

Abstract
Natural killer (NK) cells are known to be developmentally blocked and functionally inhibited in patients with acute myeloid leukemia (AML), resulting in poor clinical outcomes. Here we demonstrate that while NK cells are inhibited, closely related type 1 innate lymphoid cells (ILC1s) are enriched in the bone marrow of leukemic mice and in AML patients. Since NK cells and ILC1s share a common precursor (ILCP), we asked if AML acts on the ILCP to alter developmental potential. A combination of ex vivo and in vivo studies revealed that AML skewing of the ILCP towards ILC1s and away from NK cells represented a major mechanism of ILC1 generation. This process was driven by AML-mediated activation of the aryl-hydrocarbon receptor (AHR), a key transcription factor in ILCs, as inhibition of AHR led to decreased numbers of ILC1s and increased NK cells in the presence of AML. These results demonstrate a mechanism of ILC developmental skewing in AML and support further pre-clinical study of AHR inhibition in restoring normal NK cell development and function in the setting of AML.
Keywords: AML, ILC, Development, AHR, NK
INTRODUCTION
Acute myeloid leukemia (AML) is an aggressive and highly heterogeneous hematologic malignancy that despite decades of research has a 5-year survival rate of less than 30% (1). Natural killer (NK) cells are a type of innate lymphoid cell (ILC) that are known to possess potent anti-leukemic effects. However, our group and others have previously shown that AML is capable of inhibiting NK cell maturation and function (2–4). Utilizing AML mouse models, our group has previously demonstrated that murine NK cells fail to progress from stage 2 (CD27+CD11b−) to stage 3 (CD27+CD11b+) and that this developmental blockade is reversible (5). Chretien et al. recently demonstrated that AML patients with a more immature circulating NK cell phenotype, as determined by low killer immunoglobulin-like receptor (KIR) and CD57 expression, have worse clinical outcomes compared to patients with normal NK cell maturation (6). This suggests NK cell developmental status correlates with clinical outcomes in AML regardless of therapeutic treatment regimen.
Innate lymphoid cells (ILCs) comprise a heterogeneous family of non-T, non-B lymphocytes that share many phenotypic and functional features with antigen-specific T cells (7, 8). Unlike T cells, ILCs do not express T cell receptor genes, although they can express some germline-encoded antigen-specific receptors (7). Rather, ILCs typically respond in a non-antigen specific manner to signals such as decreased MHC class I expression or binding of ligands from the local microenvironment to activating or inhibitory receptors (9). The ILC family includes cytotoxic NK cells and non-cytotoxic “helper” ILC1s (Group 1), ILC2s (Group 2), and ILC3s (Group 3). Within the Group 1 ILC family, NK cells are defined by their expression of both eomesodermin (EOMES) and the T-box transcription factor TBET (TBX21), which drive expression of perforin/granzymes and interferon gamma (IFNγ), respectively, while ILC1s solely depend on TBET expression for development and function (10, 11). While ILC1s have been found to be important for the initial response to viral infections, including murine cytomegalovirus (MCMV), studies in solid tumor models have suggested that ILC1s promote tumor immune evasion and cancer progression (12–14). Whereas NK cells possess cytolytic functions and are potent immune surveyors, ILC1s are classically non-cytolytic and function instead to modulate the downstream immune response through secretion of cytokines such as IFNγ and tumor necrosis factor alpha (TNFα).
Studies in both mice and humans have demonstrated that NK cells and ILC1s develop from a common ILC precursor (ILCP), though the regulatory mechanisms that dictate lineage fate decisions are not yet clear (15–19). Prior mechanistic studies revealed that the NK cell developmental defect observed in AML patients results at least in part from AML-mediated activation of the aryl hydrocarbon receptor (AHR) pathway in developing NK cells (2). AHR is a ligand-activated transcription factor important in regulating genes involved in the metabolism of many endogenous ligands and environmental toxins as well as immune cell development (2, 20–22). Although AHR activation inhibits NK cell maturation, it is required for the generation and function of ILC3 populations, the maintenance of murine liver-resident Group 1 ILCs, and also serves to inhibit ILC2 function (21, 23, 24). Thus, AHR plays an important role in regulating the development of multiple ILC family members under steady state conditions. Pathological activation of AHR in disease settings such as AML creates the possibility of disrupting this balance of ILCs and driving the development of non-cytolytic, immunomodulatory ILC subsets at the expense of cytotoxic NK cells. Indeed, inhibition of AHR restored normal NK cell maturation and enhanced NK cell mediated cytotoxicity against AML targets, suggesting AHR inhibition may have clinical efficacy in AML (2).
In this report, we discovered that the Group 1 ILC compartment, consisting of both ILC1s and NK cells, is skewed away from NK cells and towards an ILC1 lineage in the bone marrow of AML patients and in mouse models of AML. This developmental perturbation was dependent on AHR signaling, as administration of an AHR agonist recapitulated these findings by increasing the number of ILC1s formed, while inhibition of AHR signaling restored NK cells and inhibited ILC1 formation. In addition, we have observed some capacity for interconversion of human NK cells to ILC1s in pre-clinical xenograft AML mouse models, suggesting ILC1s may be promoted both from early ILC precursors as well as from later developmental stages of NK cells. Our data reveal a mechanistic basis for tumor-promotion by AHR in AML via interference with ILC development. These findings offer a new rationale to target the AHR pathway in AML to restore normal ILC homeostasis, favoring mature cytotoxic NK cells to improve clinical outcomes.
MATERIALS AND METHODS
Mice
All mouse studies were conducted under an approved protocol by the Ohio State University Institutional Animal Care and Use Committee (protocol # 2009A033-R3). BoyJ Ly5.1 (CD45.1) mice were obtained from Jackson Laboratory (RRID: IMSR_JAX:002014). C57BL/6 PTD/ITD spontaneous AML mice were previously generated and characterized by our group (25). NSG mice were obtained from Jackson Laboratory (RRID:IMSR_JAX:005557). For congenic transplant studies, CD45.1 congenic mice were sublethally irradiated (400 cGy) using whole body irradiation 4 hours before being adoptively transplanted with 106 splenic cells from leukemic CD45.2 PTD/ITD mice or vehicle control (PBS). Engraftment was validated 2 weeks later via peripheral cheek bleed, and leukemic burden was monitored through cell counting and FACS analysis (5). Mice were harvested 6–8 weeks post-injection and analyzed via FACS analysis.
For the NSG studies, mice were pretreated with busulfan (intraperitoneal, 25 mg/kg) 24 hours prior to IV injection of 2*106 MV411 cells and/or human ILC precursors. For FICZ studies, following adoptive transfer, some mice were injected IP with FICZ (3 μg/mouse) daily for 4 weeks while the control arm received IP vehicle injections. All NSG studies were conducted utilizing at least 5 independent human blood donors. In all NSG experiments, mice received biweekly injections of rhIL-7 and rhIL-15 (500 ng/mouse each, NCI) (17, 26). Studies were conducted in both male and female mice at 8–15 weeks of age.
Tissue collection and isolation
All human tissue was obtained by protocols approved by the Ohio State University Internal Review Board (protocol #2009C0019). Fresh human peripheral blood (PB) was obtained from Versiti Blood Center of Wisconsin (Milwaukee, WI). AML patient samples were obtained through the Ohio State University Leukemia Tissue Bank. NK cells and ILCPs were obtained from negative enrichment followed by a magnetic CD117 positive selection (ILCP) or a CD16 depletion (for CD56bright NK). Populations were then isolated by cell sorting with a BD Aria II to >98% purity as previously described (Figure S1) (2, 17). All studies utilized at least 5 independent donors. Mouse spleen, marrow, and blood were processed as previously described (5). Livers were homogenized using a GentleMACS dissociator (Miltenyi Biotec) and lymphocytes were isolated using a 60/40 Percoll gradient spin.
Cell Culture
Human ILCPs or CD56bright NK cells were cultured as previously described from at least 5 independent donors (5, 17). The culture media for in vitro development experiments contained DMEM and F12 (2:1 ratio) supplemented with 1% antibiotic/antimycotic (Thermo Fisher Scientific), 20 mg/ml ascorbic acid, 24 μM 2-ME, 0.05 mg/ml sodium selenite (Sigma-Aldrich), and 10% heat-inactivated human AB serum (Valley Biomedical). ILCPs were plated on OP9-DL1 murine stromal cells in media supplemented with 10 ng/mL human IL-7 ± AML cells. For coculture experiments, ILCPs or NK cells were plated in 24-well plates on OP9-DL1 cells, and AML cell lines were added in a transwell insert (50,000 AML cells/well). Media were replaced, and AML cells were refreshed 2x per week (2). The AHR antagonist CH223191 was used at a concentration of 3 μM (Selleckchem), and the AHR agonist 5,11-dihydroindolo[3,2-b]carbazole-6-carboxaldehyde (FICZ) was used at 30 nM (Millipore Sigma). Where indicated, TGFβ was used at a concentration of 10 ng/mL (Miltenyi). For the switch culture experiment, ILCPs were isolated and plated as described above and treated for 2 weeks with either DMSO, FICZ, CH223191, or U937 AML cells cultured in transwells. After 2 weeks, treatment conditions changed as indicated to either remove FICZ or U937 cells for 2 more weeks. In some wells, removal of these conditions was followed with the addition of CH223191 for 2 weeks. As a control, some wells were cultured in the indicated conditions for 4 consecutive weeks prior to flow cytometric analysis. U937 and MV411 cell lines were obtained from the ATCC in the past 6 months and were used for experiments within 10 passages of thawing. ATCC validation utilizes short tandem repeat (STR) profiling. OP9-DL1 cells were obtained from the lab of Dr. J. Carlos Zuniga-Pflucker and were previously characterized, and validated (17, 26, 27). Mycoplasma testing was completed annually on cell lines.
Single Cell Clonal Assay
The single cell clonal assay was performed as previously described (17, 28). Briefly, bulk human ILCPs were sorted from the peripheral blood of 4 independent donors as described above using a BD Aria II in purity mode. Single ILCPs were then re-sorted into 96-well round bottom plates containing OP9-DL1 stromal cells using single cell mode. The outer perimeter of wells contained PBS to control evaporation. Cells were cultured at 37°C and 5% CO2 in 10 ng/mL IL-7 and IL-15 for 4 weeks before being harvested. Bulk ILCPs from each donor were also cultured in parallel as a control. Additionally, cells were cultured in conditioned media from U937 cells diluted 1:1 with fresh culture media where indicated. Conditioned media was prepared by culturing U937 at 500,000 cells/mL for 72 hours before harvesting the media and filtering through a 0.45 μm syringe filter to remove residual AML cells. Media was changed twice weekly and clonal growth was monitored using light microscopy starting after week 2 to select clones with successful growth to analyze. During flow analysis, any hCD45+Lin-CD56+CD94+ cells were included in the analysis.
