is functionally constrained by its physiological function of regulating water secretion through interactions with endogenous ligands. Indeed, the GC-C signaling axis is found in all vertebrates (Forte, 2004), a pattern associated with functional conservation. To better understand the role of toxin interactions in GC-C evolution, we compared sequence evolution in the paralogous natriuretic peptide receptors (NPR1–2), which share similar overall structure and function with GC-C. NPR1 and NPR2 are not known to interact with pathogen-derived toxins (Potter et al., 2006), and in contrast to GC-C, bear signatures of purifying selection with highly conserved sequences across primates (Tables S1,S2). Rapid evolution of GC-C is therefore associated with its unique interaction with pathogens.
To further examine how GC-C evolution in primates might be influenced by toxin interactions, we mapped rapidly evolving codons with significantly elevated dN/dS ratios to the primary structure of GC-C. Strikingly, all six positively-selected sites mapped to the extracellular ligand-binding domain of GC-C, whereas the intracellular domain is highly conserved (Figure 1B, Table S3). These patterns of diversification of both STa and the GC-C ligand-binding domain support a model of recurrent conflicts between diarrhea-causing bacteria and hosts over control of water secretion in the intestine.
In addition to primates, STa toxicity has been demonstrated in other mammalian species including rodents and livestock (Weiglmeier et al., 2010). To test whether GC-C might be subject to recurrent positive selection in other mammalian lineages, we collected sequences of GC-C from four additional groups of mammals including Bovidae (cloven-hooved ruminants), Caniformia (suborder of dog-like carnivores), Myomorpha (mouse-like rodents), and Chiroptera (bats) (Table S1). In contrast to primates, substitution patterns among Caniformia and Bovidae are consistent with purifying selection, while signals of positive selection are supported in only a subset of tests among Myomorpha GC-C (Table S2).
Patterns of positive selection were remarkably widespread in bats and strongly supported by all phylogenetic tests (Table S2). Examination of bat GC-C sequences revealed 35 codon positions with significantly elevated dN/dS values mapping exclusively to the GC-C ligand-binding domain (Figure 1B, Table S3). Sequence comparisons of bat GC-C and NPR2 ligand-binding domains revealed extensive diversification in GC-C relative to this closely related paralog (Figure S1A). Although the structural basis of the interaction between GC-C and STa has not yet been determined, a putative ligand-binding pocket has been proposed in a membrane-proximal region between residues 411–416 (Wada et al., 1996). A cluster of seven sites with elevated dN/dS values among bats is in close proximity to this region, in addition to one site identified in our analysis of GC-C in primates (Figure S1B).
Bats are known to harbor many types of viruses, including several human pathogens (Calisher et al., 2006). This widespread association with viruses is attributed to high density population structures and the ability to cover large geographic areas with long-distance flight (Brook and Dobson, 2015). Notably, frequent infection with coronaviruses likely sparked high levels of sequence diversification in the viral entry receptor ACE-2 among bats (Demogines et al., 2012), illustrating the intensity of selection pathogens place on these species. In addition to viruses, bats can carry bacteria that act as human pathogens, including genera that encode STa (Mühldorfer, 2013). Furthermore, STa-like genes have been detected in DNA isolated from the feces of wild bats (Banskar et al., 2016). Our detection of signals of intense positive selection in the GC-C ligand-binding domain of bats suggests that diarrheal pathogens may have profoundly impacted the evolution of bats, similar to their established coevolutionary relationship with viruses (Figure 1B).
Species-specific patterns of STa susceptibility in primates and bats
To test if rapid evolution of GC-C ligand-binding domains result in functional differences in STa susceptibility, we generated cell lines stably expressing GC-C variants from seven primate and five bat species. HEK293T cells were used for GC-C assays as they do not normally produce cGMP in response to STa stimulation (Figure S2A). Functional diversity in GC-C variants was assessed by measuring intracellular cGMP production in response to chemically synthesized uroguanylin and STa variants from four strains of pathogenic bacteria. Cells expressing human, chimpanzee, gorilla, gibbon and African green monkey GC-C generated similar levels of cGMP upon stimulation with STa variants from pathogenic E. coli (STp and STh), Yersinia enterocolitica (Y-ST), and Vibrio mimicus (V-ST) (Figure 2A, S2B-D). In contrast, cells expressing GC-C from orangutan and rhesus macaque were significantly more susceptible to STp, showing a large increase in cGMP production relative to other species (Figure 2A). We further characterized human and orangutan GC-C by generating dose-response curves for each toxin—measuring cGMP production across a range of toxin concentrations. Human GC-C responded similarly to each of the four toxins and reached maximal activation levels that were not statistically different (Figure 2B, Table S4). In contrast, orangutan GC-C displayed significantly higher maximal activation by STp and STh compared to the other toxins, suggesting an increased relative susceptibility to these toxin variants (Figure 2C, Table S4). Thus, variation in GC-C among primates results in differing levels of susceptibility to diverse toxins, providing evidence that ancient STa-like peptides influenced the course of GC-C evolution.
Figure 2: Species-specific patterns of STa susceptibility in primates and bats.

(A and D) (Top) cGMP levels in HEK293T cells stably expressing GC-C from seven primate species (A) and five bat species (D) after treatment with 10 μM of the indicated peptide.
* indicates a p value < 0.05 using one-way ANOVA followed by Dunnett’s T3 multiple comparison test. Means from toxin treatments in non-human primate GC-C cells were compared to the cGMP response for the same peptide in human GC-C cells (panel A) or between toxin treatments in the bat GC-C cells (panel D). (Bottom) Immunoblot of HA-tagged GC-C from indicated cell lines. (B, C, E, F) Intracellular cGMP dose-response curves for the indicated ligands are shown in HEK293T cells expressing human (B), orangutan (C), P. vampyrus (E), and M. lucifugus (F) GC-C.
