

Abstract
Aromatic amines are widely used in personal care products and human exposure to this class of chemicals is widespread. Bioanalytical methods to determine trace levels of aromatic amines in human urine are scarce. In this study, a liquid chromatography-tandem mass spectrometry (LC-MS/MS) method was developed to determine 39 primary aromatic amines (AAs) along with nicotine and cotinine in human urine. Chromatographic separation of the 41 analytes was achieved on an Ultra biphenyl (100 mm x 2.1 mm, 5 μm) column. Mass spectrometry was operated in electrospray ionization positive ion multi-reaction monitoring (MRM) mode. The method exhibited excellent linear dynamic range (0.1-50 ng/mL) with correlation coefficients (r) >0.999 for all analytes. Urine samples (2 mL) were hydrolyzed using 10 M NaOH at 95 0C for 15 h and target analytes were extracted using methyl-tert-butyl ether (MTBE). Addition of 15 μL of 0.25M HCl to the sample extracts improved the recoveries of several target analytes. The method was validated through the analysis of fortified quality control (QC) samples and a certified standard reference material (SRM). Relative recoveries (%) of target analytes fortified in QC samples were in the range of 75-114% for 37 of the 41 analytes while the other analytes exhibited lower recoveries (16-74%). The limits of detection (LOD) and limits of quantification (LOQ) of target analytes were in the range of 0.025-0.20 ng/mL and 0.1-1.0 ng/mL, respectively. Intra-day and inter-day precision of the method assessed through the analysis of fortified urine QC samples at three different concentrations were <11.7% and <15.9% (measured as RSD), respectively. The method was applied in the analysis of urine samples from the general population and known smokers; aniline, para-anisidine, para-toluidine, ortho/meta-toluidine, 3-chloroaniline, 4-chloroaniline, 3,4-dichloroaniline, and 4,4'-methylenedianiline were found in all smoker’s urine at sum concentrations ranging from 0.04 to 9.16 ng/mL.
Keywords: Aromatic amines, LC-MS/MS, Biomonitoring, Urine, Aniline
Graphical Abstract
1. Introduction
Primary aromatic amines (AAs) are chemicals containing an aromatic ring directly bonded to an amine functional group (−NH2) at ortho, meta, or para positions [1, 2]. Aniline is the parent compound from which hundreds of AA derivatives are manufactured. The estimated annual global production of aniline was 8.4 million tons in 2020 [3]. AAs are used in the production of dyes and pigments, polyurethane, rubber, pesticides, and pharmaceuticals [4-6]. AAs are major ingredients in formulations of dyes and pigments which are extensively used as colorants in hair dyes, leathers, printing inks, paints, lacquers, metal finishes, and paper products [7]. Widespread use of AAs in consumer products led to their ubiquitous environmental occurrence [8, 9]. Human exposure to AAs is prevalent because of these chemicals’ occurrence in tobacco smoke and personal care products. Besides tobacco smoke, heated cooking oil, diesel engine exhaust, hair dyes, food and water contribute to human exposure to AAs [10]. Occupational exposure to AAs has been linked to acute methemoglobinemia, hemolysis, dermatitis, and cancer, particularly, bladder cancer [11, 12]. Carcinogenic potential of hair dyes containing AAs was reported in vitro [13-15]. Many AAs have been reported as potent mutagenic, carcinogenic, and hemotoxic agents. The World Health Organization (WHO) and the International Agency for Research on Cancer (IARC) listed several AAs as known carcinogens in humans [16, 17].
Although interest in the development of reliable and sensitive analytical methods for the determination of carcinogenic AAs has increased, the polar nature of these chemicals poses challenges for trace analysis. Gas chromatography (GC) coupled with mass spectrometry (MS) has been used in the determination of AAs in hair dyes, henna, cigarette smoke, air, water, sediment, textiles, and polyamide spoons [18-21]. Nevertheless, GC-MS based methods involve a time-consuming derivatization step [22]. Recent developments in chromatography column chemistries enable sensitive analysis of AAs by liquid chromatography-tandem mass spectrometry (LC-MS/MS) methods [23]. LC-MS/MS methods have been used in the determination of AAs in kitchen utensils, textiles, cigarette smoke, mainstream water pipe smoke, polyurethane, food contact materials, and paper products [24-30].
Following exposure, AAs are metabolized in the body and excreted in urine as free, sulfated, acetylated and glucuronidated conjugates [31]. The biological half-lives of AAs are on the order of few hours to few days [32]. For instance, benzidine (i.e., p-diaminobiphenyl) and its metabolites (N-acetyl and N,N-diacetyl benzidine) were found in urine of workers from dye manufacturing industries [33]. Earlier methods of analysis of AAs in urine involved derivatization followed by GC-MS detection [33, 34]. Studies have reported the occurrence of AAs in urine and blood of occupationally exposed populations and smokers [31, 35-37]. The earlier studies on exposed populations were focused on select carcinogenic AAs such as o-toluidine, 4-aminobiphenyl, 2-naphthylamine, and benzidine [35].
Several AAs are used in the production of azodyes for application in leather, textile, and paper industries. Azo dyes can reductively break down to an array of AAs in human bodies and in the environment [38, 39]. The potential health risks of AAs has resulted in US Food and Drug Administration’s (FDA) regulations on aromatic amines in azo food dyes (21 CFR 74.705 and 21 CFR 74.706) and European Union’s (EU) listing of 22 AAs as hazardous compounds for use in consumer products (REACH legislation, Section 43 of Annex XVII) [13]. A few studies reported that azo dyes used in textiles are the sources of AAs in the environment [40-42]. There is a considerable interest in the assessment of human exposure to AAs arising from azo dyes.
The aim of this study was to develop a comprehensive analytical method capable of measuring 41 AAs simultaneously in human urine using LC-MS/MS (Table 1 and Table S1). Nicotine and cotinine, two markers of tobacco smoke exposure, were also included in the method as we envisaged that these two analytes could be determined in the same method. The chromatographic method was optimized to achieve ideal retention and resolution of all 41 analytes in a single analysis. Several sample extraction methods were examined to achieve maximum efficiency, accuracy, precision and sensitivity for simultaneous analysis of 41 analytes. To the best of our knowledge, this is the first comprehensive study to determine 39 AAs and two tobacco exposure biomarkers in urine, which can be applied in large scale human biomonitoring programs to assess exposure to these carcinogenic chemicals.
Table 1.
List of target analytes and their MRM parameters; precursor ion (Q1), product ion (Q3), declustering potential (DP), entrance potential (EP), collision energy (CE), and collision exit potential (CXP).