Flow Cytometry
All experiments were analyzed with an LSRII or Fortessa flow cytometer (BD Bioscience), followed by FlowJo software analysis (BD Bioscience). Nonspecific staining was minimized by using the appropriate isotype controls. Antibodies and phenotypes used can be found in Tables S1 and S2. Cells were stimulated with PMA/Ionomycin in the presence of Monensin/Brefeldin A for 4 hours before being stained for surface and intracellular markers and analyzed by flow cytometry. All flow analyses contained a viability dye to exclude dead cells.
Chromatin Immunoprecipitation and Quantitative Real Time PCR (qPCR)
Chromatin immunoprecipitation (ChIP) was performed against AHR, or rabbit monoclonal isotype control (Cell Signaling), as per the manufacturer’s instructions (Pierce Magnetic ChIP). The presence of binding to the EOMES promoter was determined by using 2 primer sets targeting different regions containing AHR binding motifs. Binding to the IFNG promoter was determined using a commercially available primer set (Cell Signaling) per the manufacturer instructions. qPCR was conducted using PowerUP SYBR Green master mix (Applied Biosystems) according to the manufacturer instructions and was analyzed on a ViiA 7 qPCR instrument (Life Technologies). Fold enrichment was determined through the equation 2−([Ct AHR ChIP]-[Ct Isotype ChIP]).
The primer sequences used for the EOMES promoter are as follows:
Forward Primer 1: 5’-TGAAAAAGGGCAGAAAGGCG-3’, Reverse Primer 1: 5’-GAAAGCAGGAGGTGGAAACTAACC-3’, Forward Primer 2: 5’-TCATTACGAAACAGGGCAGGTG-3’, Reverse Primer 2: 5’-CGGTGTCTACGGAGATTTATTGCG-3’.
Statistical Analysis
For experiments with independent observations, two sample t-tests or analysis of variance were used for comparisons with two or multiple groups, respectively. For experiments with correlated data, linear mixed effects models were used for analyses to take account of the correlation among measurements being taken from the same mouse or patient. Bonferroni correction was used to correct for multiple comparisons. Error bars represent ± SEM unless otherwise noted. SAS 9.4 (SAS Institute Inc., NC) was used for analysis.
RESULTS
AML skews Group 1 ILCs away from NK and towards ILC1s in a tissue specific manner
To determine whether AML is capable of disrupting Group 1 ILC homeostasis, we utilized an established mouse model of AML involving adoptive transfer of primary leukemic murine cells from Mll-PTD;Flt3-ITD double knock-in mice (CD45.2+) into immunocompetent congenic hosts (CD45.1+) (5, 25). Murine ILC1s have been well characterized, with several validated surface markers identified that distinguish ILC1s from NK cells, including CD200R and CD49a, in addition to specific transcription factor expression (11, 12). Using this model, we identified altered proportions of CD45.1+ ILC1s and NK cells within the Group 1 ILC compartment (identified as Lin-NK1.1+NKp46+Tbet+ lymphocytes) specifically in the bone marrow and liver of leukemic mice relative to controls, though no significant differences were observed in the blood or spleen (Figure 1A, B). Further analysis revealed increased absolute numbers of ILC1s in the bone marrow and liver of leukemic mice compared to littermate controls (Figure 1C). These ILC1s expressed Tbet, CD200R, CD49a, heterogeneous levels of CD127, and lacked expression of Eomes and CD49b (DX5) (data not shown). These observations were also recapitulated when directly assessing Mll-PTD;Flt3-ITD double knock-in mice (Figure S2A, B). Additionally, we observed decreased percentages of NK cells among total Group 1 ILCs in the bone marrow and liver of these mice. Interestingly, unlike the relative percentages, the absolute numbers of NK cells were decreased in the bone marrow while they were increased in the livers of leukemic mice (Figure 1C). We also assessed expression of the proliferation marker Ki67 in bone marrow NK cells and ILC1s in both leukemic mice and controls. We observed that both NK cells and ILC1s in leukemic mice had significantly higher expression of Ki67 compared to nonleukemic controls (Figure S2C). Among leukemic mice, NK cells had higher expression of Ki67 compared to ILC1s. We also observed that the Ki67 levels of NK cells were higher in both WT and leukemic mice compared with ILC1s.
Figure 1: ILC1s are expanded in the marrow and liver of leukemic mice.
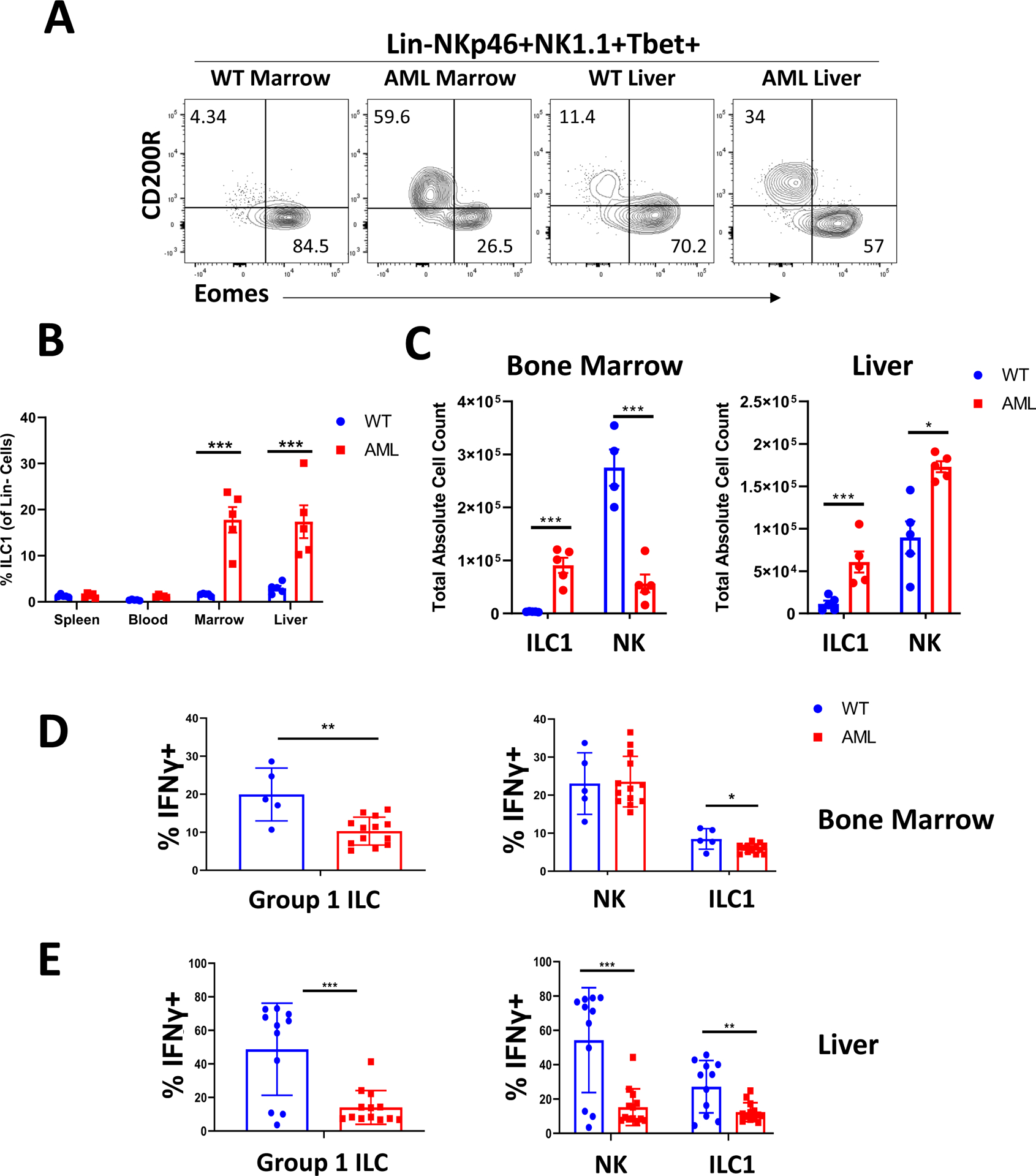
A) Representative flow plots of Eomes vs CD200R expression among Lin-NKp46+NK1.1+Tbet+ cells in the liver and bone marrow of WT and transplant leukemic mice.
B) Quantification of ILC1 frequency (among total Lin- cells) across spleen, blood, bone marrow, and liver.
C) Absolute counts for ILC1s and NK cells in the bone marrow and liver for WT and transplant leukemic mice.
n=5 WT, n=5 AML, *p<0.05, ***p<0.001. Error bars represent ±SD.
D) Representative flow plots and summary data of IFNγ expression for NK cells and ILC1s in the marrow of WT and transplant leukemic mice.
E) Summary data of IFNγ production among liver Group 1 ILCs. Data points were pooled from two independent experiments.
n=5 (BM) or 11 (Liver) WT, n=13 AML, *p<0.05, **p<0.01. Error bars represent ±SD.
Upon functional evaluation, the ILC1s isolated from the bone marrow of leukemic mice produced less IFNγ compared to bone marrow NK cells. Although NK cells retained the ability to secrete IFNγ in the setting of AML on a per cell basis, the decrease in absolute number of marrow NK cells in addition to expansion of ILC1s resulted in an overall decrease in IFNγ levels in the leukemic mice relative to WT control mice (Figure 1D). In the liver, we observed suppression of IFNγ production of both NK cells and ILC1s in the setting of AML (Figure 1E). We also observed lower production of IFNγ in ILC1s relative to NK cells even in WT mice. Of note, we did not observe significant TNFα production among bone marrow Group 1 ILCs in the leukemic mice or WT control mice (Figure S2D). Overall, these findings suggest that AML is capable of promoting the expansion of ILC1s in a tissue-specific manner while inhibiting NK cell numbers in the bone marrow. This shift in Group 1 ILC cell populations was also accompanied by a decrease in overall Group 1 ILC IFNγ production in the bone marrow and liver.