Differences in toxin susceptibility were even more pronounced among GC-C encoded by bats. We cloned GC-C from representatives of the vesper bats: Myotis lucifugus (little brown bat), Eptesicus fuscus (big brown bat), and Miniopterus natalensis (Natal long-fingered bat), as well as Old World fruit bats: Pteropus vampyrus (large flying fox) and Pteropus alecto (black flying fox) for expression in cell lines. This sampling captures roughly a 50 million year interval of divergence from the common ancestor of chiropteran bats (Agnarsson et al., 2011). We discovered a wide range of susceptibility to STa variants across bat species. GC-C from the vesper bats E. fuscus and M. natalensis responded similarly to all four toxins and produced modest levels of cGMP comparable to human GC-C. In contrast, cells expressing M. lucifugus, P. vampyrus, and P. alecto GC-C produced nearly 10-fold more cGMP in response to most toxin treatments when compared to human GC-C (Figure 2D-F). Intriguingly, both P. vampyrus and P. alecto failed to respond to treatment with 10 μM Y-ST in notable contrast to GC-C encoded by M. lucifugus (Figure 2D). Additional comparisons across a wide range of toxin concentrations revealed that P. vampyrus GC-C is only activated by Y-ST stimulation at concentrations exceeding 10 μM, well outside probable concentrations encountered during infection (Figure 2E)(Thompson et al., 1984). Thus, susceptibility to STa variants differs widely among bats with some species displaying heightened sensitivity compared to human and others resistant to activation at physiologically plausible toxin concentrations.
Intestinal organoids model physiologic outcomes of differential toxin susceptibility
We next sought to more directly measure the physiological significance of the differing toxin susceptibility we observed in our cGMP-generation assays. Cultured organoids composed of intestinal enterocytes have recently emerged as a means to directly assay symptoms of secretory diarrhea caused by STa (Pattison et al., 2016). In these cultured intestinal spheroids, GC-C mediated water secretion induces organoid swelling upon ligand stimulation, directly modeling symptoms of diarrhea. We cultured organoids derived from the small intestine of GC-C−/− mice to investigate the relationship between cGMP production and water secretion across species. We generated and compared organoids expressing GC-C from humans and the fruit bat P. vampyrus, which displayed heightened susceptibility to STp but marked resistance to Y-ST in our cell-based assays. We transduced GC-C−/− organoids with lentiviral particles encoding human or P. vampyrus GC-C conjugated to GFP via a T2A peptide and found comparable levels of GFP fluorescence in our transgenic lines, indicating similar levels of GC-C expression (Figure 3A). Organoids expressing P. vampyrus GC-C displayed considerably increased water secretion during STp treatment as measured by the total change in organoid area compared to organoids expressing human GC-C (Figure 3B, 3C). Consistent with our cGMP assays, treatment with Y-ST resulted in minimal swelling of organoids expressing P. vampyrus GC-C compared to those carrying the human variant (Figure 3D, 3E). Thus, differences in susceptibility to STa variants likely have a direct influence on the level of water secretion and symptoms of diarrhea experienced by host species during infection.
Figure 3: Intestinal organoids model physiology of toxin susceptibility.

(A) Intestinal organoids were cultured from GC-C−/− mice and transduced with lentivirus encoding GC-C variants conjugated to a cleaved GFP reporter. Mean fluorescence was calculated from individual organoids using ImageJ. *** indicates a p value < 0.0001 using one-way ANOVA followed by Dunnett’s T3 multiple comparison test in each panel. (B) Representative images are shown of individual organoids expressing GC-C from the indicated species before and after 120 min of treatment with 5 μM STp. (C) Change in individual organoid area was quantified following 120 min of STp treatment for organoids expressing GC-C from the indicated species. (D) Representative images of individual organoids expressing GC-C from the indicated species before and after 120 min of treatment with 5 μM Y-ST. (E) Change in individual organoid area was quantified following 120 min of Y-ST treatment for organoids expressing GC-C from the indicated species. Scale bars represent 50 μm.
Compensatory co-evolution of GC-C and uroguanylin in bats
In order to effectively regulate intestinal water levels, GC-C must interact with endogenous guanylin and uroguanylin peptides (Forte, 2004). To determine how rapid divergence of GC-C might influence the evolution of and interactions with its cognate ligands, we next examined sequence diversity in uroguanylin, the more potent of the two peptides (Kita et al., 1994). Sequence comparisons revealed that secreted uroguanylin peptides are generally highly conserved across mammals with little variation occurring outside the most N- and C- terminal residues (Figure S3A). In bats, however, uroguanylin sequences are highly variable, with frequent mutations occurring in core residues of the peptide (Figure 4A). Given the variability in sequence and toxin interactions we observe in bat GC-C, we hypothesized that sequence variation in bat uroguanylin might reflect compensatory mutations required to maintain affinity for its rapidly evolving receptor.
Figure 4: Coevolution of GC-C and uroguanylin in bats.

(A) Amino acid alignment of the mature uroguanylin peptide based on genome sequences of the indicated bat (top) or primate (bottom) species. Residues with mutations relative to the consensus within each clade are uniquely colored. Elipses represent stop codon loss mutations in the indicated bat species. (B) (Top) cGMP levels in HEK293T cells expressing P. vampyrus or M. lucifugus GC-C after treatment with 10 μM uroguanylin from the indicated species. * indicates a p-value < 0.05 and ** indicates a p-value<0.01 using one-way ANOVA followed by Dunnett’s T3 multiple comparison test. (Bottom) immunoblot of HA-tagged GC-C from the indicated cell lines. (C) Model for pathogen driven evolution of uroguanylin in bats. Mutations in GC-C that result in a loss of affinity for both STa and uroguanylin may provide a net fitness benefit, allowing time for compensatory mutations to arise.
To test if uroguanylin co-evolved with GC-C in bats, we synthesized active uroguanylin peptides encoded by the vesper bats M. lucifugus and E. fuscus, as well as the more distantly related P. vampyrus (Figure 4A, S3B-C). Treatment of cells expressing P. vampyrus GC-C with uroguanylin from M. lucifugus did not stimulate cGMP production, whereas treatment with P. vampyrus uroguanylin robustly activates catalytic activity (Figure 4C). Conversely, M. lucifugus GC-C responds more strongly to its species-matched uroguanylin than to the P. vampyrus peptide (Figure 4B). Intriguingly, E. fuscus GC-C responds weakly to all uroguanylin variants tested, including its own peptide (Figure 4B). These experiments are consistent with compensatory co-evolution of GC-C and uroguanylin in bats, and also reveal that some species may exist in intermediate states with sub-optimal affinity for uroguanylin.