| S.No | Analyte Name | Abbreviation | Q1 | Q3 | DP(V) | EP(V) | CE(V) | CXP(V) |
|---|---|---|---|---|---|---|---|---|
| 1 | aniline | Aniline | 94.0 | 77.1 | 80 | 8 | 25.0 | 10 |
| 2 | 2-naphthylamine | 2-NA | 144.0 | 127.1 | 80 | 8 | 32.0 | 10 |
| 3 | para-anisidine | p-Anisidine | 124.0 | 93.0 | 100 | 10 | 22.5 | 10 |
| 4 | ortho-anisidine | o-Anisidine | 124.0 | 109.0 | 80 | 10 | 23.0 | 10 |
| 5 | 2,4-dimethylaniline | 2,4-DMA | 122.0 | 107.0 | 100 | 10 | 22.5 | 10 |
| 6 | 2,6-dimethylaniline | 2,6-DMA | 122.1 | 105.0 | 95 | 10 | 22.0 | 10 |
| 7 | para-cresidine | p-CD | 138.0 | 123.0 | 102 | 10 | 25.5 | 10 |
| 8 | 2-methyl-5-nitroaniline | 5-NTD | 152.9 | 107.2 | 81 | 7 | 23.7 | 7 |
| 9 | para-toluidine | p-TD | 108.1 | 91.0 | 95 | 8 | 25.0 | 8 |
| 10 | ortho/meta-toluidine | o/m-TD | 108.0 | 91.0 | 100 | 10 | 23.0 | 8 |
| 11 | 4-chloroaniline | 4-CA | 128.1 | 93.1 | 95 | 8 | 25.8 | 8 |
| 12 | 3-chloroaniline | 3-CA | 127.8 | 93.0 | 100 | 7.5 | 25.0 | 8 |
| 13 | 2,4,5-trimethylaniline | 2,4,5-TMA | 136.0 | 121.0 | 80 | 7 | 22.4 | 8 |
| 14 | 2,4,6-trimethylaniline | 2,4,6-TMA | 136.0 | 121.0 | 80 | 7 | 22.4 | 8 |
| 15 | 3,3'-dimethylbenzidine | 3,3'-DMBD | 213.1 | 196.0 | 100 | 8 | 29.0 | 8 |
| 16 | 4-ethoxyaniline | 4-EA | 138.1 | 110.1 | 100 | 7 | 20.0 | 8 |
| 17 | 3,4-dichloroaniline | 3,4-DCA | 162.0 | 127.1 | 100 | 10 | 28.0 | 10 |
| 18 | 1,3-phenylenediamine | 1,3-PLD | 109.1 | 92.2 | 100 | 10 | 22.0 | 10 |
| 19 | 4-chloro-o-toluidine | 4-CTD | 142.1 | 107.1 | 100 | 10 | 22.5 | 10 |
| 20 | 4,4'-oxydianiline | 4,4-OD | 201.2 | 108.0 | 100 | 9 | 25.0 | 10 |
| 21 | 4-aminoacetanilide | 4-AA | 150.9 | 109.0 | 100 | 5 | 23.0 | 6 |
| 22 | 4,4'-methylenedi-o-toluidine | 4,4'-MTD | 227.1 | 120.1 | 70 | 7 | 33.5 | 8 |
| 23 | 4,4'-methylenebis(2-chloroaniline) | 4,4'-MBCA | 267.1 | 231.1 | 100 | 9 | 29.3 | 10 |
| 24 | 2-aminobiphenyl | 2-ABP | 170.0 | 153.2 | 87 | 9 | 28.1 | 10 |
| 25 | 4-aminobiphenyl | 4-ABP | 170.1 | 152.2 | 100 | 10 | 41.0 | 10 |
| 26 | benzidine | BD | 185.2 | 167.0 | 100 | 10 | 37.0 | 10 |
| 27 | ortho-dianisidine | o-DAD | 245.1 | 230.2 | 100 | 7 | 26.0 | 8 |
| 28 | 4,4'-thiodianiline | 4,4'-TD | 217.1 | 124.1 | 100 | 10 | 30.0 | 10 |
| 29 | 4,4'-methylenedianiline | 4,4'-MDA | 199.0 | 106.0 | 100 | 10 | 21.0 | 8 |
| 30 | 3,4-diaminotoluene | 3,4-DAT | 123.0 | 106.1 | 90 | 7 | 25.0 | 8 |
| 31 | 2,4-diaminotoluene | 2,4 DAT | 123.0 | 106.0 | 100 | 10 | 21.0 | 10 |
| 32 | 2,6-diaminotoluene | 2,6-DAT | 123.0 | 106.1 | 90 | 7 | 25.0 | 8 |
| 33 | 3,4/ 2,4-diaminoanisole | 3,4/2,4-DAAS | 139.0 | 124.1 | 80 | 10 | 23.0 | 10 |
| 34 | 3-(3-aminobenzyl)phenylamine | 3-3AB-PA | 199.2 | 106.1 | 110 | 10 | 24.0 | 10 |
| 35 | 2,2'-dimethyl-(1,1'-biphenyl)4,4'-diamine | DMBPDA | 213.0 | 181.1 | 100 | 10 | 32.0 | 10 |
| 36 | 2-amino-6-methoxybenzothiazole | 2-AMoBzT | 181.0 | 67.2 | 100 | 10 | 60.0 | 10 |
| 37 | 1,1-bis(4-aminophenyl)cyclohexane | 1,1-BPCH | 267.0 | 106.1 | 100 | 8 | 39.0 | 8 |
| 38 | nicotine | Nicotine | 163.1 | 13.0 | 60 | 8 | 30.0 | 10 |
| 39 | cotinine | Cotinine | 177.5 | 98.1 | 100 | 9 | 26.0 | 10 |
| 40 | ortho-aminoazotoluene | o-AAT | 226.0 | 91.1 | 95 | 8 | 29.0 | 7 |
| 41 | 4-aminoazobenzene | 4-AAB | 198.1 | 77.0 | 100 | 10 | 24.0 | 10 |
| Internal standards | ||||||||
| 45 | aniline-d5 | Aniline-D5 | 99.0 | 82.0 | 60 | 8 | 25.0 | 10 |
| 46 | 2-naphthylamine-d7 | 2-NA-D7 | 151.0 | 134.1 | 60 | 8 | 32.0 | 10 |
| 47 | ortho-anisidine-d7 | o-Anisidine-D7 | 131.0 | 113.0 | 80 | 10 | 24.0 | 8 |
| 48 | 2,3,4,5-para-anisidine-d4 | p-Anisidine-D4 | 128.0 | 97.1 | 100 | 10 | 26.0 | 8 |
| 49 | ortho-toluidine-13C6 | o-TD-13C6 | 114.0 | 97.0 | 100 | 10 | 25.0 | 10 |
| 50 | para-toluidine-d7 | p-TD-D7 | 114.9 | 98.1 | 100 | 10 | 25.5 | 10 |
| 51 | 4-chloroaniline-13C6 | 4-CA-13C6 | 134.1 | 99.1 | 85 | 7 | 24.4 | 9 |
| 52 | 1,3-benzenediamine-d4 | 1,3-PLD-D4 | 112.9 | 96.2 | 100 | 10 | 22.0 | 9 |
| 53 | 2,6-diaminotoluene-d3 | 2,6-DAT-D3 | 126.2 | 109.2 | 95 | 10 | 22.0 | 10 |
| 54 | 2,4-diaminotoluene-d3 | 2,4-DAT-D3 | 126.2 | 109.2 | 95 | 10 | 22.0 | 10 |
| 55 | 3,4-dichloroaniline-d2 | 3,4-DCA-D2 | 164.1 | 129.1 | 95 | 8 | 28.8 | 10 |
| 56 | 2,4,6-trimethylbenzeneamine-d11 | 2,4,6-TMS-D11 | 147.0 | 130.2 | 95 | 10 | 24.5 | 10 |
| 57 | benzidine-d8 | BD-D8 | 193.0 | 176.0 | 100 | 10 | 28.3 | 10 |
| 58 | 2,4-dimethylaniline-d6 | 2,4-DMA-D6 | 128.1 | 111.2 | 100 | 9 | 24.3 | 8 |
| 59 | 2,6-dimethylaniline-d6 | 2,6-DMA-D6 | 128.1 | 111.2 | 100 | 9 | 24.3 | 8 |
| 60 | 4'-aminoacetanilide-2',3',5',6'-d4 | 4-AAD-D4 | 155.1 | 113.0 | 100 | 7 | 22.4 | 8 |
| 61 | cotinine-d3 | Cotinine-D3 | 180.2 | 101.2 | 100 | 9 | 26.0 | 10 |
| 62 | nicotine -d3 | Nicotine -D3 | 166.1 | 13.0 | 60 | 8 | 30.0 | 10 |
2. Materials and Methods
2.1. Chemicals and reagents
Forty three native (unlabeled) and 18 isotope labelled analytical standards (Table 1; chemical names and their abbreviations are shown) of 95-99% purity were purchased from Toronto Research Chemicals (TRC; Toronto, ON, Canada), AccuStandard (New Haven, CT, USA) and Sigma-Aldrich (St. Louis, MO, USA). HPLC-grade water, methanol, acetone, acetonitrile, methyl tert-butyl ether (MTBE), and dichloromethane (DCM) were purchased from J. T Baker (Center Valley, PA, USA). Synthetic urine was purchased from Cerilliant (Round Rock, TX, USA). HPLC grade diethyl ether was purchased from Alfa Aesar (Tewksbury, MA, USA). Analytical grade ammonium formate (HCOONH4), acetic acid, formic acid (HCOOH), 30% ammonium hydroxide in water (NH4OH), sodium chloride (NaCl), sodium hydroxide (NaOH), hydrochloric acid (37% v/v) and dimethyl sulfoxide (DMSO) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Screw cap glass tubes (16 x 100 mm) were purchased from Fisher Scientific (Waltham, MA, USA). Solid phase extraction (SPE) cartridges, Nexus® ABS Elut 3 cc (60 mg) from Agilent Technologies (Santa Clara, CA, USA) and Oasis® HLB 3 cc (60 mg) and Oasis® WCX 3 cc (60 mg) from Waters corporation (Milford, MA, USA) were used in the study.