AML cells are capable of skewing human ILCP development away from NK cells and towards ILC1s
We next sought to determine the mechanism(s) by which AML induces expansion of ILC1 populations. Because we previously observed disruption of an NK cell precursor population in the peripheral blood of AML patients (5), and because all ILCs, including NK cells, stem from common ILCPs present in the circulation and in tissues (17, 18), we hypothesized that AML actively drives ILCP development away from NK cells and towards ILC1s. It is also possible that ILC1 expansion in AML results from conversion of mature NK cells to ILC1s. To determine whether either or both of these processes occur in AML, we isolated Lin-NKp80-CD294-KLRG1-NKp44-CD94−CD16−CD117+ ILCPs and Lin-CD56+CD94+CD16− NK cells (hereafter referred to as CD56bright NK cells, refer to Figure S1 for sorting strategy) from the peripheral blood of healthy donors as previously described (5, 17). We then cultured these cells for 4 weeks with IL-7 on OP9-DL1 stromal cells, conditions that support the development of human ILC subsets in vitro (17, 26), either alone or with the AML cell lines MV411 or U937 separated by transwells (Figure 2A). As shown in Figure 2B,C we observed a significant increase in the proportion of Lin-CD56+CD94+TBET+EOMES− ILC1s and a concomitant decrease in Lin-CD56+CD94+TBET+EOMES+ NK cells in the conditions with AML cells relative to controls. The capacity for ILCPs to generate ILC1s was significantly greater than the capacity for CD56bright NK cells to directly convert into ILC1s in vitro. To more definitively determine whether AML is capable of skewing ILCP development towards an ILC1, we performed single cell clonal assays on human ILCPs with AML conditioned media from U937 cells or control conditions for 4 weeks. Our overall cloning efficiency was 10–15%. Of the clones that successfully grew, we analyzed those containing group 1 ILCs (hCD45+Lin-CD56+CD94+, n=20 untreated clones, n=21 AML conditioned media treated clones) and saw a significant increase in ILC1 formation along with a concomitant reduction in NK potential in the AML conditioned media treated ILCPs relative to controls (Figure 2D,E and Figure S3A). Taken together, these data suggest that in the setting of AML co-culture, ILC1 expansion occurs mainly as a result of skewed ILCP development rather than from conversion of NK cells.
Figure 2: AML coculture skews ILC precursor development to promote an ILC1 phenotype in vitro.
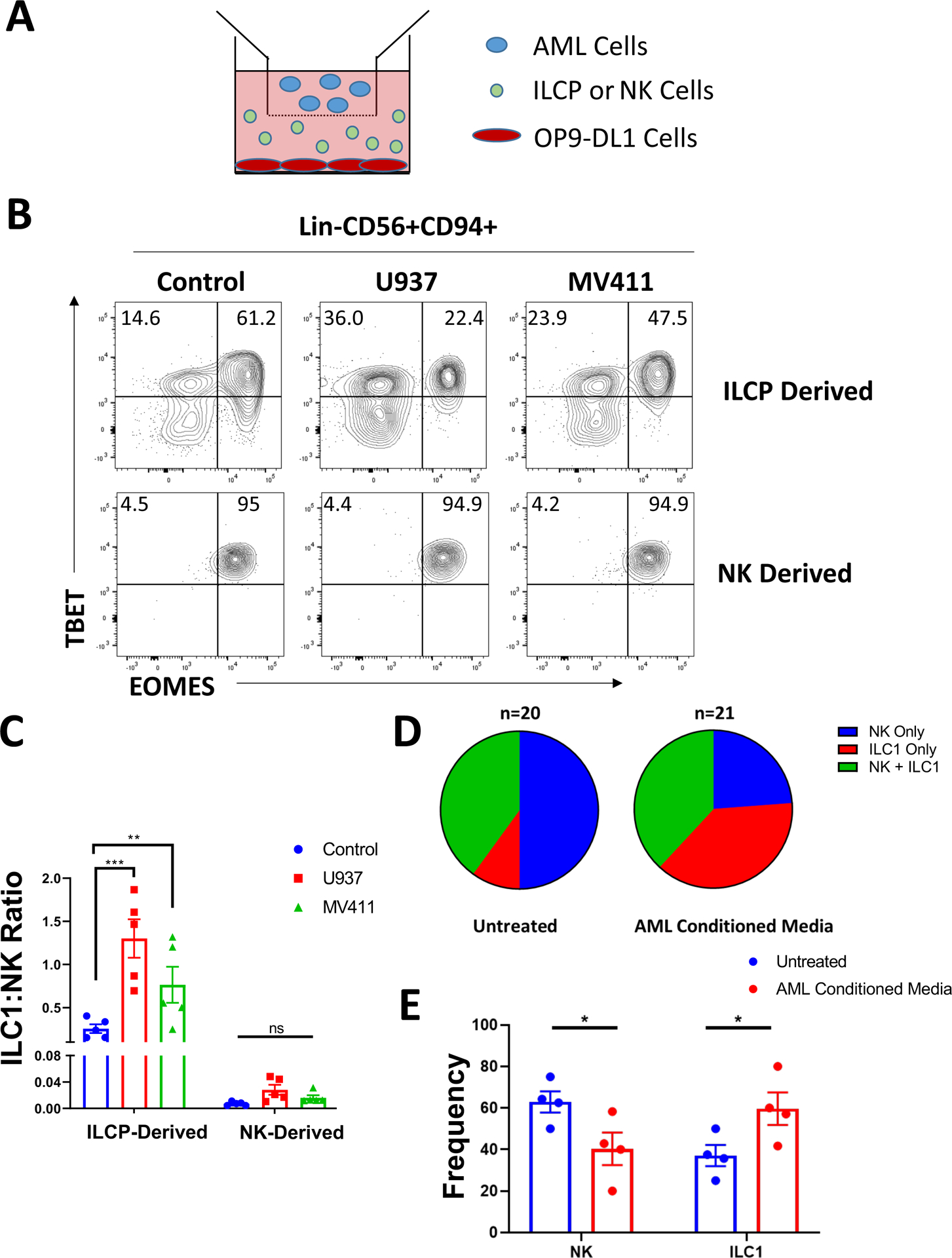
A) Schematic of coculture experiments. ILCPs (Lin-NKp80-CD294-KLRG1-NKp44-CD94−CD16−CD117+) or CD56bright NK cells (Lin-CD56+CD94+CD16−) were isolated from normal peripheral blood of 5 independent donors and cultured on OP9-DL1 cells for 4 weeks ± the AML cell lines U937 or MV411 cultured in transwells. Isolated populations were distributed evenly between conditions following initial cell sorting.
B) Post-culture analysis of TBET and EOMES expression among Lin-CD56+CD94+ cells derived from ILCP or CD56bright NK cells.
C) Summary of ILC1:NK ratios comparing control culture conditions to AML coculture.
n=5, **p<0.01, ***p<0.001. Error bars represent ±SEM.
D) Pooled summary of clonal assay data from ILCP clones isolated from the peripheral blood of 4 independent donors. For analysis, any clone containing ILC1 (hCD45+Lin-CD56+CD94+TBET+EOMES−) and/or NK cells (hCD45+Lin-CD56+CD94+TBET+EOMES+) were included in the analysis.
E) Summary of NK cell and ILC1 frequency from single ILCP clones grouped by donor for either untreated cells or those treated with AML conditioned media produced from U937 cells.
n=4 biological donors, *p<0.05. Error bars represent ±SEM.
Subsequently, we conducted similar experiments in vivo by isolating fresh human healthy donor blood ILCPs and CD56bright NK cells and adoptively transferring them with or without the AML cell line MV411 into NSG mice followed by intraperitoneal injections of recombinant human IL-7 and IL-15 (Figure 3A). Of note, the U937 cell line progressed too rapidly in vivo to allow for assessment of ILC development, even at low inoculating doses (average survival ~14 days, data not shown). After 4 weeks, we evaluated the bone marrow and liver compartments of these mice for the presence of Lin-hCD45+CD56+CD94+TBEThiEOMESLo/− ILC1s and Lin-hCD45+CD56+CD94+TBET+EOMES+ NK cells. While there were too few human cells to analyze in the livers of NSG mice in the presence or absence of AML, we observed that the bone marrow of mice co-injected with AML cells contained increased proportions of ILC1s derived from both ILCPs and NK cells compared to mice that were not co-transplanted with MV411 cells (Figure 3B,C). Functionally, we observed suppression of IFNγ secretion in the ILCP-derived ILC1s and NK cells in the setting of AML using our in vivo NSG model, similar to what was observed in the congenic AML mouse model. We also detected a concomitant suppression of TNFα production using this humanized model (Figure 3D). These in vivo data suggest that AML cells are capable of diverting both ILCPs, and to a lesser extent CD56bright NK cells, towards an ILC1 lineage with an overall suppressive effect on type 1 cytokine production.
Figure 3: In vivo co-injection of AML with ILC precursors promotes an ILC1 phenotype.