Given functionally consequential variation in GC-C that impacts both enterotoxin and uroguanylin interactions, we propose a model of compensatory coevolution triggered by bacterial enterotoxin interactions (Figure 4C). In this model, mutations in GC-C that allow escape from overstimulation by STa can provide a fitness benefit even at the cost of disrupting the interaction with uroguanylin. This intermediate state of low affinity with endogenous peptides is tolerated while toxin-susceptible variants are culled from the population, given that survival is possible when GC-C signaling is disrupted (Schulz et al., 1997). Subsequent compensatory mutations in uroguanylin that optimize signaling interactions with GC-C might then outcompete mismatched variants with compromised receptor-ligand interfaces. In this proposed scenario, a single pathogen encoded protein directly influences the evolution of the host receptor, which subsequently impacts variation in the endogenous ligand.
DISCUSSION
Since the beginnings of recorded history, infectious diarrheal diseases have plagued human populations (Barua, 1992). Outbreaks of Vibrio cholera alone account for millions of deaths, inflicted by the effects of its namesake enterotoxin. Cholera toxin, like heat-stable enterotoxins, induces secretory diarrhea by activating CFTR but uses a distinct mechanism avoiding direct interactions with host proteins (Lencer and Tsai, 2003). The discovery of CFTR as a shared target of multiple bacterial toxins led to a longstanding, though controversial, hypothesis that the high prevalence of CFTR null alleles in human populations might be explained by a heterozygote advantage for better tolerating diarrheal infections (Mowat, 2017). Under this model, balancing selection imposed by diarrheal microbes results in the maintenance of CFTR null alleles among some populations despite causing cystic fibrosis in homozygous individuals. Beyond this one hypothesis, the impact of diarrheal infections on host evolution has remained largely unexplored. This study reporting rapid diversification of the GC-C signaling pathway, which involves CFTR activation, lends support to the idea that diarrhea-causing microbes can act as a major source of selective pressure in humans and other animals.
Our evolutionary and functional analysis of bat and primate GC-C showed results consistent with ongoing evolutionary conflicts with diarrheal bacteria. Alternatively, physiological adaptations to changing dietary or environmental conditions are a potential explanation for adaptive evolution of GC-C. However, the signatures of recurrent positive selection that we observe are a hallmark of ongoing genetic conflicts. Other selective forces rarely, if ever, result in the continuous fixation of nonsynonymous mutations (Daugherty and Malik, 2012). Furthermore, we find evidence of ongoing adaptive evolution within families of bats and primates with similar dietary and environmental niches (Figure 1B). Our findings of differential susceptibility to STa variants across primates and bats, when coupled with the signatures of recurrent positive selection in the GC-C toxin-binding domain, strongly support a scenario of pathogen-driven evolution. While many examples of genetic conflicts driven by pathogenic microbes have been described for genes encoding dedicated immune functions, few consider physiologic processes like those mediated by GC-C. Our discovery of such adaptive evolution in the GC-C signaling pathway extends our knowledge of the far-reaching impact of infectious microbes on the evolution of their hosts.
Prior to this study, it was not known whether variants of STa interact differently with GC-C. We performed side by side comparisons of four different STa variants from a diverse set of bacterial pathogens that infect humans. We found that each of the toxins shared a similar ability to activate human GC-C, while other species can be significantly more susceptible or resistant to the same peptides (Figure 2). While it is tempting to speculate that resistant species have adapted in response to a particular STa variant, very little is known about the nature of STa in bacterial pathogens that infect wildlife. Diversification of GC-C in bats might therefore represent adaptation to uncharacterized STa variants. Such toxins could have unpredictable interactions with human GC-C, perhaps able to hyperstimulate cGMP production as we observe with STp in many species. Identification and characterization of unknown STa variants from other species represents an important future direction to this work.
This study set out to directly link signatures of adaptive evolution in GC-C to variation in physiological responses to bacterial toxins. Previous investigations of GC-C variation in humans relied on cellular cGMP-production assays as correlates of physiologic outcomes (Fiskerstrand et al., 2012; Müller et al., 2016). To extend and complement this approach, we generated transgenic intestinal organoids to directly model how GC-C variance directly impacts physiological response to STa (Figure 3). Our assays demonstrated a direct correlation between cellular cGMP production and organoid swelling, which supports it as an accurate model of disease severity and highlight STa as a source of potential selective pressure. While organoids represent a powerful system to study the physiology of secretory diarrhea, they are not useful for modeling disease transmission. Attempts to model ETEC infection and transmission are complicated by the synthetic conditions required to overcome the natural immunity of mice to these microbes (Bolick et al., 2018). Future work characterizing how differing vulnerabilities to STa impacts host-to-host transmission will provide useful information on how enterotoxin variation might influence the spread of pathogenic microbes.
Our results raise important questions about the biochemical interaction between STa and GC-C, which remains poorly understood. While mutational scanning has revealed a set of residues critical for ligand-stimulated cGMP production, it is unknown whether these are directly involved in STa interactions (Wada et al., 1996). Our comparisons of cGMP responses revealed surprising differences in maximal response to STa variants between and within species. For instance, orangutan GC-C displayed a near 4-fold maximal response to STp compared to V-ST, despite showing similar EC50 values (Figure 2B, Table S4). These findings suggest that increased STa susceptibility may be a result of differences in the structure of the interaction rather than ligand affinity. Using our collection of both GC-C and STa proteins to resolve a complete atomic structure of GC-C in its bound and unbound states will be important for understanding how variation at specific sites in the GC-C ligand binding domain influences susceptibility to STa.
Finally, we observed a unique pattern of GC-C-uroguanylin coevolution exclusively in bats. True compensatory coevolution between interacting proteins is theorized to be a rare phenomenon because it requires intermediate states with reduced fitness (Haag, 2007; Stephan, 1996). This constraint is likely reflected in the high degree of conservation of uroguanylin in mammals, with the exemption of bats. We propose a model where intermediate low affinity states of uroguanylin and GC-C are tolerated if they also allow for the evasion of STa, providing time for compensatory mutations to arise in uroguanylin. Indeed, we observed such a sub-optimal state in the vesper bat E. fuscus, whose GC-C was only weakly stimulated by uroguanylin encoded in its genome. These low affinity states would likely only arise when there is sufficient selective pressure from STa-producing pathogens. The signatures of positive selection we observed in bat GC-C were more intense and widespread than in other mammals, and were present in deep branches of Chiroptera. Given the appreciated frequency and intensity of bat-pathogen interactions, we posit that diarrheal pathogens triggered compensatory coevolution of this of this host signaling axis. Together these results point to the surprisingly profound impact of diarrhea-causing microbes on the evolution of bats.
