

Summary:
Adipocytes undergo intense energetic stress in obesity resulting in loss of mitochondrial mass and function. We have found that adipocytes respond to mitochondrial stress by rapidly and robustly releasing small extracellular vesicles (sEVs). These sEVs contain respiration-competent, but oxidatively-damaged mitochondrial particles, which enter circulation and are taken up by cardiomyocytes where they trigger a burst of ROS. The result is compensatory antioxidant signaling in the heart that protects cardiomyocytes from acute oxidative stress, consistent with a preconditioning paradigm. As such, a single injection of sEVs from energetically stressed adipocytes limits cardiac ischemia/reperfusion injury in mice. This study provides the first description of functional mitochondrial transfer between tissues and the first vertebrate example of “inter-organ mitohormesis”. Thus, these seemingly toxic adipocyte sEVs may provide a physiological avenue of potent cardio-protection against the inevitable lipotoxic or ischemic stresses elicited by obesity.
eTOC blurb
Crewe et. al. report that adipocytes release sEVs containing damaged mitochondria in response to energetic stress, such as that seen in chronic obesity. The sEV-associated mitochondria induce transient mitochondrial oxidative stress in cardiac tissue, resulting in an antioxidant response. Adipocyte sEVs thereby precondition the heart to protect against ischemia/reperfusion injury.
Graphical Abstract
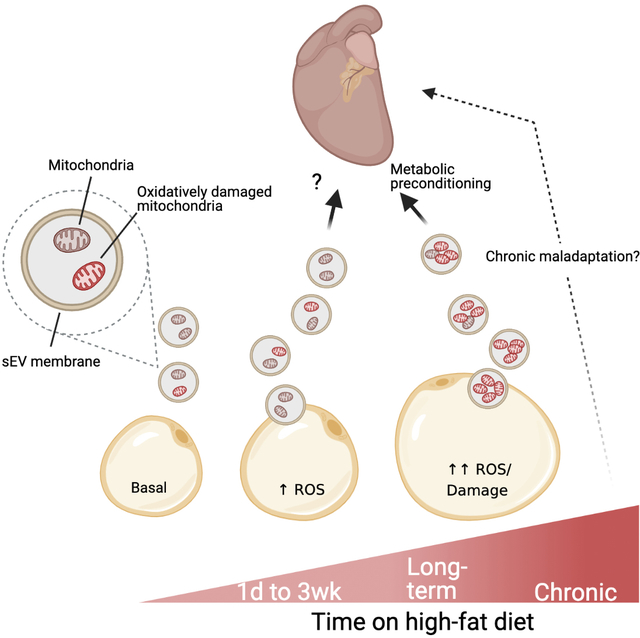
Introduction
Adipose tissue dysfunction is at the forefront of metabolic disturbances in obesity and type 2 diabetes. In the context of obesity, hypertrophied adipocytes become dysfunctional, lose their beneficial endocrine functions, and promote tissue inflammation and fibrosis (Crewe et al., 2017). It is this unhealthy expansion of the adipose tissue that triggers systemic lipotoxicity and insulin resistance and promotes the development of comorbid conditions associated with obesity, such as cardiovascular disease (CVD) (Lavie et al., 2018; Oikonomou and Antoniades, 2019). A major determinant of adipocyte function is the adequacy of mitochondrial metabolism (An et al., 2019; Kusminski et al., 2012). As in all cells, initial high fat diet (HFD)-induced mitochondrial stress in adipocytes is counteracted by mitohormesis. This is the process whereby a transient, mild, oxidative or proteotoxic challenge stimulates an adaptive cellular response that confers protection against a future lethal stress (Yun and Finkel, 2014). However, this protective pathway in adipocytes is overwhelmed in obesity. The result is severe mitochondrial dysfunction, characterized by a reduction in mitochondrial mass, high ROS production, and low cellular ATP levels (Curtis et al., 2010; Schottl et al., 2015; Wang et al., 2014).
We have recently demonstrated that in vivo overexpression of mitochondrial ferritin (FtMT) selectively in adipocytes (adipo-FtMT mouse), mimics obesity-associated impairment of mitochondria by inducing adipocyte oxidative stress and suppressing oxidative phosphorylation (Kusminski et al., 2020). The phenotype of this mouse on a HFD is striking, involving a strong effect on multiple organ systems that culminates in metabolic deterioration. Interestingly, this model displayed marked mitochondrial oxidative stress in the heart. This phenotype is of particular interest as obese mice and humans display high cardiac oxidative stress compared to their lean counterparts. The observation in the adipo-FtMT mouse suggests that a pro-oxidant signal can be transmitted from the adipocyte to the myocardium, a phenomenon that may contribute to cardiac ROS production in obesity. The pleiotropic inter-organ signaling stimulated by adipocytes in adipo-FtMT mice led us to postulate that the phenotypes were extracellular vesicle-mediated. By definition, small extracellular vesicles (sEVs) are produced in the multivesicular bodies (in the case of exosomes) or at plasma membrane budding structures (in the case of microvesicles) of parent cells and are released into the extracellular space. sEVs can have diverse signaling capabilities determined by their cell of origin, target cells and the cargo they carry, which includes miRNAs, mRNAs, signaling proteins, lipids, and enzymes (van Niel et al., 2018). Thomou et. al. have shown that adipocytes supply the majority of circulating sEV-associated miRNAs in mice, which have the ability to alter gene expression in the liver (Thomou et al., 2017). Although it is unclear which cell type contributes the majority of sEVs to circulation, the Thomou et. al. study suggests that adipocyte sEVs may contribute to whole body metabolic signaling at a level that has not yet been fully appreciated.
Here we demonstrate that adipocytes undergoing mitochondria-specific energetic stress, as in the obese condition, release small EVs (sEVs) containing mitochondrial particles that are respiration-competent but oxidatively damaged. These EV-enclosed mitochondria enter circulation and are taken up by cardiomyocytes. There they elicit transient mitochondrial dysfunction of the host network and free radical production. This is not a pathological process but, instead, results in robust adaptation of the heart to combat this adipocyte sEV-imposed stress. This adaptive mechanism is advantageous to the degree that it protects the heart from ischemia reperfusion injury, a common pathology in human obesity.
Results
Adipocyte-specific mitochondrial dysfunction induces mitochondrial oxidative stress in the heart via extracellular vesicles.
To establish the mechanism of adipocyte-to-cardiomyocyte ROS-mediated communication, we utilized our inducible, tissue-specific mouse model of FtMT over-expression. In this model, herein referred to the adipo-FtMT mouse, transgene expression is dependent on the presence of doxycycline (dox) and is restricted to mature adipocytes (Fig. 1A) (Kusminski et al., 2020). As proof-of-concept, mature adipocytes differentiated from adipo-FtMT mouse adipose stromal vascular fraction (SVF), when treated with dox, displayed robust overexpression of the FtMT protein compared to cells from control mice (Fig. S1A). FtMT over-expressing cells exhibited greater oxidative stress compared to control cells, as measured by the CellROX oxidation-sensitive dye (Fig. S1B). The level of cellular oxidative stress induced by this model was, however, not severe enough to induce apoptosis, as previously shown (Kusminski et al., 2020) and verified here by TUNEL staining (Fig. S1C). Protein carbonylation (PC) adduct formation, a measure of protein damage by ROS, was determined in adipose depots in vivo. Adipo-FtMT and control mice were supplied a high fat diet (HFD) containing 600 mg/kg dox to induce FtMT overexpression in adipocytes. Subcutaneous white adipose tissue (sWAT) of adipo-FtMT mice exhibited higher PC levels, compared to control mice, whereas the epididymal white adipose tissue (eWAT) and brown adipose tissue (BAT) showed lower levels of PC in transgenic mice (Fig. S1D). This suggests that sWAT is particularly sensitive to the pro-oxidative stress elicited by FtMT overexpression. Following 3 weeks of dox-diet, cardiac tissue was assessed for PC. Higher levels of cardiac PC were detected in adipo-FtMT mice on dox-HFD compared to controls on the same diet (Fig. 1B). No changes in PC were observed in mice on a dox-chow diet (Fig. 1B). This data suggests the dietary lipid load is necessary to promote ROS production in adipo-FtMT mouse hearts with a magnitude sufficient to induce damage to proteins. Interestingly, the cardiac ROS produced in adipo-FtMT mice on a dox-HFD is likely specific to the mitochondria. This was determined by in vivo injection of MitoB, a mitochondria-targeted probe that reacts selectively with H2O2 to form the phenol product MitoP. The ratio of MitoP/MitoB was determined by mass spectrometry (LC-MS/MS); a higher ratio is proportionate to greater oxidative stress. Cardiac tissue from adipo-FtMT mice exhibited higher levels of mitochondrial H2O2 production than control mice (Fig. 1C). The degree of cardiac oxidative stress in the adipo-FtMT mouse after just 3 weeks on dox-HFD is similar to that observed in WT mice after 16 weeks on HFD compared to their respective controls (Fig. 1B and D). Thus, acute high fat feeding in the adipo-FtMT mouse mimics the cardiac ROS effect seen in chronically obese WT mice. These data have led us to the intriguing hypothesis that there is an endocrine mechanism that relays a pro-oxidant signal from adipocytes to the myocardium in vivo.
Figure 1: Adipocyte-specific mitochondrial dysfunction induces mitochondrial oxidative stress in the heart via extracellular vesicles.

A, Mouse model schematic of adipocyte-specific, doxycycline (dox)-inducible overexpression of mitochondrial ferritin (FtMT). B, cardiac protein carbonylation (PC) assay, representative of n = 5, and quantification. Adipo-FtMT PC levels were normalized to their controls on respective diets so no comparisons can be made between dox-chow and dox-HFD. C, MitoB/P measurements in cardiac tissue following 3 weeks dox-HFD. D, PC measurements in cardiac tissue from lean and obese mice (20 wk HFD) E, Serum sEV quantification following 3 weeks of dox-HFD. F, Serum sEV count from adipo-APPβ or adipo-SOD2 shRNA mice on dox-HFD diet for 2 weeks. G-I, Control and adipo-FtMT mice were placed on dox-HFD for 4 hours and given an injection of either DMSO or GW4869. J and K, representative microscopy images and quantification (corrected total cell fluorescence; CTCF) of isolated cardiomyocytes stained with CellROX following 2 hours of sEV treatment as indicated. L, A representative confocal image and quantification of PHK26-labeled adipocyte-derived sEV uptake by mature cardiomyocytes in vitro. M, Quantification of CellROX signal in isolated cardiomyocytes 2 hours post sEV treatment as indicated. Data are presented as mean ± s.e.m. *P < 0.05, ** P < 0.01. See also Figure S1 and S2.
We tested for the possible involvement of extracellular vesicles (EVs) as mediators of the observed adipocyte-to-cardiomyocyte ROS signal. Adipo-FtMT mice on dox-HFD for 3 weeks exhibited significantly higher levels of circulating sEVs than control mice, as measured by nanoparticle tracking analysis (NTA; Fig. 1E). Furthermore, sEVs isolated from the serum of adipo-FtMT mice contained higher levels of sEV markers: Alix, CD63 and CD81 compared to sEVs isolated from the same volume of control mouse serum (Fig. S1E). Additionally, circulating sEVs from adipo-FtMT mice carried more FABP4, an adipocyte enriched protein previously used to identify adipocyte-specific sEVs (Fig. S1F)(Ferrante et al., 2015). A similar effect on circulating sEVs is seen in other mouse models of mitochondrial dysfunction: the adipocyte-specific overexpression of mitochondrially-targeted amyloid precursor protein β (adipo-APPβ) or adipocyte-specific knockdown of superoxide dismutase 2 SOD2 (adipo-SOD2 shRNA; Fig. 1F) (An et al., 2019; Cox et al., 2018). These data provide evidence that the release of sEVs is a general response of the adipocyte to mitochondrial dysfunction and not an artifact of the adipo-FtMT mouse. To assess whether serum sEVs are responsible for the observed cardiac oxidative stress in the adipo-FtMT mouse, we utilized GW4869, an inhibitor of neutral sphingomyelinase (n-SMase), which restrains exosome production in many cell types (Hessvik and Llorente, 2018). We chose an acute injection of GW4869 to avoid any compensatory responses to long term treatment. GW4869 completely abolished the rise in serum sEVs in adipo-FtMT mice following 4 hours of dox-HFD feeding (Fig. 1G). GW4869 also fully prevented the induction of cardiac oxidative stress in adipo-FtMT mice as measured by PC and thiobarbituric acid reactive substances (TBARS), a measure of lipid peroxidation (Fig. 1H and I). This data provides a line of evidence that the pro-oxidant signal is relayed from adipocytes to the heart via sEVs.
Adipocyte sEVs stimulate ROS production in isolated cardiomyocytes.
We employed an in vitro culture method as a direct approach for assessing sEV ROS signaling between adipocytes and cardiomyocytes. Serum sEVs were isolated from lean control mice or control and adipo-FtMT mice fed dox-HFD for 3 weeks. Primary wild-type cardiomyocytes treated with sEVs from lean control mice displayed significantly less oxidative stress than those cells cultured with no sEVs as measured by CellROX (Fig. 1J and K). Cardiomyocytes treated with serum sEVs from control mice on a dox-HFD exhibited more oxidative stress than treatment with lean sEVs (Fig. 1J and K). Those cells treated with sEVs from adipo-FtMT mice on a dox-HFD displayed the highest oxidative stress of any conditions examined (Fig. 1J and K). A similar, but less robust effect was seen when cardiomyocytes were treated with microvesicles (large EVs; lEV) isolated from mouse serum under the above-mentioned conditions (Fig. S1G).
To determine if adipocyte-derived sEVs are directly taken up by cardiomyocytes, we purified sEVs from the conditioned media of in vitro-differentiated adipocytes, labeled them with the fluorescent dye, PHK26, and treated cardiomyocytes with these labeled sEVs. We observed that cardiomyocytes robustly took up adipocyte sEVs by assessing the amount of transferred fluorescence (Fig. 1L). Cardiomyocytes took up sEVs to the same extent if the sEVs were isolated from control or adipo-FtMT adipocytes (Fig. 1L). However, significant oxidative stress was induced in cardiomyocytes treated with sEVs from adipo-FtMT adipocytes compared to those treated with sEVs from control adipocytes (Fig. 1M). All sEV preparations used in these experiments were within the correct size range for sEVs by NTA, carried the expected exosomal markers, and displayed acceptable purity by electron microscopy (Fig. S2A–D). Adipocytes were only used for sEV production if they reached a differentiation rate greater than 80% (Fig. S2E).
sEVs derived from palmitate-treated adipocytes stimulate ROS production and cause mitochondrial dysfunction in cardiac tissue in vivo.