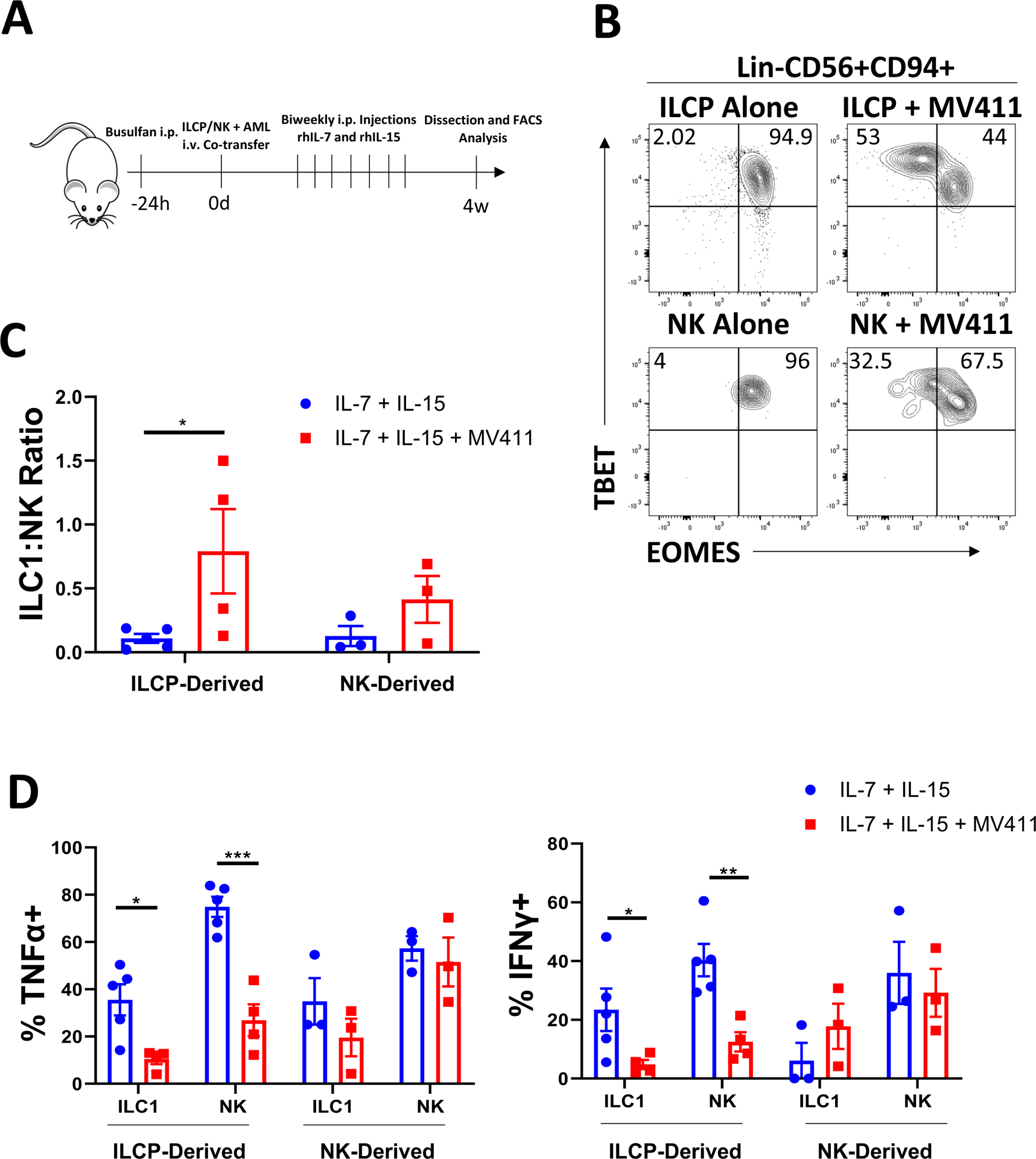
A) Schematic of in vivo studies. ILCPs (Lin-NKp80-CD294-KLRG1-NKp44-CD94−CD16−CD117+) and CD56bright NK cells (Lin-CD56+CD94+CD16−) were isolated from normal peripheral blood of 5 independent donors and adoptively transferred into NSG mice ±MV411 cells followed by biweekly injections of IL-7 and IL-15 for 4 weeks. At harvest, bone marrow was harvested and analyzed via flow cytometry for Group 1 human ILCs.
B) Representative flow plots of TBET and EOMES expression for human Lin-CD56+CD94+ cells derived from ILCPs or CD56bright NK cells ±MV411 cells.
C) Summary of ILC1:NK ratios comparing control conditions to AML co-injection.
D) Summary data of TNFα and IFNγ expression determined by flow cytometry from ILCP- or NK-derived cells and ILC1s from control or AML conditions.
n=3–5 (some mice excluded due to poor engraftment), *p<0.05, **p<0.01, ***p<0.001. Error bars represent ±SEM.
AML-derived AHR agonists are capable of promoting ILC1 generation
Our previous work has shown that soluble AML-derived AHR ligands can inhibit NK cell development from NK cell precursors through aberrant activation of the AHR pathway (2). Others have shown that AHR signaling is required for maintenance of liver resident NK cells, a tissue with active AHR signaling under normal homeostatic conditions (23). To determine whether AHR is capable of promoting development of ILC1s in vitro, we isolated human ILCPs and CD56bright NK cells from the peripheral blood of normal donors and cultured these cells on OP9-DL1 stromal cells along with IL-7 with either an AHR agonist (FICZ), an AHR inhibitor (CH223191), or vehicle control (DMSO). We also tested whether antagonism of AHR in the presence of the AML cell lines U937 or MV411 suppresses ILC1 generation. We found that activation of the AHR pathway by treating with FICZ reduced EOMES expression yet maintained TBET expression relative to controls. Furthermore, inhibition of AHR with CH223191 (AHRi) resulted in higher expression of EOMES along with co-expression of TBET (Figure 4A, B). Co-culture of ILCPs with U937 (Figure 4A,B) or MV411 cells (Figure S3B,C), which we have previously shown to secrete AHR ligands (2), in transwell recapitulated the results from the FICZ-treated conditions, demonstrating an increase in both ILC1 frequency (Figure 4B) and absolute counts (Figure S4A). This was reversed upon treatment with the AHRi. In contrast to what occurred when culturing ILCPs, CD56bright NK cells co-cultured with AML or FICZ did not show significant conversion to ILC1s in vitro.
Figure 4: In vitro AHR activation promotes an ILC1 phenotype in AML.
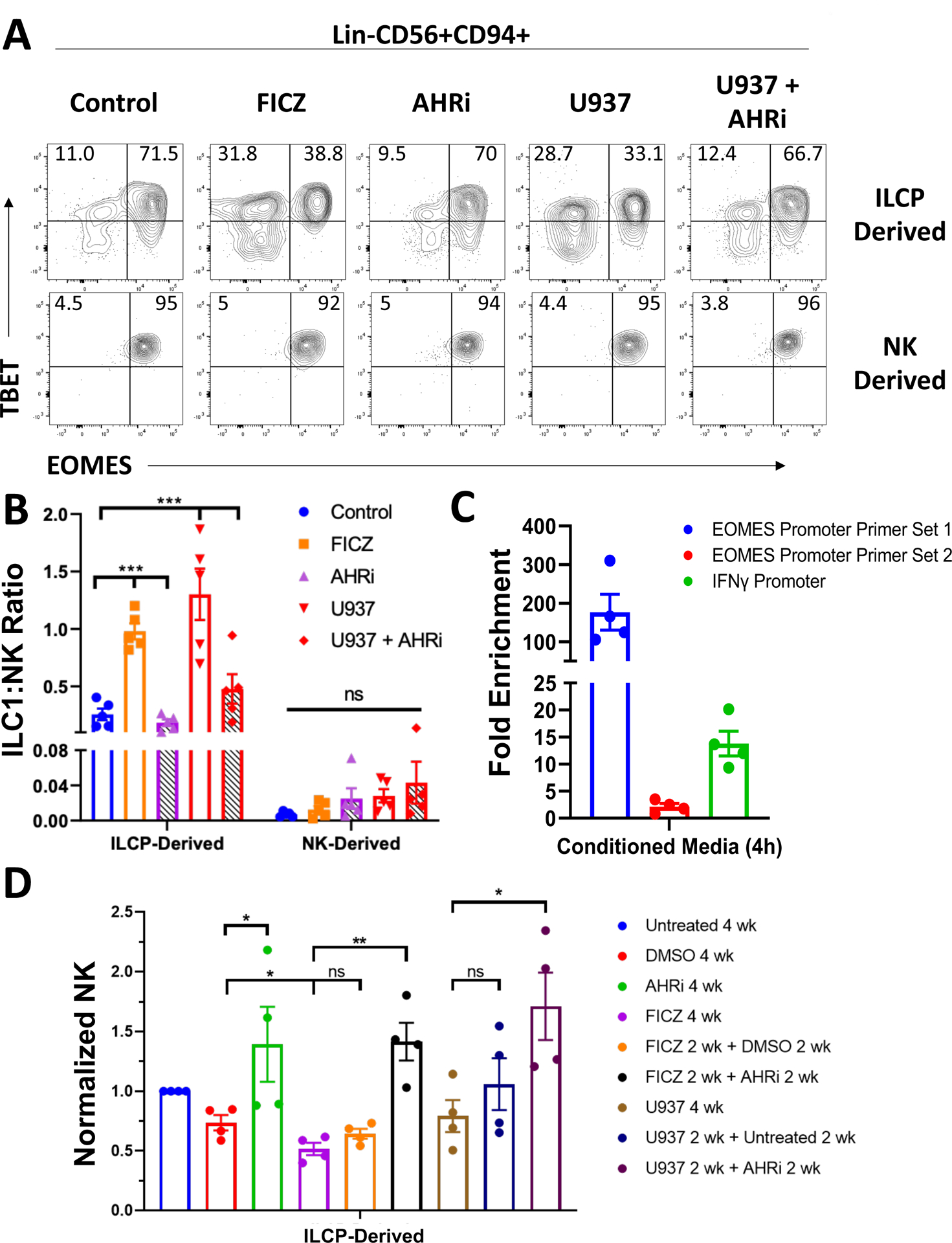
A) Post-culture analysis of TBET and EOMES expression among Lin-CD56+CD94+ cells. ILCPs (Lin-NKp80-CD294-KLRG1-NKp44-CD94−CD16−CD117+) and CD56bright NK cells (Lin-CD56+CD94+CD16−) were isolated from normal peripheral blood of 5 independent donors and cultured with IL-7 on OP9-DL1 cells for 4 weeks. Culture conditions included: 1) treatment with the AHR agonist FICZ (30 nM), 2) the AHR inhibitor (AHRi) CH223191 (3 μM), or 3) the AML cell line U937 (± AHRi). U937 were co-cultured in transwells. Isolated populations were distributed evenly between conditions following initial cell sorting.
B) Summary data of ILC1:NK ratios across all conditions for ILCP- and NK-derived Group 1 ILCs. ANOVA was used to compare AHR agonism and AHR inhibition to control conditions for the indicated comparisons.
n=5, *p<0.05, ***p<0.001. Error bars represent ±SEM. ANOVA was used for analysis in B).
C) qPCR results of AHR ChIP pull down from peripheral blood NK cells of n=4 biological donors treated for 4 hours with AML conditioned media (from U937 cells). Fold enrichment was calculated through the equation 2−([Ct AHR ChIP]-[Ct Isotype ChIP]). Two primer sets targeting different regions of the EOMES promoter containing AHR binding motifs were tested as well as a commercially available primer set targeting the IFNG promoter (Cell Signaling Technologies).
D) Summary data of normalized NK counts for the ILCP switch culture assay. ILCPs were isolated from the peripheral blood of 4 independent donors and cultured on OP9-DL1 stromal cells with IL-7 for a total of 4 weeks. Cells were either cultured for 4 consecutive weeks in the indicated conditions or had a change in treatment after 2 weeks as indicated.
n=4, *p<0.05, **p<0.01. Error bars represent ±SEM.
We also tested whether AHR was capable of directly targeting the EOMES or IFNG promoters by performing chromatin immunoprecipitation (ChIP) on NK cells from the peripheral blood of 4 donors treated for 4 hours with AML conditioned media (from U937) using a monoclonal AHR antibody or isotype control, a time point previously validated by CYP1A1 expression to indicate AHR pathway activation (2). We then performed qPCR with two primer sets targeting separate regions of the EOMES promoter containing putative AHR binding motifs (5’-GCGTG-3’ or 5’-CACGC-3’) as well as a commercially available primer set targeting the IFNG promoter (Cell Signaling Technologies). We detected amplification of all primer sets tested, albeit we detected different binding affinity of AHR to the two EOMES promoter binding motifs (Figure 4C). These data demonstrate AHR is capable of directly binding the promoters of both EOMES and IFNG in human NK cells.
