

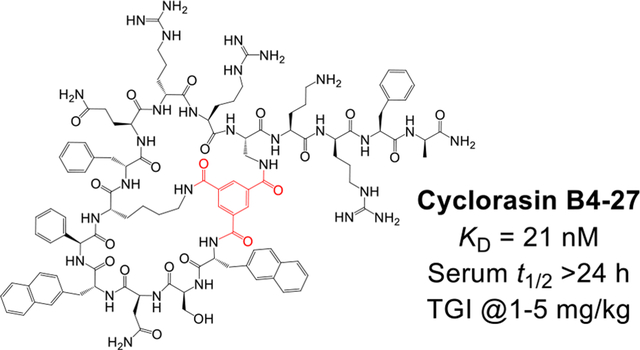
Abstract
The Ras subfamily of small GTPases is mutated in ~30% human cancers and represents compelling yet challenging anticancer drug targets owing to their flat protein surface. We previously reported a bicyclic peptidyl inhibitor, cyclorasin B3, which binds selectively to Ras-GTP with modest affinity and blocks its interaction with downstream effector proteins in vitro but lacks cell permeability or biological activity. In this study, optimization of B3 yielded a potent pan-Ras inhibitor, cyclorasin B4-27, which binds selectively to the GTP-bound forms of wild-type and mutant Ras isoforms (KD = 21 nM for KRasG12V-GppNHp) and is highly cell-permeable and metabolically stable (serum t1/2 > 24 h). B4-27 inhibits Ras signaling in vitro and in vivo by blocking Ras from interacting with downstream effector proteins and induces apoptosis of Ras mutant cancer cells. When administered systemically (i.v.), B4-27 suppressed tumor growth in two different mouse xenograft models at 1–5 mg/kg daily doses.
Keywords: Anticancer agent, bicyclic peptide, intracellular biologics, protein-protein interaction, Ras inhibitor
Graphical Abstract
INTRODUCTION
The Ras subfamily of small GTPases consists of four isoforms, HRas, NRas, KRas4A and KRas4B.1 The four isoforms share 100% sequence identity in the effector lobe (residues 186), which engages effector proteins via their Switch I and Switch II motifs, and ~86% sequence similarity within the allosteric lobe (residues 87–172) but diverge in the C-terminal hypervariable region (HVR). All isoforms possess a C-terminal CaaX motif which is farnesylated, leading to Ras accumulation on the plasma membrane.2 KRas4A and 4B also contain a polylysine stretch that interacts with the negatively charged phospholipids on the inner leaflet of the plasma membrane. The different C-terminal structures of the four isoforms result in differential membrane association and isoform-specific trafficking, impacting the biological activity of Ras isoforms.2,3
The Ras GTPases function as molecular switches during receptor signaling in mammalian cells and cycle between a GTP-bound “On” state and a GDP-bound “Off” state.4 Activation of Ras by an extracellular signal is mediated by guanine nucleotide exchange factors (GEFs), which enhance the rate of GDP dissociation by 104-fold, enabling the incorporation of GTP, as GTP is present inside the cell at a 10-fold higher concentration than GDP. The activated Ras proteins interact with downstream effector proteins, including Raf kinases and phosphoinositide 3-kinase (PI3K), and activate their signaling cascades.5 Subsequent hydrolysis of the bound GTP to GDP, which is accelerated by 103-fold in the presence of GTPase activating proteins (GAPs), inactivates the Ras proteins and terminates the signaling events.
Mutations of Ras active-site residues (e.g., Gly12, Gly13, and Gln61) frequently slow down the GTP hydrolysis by reducing the intrinsic GTPase activity of Ras proteins and/or inhibiting the binding of GAPs to Ras proteins.6 This results in an excessive Ras-GTP population, leading to uncontrolled cell proliferation and survival, which are hallmarks of cancers. Indeed, mutations in Ras (including KRas, HRas, and NRas mutations) are found in ~30% all human cancers, making Ras one of the most compelling anticancer drug targets.7 Unfortunately, Ras is also one of the most challenging targets, because it is intracellular and its surface has no major binding pocket for small molecules to bind. Nevertheless, small-molecule direct Ras inhibitors have recently been developed.8 Best results have, so far, been obtained with irreversible inhibitors that covalently modify (and inhibit) the KRas G12C mutant,9–12 with one of the inhibitors (Sotorasib) recently approved for treatment of non-small cell lung cancer (NSCLC) by the Food and Drug Administration. However, while the G12C-specific inhibitors validate direct inhibition of Ras as a viable approach to treating Ras mutant cancers, the covalent inhibition approach is unlikely to be effective for other Ras mutants (e.g., G12V, G12S, G12D, G13D, and Q61H).
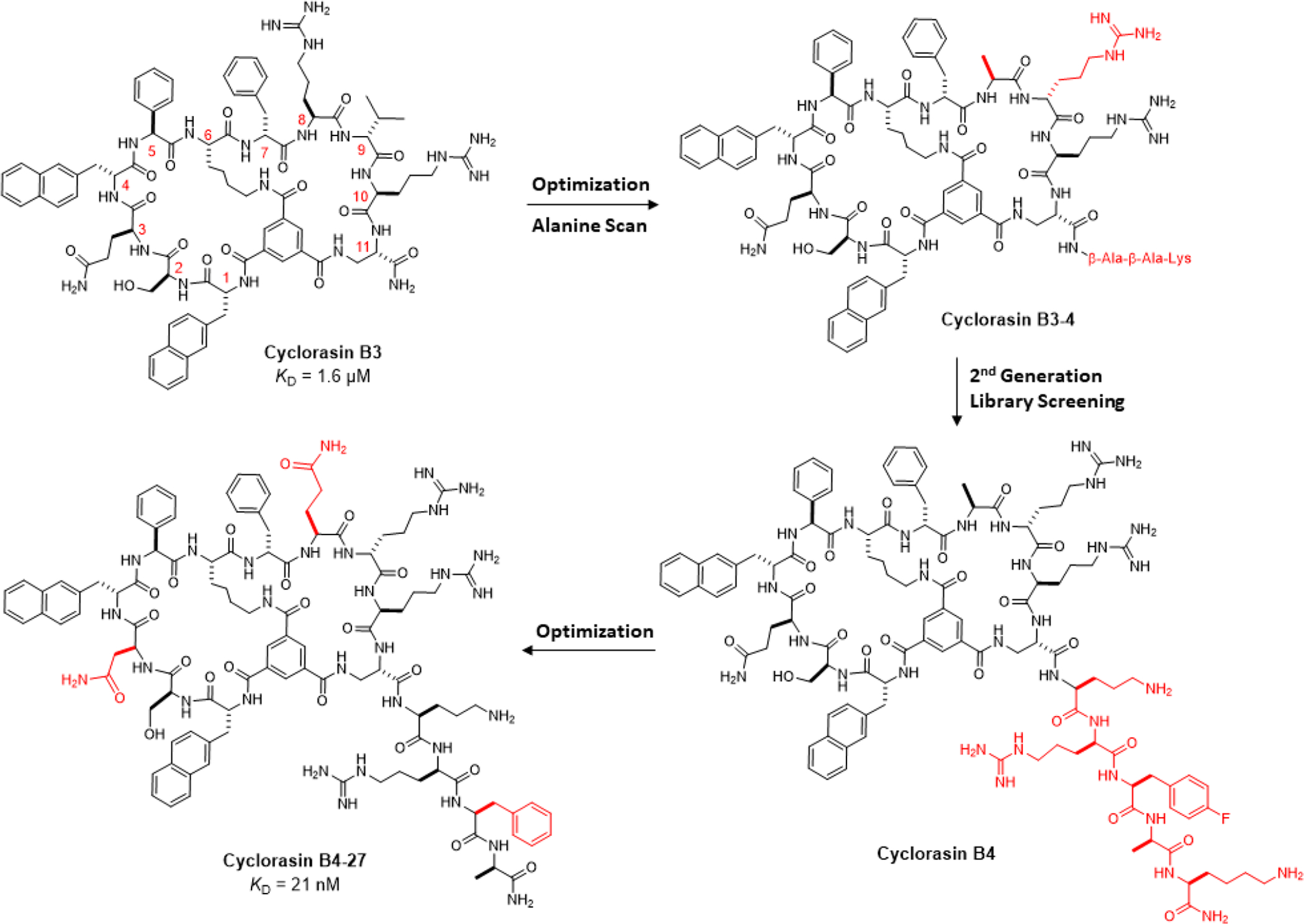
Because of their larger sizes, proteins and peptides are capable of recognizing flat binding sites and have been exploited as Ras inhibitors.13 For example, several antibodies,14–16 monobodies,17,18 and an engineered Ras-binding domain (RBD) of CRAF19 have been developed as potent and highly specific Ras inhibitors in vitro; however, intracellular delivery of these large biomolecules remains a major challenge.16 We and other investigators have explored macrocyclic peptides as Ras inhibitors. In one of the first examples of cyclic peptide inhibitors against Ras, Wu et al. screened a one bead-two compound (OBTC) library of rapamycin analogs and identified a monocyclic peptide, compound 12, which bound to KRasG12V with a KD of 0.83 μM and inhibited Ras-Raf interaction with an IC50 value of 0.70 μM.20 By screening a bicyclic peptide library, Upadhyaya et al. discovered several bicyclic peptidyl inhibitors against KRasG12V.21 One of the peptides, cyclorasin B3 (Scheme 1), bound to GTP- or GppNHp-bound KRas with KD values of 1.2 and 1.6 μM, respectively, and blocked the Ras-Raf interaction in vitro. Interestingly, cyclorasin B3 bound KRas-GDP with 8-fold lower affinity (KD = 9.3 μM). Sakamoto et al. identified from a phage-displayed library a highly potent cyclic peptide inhibitor of KRasG12D, KRpep-2d, which bound KRasG12D (KD = 1.6 nM) with 11- to 26-fold selectivity over KRasG12C (KD = 18 nM) and WT KRas (KD = 42 nM).22 More recently, Shokat and Suga groups reported a KRasG12D-selective cyclic peptide inhibitor derived from mRNA-displayed library screening.23 Unfortunately (though not surprisingly), these mono- and bicyclic peptides have poor membrane permeability and showed no to little cellular activity. To improve the cellular entry of cyclic peptide inhibitors, we previously took advantage of a cell-penetrating peptide (CPP)-like motif in compound 12, Arg-Arg-D-Nal-Arg-Fpa motif (where D-Nal is D-2-naphtylalanine and Fpa is L-4-fluorophenylalanine), and synthesized and screened a second-generation peptide library against KRasG12V.24 This effort resulted in a cell-permeable peptide, cyclorasin 9A5, which inhibited the Ras-Raf interaction in vitro and induced apoptosis in a panel of lung cancer cells by blocking the Ras signaling pathways (e.g., EC50 ≈ 3 μM against H1299 cells). In an alternative approach, we synthesized a library of cell-permeable bicyclic peptides by featuring a cyclic CPP motif in the first ring and degenerate peptide sequences in the other ring.25 Screening of the library against KRasG12V resulted in a bicyclic peptide inhibitor (peptide 49), which inhibited the Ras-Raf interaction with an IC50 value of 3.4 μM and reduced the viability of H1299 cells with an EC50 value of 8 μM. Recently, optimization of KRpep-2d generated a proteolytically stable and cell-permeable peptide KS-58 which bound KRasG12D with a KD value of 22 nM and inhibited Ras signaling and the proliferation of KRasG12Dmutant cancer cells (A427 and PANC-1).26 KS-58 inhibited the growth of PANC-1 xenografts in mice by 65% but required a high dose of 40 mg/kg. Finally, stapled and stabilized α-helical peptides have been designed to block the interaction between Ras and Son-of-Sevenless (SOS) and shown to reduce the viability of KRas mutant cell lines.27–29 Collectively, the above studies demonstrate that macrocyclic peptides can serve as potent and specific inhibitors of Ras proteins in vitro but require substantial improvement in cell permeability before being used as therapeutic agents against Ras mutant cancers.
Scheme 1.
Evolution of Ras Inhibitors with Structural Changes Highlighted in Red
In this study, we conducted a medicinal chemistry campaign on cyclorasin B321 to improve its potency as well as cell permeability. These efforts resulted in a potent and highly cell-permeable pan-Ras inhibitor, cyclorasin B4-27, which binds selectively to Ras-GTP (over Ras-GDP) blocking its interaction with effector proteins, induces apoptosis of Ras mutant cancer cells in vitro, and suppresses tumor growth in mouse xenograft models at low doses (≤5 mg/kg).
RESULTS AND DISCUSSION
Initial Optimization and Alanine Scan of Cyclorasin B3.
To improve the cell permeability of cyclorasin B3 (Scheme 1), we replaced the D-valine at position 9 with D-arginine to give B3-1 (Table S1). A flexible linker, β-Ala-β-Ala-Lys (BBK), was added to the C-terminus and the lysine side chain was labeled with fluorescein isothiocyanate (FITC). B3-1 showed modestly improved binding affinity to KRasG12V (KD = 0.58 μM) and importantly weak antiproliferative activity against lung cancer H1299 cells (EC50 = 21 μM). We next performed an “alanine scan” of B3-1, i.e., replacing each residue of B3-1 with alanine or D-alanine, to identify residues critical for Ras binding. Interestingly, substitution of alanine for arginine at position 8 produced a tighter binder in B3-4 (KD = 0.14 μM), while replacement of other residues with alanine (or D-alanine) either had no effect on Ras binding or reduced the Ras binding affinity (Table S1). B3-4 showed weak antiproliferative activity against a lung cancer cell line that carries a homozygous KRasG12C mutation (H358), with an EC50 value of 29 μM [by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay].
Design, Synthesis and Screening of 2nd-Generation Library.
A survey by Suga and colleagues revealed that potent macrocyclic peptide ligands against PPI targets have an average size of 15 ± 2 amino acids.30 Since B3-4 consists of only 11 residues (not counting the C-terminal BBK linker), we decided to extend the structure of B3-4 at the C-terminus by replacing the BBK linker with a library of peptide sequences and anticipated that a C-terminal “tail” of the proper sequence might engage in additional interactions with the Ras protein surface. On the other hand, enlargement of either ring of B3-4 might disrupt the overall interaction between the bicycle and the Ras protein surface. It would also make the rings more conformationally flexible losing some of the benefits of macrocyclization. To improve the proteolytic stability of the linear C-terminal peptide, we opted to construct the C-terminal peptide library with high contents of D- and other unnatural amino acids.