2.2. Preparation of analytical standard solutions
Individual stock solutions (1 mg/mL) of AA standards were prepared by dissolving 5 mg of each native compound in acetonitrile except for 2,4-DAT, 2,6-DAT, and 3,4-DAT, which were dissolved in water. 2,4-DAAS, and 3,4-DAAS were dissolved in methanol. Stock standard solutions were diluted in respective solvents to yield final concentrations of 100, 10, 1, 0.1 and 0.01 μg/mL. A mixture of standard containing all target analytes was prepared at 100 ng/mL in water: methanol (9:1 v/v). Isotope labelled internal standards were prepared similarly in acetonitrile. The standard solutions were stored at −20°C until further use.
2.3. Quality control samples
Laboratory quality control (QC) samples were prepared in triplicate for optimization of the sample extraction. QC samples include reagent blank (HPLC-grade water used in place of urine), matrix blank (synthetic urine) and matrix spike (synthetic urine fortified with target analytes). QC samples were prepared by fortifying native and internal standards to yield final concentrations of 5, 10 and 20 ng/mL. The QC samples were then vortex mixed and stored at 4°C until further use. Reagent blank and matrix blanks were analyzed as a check for background levels of contamination and matrix effects.
2.4. Sample extraction
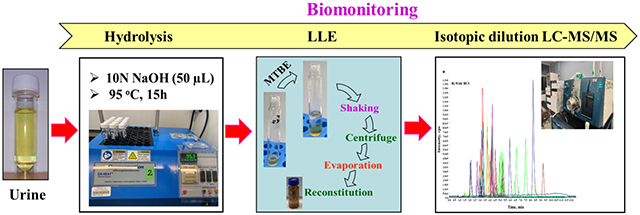
Urine samples or fortified QC samples (2 mL) were transferred into a screw cap glass tube and subjected to alkaline hydrolysis by the addition of 50 μL of 10 M NaOH solution (pH >10). After vortex mixing for 1 min, samples were placed on a hot plate at 95°C for 15 h. Samples were then brought to room temperature, and target analytes were extracted by liquid-liquid extraction (LLE) with 2 mL of MTBE. The mixture was shaken in an orbital shaker at 180 strokes per min for 10 min (Eberbach Corp., Ann Arbor, MI, USA) and centrifuged at 4000 rpm for 10 min at room temperature (24°C). MTBE extract containing the target analytes was transferred into a polypropylene tube and the extraction was repeated with 2 mL of MTBE. Both extracts were combined and 15 μL of 0.25 M HCl was added prior to evaporation to near-dryness under a gentle stream of nitrogen. The residue was re-dissolved in 200 μL of water:methanol (9:1, v/v) and transferred into a LC vial with 300 μL glass insert. We also examined week cation exchange and reverse phase SPE but those methods did not yield optimal extraction efficiency. Extraction protocol pertaining to SPE are given in the supplementary information (SI).
2.5. LC-MS/MS
An ultra-high performance liquid chromatography (UHPLC, Shimadzu LC-30 AD; Shimadzu Corporation, Kyoto, Japan) coupled with electrospray ionization triple quadrupole mass spectrometry (Sciex Triple Quad 5500, ESI-MS/MS; Applied Biosystems, Foster City, CA, USA) was used for the identification and quantification of target chemicals. Chromatographic separation was achieved on an Ultra BiPh column (100 mm x 2.1 mm, 5 μm; Restek, Bellefonte, PA, USA) connected to a Betasil C18 guard column (20 mm x 2.1 mm, 5 μm; Thermo Scientific, West Palm Beach, FL, USA). The mobile phase consisted of 0.1% formic acid in water:methanol (95:5, v/v) (A) and 0.1% formic acid in methanol (B), pumped at a flow rate of 0.3 mL/min. The gradient program was as follows: 0.0 min (95% A), 0.01-2.50 min (95-58% A), 2.50-6.50 min (58%-25% A), 6.50-8.70 min (25-5% A, hold for 1 min), and 9.70-10.0 min (5-95% A, hold for 2.50 min) with a total run time of 12.5 min. The LC column was set at room temperature (22°C) and the auto sampler temperature was set at 15 °C. The sample injection volume was 5 μL.
The mass spectrometer (MS) was operated in electrospray positive ionization mode and data acquisition was performed under multiple reaction monitoring (MRM). Compound specific MS/MS parameters, declustering potential (DP), entrance potential (EP), collision energy (CE), and collision exit potential (CXP) were optimized for each target compound by directly infusing individual analytes into the MS at 10 ng/mL using an in-built syringe pump. The optimized MS parameters for target analytes are presented in Table 1. The electrospray ionization source was operated at 4.5 kV and 500 °C. The curtain gas flow rate was set at 10 psi and collision gas, nebulizer gas and ion source gas flows were set at 8, 30, and 30 psi, respectively.
2.6. Method validation
Two different calibrations, first with standards dissolved in water:methanol (9:1, v/v) (solvent calibration curve) and second with standards fortified in urine matrix (matrix-matched calibration) were developed. The solvent calibration was used to determine the linear relationship between instrumental response and true concentration of analytes. A nine-point solvent calibration, at concentrations of 0.1, 0.2, 0.5, 1, 2, 5, 10, 20, and 50 ng/mL with 10 ng/mL of internal standards, was prepared for each analyte. A weighted linear regression was used to fit the calibration curve and the regression coefficients were >0.999 for all analytes. For the preparation of matrix-matched calibration, synthetic urine was fortified with target analytes at concentrations ranging from 0.1 to 50 ng/mL with 10 ng/mL of internal standards and passed through the entire analytical procedure (including LLE). The analytical method was validated for accuracy, precision (intra- and inter-day variances), limit of detection (LOD), and limit of quantification (LOQ) using QC samples fortified at three concentrations (5, 10 and 20 ng/mL) of analytes, as described below. Urine samples collected in 2020 from 20 healthy volunteers from Albany area of New York state were analyzed to demonstrate the feasibility of the method
3. Results and Discussion
3.1. Optimization of LC-MS/MS conditions
For the optimization of MS/MS parameters, individual analytes (prepared in water: methanol, 9:1 v/v), at a concentration of 10 ng/mL, were infused directly into the MS operated in electrospray positive ionization mode. The signal intensity of majority of the analytes was poor when the precursor ion (Q1) scan was performed. This suggested that the analytes protonated poorly under the mobile phase conditions of water:methanol mixture. Addition of 0.01% formic acid in water significantly enhanced the signal intensity of precursor ions. An earlier study showed that pH of mobile phase solvents significantly influenced the MS detection of aromatic amines [43]. Therefore, mobile phase additives such as formic acid are required to enhance ionization and to improve signal intensity of AAs. Following Q1 scan of each analyte, the most abundant product ion (Q3) was selected through the product ion scan. Optimization of MRM parameters such as CE, DP, EP, and CXP was achieved for each analyte to obtain the highest sensitivity (Table 1).