To more directly assess whether AHR blockade is capable of reversing ILCP skewing towards ILC1s, we performed a switch culture experiment with ILCPs by culturing the cells in either control conditions, FICZ, or with U937 cells cultured in transwells for 2 weeks. After 2 weeks, some wells were then switched to remove FICZ or U937 for 2 additional weeks, and where indicated, an AHRi was added. We observed that the addition of an AHRi could restore NK cell numbers compared with cells cultured in the same condition for 4 consecutive weeks or cells not receiving an AHRi following removal of FICZ or U937 (Figure 4D). This observation suggests that targeted AHR blockade can restore NK cell development from ILCPs even after being exposed to AHR-activating conditions.
Additionally, due to the established role of TGFβ in promoting ILC1-like phenotypes in cancer, we asked whether modulation of AHR activity in the presence of TGFβ activation affected the ability of ILCPs to differentiate into NK cells or ILC1s. After 4 weeks we observed that culturing of ILCPs with TGFβ completely abolished acquisition of EOMES. This effect was irrespective of modulation of AHR activity with a pharmacological AHR agonist or AHR inhibitor (Figure S4B). When assessed for functionality, the addition of TGFβ to the culture resulted in profound suppression of both IFNγ and TNFα production in the resulting ILC1s and NK cells which was not restored when co-treated with an AHRi (Figure S4C).
We also performed studies in NSG mice to assess the direct impact of AHR activation in vivo. Adoptive transfer of human ILCPs into NSG mice followed by intraperitoneal injections of IL-7, IL-15, and FICZ resulted in significant downregulation of EOMES with retention of TBET expression among the Lin-hCD45+CD56+CD94+ cells in the bone marrow (Figure 5A,B). Adoptive transfer and subsequent in vivo treatment of CD56bright human NK cells under the same conditions led to a small increase in the EOMESLo/−;TBET+ population which failed to reach statistical significance (Figure 5B,C). Interestingly, while we observed an overall suppressive effect on cytokine production when AML cells were co-transferred along with ILCP populations, we observed an overall stimulatory trend in the mice treated with FICZ relative to controls for both IFNγ and TNFα (Figure 5D). This suggests AML cells utilize additional pathways in addition to AHR to create the functional and phenotypic profiles observed. Overall, these findings show that AHR signaling is capable of skewing ILCP differentiation away from NK cells and toward ILC1s both in vitro and in vivo.
Figure 5: AHR agonism promotes an ILC1 phenotype in vivo.
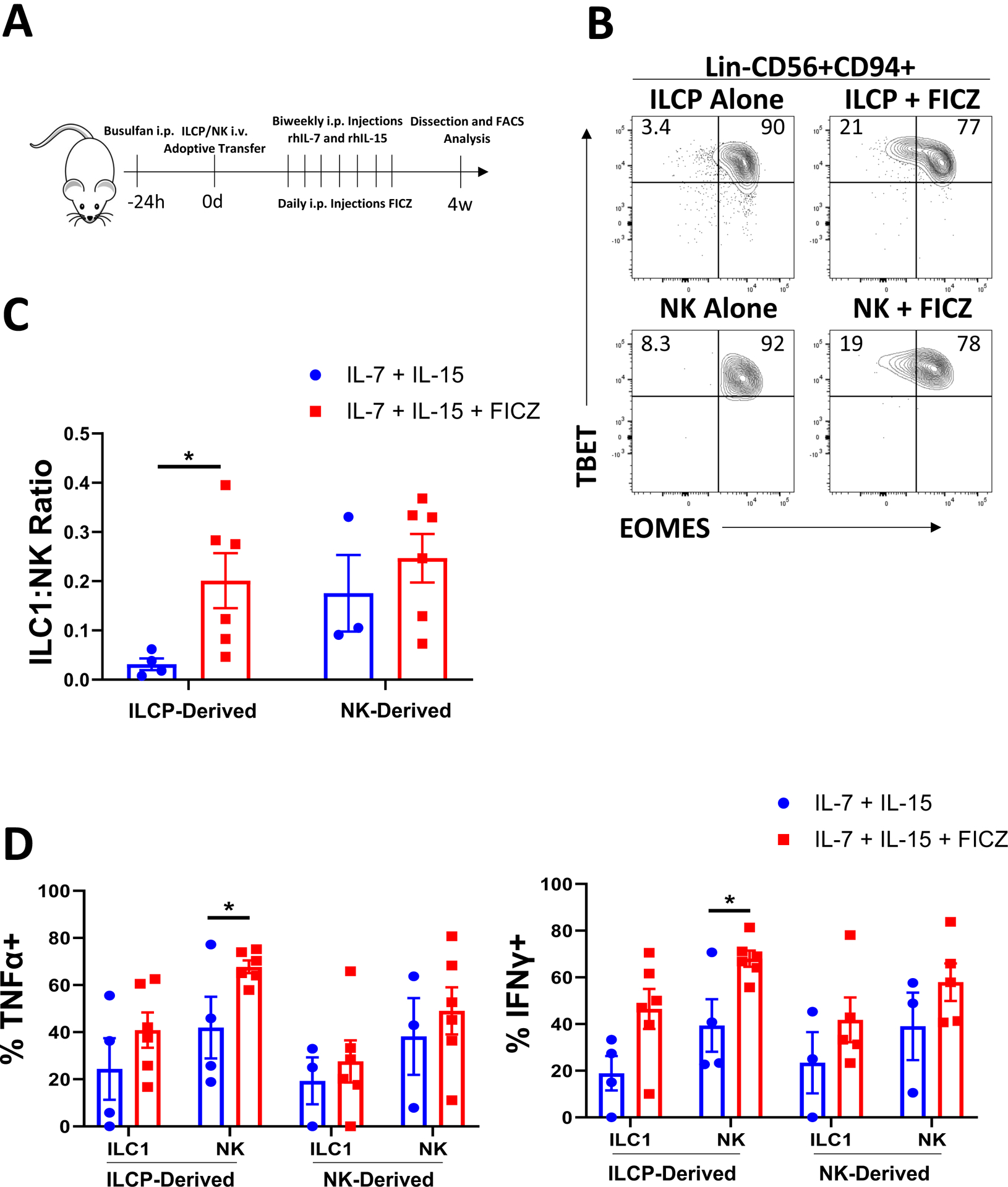
A) Schematic of in vivo studies. ILCPs (Lin-NKp80-CD294-KLRG1-NKp44-CD94−CD16−CD117+) and CD56bright NK cells (Lin-CD56+CD94+CD16−) were isolated from normal peripheral blood of 5 independent donors and adoptively transferred into NSG mice ± daily I.P. injections of FICZ (3 μg/mouse) or vehicle with biweekly injections of IL-7 and IL-15 for 4 weeks. At harvest, bone marrow was harvested and analyzed via flow cytometry for Group 1 human ILCs.
B) Representative flow plots of TBET and EOMES expression for human Lin-CD56+CD94+ cells derived from ILCPs or CD56bright NK cells ±FICZ.
C) Summary of ILC1:NK ratios comparing control conditions to FICZ-treated mice.
D) Summary data of TNFα and IFNγ expression determined by flow cytometry from ILCP- or CD56bright-derived NK cells and ILC1s from control or AML conditions.
n=3–5 (some mice excluded due to poor engraftment), *p<0.05. Error bars represent ±SEM.
Group 1 ILCs are dysregulated in the bone marrow of AML patients
After validating skewing towards an ILC1 lineage in the setting of AML using our in vivo and ex vivo models, we sought to determine whether we could identify an analogous ILC1-like population in primary untreated AML patient bone marrow samples. Recent reports suggest human ILC1s are likely found within the CD56bright population (29, 30). Upon surveying the ILC compartment, we observed a significant decrease in EOMES expression and a concomitant increase in TBET expression among the CD56bright population relative to normal marrow controls (Figure 6A,B). In contrast, we observed no significant changes in EOMES or TBET expression in the more mature CD56dim (Lin-CD56+CD94+/−CD16+) NK cell population. Taken together, these findings suggest that in the marrow of AML patients, there is skewing of Group 1 ILCs away from NK cells and towards an ILC1-like cell, similar to the observations in the murine model of AML and in our in vitro and in vivo human experimental systems.
Figure 6: Group 1 ILCs are dysregulated in the marrow of AML patients.