STAR Methods
Resource Availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Nels Elde (nelde@genetics.utah.edu).
Materials availability
Cell lines and plasmids generated in this study are available upon request without restrictions.
Data and code availability
This paper analyzes existing, publicly available data. Accession numbers for analyzed sequences are listed in Table S1.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Experimental Model and Subject Details
HEK293T Cell Culture
HEK293T cells (Female) were cultured in DMEM media (HyClone/Cytiva) supplemented with 10% heat-inactivated FBS (Gibco), 2 mM L-glutamine (HyClone/Cytiva), 1 mM Sodium Pyruvate (Gibco), 100 U/mL penicillin, and 100 μg/mL streptomycin (Gibco). Cells were grown at 37°C in a humidified incubator with 5% CO2. Cell lines were not authenticated.
Intestinal organoid culture
Small intestinal organoids derived from GC-C knockout mice were a generous gift from the laboratory of Scott Waldman and were isolated as described previously (Pattison et al., 2016). Organoids were maintained in 50 μl Matrigel droplets (Corning) in wells of a 24-well plate containing 650 μl IntestiCult mouse organoid growth medium (StemCell Technologies cat# 06005) at 37°C in a humidified incubator with 5% CO2. Organoids were passaged every 7–10 days by disruption with TrypLE (Thermo Scientific) and reseeded at a concentration of 100–500 organoids per well.
Method Details
Evolutionary analysis
GC-C and NPR1–3 nucleotide sequences were retrieved from NCBI GenBank for each species. Prior to analysis, sequences for each gene within each clade were first aligned using clustalW. In each analysis, a generally agreed upon phylogeny was used for each clade. Evidence for gene-wide positive selection was obtained using the branch-site models implemented in PAML and BUSTED software. In PAML, alignments were fitted to a F3×4 codon model and likelihood ratio tests were performed based on comparisons of NSsites model 2 to model 1 and model 8 to model 7. BUSTED analysis was performed on all branches of a user-specified tree using an online webserver with default parameters (https://www.datamonkey.org/busted). Gene trees based on the amino acid sequence of GC-C and NPR2 were generated by first aligning amino acid sequences with clustalW in each clade. Trees were generated using the PhyML plugin implemented in Geneious software using the Le Gascuel substitution model.
Sequence alignment and figure generation
Unless otherwise noted, all gene and amino acid sequences were retrieved from GenBank (Table S1). Sequence alignments were created using ClustalW and exported using Geneious. Figures were created using assets from www.BioRender.com.
Peptide synthesis
To generate a C-terminal acid with the last amino acid of the sequence pre-loaded and a density of ~0.2 mmol/g, 150 mg 2-chlorotrityl resin (ChemPep, 100–200 mesh) was washed with DMF and DCM and swelled for 30 min, 0.03 mmol of the respective amino acid was dissolved in 1 mL DMF/DCM and 0.15 mmol DIPEA was added to the amino acid solution. This mixture was added to pre-swelled resin and rotated at RT for 1 hour. Then the resin was washed with 3x DCM and capped with 20 mL 17:2:1 DCM:MeOH:DIPEA with manual mixing between washes. Finally, resin was washed 3x with ~4 mL DCM and 3x with ~4 mL DMF prior to synthesis. Remaining amino acids were chemically synthesized using solid-phase peptide synthesis (SPPS) with a standard synthesis scale of 30 μmol on a Prelude X peptide synthesizer (Gyros Protein Technologies) using Fmoc-protected amino acids (Gyros Protein Technologies). Uroguanylin peptides were synthesized using Fmoc-Cys(Acm)-OH (Aapptec) at C7/C15. The following cycles were used during SPPS: Fmoc deprotection: 3 × 3-min cycles of 2 mL 20% piperidine in DMF. Amino acid coupling: mixing Fmoc-protected amino acid in NMP (0.65 mL, 200 mM), HATU in DMF (0.65 mL, 195 mM), plus NMM in DMF (0.5 mL, 0.6 M) for 25 min with shaking at 150 rpm and nitrogen bubbling. DMF washing (2 mL) was performed between deprotection and coupling steps (3 × 30 s). Completed peptides were washed with DCM and dried prior to cleavage.
Peptide cleavage from resin
Peptides were cleaved from resin for 2 h using 3.7 mL TFA, 100 μL water, 100 μL TIS and 100 μL EDT. After cleavage, peptides were precipitated into cold diethyl ether (35 mL), stored at −20°C overnight, then washed with diethyl ether (3 × 30 mL), and finally peptides were pelleted by centrifugation and dried overnight in a desiccator prior to HPLC purification.
Crude STa peptide purification
Crude peptides were purified by reverse-phase HPLC on a Phenomenex Jupiter 4-μm Proteo C12 90 Å (250 × 21.2 mm) column with a water/ACN gradient in 0.1% TFA (Buffer A: water with 0.1% TFA, Buffer B: 90% ACN with 10% water and 0.1% TFA; for LC/MS buffer is 0.1% formic acid instead of TFA). Specific gradients over 35 min at 10 mL/min were: 29–34% Buffer B (Y-STa and STh), 38–42% Buffer B (V-ST), and 34–38% Buffer B (STp). Purified peptides were lyophilized and their purity validated by analytical HPLC on a Phenomenex Jupiter 4-μm Proteo C12 90 Å (150 × 4.6 mm) column with a linear gradient of 5–90% Buffer B over 30 min. Masses of the peptides were confirmed by LC/MS on an Agilent Technologies 6120 single-quadrupole mass spectrometer with an Agilent 1260 Infinity II HPLC using an Agilent 2.7-μm Poroshell C18 120 Å (4.6 × 50 mm) column.
Oxidation of free cysteine residues (STa)
Oxidation of free cysteine residues in purified peptides was accomplished using 677 air oxidation of solid peptide dissolved in peptide oxidation buffer (50 mM Tris Base, 2% DMSO, pH 8 at 37°C) at ≤0.4 mg/mL (to ensure intramolecular disulfide formation) for ~17 h at 37°C with shaking at 200 rpm. The reaction was quenched with glacial acetic acid (final concentration of 5%), and the pH was confirmed to be between pH 3–4. Reaction solution was then diluted 2-fold with HPLC Buffer A, spun for 10 min at 4696 xg and purified by reverse phase HPLC on a Phenomenex Jupiter 4-μm Proteo C12 column using a gradient of 36–40 % Buffer B (V-ST), 24 to 29% Buffer B (Y-STa), 30–35% Buffer B (STp), 25.5–30% Buffer B (STh) over 35 min with a flow rate of 10 mL/min. All HPLC methods started at 20% Buffer B to prevent peptide precipitation. Fractions were collected and checked for purity by LC/MS (10–90 or 10–70% Buffer B over 10 min) and lyophilized. For each peptide, eight distinct peaks were collected (Figure S2B) and tested for activity in the cellular cGMP assay. The most active peaks were: oxidized STp peak #2, oxidized STh peak #8, oxidized V-ST peak #2, and oxidized Y-ST peak #1 (Figure S2C-D).