Following optimization of MS parameters for individual analytes, chromatographic separation of the mixture of 43 analytes was examined. Because AAs require acidic conditions for efficient ionization, C18 columns were deemed unsuitable for optimal retention and separation of analytes. Biphenyl columns have been reported to offer better interaction between the aromatic group and the stationary phase and have recommended for the analysis of polar compounds [44, 45]. An Ultra BiPh column (Restek; 100 mm x 2.1 mm, 5 μm) was selected for chromatographic separation of AAs and the mobile phase conditions were optimized subsequently. Various mobile phase additives such as formic acid, acetic acid, and ammonium formate were tested for optimal chromatographic performance. Among them, 0.1% formic acid in both aqueous and organic mobile phases exhibited suitable retention and peak characteristics (shape and intensity) in comparison to acetic acid (data not shown). However, 0.1% formic acid did not provide chromatographic separation of structural isomers (e.g., ortho-TD and meta-TD). A recent study reported chromatographic separation of isomers with the mobile phase solvent containing ammonium formate buffer at pH 3.5 [42]. Nevertheless, addition of ammonium formate in the mobile phase reduced the signal intensity of several AAs targeted in our study (Fig. 1). Then, we examined a combination of water, methanol, and acetonitrile as mobile phase solvents with 0.1 % formic acid as the additive. Separation of target analytes except for two structural isomers of 2,4- and 3,4-DAAS and ortho- and meta-TD was achieved with 0.1% formic acid in water:methanol mixture (95:5, v/v) (A) and 0.1% formic acid in methanol (B). These two isomers of two AAs did not separate under various combinations of mobile phases and gradient elution. Therefore, concentrations of these two analytes were reported as the sum of two isomers. Total ion chromatogram (TIC) of target analytes under optimized chromatographic conditions (as described in section 2.2) is shown in Fig. 2.
Fig. 1.

Extracted ion chromatograms (XIC) of select primary aromatic amines injected (5 μL) at a concentration of 10 ng/mL spiked in water:methanol (9:1, v/v) analyzed with two different (ammonium formate versus formic acid) mobile phase additives.
Fig.2. LC-MS/MS total ion chromatogram (TIC, A) and extracted ion chromatogram (XIC, B-D) of target analytes (41 AA, cotinine and nicotine) injected at (5 μL) at a concentration of 10 ng/mL in water: methanol (9:1, v/v) analyzed under optimized conditions.

For the sake of distinction of analytes, extracted ion chromatograms were divided into three fractions (i.e., F1-F3) depending on their peak signal intensity and retention time. Compound number assigned on the peak are shown in Table 2.
3.2. Sample preparation
Because AAs are excreted in urine as free as well as N-acetylated, glucuronidated and sulfated conjugates, a deconjugation step was needed to determine total concentrations of analytes. Whereas enzymatic hydrolysis is used in the deconjugation of glucuronide and sulfated metabolites, acidic and alkaline hydrolysis were also used to release acetylated and glucuronidated forms of metabolites [35]. We used alkaline hydrolysis to deconjugate bound forms of AAs in urine, as reported earlier [31, 35]. The reaction involved addition of 15 μL of 10M NaOH to 2 mL of urine followed by incubation at 95° C for 15 h (details in section 2.4). A similar approach has been used in the analysis of AAs in urine by the U.S. Centers for Disease Control and Prevention (CDC) [46].
The extraction efficiencies of LLE and SPE methods were tested using fortified sample matrix. Because SPE can purify the sample extract while selectively enriching the analytes of interest, three different cartridges were tested: Oasis® WCX (3cc) cation exchange, Nexus® ABS Elut (3cc) polymeric reverse phase, and Oasis® HLB (3cc) hydrophilic-lipophilic balance stationary phases. Neutralization of basified urine sample with concentrated HCl was required prior to the passage of samples through cation exchange cartridge. The recoveries of target analytes were poor (<50%). The basified urine samples were diluted with water prior to passage through HLB and ABS Elut cartridges. Under basic conditions, the target analytes were expected to be in neutral form and thus hydrophobic interaction between the analyte and the polymeric stationary phase was envisaged [47, 48]. Although ABS Elut cartridge presented optimal recoveries (80-120%) for most analytes, noisy baseline with remarkable matrix effect was found (data not shown), which was due to the presence of residues of NaOH in the sample extract. It was found that SPE cartridges present challenges to extract all analytes from the basified urine and therefore a LLE method, as described below, was developed.
3.3. Optimization of liquid-liquid extraction
The choice of extraction solvent, volume and extraction duration were optimized to achieve maximum recoveries of target analytes from the sample matrix. For the optimization of extraction solvent, MTBE, DCM, MTBE+DCM (1:1 v/v), and acetone+DCM (1:1 v/v) were tested. Three milliliters of the solvent were added to 2 mL of the basified urine and extracted for 30 min, by shaking in an orbital shaker. All four combinations of solvents yielded recoveries >70% for eleven analytes while the remaining analytes presented a recovery in the range of 0-60%. It was also found that some analytes were lost during the nitrogen evaporation step. Fifteen microliters of concentrated 0.25 M HCl in water was added to the extract, as a keeper solvent prior to nitrogen evaporation. Following the addition of 0.25 M HCl, the recoveries of analytes in all four extraction solvents increased significantly and were in the range of 70-120%, except for 2,4-DAT, 3,4-DAT, O-AAT, and 4-AAB whose recoveries were in the range of 0-50%. Aniline showed recoveries at 140%, which was thought to be due to the conversion of certain diamines into aniline.. Although all of the solvent combinations yielded similar recoveries, MTBE was selected due to its relatively lower toxicity [49]. The enhancement in signal intensities of AAs following the addition of 0.25 M HCl into MTBE extract was significant . In further steps, extraction time was optimized by shaking sample-solvent mixture in an orbital shaker at 180 strokes per min for 5, 10, 15, 20, and 30 min. Although all target analytes were recovered within 5 min, we selected an extraction duration of 10 min to ensure complete recoveries. The volume of extraction solvent was optimized by extraction of fortified urine matrix with 1, 2, and 3 mL of MTBE. Most analytes were recovered in 2 mL of MTBE within an extraction duration of 10 min. Nevertheless, we selected two sequential extractions with 2 mL of MTBE each time.
The recoveries of 2,4-DAT, 3,4-DAT, O-AAT, 4-AAB, , and aniline were above or below the optimal range of 80-120% but they were consistent (n=3). It has been reported that 4-AAB can be reductively cleaved to aniline [42]. This explained poor recoveries for 4-AAB and higher recoveries for aniline. Therefore, aniline was analyzed in urine separately without the addition of other target analytes and recoveries of >90% was found for this compound (Table 2). 2,4-DAT and 3,4-DAT recoveries were low (30-48%), but were consistent and similar to those reported in an earlier study [50].
Table 2.