We confirmed that adipocyte sEVs home to the heart in vivo. sEVs were harvested from the culture media of control and adipo-FtMT adipocytes and labeled with PKH26 fluorescent dye. A fixed number of labeled particles were injected intravenously into wild-type mice. Whole mount confocal microscopy was used to determine the biodistribution of the fluorescent label. PKH26-labeled sEVs were detected in the myocardium to the same extent, regardless of the genotype of parent adipocytes (Fig. S3). Although, the heart is likely not the primary target of adipocyte sEVs as the liver contained substantially more fluorescent signal (Fig. S3).
sEVs from wild-type, healthy, cultured adipocytes did not produce oxidative stress in the heart when injected into mice on a chow diet (Fig. 2A). Next, we determined if sEVs produced by adipocytes under energetic stress could promote cardiac ROS production. We chose to use palmitate treatment of cultured adipocytes to simulate in vivo high fat feeding, which is a more physiological way to induce mitochondrial dysfunction compared to FtMT overexpression. We isolated sEVs from adipocytes treated with palmitate for 16 hours (sEVPA) for injection into WT mice. Mice were fasted for 3 hours to encourage feeding and then simultaneously provided with a HFD and an intravenous injection of 3 μg sEVPA (Fig. 2B). This experimental setup was used to provide the system with a lipid load, which seems to be required for the effect of adipocyte sEVs on the myocardial redox state (Fig. 1B). Each mouse consumed 0.191± 0.042g high-fat diet within the first hour, an amount that was sufficient to increase circulating triglycerides (Fig. S2F). In addition, an acute injection and diet regime is important to evaluate the specific effects of sEVPA on the heart in the absence of the confounding variables in the obese state. Mice treated with adipocyte sEVPA displayed significantly higher cardiac oxidative stress compared to PBS treatment, a transient effect that appeared at 1 hour and was resolved by 2 hours post injection (Fig. 2C). In contrast, PC adduct presence in the heart was significantly lower at 16 hours post sEVPA injection (Fig. 2C). Likewise, reduced mitochondria-specific oxidative stress in the heart was seen as early as 4 hours post sEVPA injection (Fig. 2D). This is consistent with an adaptive, antioxidant response, as catalase (CAT) protein was significantly increased 2 hours after sEVPA exposure (Fig. 2E). We determined if adipocyte sEVPA-induced ROS is an indication of mitochondrial dysfunction. Cardiac mitochondria isolated 1 hour after sEVPA injection displayed significantly reduced electron transport chain activity when utilizing fatty acids, the main energetic substrate of cardiomyocytes (Fig. 2F). 2 hours post sEVPA injection cardiac mitochondrial palmitoyl-carnitine oxidation was restored and uncoupled respiration was enhanced above that seen with PBS injection (Fig. 2G and H). Furthermore, at 2 hours, mitochondrial pyruvate oxidation was augmented in mice injected with sEVPA (Fig. S4A). Likewise, when isolated cardiomyocytes were treated in vitro with adipocyte sEVPA for 2 hours, basal and FCCP-stimulated OCRs were significantly higher than in non-treated cells when glucose and pyruvate were supplied as energetic substrates (Fig. S4B and C). Furthermore, this timepoint was also concurrent with a compensatory antioxidant effect in cardiomyocytes (Fig. S4D). These data suggest sEVPA may induce cardiac ROS production by causing mitochondrial dysfunction. However, this stress is rapidly met by cardiomyocyte adaptive mechanisms to restore energetic and redox homeostasis. sEVPA from adipo-FtMT adipocytes also displayed this adaptive response but with faster kinetic than that from WT cells. At 1 hour after injection cardiac catalase mRNA and protein were increased and at 2 hours oxidative stress was significantly decreased (Fig. S4E–G). It is important to note that the sEV dose chosen for injection (1 ×109 particles) is an estimated 0.033% of endogenous circulating sEVs in mice. Thus, the injected EVs could not be detected in tail vein serum (Fig. S4H). This physiological dose of sEVs has a surprisingly robust effect on cardiac redox state (Fig. 2).
Figure 2: sEVs from palmitate-stressed adipocytes induce cardiac ROS in vivo.

A, Protein carbonylation (PC) assay on cardiac tissue from chow-fed wild-type mice treated 2 hours after injection of sEVs from healthy adipocytes. B, Experimental design for C-H. C, PC determination in heart tissue post sEVPA injection at the indicated timepoints. D, mitoP/B ratio in cardiac tissue 4 hours post sEVPA injection. E, Catalase (CAT) protein expression in whole cardiac tissue 2 hours following a sEVPA injection. F and G, Seahorse Analysis of cardiac mitochondria isolated from mice 1 hour (F) or 2 hours (G) following the indicated injections. Palmitoyl-carnitine and malate were provided as energetic substrates. H, Palmitoyl-carnitine oxidation in isolated cardiac mitochondria 2 hours post sEVPA injection as measured by optical oxygen respirometry. Data are presented as mean ± s.e.m. *P < 0.05, ** P < 0.01, *** P < 0.001. See also Figure S3 and S4.
Mitochondrial energetic stress stimulates the release of sEVs from adipocytes.
We wanted to assess the relationship between adipocyte mitochondrial dysfunction and sEV release. We show that mature, in vitro- differentiated adipocytes overexpressing FtMT release more sEVs into the culture media (Fig. 3A). Furthermore, exposure of white or brown adipocytes to a variety of electron transport chain (ETC) inhibitors elicited a strong increase in sEV release (Fig. 3B and Fig. S5A). Enhanced release of sEVs could be detected as soon as 6 hours of antimycin A treatment, but the most robust sEV production was seen at 20 hours of antimycin A or Oligomycin treatment (Fig. 3C). All compounds were used at non-toxic concentrations. As an example, no cell death was detected in antimycin A-treated adipocytes above those treated with vehicle (Fig. S5B). Treating adipocytes with buthionine sulfoximine (BSO), a compound that causes oxidative stress by depleting cellular glutathione, did not affect sEV production (Fig. S5C), suggesting that the signal that triggers sEV release is specific to mitochondrial stress. A more physiological energetic stress can be exerted on adipocytes by treatment with palmitate, a condition that also enhanced sEV release (Fig. 3D). Likewise, the sWAT depot of wild-type mice fed a HFD for 3 days or 3 weeks produced significantly more sEVs when incubated ex vivo than those from chow-fed mice (Fig. S5D). This acute duration of HFD feeding is sufficient to induce high sWAT H2O2 production (Fig. S5E) and sWAT mitochondrial oxidative damage (Fig. S5F), with no change in total mitochondrial content (Fig. S5G). FTMT overexpression, ETC inhibition, and palmitate treatment cause mitochondrial dysfunction through different mechanisms thus, sEV release seems to be a general response to adipocyte mitochondrial energetic and oxidative stress.
Figure 3: Adipocytes undergoing energetic stress release respiration-competent but oxidatively damaged mitochondria in sEVs.

A, sEVs released from dox-treated, in vitro-differentiated, control and adipo-FtMT adipocytes. B-D, sEV production by in vitro-differentiated wild type adipocytes treated with the indicated compounds. Media was harvested for sEV quantification after 24 hours of treatment (B and D) or the timepoints indicated in C. E, Western blot was performed for mitochondrial proteins in sEVs isolated from the serum of control or adipo-FtMT mice on dox-HFD for 3 weeks (representative of n=3). F, Optiprep density gradient purification of serum sEVs isolated from adipo-FtMT mice fed dox-HFD for 3 weeks. G and H, Mitochondrial proteins were assessed in sEVs from wild type in vitro-differentiated adipocytes (G; representative of n=3) and dox-treated in vitro-differentiated control and adipo-FtMT adipocytes (H; representative of n=5). I and J, Western blot and densitometry for mitochondrial proteins in sEVs produced by primary in vitro-differentiated adipocytes treated as shown. K, Mitochondrial protein content in serum sEVs from lean and obese (20 weeks HFD) wild-type mice. L and M, Seahorse Flux Analysis of isolated serum sEVs from control or adipo-FtMT mice fed dox-HFD for 3 weeks. N, Protein carbonylation (PC) assay on isolated sEVs from control and adipo-FtMT mice on dox-HFD for 3 weeks or conditioned media from adipocytes treated as indicated. sEVs recovered from equal amounts of media or serum were loaded into the SDS-PAGE gels. Abbreviations: oligo, oligomycin; Anti A, antimycin A; PA, palmitate; Ad, adipocyte. Data are presented as mean ± s.e.m. *P < 0.05, ** P < 0.01, *** P < 0.001. See also Figure S5 and S6.
Stress-stimulated adipocyte sEVs are enriched with respiration-competent mitochondrial particles.
To determine the molecular mediators of the adipocyte sEV pro-oxidant properties, we performed an unbiased proteomics screen of serum sEVs from control and adipo-FtMT mice on dox-HFD for 3 weeks. Surprisingly, the most highly enriched proteins in adipo-FtMT serum sEVs were of mitochondrial origin (Fig. S6). We confirmed this finding by Western blot analysis. Adipo-FtMT serum sEVs contained higher levels of VDAC, HSP60 and COXIV compared to sEVs from control mice (Fig. 3E). To confirm that the observed mitochondrial proteins are enclosed in exosome-like vesicles and not simply contaminating mitochondria, we utilized an OptiPrep density gradient to purify sEVs isolated from the serum of adipo-FtMT mice on a 3 week dox-HFD. The mitochondrial markers VDAC and COXIV equilibrated in the same fractions as known exosomal markers ALIX, CD63 and CD9, at the published exosomal density range of 1.1–1.18 g/ml (Fig. 3F). Recent studies have reported the existence of mitochondrial fatty acid oxidation enzymes and mitochondrial DNA in exosome-like vesicles (Lazar et al., 2016; Sansone et al., 2017). We found that in vitro-differentiated adipocytes do indeed basally release sEVs containing mitochondrial proteins VDAC, HSP60 and COXIV (Fig. 3G). When adipocytes are under the energetic stress provoked by FtMT overexpression, more VDAC was detected per μg of sEV protein compared to control cells (Fig. 3H). A similar result was seen when adipocytes were treated with mitochondrial ETC inhibitors. All inhibitors increased the content of mitochondrial proteins in sEVs but, in general, the amount of mitochondria was proportional to the amount of sEVs released (Fig. 3I). The exception was FCCP, a treatment that resulted in a disproportionate increase in mitochondrial protein incorporation into sEVs (Fig. 3I). Likewise, palmitate treatment also resulted in more vesicle-associated mitochondrial protein release (Fig. 3J). Furthermore, sEVs isolated from obese mouse serum contain more mitochondrial proteins compared to those from lean mice, suggesting this phenomenon has physiological relevance (Fig. 3K). These data indicate that both a total increase in sEV production and inclusion of mitochondrial proteins in sEVs are characteristic responses of adipocytes to energetic stress.
As both mitochondrial DNA and protein have been reported in EVs, we tested if intact, respiration-competent mitochondria are present in serum sEVs. We developed a Seahorse protocol that enables us to measure oxygen consumption of exosome-like vesicles. We were surprised to find a detectable pyruvate-supported oxygen consumption rate (OCR) in sEVs isolated from the serum of control mice on dox-HFD for 3 weeks (Fig. 3L). sEVs from an equal volume of adipo-FtMT serum exhibited an even higher basal OCR than control samples after the same feeding regime, consistent with our observation that sEVs from these transgenic mice contain more mitochondrial proteins (Fig. 3E and L). This OCR was abolished by addition of digitonin to disrupt biological membranes (Fig. 3L). sEVs from mice of both genotypes responded to the conditions of the Seahorse mitochondrial electron flow assay as would be expected from bona fide mitochondria: decreased OCR with rotenone and antimycin A, and increased OCR with succinate (Fig. 3L). Furthermore, all sEVs displayed higher rates of oxygen consumption when supplied with ADP, suggesting these enclosed mitochondrial particles have the capacity to generate ATP (Fig. 3M). However, despite more mitochondrial proteins, sEVs from adipo-FTMT mouse serum did not display higher succinate- or ADP-stimulated OCR than control sEVs suggesting less functional mitochondria (Fig. 3L and M). We tested if the sEV components are oxidatively-damaged, as they are derived from adipocytes under a strong mitochondrial stress. sEVs isolated from the serum of adipo-FtMT mice contained a greater amount of PC-adducts compared to those sEVs from control mice (Fig. 3N). Similarly, sEVs from the culture media of in vitro differentiated adipocytes treated with antimycin A displayed more PC than those from cells treated with vehicle (Fig. 3N). Therefore, sEVs produced from adipocytes under oxidative stress likely contain oxidatively damaged, yet functional, mitochondrial components.
Mitochondria packaged into stress-induced sEVs originate as mitochondrial-derived vesicles
The properties of these mitochondrial particles are reminiscent of a class of intracellular vesicles called mitochondrial-derived vesicles (MDVs). MDVs form in response to mitochondrial oxidative stress by budding from the mitochondrial network (Fig. 4A) to selectively deliver damaged mitochondrial components to the endo-lysosomal system, in addition to initiating other inter-organellar signaling (McLelland et al., 2014; Soubannier et al., 2012a). This quality control mechanism relies on Pink and Parkin but is independent of classic mitochondrial fission or autophagy machinery like Drp1 and LC3 (Soubannier et al., 2012a). Unlike mitochondria undergoing mitophagy, MDVs retain a membrane potential (Soubannier et al., 2012a). MDVs enter multivesicular bodies to be processed for degradation, the same compartment in which exosomes are formed. Therefore, it is also possible that MDVs can escape lysosomal degradation by release through exosomes, a theory that has also been suggested by others and recently shown in mouse embryonic fibroblasts (Sugiura et al., 2014; Todkar et al., 2021). To test this model, we first determined if sWAT mitochondria from adipo-FtMT mice produced more MDVs than those from control mice. Using a previously described method of in vitro reconstitution of MDV formation (Soubannier et al., 2012b), with minor modifications, we can demonstrate that mitochondria isolated from the sWAT of adipo-FtMT mice do produce more MDVs. This is assessed by NTA (Fig. 4B) and quantification of membrane-protected mitochondrial proteins released into the assay buffer (Fig. 4C and D). Antimycin A treatment was used as a positive control (Fig. 4C and D). When mitochondria are subjected to antimycin A-induced oxidative stress, damaged components are shuttled as MDVs into the endo-lysosomal system (Soubannier et al., 2012a). This is demonstrated by greater co-localization of TOM20 and with the endosomal marker EEA-1, when adipocytes are treated with antimycin A compared to those treated with vehicle (Fig. 4E). If lysosomal degradation of these MDVs is prevented by incubation of adipocytes with chloroquine (CQ), in addition to antimycin A, sEVs are further enriched with COXIV (Fig. 4F). These data suggest the mitochondrial proteins in sEVs are of endosomal origin and likely MDVs. In addition, sEVs released from Parkin−/− adipocytes displayed reduced packaging of mitochondrial proteins in sEVs (Fig. 4G). If we inject wild type mice with sEVs from palmitate-treated Parkin−/− adipocytes we did not detect ROS production in the heart compared to that seen for wildtype-derived sEVPA (Fig. 4H), suggesting the mitochondria packaged into sEVs is a major contributor to the observed induction of cardiac ROS.
Figure 4: Mitochondrial-derived vesicles are packaged into sEVs.

A, Electron micrograph of a mitochondrion displaying a budding structure (left) and released MDVs (right). B, Nanoparticle tracking (NTA) quantification of MDVs released from isolated mitochondria from control and adipo-FtMT sWAT. C and D, VDAC and HSP60 protein content in MDVs released from isolated sWAT mitochondria under the specified conditions. E Immunofluorescent co-stain for TOM20 (mitochondria) and EEA1 (endosomes) with or without antimycin A exposure. F, COXIV content in sEVs isolated from in vitro-differentiated adipocytes treated as indicated (anti-A, antimycin A; CQ, chloroquine). G, Mitochondrial proteins in sEVs isolated from the media of in vitro-differentiated adipocytes of the respective genotypes. sEVs recovered from equal amounts of media were analyzed by Western blot. H, Cardiac PC 1 hour following sEVPA injections from adipocytes with the indicated genotypes. Data are presented as mean ± s.e.m. *P < 0.05, ** P < 0.01, *** P < 0.001.
