

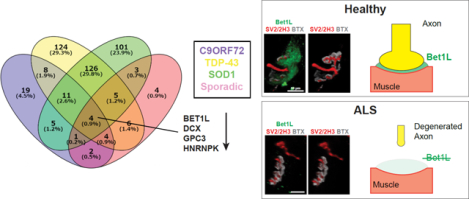
Abstract
Amyotrophic lateral sclerosis (ALS) is a fatal neuromuscular disease in which patients gradually become paralyzed due to loss of motor function. Many genetically inheritable mutations have been linked to ALS; however, the majority of ALS patients are considered sporadic. Therefore, there is a need for a common therapy that is effective for all ALS patients. Although there is evidence of the disease beginning in the periphery at the neuromuscular junction (NMJ), the specific processes involved in skeletal muscle and at the NMJ are still largely unknown. To study common disease mechanisms in ALS skeletal muscle, we performed RNA sequencing of skeletal myocytes differentiated from induced pluripotent stem cells (iPSCs) derived from familial ALS (with C9ORF72, SOD1, or TARDBP mutations) and sporadic ALS patients. Compared to healthy control lines, the myocytes from all ALS lines showed downregulation of four genes: BET1L, DCX, GPC3, and HNRNPK. We next measured the expression levels of these four genes in hind limb muscle samples from a rat model of familial ALS (SOD1G93A transgenic) and found that only the Bet1L gene, which encodes Bet1 Golgi Vesicular Membrane Trafficking Protein Like, was commonly downregulated. Bet1L protein appeared to be localized to the basal lamina of the NMJ, with decreased expression over time in SOD1G93A transgenic rats. Importantly, the expression levels began to decrease early in the disease process. Our results indicate that loss of Bet1L at the NMJ could be of interest for better understanding ALS disease progression.
Keywords: induced pluripotent stem cells, SOD1G93A transgenic rat, Bet1L, amyotrophic lateral sclerosis (ALS), sporadic, familial, neuromuscular junction
Graphical Abstract
Introduction
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disease with a widely variable genetic background. Around 90 percent of cases are considered sporadic and 10 percent are familial, or genetically inherited. Familial ALS is caused by over twenty different mutations1,2. The varied disease background has made it difficult to pinpoint an early converging pathological mechanism that could be targeted therapeutically. Despite the variability in genetic background, all ALS patients face a gradual paralysis leading to death on average 3–5 years following diagnosis2. At the cellular level, the disease is characterized by degeneration of the neuromuscular junction (NMJ), motor neuron cell death, and skeletal muscle atrophy. The mechanisms by which each of the ALS-causing mutations result in this phenotype are not completely understood. There are some common features such as protein aggregation, mitochondrial dysfunction, disrupted axonal transport, and defects in RNA processing2. However, the specific timeline of when these changes occur and which mechanisms are causative or just a downstream effect of others has yet to be elucidated. There are currently only two FDA-approved drugs that can delay the disease process for some but not all patients, and only on the scale of a few months3,4. Therefore, it is critical to better understand common disease processes in order to find biomarkers for earlier detection or therapeutic options to prolong the patient’s lifespan and quality of life.
ALS is commonly classified as a motor neuron disease. However, in recent years there has been increasing evidence that non-cell autonomous mechanisms are also involved in ALS disease pathogenesis. These mechanisms include the possible contributions of glial cells5, the immune system6, and skeletal muscle7. Specific pathology has been identified in the skeletal muscle of ALS patients, which includes fiber type grouping, metabolic changes, protein aggregation, and a decreased capacity for regeneration7–9. Interactions between skeletal muscle and motor neurons could be important, as skeletal muscle experiences pathological changes early in the disease process, and degeneration of the NMJ occurs prior to motor neuron axon degeneration10,11. This has led to the hypothesis that the disease actually begins in the periphery, causing motor neuron cell death in a retrograde manner12. Therefore, skeletal muscle and the NMJ are of interest as novel therapeutic targets. However, the pathological mechanisms occurring at the NMJ and in ALS skeletal muscle require further examination.
Human induced pluripotent stem cells (iPSCs) reprogrammed from patient somatic cells provide an opportunity to study cellular disease mechanisms in a patient-specific manner13–15. In the current study, we differentiated ALS patient iPSCs into skeletal myocytes to study common pathological changes in ALS skeletal muscle of varying genetic backgrounds. Multiple iPSC lines from ALS patients of familial (C9ORF72, SOD1, TARDBP) and sporadic backgrounds were differentiated into skeletal myocytes and used to identify common changes in gene expression by RNA sequencing. Compared to healthy control lines, all ALS lines had downregulation of four genes: BET1L, DCX, GPC3, and HNRNPK. We then characterized expression of those genes and their respective proteins in our iPSC-derived skeletal myocyte cultures as well as the skeletal muscle of a rat model of familial ALS (SOD1G93A transgenic rats). The overexpression of mutant SOD1 in rodents leads to ubiquitinated SOD1 aggregates, gliosis, motor neuron cell death, and skeletal muscle denervation, atrophy, and wasting. As with ALS patients, SOD1G93A transgenic rats present with a late onset of motor dysfunction leading to paralysis16–19. Of the four genes downregulated in human myocytes, only the Bet1L gene was downregulated in the ALS model rats. Interestingly, Bet1L protein was localized to the NMJ and appeared to be part of the basal lamina between the myofiber and motor neuron axon, as indicated by overlap with collagen IV staining. The amount of Bet1L present at the NMJs was decreased in symptomatic and end point SOD1G93A rats as indicated by both quantitative reverse-transcription polymerase chain reaction (RT-qPCR) and immunohistochemistry. Importantly, NMJ degeneration in this rat model is correlated to the loss of Bet1L expression. Therefore, NMJ degeneration and loss of Bet1L could be interconnected. The loss of Bet1L during NMJ degeneration is an intriguing phenomenon that may connect the varied genetic and sporadic backgrounds of ALS.
Materials and Methods
Human pluripotent stem cells
Human pluripotent stem cells were acquired from several sources. The ALS lines used “C9-ALS 1” (NINDS ID NDS00239 clone 3, C9ORF72 ALS, Caucasian/Ashkenazi female, age 64), “C9-ALS 2” (NINDS ID NDS00239 clone 5, C9ORF72 ALS, Caucasian/Ashkenazi female, age 64), “SOD1-ALS 1” (NINDS ID NDS00248 SOD1A4V ALS, Caucasian female, age 44), “TDP43-ALS 1” (NINDS ID NDS00245, TARDBPG892A ALS, Caucasian male, age 43), “Sporadic-ALS 1” (NINDS ID NDS00243, Sporadic ALS, Caucasian male, age 49), “Sporadic-ALS 2” (NINDS ID NDS00244, Sporadic ALS, Caucasian male, age 38) were received from Target ALS stem cell core at RUCDR Infinite Biologics (Piscataway Township, NY, USA). Healthy control pluripotent cell lines [human female embryonic stem cell line H9 (WA9), referred to as “ESC 1” and human female iPSC line IMR90, referred to as “iPSC 1”] were from WiCell, (Madison, WI, USA). Stem cell lines were cultured according to established feeder-free protocols20,21.
Differentiation of skeletal myocytes from human pluripotent stem cells
Human pluripotent stem cells were differentiated into myogenic progenitors and mature skeletal myocytes according to our lab’s previously established protocol22,23. Briefly, the stem cells were lifted using 2 mg/mL of dispase (Life Technologies, Carlsbad, CA, USA) or 0.1% collagenase (Life Technologies) and transferred to a flask coated with poly(2-hydroxyethyl methacrylate) (polyHEMA, Sigma-Aldrich, St Louis, MO, USA) in order to grow in suspension and form spheres of expanding myogenic progenitors. The expansion medium consisted of Stemline medium (S-3194, Sigma-Aldrich) with 100 ng/mL of recombinant human FGF-2 (WiCell), 5 μg/mL of heparin sulfate (Sigma-Aldrich), and 1% penicillin/streptomycin/amphotericin B (PSA; Thermo Fisher Scientific, Waltham, MA, USA). The spheres were mechanically chopped weekly using a McIlwain tissue chopper (Mickle Laboratory Engineering, Surrey, UK).
After around 6 weeks of expansion in suspension culture, the spheres were dissociated with TrypLE (Life Technologies) and plated down onto poly-L-lysine (0.1 mg/mL, Sigma-Aldrich) and laminin (5 μg/mL, Sigma-Aldrich) coated glass coverslips at a density of 200,000 cells per coverslip. At this point the cells were switched to a terminal differentiation medium consisting of DMEM/GlutaMAX (10566–016, Life Technologies), 2% B-27 serum-free supplement (Life Technologies) and 1% PSA. Myocytes became fully differentiated around 2 weeks of culture in this medium and were maintained for up to 6 weeks.
RNA Sequencing
Total RNA was isolated from cells using Direct-zol RNA Kit (Zymo Research, Irvine, CA, USA) according to the manufacturer’s instructions. RNA-seq libraries were constructed from 500 ng of total RNA using KAPA RNA HyperPrep Kits with RiboErase (KAPA Biosystems, Wilmington, MA, USA) according to the manufacturer’s instructions. Completed libraries were quantified using D1000 ScreenTape system (Agilent Technologies, Santa Clara, CA, USA), and were sequenced using Hiseq 4000 (Illumina, San Diego, CA, USA).