A 2nd generation peptide library was synthesized in the one bead-one compound (OBOC) format on TentaGel microbeads (90 μm; ~3 million beads/g; ~100 pmol peptide/bead) using the standard Fmoc/HATU chemistry (Figure S1). Library synthesis started with the installation of a common C-terminal linker sequence, β-Ala-β-Ala-Arg-Arg-Met, which permits peptide release by CNBr (after methionine) and ensures facile ionization during mass spectrometric analysis (arginines). A randomized peptide sequence of 1–4 residues (X1–X4) was then constructed by the split-and-pool synthesis method31 using the following 29 amino acids as building blocks: 7 proteinogenic L-amino acids (Gly, Ala, Ser, Ile, Asp, Gln, and His), 12 D-amino acids (D-Ala, D-Pro, D-Val, D-Thr, D-Leu, D-Asn, D-Lys, D-Glu, D-Phe, D-Arg, D-Tyr, and D-Trp), and 10 non-proteinogenic amino acids [β-Ala, D-β-homoAla, L-homoproline (Pip), cis-2-aminocyclopentylcarboxylic acid (cis-AcPc), aspartic acid α-tert-butyl ester (isoAsp), L-phenylglycine (Phg), D-2-naphthylalanine (D-Nal), L-4-fluorophenylalanine (Fpa), L-norleucine (Nle), and L-ornithine (Orn). Next, the peptide sequence of B3-4 was added. Finally, the peptides were selectively deprotected at the N-terminus (Fmoc group by piperidine) and the side chains of invariant lysine and L-2,4-diaminopropionic acid (Dap) residues within the B3-4 sequence [allyloxycarbonyl (Alloc) groups by Pd(PPh3)4] and cyclized by treatment with trimesic acid.32 The library has a theoretical diversity of 7.3 × 105 different sequences.
The library was screened for binding to biotinylated KRasG12V freshly loaded with GppNHp, a non-hydrolyzable GTP analog, in two rounds.33 In the first round of screening, the library beads were incubated with biotinylated KRas (150 nM) for 1 h, washed, and incubated with streptavidin (SA)coated magnetic beads (Dynabeads) (Figure S2a). Positive beads (~3000 beads) were isolated by magnetic sorting and subjected to a second round of screening by an enzyme-linked assay. Specifically, the ~3000 beads were again incubated with the biotinylated KRas protein (50 nM), washed, and incubated with SA-conjugated alkaline phosphatase (SA-AP). The beads were stained by the addition of 5-bromo-4-chloro-3’-indolyphosphate (BCIP) and positive beads developed turquoise color within 45 min (Figure S2b). The positive beads were isolated manually under a dissecting microscope and sorted into three groups according to their color intensity: “intensely”, “medium”, and “lightly” colored.
Screening of a total of 800 mg of the library (~2.4 × 106 beads; in two batches) produced 25 intensely colored, 60 medium colored, and 30 lightly colored beads. Sequencing analysis of the beads by mass spectrometry gave 69 unique complete sequences, including 17, 40, and 12 sequences in the “intensely”, “medium”, and “lightly” colored categories, respectively (Table S2). Inspection of the hit sequences did not reveal any obvious consensus sequence (Figure S3). We selected 17 hits for re-synthesis, because they contained: 1) positively charged residues which might improve cell permeability (e.g., D-Arg, D-Lys, and L-Orn); 2) aromatic residues which are known to enhance the cell permeability of CPPs and engage in hydrophobic interactions with protein surfaces (D-Nal, D-Phe, D-Tyr, D-Trp, L-Fpa); and 3) otherwise hydrophilic residues to improve the aqueous solubility (Table 1).
Table 1.
Sequences of Hits Derived from the 2ND Generation Library

| ||||||
|---|---|---|---|---|---|---|
| Peptide | Hit Sequence | HTRF IC50 (μM)a | MTT EC50 (μM)a | |||
| X4 | X3 | X2 | X1 | |||
| B3-4 | β-Ala | β-Ala | Lys | none | 4.4 ± 1.3 | 29 ± 6 |
| B3-4-1 | D-Tyr | Asp | Ser | D-Val | NA | NT |
| B3-4-2 | D-Glu | His | D-Pro | D-Tyr | 5.8 ± 1.1 | NA |
| B3-4-3 | D-Thr | Ser | D-Pro | Pip | 8.0 ± 1.0 | NT |
| B3-4-4 (B4) | Orn | D-Arg | Fpa | D-Ala | 1.7 ± 0.2 | 17 ± 4 |
| B3-4-5 | D-Thr | His | D-Glu | D-Arg | 6.9 ± 2.1 | NT |
| B3-4-6 | Ser | Gln | Ile | D-Arg | 9.7 ± 0.8 | NT |
| B3-4-7 | Gly | Gln | D-Leu | D-Arg | 1.7 ± 0.6 | NA |
| B3-4-8 | D-Leu | Ser | D-Nal | D-Arg | 18 ± 4 | NT |
| B3-4-9 | D-Asn | D-Val | D-Thr | D-Trp | NA | NT |
| B3-4-10 | D-Pro | D-Arg | D-Phe | D-Tyr | NA | NT |
| B3-4-11 | Nle | Gly | Nle | D-Arg | 7.2 ± 2.0 | 24 ± 7 |
| B3-4-12 | Ala | D-Arg | D-Phe | Fpa | 3.1 ± 0.6 | 19 ± 6 |
| B3-4-13 | Orn | D-Arg | D-β-homoAla | Fpa | 1.7 ± 0.1 | 30 ± 11 |
| B3-4-14 | D-Lys | His | D-Tyr | D-Glu | NA | NT |
| B3-4-15 | Orn | D-Val | D-β-homoAla | D-Trp | 2.9 ± 0.8 | NA |
| B3-4-16 | D-Pro | D-Arg | D-Nal | D-Glu | NA | NT |
| B3-4-17 | Orn | D-Asn | D-Tyr | D-Thr | 5.7 ± 0.1 | NA |
Values reported represent the mean ± SD of 3 or more independent experiments. NA, no significant activity; NT, not tested.
Characterization of Library Hits.
The 17 hits were individually re-synthesized and tested for inhibition of the Ras-Raf interaction using a homogenous time-resolved fluorescence (HTRF) assay (Figure S4).34 The peptides exhibited a wide range of IC50 values, from 1.7 μM to no significant inhibitory activity (Table 1), indicating that the exocyclic sequence indeed engages in important interactions with the Ras protein surface. Three of the compounds (B3-4-4, B3-4-7, and B3-4-13) showed ~3-fold higher potency in the HTRF assay (IC50 = 1.7 μM) than the parent compound B3-4 (IC50 = 4.4 μM). Two of the other compounds (B3-4-12 and B3-4-15) were slightly more potent than B3-4, while the remaining peptides were less active than B3-4. Next, eight of the most potent compounds in the HTRF assay were tested for their effect on the viability of H358 lung cancer cells by MTT assay. Compared to parent compound B3-4 (EC50 = 29 μM), three of the library hits showed improved cellular activity, with compound B3-4-4 being most potent (EC50 = 17 μM) (Table 1).
Further Optimization of B3-4-4.
Peptide B3-4-4 was selected for further optimization and renamed as “B4” hereafter for simplicity. Because structural modification of B4 may impact the Ras-binding affinity and cell permeability differently, both of which are critical for cellular activity, we opted to simultaneously improve several parameters, including Ras-binding potency, cell permeability, and aqueous solubility, by measuring the IC50 value in the HTRF assay (for inhibition of the Ras-Raf interaction) and the EC50 value for reduction of H358 cell viability. At this point of the project, we switched from the MTT assay to Promega’s CellTiter-Glo assay35 to assess cell viability, since CellTiter-Glo is a more reliable and sensitive viability assay. In the CellTiter-Glo assay, B3-4 and B4 exhibited EC50 values of 22 and 14 μM, respectively, against H358 cells (Table 2).
Table 2.
Sequences and Biological Activity of B4 Analogs
| Peptide | Peptide Sequencea,b | HTRF IC50 (μM)c | CellTiter-Glo EC50 (μM)c |
|---|---|---|---|
| B3-4 | ΦOSQΦΠ-K-fArR-J-BBK-NH2 | 4.4 ± 1.3 | 22 ± 4 |
| B4 | ΦSQΦΠ-K-fArR-J-ΣrΨa-K-NH2 | 1.7 ± 0.3 | 14 ± 3 |
| B4-1 | ΦSNΦΠ-K-fArR-J-ΣrΨa-K-NH2 | 0.89 ± 0.26 | 7.2 ± 0.8 |
| B4-2 | ΦSNΦΠ-K-fArR-J-ΣrΨa-NH2 | 0.70 ± 0.24 | 6.0 ± 1.6 |
| B4-3 | ΦSNΦΠ-K-fArR-J-ΣrΨ-NH2 | 0.55 ± 0.07 | 15 ± 2 |
| B4-4 | ΦSNΦΠ-K-fArR-J-Σr-NH2 | 0.70 ± 0.10 | 16 ± 4 |
| B4-5 | ΦSNΦΠ-K-fArR-J-Σ-NH2 | 1.3 ± 0.5 | 17 ± 2 |
| B4-6 | ΦSNΦΠ-K-fArR-J-ΣrFa-NH2 | 0.51 ± 0.01 | 3.2 ± 1.5 |
| B4-7 | ΦSNΦΠ-K-fArR-J-ΣrYa-NH2 | 1.1 ± 0.1 | 19 ± 10 |
| B4-8 | ΦSNΦΠ-K-fArR-J-ΣrRa-NH2 | 1.0 ± 0.1 | 20 ± 1 |
| B4-9 | ΦSNΦΠ-K-fArR-J-Σrra-NH2 | 0.70 ± 0.0 | 10 ± 8 |
| B4-10 | ΦSNΦΠ-K-fArR-J-ΣrZa-NH2 | 0.77 ± 0.03 | 9.2 ± 1.1 |
| B4-11 | ΦSNΦΠ-K-fArR-J-ΣrWa-NH2 | 2.2 ± 0.8 | NA |
| B4-12 | ΦSNΦΠ-K-fArR-J-Σrϑa-NH2 | 0.94 ± 0.33 | 11 ± 4 |
| B4-13 | ΦSNΦΠ-K-fArR-J-ΣrΠa-NH2 | 0.76 ± 0.24 | 4.5 ± 3.0 |
| B4-14 | ΦSNΦΠ-K-fArR-J-ΣrΓa-NH2 | 0.50 ± 0.04 | 4.4 ± 0.3 |
| B4-15 | ΦSNΦΠ-K-fArR-J-ΣrΛa-NH2 | 0.33 ± 0.01 | 9.0 ± 0.7 |
| B4-16 | ΦSNΦΠ-K-fArR-J-ΣrPa-NH2 | 0.41 ± 0.06 | 6.6 ± 0.4 |
| B4-17 | ΦSNΦΠ-K-fArR-J-KrΨa-NH2 | 1.1 ± 0.4 | 4.4 ± 1.5 |
| B4-18 | ΦSNΦΠ-K-fArR-J- ΘrΨa-NH2 | 1.7 ± 0.2 | NA |
| B4-19 | ΦSNΦΠ-K-fArR-J- ΞrΨa-NH2 | 0.87 ± 0.05 | 12 ± 8 |
| B4-20 | ΦSNΦΠ-K-fArR-J-NrΨa-NH2 | 0.28 ± 0.06 | 8.4 ± 5.2 |
| B4-21 | ΦSNΦΠ-K-fArR-J-QrΨa-NH2 | 0.23 ± 0.02 | 7.5 ± 4.0 |
| B4-22 | ΦSNΦΠ-K-FArR-J-ΣrFa-NH2 | 0.65 ± 0.37 | 21 ± 1 |
| B4-23 | ΦSNΦΠ-K-farR-J-ΣrFa-NH2 | 0.52 ± 0.01 | 16 ± 8 |
| B4-24 | ΦSNΦΠ-K-fζrR-J-ΣrFa-NH2 | 0.61 ± 0.09 | 7.2 ± 2.7 |
| B4-25 | ΦSNΦΠ-K-fLrR-J-ΣrFa-NH2 | 0.79 ± 0.02 | 4.8 ± 1.1 |
| B4-26 | ΦSNΦΠ-K-fSrR-J-ΣrFa-NH2 | 0.56 ± 0.17 | 11 ± 8 |
| B4-27 | ΦSNΦΠ-K-fQrR-J-ΣrFa-NH2 | 0.69 ± 0.11 | 2.1 ± 0.1 |
| B4-28 | ΦSNΦΠ-K-fHrR-J-ΣrFa-NH2 | 1.0 ± 0.2 | 8.2 ± 4.8 |
| B4-29 | ΦSNΦΠ-K-fNrR-J-ΣrFa-NH2 | 1.2 ± 0.4 | 14 ± 3 |
| B4-30 | ΦSNΦΠ-K-fΣrR-J-ΣrFa-NH2 | 0.83 ± 0.20 | 6.4 ± 0.8 |
| B4-31 | ΩSNΦΠ-K-fQrR-J-ΣrFa-NH2 | 0.64 ± 0.09 | 5.2 ± 1.1 |
| B4-32 | ΦSNΩΠ-K-fQrR-J-ΣrFa-NH2 | 0.71 ± 0.08 | 4.9 ± 1.2 |
| B4-33 | ΦNNΦΠ-K-fQrR-J-ΣrFa-NH2 | 0.86 ± 0.12 | 17 ± 5 |
| B4-34 | ΦTNΦΠ-K-fQrR-J-ΣrFa-NH2 | 1.3 ± 0.3 | 23 ± 17 |
| B4-35 | ΦςNΦΠ-K-fQrR-J-ΣrFa-NH2 | 1.2 ± 0.2 | 5.9 ± 0.5 |
| B4-36 | ΦSAΦΠ-K-fQrR-J-ΣrFa-NH2 | 1.0 ± 0.2 | NA |
| B4-37 | ΦSSΦΠ-K-fQrR-J-ΣrFa-NH2 | 0.75 ± 0.18 | 17 ± 6 |
| B4-38 | ΦSNΦΠ-K-ψQrR-J-ΣrFa-NH2 | 0.55 ± 0.03 | 15 ± 5 |
| B4-39 | ΦSNΦΠ-K- λQrR -J-ΣrFa-NH2 | 0.71 ± 0.01 | 23 ± 7 |
| B4-40 | ΦSNΦΠ-K-fQrR-J-Σrfa-NH2 | 0.56 ± 0.24 | 5.0 ± 0.4 |
| B4-41 | ΦSNΦΠ-K-fQrR-J-ΣrXa-NH2 | 0.71 ± 0.09 | 7.4 ± 0.3 |
| B4-42 | ΦSNΦΠ-K-fQrR-J-ΣrFv-NH2 | 0.61 ± 0.17 | 7.1 ± 1.9 |
| B4-43 | ΦSNΦΠ-K-fQrR-J-ΣrFt-NH2 | 0.57 ± 0.10 | 2.6 ± 1.2 |
| B4-44 | ΦSNΦΠ-K-fQrR-J-ΣrFs-NH2 | 0.75 ± 0.03 | 5.3 ± 1.4 |
| B4-45 | ΦSNΦΠ-K-fQrR-J-ΣrFn-NH2 | 2.2 ± 1.7 | 3.1 ± 0.8 |
| B4-46 | ΦSNΦΠ-K-fQrR-J-ΣrFB-NH2 | 1.1 ± 0.4 | 12 ± 3 |
L-AA’s are shown as the capitalized one letter code while D-AA’s are lowercase.