Adipocyte sEV-associated mitochondria enter circulation and are incorporated into the cardiac mitochondrial network.
To visualize MDV transfer from adipocytes to the myocardium in vivo, we generated an inducible mouse model that expresses a mitochondrially-localized Flag tag selectively in the adipocyte (adipo-mitoFlag; Fig. 5A and B). Following 11 weeks of dox-HFD diet the Flag tag was detected in both serum sEVs and whole cardiac tissue of adipo-mitoFlag mice (Fig. 5C). The Flag signal was localized to cardiomyocytes, as determined by immunofluorescent staining of heart sections for both Flag and cardiac troponin (CTN1; Fig. 5D). Moreover, adipocyte-derived mitochondria likely integrated into the cardiomyocyte network as the Flag signal was enriched in isolated cardiac mitochondria following a chronic dox-HFD (Fig. 5E). As these mitochondria are likely damaged, integration into the host mitochondrial network could explain the mitochondrial dysfunction and ROS generation detected in the heart following sEVPA injection (Fig. 2). The transfer of adipocyte mitochondrial Flag tag to the heart was not detected in mice on dox-HFD for just 3 weeks (Fig. S7A). In addition, sEVs isolated from serum of wild-type mice on a HFD for 3 days or 3 weeks did not display higher levels of mitochondrial VDAC compared to those from chow-fed mice (Fig. S7B). Thus, the systemic migration of mitochondria from the adipocyte may only occur during chronic obesity, unless this process is accelerated by a secondary insult such as FTMT overexpression or a bolus injection of sEVPA. However, even after chronic dox-HFD feeding the Flag tag signal detected in the heart is weak. This is likely because the Flag tag with its associated mitochondria are degraded soon after entry into the cardiomyocyte, as would be expected for damaged mitochondria. In keeping with this, the Flag tag was detected in the heart of WT mice just 1 hour after a bolus injection of sEVs from palmitate-stressed mitoFlag adipocytes (Fig. 5F). If mice were pre-injected with chloroquine (CQ) to inhibit lysosomal degradation, more flag accumulated in the heart after injection (Fig. 5F). Furthermore, when WT mice are injected with sEVs from palmitate-stressed primary human adipocytes, human mitochondrial DNA could be readily detected in the mouse heart, and more so if mice were pre-injected with chloroquine (Fig. 5G). Therefore, cardiac mitochondrial quality control mechanisms likely ensure that the damaged mitochondria from adipocytes do not result in sustained disruption of mitochondrial metabolism in cardiomyocytes.
Figure 5: Mitochondria released from adipocytes enter circulation and incorporate into the cardiomyocyte mitochondrial network.

A, Confocal microscopy image that demonstrates the mitochondrial localization (TOM20) of the Flag tag in dox-treated, in vitro-differentiated adipocytes from adipo-mitoFlag mice. B and C, mitoFlag detection by gene expression (B) and immunoprecipitation (C) in heart or sWAT tissue from adipo-mitoFlag mice fed dox-HFD for 11 weeks. D, Confocal microscopy images demonstrating the localization of the Flag tag in cardiomyocytes (CTN1 positive) with no primary antibody control and no mitoFlag transgene control. Arrow specifies non-specific signal and asterisks are supplied for orientation. E, Immunoprecipitated Flag tag in isolated cardiac mitochondria or whole sWAT from the adipo-mitoFlag mice on dox-HFD for 11 weeks. F, sEVs from in vitro differentiated adipocytes expressing mitoFlag and treated with palmitate were injected into WT mice. Where indicated, mice were injected with chloroquine (CQ) 3 hours prior to sEV injection. The Flag tag was immunoprecipitated from heart tissue 1 hour following sEV injection. G, sEVs from primary human (Hu) adipocytes treated with palmitate were injected into wild type mice with or without a CQ pre-injection. Human mtDNA was quantified in mouse heart tissue 1 hour following sEV injection and extensive perfusion with PBS. Data are presented as mean ± s.e.m. *P < 0.05, ** P < 0.01, *** P < 0.001. See also Figure S7.
Evidence of the adipocyte sEV-mediated stress signal in humans.
Recent studies have demonstrated the presence of mitochondrial proteins in plasma sEVs from human subjects and changes in sEV mitochondrial abundance with melanoma (Jang et al., 2019; Karimi et al., 2018). Therefore, it is possible circulating mitochondria, at least partially derive from adipocytes, and mediate a stress signal in human obesity. Human SGBS cells (Fischer-Posovszky et al., 2008; Wabitsch et al., 2001) were differentiated into adipocytes and subjected to various treatments to disrupt mitochondrial function. Palmitate treatment resulted in a strong trend toward increased sEV release, whereas antimycin A, oligomycin and FCCP significantly increased sEV release (Fig. 6A). In addition, plasma samples from metabolically unhealthy obese (MUO) patients contained significantly higher sEV levels compared to metabolically healthy lean controls (MHL; Fig. 6B). In contrast, no change in circulating sEV content was detected between lean and metabolically healthy obese individuals (MHO), although there was a trend toward higher levels in the MHO group (Fig. 6B). Furthermore, sEV-associated mitochondrial DNA (mtDNA) was robustly detected in the MUO group, whereas mtDNA was undetectable in the MHL and MHO sEVs in all but 1 subject sample (Fig. 6C). However, the majority of mtDNA was found in the lEV population and not changed between groups (Fig. 6D). Interestingly, both obese groups, regardless of metabolic health, displayed more mitochondrial protein and oxidized proteins in sEVs compared to the lean cohort (Fig. 6E and F). These data suggest there may be a physiological role for circulating sEV-associated mitochondria in obese humans.
Figure 6: Obese, metabolically unhealthy humans display high circulating sEVs that carry mitochondrial DNA.

A, sEV quantification of conditioned media from human SGBS cells differentiated into mature adipocytes under the indicated conditions (n=3–6). sEVs were isolated from the plasma of metabolically healthy lean (MHL), metabolically healthy obese (MHO) and metabolically unhealthy obese (MUO) human patients. In these samples EVs were: B, counted (n=5), C-D, mitochondrial DNA (mtDNA) was quantified (n=5), E, Mitochondrial protein was quantified (n=8) and F, oxidatively damaged proteins were assessed by the PC assay (n=4). Data are presented as mean ± s.e.m. *P < 0.05, ** P < 0.01, *** P < 0.001.
Adipocyte sEVs protect cardiac tissue during acute myocardial infarction in mice.
Obesity is a strong, independent risk factor for the development of CVD (Lavie et al., 2018; Poirier et al., 2006). Furthermore, circulating sEV have diagnostic, prognostic and therapeutic potential in CVD (Bank et al., 2015). Therefore, we went on to determine if adipocyte sEV-mediated ROS production in cardiomyocytes is pathological in the context of acute myocardial infarction, a common event in overweight and obese patients (Thomsen and Nordestgaard, 2014). Mice were injected with sEVPA under the lipid-rich conditions previously determined to stimulate mitochondrial dysfunction and free radical production in the heart (Fig. 2). 2 hours post injection, in vivo cardiac ischemia/reperfusion (IR) was performed by occlusion of the left anterior descending artery (LAD) for 45 minutes, and release of the ligature for reperfusion. Cardiac function was measured by echocardiography before IR as well as 1 day and 7 days after IR (Fig. 7A). TTC stain and plasma cardiac troponin I (CTNI) were used as measures of myocardial injury 24 hours post IR. Surprisingly, mice who received the pre-ischemia sEVPA injection displayed a significant reduction in circulating CTNI, compared to those who received PBS (Fig. 7B). TTC staining of cardiac tissue also demonstrated reduced infarct size in mice treated with sEVPA from WT adipocytes but not from parkin−/− adipocytes (Fig. 7C). This data suggests that the protective outcome correlates positively with the amount of mitochondria in the sEVsPA and the pro-oxidant effect of the sEVsPA (Fig. 4G–H). At 7 days after IR, sEVPA-treated mice presented with slightly smaller hearts, regardless of normalization to body weight, suggesting a reduction in IR-induced cardiac hypertrophy (Fig. 7D). Mice treated with sEVPA also displayed a significant reduction in infarct size 7 days post IR, measured by Masson’s Trichrome stain for scar tissue (Fig. 7E). No changes in fibrosis at the border zone were detected between treatment groups (Fig. 7F). In addition, sEVPA- treated mice exhibited less of the lipid peroxidation product 4-hydroxynonenol (4-HNE) at the site of infarction (Fig. 7G). Ejection fraction (EF) and fractional shortening (FS) were significantly preserved in sEVPA-treated mice relative to PBS-injected mice 7 days after IR (Fig. 7H). As such, the expected drop in EF from before IR to 7 days post-IR was significantly attenuated in mice pre-treated with sEVPA (Fig. 7I). Taken together, these data indicate that under the tested conditions, the adipocyte-to-cardiomyocyte ROS relay does not participate in pathology but instead fosters cardiac resilience.
Figure 7: Adipocyte sEVs produced under palmitate stress protect the heart from ischemia/reperfusion injury.

A. Experimental design for ischemia/reperfusion experiments (IR) in B-I. B, plasma cardiac troponin (CTNI) measurements and C, TTC stain of cardiac tissue 24 hours after IR. D, Absolute (left) and normalized (right) heart weight 7 days post IR. E, representative Masson’s Trichrome stain of cardiac histological sections and quantification of infarct size 7 days after IR. F, 20 x magnification of cardiac Masson’s Trichrome stain at the specified regions. G, 4-hydroxynonenal (4-HNE) stain in heart tissue at 7 days post IR. H, Cardiac functional parameters before (day 0) and at the denoted times after IR. I, Percentage drop in ejection fraction from before to 1 day and 7 days following IR. Data are presented as mean ± s.e.m. *P < 0.05, ** P < 0.01, *** P < 0.001.
Discussion
High cardiac ROS production makes a destructive contribution to the pathogenesis of cardiovascular diseases (CVD), particularly those involving ischemic injury (Senoner and Dichtl, 2019). However, low levels of ROS are appreciated to offer cardio-protection through hormesis (Antonucci et al., 2019) (Calabrese and Mattson, 2017). In fact, performing short cycles of occlusion and reperfusion at the coronary artery prior to a prolonged ischemic event (ischemic preconditioning, IPC) has a powerful cardioprotective outcome. IPC has this beneficial effect because it induces moderate levels of ROS, which stimulates adaptation of the tissue, bolstering the cellular defense system so that severe ROS production during extended ischemia is better mitigated (Dhalla et al., 2000). Here we demonstrate a natural occurrence that is reminiscent of IPC but is of metabolic, not hypoxic, origin. In the obese state, adipocytes undergo intense energetic and oxidative stress to which they respond by releasing sEVs that harbor a mitochondria-mediated pro-oxidant signal. The resulting ROS production in the cardiac muscle provokes adaptations that prevent injury during ischemia, a probable pathology in obese individuals. The sEV-stimulated cardiac adaptation includes enhanced efficiency of energy production via utilization of pyruvate (Fig. S4A), a reduction in ROS production via mitochondrial uncoupling (Fig. 2H) and enhanced antioxidant capacity via catalase upregulation (Fig. 2E), all of which are favorable conditions during IR (Dhalla et al., 2000; Ozcan et al., 2013; Ussher et al., 2012). However, like all hormetic responses, there is likely a threshold of ROS production or time of ROS exposure that, when surpassed, would promote damage instead of protection. Further work will be needed to determine if there is a point in the progression of obesity at which adipocyte sEVs indeed promote CVD. This is likely the case as others have demonstrated that adipocyte- or adipose tissue-derived sEVs promote macrophage differentiation and activation in a way that drives atherosclerotic plaque formation, and negatively impacts cardiac function (Gan et al., 2020; Kranendonk et al., 2014; Xie et al., 2018). Furthermore, adipocyte sEVs promote insulin resistance in myocytes and adipocytes and enhance fibrotic signaling in hepatocytes (Deng et al., 2009; Koeck et al., 2014; Mleczko et al., 2018; Pan et al., 2019); a systemic environment that is unfavorable for cardiovascular health.
Although adipose tissue displays signs of oxidative stress as soon as 1–3 days of HFD feeding and responds by enhancing sEV release (Fig. S5), the amount of sEV-associated mitochondria remain at basal levels (Fig. S7B). In addition, there is no sign of adipocyte-cardiomyocyte mitochondrial transfer (Fig. S7A). Only during chronic obesity, with more pronounced adipocyte energetic stress, do we detect enhanced mitochondrial packaging into sEVs, and transfer to the heart (Fig. 3K, 5C, S7A). Furthermore, the cardioprotective effects of stress-induced adipocyte sEVs is not associated with an overall increase in sEVs, but instead, the presence of damaged mitochondria in them (Fig. 4G and H, Fig. 7C). For this reason, sEV-mediated preconditioning may only be effective in mice with established obesity and not during acute high fat feeding. The content and role of sEVs released from adipocytes during acute HFD feeding are still under investigation. The adipo-FtMT mouse model allowed us to study this process in a much more acute timeframe, as circulating mitochondria were increased at just 3 weeks of dox-HFD feeding in the transgenic mice (Fig. 3E). This is likely because FtMT overexpressed induces intense adipocyte oxidative stress that mimics that seen in chronically obese WT mice.
In keeping with these findings in mice, we have shown here that human adipocytes also increase sEV production in response to mitochondrial stress (Fig. 6A). In addition, obese, metabolically unhealthy individuals have more circulating sEVs that carry more mitochondrial DNA, mitochondrial protein and oxidized proteins compared to lean individuals (Fig. 6). These data provide some evidence that circulating mitochondria may have a hormetic role in humans as we have reported in mice. These findings are in line with the clinical observation that although obese humans have a higher incidence of CVD, they have a better short-term and medium-term prognosis than lean individuals with CVD, a phenomenon called the “obesity paradox” (Lavie et al., 2014). In rodent models, the literature has provided mixed conclusions. Some demonstrate that obese mice and rats sustain less IR-induced injury than their lean counterparts ((Donner et al., 2013; du Toit et al., 2008; Edland et al., 2016; Salie et al., 2014; Thakker et al., 2006; Webster et al., 2017), whereas others report the opposite (Aoyagi et al., 2015; Clark et al., 2011; Jones et al., 1999). There are likely multiple factors that contribute to this observed “obesity paradox” in both rodents and humans. It is possible this adipocyte sEV-mediated preconditioning mechanism may contribute.