After pre-filtering the raw data by removing sequence adapters and low-quality reads, paired-end 100 bp sequences were conducted to align the human genome (hg38) by HISAT2 software in a Galaxy browser (www.galaxy.psu.edu). Transcript assembly, abundance and evaluation of differential expression were accomplished using the Cufflinks and DEseq software in a Galaxy browser. Genes exhibiting a fold change ≥± 1.5 and FDR < 0.05 were considered differentially expressed in the cells derived from ALS patients compared to in control cells (H9 and IMR90). Gene ontology analysis, protein-protein interaction analysis, and Venn diagram visualization were performed with Metascape (http://metascape.org/), STRING (https://string-db.org/) and Venny (https://bioinfogp.cnb.csic.es/tools/venny/) software, respectively. The raw RNA sequencing data file can be found in the Supplemental Material.
Quantitative reverse-transcription polymerase chain reaction (RT-qPCR) for iPSC-derived skeletal myocytes
First-strand cDNA was synthesized from 1 μg of total RNA using ReverTra Ace qPCR RT Master Mix with gDNA Remover (Toyobo Co., Ltd., Osaka, Japan). qPCR was performed with FastStart Essential DNA Green Master (Roche, Basel, Switzerland) using Light Cycler 96 (Roche). PCR was performed with the following thermocycling conditions: denaturation at 95°C for 10 minutes and 45 cycles of denaturation at 95°C for 10 seconds, annealing at 58 °C for 10 seconds and elongation at 72 °C for 10 seconds. Data were normalized to the expression of ACTB. Primer sequences can be found in Supplemental Table 1.
Immunocytochemistry
Cells were fixed with ice cold methanol then permeabilized and blocked for 20 minutes with 0.1% Triton X-100 (Sigma-Aldrich) and 5% normal donkey serum (NDS; Jackson ImmunoResearch, West Grove, PA, USA) in a phosphate buffered saline (PBS) solution. Primary antibodies were added according to the dilutions in Supplemental Table 2 and incubated at room temperature for one hour. Secondary antibodies (listed in Supplemental Table 3) were added at a dilution of 1:1000 and incubated at room temperature for 30 minutes. Hoechst 33258 (0.5 μg/ml in PBS, Sigma-Aldrich) was added to label nuclei. Coverslips were mounted to slides using Fluoromount-G mounting medium (SouthernBiotech, Birmingham, AL, USA).
SOD1G93A transgenic rats
SOD1G93A transgenic rats used in this study were maintained as previously described16,18,19,24–27. All rats were housed, bred, and sacrificed in accordance with UW-Madison and NIH standards of animal care. Breeder male SOD1G93A rats were crossed with female Sprague Dawley rats (Taconic Biosciences, Germantown, NY, USA). Genotyping of SOD1G93A positive rats and wild-type (WT) controls was determined from ear punches using real-time PCR, which was performed by Transnetyx (Cordova, TN, USA). Rats were sacrificed at various disease stages including presymptomatic (Day 90), symptomatic, and end point. Disease stage was determined using the Basso-Beattie-Bresnahan (BBB) locomotor rating scale between 0 (no movement) to 21 (normal locomotion)28. Rats were considered symptomatic with a BBB score between 17 and 1218,19,27. The average age at which rats became symptomatic was at 179.5 ± 8 days (n=8 total; 188 ± 7 days in female rats (n=4) and 171 ± 6 days in male rats (n=4), mean ± SEM). Rats were considered to be at end point when they no longer had the reflexes to right themselves from a sideways position within 30 seconds. For each time point, an age-matched wild-type control was also sacrificed, and hindlimb (tibialis anterior, TA) muscles were harvested from each.
RT-qPCR on homogenized hindlimb muscles
TA muscles from each disease stage were homogenized using the Silent Crusher M homogenizer (Heidolph, Schwabach, Germany) and RNA was extracted using the RNeasy Fibrous Tissue Mini Kit (Qiagen, Hilden, Germany). DNase treatment and reverse transcription were performed according to product instructions (Promega, Madison, WI, USA). qPCR was performed with Fast SYBR Green Master Mix (Thermo Fisher Scientific) using the DNA Engine Opticon 2 System (Bio-Rad, Hercules, CA, USA). The following thermocycling conditions were used: denaturation at 95 °C for 10 minutes and 40 cycles of denaturation at 95 °C for 10 seconds and annealing at 60 °C for 45 seconds. Data were normalized to the expression of HPRT1 and displayed as relative to the age-matched WT animals corresponding to presymptomatic SOD1G93A rats. Primer sequences can be found in Supplemental Table 4.
Immunohistochemistry
TA muscles were sectioned at 20-micron thickness using a cryostat. The muscle sections were placed on a glass slide and fixed with 4% paraformaldehyde (PFA; Electron Microscopy Sciences, Hatfield, PA, USA) in PBS for 20 minutes at room temperature. Sections were then blocked in 5% NDS in PBS for 2 hours at room temperature followed by primary antibody incubation in 5% NDS and 0.3% Triton-X 100 overnight at 4°C. Next, the sections were washed five times for 5 minutes each with PBS before adding secondary antibodies diluted in 5% NDS in PBS. The wash step was repeated, then the sections were incubated in Hoechst 33258 for 15 minutes at room temperature, followed by additional washes. The last PBS wash was removed from the slide and Fluoromount-G mounting medium was added and a glass coverslip was placed on top. NMJs were labeled using α-bungarotoxin (BTX) conjugated to Alexa Fluor 647, which was added at the same time as the primary antibodies. The information and dilutions of primary and secondary antibodies can be found in Supplemental Table 2 and Supplemental Table 3. While rabbit monoclonal anti-Bet1L antibody (Thermo Fisher Scientific/Invitrogen, Waltham, MA, USA) was used for most of the Bet1L immunohistochemistry, mouse monoclonal antibody (Santa Cruz Biotechnology, Dallas, TX, USA) was used for co-labeling with anti-NG2 antibody. In this case, the mouse antibody was identified by an antibody detection kit (Vector Fluor™ Excel Amplified anti-mouse IgG DyLight 488® Antibody Kit, Vector Laboratories, Burlingame, CA, USA). Additionally, the specificity of rabbit polyclonal anti-Bet1L antibody was tested by antigen blocking. Before staining with muscle sections, the antibody was incubated with 100x molar excess protein containing recombinant Bet1L (human Bet1L 293T cell lysate, sc-115664; Santa Cruz Biotechnology) at 4 °C overnight.
Image acquisition and analysis
Fluorescent images were taken using a Nikon Eclipse 80i fluorescent microscope (Nikon, Tokyo, Japan) with a DS-QilMC CCD camera (Nikon). The three-dimensional renderings of rat NMJ immunohistochemistry were created using the z-stack feature of a Leica TCS SP8 confocal microscope (Leica, St Galen, Switzerland). Skeletal myocyte differentiation was quantified as previously described9. Day 14 skeletal myocytes were stained for Pax7, MyoD, Myogenin, and Myosin heavy chain (MHC). Additionally, beta-Tubulin III (TuJ1) was stained to identify the presence of neuronal cells in terminally differentiated cells. Stained cells were imaged at 6 randomly selected fields of view. The total number of nuclei and the number of nuclei within positively labeled myocytes were counted to obtain a percentage for each image.
To quantify the number of rat NMJs positive for Bet1L, muscle sections were labeled with BTX and a Bet1L antibody. Muscle sections were scanned using the Nikon Eclipse 80i fluorescent microscope and the number of NMJs positive or negative for Bet1L were analyzed. The average number of total NMJs counted per animal was 81. Four WT and four SOD1G93A rats were counted for each time point (presymptomatic, symptomatic, and end point), leading to approximately 250 NMJs counted per condition. When analyzing Bet1L-stained NMJs, the person performing the quantification was blinded to the sample genotype and age to avoid bias.
Statistical Analysis
Statistical analyses were conducted using GraphPad Prism software (La Jolla, CA, USA). Graphs were presented as means ± SEM. One-way ANOVA was used to compare data points across cell lines or time points. A statistically significant one-way ANOVA was followed up with Tukey’s post-hoc test for multiple comparisons. An unpaired Student’s t-test was used when comparing only two groups. Gaussian distributions were assumed, and differences were considered significant when P<0.05.
Results
RNA Sequencing reveals four commonly down-regulated genes in ALS patient iPSC-derived skeletal myocytes
We first determined common changes in the gene expression profile of skeletal muscle across familial and sporadic ALS disease backgrounds. iPSC lines from ALS patients (familial cases with C9ORF72, SOD1, or TARDBP mutations; and two sporadic lines) and two healthy controls were differentiated into skeletal myocytes. Differentiation was confirmed through immunostaining for muscle markers (Pax7, MyoD, Myogenin, and MHC) and a non-muscle and neuronal marker (beta-Tubulin III, TuJ1). The percentages of positively stained cells were quantified for each cell line (Supplemental Fig. 1). While the ESC 1 line had significantly higher levels of MHC expression compared to two of the six ALS lines (Sporadic-ALS 1 and TDP43 -ALS 1), there were no significant differences between these lines when quantifying the other markers, and none of the ALS lines were statistically different from the other control line iPSC 1. Therefore, the level of muscle differentiation was generally consistent between control and ALS lines.