J = L-2,3-diaminopropionic acid; Π = L-phenylglycine; Φ = D-2-napthylalanine; Σ = L-ornithine; Γ = L-3,4-difluorophenylalanine; ϧ = 3-cyclohexyl-L-alanine; Λ = L-3-chlorophenylalanine; λ = D-3-chlorophenylalanine; P = 3-(2-pyridyl)-L-alanine; Ω = 3-(3-benzo(b)thienyl)-D-alanine; Ψ = L-4-fluorophenylalanine; ψ = D-4-fluorophenylalanine; Z = L-homophenylalanine; Θ = L-2,3-dia-minopropionic acid; Ξ = L-2,4-diaminobutyric acid; ζ = L-α-aminobutyric acid; ς = L-homoserine; B = β-alanine; X = L-β-homo-phenylalanine.
Values reported represent the mean ± SD of 3 or more independent experiments. NA, no significant activity; NT, not tested.
During our initial optimization of B3, we had discovered that replacement of L-Gln at position 3 with L-Asn improved the Ras-binding affinity by ~2-fold (data not shown). The same substitution in B4 also resulted in ~2-fold improvement in both Ras-binding affinity (IC50 = 0.89 μM for B4-1 in the HTRF assay) and antiproliferative activity against H358 cells (EC50 = 7.2 μM; Table 2).
We next evaluated the importance of the exocyclic C-terminal residues, which were derived from the library screening, to minimize the size of the inhibitor. Truncation of the C-terminal L-Lys, which was originally added for the purpose of fluorescent labeling, slightly improved the Ras-binding affinity (IC50 = 0.70 μM for B4-2) and cellular activity (EC50 = 6.0 μM for B4-2). Truncation of the D-alanine (B4-3) slightly improved KRas binding but substantially decreased cellular activity, suggesting the C-terminal D-alanine is beneficial for cellular entry, proteolytic stability, and/or reducing nonspecific binding to off-target proteins. Further truncation of the C-terminal L-Fpa and D-Arg decreased both KRas binding and antiproliferative activity, indicating that these two residues engage in important interactions with the Ras protein (Table 2, peptides B4-4 and B4-5). Thus, the four C-terminal residues derived from library screening (Orn-D-Arg-Fpa-D-Ala) are critical for inhibitor activity.
To optimize the C-terminal tetrapeptide, we replaced Fpa at position 14 with different aromatic amino acids including L-Phe, L-Tyr, L-homoPhe, L-Trp, L-3-chlorophenylalanine (L-3Cpa), L-phenylglycine (L-Phg), L-3,4-difluorophenylalanine (L-F2pa), L-3-cyclohexylalanine (L-Cha), L-3-(2-pyridyl)-alanine (L-Pya), and L- or D-arginine (Table 2, peptides B4-6 to B4-16). All substitutions were well tolerated with respect to KRas binding (IC50 = 0.3 to 1.0 μM) except for L-Trp, which decreased the binding affinity by >3-fold. On the other hand, most of these substitutions decreased the activity in the CellTiter-Glo assay, presumably due to reduced cellular entry efficiency. Peptide B4-6, which contains an L-phenylalanine at position 14, was most active, with an IC50 value of 0.51 μM in the HTRF assay and an EC50 value of 3.2 μM in the cell viability assay. We also replaced L-Orn at position 12 with other positively charged amino acids including L-Lys, L-Dap, and L-2,4diaminobutyric acid (L-Dab) or neutral residues L-Asn or L-Gln (Table 2, peptides B4-17 to B4-21). Substitution of other positively charged residues for Orn all decreased the KRas binding affinity. Interestingly, substitution of L-Asn or L-Gln substantially increased the KRas binding affinity (by ~3-fold) but not the cellular activity, suggesting that a positively charge at position 12 facilitates cellular entry. No attempt was made to replace D-Arg at position 13 because of its importance for KRas binding, aqueous solubility, and likely cellular uptake (as well as the lack of suitable commercially available analogs).
Peptide B4-6 was chosen for further optimization within the bicyclic ring system. Inversion of the stereochemistry of D-Phe at position 7 (Table 2, B4-22) had a very minor effect on KRas binding but greatly decreased the cellular activity, highlighting the importance of D-Phe for cellular uptake. Next, the L-Ala residue at position 8 was replaced with various amino acids including D-Ala, L-Leu, L-Ser, L-Orn, L-Abu, L-His, L-Asn, and L-Gln (Table 2, peptides B4-23 to B4-30). We found that L-Gln at this position gave the highest potency in the cellular assay (EC50 = 2.1 μM for B4-27) and an IC50 value of 0.69 μM in the HTRF assay (Figure 1a); therefore, L-glutamine was kept at position 8. Replacement of the D-Nal residues at positions 1 and 4 with structurally similar 3-(3-benzo(b)thienyl)D-alanine (D-Bta) residues did not impact the binding affinity but reduced the cellular activity by ~2-fold (Table 2, peptides B4-31 and B4-32). Modification of L-Ser at position 2 or L-Asn at position 3 also failed to improve either the KRas binding affinity or the antiproliferative activity (Table 2, peptides B4-33 to B4-37). Surprisingly, even minor structural changes at position 7, e.g., replacement of D-Phe with D-Fpa (B4-38) or D-Cpa (B4-39), resulted in ≥7-fold decrease in cellular activity. During the final round of optimization, we substituted D-Phe or L-β-homophenylalanine for the L-Phe at position 14 of B4-27 (Table 2, peptides B4-40 and B4-41). In addition, we replaced D-Ala at position 15 with D-Val, D-Ser, D-Thr, D-Asn, or β-Ala to give peptides B4-42 to B4-46. However, none of these modifications led to any improvement in either KRas binding or antiproliferative activity. Thus, peptide B4-27 remains the most potent inhibitor of the series and was carried forward for further investigation.
Figure 1.
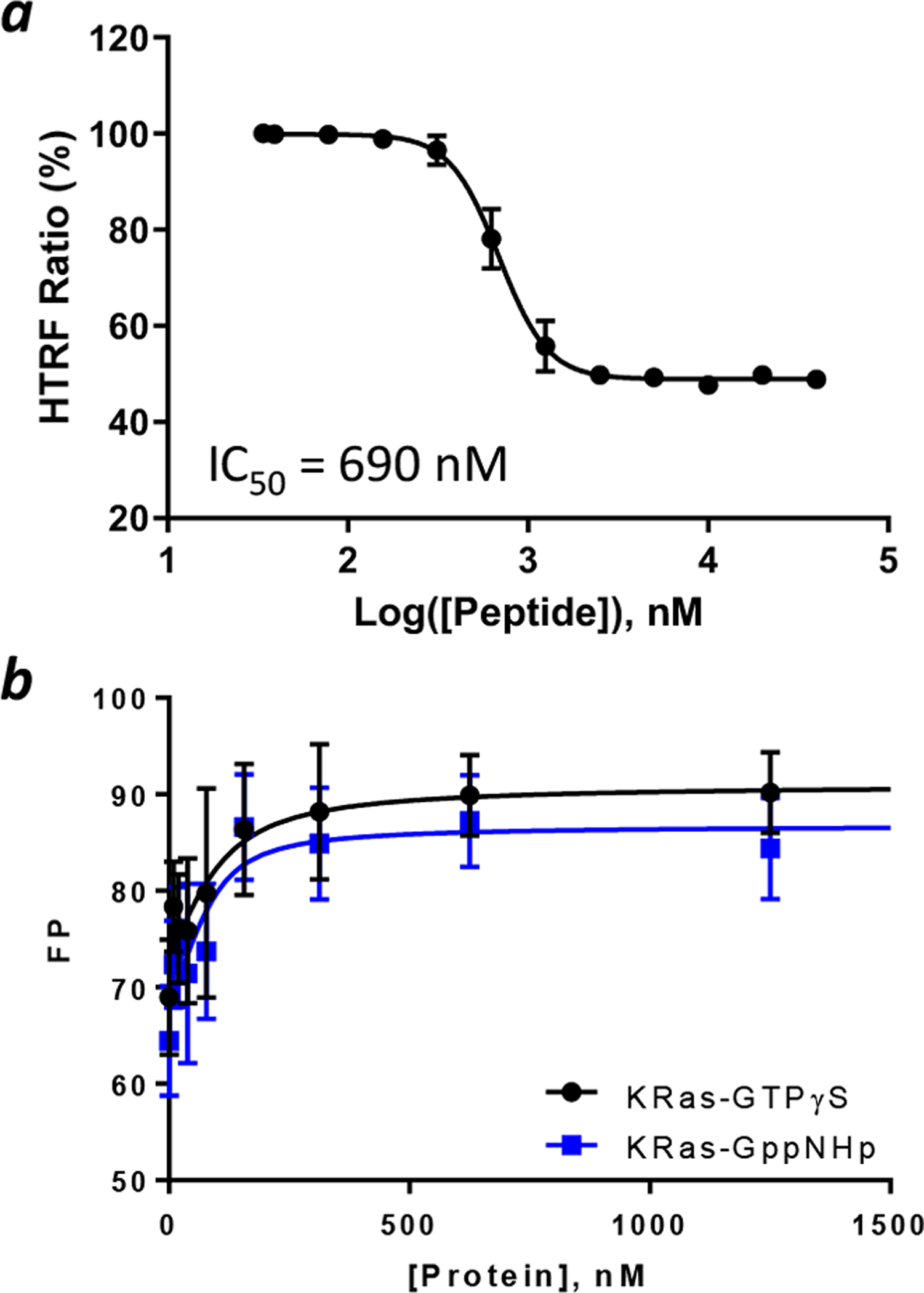
Binding affinity of B4-27 to KRas. (a) HTRF assay showing inhibition of the interaction between KRasG12V-GppNHp (50 nM) and CRAF RBD (50 nM) by B4-27. (b) Binding of B4-27FAM (100 nM) to KRasG12V loaded with GTPγS, GppNHp, or GDP as monitored by FP. Data reported are presented as mean ± SD of n ≥ 3 independent experiments.
B4-27 Is a Selective Inhibitor of Ras-GTP.
B4-27 was labeled with a fluorescein at its C-terminus through a (mini-PEG)2-Lys linker (B4-27FAM) and tested for binding to GTPγS, GppNHp-, or GDP-loaded KRasG12V by fluorescence polarization (FP). B4-27FAM bound KRas-GTPγS, KRas-GppNHp, and KRas-GDP with KD values of 42 ± 29, 21 ± 15, and ~230 nM, respectively (Figure 1b). Note that because of the small change in FP value upon binding of B4-27FAM to KRas-GDP, the KD value of the latter complex could not be accurately determined. Compared to the parent compound B3, which has KD values of 1.6 and 9.3 μM for KRas-GppNHp and KRas-GDP, respectively,21 B4-27 represents a 76-fold improvement in the Ras binding affinity but retains the ~10-fold selectivity for the active vs inactive form of KRas. We also tested B4-27FAM for binding to five arbitrarily selected control proteins available in our laboratory, including bovine serum albumin (BSA), glutathione-S-transferase (GST)-RBD, purine nucleoside phosphorylase (PNP), protein-tyrosine phosphatase 1B (PTP1B), and mouse double minute 2 homolog (MDM2). B4-27FAM showed weak binding to BSA (KD = 57 μM) but no significant binding to GST-RBD, PNP, PTP1B, or MDM2 (Figure S5). B4-27 is therefore a selective ligand of the active form of KRas.
B4-27 Efficiently Enters the Mammalian Cell and Colocalizes with KRas.
The cellular entry efficiency of B4-27 was assessed by flow cytometry analysis of human cervical cancer (HeLa) cells treated with B4-27FAM and compared to that of CPP12FITC, one of the most active cyclic cell-penetrating peptides,36 as well as parent compounds B3-4FAM and B4FAM. HeLa cells treated with 5 μM B4-27FAM showed a mean fluorescence intensity (MFI) value of 226% relative to that of CPP12 (100%) (Figure 2a). Under similar conditions, CPP12 has a cytosolic entry efficiency of 120% (defined as the ratio of cytosolic over extracellular concentration).36 In comparison, parent compounds B3-4 and B4 exhibited 4- and 2.5-fold lower cellular entry efficiency, respectively, than B4-27 (Figure 2b). To determine the intracellular distribution of B4-27, we examined HeLa, H358, and A549 (human lung cancer) cells after treatment with 5 μM B4-27FAM for 2 h by live-cell confocal microscopy. All cells became intensely fluorescent, exhibiting a combination of diffuse fluorescence throughout the entire cell volume and highly fluorescent puncta in the cytoplasmic region (Figure S6). The presence of diffuse fluorescence indicates that a significant fraction of the internalized B4-27FAM had reached the cytosol, while the fluorescence puncta likely represent the B4-27FAM molecules that were still inside the endosomes. In contrast, cells treated with B3-4FAM and B4FAM showed weaker and predominantly punctate fluorescence inside the cytoplasm (Figure S7). In H358 and A549 cells, some of the B4-27FAM molecules were concentrated at the plasma membrane (Figure 2c and S6), suggesting that they were bound to the membrane-associated Ras proteins. To test this notion, we treated Madin-Darby canine kidney (MDCK) cells stably expressing a green fluorescent protein (GFP)-KRasG12V fusion with 5 μM tetramethylrhodamine-labeled B4-27 (B4-27TMR) and imaged the cells by live-cell confocal microscopy. The plasma membrane-bound B4-27TMR co-localized with the GFP-KRasG12V fusion protein, especially at the interfaces between adjacent cells (Figure 2d). Interestingly, for membrane regions that are not in direct contact with neighboring cells, there is less accumulation of GFP-KRasG12V or B4-27TMR (Figure S8). These observations demonstrate that that B4-27 efficiently enters the mammalian cell and a fraction of the inhibitors are bound to Ras proteins at the plasma membrane.