Retention time (RT), regression coefficient (r) of the calibration curve, recovery %, intra-day and inter-day precision (%RSD), limit of detection (LOD), and limit of quantification (LOQ) of aromatic amines under optimized LLE and LC-MS/MS conditions.
| S. No | Analyte | RT (min) |
Regression coefficient (r) |
5 ng/mL; n=3 | 10 ng/mL; n=3 | 20 ng/mL; n=3 | LOD (ng/mL) |
LOQ (ng/mL) |
||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Recovery % | %RSD | Recovery % | %RSD | Recovery % | %RSD | |||||||||
| Intra-day | Inter-day | Intra-day | Inter-day | Intra-day | Inter-day | |||||||||
| 1 | Aniline | 2.07 | 0.9999 | 95.4 | 2.16 | 2.78 | 91.2 | 3.89 | 2.88 | 92.4 | 2.54 | 3.12 | 0.100 | 0.2 |
| 2 | 2-NA | 5.13 | 0.9997 | 94.6 | 1.59 | 2.15 | 95.5 | 5.96 | 4.61 | 93.1 | 2.12 | 2.89 | 0.025 | 0.1 |
| 3 | p-Anisidine | 2.97 | 0.9991 | 96.4 | 3.46 | 4.29 | 93.9 | 4.18 | 6.19 | 101 | 4.67 | 5.14 | 0.050 | 0.1 |
| 4 | o-Anisidine | 3.21 | 0.9993 | 99.1 | 2.14 | 2.96 | 109 | 8.21 | 5.04 | 105 | 4.23 | 3.48 | 0.025 | 0.1 |
| 5 | 2,4-DMA | 4.17 | 0.9992 | 109 | 4.43 | 3.73 | 113 | 3.39 | 4.18 | 99.8 | 4.28 | 6.03 | 0.050 | 0.1 |
| 6 | 2,6-DMA | 4.47 | 0.9998 | 93.9 | 5.42 | 5.27 | 104 | 4.50 | 2.99 | 98.3 | 4.67 | 4.78 | 0.050 | 0.1 |
| 7 | P-CD | 4.19 | 0.9995 | 110 | 4.38 | 3.87 | 105 | 6.16 | 5.95 | 113 | 8.49 | 10.3 | 0.025 | 0.1 |
| 8 | 5-NTD | 6.36 | 0.9995 | 54.9 | 7.60 | 9.20 | 61.0 | 8.22 | 11.3 | 58.2 | 6.10 | 9.89 | 0.500 | 1.0 |
| 9 | p-TD | 3.31 | 0.9992 | 101 | 3.68 | 5.20 | 101 | 6.12 | 6.90 | 108 | 7.80 | 7.96 | 0.025 | 0.1 |
| 10 | o/m-TD | 3.14 | 0.9990 | 103 | 3.26 | 3.30 | 107 | 5.86 | 3.44 | 109 | 5.51 | 4.60 | 0.050 | 0.1 |
| 11 | 4-CA | 4.00 | 0.9996 | 105 | 6.96 | 7.26 | 107 | 5.65 | 6.09 | 114 | 3.43 | 4.11 | 0.050 | 0.1 |
| 12 | 3-CA | 4.84 | 0.9995 | 78.4 | 5.04 | 4.37 | 80.6 | 3.95 | 6.43 | 81.3 | 4.56 | 9.98 | 0.050 | 0.1 |
| 13 | 2,4,5-TMA | 4.94 | 0.9997 | 86.6 | 3.58 | 2.17 | 82.6 | 3.74 | 3.29 | 84.9 | 7.07 | 7.38 | 0.050 | 0.1 |
| 14 | 2,4,6-TMA | 5.11 | 0.9999 | 100 | 2.48 | 2.23 | 99.1 | 6.41 | 3.14 | 101 | 8.35 | 8.35 | 0.050 | 0.2 |
| 15 | 3,3'-DMBD | 4.32 | 0.9993 | 84.8 | 7.40 | 6.03 | 99.5 | 4.60 | 6.64 | 103 | 5.16 | 9.44 | 0.050 | 0.2 |
| 16 | 4-EA | 3.89 | 0.9995 | 92.7 | 7.95 | 4.81 | 111 | 5.11 | 4.51 | 103 | 5.92 | 7.03 | 0.025 | 0.1 |
| 17 | 3,4-DCA | 6.72 | 0.9991 | 105 | 4.56 | 4.08 | 114 | 3.45 | 2.84 | 115 | 3.46 | 5.11 | 0.100 | 0.2 |
| 18 | 1,3-PLD | 1.29 | 0.9992 | 75.5 | 4.50 | 5.50 | 85.3 | 4.90 | 5.64 | 82.2 | 5.20 | 5.40 | 0.200 | 0.5 |
| 19 | 4-CTD | 5.27 | 0.9999 | 85.9 | 11.7 | 7.36 | 95.9 | 3.60 | 5.74 | 83.2 | 8.89 | 11.1 | 0.025 | 0.1 |
| 20 | 4,4'-OD | 2.55 | 0.9997 | 71.6 | 3.43 | 4.50 | 79.4 | 11.1 | 7.57 | 78.5 | 7.22 | 9.93 | 0.020 | 0.1 |
| 21 | 4-AA | 1.59 | 0.9997 | 108 | 4.50 | 4.54 | 102 | 2.40 | 4.95 | 105 | 5.43 | 6.15 | 0.100 | 0.2 |
| 22 | 4,4'-MTD | 4.11 | 0.9995 | 80.7 | 2.41 | 2.78 | 86.4 | 4.88 | 4.86 | 82.8 | 5.91 | 6.85 | 0.025 | 0.1 |
| 23 | 4,4'-MBCA | 8.34 | 0.9998 | 75.6 | 5.60 | 6.20 | 81.4 | 7.57 | 11.4 | 79.9 | 4.60 | 5.42 | 0.100 | 0.2 |
| 24 | 2-ABP | 7.20 | 0.9993 | 83.0 | 5.19 | 8.18 | 82.2 | 5.97 | 6.71 | 84.6 | 5.08 | 7.21 | 0.050 | 0.2 |
| 25 | 4-ABP | 6.42 | 0.9995 | 97.1 | 3.67 | 5.88 | 94.0 | 6.03 | 4.61 | 96.2 | 6.68 | 8.57 | 0.050 | 0.2 |
| 26 | BD | 3.12 | 0.9996 | 94.6 | 3.43 | 3.31 | 98.7 | 8.85 | 6.26 | 103 | 5.35 | 7.47 | 0.050 | 0.1 |
| 27 | o-DAD | 4.37 | 0.9995 | 62.8 | 7.69 | 10.4 | 64.8 | 9.52 | 5.44 | 63.4 | 10.97 | 13.8 | 0.025 | 0.1 |
| 28 | 4,4'-TD | 5.04 | 0.9996 | 74.0 | 5.84 | 8.83 | 72.5 | 5.90 | 6.68 | 73.8 | 10.60 | 11.3 | 0.025 | 0.1 |
| 29 | 4,4'-MDA | 2.99 | 0.9996 | 96.2 | 4.81 | 2.74 | 94.9 | 3.80 | 2.89 | 92.9 | 7.10 | 6.32 | 0.025 | 0.1 |
| 30 | 3,4-DAT | 3.14 | 0.9995 | 31.2 | 5.41 | 6.89 | 28.9 | 3.45 | 7.29 | 30.1 | 5.79 | 8.15 | 0.050 | 0.1 |
| 31 | 2,4 DAT | 2.02 | 0.9997 | 36.7 | 4.96 | 3.89 | 48.5 | 2.71 | 9.70 | 45.2 | 5.92 | 7.89 | 0.100 | 0.2 |
| 32 | 2,6-DAT | 1.44 | 0.9997 | 68.4 | 4.89 | 6.78 | 75.3 | 5.08 | 12.0 | 70.2 | 9.21 | 6.78 | 0.025 | 0.1 |
| 33 | 3,4 /2,4 -DAAS | 4.18 | 0.9994 | 100 | 4.46 | 2.03 | 105 | 8.60 | 9.81 | 111 | 9.91 | 10.4 | 0.100 | 0.2 |
| 34 | 3-3AB-PA | 3.64 | 0.9995 | 102 | 3.85 | 2.19 | 101 | 5.20 | 4.42 | 112 | 6.49 | 9.80 | 0.025 | 0.1 |
| 35 | DMBPDA | 3.27 | 0.9996 | 99.3 | 4.13 | 5.18 | 94.9 | 9.66 | 6.42 | 104 | 5.68 | 6.94 | 0.025 | 0.1 |
| 36 | 2-AMoBzT | 5.15 | 0.9995 | 92.1 | 5.62 | 3.97 | 94.3 | 6.74 | 8.74 | 102 | 9.02 | 8.64 | 0.050 | 0.1 |
| 37 | 1,1-BPCH | 5.77 | 0.9993 | 102 | 3.00 | 8.17 | 107 | 4.06 | 2.14 | 109 | 6.69 | 10.6 | 0.025 | 0.1 |
| 38 | Nicotine | 2.68 | 0.9991 | 100 | 4.89 | 6.89 | 109 | 4.86 | 3.33 | 111 | 5.42 | 4.12 | 0.100 | 0.2 |
| 39 | Cotinine | 3.32 | 0.9998 | 82.6 | 5.66 | 3.45 | 91.8 | 6.39 | 3.59 | 92.3 | 3.78 | 5.16 | 0.025 | 0.1 |
| 40 | o-AAT | 9.15 | 0.99994 | 12.0 | 7.89 | 11.2 | 15.8 | 7.70 | 15.9 | 32.3 | 7.00 | 5.30 | 0.025 | 0.1 |
| 41 | 4-AAB | 8.14 | 0.9994 | 24.9 | 11.2 | 9.58 | 45.4 | 6.59 | 11.8 | 43.9 | 7.70 | 9.48 | 0.025 | 0.1 |
3.4. Method validation