Differentiated skeletal myocytes were collected at day 14 for transcriptome analysis using RNA sequencing. The full RNA sequencing data can be found in the Supplemental Material. Principal Component Analysis (PCA) plots demonstrate the transcriptional differences between the control and ALS lines, as well as the variation within ALS lines (Fig. 1A). There were only 4 genes that were commonly downregulated in all ALS lines compared to healthy controls (Fig. 1B). These were BET1L (Bet1 Golgi Vesicular Membrane Trafficking Protein Like), DCX (Doublecortin), GPC3 (Glypican 3), and HNRNPK (Heterogeneous Nuclear Ribonucleoprotein K). Interestingly, gene ontology analysis classified these genes as being involved in vesicle-mediated transport (Fig. 1B), as is supported by their protein-protein interactomes (Fig. 1C–F).
Figure 1. RNA-Sequencing identifies four commonly down-regulated ALS genes.
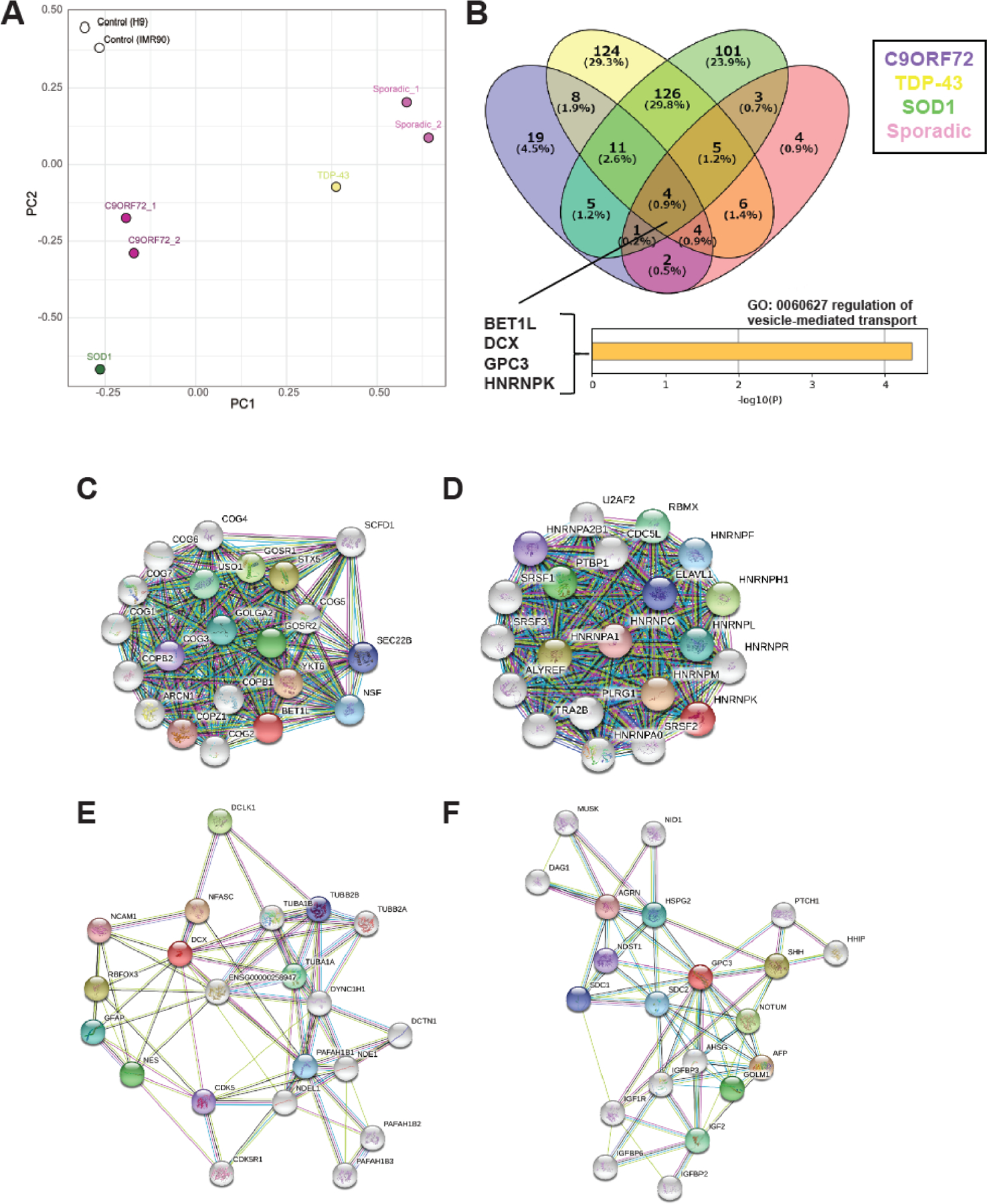
ALS patient iPSCs were collected for RNA sequencing after 14 days of terminal differentiation. (A) Principal Component Analysis (PCA) plot showing the clustering of control lines compared to the variation in ALS lines. (B) Compared to healthy control lines, only four genes were commonly down-regulated across all ALS lines: BET1L, DCX, GPC3, and HNRNPK. (C-F) Interactomes for each of the genes of interest.
To confirm the RNA sequencing results and further investigate the four genes of interest, each cell line was again terminally differentiated into skeletal myocytes and analyzed by RT-qPCR and immunostaining. The results from RT-qPCR confirmed that expression levels of each of the four genes were significantly lower in all ALS lines compared to healthy controls (P<0.01, Fig. 2A). While immunostaining with antibodies against each of the four proteins of interest did not show an appreciable difference in protein expression levels between ALS lines and controls, we were able to visualize the localization of each protein within skeletal myocytes (Fig. 2B, Supplemental Fig. 2). It was valuable to confirm that the proteins are in fact being expressed in our myocytes and not other cell types in culture because there is very limited information about the role of these proteins in skeletal muscle. Bet1L was primarily localized in the periphery of the nuclei (Fig. 2B, Supplemental Fig. 2), which correlates with its reported function as a SNARE protein for transport between the endoplasmic reticulum (ER) and Golgi apparatus29–31. Doublecortin (DCX), a microtubule-associated protein, is located throughout the length of the myocytes (Fig. 2B, Supplemental Fig. 2). Doublecortin is typically studied in the context of its role in neuronal development and microtubule stabilization32,33. However, it is also involved in myogenic development and regeneration34. Next, the proteoglycan Glypican 3 (GPC3) seemed to be diffusely expressed in myocytes as well as surrounding cells (Fig. 2B, Supplemental Fig. 2). Glypican 3 is commonly studied for its upregulation during hepatocellular carcinoma35, but its role in other cell types such as skeletal muscle has not been determined. Finally, hnRNP K was expressed primarily in the myonuclei (Fig. 2B, Supplemental Fig. 2), which is expected from its function as an RNA-binding protein that regulates several aspects of gene expression36. There was no difference in the localization of the proteins of interest when comparing control and ALS lines. Representative images of the four markers in ALS lines can be found in Supplemental Fig. 2. Interestingly, we found that Bet1L localization in iPSC-derived myocytes had changed with time. In more matured myocytes at 6 weeks of terminal differentiation, Bet1L primarily had a linear localization along the length of the myocytes (Fig. 2C).
Figure 2. Expression of the four proteins of interest in iPSC-derived skeletal myocytes.