We determined that the pro-oxidant signal relayed from the adipocyte to the cardiomyocyte is the physical transfer of damaged mitochondria (MDVs) via sEVs. The imposed oxidative stress likely occurs through incorporation of sEV mitochondria into the cardiomyocyte mitochondrial network, inducing temporary dysfunction of the host network. Because these MDVs are derived from the endosome of the adipocyte, they are likely incorporated into bona fide exosomes. The MDVs produced in the reconstitution assay (Fig. 4B) had a varied size distribution of 30–500 nm. It is expected that only the smaller range of MDVs (30–150 nm) is packaged into exosomes due to the physical size restraints. MDVs may also exist in other larger forms of EVs, like microvesicles, which could accommodate the entire MDV size range. The physiological role of MDV-release from cells is likely one of cell autonomous protection. It has recently been reported that cardiomyocytes expel damaged mitochondria in an autophagy-dependent manner via a form of EVs called exophers (Nicolas-Avila et al., 2020). These exophers are taken up and degraded by tissue macrophages. The ability of the cardiomyocytes to “outsource” mitophagy is essential for these long-lived cells to preserve metabolic homeostasis under intense mechanical and energetic stress. Therefore, in the context of the adipocyte, it is possible a similar mechanism is employed, where damaged mitochondria are released to be degraded by tissue macrophages. In fact, it has been shown that adipocyte derived EVs and mitochondria are actively taken up by adipose tissue macrophages (Brestoff et al., 2021; Flaherty et al., 2019). Therefore, this mechanism likely evolved as a protective pathway but, in the case of adipocytes, the EVs that evade tissue macrophages can enter circulation and moonlight as a potent stress warning signal. In obesity this warning signal may be, at least acutely, important to protect other organs from lipotoxic stress as adipose tissue function declines.
This is, to our knowledge, the first vertebrate example of what could be considered “inter-organ mitohormesis”, where an exogenously imposed mitochondrial stress is sensed by the adipocyte, which responds in a way that propagates this signal to other organ systems. A similar phenomenon has been shown in C. elegans where Durieux et. al. knocked down cytochrome c oxidase-1 (cco-1) to reduce mitochondrial function. This is a well-known condition that extends the lifespan of c. elegans through activation of the mitochondrial unfolded protein response (UPRmt) (Durieux et al., 2011). Interestingly, knocking down cco-1 specifically in the brain increased longevity of the whole organism, which was the result of brain-derived, wnt ligand/EGL-20-mediated activation of the UPRmt in peripheral organs (Durieux et al., 2011; Zhang et al., 2018). In contrast to this work in C. elegans, the evidence in our study does not point to a “mitokine” that activates a protective pathway in a distal cell. Instead, we discovered that the direct transfer of damaged mitochondria, evokes adaptation in the receiving cell to provide a cell-autonomous mechanism of protection. Cardiac ROS production, and protection from IR injury was not seen when sEVs from Parkin−/− adipocytes were injected into mice, a condition we found reduces mitochondrial packaging into sEVs (Fig. 4G and H). This suggests the adaptive response is mediated by sEV-associated mitochondria. However, because sEVs contain many factors, we cannot rule out the possibility that mitochondria-independent signaling pathways also contribute to EV-mediated cardio protection. Future work will elucidate adipocyte sEV-induced signaling events in the heart and determine how they may be manipulated for pharmacological intervention in obesity-associated CVD.
Limitations of study:
There are noteworthy limitations of our study. The first is the lack of genetic tools to specifically and inducibly reduce EV release from adipocytes. Future work will focus on developing such a mouse model, which would allow us to test the impact of endogenously produced sEVs, specifically from adipocytes, on cardiovascular outcomes in the obese state. Additionally, we have found that pre-ischemic injection of adipocyte sEVs reduces the initial infarct size during IR, consistent with a preconditioning model. However, this confounds any analysis of potential effects of sEVs on tissue remodeling following IR, as initial infarct size is a major determinant of remodeling. Therefore, further work will be aimed at understanding whether the strongest effects of adipocyte sEVs are on initial infarct size or subsequent remodeling.
STAR Methods
RESOURCE AVAILABILITY
Lead Contact
Information and requests for reagents and mice should be directed to the Lead Contact, Philipp Scherer (Philipp.Scherer@UTSouthwestern.edu)
Materials Availability
All data or other materials/mouse models are available from the corresponding author upon request. Correspondence to Philipp E. Scherer.
Data and Code Availability
Proteomics data can be accessed at MassIVE, massive.ucsd.edu; accession #: MSV000085896
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals –
Transgenes were inducibly and specifically overexpressed in adipocytes using a Tet-On approach. To this end, mouse lines carrying an adiponectin promoter-driven reverse tetracycline transactivator (rtTA) and a tetracycline response element (TRE) promoter-driven transgene of interest were crossed. The TRE-FtMT and TRE-mitoAPPβ mouse lines were previously published by Kusminski et al. (Kusminski et al., 2020) and An et al., (An et al., 2019)respectively. The TRE-SOD2 shRNA mouse line was a generous gift from Gerald Shadel (Salk Institute for Biological Studies, La Jolla, CA, USA). All mice used I n this study were males on a C57Bl/6 background. Control mice were littermates containing only the adiponectin-rtTA transgene.
Generation of TRE-mitoFlag mice –
A mitoFlag DNA cassette encoding an mRuby2-based, FLAG-tagged ‘spaghetti monster’ fluorescent protein (Viswanathan et al., 2015), triple glycine linker, and mitochondria-targeting fragment of rat synaptojanin-2-binding protein (amino acids 109–145)(Nemoto and De Camilli, 1999) was synthetized by GENEWIZ (South Plainfield, NJ, USA) and cloned into the pTRE WPRE vector (sequence available upon request). The TRE-mitoFlag transgene was liberated from this vector, purified, and injected into fertilized C57Bl/6 oocytes. Injected oocytes were transplanted into pseudo-pregnant foster mothers and offspring was screened for transgene insertion using the following primers: 5’-ATA GAA GAC ACC GGG ACC GA-3’ and 5’-GGC CAG GAT ATC GAA AGC GA-3’. Specific induction of mitoFlag expression was screened for by crossing TRE-mitoFlag positive offspring to adiponectin-rtTA mice.
Human Subjects –
26 men and women participated in this study, which was approved by the Human Research Protection Office at Washington University School of Medicine in St. Louis, MO (ClinicalTrials.gov number NCT02706262). All subjects provided written consent, informed consent before their participation. A comprehensive medical evaluation was completed by all subjects to determine eligibility, which included a history and physical examination, standard blood tests, hemoglobin A1C (HbA1c), an oral glucose tolerance test (OGTT) and magnetic resonance imaging (MRI) (3-T MAGNETOM Vida; Siemens) to determine intrahepatic triglyceride (IHTG) content. The subjects were divided into 3 groups:
metabolically healthy lean (MHL), defined as a BMI 18.5–24.9 kg/m2; IHTG content ≤5%, serum triglyceride <150 mg/dL, fasting plasma glucose <100 mg/dL, 2-h OGTT plasma glucose ≤140 mg/dL, and HbA1c ≤5.6.
metabolically healthy obese (MHO), defined as BMI 30–49.9 kg/m2; IHTG content ≤5%, serum triglyceride <150 mg/dL, fasting plasma glucose <100 mg/dL, 2-h OGTT plasma glucose ≤140 mg/dL, and HbA1c ≤5.6.
metabolically unhealthy obese (MUO), defined as BMI 30–49.9 kg/m2, IHTG content ≥5.6% and HbA1c ≥5.7% or fasting plasma glucose ≥100 mg/dL or 2-h OGTT plasma glucose ≥140 mg/dL.
No subject had a history of diabetes, took medications known to affect the study outcomes, or consumed excessive amounts of alcohol (>21 units of alcohol/week for men and >14 units for women).
SGBS Cell Culture and Differentiation –
Human SGBS cells were cultured in culture media (DMEM-F12, 1X penicillin-streptomycin, 33μM biotin, 17μM pantothenate and 10% FBS). Cells were grown to 80–90% confluence at 37°C and 5% CO2. SGBS cells were differentiated into adipocytes with differentiation media (culture media supplemented with 0.01 mg/ml transferrin, 20 nM insulin, 100 nM cortisol, 0.2 nM T3, 25 nM dexamethasone, 250 μM 3-isobutyl-1-methylxanthine [IBMX] and 2μM rosiglitazone) for 4 days. After which, the media was changed to maintenance media (culture media supplemented with 0.01 mg/ml transferrin, 20 nM insulin, 100 nM cortisol, 0.2 nM triiodothyronine [T3]). Cells were allowed to differentiate for a further 10 days before experiments were conducted
Isolation of stromal vascular cells (SVF) and generation of adipocytes –
Inguinal fat pads were dissected from 4–6 week-old mice between. Fat tissues were minced and digested for 1 hour at 37°C in buffer containing 100 mM HEPES, 120 mM NaCl, 50 mM KCl, 1.5% BSA, 5 mM glucose, 1 mM calcium and 1 mg/ml collagenase D. The dispersed tissue was then filtered through a 100 μm cell strainer, and centrifuged at 600 g for 5 minutes at 4°C. The pelleted SVF cells were resuspended in culture media (DMEM/F12 media containing 10% FBS, GlutaMax, 1X penicillin-streptomycin and gentamicin) and filtered through a 45 μm cell strainer. Cells were centrifuged again as described above. The pelleted cells were resuspended in culture media and grown at 37°C, 5% CO2. For in vitro differentiation experiments, SVF cells were allowed to grow to ~95% confluency. Adipogenesis was induced by culture media supplemented with 500 μM 3-isobutyl-1-methylxanthine (IBMX), 1 μM dexamethasone, 5 μg/ml Insulin and 1 μm rosiglitazone for 2 days. Following the 2 days of induction, cells were maintained in media containing only 5 μg/ml Insulin. All doxycycline treatments were performed for 24 hours at 1 μg/ml before harvest at day 8 of differentiation. Isolation of adipocyte precursors from the interscapular brown adipose tissue was done as described above for white fat pads but with a digestion buffer containing: 100 mM HEPES, 120 mM NaCl, 5 mM KCl, 4% BSA, 5 mM glucose, 1.3 mM calcium and 0.75 mg/ml collagenase B. Adipocyte differentiation was induced by the addition of 500 μM IBMX, 1 nm T3, 1μM dexamethasone, 0.116 μg/ml Insulin, 125 μM indomethacin in culture media (DMEM supplemented with 10% FBS, penicillin-streptomycin and 20 mM HEPES). Following 2 d of induction, cells were maintained in media containing only 1 nM T3 and 116 ng/ml insulin. Cells were used for sEV secretion assays at 8 days of differentiation.
Isolation and culture of primary human adipocytes –
Subcutaneous adipose tissue was acquired via abdominoplasty from a 30 year-old, non-diabetic female. Adipose tissue lobules were carefully liberated from fibrous tissue, washed extensively in HBSS and digested in a collagen solution, shaking at 37°C (DMEM/F-12, 17μM pantothenate, 33μM biotin, 1x penicillin-streptomycin, 50 mg/ml BSA and 1.5 mg/ml collagenase, type II). Following digestion samples were centrifuged at 600 g for 5 minutes at room temperature. Floated adipocyte were aspirated and moved to a fresh tube with HBSS. Adipocytes were washed 3x with HBSS. Adipocytes were re-suspended in culture media (DMEM/F-12 with 5% FBS and 1x, penicillin-streptomycin and 400 μM BSA-conjugated palmitate) at the ratio of 1 ml adipocyte to 10ml media. The conditioned media was recovered after 16 hours of culture, and sEVs were isolated as described below.
Cardiomyocyte isolation and culture –
Cardiomyocytes were isolated using the previously published Langendorff-free method (Ackers-Johnson et al., 2016). Mice were anaesthetized with isoflurane. The chest cavity was opened, and the inferior vena cava was cut. 7 ml EDTA buffer (10 mM HEPES, 0.5 mM NaH2PO4, 130 mM NaCl, 5 mM KCl, 10 mM glucose, 10 mM 2,3-butanedione monoxime (BDM), 10 mM Taurine, and 5 mM EDTA pH 7.4) was injected into the right ventricle. Lahey forceps were used to clamp the aorta and facilitate clean removal of the heart. The heart was immediately transferred to a petri dish with EDTA buffer at 37°C and 10 ml EDTA buffer was slowly injected into the left ventricle. The heart was moved to a petri dish with warm perfusion buffer (10 mM HEPES, 0.5 mM NaH2PO4, 130 mM NaCl, 5 mM KCl, 10 mM glucose, 10 mM 2,3-butanedione monoxime (BDM), 10 mM Taurine, and 1 mM MgCl2 pH 7.4) and perfused with 3 ml perfusion buffer through the left ventricle. The heart was then transferred to a final petri dish containing collagenase buffer. The cardiac muscle was digested by perfusion of 40–50 ml collagenase buffer (perfusion buffer containing 0.5 mg/ml collagenase 2, 0.5 mg/ml collagenase 4 and 0.05 mg/ml protease XIV) through the left ventricle pre-warmed to 37°C. Once sufficiently dissociated, the atria were removed, and the ventricular tissue was placed into a petri dish with 3 ml collagenase buffer. The tissue was pulled apart with fine forceps to produce ~1 mm3 pieces, which were then further dissociated with gentle pipetting. Perfusion buffer containing 5% FBS was added to the cell suspension to stop further digestion, and the sample was then passed through a 100 μm filter. Myocytes were allowed to settle by gravity for 20 minutes. The pellet was resuspended and 3 sequential rounds of gravity settling were performed in buffers containing 0.34, 0.68, and 1.02 mM Ca2+, for calcium re-introduction. Following the final gravity settling step cells were resuspended in plating medium (M199 media supplemented with 10% FBS, penicillin-streptomycin and 10 mM BDM) and visualized to ensure the presence of viable, rod-shaped myocytes. The cells were then plated on laminin-coated plates for 1 hour. The media was then replaced with culture media (M199 media supplemented with 5% BSA, penicillin-streptomycin, 10 mM BDM, 1X ITS [Sigma I3146; 100x] and 1X chemically defined lipid [Thermo Scientific 11905–031; 100x]) and cardiomyocytes were maintained in a humidified cell culture incubator at 37°C, 5% CO2. All experiments were conducted on cardiomyocytes in culture media within 1 day after isolation.
METHOD DETAILS
Mouse treatments –
Transgene expression was induced with mouse chow or HFD- containing 600 mg/kg doxycycline. GW4869 was dissolved in DMSO at 5 mg/ml then diluted to 0.3 mg/ml in PBS. Each mouse received 200 μl injection solution (60 μg/mouse). Control mice were injected with a 6% DMSO solution in PBS. Where indicated, mice were injected retro-orbitally with ~3 μg sEV protein, which corresponded to 1 ×109 sEV particles diluted in sterile PBS. For biodistribution studies mice received 30 ug of PHK26-labeled sEVs via retro-orbital injection every 12 hours for a 24 hour period. Where indicated mice were injected with 60 mg/kg chloroquine (CQ) diphosphate salt dissolved in PBS.
Chemicals and reagents –
Mature adipocytes were treated for 20 hours with the following chemicals: 1 μg/ml doxycycline, 1 μM rotenone, 10 μM antimycin A, 1 μM oligomycin, 3.5 μM FCCP, 2 μM mitoQ, 0.2 mg/ml buthionine sulfoximine (BSO), 400 μM palmitate-BSA, or 50 μM chloroquine. In experiments where mitoQ was used to test suppression of oligomycin- or palmitate-induced sEV secretion, adipocytes were pre-treated with mitoQ 3 hours prior to oligomycin or palmitate were added and treatment was maintained for the duration of the 20-hour experiment. All palmitate-BSA treated cells received 500 μM carnitine and were compared to the control cells which were treated with the same amount of BSA and carnitine.
sEV biodistribution studies –
sEVs were stained with PHK26 per the manufacturer’s instructions and injected into C57BL/6 mice as specified in the Mouse treatments section. Mice were euthanized by cervical dislocation under isoflurane. The heart was perfused with ice cold PBS to remove blood. Cardiac tissue was separated into the left ventricle, right ventricle and atria and sliced into pieces ~10 mm thick. Pieces were mounted onto a microscope slide with Dako fluorescent mounting medium and PHK26 fluorescence was detected using a Zeiss LSM8800 Airyscan confocal microscope.