Linear dynamic range, selectivity, accuracy, precision, and sensitivity of the method were determined by following validation guidelines [51]. In this study, 18 of the 41 target analytes had corresponding isotope labelled standards. We used alternative IS for remining analytes that did not have labelled IS, but with properties similar to those of target compounds. To determine the selectivity of the method, reagent blank and matrix blank samples were analyzed along with fortified urine samples at all calibration levels (from 0.1 to 50 ng/g). No interference was found at the retention times of target analytes except for O-AAT, 3,4-DCA, and nicotine which were found at concentrations of 0.114, 0.081, and 0.102 ng/mL, respectively.. These three analytes were present in the background at trace levels, as described earlier. A nine-point matrix matched calibration curve (0.1 - 50 ng/mL, n=3) was analyzed by following the method developed here to determine the linearity range of each analyte. Excellent linearity (correlation coefficient (r) range 0.9990-0.9999) was found for all analytes (Table 2). Accuracy and precision of the method were determined by analyzing fortified QC samples (matrix spike) at three concentrations (5, 10, and 20 ng/mL). Triplicate extractions were performed and the extracts were injected in duplicate daily for 3 days. At all three levels, the recoveries of 2,4 and 3,4-DAT were from 28.9% to 48.5% with an intra-day RSD of 2.71%-5.92% and inter-day RSD of 3.89%-9.70%. O-AAT and 4-AAB recoveries ranged from 12.1% to 45.4% with RSD values <16%. For the remaining 37 analytes, recoveries were in the range of 55% to 115% and RSD values <14% (Table 2). Isotopic dilution method of quantification accounted for the low recoveries encountered for some analytes. The LOD and LOQ values were calculated from the matrix matched calibration that yielded signal to noise ratio of 3 and 10, respectively. The LODs and LOQs of targeted chemicals were in the range of 0.025-0.20 ng/mL and 0.1-1.0 ng/mL, respectively (Table 2).
Matrix effect (ME) was assessed by post extraction spiking method and calculated by comparing the analyte peak area in pure solvent to that of an urine extract spiked with analytes. The ME (%) was calculated using the following formula:
Where A is the peak area of an analyte fortified in water:methanol mixture (95:5, v/v) and B is the peak area of an analyte fortified in a real urine extract that passed through all analytical steps. The results are presented in Table S2 (Supplementary Information). ME for the majority of the analytes fell between 20% and 50%. However, quantification of analytes was through isotopic dilution and matrix matched calibration curves, which allowed correction for ME in real urine samples.
3.5. Quantification of AAs and tobacco components in urine
The developed method was applied in the analysis of AAs in 20 urine samples from healthy volunteers in the Albany area of New York state. Similarly, urine samples from three known smokers, and a certified smoker’s urine reference material (NIST SRM 3672) were analyzed. Out of 39 AAs, ten AAs namely, aniline, p-anisidine, o-anisidine, p-TD, o/m-TD, 4-CA, 3-CA, 3,4-DCA, MDA, 2,4-DAT were found in urine from healthy volunteers at a mean concentration ranging from 0.03 to 2.11 ng/mL. Two tobacco biomarkers (nicotine mean: 69 ng/mL (SD: ± 133 and cotinine mean: 245 ng/mL (SD: ± 473.6) were also detected at notable concentrations in urine samples from healthy volunteers. It is likely that some of the urine donors were smokers. Among all AAs analyzed, 2,4-DAT was found at the highest concentration (9.10 ng/mL) (Table 3). Aniline, p-anisidine, p-TD, o/m-TD, 4-CA, 3-CA, and 4,4'-MDA were found in certified smoker’s urine reference material, along with two tobacco biomarkers (Table 3). o/m-TD concentration in (known) smoker’s urine was 4.41 ng/mL whereas that in the general population was 0.63 ng/mL.
Table 3.
Concentrations of primary aromatic amines measured in real urine samples by following the method developed in this study.
| Sample type | Aromatic amines (ng/mL) | Tobacco components | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Aniline | p-anisidine | o-anisidine | p-TD |
o/m- TD |
4-CA | 3-CA | 3,4- DCA |
4,4'- MDA |
2,4 - DAT |
Nicotine (ng/mL) |
Cotinine (ng/mL) |
||
| Urine samples from healthy adults (n =20) | max | 1.50 | 1.01 | 0.21 | 0.08 | 0.63 | 0.28 | 1.01 | 0.72 | 0.42 | 9.10 | 446 | 1890 |
| mean | 0.65 | 0.13 | 0.06 | 0.03 | 0.16 | 0.10 | 0.29 | 0.20 | 0.13 | 2.11 | 69 | 245 | |
| median | 0.53 | 0.01 | 0.04 | 0.03 | 0.08 | 0.07 | 0.27 | 0.11 | 0.08 | 0.47 | 0.38 | 0.43 | |
| Known smoker’s urine 1 | 2.74 | 0.37 | <LOD | 0.06 | 2.26 | 0.11 | 0.05 | <LOD | <LOD | <LOD | 807 | 127 | |
| Known smoker’s urine 2 | 3.47 | 0.39 | <LOD | 0.07 | 2.32 | 0.11 | 0.04 | <LOD | <LOD | <LOD | 720 | 123 | |
| Known smoker’s urine 3 | 1.16 | 0.83 | <LOD | 0.10 | 4.41 | 0.82 | 9.16 | 1.12 | 0.26 | <LOD | 1210 | 357 | |
| NIST SRM 3672* | 3.32 | 0.44 | <LOD | 0.27 | 4.57 | 0.16 | 0.26 | <LOD | 0.46 | <LOD | 725 | 1120 | |
values are average of triplicate analysis.
4. Conclusions
Here we describe an isotopic dilution, LC-MS/MS method to quantify 44 primary aromatic amines and two tobacco components simultaneously in human urine. Alkaline hydrolysis followed by LLE with MTBE yielded acceptable recoveries for 37 target analytes. The developed method is selective and sensitive with acceptable levels of accuracy and precision. The method was successfully applied in the determination of AAs in real urine samples and a NIST SRM 3672. Concentrations of select AA were significantly higher in smokers than in non-smokers. The method can be applied in large scale biomonitoring studies to assess human exposure to primary aromatic amines. In addition, we also examined for the presence of acetylated and hydroxylated metabolites of select AAs in smoker’s urine extract using predicted MRM transitions. Acetylated and hydroxylated metabolites were found with marked peak intensities. In an earlier study, we reported the occurrence of hydroxylatted metabolites such as N-acetyl-para-aminophenol and p-aminophenol, which are non-specific metabolites of AAs [52,53]. Further studies are needed for the determination of acetylated and hydroxylated metabolites of AAs in human urine.