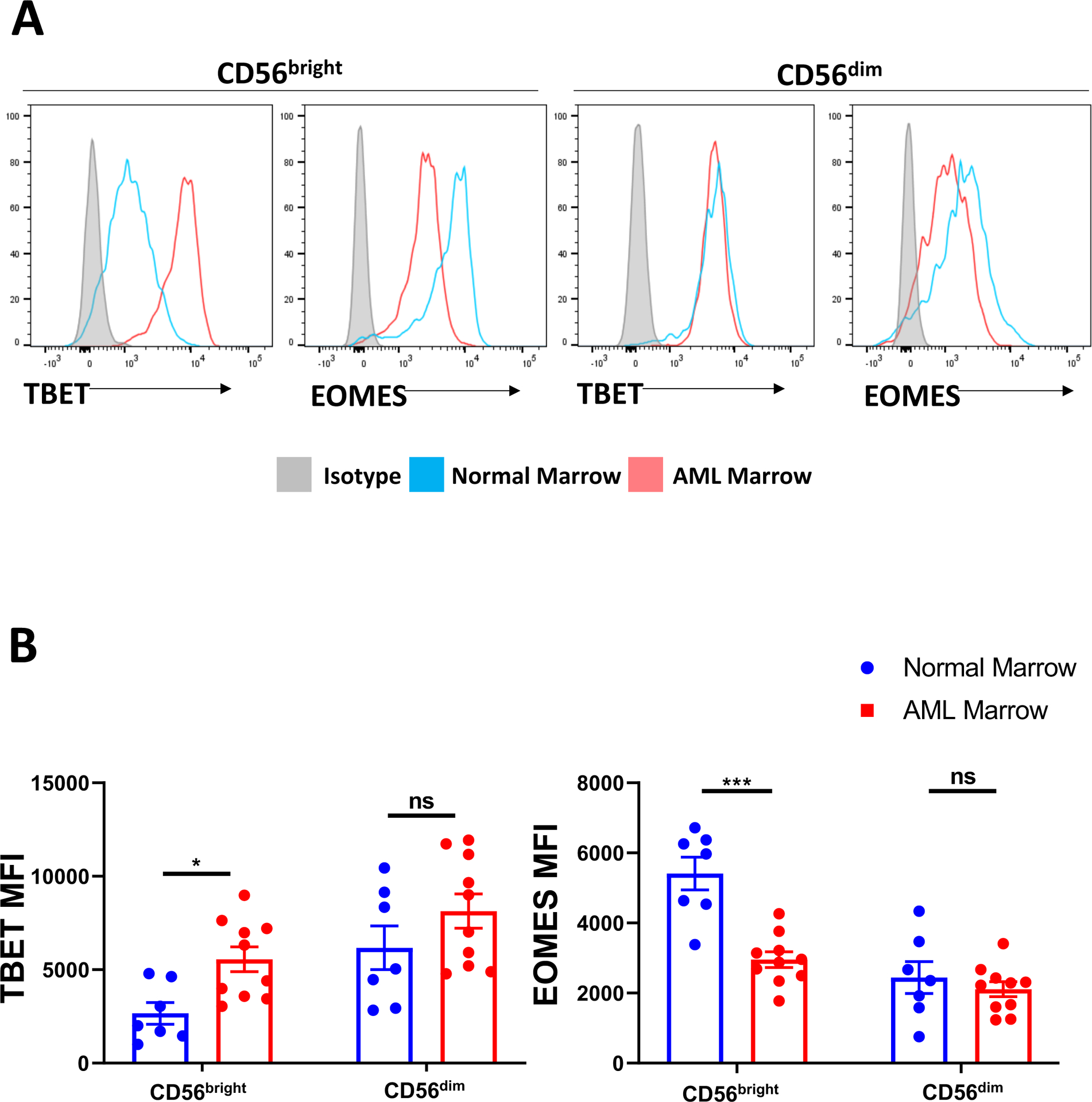
A) Representative flow plots comparing TBET and EOMES expression in CD56bright (Lin-CD56+CD94+CD16−) or CD56dim (Lin-CD56+CD94+/−CD16+) between AML and normal bone marrow.
B) Summary data for TBET and EOMES expression in CD56bright and CD56dim populations across all donors analyzed.
n=7 normal donors, n=10 AML donors, *p<0.05, **p<0.01, ***p<0.001, error bars represent ±SEM. MFI data were log transformed prior to statistical analysis.
DISCUSSION
The role of ILCs in normal physiology and pathophysiology is a burgeoning area in the field. Despite this, the role of non-NK ILCs in controlling or promoting cancer progression is a relatively understudied area to date, especially in hematologic malignancies. NK cells are important immune surveyors that slow progression of multiple cancer types, including AML (9, 31, 32). In contrast, the role of the closely related ILC1 subset is currently being dissected in several cancer types. Recent reports have demonstrated that ILCs typically only detected in tissues at steady state can be detected in the circulation and in tissues in the setting of AML and other diseases where they are normally not found (33, 34). The mechanisms by which this occurs is currently not completely understood. In this study, we identified tissue specific expansion of ILC1s in AML mice. Through a combination of ex vivo and in vivo modeling utilizing human cells, we also showed that ILC1s are expanded in vitro and in AML patients. We demonstrated that ex vivo or in vivo exposure of ILCPs to AML or AHR agonists increased production of ILC1s. We also showed that AML is capable of skewing human ILCP development towards ILC1s and away from NK cells at the clonal level. In contrast, when starting with normal healthy donor CD56bright NK cells, we observed no increase in ILC1s generated in vitro and only a mild increase in ILC1s in vivo that did not reach statistical significance. These observations lead us to conclude that skewing of ILCP development towards ILC1s and away from NK cells represents a major mechanism leading to localized increases in ILC1 production in AML and likely accounting at least in part to the loss of peripheral CD56bright NK cells in AML patients that we observed previously (5). We also found that pharmacologic AHR blockade was capable of restoring ILCP-derived NK cell levels even after exposure to pharmacologic AHR agonists or exposure to AML cells, suggesting AHR inhibition may be a beneficial treatment option in AML by restoring the functional NK cell pool. Finally, we identified skewing of Group 1 ILCs in the bone marrow of primary human AML patient samples towards an ILC1-like phenotype, as was observed in our experimental models. To the best of our knowledge, this is the first demonstration of AHR driving ILC1 generation and skewing human ILCPs towards the ILC1 lineage at the expense of NK cell differentiation in the setting of malignancy.
Functionally, we observed suppression of IFNγ production in our congenic AML mouse model as well as in our humanized NSG model, which also showed concomitant suppression of TNFα. While we did not observe differences in IFNγ production among bone marrow NK cells in leukemic mice, we did observe a significant decrease in IFNγ production from the total Group 1 ILC compartment in the bone marrow, resulting from loss of the total number of NK cells and a concomitant expansion of hypofunctional ILC1s. In contrast, we observed suppression of IFNγ production in both NK cells and ILC1s in the liver of leukemic mice, underscoring the importance of the tissue microenvironment in determining immune cell function.
Decreased amounts of type 1 cytokines could reduce the downstream immune response required for effective control of AML blasts, as IFNγ is associated with enhancing immune responses and TNFα possesses both pro- and anti-cancer effects (35, 36). Others have demonstrated that ILC1s are capable of limiting T cell responses and secrete lower amounts of T cell chemoattractants than NK cells (9, 37). T cell suppression in AML has been documented, suggesting our findings may contribute to this dysfunction (38). This effect could promote tumor growth through inhibition of the adaptive immune system by subsets of the innate immune system. Furthermore, loss of mature NK cells eliminates a mechanism of immune surveillance that is vital for controlling cancer progression. Indeed, we have previously demonstrated a decrease in cytotoxic potential among NK cells in AML (5). Thus, this developmental shunting towards ILC1s may serve as a mechanism of immune evasion in AML through a decrease in direct NK cell-mediated killing. We also noted differences in proliferation, as determined by Ki67 expression, of NK cells and ILC1s in the bone marrow compartment of leukemic mice compared to nonleukemic controls. Among leukemic mice, NK cells had higher expression of Ki67 compared to ILC1s, suggesting the increase in ILC1s observed in leukemic mice is not solely due to increased proliferation of ILC1s over NK cells. It is possible these increases in Ki67 expression are indicative of faster cell turnover in the setting of AML. Thus, ILC1 expansion in AML could be the result of both skewed ILC development in addition to increased cell death of mature NK cells relative to ILC1s. The effects of AML on NK and ILC1 turnover will be the subject of further investigation.
The functional differences observed between direct AHR agonism with FICZ (which resulted in increased IFNγ and TNFα production), and exposure to AML cells (which led to suppression of these cytokines) suggest that AML may utilize pathways in addition to AHR to alter the function of Group 1 ILCs. The observed stimulatory effects of FICZ on mature NK cells are consistent with previous reports (39). While FICZ is a high affinity AHR agonist, other molecules that act as AHR ligands, such as the tryptophan degradation product kynurenine or the compound TCDD, have been shown to elicit different downstream immune phenotypes compared to FICZ (40–43). In addition, a subset of AML patients have overactive indoleamine 2,3-dioxygenase 1 (IDO1), the first enzyme involved in the tryptophan degradation pathway leading to kynurenine production. These patients have worse clinical outcomes than patients with normal activity, presumably due to increased signaling through the AHR pathway leading to immune dysregulation (44, 45). Thus, kynurenine may be a particularly relevant AHR agonist to assess using these models. Indeed, testing the effects of different AHR ligands will be an area of future investigation. It is possible these differences are due at least in part to differential activation of downstream effector molecules such as microRNAs like miR-29b (2, 5), which in turn lead to regulation of target genes such as EOMES. Notably, ILCPs do not express EOMES and the ability to secrete IFNγ is not detected until cells commit to the group 1 ILC lineage (26, 46). These observations suggest AHR directly or indirectly prevents acquisition of EOMES rather than suppressing EOMES after it is expressed. In support of this hypothesis, we did not observe decreased EOMES expression upon direct pharmacological AHR agonism in mature NK cells ex vivo, which have already acquired EOMES expression. Our ChIP results demonstrate that AHR is capable of directly binding the promoters of EOMES and IFNG, although the downstream consequences of this binding on expression levels remain to be determined.
In addition to AHR agonists, leukemic blasts have been shown to secrete several cytokines, such as IL-1β, GM-CSF, IL-6, IL-8, TNFα, and TGFβ that could synergize with AHR signaling to produce the functional and phenotypic profiles observed (47–50). In particular, IL-1β has been heavily implicated in promoting plasticity of mature ILC subsets (51). Furthermore, previous studies in solid tumors have demonstrated TGFβ-dependent conversion of NK cells to ILC1s intratumorally, leading to poor tumor control (13, 14). However, we did not observe significant conversion from NK cells to ILC1s in the setting of AML. We tested the effects of TGFβ on ILCPs using our co-culture model and observed complete failure of the cells to acquire EOMES expression along with concomitant functional suppression of type 1 cytokines. These findings, together with our additional cloning experiments and in vivo modeling, suggest that AHR activation likely plays a role in promoting ILC1s, while additional factors (including potentially TGFβ) may play a role in inhibiting mature ILC function. Testing the synergistic effects of these cytokines with AHR activation in our models may better recapitulate the observations observed in AML patients. In addition to cytokine signaling, it is also possible that AML may directly modulate other key factors driving NK cell differentiation, including NOTCH (28, 52) or WNT (53) signaling pathways which could contribute to this phenotype.
Our observations of increased ILC1s in AML support a growing body of evidence that ILC1 phenotypes can be induced in the disease setting. In addition to fibrosarcoma mouse models (13), increased ILC1s have also been found in patients and mice with nonalcoholic fatty liver disease (NAFLD) (54) as well as in some murine models of infection (55). In contrast to these previous reports, which demonstrate conversion from mature NK cells to ILC1s as the dominant mechanism, our findings support that increased ILC1s in AML is primarily driven by skewed development of ILC precursors. In addition, previous work by Salome et al. described an ILC1-like population functionally inhibited in the peripheral blood of AML patients (29), however the mechanism by which this expansion occurs is not known. While we did not observe significant levels of ILC1s in the blood of leukemic mice, further work dissecting the localization and function of ILC1s in AML is clearly warranted. Our in vivo models and mechanistic insight into how ILC1s are likely promoted in AML provide a tool to elucidate additional mechanisms of evasion and what role ILC1s play in promoting disease progression.