Isolation of sEVs from cell culture conditioned media and mouse serum –
Adipocytes were cultured on gelatin-coated plates under the indicated conditions in growth media containing sEV-free FBS for 24 hours at day 8 of differentiation. sEVs used for Western blot analysis were isolated from the media by differential centrifugation. Freshly harvested media were centrifuged for 15 minutes at 600 g, 4°C to remove whole cells. The supernatant was then centrifuged for 20 minutes at 1,200 g, 4°C to remove cell debris and apoptotic bodies. The resulting supernatant was centrifuged for 30 minutes at 10,000 g, 4°C to remove large EVs. The final supernatant was centrifuged in a swinging bucket rotor at 100 000 g for 1 hour to pellet sEVs. The pellet was resuspended in PBS and centrifuged again at 100 000 g for 1 hour. Serum sEVs were isolated in a similar manner. Sera from like conditions were pooled to reach 500 μl. Serum samples were diluted to 8 ml in PBS and filtered through a 0.2 μm filter. The filtrate was centrifuged in a swinging bucket rotor for 1 hour at 100 000 g to pellet sEVs. The pellet was resuspended in PBS and centrifuged again at 100 000 g for 1 hr. The final pellet was resuspended in PBS for cell culture treatment or in RIPA buffer (10 mM Tris-HCl, 2 mM EDTA, 0.3% NP40, 0.3% deoxycholate, 0.1% SDS and 140 mM NaCl pH 7.4) for Western blot. Microvesicles used for cell culture treatment were prepared from plasma samples that were cleared by the first 2 centrifugation steps described above for isolation of sEVs from culture media. The microvesicles were pelleted by the subsequent 10, 000xg spin and resuspended in PBS. For mouse injections sEVs were isolated from large volumes of media by the ExtraPEG method (Rider et al., 2016). Large volumes of media were cleared step-wise as described above to the 10, 000xg centrifugation spin step. A polyethylene glycol 600 (PEG 600)/NaCl solution was added to the supernatant to a final concentration of 8% PEG 600 and 500 mM NaCl to precipitate sEVs. Samples were incubated overnight at 4°C, and subsequently centrifuged for 45 minutes at 3,400 xg. The pellet containing the sEV fraction was resuspended in 10 ml sterile PBS and centrifuged for 1 hour at 100, 000 xg. The washed pellet was then resuspended in sterile PBS, aliquoted and stored at − 80°C. Aliquots were freshly defrosted for each injection, and never freeze-thawed a second time. In all conditions, adipocyte cultures were only used for sEV isolation if the differentiation rate reached 80% or higher.
OptiPrep density gradient for sEV purification –
The protocol was carried out as previously described with minor adjustments (Greening et al., 2015). A crude sEV fraction was isolated from 3 ml mouse serum using the PEG-precipitation method as described above and resuspended in 420 μl 10 mM Tris HCl, 0.25 M sucrose, pH 7.5. The stock solution of OptiPrep (60%w/v Iodixanol) was diluted in buffer containing 10 mM Tris-HCl, 250 mM sucrose, pH 7.5 to produce gradient solutions of 40%w/v, 20%w/v, 10%w/v, and 5%w/v. A discontinuous gradient was formed by placing 2.5 ml 40%w/v OptiPrep solution at the bottom of an ultracentrifuge tube and layering 2.5 ml of each solution on top in from the highest to lowest concentration. The sEV pellet was layered on top. The gradient was centrifuged for 16 hours at 100, 000 xg. Twelve, 830 μl fractions were collected from the meniscus, diluted to 10 ml in PBS and centrifuged at 100,000 xg for 1.5 hours. The pellet of each fraction was resuspended in 30 μl RIPA buffer (10 mM Tris-HCl, 2 mM EDTA, 0.3% NP40, 0.3% deoxycholate, 0.1% SDS and 140 mM NaCl pH 7.4). 10 ul of each lysate was prepped for a non-reducing Western blot to detect the tetraspanin exosome markers. The remaining 20 μl of each sample was denatured and reduced for Western blot analysis of mitochondrial proteins. A second gradient was run simultaneously with the top layer of 420 μl 10 mM Tris HCl, 25 mM sucrose, pH 7.5. Identical fractions were collected and diluted 1:10, 000. Absorbance at 244 nm was measured for the diluted samples and the iodixanol concentration was determined using the extinction coefficient of 3 201g−1cm−1.
Quantification of sEVs in culture media and serum –
SVF cells were differentiated into mature adipocytes. At day 8 of differentiation, adipocytes were treated as indicated in FluoroBrite DMEM containing 1X penicillin-streptomycin and 10% EV-depleted FBS. FBS was depleted of EVs by centrifugation FBS for 2 hours at 120, 000xg. Following 20 hours of treatment, 500 μl of media was cleared through sequential centrifugation: 600 xg for 15 min, then 1, 200 xg for 20 min. The cleared supernatant was diluted 1:100 in PBS and analyzed using the Particle Metrix ZetaView particle tracking analyzer. For sEV quantification in serum samples, 10 μl serum was diluted to 1 ml in PBS. Samples were filtered through a 0.2 μm syringe-driven filter, followed by a 1:100 dilution for nanoparticle tracking analysis (NTA) with the ZetaView. sEVs and lEVs were taken to have the size distribution of 45 – 200 nm and > 200 nm respectively.
Flow cytometry for FABP4 expression –
Bead-assisted flow cytometric analysis of sEVs was conducted as previously described (Suarez et al., 2017). Briefly, purified sEVs were bound to 4 μm aldehyde/sulfate latex beads in PBS pH 7.4 overnight. sEV-bound beads were blocked in 100 mM glycine for 30 minutes at room temperature then washed twice with BCB buffer (PBS, 0.1% BSA pH 7.4). Primary antibody against FABP4 was added to the bead-linked sEVs and incubated for 30 minutes at room temperature. After washing, the bead-sEVs were incubated with fluorescent secondary antibody (BV421) for 30 minutes at room temperature. The beads were washed once and sEVs were stained with PKH26 cell membrane dye. The bead-sEVs were washed 3 times and analyed by Flow Cytometry. The bead-bound sEVs were selected on a forward scatter versus side scatter plot and gated to exclude the debris, which are found in the left lower corner. EVs were selected based on PE fluorescence from PKH26 dye and the mean fluorescence (MFI) was measured to evaluate the strength of the signal that represents the amount of sEVs linked to beads.
Immunofluorescence and fluorescent dyes –
Cells were washed with PBS and fixed for 20 minutes with fresh 4% paraformaldehyde. Cells were permeabilized post fixation with 0.1% Triton X-100 for 10 min. See Key Resources Table for a list of primary and secondary antibodies. For CellROX images, cells were treated with 5 μM CellROX reagent for 45 minutes. Cell were washed 3 times with PBS and imaged live per the manufacturer’s instructions. TUNEL staining was performed using the In Situ Cell Death Detection Kit (Roche). Where cardiac tissue was stained, hearts were fixed for 24 hours in fresh 4% paraformaldehyde. Tissues were embedded in paraffin blocks and cut into 5 μm sections. Confocal images were acquired on a Zeiss LSM8800 Airyscan confocal microscope. Other fluorescence pictures were taken with a Keyence BZ-X700 microscope.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Alix | Santa Cruz | Cat#: sc-53540 RRID:AB_673819 |
| CALR; Calreticulin | Cell Signaling Technology | Cat#: 12238 RRID:AB_2688013 |
| Catalase | Santa Cruz | Cat #: sc-271803 RRID:AB_10708550 |
| CD9 | BD Biosciences | Cat #: 553758 RRID:AB_395032) |
| CD81 | Santa Cruz | Cat #: sc-7637 RRID:AB_627190 |
| CD63 | Santa Cruz | Cat #: sc-5275 RRID:AB_627877 |
| COXIV | Abcam | Cat #: ab14744 RRID:AB_301443 |
| DYKDDDDK Tag (D6W5B); Flag | Cell Signaling Technology | Cat #: 14793 RRID:AB_2572291 |
| EEA1 (C45B10) | Cell Signaling Technology | Cat #: 3288 RRID:AB_2096811 |
| FABP4 | Bioss Antibodies | Cat#: bs-4059R RRID:AB_10856161 |
| GAPDH; Glyceraldehyde 3-phosphate Dehydrogenase | Cell Signaling Technology | Cat #: 5174 RRID:AB_10622025 |
| H3; Histone 3 | Cell Signaling Technology | Cat #: 4499 RRID:AB_10544537 |
| HSP60 | Santa Cruz | Cat #: sc-1052 RRID:AB_631683 |
| anti-Mitochondria, human | Abcam | Cat #: ab92824 RRID:AB_10562769 |
| FtMT; mitochondrial ferritin | Sigma | SAB2700108 |
| PDH E1α; Pyruvate dehydrogenase E1α | Abcam | Cat #: ab110330 RRID:AB_10858459 |
| TOM20 (F-10) | Santa Cruz | Cat #: sc-17764 RRID:AB_628381 |
| Total OXPHOS cocktail | Abcam | Cat #: ab110413 RRID:AB_2629281 |
| VDAC | Millipore Sigma | Cat #: ab10527 RRID:AB_10806766 |
| Donkey anti-rabbit IRDye 680RD | Li-cor | Cat #: 926-68073 RRID:AB_10954442 |
| Donkey anti-mouse IRDye 680RD | Li-cor | Cat #: 926-68072 RRID:AB_10953628 |
| Goat anti-rat IRDye 680RD | Li-cor | Cat #: 925-68076 RRID:AB_2814913 |
| Goat anti-rabbit IRDye 800CW | Li-cor | Cat #: 925-32211 RRID:AB_2651127 |
| Goat anti-mouse IRDye 800CW | Li-cor | Cat #: 926-32210 RRID:AB_621842 |
| Goad anti-mouse HRP | Abcam | Cat #: ab97023 RRID:AB_10679675 |
| Chicken anti-mouse Alexa Fluor 488 | ThermoFisher | Cat #: A-21200 RRID:AB_2535786 |
| Donkey anti-rabbit Alexa Fluor 594 | ThermoFisher | Cat #: A-21207 RRID:AB_141637 |
| BV421 Goat Anti-Rabbit IgG (Flow Cytometry) | BD Biosciences | Cat #: 565014 RRID:AB_2716308 |
| FITC Rat Anti-Mouse Ig, κ Light Chain (Flow Cytometry) | BD Biosciences | Cat #: 550003 RRID:AB_393527 |
| Chemicals, peptides, and recombinant proteins | ||
| 3-isobutyl-1-methylxanthine (IBMX) | Sigma | Cat#: I7018 |
| 800W labeled streptavidin | Li-cor | Cat#: 926-32230 |
| ADP, Adenosine 5′-diphosphate sodium salt | Sigma | Cat#: A2754 |
| Aldehyde/sulfate latex beads (4μm) | ThermoFisher | Cat#: A37304 |
| Amicon Ultra-15 centrifugal filter units (100KD) | Millipore | Cat#: UFC910024 |
| Antimycin A from Streptomyces sp. | Sigma | Cat#: A8674 |
| BDM, 2,3-Butanedione monoxime | Sigma | Cat#: B0753 |
| Biotin | Sigma | Cat#: B4639 |
| BSA (Fatty acid free, low endotoxin) | Sigma | Cat#: A8806 |
| BSO (L-Buthionine-sulfoximine) | Sigma | Cat#: B2515 |
| L-Carnitine inner salt | Sigma | Cat#: C0158 |
| CellROX Deep Red Reagent | ThermoFisher | Cat#: C10422 |
| Chloroquine diphosphate salt (CQ) | Sigma | Cat#: C6628 |
| Chemically defined lipid Concentrate | ThermoFisher | Cat#: 11905-031 |
| Collagenase D | Roche | Cat#: 11088882001 |
| Collagenase 2 | Worthington | Cat#: LS004176 |
| Collagenase 4 | Worthington | Cat#: LS004188 |
| Dapi | Sigma | Cat#: D9542 |
| Dexamethasone | Sigma | Cat#: D4902 |
| Diluent C for general membrane labeling | Sigma | Cat#: CGLDIL |
| Dispase II | Roche | Cat#: 04942078001 |
| DNase I | Qiagen | Cat #: 79254 |
| DMEM high glucose | ThermoFisher | Cat#: 11965092 |
| DMEM F12 with GlutaMAX | ThermoFisher | Cat#: 10565-042 |
| Doxycycline chow diet | Bio Serve | Cat#: S4107 |
| DSPE-PEG-biotin | Nanocs | Cat#: PG2-BNDS-2k |
| Dynabeads | Invitrogen | Cat#: 110.35 |
| EZ-Link hydrazide-biotin | ThermoFisher | Cat#: 21339 |
| Fc Block | BD Biosciences | Cat#: 553142 |
| FCCP (Carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone) | Sigma | Cat#: C2920 |
| Fetal Bovine Serum (FBS) | Fisher Scientific | Cat#: 03-600-511 |
| Fetal Bovine Serum (FBS) Exosome-Free | System Biosciences | Cat#: EXO-FBSHI-50A-1 |
| Fluorescent mounting medium | Dako | Cat#: S3023 |
| FluoroBright DMEM | ThermoFisher | Cat#: A1896701 |
| Fluorophenyl column (2.7 micron, 2.1×100 mm) | Restek Corporation | Cat# 9319A12 |
| Gelatin (2%) | Sigma | Cat#: G1393 |
| GelCode blue | ThermoFisher | Cat#: 24590 |
| Gentamycin | Gibco | Cat#: 15750-060 |
| Glucose | Gibco | Cat#: 15023-021 |
| GW4869 | Sigma | Cat#: 567715 |
| HEPES | Gibco | Cat#: 15630-080 |
| Heart slicer matrix | Zinc Instruments | Cat#: HSMA001-1 |
| In Situ Cell Death Detection Kit | Roche | Cat#: 12156792910 |
| Insulin | Sigma | Cat#: I6634 |
| Iodixanol, 60%w/v | BioVision | Cat#: M1248 |
| iScript cDNA Synthesis Kit | BIORAD | Cat#: 1708890 |
| Laminin | ThermoFisher | Cat#: 23017015 |
| ITS Liquid Media Supplement | Sigma | Cat#: I3146 |
| NaF | Sigma | Cat#: S6776 |
| Na3VO4 (Orthovanadate) | Sigma | Cat#: S6508 |
| NTA 488nm Fluorescence Standard Beads | Malvern | Cat#: NTA4095 |
| NuPAGE 4–12% Bis-Tris Gel | ThermoFisher | Cat#: NP0335BOX |
| L-(–)-Malic acid | Sigma | Cat#: M6413 |
| MDA, Malonaldehyde bis(dimethyl acetal) | Fisher Scientific | Cat#: AC 14861-1000 |
| M199 medium | Sigma | Cat#: M4530 |
| MatTek 8 well culture slides | MatTek | Cat#: CCS-8 |
| MitoB | Cayman Chemical | Cat#: 17116 |
| MitoQ | a generous gift from Mike Murphy | |
| Mouse diet, doxycycline (600mg/kg) chow | Bio Serve | Cat#: S4107 |
| Mouse diet, High fat diet (60%) | Bio Serve | Cat#: S5867 |
| Mouse diet, High fat diet (60%), doxycycline (600mg/kg) | Bio Serve | custom order |
| Oligomycin from Streptomyces diastatochromogenes | Sigma | Cat#: O4876 |
| Palmitate, sodium salt | Sigma | Cat#: P9767 |
| Palmitate, sodium salt (alternative source) | Santa Cruz | Cat#: sc-215881 |
| Pantothenate | Sigma | Cat#: P5155 |
| Penicillin-Streptomycin (10, 000U/mL) | ThermoFisher | Cat#: 15140122 |
| PKH26 Red Fluorescent Cell Linker kit | Sigma | Cat#: PKH26GL-1KT |
| Protease inhibitor cocktail | Millipore | Cat#: 539137 |
| Protease XIV | Sigma | Cat#: P5147 |
| Protein sample loading buffer (4x) | Li-cor | Cat#: 928-40004 |
| Pyruvate, sodium salt | Sigma | Cat#: P4562 |
| QIAamp DNA micro kit | Qiagen | Cat#: 56304 |
| Quick Western Kit | Li-cor | Cat#: 926-69100 |
| Rotenone | Sigma | Cat#: R8875 |