Supplementary Material
Highlights.
An LC-MS/MS method was developed for simultaneously analysis of 39 aromatic amines in urine.
Alkaline hydrolysis followed by liquid-liquid extraction offered optimal accuracy and precision.
A polar biphenyl column and mobile additive enhanced chromatographic separation and ionization.
Eight aromatic amines were commonly found in urine of smokers at 0.04-9.16 ng/mL.
Acknowledgements
Research reported here was supported by the National Institute of Environmental Health Sciences (NIEHS) under the award numbers U2CES026542 (KK). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIEHS.
Footnotes
Supplementary information (SI)
The SI contains information on target analytes and their CAS (chemical abstracts service) numbers, structures and carcinogenicity grouping (Table S1.), Weak anion exchange and reverse phase solid phase extraction protocols, and the recoveries (%) of targeted analytes extracted without alkaline hydrolysis (Fig. S1).
Conflict of interest
The authors declare that they have no known competing financial interest or personal relationships that could have appeared to influence the work reported in this paper.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Marand A, Karlsson D, Dalene M, Skarping G, Determination of amines as pentafluoropropionic acid anhydride derivatives in biological samples using liquid chromatography and tandem mass spectrometry, Analyst 129 (2004) 522–528. [DOI] [PubMed] [Google Scholar]
- [2].Cramer GM, Ford RA, Hall RL, Estimation of toxic hazard—a decision tree approach, Food and Cosmetics Toxicology 16 (1976) 255–276. [DOI] [PubMed] [Google Scholar]
- [3].Mohammed M, Mekala LP, Chintalapati S, Chintalapati VR, New insights into aniline toxicity: Aniline exposure triggers envelope stress and extracellular polymeric substance formation in Rubrivivax benzoatilyticus JA2, J Hazard Mater 385 (2020) 121571. [DOI] [PubMed] [Google Scholar]
- [4].Fishbein L, Aromatic-amines of major industrial importance - use and occurrence, IARC Sci Publ DOI (1981) 51–74. [PubMed] [Google Scholar]
- [5].Gnanarajan TP, Nasar AS, Iyer NP, Radhakrishnan G, Synthesis of poly(urethane-imide) using aromatic secondary amine-blocked polyurethane prepolymer, J Polym Sci Pol Chem 38 (2000) 4032–4037. [Google Scholar]
- [6].Kruger RH, Boissiere C, Klein-Hartwig K, Kretzschmar HJ, New phenylenediamine antiozonants for commodities based on natural and synthetic rubber, Food Addit Contam 22 (2005) 968–974. [DOI] [PubMed] [Google Scholar]
- [7].Garrigos MC, Reche F, Pernias K, Sanchez A, Jimenez A, Determination of some aromatic amines in finger-paints for children's use by supercritical fluid extraction combined with gas chromatography, J Chromatogr A 819 (1998) 259–266. [DOI] [PubMed] [Google Scholar]
- [8].Muz M, Ost N, Kuhne R, Schuurmann G, Brack W, Krauss M, Nontargeted detection and identification of (aromatic) amines in environmental samples based on diagnostic derivatization and LC-high resolution mass spectrometry, Chemosphere 166 (2017) 300–310. [DOI] [PubMed] [Google Scholar]
- [9].Doronin SY, Chernova RK, Gusakova NN, Analytical aspects of reactions of primary aromatic amines with p-dimethylaminocinnamic aldehyde in the presence of surfactant ions and micelles, J Anal Chem 60 (2005) 412–419. [Google Scholar]
- [10].Skipper PL, Tannenbaum SR, Ross RK, Yu MC, Nonsmoking-related arylamine exposure and bladder cancer risk, Cancer Epidem Biomar 12 (2003) 503–507. [PubMed] [Google Scholar]
- [11].Hjelm M, Holmdahl MH, Biochemical effects of aromatic amines. 2. Cyanosis methaemoglobinaemia and heinz-body formation Induced by a local anaesthetic agent (Prilocaine), Acta Anaesth Scand 9 (1965) 99–120. [DOI] [PubMed] [Google Scholar]
- [12].Talaska G, Aromatic amines and human urinary bladder cancer: Exposure sources and epidemiology, J Environ Sci Heal C 21 (2003) 29–43. [DOI] [PubMed] [Google Scholar]
- [13].Crettaz S, Kampfer P, Bruschweiler BJ, Nussbaumer S, Deflorin O, Survey on hazardous non-regulated aromatic amines as cleavage products of azo dyes found in clothing textiles on the Swiss market, J Consum Prot Food S 15 (2020) 49–61. [Google Scholar]
- [14].Andrew AS, Schned AR, Heaney JA, Karagas MR, Bladder cancer risk and personal hair dye use, Int J Cancer 109 (2004) 581–586. [DOI] [PubMed] [Google Scholar]
- [15].Castelao JE, Yuan JM, Skipper PL, Tannenbaum SR, Gago-Dominguez M, Crowder JS, Ross RK, Yu MC, Gender- and smoking-related bladder cancer risk, JNCI-J Natl Cancer I 93 (2001) 538–545. [DOI] [PubMed] [Google Scholar]
- [16].Baan R, Straif K, Grosse Y, Secretan B, El Ghissassi F, Bouvard V, Benbrahim-Tallaa L, Cogliano V, W.I.A.R.C.M. W, Special Report: Policy: Carcinogenicity of some aromatic amines, organic dyes, and related exposures, Lancet Oncol 9 (2008) 322–323. [DOI] [PubMed] [Google Scholar]
- [17].Mattarozzi M, Lambertini F, Suman M, Careri M, Liquid chromatography-full scan-high resolution mass spectrometry-based method towards the comprehensive analysis of migration of primary aromatic amines from food packaging, J Chromatogr A 1320 (2013) 96–102. [DOI] [PubMed] [Google Scholar]
- [18].Rubio L, Sanllorente S, Sarabia LA, Ortiz MC, Optimization of a headspace solid-phase microextraction and gas chromatography/mass spectrometry procedure for the determination of aromatic amines in water and in polyamide spoons, Chemometr Intell Lab 133 (2014) 121–135. [Google Scholar]
- [19].Stabbert R, Schafer KH, Biefel C, Rustemeier K, Analysis of aromatic amines in cigarette smoke, Rapid Commun Mass Sp 17 (2003) 2125–2132. [DOI] [PubMed] [Google Scholar]
- [20].Yang L, Yiwei W, Caiying L, Yan Z, Determination of aromatic amines from textiles using dispersive liquid-liquid microextraction, J Sep Sci 36 (2013) 947–952. [DOI] [PubMed] [Google Scholar]
- [21].Akyuz M, Simultaneous determination of aliphatic and aromatic amines in indoor and outdoor air samples by gas chromatography-mass spectrometry, Talanta 71 (2007) 486–492. [DOI] [PubMed] [Google Scholar]
- [22].Kataoka H, Derivatization reactions for the determination of amines by gas chromatography and their applications in environmental analysis, J Chromatogr A 733 (1996) 19–34. [DOI] [PubMed] [Google Scholar]
- [23].Perez-Fernandez V, Rocca LM, Tomai P, Fanali S, Gentili A, Recent advancements and future trends in environmental analysis: Sample preparation, liquid chromatography and mass spectrometry, Anal Chim Acta 983 (2017) 9–41. [DOI] [PubMed] [Google Scholar]
- [24].Saha S, Mistri R, Ray BC, Rapid and sensitive method for simultaneous determination of six carcinogenic aromatic amines in mainstream cigarette smoke by liquid chromatography/electrospray ionization tandem mass spectrometry, J Chromatogr A 1216 (2009) 3059–3063. [DOI] [PubMed] [Google Scholar]
- [25].Garcia-Lavandeira J, Salgado-Petinal C, Blanco E, Cela R, A sensitive and efficient procedure for the high throughput determination of banned aromatic amines in textiles and leather products aided by advanced sample composition, Anal Bioanal Chem 397 (2010) 751–763. [DOI] [PubMed] [Google Scholar]