Oxidation of free cysteines (uroguanylins)
Crude peptides were first validated by LC/MS and analytical HPLC, as described peptide dissolved in peptide oxidation buffer at ≤0.4 mg/mL for 14–18 h at 37°C with shaking at 200 rpm. The reaction was quenched with glacial acetic acid (5% final concentration) and the pH was confirmed to be between 3–4. Reaction solution was then diluted 2-fold with HPLC Buffer A, spun for 10 min at 4696 xg and purified by reverse-phase HPLC on a Phenomenex Jupiter 4-μm Proteo C12 column (see specs above) using a gradient of 28–35 % Buffer B (Human), 23–35 % Buffer B (P. vampyrus), 22–40% Buffer B (M. lucifigus), or 32–50% Bufer B (E. fuscus) over 35 min, all HPLC methods starting at 20% Buffer B to prevent peptide precipitation. Fractions were collected and checked for 702 purity by LC/MS (10–90 or 10–70% Buffer B over 10 min, 1 mL/min).
Acm removal and formation of second disulfide
Dry purified uroguanylin peptides with a single formed disulfide and Cys(Acm) protecting groups at positions C7 and C15 (1–5 mg) were dissolved in Buffer A to a concentration of ~400 μM. Fresh I2-TFA mixture was prepared (~10 mg I2 dissolved in 5 mL ACN, then added to a solution of 15 mL H2O with 0.6 mL TFA), and the I2-TFA mix was added (2x volume) to the peptide-Buffer A solution. This mixture reacted on a rotator at RT for 20 min before quenching with 1 M ascorbic acid added drop-wise until solution changes from rust color to colorless, clear solution (typically less than 30 μL). Quenched solution was diluted with one volume Buffer A, spun 10 min at 4696 xg, and transferred to a new tube for purification. Same-day purification proceeded via reverse-phase HPLC using a Phenomenex 5-μm C18-Kinetex column C10 100Å (150 × 4.6 mm) with a gradient of 20–52% Buffer B (human uroguanylin), 14–23% Buffer B (P. vampyrus), 10–30% Buffer B (M. lucifugus), or 22–30% Buffer B (E. fuscus) over 30 min at 2 mL/min to achieve baseline separation of peptide topoisomers. Two individual peaks were collected separately and their mass confirmed by LC/MS. Generally the larger peak was determined to be the active isomer (consistent with previous studies(Chino et al., 1996)). The uroguanylin topoisomers were found to interconvert at RT (as seen previously for human uroguanylin(Chino et al., 1996, 1998; Klodt et al., 1997)), with M. lucifigus converting the fastest—within 30 min of peak purification). Therefore, fractions were placed on ice immediately after purification until confirmation by LC/MS, then pooled and lyophilized. All dry, purified peptides were stored in parafilmed containers in the dark at −20°C. We tested the order of disulfide bond formation and found that forming the C7-C15 disulfide second yielded more of the active isomer compared to forming the C4-C12 disulfide second.
GC-C cloning and lentivirus generation
GC-C sequences were downloaded from GenBank based on whole genome sequences from each species tested. Each C-terminally HA tagged GC-C variant was synthesized via gene synthesis (Life Technologies). GC-C was then cloned into the lentiviral transfer vector pUltra (Addgene #24129) between the XbaI and BamHI restriction sites by Gibson Assembly, in frame with GFP and the T2A linker sequence. To generate lentiviral particles, 10 cm dishes were seeded with 3 × 106 293T cells 24 hours prior to transfection. Cells were then transfected with 7.6 μg pUltra-GC-C, 7.6 μg psPAX2 packaging plasmid (Addgene # 12260), and 3.8 μg pMD2.G envelope plasmid (Addgene # 12259) with 56 μl FuGene HD transfection reagent according to the manufacturer’s specifications. Media was replaced 24 hours post-transfection and replaced with 10 mL media. Viral supernatants were collected 48 hours-post transfection and passed through a 0.4 μm followed by overnight incubation with 1X PEG-IT solution (System Biosciences) at 4°C. Precipitated viral particles were centrifuged at 1500xg for 30 min at 4°C and resuspended in PBS at a final volume of 500 μl before storage at −80°C.
Construction of transgenic 293T cell lines
To generate each GC-C expressing cell line 2.5 × 105 293T cells were seeded into a single well of a 6 well plate. After 24 hours of growth, 50 μL concentrated lentivirus encoding GC-C linked to a GFP reporter was added to a total of 1 mL growth medium containing 8 μM polybrene (Sigma). 24 hours post transduction, cells were transferred to a T75 flask and grown for 5–7 days. Transgenic lines were established by FACS sorting the top 25% of GFP expressing cells from each transduction.
Cellular cGMP assay
293T cells expressing GC-C variants were grown to confluence in 24-well plates before ligand stimulation. Toxin and uroguanylin solutions were diluted in reduced serum media (Opti-MEM, Thermo Scientific) to the appropriate experimental concentration. For each measurement, cell culture media was then aspirated and replaced with the ligand-containing solution in 3–6 replicate wells. Following incubation at 37°C/5% CO2 for 20 min, the ligand solution was aspirated and cells lysed directly in 0.1 M HCl. Intracellular cGMP was then measured using an Enzyme Linked Immunosorbent Assay kit (Enzo Scientific) according to the manufacturer’s specifications. Absorbance was measured using a plate reader (BioTek), and cGMP concentrations calculated based on a standard curve. Four-parameter variable slope dose-response curves were generated in Prism 8 (GraphPad).