| Streptavidin coated plates (Pierce) | ThermoFisher | Cat#: 15501 |
| Stop solution (Elisa) | ThermoFisher | Cat#: N600 |
| Succinate, Sodium dibasic hexahydrate | Sigma | Cat#: S2378 |
| SYBR Green PCR Master Mix | Applied Biosystems | Cat#: 4309155 |
| Syringe-driven filter (0.22 μm) | Millipore | Cat#: SLGP033NS |
| TMB Substrate (Elisa) | ThermoFisher | Cat#: N301 |
| Trans-blot Turbo Nitrocellulose Transfer pack | BioRad | Cat#: 1704158 |
| Trans-blot Turbo PVDF Transfer pack | BioRad | Cat#: 1704156 |
| Transferrin (human) | Sigma | Cat#: T2252 |
| Triiodothyronine (T3) | Sigma | Cat#: T6397 |
| Taurine | Sigma | Cat#: T8691 |
| Trehalose | Sigma | Cat#: T0167 |
| Trichloroacetic Acid | Sigma | Cat#: T4885 |
| Thiobarbituric Acid (TBA) | Caymen Chemical | Cat#: 10009199 |
| Triton-X100 | Sigma | Cat#: T9284 |
| TRIzol reagent | Fisher Scientific | Cat#: 12034977 |
| Rosiglitazone | Sigma | Cat#: R2408 |
| Critical commercial assays | ||
| Cardiac troponin I (CTNI) Elisa kit | Life Diagnostics | Cat#: CTNI-1-HSP |
| QIAamp DNA micro Kit | Qiagen | Cat#: 56304 |
| Deposited data | ||
| Adipo-FtMT Proteomics: MassIVE, massive.ucsd.edu; accession #: MSV000085896. | ||
| Experimental models: cell lines | ||
| SGBS cells: human adipocyte precursor. | (Fischer-Posovszky et al., 2008) | |
| Experimental models: organisms/strains | ||
| mouse: WT C57BL6/J | Jackson Laboratory | No. 000664 |
| mouse: B6-Tg TRE-(FtMT) Mitoferritin | ||
| mouse: B6-Tg adiponectinP-rtTA | (Wang et. al., 2010) | N/A |
| mouse: TRE-SOD2-shRNA-835 | Jackson Laboratory | No. 032649 |
| Mouse: TRE-mito-APP | (An et. al., 2019) | N/A |
| Oligonucleotides | ||
| human mtDNA, Fwd: 5’CTAGAAACCCCGAAACCAAA3’ | ||
| human mtDNA,Rev: 5’CCAGCTATCACCAAGCTCGT3’ | ||
| human mtDNA gene mitochondrially encoded tRNA leucine 1 (MT-TL1), Fwd: 5’CACCCAAGAACAGGG TTTGT3’ | ||
| human mtDNA gene mitochondrially encoded tRNA leucine 1 (MT-TL1), Rev: 5’TGGCCATGGGTATGTTGTTA3’ | ||
| Mouse β-2 microglobulin (B2M), Fwd: 5’ATGGGAAGCCGAACATACTG3’ | ||
| Mouse β-2 microglobulin (B2M), Rev: 5’CAGTCTCAGTGGGGGTGAA3’ | ||
| Human B2M nuclear DNA, Fwd: 5’TGCTGTCTCCATGTTTGATGTATCT3’ | ||
| Human B2M nuclear DNA, Rev: 5’TCTCTGCTCCCCACCTCTAAG T3’ | ||
| qPCR TRE-mitoFlag, Fwd: AGTTCAAGTGCACCGGAGAG | ||
| qPCR TRE-mitoFlag, Rev: GGCCAGGATATCGAAAGCGA | ||
| Software and algorithms | ||
| Excel | Microsoft | N/A |
| Fiji (ImageJ) | Fiji | https://fiji.sc/ |
| FlowJo | FlowJo | https://www.flowjo.com/ |
| Image J | NIH | https://imagej.nih.gov/ij/ |
| LabSolutions V 5.82 | Shimadzu Scientific Instruments | N/A |
| LabSolutions Insight V 2.0 program packages | Shimadzu Scientific Instruments | N/A |
| Prism | GraphPad Software | GraphPad Software |
| Other | ||
| Zeiss LSM8800 Airyscan Confocal Microscope | Zeiss | |
| Keyence BZ-X700 Fluorescence Microscope | Keyence | |
| 1200EX Transmission Electron Microscope | JEOL | |
| LSRFortessa SORP | BD Bioscience | |
| FACS Aria Fusion | BD Bioscience | |
| MagNA Lyser | Roche | |
| ZetaView | Particle Metrix | |
| Odyssey Infrared Imager | Li-cor | |
| Seahorse XFe24 Analyzer | Agilent | |
Western blot –
At the time of euthanasia, 3 ml ice cold PBS was perfused through the left ventricle of the heart, followed by tissue harvest. Protein was extracted from tissues by homogenization in PBS supplemented with 1 mM EDTA, 20 mM NaF, 2 mM Na3VO4, and protease inhibitor cocktail. 5X RIPA buffer was added to the homogenate for a final concentration of 1X (10 mM Tris-HCl, 2 mM EDTA, 0.3% NP40, 0.3% deoxycholate, 0.1% SDS and 140 mM NaCl pH 7.4). Cell culture samples were harvested directly in 1 X RIPA buffer containing 20 mM NaF, 2 mM Na3VO4, and 1X protease inhibitor cocktail. The samples were then cleared by centrifugation at 10, 000 xg for 5 minutes. 20–50 μg/lane of supernatant protein was separated by SDS-PAGE. For EV blots 20 μg of total EV protein dissolved in 1X RIPA buffer was loaded in each gel lane. Where indicated EV protein was loaded into the gel by yield from equal volumes of media (15 ml). Following electrophoresis proteins were transferred to nitrocellulose membrane. The blots were then incubated overnight at 4°C with primary antibodies in a 1% BSA TBST blocking solution. An Odyssey Infrared Imager was used to visualize Western blots with Li-cor IRdye secondary antibodies. See Key Resources Table for a list of primary and secondary antibodies.
Flag-tag immunoprecipitation–
Cardiac tissue lysates were prepared as described in the Western blot section. The anti-Flag antibody (Cell Signaling Technology, cat# 14793) was bound to protein A coated Dynabeads (ThermoFisher Scientific, cat#: SM0243) in TBST (50mM Tris-HCl, 0.1 mM EDTA, 150 mM NaCl and 0.5% Tween-20, pH 7.5). 100 μg protein were incubated with the anti-Flag beads overnight at 4°C. Beads were washed 3 times with TBST and eluted directly in Western blot sample buffer with 100 mM DTT at 95°C for 5 minutes. Proteins were then separated by SDS-PAGE. Secondary antibodies described in the Western blot section or the Quick Western Kit (LI-COR), which does not detect denatured IgG, were used for detection.
Protein carbonylation (PC) and thiobarbituric acid reactive substances (TBARS) assays –
Tissue was homogenized in cold buffer containing 100 mM sodium acetate, 20 mM NaCl, 0.1 mM EDTA and 0.1 mM PMSF, pH 5.5. Following tissue disruption Triton X-100 was added to each sample to a final concentration of 1% and incubated for 20 minutes on ice. Homogenates were cleared for 10 min at 16, 000 xg, 4°C. Protein concentrations of the supernatants were determined. For protein carbonylation detection, 50 μg of protein was combined with 0.5 mM EZ-Link hydrazide-biotin. The samples were incubated at room temperature for 2 hours, after which, the reaction was stopped by addition of protein loading buffer (Licor) in the absence of a reducing agent. Proteins were allowed to denature at room temperature for 20 minutes and separated by SDS-PAGE and transferred to a PVDF membrane. Detection of biotin-modified protein was accomplished using 800W labeled streptavidin. Membranes were re-probed with anti-GAPDH antibodies for a loading control as described in the Western blot section. For the TBARS assay, 100 μl of the cleared supernatant used for the protein carbonylation assay was neutralized by the addition of 10 μl 10X PBS. The resulting sample was combined with 200 μl of cold 10% trichloroacetic acid to precipitate protein. Samples were allowed to incubate on ice for 20 minutes and then centrifuged at 2, 200 xg for 15 min at 4°C. The supernatants were transferred to a new tube and an equal volume of 0.67% w/v thiobarbituric acid (TBA) was added. Samples were boiled for 10 minutes at 95°C following which the absorbance of each sample was measured at 540 nm on a TECAN Sunrise plate reader. The concentration of lipid peroxides was calculated from an 1,1,3,3-tetramethoxypropane (MDA) standard curve prepared in parallel to the experimental samples.
Mitochondrial DNA (mtDNA) determination –
Mitochondria DNA content (mtDNA) was assessed by absolute quantification using real-time PCR. DNA was isolated from tissue samples using QIAamp DNA Micro Kit (Qiagen). Primers for mouse mtDNA were adopted from Malik et. al. (Malik et al., 2016) to assess the DNA quality of the samples before assessment of human mtDNA, Fwd: 5’CTAGAAACCCCGAAACCAAA3’ and Rev: 5’ CCAGCTATCACCAAGCTCGT3’. Mouse ß-2 microglobulin (B2M) primers were used to amplify mouse nuclear DNA Fwd: 5’ ATGGGAAGCCGAACATACTG3’ and Rev: 5’ CAGTCTCAGTGGGGGTGAA3’. Primers for the human mtDNA gene mitochondrially encoded tRNA leucine 1 (MT-TL1), were adopted from Venegas and Halberg (Venegas and Halberg, 2012) Fwd: 5’CACCCAAGAACAGGG TTTGT3’ and Rev: 5’TGGCCATGGGTATGTTGTTA3’. Human B2M nuclear DNA as assessed with the primers, Fwd: 5’TGCTGTCTCCATGTTTGATGTATCT3’ and Rev: 5’TCTCTGCTCCCCACCTCTAAG T3’. No human nuclear DNA was detected in mouse tissues. PCR products were purified, and dilution standards were prepared with a range of 10–108 copies per 2 μl for absolute quantification. mtDNA and nuclear DNA copy number was determined from template DNA by qPCR using a Quant Studio 6 Flux (Applied Biosystems) and PowerUP SYBR Master Mix (Applied Biosystems) using the following protocol: preincubation at 95°C for 20s; desaturation at 95°C for 1s; annealing and extension at 60°C for 20s (repeated denaturation and extension for 40 cycles). Specificity of the primers were confirmed by a single melt peak after melt curve analysis. For mtDNA quantification in human plasma sEVs, sEVs were isolated from 500ul plasma and incubated with 1 unit DNase I for 1 hour at 37°C to digest contaminating DNA. DNase I was inactivated by heating samples to 65°C for 10 min. sEV DNA was extraction with the QIAamp DNA micro kit and mtDNA was quantified as described above.
Lipid Capture Elisa for sEV Cargo in Human Plasma –
This assay was adapted from a previously described protocol for a lipid-based EV capture system (Wan et al., 2017). DSPE-PEG-biotin was used as the lipid capture probe. DSPE-PEG-biotin was dissolved in 50% ethanol to make a 5 mM stock solution, aliquoted and stored at −80°C. The day of the assay DSPE-PEG-biotin was diluted to 200 μM in Elisa wash buffer (PBS pH 7.4, 0.1% BSA, 0.05% Tween-20). 100μl of this working stock was added to the wells of a streptavidin-coated 96 well plate and incubated for 30 min, shaking at room temperature to immobilize the probe. Following washing, 40 μl human plasma was added to each well containing 80 μl Diluent C (Sigma). Model human plasma with a known EV concentration was serially diluted and used for a standard curve. The plate was incubated for 1 hour at room temperature, shaking, to capture lipid-particles. The plate was washed, and captured lipid particles were permeabilized with 0.001% Triton X-100 for 5 minutes at room temperature. Wells were than incubated with anti-mitochondria primary antibody (Abcam) and anti-mouse HRP secondary antibody in sequence. After extensive washing 100 μl of TMB substrate solution (ThermoFisher) was added to each well and after acceptable development, the reaction was stopped with Stop Solution (ThermoFisher). Absorbance was read at 450nm.
qPCR –
Tissues were homogenized in TRIzol (ThermoFisher) using the Qiagen TissueLyser II. RNA was isolated per the manufacturer’s protocol. RNA yield and quality were determined using a NanoDrop. cDNA was prepared by reverse transcribing 1 μg of RNA with the iScript cDNA Synthesis Kit (BioRad). Gene expression was calculated by the standard threshold cycle method using B2M for normalization. Primers for the TRE-mitoFlag transcript are, Fwd: AGTTCAAGTGCACCGGAGAG and Rev: GGCCAGGATATCGAAAGCGA.
Transmission electron microscopy (TEM) –
For visualization of MDVs and budding mitochondria by TEM, 5 μl of the either the MDV fraction or isolated mitochondria were fixed overnight with fresh 4% paraformaldehyde. 5 μl of the fixed MDV solution was transferred to each Formvar-carbon coated electron microscopy grid (200μm Cu). The grids were covered and incubated for 10 minutes at room temperature. The grids were washed with distilled water, stained with 2% uranyl acetate, and imaged on a JEOL 1200EX transmission electron microscope.
Serum sEV and cardiomyocyte seahorse analysis –
sEV oxygen consumption rates (OCRs) were determined using an Agilent Seahorse XFe24 Analyzer. Control and adipo-FtMT mice were fed dox-HFD for 3 weeks, sera were collected and samples from like genotypes were pooled, if need be, to reach 400 μl. Serum samples were diluted to 1 ml in PBS and filtered through a 0.2 μm filter. 1 ml polyethylene glycol 600 (PEG600)/NaCl solution were added to the filtrate to final concentrations of 8% PEG600 and 500 mM NaCl to precipitate sEVs. Samples were rocked for 2 hours a at 4°C and then centrifuged for 30 min at 3,500 g, 4°C. Each sEV pellet was resuspended in 50 μl MAS1 buffer (70 mM sucrose, 220 mM mannitol, 10 mM KH2PO4, 5 mM MgCl2, 2 mM HEPES, 1 mM EGTA, and 0.2% FA-free BSA, pH 7.2) containing 8% PEG. Samples were transferred to XF24 V7 cell culture microplates and centrifuged for 30 minutes at 3,500 g, 4°C to pellet the sEVs at the bottom of the plate. The plate was then subjected to a 10 min equilibration period in a 37°C incubator. 450 μl starting buffer containing 8% PEG600 was carefully added to each well without disturbing the pellet. The plate was then transferred to the Seahorse Analyzer. For the electron flow (EF) experiments, the starting buffer was MAS1 containing 10 mM pyruvate, 2 mM malate, and 4 μM FCCP. OCR measurements were obtained following sequential additions of rotenone (2 μM final), succinate (10 mM final), and antimycin A (4 μM final). For the electron-coupling experiments (EC), the starting buffer was MAS1 containing 10 mM succinate and 2 μM rotenone. OCR measurements were obtained following sequential additions of ADP (4 mM final), oligomycin (2 μM final), and FCCP (4 μM final). For cardiomyocyte Seahorse assays, primary cardiomyocytes were plated on laminin-coated Seahorse cell culture plates. 3 hours after plating, the cells were treated with 1×108 adipocyte-derived sEVs as indicated. OCRs were obtained following sequential addition of oligomycin (1.2 μM final), FCCP (4 μM final), and rotenone/antimycin A (1 μM final each). All chemicals were purchased from Sigma-Aldrich.