Figure 2.
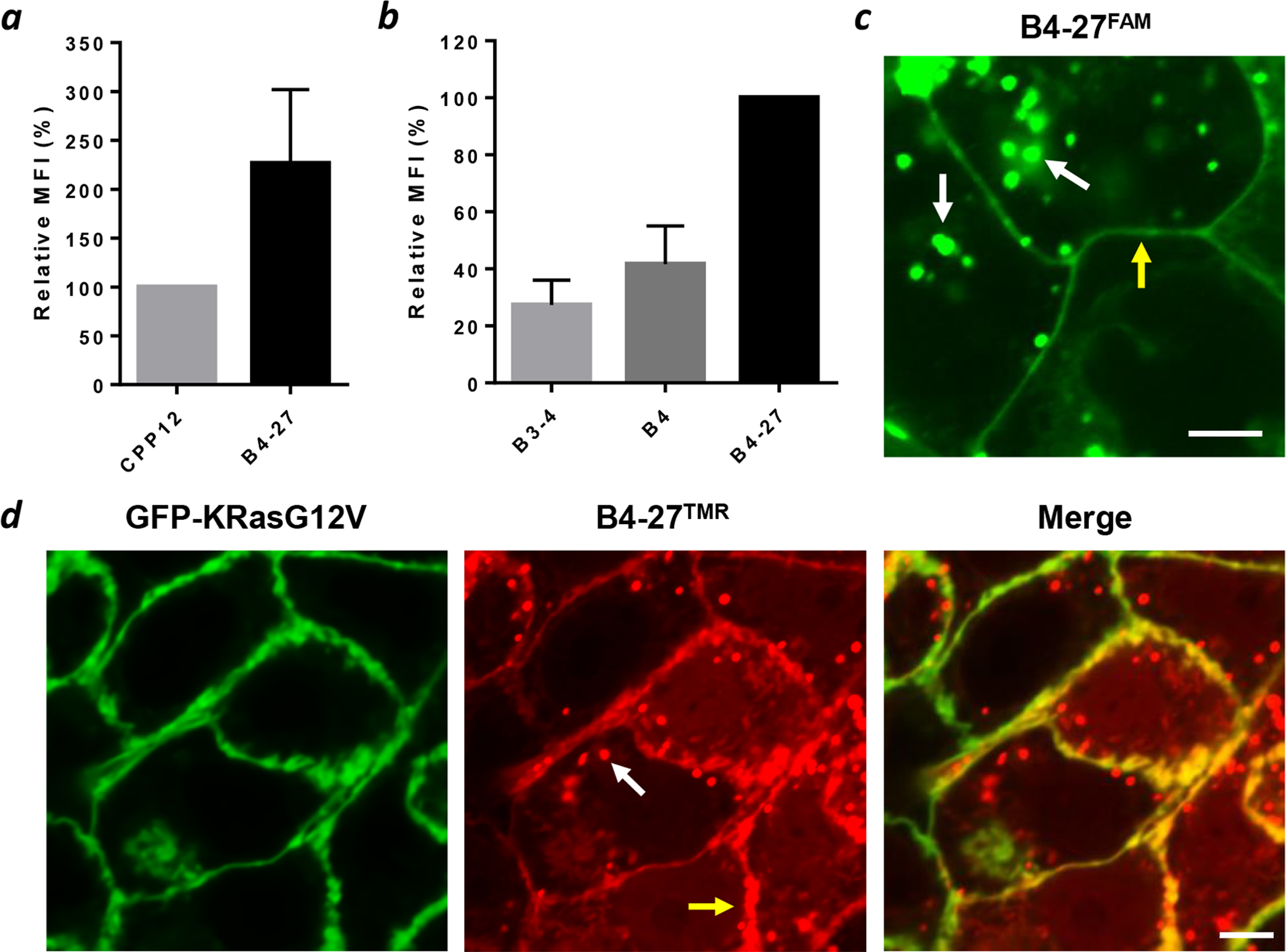
Cellular entry and intracellular distribution of B4-27. (a) Flow cytometry analysis of HeLa cells after treatment with 5 μM CPP12FITC or B4-27FAM for 2 h in the presence of 1% FBS. Values reported represent the mean ± SD from 3 independent experiments (n = 3) and are relative to that of CPP12FITC (100%). (b) Flow cytometry analysis of HeLa cells after treatment with 5 μM B3-4FAM, B4FAM or B4-27FAM for 2 h in the presence of 1% FBS. Values reported represent the mean ± SD from 3 independent experiments (n = 3) and are relative to that of B4-27FAM (100%). (c) Live-cell confocal microscopic images of H358 cells after treatment with 3 μM B4-27FAM for 2 h in the presence of 1% FBS. (d) Co-localization of B4-27TMR (red) with GFP-KRasG12V (green) in MDCK cells stably expressing GFP-KRasG12V after treatment with 5 μM B4-27TMR for 2 h in the presence of 1% FBS. White arrows, putative endosomes; yellow arrows, plasma membrane; scale bars, 5 μm.
B4-27 Inhibits Intracellular Ras-Effector Interaction and Ras Signaling Pathways.
We tested the ability of B4-27 to inhibit the intracellular Ras-effector interactions by using a Ras-Raf BRET2 assay.37 Human embryonic kidney (HEK293T) cells were transfected with plasmids expressing a Renilla luciferase variant-KRasG12V (or G12D) fusion protein (RLuc8KRas) as the BRET donor and a GFP-CRAF RBD fusion protein as the acceptor. Under normal conditions, interaction between RLuc8-KRas and GFP-CRAF RBD produces a BRET signal in the presence of a luciferase substrate, Coelenterazine 400a. On the other hand, inhibition of the Ras-Raf interaction would decrease the BRET signal. Treatment of HEK293T expressing RLuc8-KRasG12V (or RLuc8-KRasG12D) and GFP-CRAF RBD with B4-27 (0–15 μM) dose-dependently reduced the BRET signal (Figure 3a and 3b), indicating that B4-27 is capable of blocking KRasG12V and KRasG12D from binding to their downstream effector proteins in a cellular context. In contrast, B3-4 did not decrease the BRET signal over the same concentration range (0–15 μM).
Figure 3.
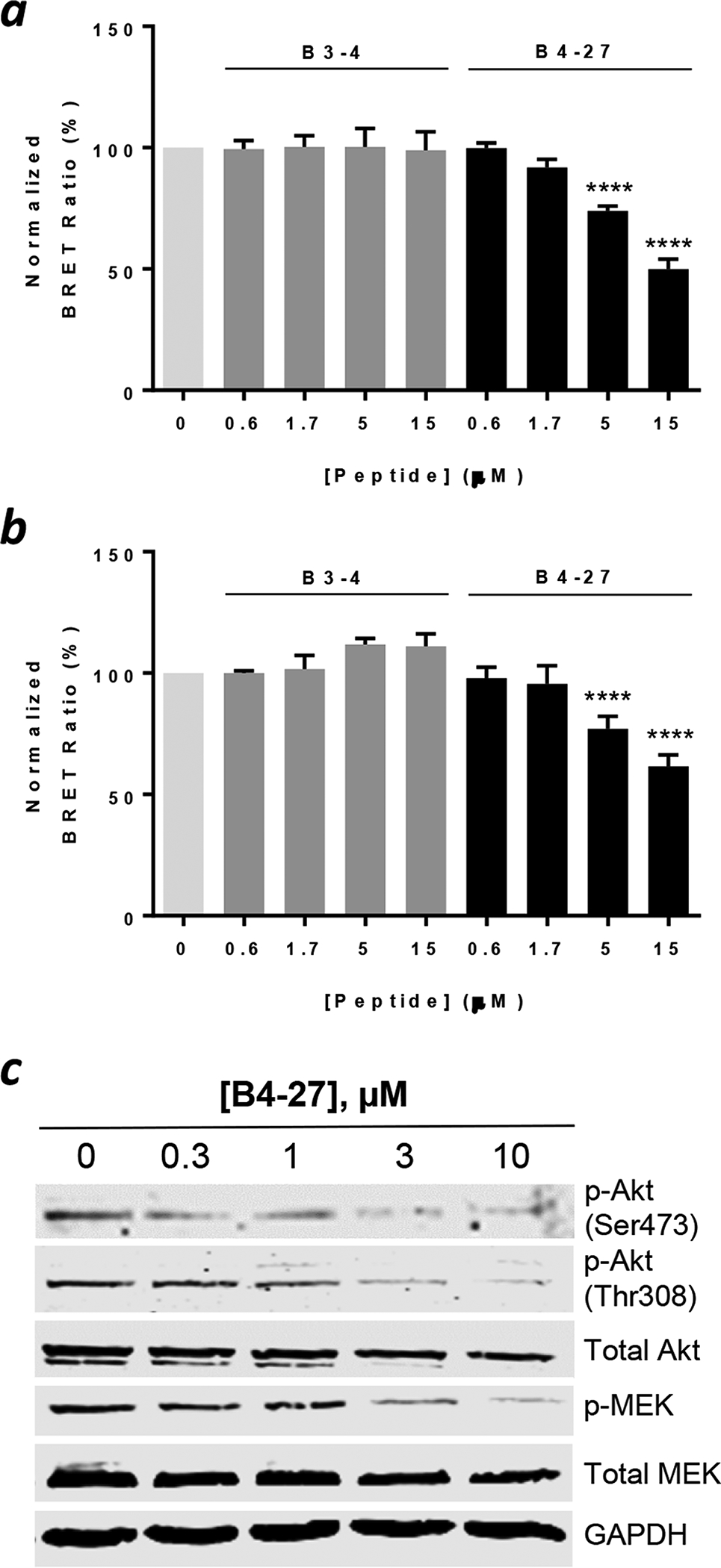
Inhibition of Ras signaling by B4-27. (a, b) Dose-de-pendent inhibition of the interaction between KRasG12V (a) or KRasG12D (b) and CRAF RBD by B4-27 or B3-4 in HEK293T cells as monitored by the BRET2 assay.37 BRET signals reported represent the mean ± SD from 4 independent experiments and are relative to that of vehicle (DMSO)-treated cells (100%). ****, p < 0.0001. (c) Western blots showing the effect of B4-27 on the phosphorylation levels of signaling proteins downstream of Ras (Akt, MEK) in H358 cells. GAPDH was used as a loading control. Representative images from 4 independent experiments are shown.
We next examined the ability of B4-27 to inhibit the Ras/Raf/MEK/ERK and Ras/PI3K/Akt signaling pathways. H358 cells were treated with varying concentrations of B4-27 (0–10 μM) for 4 h and stimulated for 10 min with epidermal growth factor (EGF; 50 ng/mL). The cells were lysed and the phosphorylation levels of MEK and Akt were monitored by Western blot analysis using antibodies specific for phosphorylated Akt [anti-p-Akt (Ser473) and anti-p-Akt (Thr308)] and MEK (anti-p-MEK). B4-27 dose-dependently inhibited the phosphorylation of Akt and MEK (IC50 ≈ 3 μM), while the total Akt and MEK levels remained relatively constant (Figure 3c). Thus, B4-27 effectively inhibits the signaling pathways downstream of Ras.
B4-27 Is a Pan-Ras Inhibitor and Induces Apoptosis of Ras Mutant Cancer Cells.
Given its ability to block the Ras-Raf interaction (and likely Ras-PI3K interaction as well), B4-27 likely binds at or near the effector-binding site on Ras. Because the effector-binding site is identical for all four Ras isoforms, we expected B4-27 to be a pan-Ras inhibitor, capable of inhibiting all 4 Ras isoforms, regardless of the mutational status (i.e., WT and G12, G13, and Q61 mutants). To test this notion, we treated a panel of human cancer cell lines carrying different Ras mutations, including non-small cell lung cancer cell lines (H358, heterozygous KRasG12C and A549, homozygous KRasG12S), colorectal cancer cell lines (DLD-1, heterozygous KRasG13D and SW480, homozygous KRasG12V), a lung cancer cell line carrying mutant HRas (H1915, HRasQ61L), and a lung cancer cell line carrying mutant NRas (H1299, NRasQ61K), with B4-27. B4-27 reduced the viability of all of the tested Ras mutant cancer cell lines with EC50 values of 0.5 ± 0.3 μM for A549 cells, 2.1 ± 0.2 μM for H358 cells, 4.9 ± 0.7 μM for DLD-1 cells, 8.7 ± 3.4 μM for SW480 cells, 6.8 ± 2.2 μM for H1915 cells, and 12 ± 2 μM for H1299 cells (Figure 4a). We also tested B4-27 against two non-cancerous cell lines that carry WT Ras, including a murine fibroblast cell line (NIH 3T3) and a human embryonic kidney (HEK293T) cell line. B4-27 reduced the viability of the non-cancerous cell lines, but only at high concentrations (EC50 ≥ 20 μM). Several factors may contribute to the greater sensitivity of Ras mutant cancer cells to B4-27, including more robust endocytic uptake of the inhibitor,38 reduced binding affinity of Ras mutants to effector proteins,39 and “addiction” to Ras activity for signaling/survival by cancer cells.40
Figure 4.