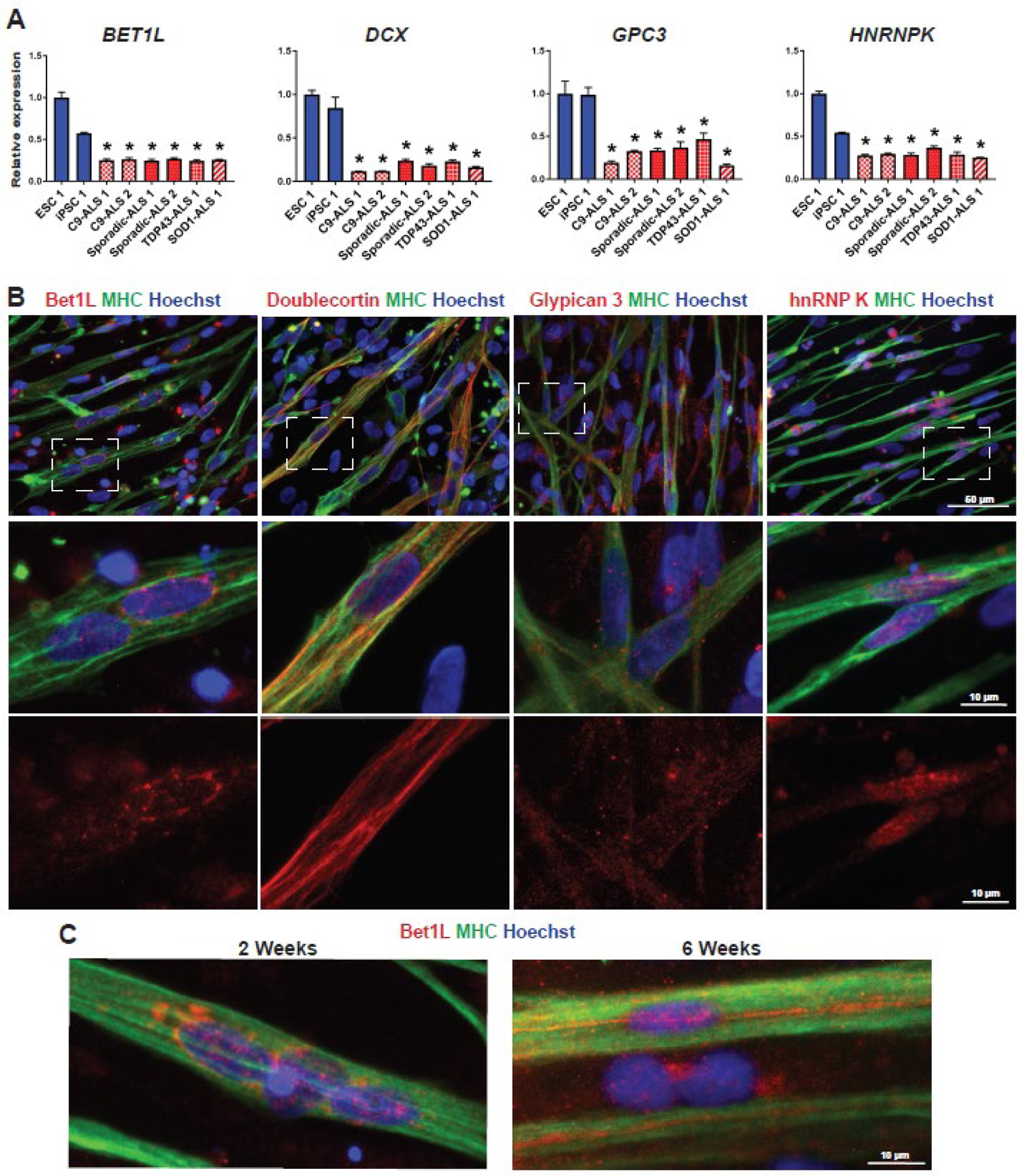
(A) RT-qPCR of day 14 skeletal myocytes was used to confirm down-regulation of each of the four genes identified from RNA sequencing. Data was normalized to control line ESC 1 and analyzed using one-way ANOVA followed by Tukey’s post-hoc multiple comparisons test. *P<0.01 compared to ESC 1 and iPSC 1 (B) Representative images of each protein of interest in healthy control myocytes at day 14. Skeletal myocytes are indicated by positive expression of myosin heavy chain (MHC). The middle row is enlarged from the dashed squares, and the bottom row shows red channel only. (C) The localization of Bet1L protein changed over time, with a more aligned expression pattern in mature myocytes around 6 weeks.
SOD1G93A rat muscle shows Bet1L localization at the NMJ and decreased expression over time
Next, we used a rat model of familial ALS (SOD1G93A transgenic rat)16,18,19,24–26 to see if the four genes of interest were also downregulated in an additional model of ALS pathology. Hindlimb muscles (tibialis anterior, TA) were harvested from SOD1G93A rats at the presymptomatic, symptomatic, and end point stages of the disease. We performed immunohistochemistry of muscle cross-sections and determined the localization of each of the four markers in ALS rat muscles (Fig. 3A, Supplemental Fig. 3A). Each marker was co-labeled with fluorescently-conjugated bungarotoxin (BTX) to indicate muscle endplates. Doublecortin and Glypican 3 had mostly diffuse localization in the myofibers that was slightly stronger in the sarcolemma, including regions where NMJs were located (Supplemental Fig. 3A). hnRNP K was expressed in cell nuclei, including some that were located near the NMJ (Supplemental Fig. 3A).
Figure 3. Focal expression of Bet1L at NMJs in the skeletal muscle of ALS model rats.
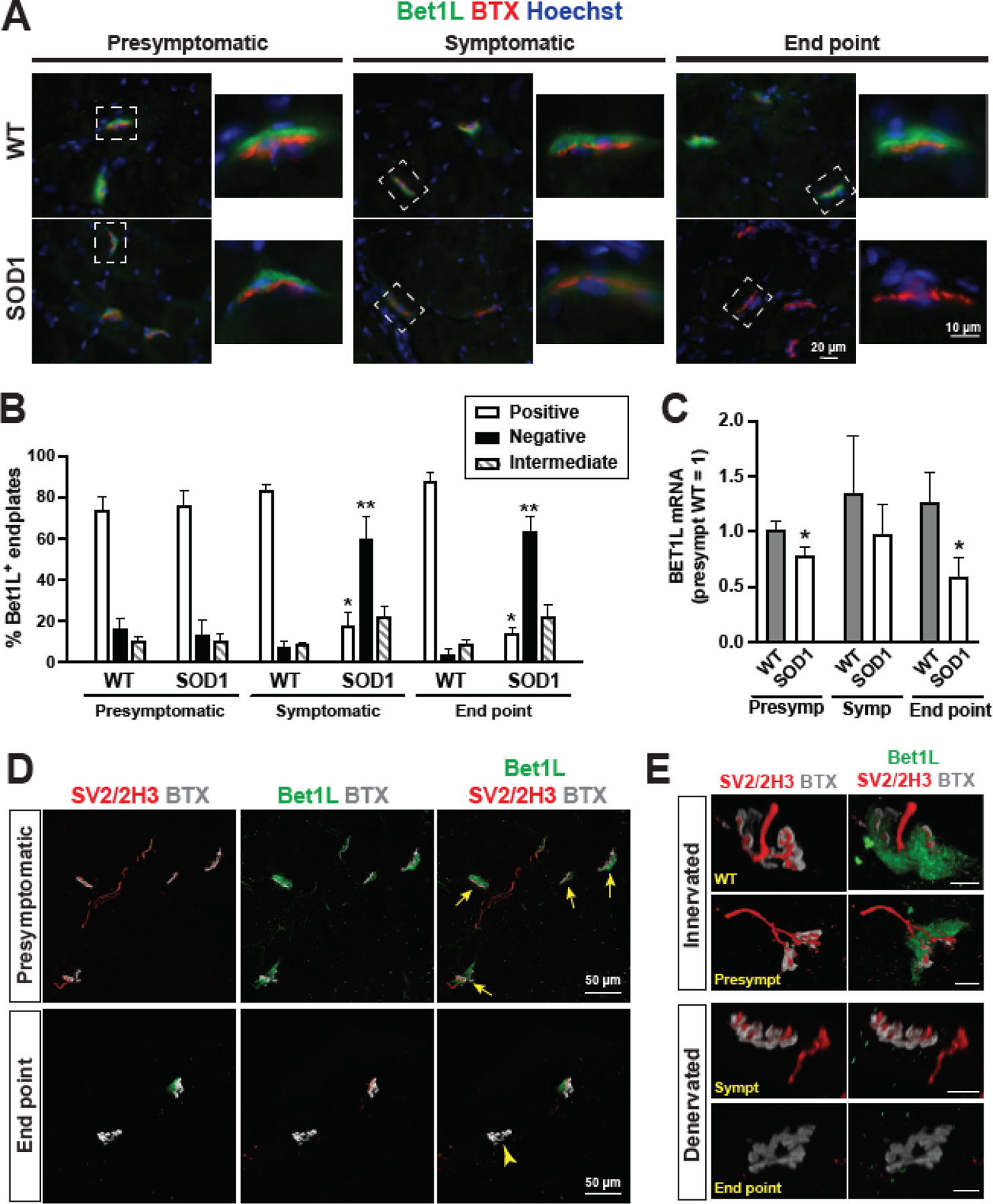
(A) Immunohistochemistry in cross-sections of SOD1G93A tibialis anterior muscles showed that Bet1L (green) was localized to the NMJ, as indicated by BTX (red). Expression of Bet1L was decreased in symptomatic and end point SOD1G93A rats compared to presymptomatic and WT controls. (B) Immunostained muscle sections were counted for Bet1L-positive, intermediate, or negative NMJs at each time point according to representative images in (A). By the symptomatic time point, the amount of Bet1L-positive NMJs was significantly reduced. Muscle sections from 8 WT and 8 SOD1G93A rats were counted at each time point, with an average of 81 NMJs counted per animal. *P<0.05 vs. other positive groups. **P<0.05 vs. other negative groups. (C) Tibialis anterior muscles were harvested at presymptomatic, symptomatic, and end point disease stages and homogenized to collect RNA for RT-qPCR to detect Bet1L gene expression. Results were normalized to the mean value of the WT presymptomatic rats. N = 8 rats per time point (4 male and 4 female). *P<0.05 vs. counterpart WT, according to unpaired Student’s t test. (D) Immunohistochemistry for Bet1L co-localization with axonal markers SV2 and 2H3 for comparison of Bet1L expression at innervated NMJs (yellow arrows) in presymptomatic rats and denervated NMJs (a yellow arrow head) in end stage SOD1G93A rats. (E) Representative images of Bet1L staining at innervated and denervated NMJs along disease progression compared to a WT control NMJ. Scale bars = 10 μm.