Western blotting
293T cells expressing GC-C were grown to confluence in 24-well plates following immunoblot analysis. To avoid aggregation, cells were lysed directly in 2x Laemmli buffer containing 8 M urea (Sigma), 3 M thiourea (Sigma), and β-mercaptoethanol for 30 min at room temperature prior to analysis. Total protein was resolved by Mini-PROTEAN GTX polyacrylamide gel electrophoresis (Bio-rad). Proteins were detected using anti-HA (Sigma Cat# H6908, 1:1000) and anti-Actin (Sigma Cat#A2066, 1:400) antibodies. Blots were visualized using film or C-DiGit chemiluminescent imager (LI-COR).
Intestinal organoid transduction
For each transduction approximately 500 organoids were mechanically disrupted by vigorous pipetting using a 20 μl pipette tip. Disrupted organoids were then resuspended in 100 μl concentrated lentivirus solution containing 8 μM polybrene (Sigma), 10 μM Y-27632 (Sigma) and 10 μM SB431542 (Sigma). Lentiviral infection was allowed to proceed at 37°C/5% CO2 for 6 h prior to resuspension in Matrigel and seeding into a new well. Transduced organoids were monitored for GFP expression and 20–50 organoids with uniform fluorescence were selected manually 72 h post-transduction and transferred to a new well to establish transgenic lines.
Organoid swelling assay
Organoids were grown in a 24 well plate for 5–6 days after passaging prior to swelling assays at a concentration of 100–500 organoids per well. Organoids were imaged on an ImageXPress Pico Automated Cell Imaging System (Molecular Devices) using the live cell imaging cassette with temperature set to 37˚C, 5% CO2, and humidity levels 85–95%. These parameters were monitored and remained constant throughout each experiment. For swelling analysis, ST toxins were added directly to the organoid culture media to a final concentration of 5 μM. Following addition of toxin, a 24 mm2 area of each well (corresponding to ~13% of the total well area) was imaged every 10 min at 4x magnification for the full 2 hours of the experiment. The imaged area was constant across the experiment and analogous between wells and contained at least 50 organoids for each analyzed strain. 20 stacked transmitted light images were collected for each well with a focus step of 25 μm and the Best Plane image was calculated by the ImageXPress software. GFP images were collected at the beginning and end of the experiment using the same imaging parameters, and both the best focus and maximum fluorescence images for each stack were calculated and output by the ImageXPress software.
Organoids used for analysis were identified in ImageJ and assigned ROI identifiers so that the same organoids could be easily assessed across timepoints. 30 organoids from each strain were selected randomly to include a variety of starting sizes and were checked for GC-C expression by comparison with GFP images. GC-C expressing organoids that ruptured during the experiment were excluded from analysis. Swelling was assessed by measuring the area of each organoid in ImageJ at t=0 and t=120 min. The percent change in organoid area from 0 to 120 min was calculated for each organoid and plotted in Prism.
Quantification and statistical analysis
All statistical tests were performed using Prism 8. Specific statistical comparisons are described in the accompanying figure legends and replicate numbers and sample sizes are described in the above methods. For cGMP-generation assays, mean cGMP concentrations were compared across cell lines or within cell lines using one-way ANOVA followed by Dunnett’s Type 3 multiple comparisons test to assess significance between specific treatment pairings. For dose response curves, data were fit by nonlinear regression using the least squares method to a log(agonist) vs. response equation with variable slope. Best fit values and SEM (see Table S4) from this regression were used to compare between curves using one-way ANOVA followed by Dunnett’s Type 3 multiple comparisons test. For organoid assays, significance in organoid swelling was similarly assessed by one-way ANOVA followed by Dunnett’s T3 multiple comparisons test. For all statistical analysis, significance was defined as p<0.05.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit Anti-HA | Sigma | Sigma-Aldrich Cat# H6908, RRID:AB_260070 |
| Rabbit Anti-Actin | Sigma | Sigma-Aldrich Cat# A2066, RRID:AB_476693 |
| Goat Anti-Rabbit IgG, Peroxidase Conjugated | Sigma | Sigma-Aldrich Cat# AP132P, RRID:AB_90264 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| STp | This paper | N/A |
| STh | This paper | N/A |
| Y-ST | This paper | N/A |
| V-ST | This paper | N/A |
| Uroguanylin peptides (Human, P. vampyrus, and M.lucifugus variants) | This paper | N/A |
| Polybrene | Sigma | Cat# TR-1003 |
| FuGene HD Transfection Reagent | Promega | Cat# E2311 |
| PEG-it Virus Precipitation Solution (1x) | System Biosciences | Cat# LV810A-1 |
| Matrigel Basement Membrane Matrix | Corning | Cat# 356231 |
| IntestiCult Intestinal Organoid Growth Media (Mouse) | Stem Cell Technologies | Cat# 06005 |
| Y-27632 dihydrochloride | Sigma | Cat# Y0503-1 MG |
| SB 431542 hydrate | Sigma | Cat# S4317-5MG |
| Critical Commercial Assays | ||
| Direct cGMP ELISA Kit | Enzo | Cat# ADI-901-014 |
| Deposited Data | ||
| GC-C and NPR1-3 Gene Sequences | NCBI Genbank | See Table S1 for accession numbers. |
| Experimental Models: Cell Lines | ||
| HEK 293T Cells | Lab of Wes Sundquist | N/A |
| GC-C −/− Mouse Intestinal Organoids | Lab of Scott Waldman; Pattison et al. (2016). | N/A |
| Oligonucleotides | ||
| F primer for primate GC-C cloning (all except Orangutan): ggaggagaatcccgggccttctagaATGAAGACGTTGCTGTTG | This study | N/A |
| F primer for Orangutan GC-C cloning: ggaggagaatcccgggccttctagaATGAACACGTTGCTGTTG | This study | N/A |
| R primer for all primate GC-C cloning: ccctgccctcgccggagccggatccttatgcataatccggaacatcatacggataAAAATAGGTGCTCTCCTTG | This study | N/A |
| R primer for M. lucifugus GC-C cloning: ccctgccctcgccggagccggatccttatgcataatccggaacatcatacggataGAAATAGGTGGTCTCCTTG | This study | N/A |
| F primer for M. lucifugus and E. fuscus GC-C cloning: ggaggagaatcccgggccttctagaATGAAGAAACCACTGTTG | This study | N/A |
| R primer for E. fuscus GC-C cloning: ccctgccctcgccggagccggatccttatgcataatccggaacatcatacggataAAAATGGGTGTTCTCCTC | This study | N/A |
| F primer for M. natalensis GC-C cloning: ggaggagaatcccgggccttctagaATGAAGACACCACGCTTG | This study | N/A |
| R primer for M. natalensis GC-C cloning: ccctgccctcgccggagccggatccttatgcataatccggaacatcatacggataAAAATGGGTGTTCTCCTTG | This study | N/A |
| F primer for Pteropus spp. GC-C cloning: ggaggagaatcccgggccttctagaATGACGATACCTCTGTTG | This study | N/A |
| R primer for Pteropus spp. GC-C cloning: ccctgccctcgccggagccggatccttatgcataatccggaacatcatacggataAAAATGGGTGCTCTCCTTG | This study | N/A |
| Recombinant DNA | ||
| pMD2.G | Gift from Didier Trono (unpublished) | Addgene #12259 |
| pULTRA | Lou et al. (2012) | Addgene # 24129 |
| psPAX2 | Gift from Didier Trono (unpublished) | Addgene #12260 |
| Software and Algorithms | ||
| PAML | Yang, Z. (2007) | RRID:SCR_014932: http://abacus.gene.ucl.ac.uk/software/paml.html |
| BUSTED | Murrell et al. (2015) | https://www.datamonkey.org/busted |
| Geneious | Geneious | RRID:SCR_010519: https://www.geneious.com/ |
| Prism 8 | Graphpad | RRID:SCR_002798: https://www.graphpad.com/ |
| FIJI | ImageJ | RRID:SCR_002285: https://imagej.net/Fiji |
| Other | ||
| Image XPress Pico Automated Cell Imaging System | Molecular Devices | N/A |
| Synergy HT Plate Reader | BioTek | N/A |
| C-DiGit Blot Scanner | LI-COR | N/A |
Highlights.