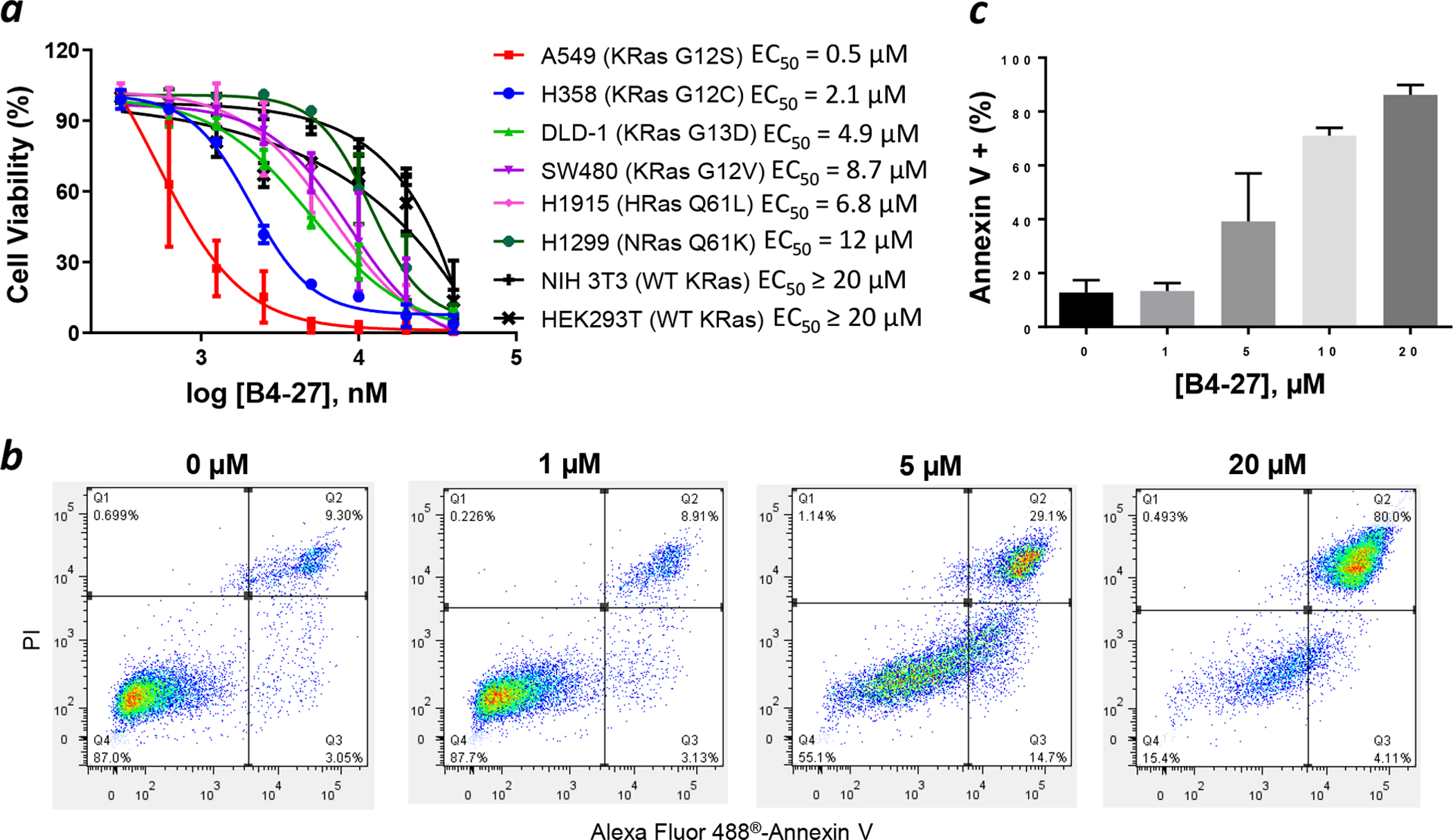
B4-27 causes apoptotic death of Ras mutant cancer cell lines. (a) Effect of B4-27 on the viability of various cancer and non-cancerous cell lines as monitored by the Cell-Titer Glo® 2.0 Viability Assay. Cells were incubated with indicated concentrations of B4-27 for 96 h in the presence of 10% FBS. (b) Annexin V/PI staining of H358 cells after treatment with indicated concentrations of B4-27 for 24 h in the presence of 10% FBS. Cytometry data were gated so that Q1 contained necrotic cells, Q2 contained late apoptotic cells, Q3 represented early apoptotic cells and Q4 were live cells. (c) Percentage of annexin V-positive cells (in Q2 and Q3) as a function of B4-27 concentration. Data shown represent the mean ± SD from at least three independent experiments.
Since simultaneous inhibition of the MEK/ERK and PI3K/Akt signaling pathways is known to cause apoptosis of cancer cells,41,42 B4-27 is expected to induce apoptosis of Ras mutant cancer cells. To test this hypothesis, H358 cells were treated with varying concentrations of B4-27 for 24 h, stained with Alexa Fluor® 488-annexin V and propidium iodide (PI), and analyzed by flow cytometry. The presence of a significant population of annexin V-positive but PI-negative cells at intermediate B4-27 concentrations (5 and 10 μM) and the absence of annexin V-negative and PI-positive cells are consistent cell death by apoptotic mechanisms (Figure 4b). At 10 μM B4-27, >70% of the cell population had already undergone or was undergoing apoptosis (Figure 4c). To determine whether B4-27 caused any cell death by nonspecific mechanisms, e.g., membrane disruption, we examined the cell culture medium of H358 cells after B4-27 treatment for the presence of lactate dehydrogenase (LDH) activity. B4-27 did not cause significant LDH release until concentrations at ≥ 20 μM (Figure S9). Taken together, our results strongly suggest that B4-27 efficiently enters the cytosol of mammalian cells, binds selectively to the active forms of WT and mutant Ras proteins, blocks Ras proteins from engaging their downstream effector proteins, and induces apoptosis of Ras mutant cancer cells. However, we cannot rule out the possibility that B4-27 may also engage other intracellular protein(s).
B4-27 Is Metabolically Stable.
The proteolytic stability of B4-27 was assessed by incubating it in human serum at 37 °C for up to 24 h and analyzing the reaction mixture by analytical HPLC. Approximately 85% of the B4-27 remained intact after 24 h of incubation, indicating a serum t1/2 of > 24 h (Figure S9).
B4-27 Inhibits Tumor Growth in Mouse Xenograft Models.
To test whether B4-27 can suppress tumor growth in vivo, we generated an A549 xenograft model. Approximately 1.5 million A549 cells were subcutaneously injected into nude mice. When the tumors grew to ~40 mm3 in volume, the mice (n = 6 in each group) were dosed with B4-27 at 1 or 5 mg/kg per day or vehicle (PBS) through tail vein injection for a total of 9 days. B4-27 significantly inhibited tumor growth at both tested concentrations, with an average tumor volume of ~50 mm3 for the 1 mg/mL group and ~35 mm3 for the 5 mg/mL group, while the vehicle-treated group had an average tumor volume of ~100 mm3 on day 9 after injection (Figure 5a). Importantly, the 5-mg/kg treatment completely halted tumor growth during the 9-day period. The average tumor weight on the day of the harvest was also 39% and 46% less for the 1- and 5-mg/kg treatment groups, respectively, than that of the control group (Figure 5b,c). B4-27 treatment did not result in any significant body weight loss or any apparent toxicity (Figure 5d). B4-27 was also tested in an H358 xenograft model and a daily dose of 5 mg/kg largely suppressed the tumor growth after 9 days (Figure S10).
Figure 5.
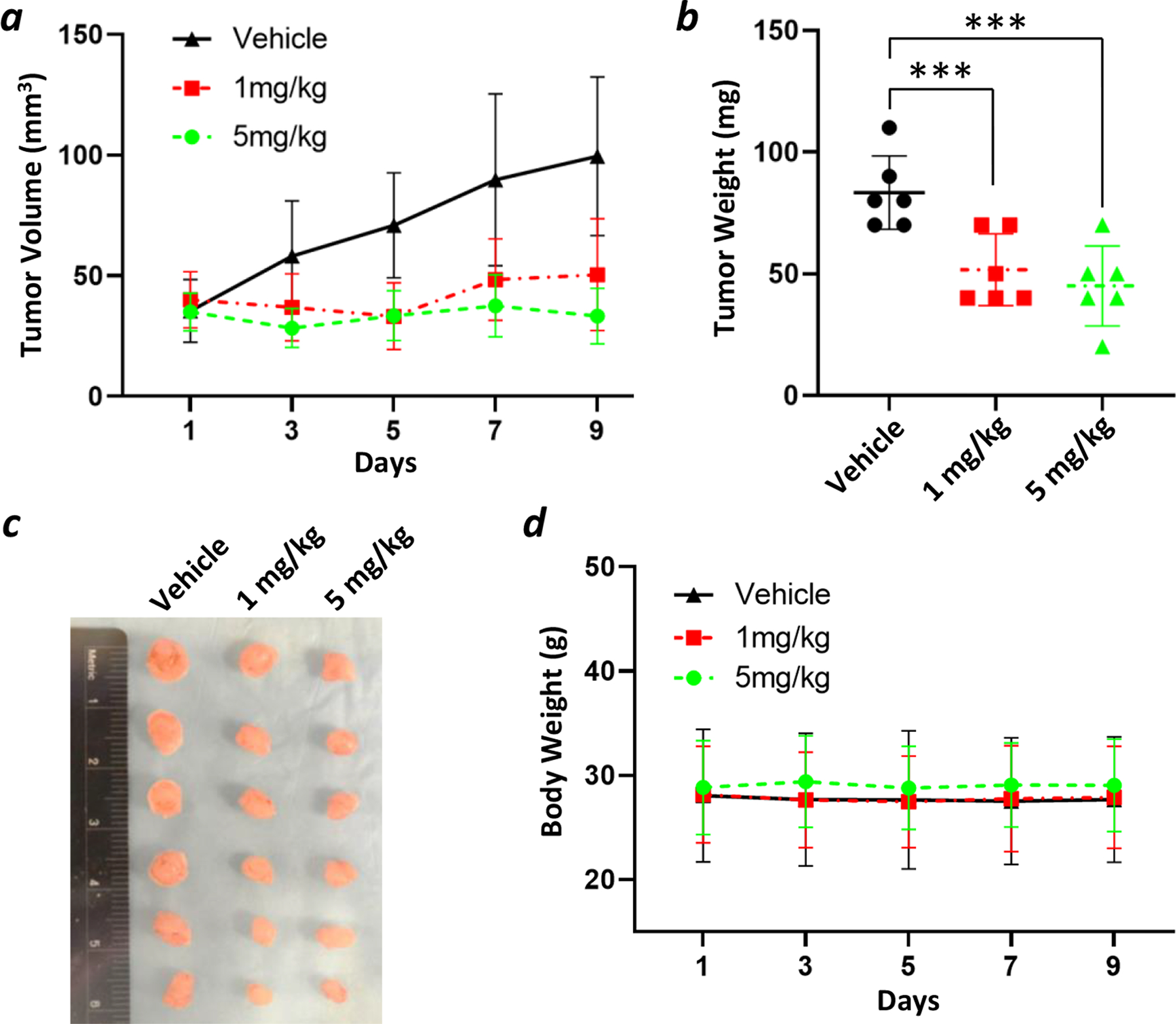
Suppression of tumor growth by B4-27 in A549 mouse xenografts (n = 6). (a) Tumor volume of B4-27- and vehicle-treated groups over 9 days. (b) The average tumor weight of B4-27- and vehicle-treated groups on the day of harvest. ***, p < 0.001. (c) Macroscopic images of the tumors resected from mice on the day of harvest. (d) The average mouse body weight of B4-27- and vehicle-treated groups over 9 days.
Representative samples of tumors were analyzed by immunohistochemistry (IHC) using hematoxylin and eosin (H&E), pERK, and Ki-67 staining. Immunohistochemical analysis showed that B4-27 dramatically decreased the levels of ERK phosphorylation in both xenograft models (Figure 6), confirming that tumor growth inhibition was caused by suppression of Ras-mediated cell signaling. In addition, IHC revealed a large decrease in cell proliferation as indicated by Ki-67 staining.
Figure 6.
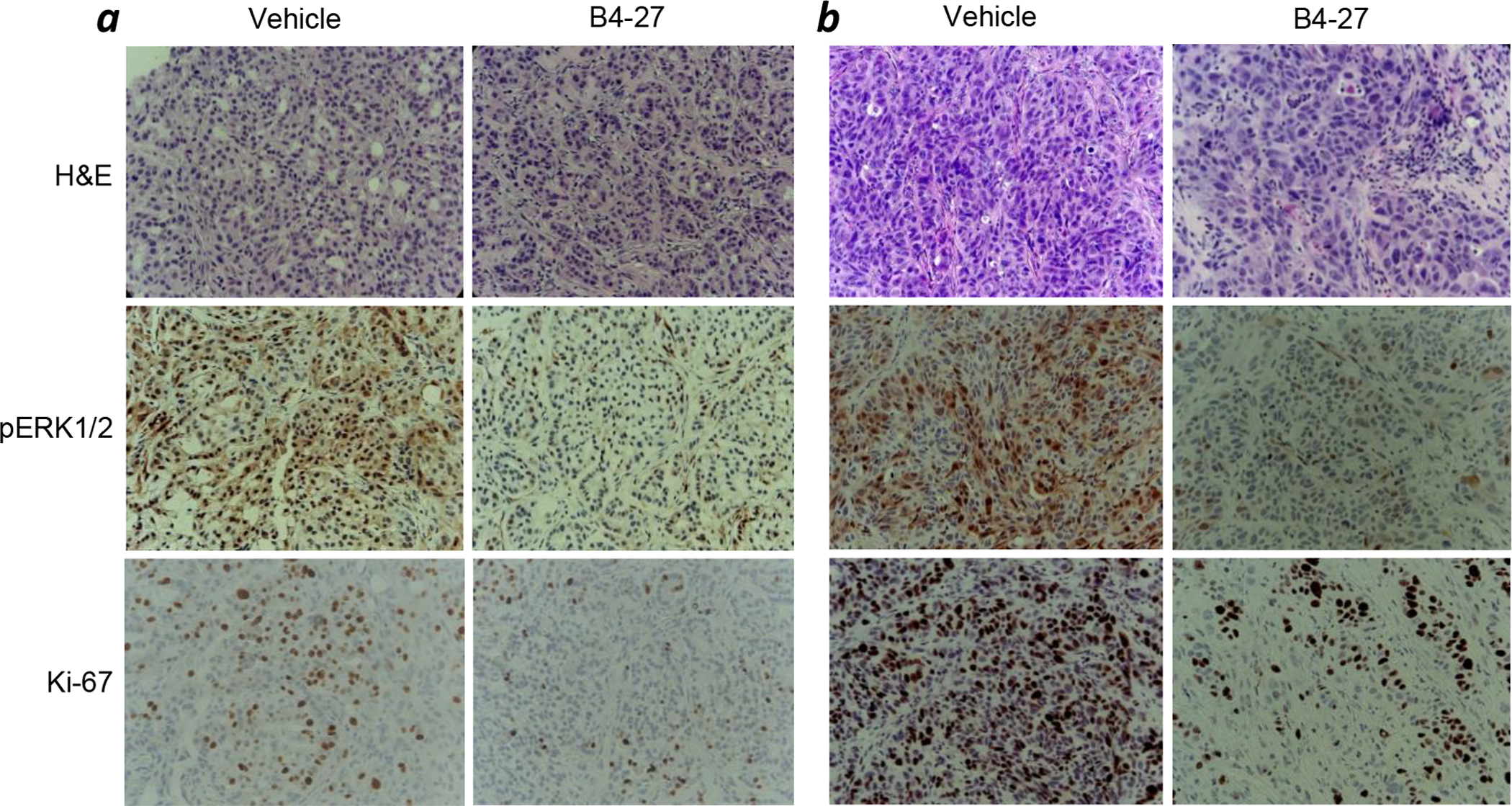
Representative images of H&E staining and immunohistochemical staining for p-ERK and Ki-67 in A549 (a) and H358 xenografts (b).