Cardiac mitochondria oxygen consumption –
Wild-type mice were injected with 1×109 adipocyte-derived sEVs as indicated and euthanized 2 hours later. Cardiac tissue was perfused with 3 ml cold isolation buffer (10 mM MOPS, 1 mM EDTA, 210 mM mannitol, and 70 mM sucrose, pH 7.4). The tissue was disrupted in ice cold isolation buffer using a Potter-Elvehjem homogenizer. The homogenate was centrifuged at for 10 minutes at 600 g, 4°C. The resulting supernatant was filtered through cheese cloth and centrifuged at for 15 minutes at 10,000 to pellet mitochondria. Mitochondria were resuspended in isolation buffer to a final concentration of 15 mg/ml. Mitochondria were diluted to 250 μg/ml room temperature respiration buffer (10 mM MOPS, 5 mM K2HPO4, 210 mM Mannitol, and 70 mM sucrose, pH 7.4) containing 100 μM pyruvate and 1 mM malate or 25 μM palmitoyl-carnitine and 1 mM malate. OCRs were determined at room temperature using a Neofox oxygen chamber with 175 μl capacity or using the Seahorse Flux Analyzer. For the Neofox experiments state 3 respiration was initiated at 2 minutes after transfer of sample to the oxygen chamber by addition of 250 μM ADP. For the Seahorse electron flow experiment, 5 μg of mitochondria were transferred to XF24 V7 cell culture microplates and centrifuged for 20 minutes at 3,500 g, 4°C to pellet the mitochondria at the bottom of the plate. The plate was then subjected to a 5 min equilibration period in a 37°C incubator. 450 μl starting buffer (MAS1 containing 10 mM pyruvate, 2 mM malate, and 4 μM FCCP) was carefully added to each well without disturbing the pellet. the starting buffer was. OCR measurements were obtained following sequential additions of rotenone (2 μM final), succinate (10 mM final), antimycin A (4 μM final) and ascorbate (10 mM final)/TMPD (100 μM final).
Mitochondria-derived vesicle (MDV) reconstitution assay –
The protocol used for In vitro MDV production was adapted from that described by Soubannier et. al.(Soubannier et al., 2012b). Inguinal fat pads were freshly harvested from control and adipo-FtMT mice, which had both been on dox-HFD for 3 weeks. Mitochondria were isolated as described above in the Cardiac mitochondria oxygen consumption section. Mitochondria were diluted to 1 mg/ml in respiration buffer containing with 10 mM pyruvate, 2 mM malate and 2.5 mM ADP at 37°C to stimulate ATP production. Where indicated 50 μM antimycin A was added to the incubation. Following 1 hour incubation, mitochondria were pelleted by centrifugation for 5 minutes at 10, 000 xg. The supernatant, containing released MDVs, was digested with proteinase K to remove contaminating proteins that were not protected by vesicle membrane. Following addition of 2 mM PMSF, the membrane-protected proteins were used for Western blot analysis, electron microscopy or NTA.
MitoB/MitoP mass spectrometry assay –
Measurement of in vivo mitochondrial H2O2 production was performed according to the previously published protocol with modifications to the LC-MS/MS method(Cocheme et al., 2012). In brief, mice were injected retro-orbitally with 0.8 μmol/kg MitoB (Cayman Chemical, cat# 17116). Mice were euthanized 3 hours after injection and 100 mg of tissues were clamp-frozen. Tissues were processed as previously described previously (Cocheme et al., 2012). MitoB and MitoP compounds were analyzed by injecting 1 μL of sample into the LC/MS/MS system consisting of a Shimadzu LCMS-8060 triple quadrupole mass spectrometer operating the DUIS ion source in electrospray positive mode coupled to a Nexera X2 UHPLC (Shimadzu Scientific Instruments, Columbia, MD). Compounds were resolved on a Fluorophenyl column (2.7 micron, 2.1×100 mm, Restek Corporation, Bellefonte, PA) maintained at 30°C. The mobile phase consisted of water containing 0.1% formic acid (eluent A) and acetonitrile containing 0.1% FA (eluent B). Gradient elution was performed starting at 20% eluent B, a linear increase to 75% eluent B until 2.0 minutes, a step to 100% eluent B until 2.1 minutes, 100% eluent B until 5.6 minutes and re-equilibration from 5.7 until 7 minutes. The flow rate was set to 0.5 mL/minute.
Proteomics –
Samples were run ~ 5 mm into an SDS-PAGE gel. Proteins were stained with GelCode Blue. All proteins were excised from the gel and submitted to the UT Southwestern Proteomics Core. Samples were processed as previously described (Kalantari et al., 2016).
Cardiac ischemia/reperfusion –
14 week-old male mice were subjected to cardiac ischemia by totally occluding the left anterior descending (LAD) coronary artery for 45 minutes. In brief, mice were anaesthetized in an airtight chamber using 2.5% isoflurane, endotracheally intubated, and ventilated using a Harvard Apparatus volume control ventilator with 100% O2, supplemented with 2.5% vaporized isoflurane. Following lateral thoracotomy (from the 4th intercostal space) and pericardiectomy, the LAD coronary artery was visualized and ligated with 6–0 prolene suture mounted on a 2–3 mm long piece of PE-10 tubing. Occlusion of the LAD coronary artery resulted in immediate blanching of the left ventricular anterior wall indicative of myocardial ischemia. Thereafter, deep anesthesia was maintained with 1.5% isoflurane. After 45 minutes of LAD coronary artery occlusion, the ligation was removed and reperfusion was visually confirmed. Subsequently, the thoracic cavity and skin were closed in layers using 6–0 absorbable Vicryl sutures. Mice were extubated and moved to cages on a heating pad for 2 hours. Mice were subjected to echocardiography 24 hour and 7 days after surgery. Infarct size was measured by TTC stain at 24 hr of reperfusion and histological assessment at 7 days of reperfusion.
Histological assessment of infarct size –
Mice were sacrificed 7 days after I/R. Hearts were harvested and immediately washed with PBS supplemented with 1 M KCl. Hearts were fixed for 24 hour in fresh 4% paraformaldehyde. Each heart was cut into 2 mm sections using a mouse heart slicer matrix (Zinc Instruments) to ensure identical regions are sectioned for histology between hearts. Cut samples were submitted to the University of Texas Southwestern Molecular Pathology Core for paraffin embedding, sectioning, and Masson’s Trichrome stain.
Cardiac TTC stain –
The staining protocol was adapted from a previously publish protocol (Kim et al., 2012). For the TTC stain the PE-10 tubing was removed during reperfusion but the suture loop was left intact. 24 hours after the surgery the mice were anesthetized, intubated and ventilated. After opening the chest cavity, the LAD was re-ligated by forcing the tip of a cut pipet tip through the loop. A small piece of __ tubing was used to wrap the pipet tip to provide friction so that the suture did not slip. The heart was then immediately perfused with a 2% Evan’s blue dye solution dissolved in PBS, pH 7.4 by injection into the upper left ventricle. The heart was subsequently perfused with 1ml of 1M KCl in PBS, pH 7.4 by injection at the same location. The heart was then removed rinsed in PBS and frozen at − 80°C for 15 minutes. Each heart was cut into 1 mm sections using a mouse heart slicer matrix (Zinc Instruments) and each piece was placed in a well of a 96 well plate containing a 1% (w/v) 2,3,5-Triphenyltetrazolium chloride (TTC) solution in a phosphate buffer, pH 7.4. Heart slices were incubated in TTC at 37°C for 20 min. Each slice was transferred into another well containing 10% formalin buffered with PBS, pH 7.4. Images were taken with a Zeiss SteREO Discovery stereoscope. ImageJ software was used for planimetry. Only images that had a clearly defined area at risk (red only) and infarct (white) were quantified.
Echocardiography-
Assessment of left ventricular systolic function was performed on conscious mice using a VisualSonics Vevo 2100 micro ultrasound system. Echocardiographic M-mode images were obtained from a parasternal short axis view. The echocardiography and data analysis were performed by a single, blinded individual. The left ventricular ejection fraction (LVEF) and left ventricular fractional shortening (LVFS) were calculated.
Cardiac troponin I measurements –
Plasma was collected from mice 1 day after ischemia/reperfusion and cardiac troponin I was determined using a Cardiac Troponin I (CTNI) ELISA Kit (Life Diagnostics Inc).
QUANTIFICATION AND STATISTICAL ANALYSIS
All data is presented as mean ± s.e.m. Statistical significance was determined by a one or two-sided Student’s t-tests or, where indicated, two-way ANOVAs. A p value < 0.05 was considered to be statistically significant (*p<0.05, ** p<0.01 and ***p<0.001). All qPCR data points are the average of technical duplicates. For all mouse studies, the n value corresponds to individual mice of a given treatment. For in vitro-derived adipocyte culture experiments, the n values correspond to separate SVF preparations from separate mice or a different passage number of SVF from the same mouse. For cardiomyocyte experiments and isolated cardiac mitochondrial oxygen consumption experiments, the n values correspond to separate preparations from separate mice. Data was analyzed using GraphPad Prism Software version 8.4.3 and Microsoft Excel version 16.38.
Supplementary Material
Highlights.
Mitochondrial stress stimulates adipocyte sEV release
Adipocyte stress-induced sEVs are enriched with oxidatively damaged mitochondria
Mitochondria in sEVs from stressed adipocytes induce a burst of ROS in cardiac tissue
The adipocyte-derived pro-oxidant signal protects the heart through hormesis
Acknowledgements:
We thank Charlotte E. Lee and Histology Core at UT Southwestern for assistance in embedding and processing of histological samples. Additionally, we kindly thank the Metabolic Phenotyping Core for mitoB/P measurements, the Proteomics Core for protein identification, the Transgenic Core for their help in the generation of mice and the Live Cell Imaging Core Facility and Electron Microscopy Core Facility. We thank Shimadzu Scientific Instruments for the collaborative efforts in mass spectrometry technology resources. We also thank Dr. Gerald Shadel for supplying us with the TRE-SOD2 shRNA mouse. We thank Dr. Martin Wabitsch (Division of Pediatric Endocrinology and Diabetes, Department of Pediatrics and Adolescent Medicine, Ulm University Medical Center) for permission to use SGBS cells. The graphical abstract was made using BioRender.com. This study was supported by US National Institutes of Health (NIH) grants R01-DK55758, R01-DK127274, R01-DK099110, RC2 DK118620 and P01-AG051459 (P.E.S.). C.C is supported by grant K99DK122019. J.F. is supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) grant 414232833. Y.A.A. is supported by NIH grant K01-DK125447. P.F.P. is supported by a DFG Heisenberg professorship (Fi1700/7-1).
Footnotes
Declaration of Interests: The authors declare no competing interests.