Among these molecules, we are specifically interested in the expression and distribution of Bet1L protein in the muscle of SOD1G93A rats. Immunohistochemistry showed that Bet1L was strongly localized to the NMJ and the localization was primarily on the presynaptic side (Fig. 3A, Supplemental Fig. 3A). Although, it appears that there could also be some Bet1L on the post-synaptic muscle membrane. Notably, we found that Bet1L protein expression at the NMJ was decreased in symptomatic and end point ALS rats. To quantify this, cohorts of 8 SOD1G93A rats and 8 age-matched WT controls (4 male and 4 female each) were sacrificed at presymptomatic, symptomatic, and end point stages. The hind limb muscles were sectioned and immunostained for Bet1L and BTX. The muscle sections were then scanned and NMJs were counted as either positive for Bet1L (similar to the presymptomatic example images in Figure 3A), intermediate Bet1L expression (similar to the symptomatic SOD1G93A image in Figure 3A), or no Bet1L expression (similar to the end point SOD1G93A image in Figure 3A). We found that the amount of Bet1L protein at the NMJ was significantly decreased in symptomatic and end point ALS rats compared to presymptomatic rats and age-matched WT controls (Fig. 3B, P<0.01). There was a dramatic decrease in the number of Bet1L-positive NMJs of SOD1G93A rats between the presymptomatic and symptomatic stages, and the number of Bet1L-positive NMJs remained low at end point. To determine whether sex differences in the ALS rat model may influence the loss of Bet1L protein expression, we compared the Bet1L expression levels at the NMJs of 4 male and 4 female rats from each group and observed the same patterns of expression loss (Supplemental Fig. 4). As skeletal muscle sections often exhibit high background or non-specific signals, we stained the sections following antibody blocking to confirm the specificity of the Bet1L antibody (Supplemental Fig. 5).
In order to see if the four genes of interest were downregulated transcriptionally in the SOD1G93A rats, TA muscles were homogenized and total RNA was extracted for RT-qPCR. Of the four genes of interest, only Bet1L gene showed a clear decrease in SOD1G93A rats compared to non-transgenic wild type (WT) controls. The difference was statistically significant (P<0.05) in presymptomatic and end point rats (Fig. 3C, Supplemental Fig. 3B and C). The combined RT-qPCR and immunohistochemistry quantification suggest that loss of Bet1L expression may begin early in the disease process.
We next investigated the relationship between loss of Bet1L protein and NMJ denervation. Our previous study indicated that NMJ denervation initiates in presymptomatic SOD1G93A rats and almost all NMJs become denervated in end point rats26. Immunostaining for Bet1L with synaptic marker SV2 and neurofilament marker NF-M (clone 2H3) in symptomatic SOD1G93A rats shows loss of Bet1L in some cases prior to loss of innervation indicating that these events were occurring close in time, although further experiments would be required to determine their relationship (Fig. 3D–E).
Bet1L is not expressed in motor neuron axons, terminal Schwann cells, or kranocytes
In order to determine if a specific cell type was expressing Bet1L at NMJs, we used immunohistochemistry on the TA muscle sections of WT rats. Based on the apparent presynaptic localization of Bet1L, we initially hypothesized that it was expressed in presynaptic axon terminals. However, co-staining with synaptic terminal and motor axons (SV2 and 2H3) and BTX indicated that Bet1L did not appear to be expressed within motor neuron axons and terminals (Fig. 4A). Given that Bet1L is known as a component of SNARE complexes30,31, we next checked for co-localization with SNAP-25 and Syntaxin 1, SNARE proteins involved in synaptic vesicle exocytosis37. The expression of these markers did not overlap with Bet1L either (Fig. 4B and C). We also considered whether Bet1L could be expressed by terminal Schwann cells, a non-myelinating glial cell important for NMJ development and maintenance38,39. However, immunostaining with Schwann cell marker S100 and terminal Schwann cell marker NG2 showed that Bet1L was closely localized, but not co-expressed (Fig. 4D–E). A rotating 3-dimensional view of Bet1L expression relative to S100 can be seen in Supplemental Video 1. Based on the cap-like shape of Bet1L expression, we next hypothesized that Bet1L could be expressed by kranocytes, another type of NMJ-specific glial cell that is sometimes referred to as NMJ-capping cells. The role of kranocytes at the NMJ has not been well characterized yet, but one study showed that they become activated upon axonal injury and facilitate nerve sprouting before terminal Schwann cells become activated40. To test if Bet1L could be expressed in kranocytes, we co-stained for Bet1L and CD34, a kranocyte marker (Fig. 4F). However, again Bet1L did not seem to co-localize with CD34. Based on these results, it did not seem that a specific cell type was expressing Bet1L at the NMJ.
Figure 4. Characterizing Bet1L expression at the NMJ.
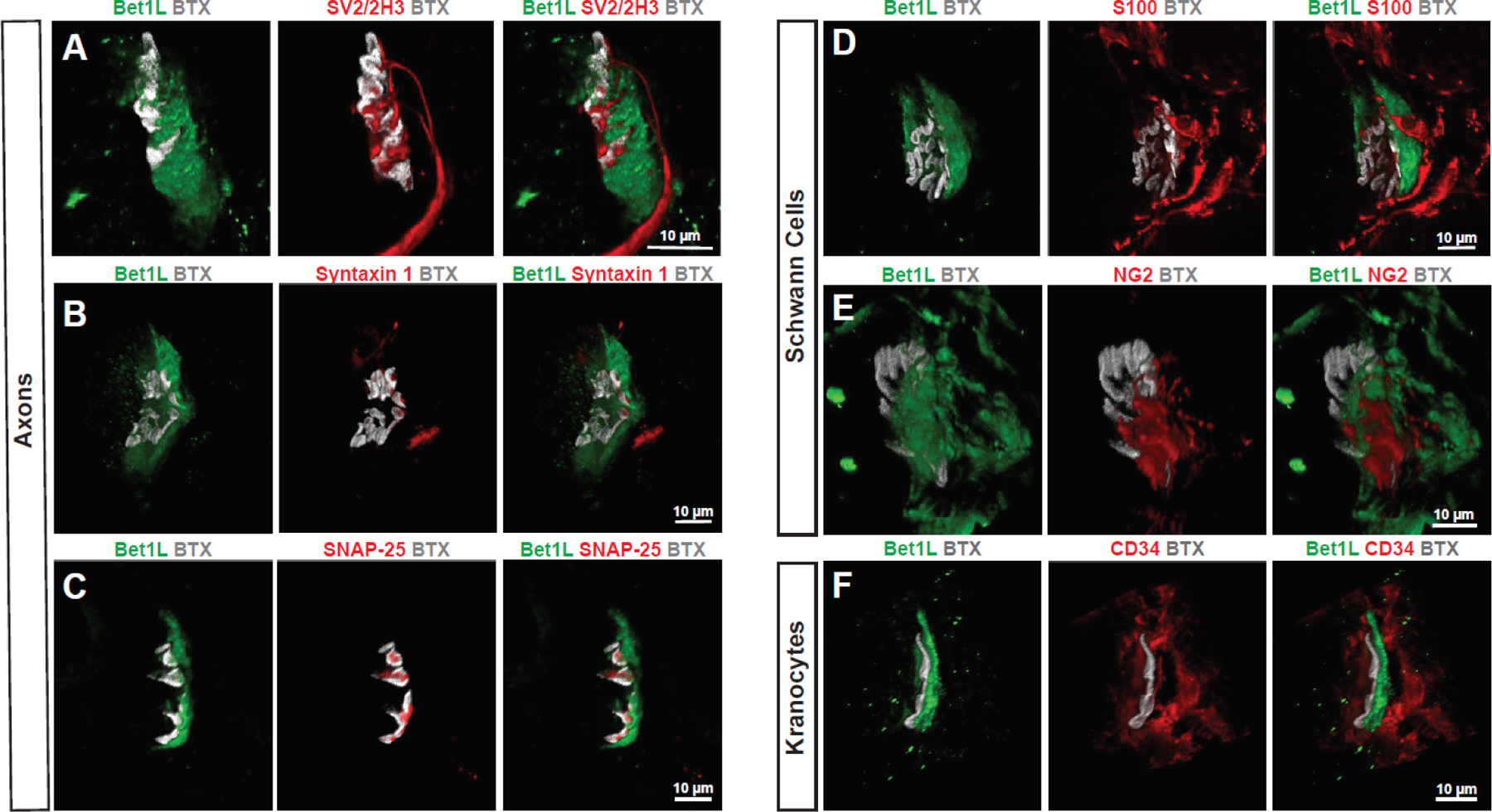
Confocal z-stack images of Bet1L (green) at WT rat NMJs as indicated by BTX labeling of acetylcholine receptors (grey). Bet1L expression did not colocalize with axonal markers (A), synaptic SNARE proteins (B-C), terminal Schwann cells (D-E), or kranocytes (F).
Bet1L is localized to the basal lamina of the NMJ
Bet1L did not appear to be expressed in any of the common cell types of the NMJ and was primarily located between the post-synaptic membrane and the motor neuron axons. Therefore, we next hypothesized that Bet1L could be a protein that is secreted into the basal lamina, the extracellular matrix of the NMJ41. To address this possibility, we compared Bet1L localization with Laminin, a known component of the NMJ basal lamina. While not a direct co-stain, the localization of Laminin in relation to BTX appears very similar to Bet1L (Fig. 5A). We then investigated whether Bet1L interacts with Dystrophin and Utrophin, members of the dystroglycan complex which connects the skeletal muscle membrane with the basal lamina42,43. While there was some potential overlap, Bet1L seemed to be primarily peripheral to these proteins (Fig. 5B). However, a co-stain with Collagen IV, a major protein component to the basal lamina44 did show overlap with Bet1L, supporting our hypothesis that Bet1L is a component of the basal lamina (Fig. 5C).
Figure 5. Bet1L expression indicates a possible muscle-secreted component of the basal lamina.