The results from our studies reveal ILCP developmental skewing away from NK cells and towards ILC1s as a key mechanism for ILC1 expansion in AML. We show this developmental skewing is driven by AHR activation, and thus our data provide a novel rationale for targeting the AHR pathway for additional pre-clinical development as a potential therapy in AML. Our findings have broad implications for the importance of ILCP differentiation in tumor immune evasion and targeting of this developmental axis for the treatment of AML and other cancers. With the identification of ILC1 promotion, we further provide a novel perspective for the study of this phenomenon in other cancer types, and possibly other states of NK cell deregulation.
Supplementary Material
KEY POINTS.
ILC1s are enriched in the bone marrow of AML patients and leukemic mice
AML promotes ILC1s in part via AHR-mediated skewing of ILC precursor development
ACKNOWLEDGMENTS
Research reported in this publication was supported by The Ohio State University Comprehensive Cancer Center and the National Institutes of Health under grant number P30 CAO16058. We would like to thank the Leukemia Tissue Bank at The Ohio State University Comprehensive Cancer Center, Columbus, OH, for sample processing and storage services and The Ohio State University Comprehensive Cancer Center’s Genomics Shared Resources for technical support. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
We would also like to thank Victoria Sellers for her managerial support of this study as well as Jesse Kowalski, Charlene Mao, and the Flow Cytometry Shared Resource for flow cytometry and cell sorting technical support. We are grateful to the AML patients for their contribution of samples, making all this work possible. Finally, we would like to thank Dr. Eugene Oltz for his feedback and fruitful discussion of the manuscript.
This work was supported by grants from the National Institutes of Health/National Cancer Institute (5K22CA218466-02 to B.L.M.-B., CA199447 and CA208353 to A.G.F. and CA068458 and CA210087 to M.A.C.), American Association for Cancer Research award (17-20-46-MUND) (B.L.M.-B.), and from the Pelotonia Organization (OSU-CCC; M.R.L., N.S., A.P.N., B.L.M.-B., and A.G.F.).
Footnotes
DECLARATION OF INTERESTS
The authors declare no potential conflicts of interest.
REFERENCES
- 1.Howlader N, Noone AM, Krapcho M, Miller D, Brest A, Yu M, Ruhl J, Tatalovich Z, Mariotto A, Lewis DR, Chen HS, Feuer EJ, Cronin KA (eds). SEER Cancer Statistics Review, 1975–2017, National Cancer Institute. Bethesda, MD, https://seer.cancer.gov/csr/1975_2017/, based on November 2019 SEER data submission, posted to the SEER web site, April 2020. [Google Scholar]
- 2.Scoville SD, Nalin AP, Chen L, Chen L, Zhang MH, McConnell K, Beceiro Casas S, Ernst G, Traboulsi AA, Hashi N, Williams M, Zhang X, Hughes T, Mishra A, Benson DM, Saultz JN, Yu J, Freud AG, Caligiuri MA, and Mundy-Bosse BL. 2018. Human AML activates the aryl hydrocarbon receptor pathway to impair NK cell development and function. Blood 132: 1792–1804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stringaris K, Sekine T, Khoder A, Alsuliman A, Razzaghi B, Sargeant R, Pavlu J, Brisley G, de Lavallade H, Sarvaria A, Marin D, Mielke S, Apperley JF, Shpall EJ, Barrett AJ, and Rezvani K. 2014. Leukemia-induced phenotypic and functional defects in natural killer cells predict failure to achieve remission in acute myeloid leukemia. Haematologica 99: 836–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Costello RT, Sivori S, Marcenaro E, Lafage-Pochitaloff M, Mozziconacci MJ, Reviron D, Gastaut JA, Pende D, Olive D, and Moretta A. 2002. Defective expression and function of natural killer cell-triggering receptors in patients with acute myeloid leukemia. Blood 99: 3661–3667. [DOI] [PubMed] [Google Scholar]
- 5.Mundy-Bosse BL, Scoville SD, Chen L, McConnell K, Mao HC, Ahmed EH, Zorko N, Harvey S, Cole J, Zhang X, Costinean S, Croce CM, Larkin K, Byrd JC, Vasu S, Blum W, Yu J, Freud AG, and Caligiuri MA. 2016. MicroRNA-29b mediates altered innate immune development in acute leukemia. The Journal of clinical investigation 126: 4404–4416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chretien AS, Fauriat C, Orlanducci F, Galseran C, Rey J, Bouvier Borg G, Gautherot E, Granjeaud S, Hamel-Broza JF, Demerle C, Ifrah N, Lacombe C, Cornillet-Lefebvre P, Delaunay J, Toubert A, Gregori E, Luche H, Malissen M, Arnoulet C, Nunes JA, Vey N, and Olive D. 2017. Natural Killer Defective Maturation Is Associated with Adverse Clinical Outcome in Patients with Acute Myeloid Leukemia. Frontiers in immunology 8: 573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vivier E, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, Koyasu S, Locksley RM, McKenzie ANJ, Mebius RE, Powrie F, and Spits H. 2018. Innate Lymphoid Cells: 10 Years On. Cell 174: 1054–1066. [DOI] [PubMed] [Google Scholar]
- 8.Cherrier DE, Serafini N, and Di Santo JP. 2018. Innate Lymphoid Cell Development: A T Cell Perspective. Immunity 48: 1091–1103. [DOI] [PubMed] [Google Scholar]
- 9.Chiossone L, Dumas PY, Vienne M, and Vivier E. 2018. Natural killer cells and other innate lymphoid cells in cancer. Nature reviews. Immunology 18: 671–688. [DOI] [PubMed] [Google Scholar]
- 10.Pikovskaya O, Chaix J, Rothman NJ, Collins A, Chen YH, Scipioni AM, Vivier E, and Reiner SL. 2016. Cutting Edge: Eomesodermin Is Sufficient To Direct Type 1 Innate Lymphocyte Development into the Conventional NK Lineage. Journal of immunology (Baltimore, Md. : 1950) 196: 1449–1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Riggan L, Freud AG, and O’Sullivan TE. 2019. True Detective: Unraveling Group 1 Innate Lymphocyte Heterogeneity. Trends in immunology 40: 909–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Weizman OE, Adams NM, Schuster IS, Krishna C, Pritykin Y, Lau C, Degli-Esposti MA, Leslie CS, Sun JC, and O’Sullivan TE. 2017. ILC1 Confer Early Host Protection at Initial Sites of Viral Infection. Cell 171: 795–808.e712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gao Y, Souza-Fonseca-Guimaraes F, Bald T, Ng SS, Young A, Ngiow SF, Rautela J, Straube J, Waddell N, Blake SJ, Yan J, Bartholin L, Lee JS, Vivier E, Takeda K, Messaoudene M, Zitvogel L, Teng MWL, Belz GT, Engwerda CR, Huntington ND, Nakamura K, Holzel M, and Smyth MJ. 2017. Tumor immunoevasion by the conversion of effector NK cells into type 1 innate lymphoid cells. Nature immunology 18: 1004–1015. [DOI] [PubMed] [Google Scholar]
- 14.Cortez VS, Ulland TK, Cervantes-Barragan L, Bando JK, Robinette ML, Wang Q, White AJ, Gilfillan S, Cella M, and Colonna M. 2017. SMAD4 impedes the conversion of NK cells into ILC1-like cells by curtailing non-canonical TGF-beta signaling. Nature immunology 18: 995–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Constantinides MG, McDonald BD, Verhoef PA, and Bendelac A. 2014. A committed precursor to innate lymphoid cells. Nature 508: 397–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xu W, Cherrier DE, Chea S, Vosshenrich C, Serafini N, Petit M, Liu P, Golub R, and Di Santo JP. 2019. An Id2(RFP)-Reporter Mouse Redefines Innate Lymphoid Cell Precursor Potentials. Immunity 50: 1054–1068.e1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Scoville SD, Mundy-Bosse BL, Zhang MH, Chen L, Zhang X, Keller KA, Hughes T, Chen L, Cheng S, Bergin SM, Mao HC, McClory S, Yu J, Carson WE 3rd, Caligiuri MA, and Freud AG. 2016. A Progenitor Cell Expressing Transcription Factor RORgammat Generates All Human Innate Lymphoid Cell Subsets. Immunity 44: 1140–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lim AI, Li Y, Lopez-Lastra S, Stadhouders R, Paul F, Casrouge A, Serafini N, Puel A, Bustamante J, Surace L, Masse-Ranson G, David E, Strick-Marchand H, Le Bourhis L, Cocchi R, Topazio D, Graziano P, Muscarella LA, Rogge L, Norel X, Sallenave JM, Allez M, Graf T, Hendriks RW, Casanova JL, Amit I, Yssel H, and Di Santo JP. 2017. Systemic Human ILC Precursors Provide a Substrate for Tissue ILC Differentiation. Cell 168: 1086–1100.e1010. [DOI] [PubMed] [Google Scholar]
- 19.Daussy C, Faure F, Mayol K, Viel S, Gasteiger G, Charrier E, Bienvenu J, Henry T, Debien E, Hasan UA, Marvel J, Yoh K, Takahashi S, Prinz I, de Bernard S, Buffat L, and Walzer T. 2014. T-bet and Eomes instruct the development of two distinct natural killer cell lineages in the liver and in the bone marrow. The Journal of experimental medicine 211: 563–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Denison MS, and Nagy SR. 2003. Activation of the aryl hydrocarbon receptor by structurally diverse exogenous and endogenous chemicals. Annual review of pharmacology and toxicology 43: 309–334. [DOI] [PubMed] [Google Scholar]