Inclusion and Diversity: One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in science. One or more of the authors of this paper self-identifies as a member of the LGBTQ+ community.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References:
- Ackers-Johnson M, Li PY, Holmes AP, O’Brien SM, Pavlovic D, and Foo RS (2016). A Simplified, Langendorff-Free Method for Concomitant Isolation of Viable Cardiac Myocytes and Nonmyocytes From the Adult Mouse Heart. Circ Res 119, 909–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An YA, Crewe C, Asterholm IW, Sun K, Chen S, Zhang F, Shao M, Funcke JB, Zhang Z, Straub L, et al. (2019). Dysregulation of Amyloid Precursor Protein Impairs Adipose Tissue Mitochondrial Function and Promotes Obesity. Nat Metab 1, 1243–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonucci S, Mulvey JF, Burger N, Di Sante M, Hall AR, Hinchy EC, Caldwell ST, Gruszczyk AV, Deshwal S, Hartley RC, et al. (2019). Selective mitochondrial superoxide generation in vivo is cardioprotective through hormesis. Free Radic Biol Med 134, 678–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoyagi T, Higa JK, Aoyagi H, Yorichika N, Shimada BK, and Matsui T (2015). Cardiac mTOR rescues the detrimental effects of diet-induced obesity in the heart after ischemia-reperfusion. Am J Physiol Heart Circ Physiol 308, H1530–1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bank IE, Timmers L, Gijsberts CM, Zhang YN, Mosterd A, Wang JW, Chan MY, De Hoog V, Lim SK, Sze SK, et al. (2015). The diagnostic and prognostic potential of plasma extracellular vesicles for cardiovascular disease. Expert Rev Mol Diagn 15, 1577–1588. [DOI] [PubMed] [Google Scholar]
- Brestoff JR, Wilen CB, Moley JR, Li Y, Zou W, Malvin NP, Rowen MN, Saunders BT, Ma H, Mack MR, et al. (2021). Intercellular Mitochondria Transfer to Macrophages Regulates White Adipose Tissue Homeostasis and Is Impaired in Obesity. Cell Metab 33, 270–282 e278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabrese EJ, and Mattson MP (2017). How does hormesis impact biology, toxicology, and medicine? NPJ Aging Mech Dis 3, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark C, Smith W, Lochner A, and du Toit EF (2011). The effects of gender and obesity on myocardial tolerance to ischemia. Physiol Res 60, 291–301. [DOI] [PubMed] [Google Scholar]
- Cocheme HM, Logan A, Prime TA, Abakumova I, Quin C, McQuaker SJ, Patel JV, Fearnley IM, James AM, Porteous CM, et al. (2012). Using the mitochondria-targeted ratiometric mass spectrometry probe MitoB to measure H2O2 in living Drosophila. Nat Protoc 7, 946–958. [DOI] [PubMed] [Google Scholar]
- Cox CS, McKay SE, Holmbeck MA, Christian BE, Scortea AC, Tsay AJ, Newman LE, and Shadel GS (2018). Mitohormesis in Mice via Sustained Basal Activation of Mitochondrial and Antioxidant Signaling. Cell Metab 28, 776–786 e775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crewe C, An YA, and Scherer PE (2017). The ominous triad of adipose tissue dysfunction: inflammation, fibrosis, and impaired angiogenesis. J Clin Invest 127, 74–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis JM, Grimsrud PA, Wright WS, Xu X, Foncea RE, Graham DW, Brestoff JR, Wiczer BM, Ilkayeva O, Cianflone K, et al. (2010). Downregulation of adipose glutathione S-transferase A4 leads to increased protein carbonylation, oxidative stress, and mitochondrial dysfunction. Diabetes 59, 1132–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng ZB, Poliakov A, Hardy RW, Clements R, Liu C, Liu Y, Wang J, Xiang X, Zhang S, Zhuang X, et al. (2009). Adipose tissue exosome-like vesicles mediate activation of macrophage-induced insulin resistance. Diabetes 58, 2498–2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhalla NS, Elmoselhi AB, Hata T, and Makino N (2000). Status of myocardial antioxidants in ischemia–reperfusion injury. Cardiovascular Research 47, 446–456. [DOI] [PubMed] [Google Scholar]
- Donner D, Headrick JP, Peart JN, and du Toit EF (2013). Obesity improves myocardial ischaemic tolerance and RISK signalling in insulin-insensitive rats. Dis Model Mech 6, 457–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- du Toit EF, Smith W, Muller C, Strijdom H, Stouthammer B, Woodiwiss AJ, Norton GR, and Lochner A (2008). Myocardial susceptibility to ischemic-reperfusion injury in a prediabetic model of dietary-induced obesity. Am J Physiol Heart Circ Physiol 294, H2336–2343. [DOI] [PubMed] [Google Scholar]
- Durieux J, Wolff S, and Dillin A (2011). The cell-non-autonomous nature of electron transport chain-mediated longevity. Cell 144, 79–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edland F, Wergeland A, Kopperud R, Asrud KS, Hoivik EA, Witso SL, R AE, Madsen L, Kristiansen K, Bakke M, et al. (2016). Long-term consumption of an obesogenic high fat diet prior to ischemia-reperfusion mediates cardioprotection via Epac1-dependent signaling. Nutr Metab (Lond) 13, 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrante SC, Nadler EP, Pillai DK, Hubal MJ, Wang Z, Wang JM, Gordish-Dressman H, Koeck E, Sevilla S, Wiles AA, et al. (2015). Adipocyte-derived exosomal miRNAs: a novel mechanism for obesity-related disease. Pediatr Res 77, 447–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer-Posovszky P, Newell FS, Wabitsch M, and Tornqvist HE (2008). Human SGBS cells - a unique tool for studies of human fat cell biology. Obes Facts 1, 184–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flaherty SE 3rd, Grijalva A, Xu X, Ables E, Nomani A, and Ferrante AW Jr. (2019). A lipase-independent pathway of lipid release and immune modulation by adipocytes. Science 363, 989–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan L, Xie D, Liu J, Bond Lau W, Christopher TA, Lopez B, Zhang L, Gao E, Koch W, Ma XL, et al. (2020). Small Extracellular Microvesicles Mediated Pathological Communications Between Dysfunctional Adipocytes and Cardiomyocytes as a Novel Mechanism Exacerbating Ischemia/Reperfusion Injury in Diabetic Mice. Circulation 141, 968–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greening DW, Xu R, Ji H, Tauro BJ, and Simpson RJ (2015). A protocol for exosome isolation and characterization: evaluation of ultracentrifugation, density-gradient separation, and immunoaffinity capture methods. Methods Mol Biol 1295, 179–209. [DOI] [PubMed] [Google Scholar]
- Hessvik NP, and Llorente A (2018). Current knowledge on exosome biogenesis and release. Cell Mol Life Sci 75, 193–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang SC, Crescitelli R, Cvjetkovic A, Belgrano V, Olofsson Bagge R, Sundfeldt K, Ochiya T, Kalluri R, and Lotvall J (2019). Mitochondrial protein enriched extracellular vesicles discovered in human melanoma tissues can be detected in patient plasma. J Extracell Vesicles 8, 1635420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones SP, Girod WG, Granger DN, Palazzo AJ, and Lefer DJ (1999). Reperfusion injury is not affected by blockade of P-selectin in the diabetic mouse heart. Am J Physiol 277, H763–769. [DOI] [PubMed] [Google Scholar]
- Kalantari R, Hicks JA, Li L, Gagnon KT, Sridhara V, Lemoff A, Mirzaei H, and Corey DR (2016). Stable association of RNAi machinery is conserved between the cytoplasm and nucleus of human cells. RNA 22, 1085–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karimi N, Cvjetkovic A, Jang SC, Crescitelli R, Hosseinpour Feizi MA, Nieuwland R, Lotvall J, and Lasser C (2018). Detailed analysis of the plasma extracellular vesicle proteome after separation from lipoproteins. Cell Mol Life Sci 75, 2873–2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SC, Boehm O, Meyer R, Hoeft A, Knufermann P, and Baumgarten G (2012). A murine closed-chest model of myocardial ischemia and reperfusion. J Vis Exp, e3896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koeck ES, Iordanskaia T, Sevilla S, Ferrante SC, Hubal MJ, Freishtat RJ, and Nadler EP (2014). Adipocyte exosomes induce transforming growth factor beta pathway dysregulation in hepatocytes: a novel paradigm for obesity-related liver disease. J Surg Res 192, 268–275. [DOI] [PubMed] [Google Scholar]
- Kranendonk ME, Visseren FL, van Balkom BW, Nolte-’t Hoen EN, van Herwaarden JA, de Jager W, Schipper HS, Brenkman AB, Verhaar MC, Wauben MH, et al. (2014). Human adipocyte extracellular vesicles in reciprocal signaling between adipocytes and macrophages. Obesity (Silver Spring) 22, 1296–1308. [DOI] [PubMed] [Google Scholar]
- Kusminski CM, Ghaben AL, Morley TS, Samms RJ, Adams AC, An Y, Johnson JA, Joffin N, Onodera T, Crewe C, et al. (2020). A Novel Model of Diabetic Complications: Adipocyte Mitochondrial Dysfunction Triggers Massive beta-Cell Hyperplasia. Diabetes 69, 313–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusminski CM, Holland WL, Sun K, Park J, Spurgin SB, Lin Y, Askew GR, Simcox JA, McClain DA, Li C, et al. (2012). MitoNEET-driven alterations in adipocyte mitochondrial activity reveal a crucial adaptive process that preserves insulin sensitivity in obesity. Nat Med 18, 1539–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavie CJ, Arena R, Alpert MA, Milani RV, and Ventura HO (2018). Management of cardiovascular diseases in patients with obesity. Nat Rev Cardiol 15, 45–56. [DOI] [PubMed] [Google Scholar]
- Lavie CJ, McAuley PA, Church TS, Milani RV, and Blair SN (2014). Obesity and cardiovascular diseases: implications regarding fitness, fatness, and severity in the obesity paradox. J Am Coll Cardiol 63, 1345–1354. [DOI] [PubMed] [Google Scholar]
- Lazar I, Clement E, Dauvillier S, Milhas D, Ducoux-Petit M, LeGonidec S, Moro C, Soldan V, Dalle S, Balor S, et al. (2016). Adipocyte Exosomes Promote Melanoma Aggressiveness through Fatty Acid Oxidation: A Novel Mechanism Linking Obesity and Cancer. Cancer Res 76, 4051–4057. [DOI] [PubMed] [Google Scholar]
- Malik AN, Czajka A, and Cunningham P (2016). Accurate quantification of mouse mitochondrial DNA without co-amplification of nuclear mitochondrial insertion sequences. Mitochondrion 29, 59–64. [DOI] [PubMed] [Google Scholar]
- McLelland GL, Soubannier V, Chen CX, McBride HM, and Fon EA (2014). Parkin and PINK1 function in a vesicular trafficking pathway regulating mitochondrial quality control. EMBO J 33, 282–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mleczko J, Ortega FJ, Falcon-Perez JM, Wabitsch M, Fernandez-Real JM, and Mora S (2018). Extracellular Vesicles from Hypoxic Adipocytes and Obese Subjects Reduce Insulin-Stimulated Glucose Uptake. Mol Nutr Food Res 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemoto Y, and De Camilli P (1999). Recruitment of an alternatively spliced form of synaptojanin 2 to mitochondria by the interaction with the PDZ domain of a mitochondrial outer membrane protein. EMBO J 18, 2991–3006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolas-Avila JA, Lechuga-Vieco AV, Esteban-Martinez L, Sanchez-Diaz M, Diaz-Garcia E, Santiago DJ, Rubio-Ponce A, Li JL, Balachander A, Quintana JA, et al. (2020). A Network of Macrophages Supports Mitochondrial Homeostasis in the Heart. Cell 183, 94–109 e123. [DOI] [PubMed] [Google Scholar]
- Oikonomou EK, and Antoniades C (2019). The role of adipose tissue in cardiovascular health and disease. Nat Rev Cardiol 16, 83–99. [DOI] [PubMed] [Google Scholar]
- Ozcan C, Palmeri M, Horvath TL, Russell KS, and Russell RR 3rd (2013). Role of uncoupling protein 3 in ischemia-reperfusion injury, arrhythmias, and preconditioning. Am J Physiol Heart Circ Physiol 304, H1192–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Y, Hui X, Hoo RLC, Ye D, Chan CYC, Feng T, Wang Y, Lam KSL, and Xu A (2019). Adipocyte-secreted exosomal microRNA-34a inhibits M2 macrophage polarization to promote obesity-induced adipose inflammation. J Clin Invest 129, 834–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poirier P, Giles TD, Bray GA, Hong Y, Stern JS, Pi-Sunyer FX, and Eckel RH (2006). Obesity and cardiovascular disease: pathophysiology, evaluation, and effect of weight loss. Arterioscler Thromb Vasc Biol 26, 968–976. [DOI] [PubMed] [Google Scholar]
- Rider MA, Hurwitz SN, and Meckes DG Jr. (2016). ExtraPEG: A Polyethylene Glycol-Based Method for Enrichment of Extracellular Vesicles. Sci Rep 6, 23978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salie R, Huisamen B, and Lochner A (2014). High carbohydrate and high fat diets protect the heart against ischaemia/reperfusion injury. Cardiovasc Diabetol 13, 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sansone P, Savini C, Kurelac I, Chang Q, Amato LB, Strillacci A, Stepanova A, Iommarini L, Mastroleo C, Daly L, et al. (2017). Packaging and transfer of mitochondrial DNA via exosomes regulate escape from dormancy in hormonal therapy-resistant breast cancer. Proc Natl Acad Sci U S A 114, E9066–E9075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schottl T, Kappler L, Fromme T, and Klingenspor M (2015). Limited OXPHOS capacity in white adipocytes is a hallmark of obesity in laboratory mice irrespective of the glucose tolerance status. Mol Metab 4, 631–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senoner T, and Dichtl W (2019). Oxidative Stress in Cardiovascular Diseases: Still a Therapeutic Target? Nutrients 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soubannier V, McLelland GL, Zunino R, Braschi E, Rippstein P, Fon EA, and McBride HM (2012a). A vesicular transport pathway shuttles cargo from mitochondria to lysosomes. Curr Biol 22, 135–141. [DOI] [PubMed] [Google Scholar]
- Soubannier V, Rippstein P, Kaufman BA, Shoubridge EA, and McBride HM (2012b). Reconstitution of mitochondria derived vesicle formation demonstrates selective enrichment of oxidized cargo. PLoS One 7, e52830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suarez H, Gamez-Valero A, Reyes R, Lopez-Martin S, Rodriguez MJ, Carrascosa JL, Cabanas C, Borras FE, and Yanez-Mo M (2017). A bead-assisted flow cytometry method for the semi-quantitative analysis of Extracellular Vesicles. Sci Rep 7, 11271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiura A, McLelland GL, Fon EA, and McBride HM (2014). A new pathway for mitochondrial quality control: mitochondrial-derived vesicles. EMBO J 33, 2142–2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakker GD, Frangogiannis NG, Bujak M, Zymek P, Gaubatz JW, Reddy AK, Taffet G, Michael LH, Entman ML, and Ballantyne CM (2006). Effects of diet-induced obesity on inflammation and remodeling after myocardial infarction. Am J Physiol Heart Circ Physiol 291, H2504–2514. [DOI] [PubMed] [Google Scholar]
- Thomou T, Mori MA, Dreyfuss JM, Konishi M, Sakaguchi M, Wolfrum C, Rao TN, Winnay JN, Garcia-Martin R, Grinspoon SK, et al. (2017). Adipose-derived circulating miRNAs regulate gene expression in other tissues. Nature 542, 450–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomsen M, and Nordestgaard BG (2014). Myocardial infarction and ischemic heart disease in overweight and obesity with and without metabolic syndrome. JAMA Intern Med 174, 15–22. [DOI] [PubMed] [Google Scholar]
- Todkar K, Chikhi L, Desjardins V, El-Mortada F, Pepin G, and Germain M (2021). Selective packaging of mitochondrial proteins into extracellular vesicles prevents the release of mitochondrial DAMPs. Nat Commun 12, 1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ussher JR, Wang W, Gandhi M, Keung W, Samokhvalov V, Oka T, Wagg CS, Jaswal JS, Harris RA, Clanachan AS, et al. (2012). Stimulation of glucose oxidation protects against acute myocardial infarction and reperfusion injury. Cardiovasc Res 94, 359–369. [DOI] [PubMed] [Google Scholar]
- van Niel G, D’Angelo G, and Raposo G (2018). Shedding light on the cell biology of extracellular vesicles. Nat Rev Mol Cell Biol 19, 213–228. [DOI] [PubMed] [Google Scholar]
- Venegas V, and Halberg MC (2012). Measurement of mitochondrial DNA copy number. Methods Mol Biol 837, 327–335. [DOI] [PubMed] [Google Scholar]
- Viswanathan S, Williams ME, Bloss EB, Stasevich TJ, Speer CM, Nern A, Pfeiffer BD, Hooks BM, Li WP, English BP, et al. (2015). High-performance probes for light and electron microscopy. Nat Methods 12, 568–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wabitsch M, Brenner RE, Melzner I, Braun M, Moller P, Heinze E, Debatin KM, and Hauner H (2001). Characterization of a human preadipocyte cell strain with high capacity for adipose differentiation. Int J Obes Relat Metab Disord 25, 8–15. [DOI] [PubMed] [Google Scholar]
- Wan Y, Cheng G, Liu X, Hao SJ, Nisic M, Zhu CD, Xia YQ, Li WQ, Wang ZG, Zhang WL, et al. (2017). Rapid magnetic isolation of extracellular vesicles via lipid-based nanoprobes. Nat Biomed Eng 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang PW, Kuo HM, Huang HT, Chang AY, Weng SW, Tai MH, Chuang JH, Chen IY, Huang SC, Lin TK, et al. (2014). Biphasic response of mitochondrial biogenesis to oxidative stress in visceral fat of diet-induced obesity mice. Antioxid Redox Signal 20, 2572–2588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster I, Salie R, Marais E, Fan WJ, Maarman G, Huisamen B, and Lochner A (2017). Myocardial susceptibility to ischaemia/reperfusion in obesity: a re-evaluation of the effects of age. BMC Physiol 17, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z, Wang X, Liu X, Du H, Sun C, Shao X, Tian J, Gu X, Wang H, Tian J, et al. (2018). Adipose-Derived Exosomes Exert Proatherogenic Effects by Regulating Macrophage Foam Cell Formation and Polarization. J Am Heart Assoc 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun J, and Finkel T (2014). Mitohormesis. Cell Metab 19, 757–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Wu X, Chen P, Liu L, Xin N, Tian Y, and Dillin A (2018). The Mitochondrial Unfolded Protein Response Is Mediated Cell-Non-autonomously by Retromer-Dependent Wnt Signaling. Cell 174, 870–883 e817. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Proteomics data can be accessed at MassIVE, massive.ucsd.edu; accession #: MSV000085896
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.