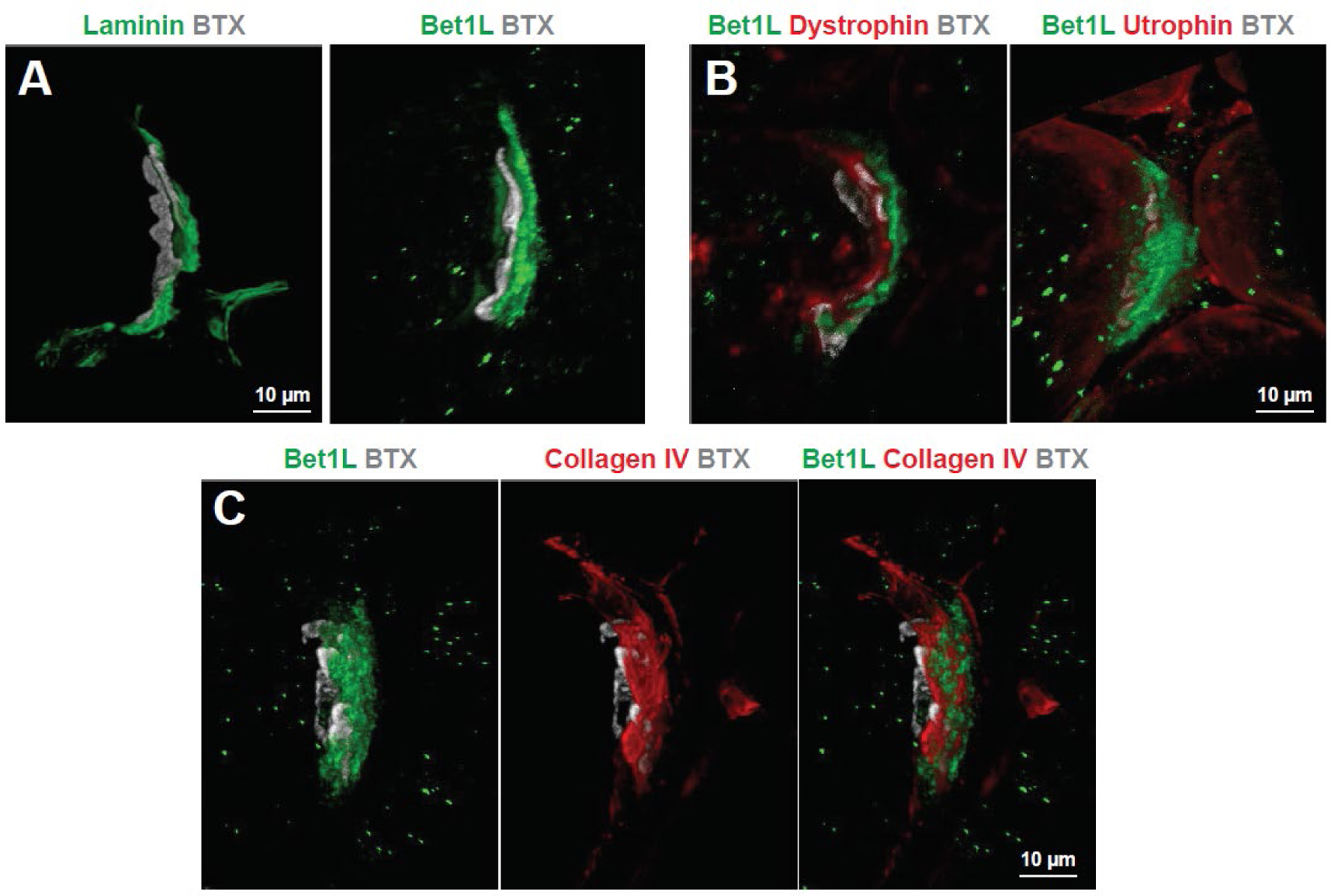
(A) Laminin staining (green) at the NMJ of a WT rat shows similar localization as Bet1L in relation to acetylcholine receptors (grey). Based on this, we further investigated the localization of Bet1L at the NMJ in relation to components of the dystroglycan complex (B), and the basal lamina protein Collagen IV (C). While there was some possible overlap, Bet1L seemed mostly peripheral to Dystrophin and Utrophin. However, it appeared to be closely expressed with collagen IV suggesting that Bet1L is a component of the synaptic basal lamina.
At first it seemed that there were conflicting results as to the localization of Bet1L in vitro versus in vivo. While human iPSC-derived myocytes expressed Bet1L intracellularly (Fig. 2B), Bet1L was primarily localized to the extracellular basal lamina of SOD1G93A rat muscles (Fig. 5C). However, it was noted that although young iPSC-derived myocytes primarily expressed Bet1L adjacent to the nuclei, older myocytes showed a migration of Bet1L to the periphery (Fig. 2C). Together, this in vitro data may also support a possibility that Bet1L could be produced in skeletal muscle and secreted into the basal lamina.
Discussion
In this study, we differentiated iPSCs from ALS patients with C9ORF72, TARDBP, SOD1, and sporadic backgrounds into skeletal myocytes and analyzed their gene expression using RNA sequencing. We found that all ALS lines had downregulation of four specific genes: BET1L, DCX, GPC3, and HNRNPK. Interestingly, all four genes were categorized by gene ontology as being related to vesicle-mediated transport. Defects in vesicle transport have been increasingly linked to many different ALS-causing mutations45, including a recent RNA sequencing which revealed that patients with the C9ORF72 repeat expansion had decreased expression of genes related to ER-Golgi transport, vacuolar transport, vesicle-mediated transport, and lysosomes46. RNA sequencing of additional patient cell lines should be completed to further support our results and see if they can be expanded to other forms of familial ALS, such as FUS, SQSTM1, TBK1, and OPTN1 among others1. The project minE ALS Sequencing Consortium will reveal more commonalities in the near future as they continue to sequence large numbers of ALS patients and controls47.
When investigating disease mechanisms, a combination of both human cells and animal models is often required. Animal models are useful to study aspects of pathology that are difficult to observe in human patients such as NMJ degeneration. However, physiological differences between humans and rodent models should be considered. In this case, of the four down-regulated genes identified in iPSC-derived skeletal myocytes, BET1L was the only gene that was also down-regulated in skeletal muscle samples from a rat model of familial ALS (SOD1G93A transgenic). Notably, immunohistochemistry for Bet1L in muscle cross-sections showed that Bet1L proteins were strongly localized to the NMJ and had decreased expression over the disease course in SOD1G93A but not WT rats. Furthermore, the loss of Bet1L at the NMJ of SOD1G93A rats appeared to begin early in the disease process. We found a decrease in Bet1L gene expression by RT-qPCR in presymptomatic rats compared to WT, and immunostaining showed a dramatic decrease in the number of Bet1L-positive NMJs in symptomatic ALS rats. The discrepancy between the dramatic loss of Bet1L protein at the NMJ via immunohistochemistry and the more modest decrease via RT-qPCR could be explained by possible Bet1L expression in other regions of muscle tissue, as the quantification of Bet1L immunohistochemistry focused specifically on the NMJs but the qPCR was performed from whole-tissue lysate. While we cannot say definitively whether Bet1L loss comes before or after NMJ denervation, the two phenomena occurred around the same timeline in SOD1G93A rats26. Therefore, Bet1L loss should be studied more closely as relevant to the NMJ degeneration process. To supplement the immunohistochemical data, several antibodies for Bet1L had been tested for western blot analysis but this still requires further optimization as a future direction.
The data from our rat studies are consistent with other rodent and human studies which have found early changes to the NMJ7,10–12. In order to investigate the importance of Bet1L expression at the NMJ, several future directions would be helpful. First, rodent knock out models of Bet1L could be used to confirm if loss of Bet1L protein is sufficient to cause denervation in WT rodents, or if it would increase disease severity in ALS models. It would then be interesting to see if Bet1L loss is more common at specific fiber types and if increasing Bet1L expression could reverse the ALS phenotype in SOD1G93A rats. Furthermore, use of acute injury models like sciatic nerve damage48 will be helpful to determine whether Bet1L loss is specific to the ALS disease process.
Bet1L protein is commonly associated with SNARE proteins and Golgi-related vesicle transport29–31 but its specific function in skeletal muscle has not yet been determined. In this study, we found some differences in the localization of Bet1L in iPSC-derived myocytes compared to the SOD1G93A rat model. At early time points in iPSC-derived skeletal myocytes, the localization of Bet1L seemed in line with the role of Bet1L in vesicle transport between the ER and Golgi. However, at later time points Bet1L expression seemed to be localized to the periphery of the myocytes. This change in localization is similar to that of acetylcholinesterase, a muscle-secreted enzyme that regulates neurotransmission through hydrolysis of excess acetylcholine at the NMJ49. In cultured myocytes, acetylcholinesterase is assembled in the Golgi apparatus and then becomes localized to the muscle membrane around the time point that myocytes begin to spontaneously contract50,51. Bet1L expression in the periphery was noted in myocytes around 4–6 weeks old, which correlates with the onset of spontaneous contractions in our iPSC-derived myocytes9,23,52. However, we did not observe any significant differences in contraction dynamics between control and ALS lines. Thus, it is unlikely that Bet1L has a significant role in muscle contraction. An interesting future direction would be to co-culture iPSC-derived skeletal myocytes with motor neurons in order to see how Bet1L is localized in the presence of motor neurons, and if it is required for the formation and maintenance of NMJs in vitro.