- 21.Hughes T, Briercheck EL, Freud AG, Trotta R, McClory S, Scoville SD, Keller K, Deng Y, Cole J, Harrison N, Mao C, Zhang J, Benson DM, Yu J, and Caligiuri MA. 2014. The transcription Factor AHR prevents the differentiation of a stage 3 innate lymphoid cell subset to natural killer cells. Cell reports 8: 150–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee JS, Cella M, McDonald KG, Garlanda C, Kennedy GD, Nukaya M, Mantovani A, Kopan R, Bradfield CA, Newberry RD, and Colonna M. 2011. AHR drives the development of gut ILC22 cells and postnatal lymphoid tissues via pathways dependent on and independent of Notch. Nature immunology 13: 144–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang LH, Shin JH, Haggadone MD, and Sunwoo JB. 2016. The aryl hydrocarbon receptor is required for the maintenance of liver-resident natural killer cells. The Journal of experimental medicine 213: 2249–2257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li S, Bostick JW, Ye J, Qiu J, Zhang B, Urban JF Jr., Avram D, and Zhou L. 2018. Aryl Hydrocarbon Receptor Signaling Cell Intrinsically Inhibits Intestinal Group 2 Innate Lymphoid Cell Function. Immunity 49: 915–928.e915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zorko NA, Bernot KM, Whitman SP, Siebenaler RF, Ahmed EH, Marcucci GG, Yanes DA, McConnell KK, Mao C, Kalu C, Zhang X, Jarjoura D, Dorrance AM, Heerema NA, Lee BH, Huang G, Marcucci G, and Caligiuri MA. 2012. Mll partial tandem duplication and Flt3 internal tandem duplication in a double knock-in mouse recapitulates features of counterpart human acute myeloid leukemias. Blood 120: 1130–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen L, Youssef Y, Robinson C, Ernst GF, Carson MY, Young KA, Scoville SD, Zhang X, Harris R, Sekhri P, Mansour AG, Chan WK, Nalin AP, Mao HC, Hughes T, Mace EM, Pan Y, Rustagi N, Chatterjee SS, Gunaratne PH, Behbehani GK, Mundy-Bosse BL, Caligiuri MA, and Freud AG. 2018. CD56 Expression Marks Human Group 2 Innate Lymphoid Cell Divergence from a Shared NK Cell and Group 3 Innate Lymphoid Cell Developmental Pathway. Immunity 49: 464–476.e464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ramsdell F, Zúñiga-Pflücker JC, and Takahama Y. 2006. In vitro systems for the study of T cell development: fetal thymus organ culture and OP9-DL1 cell coculture. Curr Protoc Immunol Chapter 3: Unit 3.18. [DOI] [PubMed] [Google Scholar]
- 28.Nalin AP, Kowalski JJ, Sprague AC, Schumacher BK, Gerhardt AG, Youssef Y, Vedantam KV, Zhang X, Siebel CW, Mace EM, Caligiuri MA, Mundy-Bosse BL, and Freud AG. 2020. Notch Regulates Innate Lymphoid Cell Plasticity during Human NK Cell Development. Journal of immunology (Baltimore, Md. : 1950) 205: 2679–2693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Salome B, Gomez-Cadena A, Loyon R, Suffiotti M, Salvestrini V, Wyss T, Vanoni G, Ruan DF, Rossi M, Tozzo A, Tentorio P, Bruni E, Riether C, Jacobsen EM, Jandus P, Conrad C, Hoenig M, Schulz A, Michaud K, Della Porta MG, Salvatore S, Ho PC, Gfeller D, Ochsenbein A, Mavilio D, Curti A, Marcenaro E, Steinle A, Horowitz A, Romero P, Trabanelli S, and Jandus C. 2019. CD56 as a marker of an ILC1-like population with NK cell properties that is functionally impaired in AML. Blood Adv 3: 3674–3687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Allan DSJ, Cerdeira AS, Ranjan A, Kirkham CL, Aguilar OA, Tanaka M, Childs RW, Dunbar CE, Strominger JL, Kopcow HD, and Carlyle JR. 2017. Transcriptome analysis reveals similarities between human blood CD3(−) CD56(bright) cells and mouse CD127(+) innate lymphoid cells. Sci Rep 7: 3501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ruggeri L, Capanni M, Urbani E, Perruccio K, Shlomchik WD, Tosti A, Posati S, Rogaia D, Frassoni F, Aversa F, Martelli MF, and Velardi A. 2002. Effectiveness of donor natural killer cell alloreactivity in mismatched hematopoietic transplants. Science (New York, N.Y.) 295: 2097–2100. [DOI] [PubMed] [Google Scholar]
- 32.Smyth MJ, Crowe NY, and Godfrey DI. 2001. NK cells and NKT cells collaborate in host protection from methylcholanthrene-induced fibrosarcoma. Int Immunol 13: 459–463. [DOI] [PubMed] [Google Scholar]
- 33.Trabanelli S, Curti A, Lecciso M, Salomé B, Riether C, Ochsenbein A, Romero P, and Jandus C. 2015. CD127+ innate lymphoid cells are dysregulated in treatment naïve acute myeloid leukemia patients at diagnosis. Haematologica 100: e257–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Munneke JM, Björklund AT, Mjösberg JM, Garming-Legert K, Bernink JH, Blom B, Huisman C, van Oers MH, Spits H, Malmberg KJ, and Hazenberg MD. 2014. Activated innate lymphoid cells are associated with a reduced susceptibility to graft-versus-host disease. Blood 124: 812–821. [DOI] [PubMed] [Google Scholar]
- 35.Ersvaer E, Skavland J, Ulvestad E, Gjertsen BT, and Bruserud Ø. 2007. Effects of interferon gamma on native human acute myelogenous leukaemia cells. Cancer Immunol Immunother 56: 13–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tian T, Wang M, and Ma D. 2014. TNF-α, a good or bad factor in hematological diseases? Stem Cell Investig 1: 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schuster IS, Wikstrom ME, Brizard G, Coudert JD, Estcourt MJ, Manzur M, O’Reilly LA, Smyth MJ, Trapani JA, Hill GR, Andoniou CE, and Degli-Esposti MA. 2014. TRAIL+ NK cells control CD4+ T cell responses during chronic viral infection to limit autoimmunity. Immunity 41: 646–656. [DOI] [PubMed] [Google Scholar]
- 38.Knaus HA, Berglund S, Hackl H, Blackford AL, Zeidner JF, Montiel-Esparza R, Mukhopadhyay R, Vanura K, Blazar BR, Karp JE, Luznik L, and Gojo I. 2018. Signatures of CD8+ T cell dysfunction in AML patients and their reversibility with response to chemotherapy. JCI Insight 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Moreno-Nieves UY, Mundy DC, Shin JH, Tam K, and Sunwoo JB. 2018. The aryl hydrocarbon receptor modulates the function of human CD56(bright) NK cells. European journal of immunology 48: 771–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Quintana FJ, Basso AS, Iglesias AH, Korn T, Farez MF, Bettelli E, Caccamo M, Oukka M, and Weiner HL. 2008. Control of T(reg) and T(H)17 cell differentiation by the aryl hydrocarbon receptor. Nature 453: 65–71. [DOI] [PubMed] [Google Scholar]
- 41.Mezrich JD, Fechner JH, Zhang X, Johnson BP, Burlingham WJ, and Bradfield CA. 2010. An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. Journal of immunology (Baltimore, Md. : 1950) 185: 3190–3198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Murray IA, Morales JL, Flaveny CA, Dinatale BC, Chiaro C, Gowdahalli K, Amin S, and Perdew GH. 2010. Evidence for ligand-mediated selective modulation of aryl hydrocarbon receptor activity. Molecular pharmacology 77: 247–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Adams S, Teo C, McDonald KL, Zinger A, Bustamante S, Lim CK, Sundaram G, Braidy N, Brew BJ, and Guillemin GJ. 2014. Involvement of the kynurenine pathway in human glioma pathophysiology. PLoS One 9: e112945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Arandi N, Ramzi M, Safaei F, and Monabati A. 2018. Overexpression of indoleamine 2,3-dioxygenase correlates with regulatory T cell phenotype in acute myeloid leukemia patients with normal karyotype. Blood Res 53: 294–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Folgiero V, Goffredo BM, Filippini P, Masetti R, Bonanno G, Caruso R, Bertaina V, Mastronuzzi A, Gaspari S, Zecca M, Torelli GF, Testi AM, Pession A, Locatelli F, and Rutella S. 2014. Indoleamine 2,3-dioxygenase 1 (IDO1) activity in leukemia blasts correlates with poor outcome in childhood acute myeloid leukemia. Oncotarget 5: 2052–2064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Freud AG, Keller KA, Scoville SD, Mundy-Bosse BL, Cheng S, Youssef Y, Hughes T, Zhang X, Mo X, Porcu P, Baiocchi RA, Yu J, Carson WE 3rd, and Caligiuri MA. 2016. NKp80 Defines a Critical Step during Human Natural Killer Cell Development. Cell reports 16: 379–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bradbury D, Bowen G, Kozlowski R, Reilly I, and Russell N. 1990. Endogenous interleukin-1 can regulate the autonomous growth of the blast cells of acute myeloblastic leukemia by inducing autocrine secretion of GM-CSF. Leukemia 4: 44–47. [PubMed] [Google Scholar]
- 48.Sepehrizadeh Z, Mohammadi M, Emami A, Yazdi MT, Bozchlou SH, Khorramizadeh MR, Shapourabadi MB, Jaberi E, Rajaei N, and Setayesh N. 2014. Assessment of cytokine expression profile in acute myeloid leukemia patients before and after chemotherapy. Turk J Haematol 31: 149–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tobler A, Moser B, Dewald B, Geiser T, Studer H, Baggiolini M, and Fey MF. 1993. Constitutive expression of interleukin-8 and its receptor in human myeloid and lymphoid leukemia. Blood 82: 2517–2525. [PubMed] [Google Scholar]
- 50.Szczepanski MJ, Szajnik M, Welsh A, Whiteside TL, and Boyiadzis M. 2011. Blast-derived microvesicles in sera from patients with acute myeloid leukemia suppress natural killer cell function via membrane-associated transforming growth factor-beta1. Haematologica 96: 1302–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bal SM, Golebski K, and Spits H. 2020. Plasticity of innate lymphoid cell subsets. Nature reviews. Immunology 20: 552–565. [DOI] [PubMed] [Google Scholar]
- 52.Bill M, Pathmanathan A, Karunasiri M, Shen C, Burke MH, Ranganathan P, Papaioannou D, Zitzer NC, Snyder K, LaRocco A, Walker AE, Brannan ZJ, Nalin AP, Freud AG, Dikov MM, Zhang X, Bloomfield CD, Garzon R, and Dorrance AM. 2020. EGFL7 Antagonizes NOTCH Signaling and Represents a Novel Therapeutic Target in Acute Myeloid Leukemia. Clin Cancer Res 26: 669–678. [DOI] [PubMed] [Google Scholar]
- 53.Gruszka AM, Valli D, and Alcalay M. 2019. Wnt Signalling in Acute Myeloid Leukaemia. Cells 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cuff AO, Sillito F, Dertschnig S, Hall A, Luong TV, Chakraverty R, and Male V. 2019. The Obese Liver Environment Mediates Conversion of NK Cells to a Less Cytotoxic ILC1-Like Phenotype. Frontiers in immunology 10: 2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Park E, Patel S, Wang Q, Andhey P, Zaitsev K, Porter S, Hershey M, Bern M, Plougastel-Douglas B, Collins P, Colonna M, Murphy KM, Oltz E, Artyomov M, Sibley LD, and Yokoyama WM. 2019. Toxoplasma gondii infection drives conversion of NK cells into ILC1-like cells. Elife 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.