Intestinal enterotoxin receptor GC-C bears signatures of a host-pathogen arms race
Primate and bat GC-C show species-specific susceptibility to enterotoxin variants
Bats exhibit unique compensatory evolution of the GC-C/uroguanylin signaling axis
ACKNOWLEDGEMENTS
The authors thank Scott Waldman and Amanda Pattison for generously providing GC-C knockout organoids.
Funding:
N.C.E. is supported by the National Institute of Health, United States (R35GM134936) and the Burroughs Wellcome Fund Investigators in the Pathogenesis of Infectious Disease Program, United States. M.S.K. is supported by the National Institute of Health, United States (P50AI150464). N.C.E and M.S.K. are H.A. & Edna Benning Presidential Endowed Chairs (University of Utah, United States). C.M.C is supported by the National Institute of Health, United States (T32AI055434). Z.A.H. is supported by a postdoctoral fellowship from the Helen Hay Whitney Foundation.
Footnotes
Competing interests: The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Agnarsson I, Zambrana-Torrelio CM, Flores-Saldana NP, and May-Collado LJ (2011). A time-calibrated species-level phylogeny of bats (Chiroptera, Mammalia). PLOS Curr. Tree Life. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banskar S, Bhute SS, Suryavanshi MV, Punekar S, and Shouche YS (2016). Microbiome analysis reveals the abundance of bacterial pathogens in Rousettus leschenaultii guano. Sci. Rep. 6, 36948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barber MF, and Elde NC (2014). Nutritional immunity. Escape from bacterial iron piracy through rapid evolution of transferrin. Science 346, 1362–1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartfeld S (2016). Modeling infectious diseases and host-microbe interactions in gastrointestinal organoids. Dev. Biol. 420, 262–270. [DOI] [PubMed] [Google Scholar]
- Barua D (1992). History of Cholera. In Cholera, Barua D, and Greenough WB, eds. (Boston, MA: Springer US; ), pp. 1–36. [Google Scholar]
- Basu N, Arshad N, and Visweswariah SS (2010). Receptor guanylyl cyclase C (GC-C): regulation and signal transduction. Mol. Cell. Biochem. 334, 67–80. [DOI] [PubMed] [Google Scholar]
- Bolick DT, Medeiros PHQS, Ledwaba SE, Lima A. a. M., Nataro JP, Barry EM, and Guerrant RL (2018). Critical Role of Zinc in a New Murine Model of Enterotoxigenic Escherichia coli Diarrhea. Infect. Immun. 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brook CE, and Dobson AP (2015). Bats as ‘special’ reservoirs for emerging zoonotic pathogens. Trends Microbiol. 23, 172–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calisher CH, Childs JE, Field HE, Holmes KV, and Schountz T (2006). Bats: Important Reservoir Hosts of Emerging Viruses. Clin. Microbiol. Rev. 19, 531–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chino N, Kubo S, Miyazato M, Nakazato M, Kangawa K, and Sakakibara S (1996). Generation of two isomers with the same disulfide connectivity during disulfide bond formation of human uroguanylin. Lett. Pept. Sci. 3, 45–52. [Google Scholar]
- Chino N, Kubo S, Kitani T, Yoshida T, Tanabe R, Kobayashi Y, Nakazato M, Kangawa K, and Kimura T (1998). Topological isomers of human uroguanylin: interconversion between biologically active and inactive isomers. FEBS Lett. 421, 27–31. [DOI] [PubMed] [Google Scholar]
- Daugherty MD, and Malik HS (2012). Rules of engagement: molecular insights from host-virus arms races. Annu. Rev. Genet. 46, 677–700. [DOI] [PubMed] [Google Scholar]
- Demogines A, Farzan M, and Sawyer SL (2012). Evidence for ACE2-Utilizing Coronaviruses (CoVs) Related to Severe Acute Respiratory Syndrome CoV in Bats. J. Virol. 86, 6350–6353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta D, and Clevers H (2017). Organoid culture systems to study host–pathogen interactions. Curr. Opin. Immunol. 48, 15–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiskerstrand T, Arshad N, Haukanes BI, Tronstad RR, Pham KD-C, Johansson S, Håvik B, Tønder SL, Levy SE, Brackman D, et al. (2012). Familial Diarrhea Syndrome Caused by an Activating GUCY2C Mutation. N. Engl. J. Med. 366, 1586–1595. [DOI] [PubMed] [Google Scholar]
- Fleckenstein JM, Hardwidge PR, Munson GP, Rasko DA, Sommerfelt H, and Steinsland H (2010). Molecular mechanisms of enterotoxigenic Escherichia coli infection. Microbes Infect. 12, 89–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forte LR (2004). Uroguanylin and guanylin peptides: pharmacology and experimental therapeutics. Pharmacol. Ther. 104, 137–162. [DOI] [PubMed] [Google Scholar]
- Gonzales-Siles L, and Sjöling Å (2016). The different ecological niches of enterotoxigenic Escherichia coli. Environ. Microbiol. 18, 741–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haag ES (2007). Compensatory vs. pseudocompensatory evolution in molecular and developmental interactions. Genetica 129, 45–55. [DOI] [PubMed] [Google Scholar]
- Kita T, Smith CE, Fok KF, Duffin KL, Moore WM, Karabatsos PJ, Kachur JF, Hamra FK, Pidhorodeckyj NV, Forte LR, et al. (1994). Characterization of human uroguanylin: a member of the guanylin peptide family. Am. J. Physiol.-Ren. Physiol. 266, F342–F348. [DOI] [PubMed] [Google Scholar]