CONCLUSION
We have discovered a bicyclic peptidyl pan-Ras inhibitor, B4-27, by screening a 2nd-generation peptide library followed by medicinal chemistry efforts. Compared to the parent compound (cyclorasin B3), B4-27 is 76-fold more potent for KRas binding and has vastly improved cell permeability, while maintain excellent proteolytic stability. B4-27 binds selectively to the active form of Ras proteins and inhibits their interaction with downstream effector proteins causing growth inhibition and apoptosis of Ras mutant cancer cells in vitro and in vivo. Our data suggest that B4-27 is a promising agent for the treatment of a wide spectrum of Ras mutant-driven cancers.
EXPERIMENTAL METHODS
Materials.
Reagents for peptide synthesis were purchased from Chem-Impex (Wood Dale, IL), NovaBiochem (La Jolla, CA), or Anaspec (San Jose, CA). Rink amide resin (100–200 mesh, 0.43 mmol/g) was from Chem-Impex (Wood Dale, IL). All solvents and other chemical reagents were obtained from Sigma-Aldrich, Fisher Scientific (Pittsburgh, PA), or VWR (West Chester, PA) and were used without further purification unless noted otherwise. Dynabeads M-280 streptavidin were purchased from Invitrogen (Carlsbad, CA). Cell culture media, fetal bovine serum (FBS), penicillin-streptomycin, 0.25% trypsin-EDTA, DPBS, isopropyl β-D-1-thiogalactopyranoside (IPTG), guanosine 5′-[β,γ-imido]triphosphate (GppNHp), guanosine 5′-O-(3-thiotriphosphate) (GTPγS), guanosine 5′diphosphate (GDP), 2,2,2-trifluoroethanol (TFE), streptavidin-alkaline phosphatase (SA-AP), protease inhibitor cocktail tablets, ampicillin, 5(6)-carboxyfluorescein, and 5(6)-carboxytetramethylrhodamine were purchased from Sigma-Aldrich (St. Louis, MO). Biotin-(PEG)4-NHS ester, His-Pur™ Cobalt Resin, RIPA lysis buffer, CyQUANT™ LDH cytotoxicity assay, Lipofectamine® 2000, and Alexa Fluor® 488 Annexin V/Dead Cell apoptosis kit were purchased from Thermo Fisher Scientific. Micro Bio-Spin™ 6 desalting columns were purchased from Bio-Rad (Hercules, CA). Anti-GST-Tb cryptate and anti-HA-d2 monoclonal antibodies were purchased from CisBio (Bedford, MA). The cell proliferation kit (MTT) was purchased from Roche (Indianapolis, IN). The CellTiter-Glo® reagent was purchased from Promega (Madison, WI). DNA plasmids encoding RLuc8-KRas and GFP-CRAF RBD fusion proteins were kindly provided by Dr. T. H. Rabbitts (Institute of Cancer Research, United Kingdom).
H358, NIH 3T3, A549, DLD-1, H1915, H1299, SW480 and HeLa cell lines were purchased from American Type Culture Collection (Manassas, VA). HEK293T cell line was a generous gift from Dr. K. Nakanishi’s group (The Ohio State University). MDCK cell line stably expressing GFP-KRasG12V was a generous gift from Drs. J. F. Hancock and Y. Zhou (University of Texas, Houston, TX). All cell lines were maintained in a humidified chamber with 5% CO2 at 37 °C. Antibodies against GAPDH (Cat. No. 5174), phospho-Akt (T308) (Cat. No. 9275), phospho-Akt (S473) (Cat. No. 9271), phospho-MEK1/2(S217/221) (Cat. No. 9121), Akt (Cat. No. 9272), and MEK1/2(Cat. No. 9122) were from Cell Signaling Technology (Danvers, MA). Anti-rabbit IRDye 800CW (Cat. No. 926–32211) and antimouse IRDye 800CW (Cat. NO. 926–32210) were from LI-COR (Lincoln, NE).
Protein Expression and Purification.
His6-KRasG12V(1–186)-HA was expressed in Escherichia coli BL21(DE3) cells in 2 L of Luria broth (LB) containing 50 μg/mL kanamycin sulfate. The cells were grown in a shaker at 37 °C to an OD600 of 0.6 and protein expression was induced by the addition of 0.3 mM IPTG at 30 °C for 5 h. The cells were pelleted by centrifugation at 5000 rpm in Sorvall SLA-3000 rotor, and the cell pellets were stored at −80 °C. For lysis, the cell pellet was thawed at RT and re-suspended in 50 mL of lysis buffer [50 mM Tris pH 7.5, 300 mM NaCl, 10 mM imidazole, 5 mM MgCl2, 5 mM β-mercaptoethanol, 2 cOmplete™ protease inhibitor tablets (Roche), and lysozyme (100 μg/mL)] and stirred at 4 °C for 10 min. Protamine sulfate (250 mg) was added to precipitate nucleic acids, and the lysis solution was stirred at 4 °C for 20 min. For complete lysis, the mixture was sonicated at 70% amplitude on ice for 1 min (2-s pulses with an 8-s pause in between to maintain low temperature). The lysed solution was centrifuged at 15000 rpm in Sorvall SS-34 rotor for 30 min yielding a clear supernatant which was directly loaded onto 2 mL of pre-equilibrated His-Pur™ Cobalt resin (ThermoFisher) and incubated at 4 °C for 1 h to ensure complete binding. Pre-equilibration of the resin was performed by washing with 50 mM Tris pH 7.5, 300 mM NaCl, 10 mM imidazole, 5 mM MgCl2, and 5 mM β-mercaptoethanol. The flow-through was discarded, and the resin was washed with 50 mL of 50 mM Tris, pH 7.5, 300 mM NaCl, 20 mM imidazole, 5 mM MgCl2, and 5 mM β-mercaptoethanol and 100 mL of the same buffer containing 30 mM imidazole. Elution was performed by adding 15 mL of 50 mM HEPES, pH 7.4, 150 mM NaCl, 150 mM imidazole, 2 mM β-mercaptoethanol to the resin and collecting the eluted protein by gravity filtration after 5 min of stationary incubation. The eluted protein was dialyzed into HTRF assay buffer (50 mM HEPES, pH 7.4, 150 mM NaCl, 5 mM MgCl2, and 2 mM β-mercaptoethanol) using Amicon Ultra-15 centrifugal filter units (MWCO: 10 kDa) and concentrated to ~5 mg/mL. Protein concentration was determined by the Bradford assay (Bio-Rad) and the protein was aliquoted and frozen at −80 °C after the addition of 20% (v/v) glycerol.
GST-RBD was expressed in E. coli BL21(DE3) cells in 2 L of LB containing 50 μg/mL ampicillin. The cells were grown in a shaker at 37 °C to an OD600 of 0.6 and protein expression was induced by the addition of 0.1 mM IPTG at 30 °C for 5 h. The cells were pelleted by centrifugation at 5000 rpm in Sorvall SLA-3000 rotor, and the cell pellets were stored at −80 °C. The cell pellet was thawed at RT and re-suspended in 50 mL of lysis buffer (10 mM Na2HPO4, pH 7.4, 137 mM NaCl, and 2.7 mM KCl) supplemented with 5 mM β-mercaptoethanol, 5 mM EDTA, 0.1% Triton-X-100, 200 μg/mL lysozyme, and 2 cOmplete™ protease inhibitor tablets (Roche). This mixture was stirred at 4 °C for 30 min and sonicated at 70% amplitude on ice for 2 min (in 2-s pulses with 8-s pauses in between to maintain low temperature). After sonication, 500 mg of protamine sulfate was added to the lysate and the solution was stirred for an additional 15 min at 4 °C. The lysate was centrifuged at 15000 rpm in in Sorvall SS-34 rotor at 4 °C for 30 min to yield a clear supernatant, which was directly loaded onto 2 mL of pre-equilibrated glutathione-Sepharose 4B resin (GE Healthcare) and incubated at 4 °C for 1 h to ensure complete binding. Pre-equilibration of the resin was performed by washing with 1x PBS (pH 7.4) supplemented with 5 mM β-mercaptoethanol and 5 mM EDTA. The flow-through was discarded and the resin was washed with 100 mL of 1x PBS (pH 7.4) supplemented with 5 mM β-mercaptoethanol, 5 mM EDTA and 0.5% Triton-X-100 and 100 mL of the same buffer without Triton-X-100. The bound protein was eluted with 1x PBS (pH 7.4) containing 10 mM glutathione and 5 mM β-mercaptoethanol, dialyzed into 1x PBS, pH 7.4, 5 mM β-mercaptoethanol using Amicon Ultra-15 centrifugal filter units (MWCO: 10 kDa), and concentrated to ~10 mg/mL. The concentration of the protein was determined using the Bradford assay (Bio-Rad) and the protein was aliquoted and frozen at −80 °C after the addition of 20% glycerol.
Protein Biotinylation.
To biotinylate His6-KRasG12V(1–186)-HA, the freshly thawed protein solution (~200 μM) was incubated with 3 eq. of biotin-(PEG)4-NHS (20 mM stock) for 2 h on ice. The mixture was desalted using Micro Bio-Spin™ 6 Desalting Columns (Bio-Rad, USA, MWCO: 6 kDa) to remove any free biotin. The protein concentration was determined using the Bradford assay and the protein was stored at −80 ⁰C after the addition of 20% (v/v) glycerol.
Nucleotide Exchange of Ras Protein.
Nucleotide exchange was performed by incubating purified KRas (~100 μM) with 200 eq. of EDTA and 40 eq. of GppNHp, GTPγS, or GDP for 1 h on ice.43 After 1 h, the reaction was quenched by the addition of 800 eq. of MgCl2 and incubating the solution for 15 min. This mixture was desalted using Micro Bio-Spin™ 6 Desalting Columns (Bio-Rad, USA, MWCO: 6 kDa) preequilibrated with the proper assay buffer. Protein concentration was re-measured by the Bradford assay and the freshly exchanged protein was used for the assay.
Synthesis of 2nd Generation Peptide Library.
The bicyclic peptide library was synthesized on 800 mg of TentaGel S NH2 resin (90 μm diameter, 0.24 mmol/g; Figure S1). All chemical steps were performed at RT unless otherwise noted. The linker sequence (β-Ala-β-Ala-Arg-Arg-Met) was synthesized with 4 eq. of Fmoc-amino acids and HATU/HOAt/DIPEA (4/4/8 eq.) as coupling agents. A pre-activated mixture was quickly added to the resin and the coupling reaction was allowed to proceed for 40 min, and the resin was washed with DMF and DCM. The Fmoc group was removed by treatment with 20% piperidine (2 × 5 min), followed by washing with DMF. The randomized positions (X4-X1) were synthesized by the split-and-pool method.31 Briefly, the resin was split into 29 equal portions and placed into 29 individual reaction vessels; to each vessel a pre-activated solution of 3.6 eq. of Fmoc-amino acid, 0.4 eq. of CD3CO2D, and the aforementioned coupling agents were added and the reaction was allowed to proceed for 1 h. For coupling reactions of D-Ala, D-Leu, Orn, and D-Lys, the mixture contained 3.6 eq. of Fmoc-amino acid, 0.2 eq. of CD3CO2D, and 0.2 eq. of CH3CO2H. For coupling reactions of β-Ala, Nle, isoAsp, and cis-AcPc, the mixture contained 3.6 eq. of Fmoc-amino acid, 0.2 eq. of CD3CO2D, and 0.2 eq. of CH3CD2CO2H. After the coupling reactions were complete, the resin from the 29 reactions was combined into one reaction vessel and washed with DMF and DCM. Next, a small amount of resin was removed and set aside (to generate the truncation products) while the remaining resin was treated with piperidine as described. After exhaustive washing, the resin was again split into 29 equal aliquots and the split-and-pool process was repeated three more times. After the installation of the X4 residue, all resin aliquots including those previously set aside were combined into a single reaction vessel, washed with DMF, and treated with piperidine. Next, the resin was treated with 2 eq. of Fmoc-Dap(Alloc)-OH, 1 eq. of CD3CO2D, 1 eq. of CH3CD2CO2H, and the coupling agents as previously described. Coupling of the remaining residues in the bicyclic region was performed as described above with the exception of phenylglycine (Phg), for which the coupling reaction contained 3 eq. of Fmoc-L-Phg, 3 eq. of 1-cyano2-ethoxy-2-oxoethylidenaminooxy)dimethylamino-morpholino-carbenium hexafluorophosphate (COMU), and 4 eq. of 2,6-lutidine (30 min). The N-terminal Fmoc group was removed with 10% piperidine (2 × 5 min) and the resin was treated twice with a mixture of trimesic acid/HATU/DIPEA (10/3/15 eq.) (30 min each). The resin was incubated three times with 0.25 eq. of Pd(PPh3)4 and 20 eq. of phenylsilane in DCM to remove the Alloc groups on the C-terminal Dap and an internal Lys (15 min each time). The resin was washed with 2% sodium dimethyldithiocarbamate hydrate in DMF (2 × 10 min), DMF, and DCM and incubated with 1 M HOBt in DMF for 30 min. The resin was treated twice with PyBOP/HOBt/DIPEA (5/5/10 eq.) for 45 min each time to bicyclize the peptide. The peptides were deprotected by treatment with a cocktail containing 92.5/7.5/5/5/2.5/2.5/1.25 (v/v) TFA/phenol/water/thioanisole/triisopropylsilane/2,2’ - (ethylenedioxy)diethanethiol/anisole for 4 h at RT. The resin was exhaustively washed with TFA, DCM, DMF, and H2O before drying under vacuum and storage at −20 °C.
Library Screening.