In rats, Bet1L appeared to have a pre-synaptic localization but was not expressed in motor neuron axons, terminal Schwann cells, or kranocytes. Bet1L seemed to be localized very close to acetylcholine receptor-positive endplates, often closer to the synapse than terminal Schwann cells and even approaching axons. We found that Bet1L expression overlapped with the basal lamina protein Collagen IV, leading to the hypothesis that Bet1L is a secreted component of the NMJ basal lamina. Further studies such as immuno-electron microscopy and protein immunoprecipitation would be helpful to determine whether skeletal muscle secretes Bet1L, and to identify how Bet1L proteins interact with other components of the NMJ basal lamina.
In addition to studying the role of Bet1L in human myocytes in vitro, an important future direction is to confirm whether Bet1L is localized to the NMJ in humans and if its expression level changes in ALS patients. Expression of the Bet1L gene has been confirmed in human skeletal muscle according to GTEx portal (accession number phs000424.v8.p2), but its specific role in skeletal muscle is unknown and has not yet been studied in the context of ALS patient tissue. Staining for Bet1L protein in human ALS patient skeletal muscle would confirm our results as well as validate the use of iPSC-derived skeletal myocytes for modeling ALS skeletal muscle pathology. To see if loss of Bet1L at the NMJ is specific to ALS, we plan to use blinded human muscle samples that are from additional neuromuscular diseases in addition to ALS and healthy controls.
While this paper mainly explored BET1L expression, future work could also investigate the role of DCX, GPC3, and HNRNPK in ALS disease progression. Despite not showing downregulation in the SOD1G93A rats, they could still be of interest to test on human muscle samples as there are many physiological differences between humans and rat models53. Doublecortin is expressed in developing mouse skeletal muscle and is important for myofiber maturation34, as well as embryonic NMJ development54. The role of Glypican 3 has not been explored in skeletal muscle or at the NMJ, but it has been shown to interact with the glucose transporter GLUT4 in mouse adipocytes as well as in vitro with GLUT4 purified from human HEK293 cells55. It would be interesting to see if Glypican 3 also interacts with GLUT4 in skeletal muscle, as glucose metabolism is often affected in ALS56. Finally, down-regulation of HNRNPK may influence vesicle-mediated transport through changes in the regulation of other vesicle transport-related genes. For example, hnRNP K protein binds to and affects the processing of RNA important for the organization and function of the axonal cytoskeleton57. HNRNPK has interesting ties to familial ALS, including TDP-43 and C9ORF72-related pathology. The presence of hnRNP K in stress granules is required for the recruitment of TDP-43 which leads to the formation of cytosolic TDP-43 aggregates58. Several types of hnRNP proteins including hnRNP K have been shown to interact with repeat RNA foci in ALS patients with the C9ORF72 mutation59–61. Interestingly, from immunostaining of SOD1G93A rats, we found that the levels of hnRNP K expression varied between nuclei in the muscle tissue. One future direction could be to compare expression levels between the different cell types present.
Conclusions
Based on the results of this study, Bet1L seems to be a component of the NMJ basal lamina that is decreased with ALS disease progression. Importantly, immunostaining for Bet1L at the NMJ of symptomatic rats showed that Bet1L expression was decreased early in the disease process, concurrently with NMJ denervation. This makes Bet1L of great interest for understanding the process of NMJ degeneration in ALS patients. Future directions will work to confirm if Bet1L is also decreased in ALS patient muscle tissue and if preventing loss of Bet1L can help preserve NMJ innervation.
Supplementary Material
Highlights:
Four common genes were downregulated in human myocytes across ALS backgrounds
Bet1L is decreased with disease progression in a rat model of familial ALS
Bet1L is localized to the basal lamina of rat neuromuscular junctions
Acknowledgements
We would like to thank Dr. Hiroshi Nishimune (Tokyo Metropolitan Institute of Gerontology, Tokyo Japan) for lending his expertise on NMJ structure. The monoclonal antibodies for Pax7 (developed by A. Kawakami at University of Tokyo), myogenin (F5D, developed by W.E. Wright at University of Texas Southwestern Medical Center), and MHC (MF-20, developed by D.A. Fischman at Weill Cornell Medical College) were obtained from the Developmental Studies Hybridoma Bank, created by the NICHD of the NIH and maintained at The University of Iowa, Department of Biology, Iowa City, IA 52242.
Funding
This work was supported by grants from the Amyotrophic Lateral Sclerosis Association (15-IIP-201, M.S.), National Institutes of Health (R01NS091540 and R01AR077191, M.S.), the University of Wisconsin Foundation (M.S.), and UW Stem Cell & Regenerative Medicine Center (M.S. and E.L.)
Footnotes
Declarations of interest
The authors declare no conflicts of interest.
References
- 1.Corcia P, Couratier P, Blasco H, et al. Genetics of amyotrophic lateral sclerosis. Rev Neurol (Paris). 2017;173(5):254–262. [DOI] [PubMed] [Google Scholar]
- 2.Brown RH, Al-Chalabi A. Amyotrophic Lateral Sclerosis. N Engl J Med. 2017;377(16):1602. [DOI] [PubMed] [Google Scholar]
- 3.Sawada H. Clinical efficacy of edaravone for the treatment of amyotrophic lateral sclerosis. Expert Opin Pharmacother. 2017;18(7):735–738. [DOI] [PubMed] [Google Scholar]
- 4.Cetin H, Rath J, Fuzi J, et al. Epidemiology of amyotrophic lateral sclerosis and effect of riluzole on disease course. Neuroepidemiology. 2015;44(1):6–15. [DOI] [PubMed] [Google Scholar]
- 5.Filipi T, Hermanova Z, Tureckova J, Vanatko O, Anderova AM. Glial Cells-The Strategic Targets in Amyotrophic Lateral Sclerosis Treatment. J Clin Med. 2020;9(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hovden H, Frederiksen JL, Pedersen SW. Immune system alterations in amyotrophic lateral sclerosis. Acta Neurol Scand. 2013;128(5):287–296. [DOI] [PubMed] [Google Scholar]
- 7.Loeffler JP, Picchiarelli G, Dupuis L, Gonzalez De Aguilar JL. The Role of Skeletal Muscle in Amyotrophic Lateral Sclerosis. Brain Pathol. 2016;26(2):227–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pansarasa O, Rossi D, Berardinelli A, Cereda C. Amyotrophic lateral sclerosis and skeletal muscle: an update. Mol Neurobiol. 2014;49(2):984–990. [DOI] [PubMed] [Google Scholar]
- 9.Lynch E, Semrad T, Belsito VS, et al. C9ORF72-related cellular pathology in skeletal myocytes derived from ALS-patient induced pluripotent stem cells. Dis Model Mech. 2019;12(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fischer LR, Culver DG, Tennant P, et al. Amyotrophic lateral sclerosis is a distal axonopathy: evidence in mice and man. Exp Neurol. 2004;185(2):232–240. [DOI] [PubMed] [Google Scholar]
- 11.Moloney EB, de Winter F, Verhaagen J. ALS as a distal axonopathy: molecular mechanisms affecting neuromuscular junction stability in the presymptomatic stages of the disease. Front Neurosci. 2014;8:252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Krakora D, Macrander C, Suzuki M. Neuromuscular junction protection for the potential treatment of amyotrophic lateral sclerosis. Neurol Res Int. 2012;2012:379657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yu J, Vodyanik MA, Smuga-Otto K, et al. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318(5858):1917–1920. [DOI] [PubMed] [Google Scholar]
- 14.Takahashi K, Tanabe K, Ohnuki M, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131(5):861–872. [DOI] [PubMed] [Google Scholar]
- 15.Liu C, Oikonomopoulos A, Sayed N, Wu JC. Modeling human diseases with induced pluripotent stem cells: from 2D to 3D and beyond. Development. 2018;145(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Suzuki M, Tork C, Shelley B, et al. Sexual dimorphism in disease onset and progression of a rat model of ALS. Amyotroph Lateral Scler. 2007;8(1):20–25. [DOI] [PubMed] [Google Scholar]
- 17.Howland DS, Liu J, She Y, et al. Focal loss of the glutamate transporter EAAT2 in a transgenic rat model of SOD1 mutant-mediated amyotrophic lateral sclerosis (ALS). Proc Natl Acad Sci U S A. 2002;99(3):1604–1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Van Dyke JM, Smit-Oistad IM, Macrander C, Krakora D, Meyer MG, Suzuki M. Macrophage-mediated inflammation and glial response in the skeletal muscle of a rat model of familial amyotrophic lateral sclerosis (ALS). Exp Neurol. 2016;277:275–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Krakora D, Mulcrone P, Meyer M, et al. Synergistic effects of GDNF and VEGF on lifespan and disease progression in a familial ALS rat model. Mol Ther. 2013;21(8):1602–1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ludwig TE, Bergendahl V, Levenstein ME, Yu J, Probasco MD, Thomson JA. Feeder-independent culture of human embryonic stem cells. Nat Methods. 2006;3(8):637–646. [DOI] [PubMed] [Google Scholar]