- Klodt J, Kuhn M, Marx UC, Martin S, Röusch P, Forssmann W-G, and Adermann K (1997). Synthesis, biological activity and isomerism of guanylate cyclase C-activating peptides guanylin and uroguanylin. J. Pept. Res. 50, 222–230. [DOI] [PubMed] [Google Scholar]
- Lencer WI, and Tsai B (2003). The intracellular voyage of cholera toxin: going retro. Trends Biochem. Sci. 28, 639–645. [DOI] [PubMed] [Google Scholar]
- Lou E, Fujisawa S, Morozov A, Barlas A, Romin Y, Dogan Y, Gholami S, Moreira AL, Manova-Todorova K, Moore MAS, 2012. Tunneling Nanotubes Provide a Unique Conduit for Intercellular Transfer of Cellular Contents in Human Malignant Pleural Mesothelioma. Plos One 7, e33093. 10.1371/journal.pone.0033093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mowat A (2017). Why does cystic fibrosis display the prevalence and distribution observed in human populations? Curr. Pediatr. Res [Google Scholar]
- Mühldorfer K (2013). Bats and Bacterial Pathogens: A Review. Zoonoses Public Health 60, 93–103. [DOI] [PubMed] [Google Scholar]
- Müller T, Rasool I, Heinz-Erian P, Mildenberger E, Hülstrunk C, Müller A, Michaud L, Koot BGP, Ballauff A, Vodopiutz J, et al. (2016). Congenital secretory diarrhoea caused by activating germline mutations in GUCY2C. Gut 65, 1306–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murrell B, Weaver S, Smith MD, Wertheim JO, Murrell S, Aylward A, Eren K, Pollner T, Martin DP, Smith DM, et al. (2015). Gene-wide identification of episodic selection. Mol. Biol. Evol. 32, 1365–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson EJ, Harris JB, Glenn Morris J, Calderwood SB, and Camilli A (2009). Cholera transmission: the host, pathogen and bacteriophage dynamic. Nat. Rev. Microbiol. 7, 693–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa H, Qiu Y, Ogata CM, and Misono KS (2004). Crystal structure of hormone-bound atrial natriuretic peptide receptor extracellular domain: rotation mechanism for transmembrane signal transduction. J. Biol. Chem. 279, 28625–28631. [DOI] [PubMed] [Google Scholar]
- Pattison AM, Blomain ES, Merlino DJ, Wang F, Crissey MAS, Kraft CL, Rappaport JA, Snook AE, Lynch JP, and Waldman SA (2016). Intestinal Enteroids Model Guanylate Cyclase C-Dependent Secretion Induced by Heat-Stable Enterotoxins. Infect. Immun. 84, 3083–3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potter LR, Abbey-Hosch S, and Dickey DM (2006). Natriuretic Peptides, Their Receptors, and Cyclic Guanosine Monophosphate-Dependent Signaling Functions. Endocr. Rev. 27, 47–72. [DOI] [PubMed] [Google Scholar]
- Rivera-Chávez F, and Mekalanos JJ (2019). Cholera toxin promotes pathogen acquisition of host-derived nutrients. Nature 572, 244–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz S, Lopez MJ, Kuhn M, and Garbers DL (1997). Disruption of the guanylyl cyclase-C gene leads to a paradoxical phenotype of viable but heat-stable enterotoxin-resistant mice. J. Clin. Invest. 100, 1590–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephan W (1996). The Rate of Compensatory Evolution. Genetics 144, 419–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takao T, Shimonishi Y, Kobayashi M, Nishimura O, Arita M, Takeda T, Honda T, and Miwatani T (1985a). Amino acid sequence of heat-stable enterotoxin produced by Vibrio cholerae non-01. FEBS Lett. 193, 250–254. [DOI] [PubMed] [Google Scholar]
- Takao T, Tominaga N, Yoshimura S, Shimonishi Y, Hara S, Inoue T, and Miyama A (1985b). Isolation, primary structure and synthesis of heat-stable enterotoxin produced by Yersinia enterocolitica. Eur. J. Biochem. 152, 199–206. [DOI] [PubMed] [Google Scholar]
- Thompson MR, Brandwein H, LaBine-Racke M, and Giannella RA (1984). Simple and reliable enzyme-linked immunosorbent assay with monoclonal antibodies for detection of Escherichia coli heat-stable enterotoxins. J. Clin. Microbiol. 20, 59–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troeger C, Blacker BF, Khalil IA, Rao PC, Cao S, Zimsen SR, Albertson SB, Stanaway JD, Deshpande A, Abebe Z, et al. (2018). Estimates of the global, regional, and national morbidity, mortality, and aetiologies of diarrhoea in 195 countries: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Infect. Dis. 18, 1211–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viswanathan VK, Hodges K, and Hecht G (2009). Enteric infection meets intestinal function: how bacterial pathogens cause diarrhoea. Nat. Rev. Microbiol. 7, 110–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wada A, Hirayama T, Kitaura H, Fujisawa J, Hasegawa M, Hidaka Y, and Shimonishi Y (1996). Identification of ligand recognition sites in heat-stable enterotoxin receptor, membrane-associated guanylyl cyclase C by site-directed mutational analysis. Infect. Immun. 64, 5144–5150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiglmeier PR, Rösch P, and Berkner H (2010). Cure and curse: E. coli heat-stable enterotoxin and its receptor guanylyl cyclase C. Toxins 2, 2213–2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z (2007). PAML 4: Phylogenetic Analysis by Maximum Likelihood. Mol. Biol. Evol. 24, 1586–1591. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This paper analyzes existing, publicly available data. Accession numbers for analyzed sequences are listed in Table S1.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.