- [26].Xiao HL, Qian F, Xu Y, Pan JH, Tu HY, Wang HQ, Lin SJ, Han JC, A rapid and sensitive method for the detection of aromatic amines in cosmetics, J Chromatogr Sci 52 (2014) 115–119. [DOI] [PubMed] [Google Scholar]
- [27].Sanchis Y, Coscolla C, Roca M, Yusa V, Target analysis of primary aromatic amines combined with a comprehensive screening of migrating substances in kitchen utensils by liquid chromatography-high resolution mass spectrometry, Talanta 138 (2015) 290–297. [DOI] [PubMed] [Google Scholar]
- [28].Devreux V, Combet S, Clabaux E, Gueneau ED, From pigments to coloured napkins: comparative analyses of primary aromatic amines in cold water extracts of printed tissues by LC-HRMS and LC-MS/MS, Food Addit Contam A 37 (2020) 1985–2010. [DOI] [PubMed] [Google Scholar]
- [29].Schubert J, Kappenstein O, Luch A, Schulz TG, Analysis of primary aromatic amines in the mainstream waterpipe smoke using liquid chromatography-electrospray ionization tandem mass spectrometry, J Chromatogr A 1218 (2011) 5628–5637. [DOI] [PubMed] [Google Scholar]
- [30].Johnson JR, Karlsson D, Dalene M, Skarping G, Determination of aromatic amines in aqueous extracts of polyurethane foam using hydrophilic interaction liquid chromatography and mass spectrometry, Anal Chim Acta 678 (2010) 117–123. [DOI] [PubMed] [Google Scholar]
- [31].Mazumder S, Ahamed RA, McGahee E, Wang LQ, Seyler TH, A new automated method for the analysis of aromatic amines in human urine by GC-MS/MS, J Anal Toxicol 43 (2019) 25–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].IARC. IARC Monographs on the evalution of carcinogenicc risks to humans: some aromatic amines, organic dyes, and related exposure, 99 (2010) 706. [PMC free article] [PubMed] [Google Scholar]
- [33].Hsu FF, Lakshmi V, Rothman N, Bhatnager VK, Hayes RB, Kashyap R, Parikh DJ, Kashyap SK, Turk J, Zenser T, Davis B, Determination of benzidine, N-acetylbenzidine, and N,N'-diacetylbenzidine in human urine by capillary gas chromatography negative ion chemical ionization mass spectrometry, Anal Biochem 234 (1996) 183–189. [DOI] [PubMed] [Google Scholar]
- [34].Weiss T, Angerer J, Simultaneous determination of various aromatic amines and metabolites of aromatic nitro compounds in urine for low level exposure using gas chromatography-mass spectrometry, J Chromatogr B 778 (2002) 179–192. [DOI] [PubMed] [Google Scholar]
- [35].Hui BY, Zain NNM, Mohamad S, Fauzi HM, Alias Y, Chandrasekaram K, Rahim NY, Yahaya N, Raoov M, Determination of aromatic amines in urine using extraction and chromatographic analysis: A minireview, Anal Lett 52 (2019) 2974–2992. [Google Scholar]
- [36].DeBruin LS, Josephy PD, Pawliszyn JB, Solid-phase microextraction of monocyclic aromatic amines from biological fluids, Anal Chem 70 (1998) 1986–1992. [DOI] [PubMed] [Google Scholar]
- [37].Lamani X, Horst S, Zimmermann T, Schmidt TC, Determination of aromatic amines in human urine using comprehensive multi-dimensional gas chromatography mass spectrometry (GCxGC-qMS), Anal Bioanal Chem 407 (2015) 241–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Chung KT, Azo dyes and human health: A review, J Environ Sci Heal C 34 (2016) 233–261. [DOI] [PubMed] [Google Scholar]
- [39].Voyksner RD, Straub R, Keever JT, Freeman HS, Hsu WN, Determination of aromaticamines originating from azo dyes by chemical-reduction combined with liquid-chromatography mass-spectrometry, Environ Sci Technol 27 (1993) 1665–1672. [Google Scholar]
- [40].Kawakami T, Isama K, Nakashima H, Tsuchiya T, Matsuoka A, Analysis of primary aromatic amines originated from azo dyes in commercial textile products in Japan, J Environ Sci Heal A 45 (2010) 1281–1295. [DOI] [PubMed] [Google Scholar]
- [41].Balcik U, Chormey DS, Ayyildiz MF, Bakirdere S, Liquid phase microextraction based sensitive analytical strategy for the determination of 22 hazardous aromatic amine products of azo dyes in wastewater and tap water samples by GC-MS system, Microchem J 155 (2020) 104712. [Google Scholar]
- [42].Kampfer P, Crettaz S, Nussbaumer S, Scherer M, Krepich S, Deflorin O, Quantitative determination of 58 aromatic amines and positional isomers in textiles by high-performance liquid chromatography with electrospray ionization tandem mass spectrometry, J Chromatogr A 1592 (2019) 71–81. [DOI] [PubMed] [Google Scholar]
- [43].Kiontke A, Oliveira-Birkmeier A, Opitz A, Birkemeyer C, Electrospray ionization efficiency Is dependent on different molecular descriptors with respect to solvent pH and instrumental configuration, Plos One 11 (2016) e10167502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Lindner JM, Vogeser M, Grimm SH, Biphenyl based stationary phases for improved selectivity in complex steroid assays, J Pharmaceut Biomed 142 (2017) 66–73. [DOI] [PubMed] [Google Scholar]
- [45].Ferro MD, Santos SAO, Silvestre AJD, Duarte MF, Chromatographic separation of phenolic compounds from extra virgin olive oil: Development and validation of a new method Based on a biphenyl HPLC column, Int J Mol Sci 20 (2019) 30625994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Aromatic amines: laboratory procedure manual (2020) Centers for Disease Control and Prevention. https://wwwn.cdc.gov/nchs/data/nhanes/2013-2014/labmethods/AA-AAS-H-MET-508. March 2021.
- [47].Colas C, Garcia P, Popot MA, Bonnaire Y, Bouchonnet S, Optimization of solid-phase extraction for the liquid chromatography-mass spectrometry analysis of harpagoside, 8-para-coumaroyl harpagide, and harpagide in equine plasma and urine, J Chromatogr Sci 46 (2008) 174–183. [DOI] [PubMed] [Google Scholar]
- [48].Dias NC, Poole CF, Mechanistic study of the sorption properties of OASIS((R)) HLB and its use in solid-phase extraction, Chromatographia 56 (2002) 269–275. [Google Scholar]
- [49].Joshi DR, Adhikari N, An Overview on common organic solvents and their toxicity, J Pharm Res Int 28 (2019) 49840. [Google Scholar]
- [50].Shahrestani M, Tehrani MS, Shoeibi S, Azar PA, Husain SW, Comparison between different extraction methods for determination of primary aromatic amines in food simulant, J Anal Methods Chem 2018 (2018) 1651629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Gonzalez O, Blanco ME, Iriarte G, Bartolome L, Maguregui MI, Alonso RM, Bioanalytical chromatographic method validation according to current regulations, with a special focus on the non-well defined parameters limit of quantification, robustness and matrix effect, J Chromatogr A 1353 (2014) 10–27. [DOI] [PubMed] [Google Scholar]
- [52].Smarr MM, Kannan K, Chen Z, Kim S, Buck Louis GM, Male urinary paracetamol and semen quality, Andrology 5 (2017) 1082–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Smarr MM, Grantz KL, Sundaram R, Maisog JM, Honda M, Kannan K, Buck Louis GM, Urinary paracetamol and time-to-pregnancy. Human Reproduc 31 (2016) 2119–2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.