For the first round of screening (magnetic screening), 800 mg of library resin was washed thoroughly with distilled H2O and incubated with blocking buffer (1x PBS, pH 7.4, 0.05% Tween 20) supplemented with 0.1% BSA for 2 h at 4 °C. The resin was drained, washed once with blocking buffer, and incubated with blocking buffer containing biotinylated His6-KRas(1–186)-HA (150 nM) for 1 h at 4 °C. The resin was drained and washed briefly with blocking buffer to remove any unbound protein. Thirty μL of streptavidin-conjugated Dynabeads (Invitrogen) was mixed with 5 mL of blocking buffer and the mixture was added to and incubated with the library resin for 30 min at 4 °C. Positive beads containing Dynabeads on their surfaces were isolated by placing a magnetic particle concentrator (TA Dynal MPC-1) next to a 15-ml Falcon tube. Positive beads were attracted to the wall of the tube (where the magnet was placed), whereas negative beads settled to the bottom of the tube and were removed by a pipette. Positive beads were transferred to a 2-mL BioRad column and washed with blocking buffer (3x) to remove the magnetic particles, 6 M guanidinium-HCl (3x) to denature and remove any bound protein, and again blocking buffer (3x). For the second round of screening, the positive beads from magnetic screening were incubated with blocking buffer containing 0.1% BSA for 30 min at 4 °C. After washing with blocking buffer, the beads were incubated with biotinylated His6-KRas(1–186)-HA (50 nM) for 30 min at 4 °C. The beads were briefly washed with blocking buffer to remove unbound protein and incubated with 1 μg/mL SA-AP for 10 min at 4 °C. The beads were drained and washed with blocking buffer (3x) and then staining buffer (30 mM Tris-HCl, pH 8.5, 100 mM NaCl, 5 mM MgCl2, 20 μM ZnCl2) (3x). The resin was re-suspended in 5 mL of staining buffer in a Petri dish and 100 μL of a 5bromo-4-chloro-3-indolylphosphate (BCIP) solution (5 mg/mL) was added. The dish was placed on a rocking shaker and the beads were monitored for color changes over 45 min. Turquoise-colored beads were manually isolated under a dissecting microscope and sorted into ‘intensely’, ‘medium’, and ‘lightly’ colored groups.
Sequencing of Library Hits.
Positive beads from library screening were placed in individual microcentrifuge tubes and each treated with 25 μL of cyanogen bromide (40 mg/mL in 70% TFA/H2O) for 12 h in the dark. The solution was dried in a SpeedVac vacuum concentrator (ThermoFisher, USA) and the peptide from each bead was dissolved in 10 μL of 1:1 CH3CN/H2O, sonicated and spun down. A small aliquot of the peptide solution (1.5 μL) was mixed with 0.5 μL of 4-hydroxy-α-cinnamic acid and 1 μL of the mixture was spotted onto a MALDI sample plate. MS analysis was performed on a Bruker Microflex MALDI-TOF instrument, and the data was analyzed by Bruker Daltonics FlexAnalysis 3.3 (Bruker Daltonic Gmb, Germany).
Individual Peptide Synthesis.
Bicyclic peptides were synthesized manually on Rink Amide resin (50–100 mesh, 0.43 mmol/g, Chem-Impex). Typical Fmoc deprotection was performed using 20% piperidine in DMF (v/v) twice for 5 min at RT while mixing. The typical coupling reaction involved 5 eq. of Fmoc-amino acid, 5 eq. HATU and 10 eq. diisopropylethylamine (DIPEA) in DMF for 30 min at RT while mixing. Coupling of L-phenylglycine (Phg) contained 3 eq. of Fmoc-L-Phg, 3 eq. of 1-COMU and 4 eq. of 2,6-lutidine in DMF and was repeated twice (each 30 min incubation). Following Phg coupling, the resin was treated with 15 eq. of acetic anhydride and 15 eq. of DIPEA in DCM for 30 min at RT to acetylate any unreacted amine and the Fmoc group was removed with 10% piperidine (2 × 5 min). After all desired residues were coupled and the N-terminal Fmoc group was removed, the peptide was treated (3x) with 10 eq. of trimesic acid, 3 eq. of HATU, and 15 eq. of DIPEA in DMF for 1 h. The allyl protecting group was removed by treatment in the dark with 0.3 eq. of Pd(PPh3)4 and 10 eq. of phenylsilane in dry DCM (3 × 15 min). The resin was incubated with 1 M HOBt in DMF for 30 min and the peptide was cyclized by using 10 eq. of PyBOP, 10 eq. of HOBt, and 20 eq. of DIPEA for 1 h at RT. Cleavage and deprotection of the bicyclic peptide was performed by incubating the resin with 92.5/2.5/2.5/2.5 (v/v) TFA/TIPS/DMB/H2O for 3 h at RT. The crude peptide was triturated with cold ethyl ether (3x) and purified by reversed-phase HPLC equipped with a semipreparative C18 column. The purity of all peptides used in this work was judged to be ≥95% by analytical HPLC (monitored at 214 nm) and their authenticity was confirmed by high-resolution MALDI-TOF mass spectrometry (Figure S11).
Fluorescent Labeling of Peptides.
To prepare fluorescently labeled B4-27, Fmoc-Lys(Mtt)-OH and two Fmoc-miniPEG residues were added at its C-terminus during peptide synthesis. After the bicyclic peptide synthesis was complete (while peptide still on resin), the 4-methyltrityl (Mtt) group on the C-terminal Lys was removed by treatment with 2% TFA and 1% triispopropylsilane in DCM (6 × 5 min). The peptide was incubated overnight with 2 eq. of 5(6)-carboxyfluorescein or 5(6)-carboxytetramethylrhodamine, 5 eq. of PyBOP, 5 eq. of HOBT, and 10 eq. of DIPEA in DMF at RT. Peptide cleavage, deprotection, purification, and quality assessment were performed as previously described.
HTRF Assay.
Recombinant GST-RBD (50 nM), His6 -KRasG12V(1–186)-HA (50 nM), anti-HA monoclonal antibody labeled with d2 acceptor (CisBio, USA, 2 μg/mL), anti-GST monoclonal antibody labeled with Tb cryptate donor (CisBio, USA, 2.5 μg/mL), and varying concentrations of a peptide (040 μM) were mixed in HTRF assay buffer (total volume 20 μL) in a 384-well plate (Greiner). The plate was incubated for 1 h at RT and the HTRF signals (the donor/acceptor ratio) were measured on a Tecan Infinite M1000 Pro microplate reader using HTRF Terbium program and plotted as a function of the peptide concentration. The data was analyzed using GraphPad Prism 6.0 and IC50 values were obtained by fitting the data to the dose-response inhibition curves.
Fluorescence Polarization.
FP experiments were performed by incubating 100 nM B4-27FAM with varying concentrations of KRas loaded with GTPγS, GDP, or GppNHp in HBS buffer (50 mM HEPES, pH 7.4, 150 mM NaCl) supplemented with 5 mM MgCl2 and 1% 2,2,2-trifluoroethanol (TFE). TFE stabilizes the Switch I region of HRas in a physiologically relevant conformation.44 The solution was incubated for 1 h at RT with gentle mixing and transferred into black 384-well microplates. Fluorescence polarization was measured on Tecan Infinite M1000 plate reader, and titration curves were fitted using GraphPad Prism 6.0 to the following equation:
where FP is the measured polarization, Amin is the minimum FP value, Amax is the maximum FP value, Qb is the quantum yield of the bound fluorophore, Qf is the quantum yield of the free fluorophore, L is the ligand concentration, KD is the dissociation constant, and x is the protein concentration.
Cell Culture.
NIH 3T3, HEK293T, HeLa, and MDCK-GFPKRasG12V cells were cultured in Dulbecco’s modified eagle’s medium (DMEM) supplemented with 10% FBS and 1% penicillin-streptomycin sulfate. H358, H1299, H1915, and DLD-1 cells were cultured in RPMI-1640 medium supplemented with 10% FBS and 1% penicillin-streptomycin sulfate. A549 cells were cultured in F-12K medium supplemented with 10% FBS and 1% penicillin-streptomycin sulfate. Cells were cultured in a humidified incubator at 37 °C in the presence of 5% CO2.
MTT Cell Viability Assay.
H358 cells were seeded in a transparent 96-well plate at a density of 5,000 cells/well in 100 μL of full growth medium and grown overnight. Next day, cells were treated with varying concentrations of a serially diluted peptide (0–40 μM) in 10 μL of assay media containing 10% FBS and incubated at 37 °C with 5% CO2 for 96 h. After incubation, 10 μL of MTT stock solution (Roche) was added to each well. After an additional 4 h incubation at 37 °C, 100 μL of SDS-HCl solubilizing solution was added to each well and the plate incubated overnight at 37 °C. The absorbance of the formazan product was measured at 570 nm on a Tecan M1000 plate reader.
CellTiter-Glo® 2.0 Viability Assay.
Cells (H358, DLD-1, A549, SW480, H1915, H1299, NIH 3T3, or HEK293T) were seeded in an opaque 96-well microplates plate at a density of 5,000 cells/well in 100 μL of full growth medium and grown overnight. Next day, cells were treated with varying concentrations of a serially diluted peptide (0–40 μM) in 10 μL of assay media containing 10% FBS and incubated at 37 °C with 5% CO2 for 96 h. After incubation, the plate was removed and pre-equilibrated to RT before the addition of 100 μL of CellTiter-Glo® 2.0 reagent (Promega, WI, USA) to each well. The plate was incubated for 15 min on a rotary shaker in the dark and the luminescence was detected on a Tecan Infinite M1000 Pro microplate reader.
Annexin-V/PI Cell Apoptosis Assay.
H358 cells were seeded into a 12-well microplate (100,000 cells/well) in 1 mL of RPMI containing 10% FBS and 1% penicillin-streptomycin sulfate. The next day, the media was removed, and each well was washed twice with DPBS before the addition of peptide B4-27 at the desired concentration in 1 mL of RPMI media containing 10% FBS. After 24 h of treatment, the media was collected into a 15-mL falcon tube. Adherent cells were washed with DPBS and removed from each well by treating with 300 μL of 0.25% (w/v) trypsin for 5 min at 37 °C and added to the corresponding falcon tube. Following centrifugation and resuspension in DPBS (repeated twice) to remove any remaining trypsin, the pelleted cells were re-suspended in 100 μL of 1x annexin-binding buffer. Five L of Alexa Fluor® 488 annexin V and 1 μL of propidium iodide (PI, μ100 μg/ml) were added to the cell suspension. The tubes were incubated on a rotary shaker for 15 min at RT to allow for staining, and 400 μL of annexin-binding buffer was added to each sample. The stained cells were immediately analyzed on a BD LSR Fortessa flow cytometer, measuring the emission at both 530 nm and 575 nm.
Intracellular Ras-Raf BRET Assay.
The BRET assay was performed as previously described elsewhere.37 HEK293T cells were seeded into a 6-well microplate (650,000 cells/well) in 1 mL of DMEM containing 10% FBS and 1% penicillin-streptomycin sulfate. The next day, cells were transfected with an appropriate amount of BRET-based Ras biosensor using Lipofectamine 2000 transfection reagent. After 24 h of incubation, cells were re-seeded in a white 96-well plate with clear bottom in OptiMEM medium supplemented with 4% FBS. Cells were incubated for 4 h at 37 °C and then treated with peptide B4-27 (or B3-4) at 0, 0.56, 1.67, 5, or 15 μM in OptiMEM + 4% FBS (final 0.5% DMSO in each well) in quadruplicates. Cells were incubated for 20 h at 37 °C and Coelenterazine 400a (10 μM final concentration) was added to cells right before the measurement. BRET2 reading was measured on a Tecan M1000 plate reader using Dual Luminescence module.
Western Blot Analysis.
H358 cells were maintained in RPMI-1640 supplemented with 10% FBS and 1% penicillin-streptomycin sulfate. Cells (1 × 106 cells/well) were seeded in a 6-well plate overnight, washed with DPBS once, and treated with indicated concentrations of peptide B4-27 in full growth media for 4 h. DMSO was kept at 0.5% (v/v) in all wells. Before harvesting, cells were stimulated with EGF (50 ng/mL) for 10 min. The cells were washed twice with cold DPBS, detached by treatment with 0.25% Trypsin-EDTA solution (0.3 mL/well), and all fractions were collected. After centrifugation in a microcentrifuge (500 g, 5 min), cell pellets were lysed on ice for 30 min in IP lysis buffer (150 μL/sample) supplemented with protease and phosphatase inhibitors. Cell lysates were centrifuged at 15000 rpm in a microcentrifuge equipped with Eppendorf FA-45-24-11 rotor for 10 min, and the extracted proteins in the supernatant were collected. The total protein concentration was measured by using BCA Protein Assay Kit (Thermo, #23235), and equal amounts of protein were loaded onto different lanes of a 12% SDS-PAGE gel. After separation by electrophoresis, the proteins were transferred electrophoretically to a 0.45 μm nitrocellulose membrane at 4 °C. The membrane was first blocked with 10% nonfat dry milk in TBST (20 mM Tris, pH 7.5, 150 mM NaCl, 0.1% (v/v) Tween-20) at 4 °C for 1 h, and then incubated with the proper primary antibody at 4 °C overnight. The antibody sources and conditions used were as follows: anti-MEK and anti-pMEK monoclonal antibodies (1:1000 dilution, Cell Signaling Technologies, 9122 and 9121), anti-Akt and anti-pAkt monoclonal antibodies (1:200 dilution, Cell Signaling Technologies, 9272, 9271 and 9275), anti-GAPDH monoclonal antibodies (1:1000 dilution, Cell Signaling Technologies, 5174). The membrane was washed three times with TBST and incubated with IRDye secondary antibodies (LI-COR, 1:10000 dilution) at RT for 2 h. The membrane was again washed with TBST three times, and fluorescent signals were recorded using a LICOR Odyssey CLx instrument.
Serum Stability Assay.
Diluted human serum (25%; H4522 human serum, Sigma) was incubated at 37 °C for 15 min and added to peptide stock in DMSO to ~100 μM final peptide concentration. The solution was incubated at 37 °C and 50-μL aliquots were withdrawn at various time points. This solution was mixed with 50 μL of 15% trichloroacetic acid (TCA) in MeOH and 50 μL of acetonitrile, and the mixture was stored at 4 °C overnight. Finally, the samples were centrifuged at 15,000 rpm for 10 min in a microcentrifuge equipped with Eppendorf FA-45-24-11 rotor, and the supernatant was analyzed by reversed-phase HPLC equipped with an analytical C18 column (Waters). The amount of peptide remaining at each time point was determined by integrating the area under the peptide peak in the resulting HPLC chromatogram (monitored at 214 nm) and comparing to the peptide amount at time zero.