- 21.Ludwig TE, Levenstein ME, Jones JM, et al. Derivation of human embryonic stem cells in defined conditions. Nat Biotechnol. 2006;24(2):185–187. [DOI] [PubMed] [Google Scholar]
- 22.Hosoyama T, McGivern JV, Van Dyke JM, Ebert AD, Suzuki M. Derivation of myogenic progenitors directly from human pluripotent stem cells using a sphere-based culture. Stem Cells Transl Med. 2014;3(5):564–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jiwlawat S, Lynch E, Glaser J, et al. Differentiation and sarcomere formation in skeletal myocytes directly prepared from human induced pluripotent stem cells using a sphere-based culture. Differentiation. 2017;96:70–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Klein SM, Behrstock S, McHugh J, et al. GDNF delivery using human neural progenitor cells in a rat model of ALS. Hum Gene Ther. 2005;16(4):509–521. [DOI] [PubMed] [Google Scholar]
- 25.Suzuki M, McHugh J, Tork C, et al. Direct muscle delivery of GDNF with human mesenchymal stem cells improves motor neuron survival and function in a rat model of familial ALS. Mol Ther. 2008;16(12):2002–2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Suzuki M, McHugh J, Tork C, et al. GDNF secreting human neural progenitor cells protect dying motor neurons, but not their projection to muscle, in a rat model of familial ALS. PLoS One. 2007;2(8):e689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hayes-Punzo A, Mulcrone P, Meyer M, McHugh J, Svendsen CN, Suzuki M. Gonadectomy and dehydroepiandrosterone (DHEA) do not modulate disease progression in the G93A mutant SOD1 rat model of amyotrophic lateral sclerosis. Amyotroph Lateral Scler. 2012;13(3):311–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Basso DM, Beattie MS, Bresnahan JC. A sensitive and reliable locomotor rating scale for open field testing in rats. J Neurotrauma. 1995;12(1):1–21. [DOI] [PubMed] [Google Scholar]
- 29.Xu Y, Wong SH, Zhang T, Subramaniam VN, Hong W. GS15, a 15-kilodalton Golgi soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) homologous to rbet1. J Biol Chem. 1997;272(32):20162–20166. [DOI] [PubMed] [Google Scholar]
- 30.Xu Y, Martin S, James DE, Hong W. GS15 forms a SNARE complex with syntaxin 5, GS28, and Ykt6 and is implicated in traffic in the early cisternae of the Golgi apparatus. Mol Biol Cell. 2002;13(10):3493–3507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tai G, Lu L, Wang TL, et al. Participation of the syntaxin 5/Ykt6/GS28/GS15 SNARE complex in transport from the early/recycling endosome to the trans-Golgi network. Mol Biol Cell. 2004;15(9):4011–4022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Francis F, Koulakoff A, Boucher D, et al. Doublecortin is a developmentally regulated, microtubule-associated protein expressed in migrating and differentiating neurons. Neuron. 1999;23(2):247–256. [DOI] [PubMed] [Google Scholar]
- 33.Moores CA, Perderiset M, Kappeler C, et al. Distinct roles of doublecortin modulating the microtubule cytoskeleton. EMBO J. 2006;25(19):4448–4457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ogawa R, Ma Y, Yamaguchi M, et al. Doublecortin marks a new population of transiently amplifying muscle progenitor cells and is required for myofiber maturation during skeletal muscle regeneration. Development. 2015;142(4):810. [DOI] [PubMed] [Google Scholar]
- 35.Montalbano M, Rastellini C, McGuire JT, et al. Role of Glypican-3 in the growth, migration and invasion of primary hepatocytes isolated from patients with hepatocellular carcinoma. Cell Oncol (Dordr). 2018;41(2):169–184. [DOI] [PubMed] [Google Scholar]
- 36.Wang Z, Qiu H, He J, et al. The emerging roles of hnRNPK. J Cell Physiol. 2020;235(3):1995–2008. [DOI] [PubMed] [Google Scholar]
- 37.Sollner T, Whiteheart SW, Brunner M, et al. SNAP receptors implicated in vesicle targeting and fusion. Nature. 1993;362(6418):318–324. [DOI] [PubMed] [Google Scholar]
- 38.Santosa KB, Keane AM, Jablonka-Shariff A, Vannucci B, Snyder-Warwick AK. Clinical relevance of terminal Schwann cells: An overlooked component of the neuromuscular junction. J Neurosci Res. 2018;96(7):1125–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Castro R, Taetzsch T, Vaughan SK, et al. Specific labeling of synaptic schwann cells reveals unique cellular and molecular features. Elife. 2020;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Court FA, Gillingwater TH, Melrose S, et al. Identity, developmental restriction and reactivity of extralaminar cells capping mammalian neuromuscular junctions. J Cell Sci. 2008;121(Pt 23):3901–3911. [DOI] [PubMed] [Google Scholar]
- 41.Patton BL. Basal lamina and the organization of neuromuscular synapses. J Neurocytol. 2003;32(5–8):883–903. [DOI] [PubMed] [Google Scholar]
- 42.Blake DJ, Tinsley JM, Davies KE. Utrophin: a structural and functional comparison to dystrophin. Brain Pathol. 1996;6(1):37–47. [DOI] [PubMed] [Google Scholar]
- 43.Jacobson C, Cote PD, Rossi SG, Rotundo RL, Carbonetto S. The dystroglycan complex is necessary for stabilization of acetylcholine receptor clusters at neuromuscular junctions and formation of the synaptic basement membrane. J Cell Biol. 2001;152(3):435–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sanes JR. The basement membrane/basal lamina of skeletal muscle. J Biol Chem. 2003;278(15):12601–12604. [DOI] [PubMed] [Google Scholar]
- 45.Burk K, Pasterkamp RJ. Disrupted neuronal trafficking in amyotrophic lateral sclerosis. Acta Neuropathol. 2019;137(6):859–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dickson DW, Baker MC, Jackson JL, et al. Extensive transcriptomic study emphasizes importance of vesicular transport in C9orf72 expansion carriers. Acta Neuropathol Commun. 2019;7(1):150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.van der Spek RAA, van Rheenen W, Pulit SL, et al. The project MinE databrowser: bringing large-scale whole-genome sequencing in ALS to researchers and the public. Amyotroph Lateral Scler Frontotemporal Degener. 2019;20(5–6):432–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Geuna S. The sciatic nerve injury model in pre-clinical research. J Neurosci Methods. 2015;243:39–46. [DOI] [PubMed] [Google Scholar]
- 49.Rotundo RL, Rossi SG, Kimbell LM, Ruiz C, Marrero E. Targeting acetylcholinesterase to the neuromuscular synapse. Chem Biol Interact. 2005;157–158:15–21. [DOI] [PubMed] [Google Scholar]
- 50.Rossi SG, Rotundo RL. Transient interactions between collagen-tailed acetylcholinesterase and sulfated proteoglycans prior to immobilization on the extracellular matrix. J Biol Chem. 1996;271(4):1979–1987. [DOI] [PubMed] [Google Scholar]
- 51.Rossi SG, Rotundo RL. Cell surface acetylcholinesterase molecules on multinucleated myotubes are clustered over the nucleus of origin. J Cell Biol. 1992;119(6):1657–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jiwlawat N, Lynch EM, Napiwocki BN, et al. Micropatterned substrates with physiological stiffness promote cell maturation and Pompe disease phenotype in human induced pluripotent stem cell-derived skeletal myocytes. Biotechnol Bioeng. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jones RA, Harrison C, Eaton SL, et al. Cellular and Molecular Anatomy of the Human Neuromuscular Junction. Cell Rep. 2017;21(9):2348–2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bourgeois F, Messeant J, Kordeli E, et al. A critical and previously unsuspected role for doublecortin at the neuromuscular junction in mouse and human. Neuromuscul Disord. 2015;25(6):461–473. [DOI] [PubMed] [Google Scholar]
- 55.Taguchi A, Emoto M, Okuya S, et al. Identification of Glypican3 as a novel GLUT4-binding protein. Biochem Biophys Res Commun. 2008;369(4):1204–1208. [DOI] [PubMed] [Google Scholar]
- 56.Kirk SE, Tracey TJ, Steyn FJ, Ngo ST. Biomarkers of Metabolism in Amyotrophic Lateral Sclerosis. Front Neurol. 2019;10:191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liu Y, Szaro BG. hnRNP K post-transcriptionally co-regulates multiple cytoskeletal genes needed for axonogenesis. Development. 2011;138(14):3079–3090. [DOI] [PubMed] [Google Scholar]
- 58.Moujalled D, James JL, Yang S, et al. Phosphorylation of hnRNP K by cyclin-dependent kinase 2 controls cytosolic accumulation of TDP-43. Hum Mol Genet. 2015;24(6):1655–1669. [DOI] [PubMed] [Google Scholar]
- 59.Zamiri B, Mirceta M, Bomsztyk K, Macgregor RB Jr., Pearson CE. Quadruplex formation by both G-rich and C-rich DNA strands of the C9orf72 (GGGGCC)8*(GGCCCC)8 repeat: effect of CpG methylation. Nucleic Acids Res. 2015;43(20):10055–10064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cooper-Knock J, Higginbottom A, Stopford MJ, et al. Antisense RNA foci in the motor neurons of C9ORF72-ALS patients are associated with TDP-43 proteinopathy. Acta Neuropathol. 2015;130(1):63–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Farg MA, Sundaramoorthy V, Sultana JM, et al. C9ORF72, implicated in amytrophic lateral sclerosis and frontotemporal dementia, regulates endosomal trafficking. Hum Mol Genet. 2014;23(13):3579–3595. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.