LDH Release Assay.
H358 cells were seeded into a transparent 96-well microplate (5,000 cells/well) in 100 μL of RPMI containing 10% FBS and 1% penicillin-streptomycin sulfate. The next day, cells were treated with varying concentrations of serially diluted peptide B4-27 (0–20 μM) in 10 μL of assay media containing 10% FBS. Ten μL of sterile H2O was added to the media for the spontaneous LDH release control and 10 μL of 10x lysis buffer was added to the media for the maximum LDH release control. After 45 min of treatment at 37 °C, 50 μL of each sample, 1x LDH positive control, and RPMI containing 10% FBS and 1% penicillin-streptomycin sulfate were transferred to a 96-well flat-bottom plate. Fifty μL of LDH assay solution was added to each well and the plate was incubated on a rotary shaker for 15 min at RT in the dark. The LDH reaction was quenched by the addition of 50 μL of stop solution and the absorbances at 490 and 670 nm were measured on a Tecan M1000 plate reader. % LDH release was calculated according to the manufacturer’s protocol.
Confocal Microscopy.
HeLa and A549 cells were seeded in a 35-mm glass-bottomed microwell dish with 4 compartments (Greiner) at a density of 5 × 104 cells/mL (300 μL in each compartment) and cultured overnight. H358 cells were seeded similarly at a density of 15 × 104 cells/mL and cultured overnight. The cells were gently washed with DPBS and treated for 2 h with fluorescein-labeled B4-27 (3 or5 μM) in phenol-red free DMEM for HeLa and A549 cells or RPMI for H358 cells containing 1% FBS and 1% penicillin-streptomycin sulfate. After removal of the medium, the cells were gently washed with DPBS twice. Cells were imaged immediately on a Nikon A1R live-cell confocal laser scanning microscope (ECLIPSE Ti-E automated, inverted) equipped with a 20x or 100x oil objective (1.45 N.A.) and a heated (37 °C) chamber supplied with 5% CO2. The data were analyzed using NIS-Elements AR.
For co-localization analysis, MDCK cells stably expressing GFP-KRasG12V were seeded in a 35-mm glass-bottomed microwell dish with 4 compartments (Greiner) at a density of 5 × 104 cells/mL (300 μL in each compartment) and cultured overnight. The cells were gently washed with DPBS and treated for 2 h with TMR-labeled B4-27 (5 μM) in phenol red-free DMEM containing 1% FBS and 1% penicillin-streptomycin sulfate. After incubation, the media was aspirated, and the cells were gently washed with DPBS twice before addition of fresh phenol red-free media. Cells were imaged immediately on a Nikon A1R live-cell confocal laser scanning microscope (ECLIPSE Ti-E automated, inverted) equipped with a 100x oil objective (1.45 N.A.) and a heated (37 °C) chamber supplied with 5% CO2. For the red channel (TMR), the laser line with λEx 561 nm was set at 0.5% laser power. For the green channel (GFP), the laser line with λEx 487 nm was set at 0.4 % laser power. Images were acquired in Channel Series mode (i.e., each fluorescent channel was imaged independently, rather than simultaneously) to eliminate spectral bleed through. Images were denoised and analyzed using NIS-Elements AR. Colocalization on the membrane was analyzed inside the regions of interest (ROIs) using Pearson’s correlation constant.
Flow Cytometry.
HeLa, A549 and H358 cells were seeded in a 12-well plate at a density 1.5 × 105 cells per well and cultured overnight. Next day, 5 μM fluorescein-labeled B4-27, CPP12, B3-4 or B4 peptides were added in DMEM or RPMI supplemented with 1% FBS and 1% penicillin-streptomycin sulfate and incubated at 37 °C for 2 h. The cells were washed with cold DPBS twice, detached from the plate with 0.25% trypsin, diluted into cold DPBS and pelleted at 300 g for 5 min at 4 °C. The supernatant was discarded and the cells were washed twice with cold DPBS and re-suspended in 200 μL of cold DPBS. The samples were analyzed on a BD FACS LSR II flow cytometer. For the fluorescein-labelled peptides, a 488-nm laser was used for excitation, and the fluorescence was analyzed in the FITC channel.
Mouse Xenograft Models.
All animal experiments were performed in compliance with institutional animal care guidelines and according to committee-approved protocols. For A549 xenografts, ~1.5 × 106 A549 cells were injected subcutaneously into 8-week-old male nude mice. Tumors were allowed to grow to a size of ~40 mm3, and six mice per group were treated with 100 μL injections of vehicle (1.5% (vol/vol) DMSO in saline) or B4-27 (1 or 5 mg/kg in 1.5% (vol/vol) DMSO in saline) via tail vein injection daily for 9 days and tumor volumes were measured every other day. After 9 days, mice were sacrificed and tumors were collected. Pictures of tumors were taken, and tumor weight was measured.
For H358 xenografts, ~2 × 106 H358 cells were injected subcutaneously into 8-week-old male nude mice. Tumors were allowed to grow to a size of ~35 mm3, and seven mice per group were treated with 100-μL injections of vehicle (1.5% (vol/vol) DMSO in saline) or B4-27 (5 mg/kg in 1.5% (vol/vol) DMSO in saline) via tail vein injection daily for 9 days. Body weights were recorded and monitored for any signs of toxicity. Tumors were measured every other day and excised and retained for further analysis on day 9.
Histology and Immunohistochemistry.
Tumors were harvested from mice and fixed with 4% formaldehyde buffer for 24 h at RT. Paraffinized specimens were then sectioned (4-μm thick slices), and deparaffinized by xylenes (3 × 10 min) each followed by dipping in graded alcohols (100%, 95%, 80% and 70%) 5 min each. The slices were quenched for endogenous peroxidase activity with 3% H2O2 solution for 15 min and autoclaved at 100 °C for 30 min for antigen retrieval in citric acid buffer, pH 6.0. Sections were blocked with 10% normal goat serum for 30 min at RT and incubated with specific rabbit primary antibodies against Ki-67 (1:200 dilution, Abcam, ab16667) and p-ERK1/2 (1:200 dilution, Cell Signaling Technology, 4376) overnight at 4 °C. Subsequently, the sections were incubated with Horse anti-Rabbit IgG secondary antibody (1:500 dilution, BA-1100, Vector labs, Burlingame, CA.) for 30 min at RT. After incubation with avidin-biotin ABC complex (PK-4000, Vector labs, Burlingame, CA) followed by staining with DAB solution (SK-4105, Vector labs, Burlingame, CA), slides were washed thoroughly with tap water, counterstained with hematoxylin (H-3401, Vector labs, Burlingame, CA) for 5 s and dipped briefly in graded alcohols (70%, 80%, 95% and 100%), and in xylenes (2 × 5 min). Finally, slides were mounted and imaged.
Statistical Analysis.
Statistical calculations were performed using GraphPad Prism® 6.0 (GraphPad Software, Inc.) and data are expressed as mean ± SD. Statistical significance was determined using One-way ANOVA. P values ≤0.05 were considered significant.
Supplementary Material
ACKNOWLEDGMENT
We thank Drs. J. F. Hancock and Y. Zhou (University of Texas, Houston, TX) for MDCK cell line stably expressing GFPKRasG12V, Dr. T. H. Rabbitts (Institute of Cancer Research, United Kingdom) for plasmids encoding RLuc8-KRas and GFP-CRAF RBD fusion proteins, and The Ohio State University CCIC for assistance with high-resolution mass spectrometry and flow cytometry.
Funding Sources
Financial support from the National Institutes of Health (GM122459 and CA234124) is gratefully acknowledged.
ABBREVIATIONS
- BRET
bioluminescence resonance energy transfer
- CPP
cell-penetrating peptide
- FAM
5(6)-carboxyfluorescein
- FBS
fetal bovine serum
- FITC
fluorescein isothiocyanate
- FP
fluorescence polarization
- GFP
green fluorescent protein
- GST
glutathione-S-transferase
- HTRF
homogenous time-resolved fluorescence
- MDCK
Madin-Darby canine kidney
- PPI
protein-protein interaction
- RBD
Ras-binding domain
- TMR
tetramethylrhodamine
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/
Table S1, Sequences and Ras-binding affinities of Cyclorasin B3 analogs; Table S2, Sequences of hit peptides derived from 2nd generation library screening; Figure S1, Synthesis of the 2nd generation bicyclic linker library; Figure S2, Library screening scheme; Figure S3, Histogram with the abundance of amino acids in library screening hits; Figure S4, Schematic representation of HTRF assay; Figure S5, Binding of B4-27FAM to five arbitrarily selected proteins by FP; Figures S6–S8, Confocal microscopy images of fluorescently-labeled B4-27; Figure S9, LDH release and serum stability assays of B4-27; Figure S10, In vivo characterization of B4-27 in H358 mice xenograft; Figure S11, Analytical data (HPLC purity and HRMS data) for all compounds.
Molecular formula strings of peptides.
The authors declare the following competing financial interests: A patent application has been filed on the findings of this work. D.P. is a co-founder and shareholder of Entrada Therapeutics, Inc.
REFERENCES
- (1).Papke B; Der CJ Drugging RAS: Know the enemy. Science 2017, 355, 1158–1163. [DOI] [PubMed] [Google Scholar]
- (2).Wright LP; Philips MR Thematic review series: Lipid posttranslational modifications CAAX modification and membrane targeting of Ras. J. Lipid Res 2006, 47, 883–891. [DOI] [PubMed] [Google Scholar]
- (3).Parker JA; Mattos C The Ras–membrane interface: Isoform-specific differences in the catalytic domain. Mol. Cancer Res 2015, 13, 595–603. [DOI] [PubMed] [Google Scholar]
- (4).Simanshu DK; Nissley DV; McCormick F RAS proteins and their regulators in human disease. Cell 2017, 170, 17–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Keeton AB; Salter EA; Piazza GA The RAS–effector interaction as a drug target. Cancer Res. 2017, 77, 221–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Barbacid M Ras genes. Annu. Rev. Biochem 1987, 56, 779–827. [DOI] [PubMed] [Google Scholar]
- (7).Prior IA; Hood FE; Hartley JL The frequency of Ras mutations in cancer. Cancer Res. 2020, 80, 2969–2974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Moore AR; Rosenberg SC; McCormick F; Malek S RAS-targeted therapies: is the undruggable drugged? Nat. Rev. Drug Discov 2020, 19, 533–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Ostrem JM; Peters U; Sos ML; Wells JA; Shokat KM KRas(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 2013, 503, 548–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Janes MR; Zhang J; Li L-S; Hansen R; Peters U; Guo X; Chen Y; Babbar A; Firdaus SJ; Darjania L; Feng J; Chen JH; Li S; Li S; Long YO; Thach C; Liu Y; Zarieh A; Ely T; Kucharski JM; Kessler LV; Wu T; Yu K; Wang Y; Yao Y; Deng X; Zarrinkar PP; Brehmer D; Dhanak D; Lorenzi MV; Hu-Lowe D; Patricelli MP; Ren P; Liu Y Targeting KRAS mutant cancers with a covalent G12C-specific inhibitor. Cell 2018, 172, 578–589. [DOI] [PubMed] [Google Scholar]
- (11).Lanman BA; Allen JR; Allen JG; Amegadzie AK; Ashton KS; Booker SK; Chen JJ; Chen N; Frohn MJ; Goodman G; Kopecky DJ; Liu L; Lopez P; Low JD; Ma V; Minatti AE; Nguyen TT; Nishimura N; Pickrell AJ; Reed AB; Shin Y; Siegmund AC; Tamayo NA; Tegley CM; Walton MC; Wang H-L; Wurz RP; Xue M; Yang KC; Achanta P; Bartberger MD; Canon J; Hollis LS; McCarter JD; Mohr C; Rex K; Saiki AY; San Miguel T; Volak LP; Wang KH; Whittington DA; Zech SG; Lipford JR; Cee VJ Discovery of a covalent inhibitor of KRAS G12C (AMG 510) for the treatment of solid tumors. J. Med. Chem 2020, 63, 52–65. [DOI] [PubMed] [Google Scholar]
- (12).Fell JB; Fischer JP; Baer BR; Blake JF; Bouhana K; Briere DM; Brown KD; Burgess LE; Burns AC; Burkard MR; Chiang H; Chicarelli MJ; Cook AW; Gaudino JJ; Hallin J; Hanson L; Hartley DP; Hicken EJ; Hingorani GP; Hinklin RJ; Mejia MJ; Olson P; Otten JN; Rhodes SP; Rodriguez ME; Savechenkov P; Smith DJ; Sudhakar N; Sullivan FX; Tang TP; Vigers GP; Wollenberg L; Christensen JG; Marx MA Identification of the clinical development candidate MRTX849, a covalent KRAS G12C inhibitor for the treatment of cancer. J. Med. Chem 2020, 63, 6679–6693. [DOI] [PubMed] [Google Scholar]
- (13).Pei D; Chen K; Liao H Targeting Ras with macromolecules. Cold Spring Harb. Perspect. Med 2018, 8, a031476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Cochet O; Kenigsberg M; Delumeau I; Virone-Oddos A; Multon MC; Fridman WH; Schweighoffer F; Teillaud JL; Tocqué B Intracellular expression of an antibody fragment-neutralizing p21 ras promotes tumor regression. Cancer Res. 1998, 58, 1170–1176. [PubMed] [Google Scholar]
- (15).Tanaka T; Rabbitts TH Interfering with RAS-effector protein interactions prevent RAS-dependent tumour initiation and causes stop-start control of cancer growth. Oncogene 2010, 29, 6064–6070. [DOI] [PubMed] [Google Scholar]
- (16).Shin S-M; Kim J-S; Park S-W; Jun S-Y; Kweon H-J; Choi D-K; Lee D; Cho Y-B; Kim Y-S Direct targeting of oncogenic RAS mutants with a tumor-specific cytosol-penetrating antibody inhibits RAS mutant–driven tumor growth. Sci. Adv 2020, 6, eaay2174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Cetin M; Evenson WE; Gross GG; Jalali-Yazdi F; Krieger D; Arnold D; Takahashi TT; Roberts RW RasIns: Genetically encoded intrabodies of activated Ras proteins. J. Mol. Biol 2017, 429, 562–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Spencer-Smith R; Koide A; Zhou Y; Eguchi RR; Sha F; Gajwani P; Santana D; Gupta A; Jacobs M; Herrero-Garcia E; Cobbert J; Lavoie H; Smith M; Rajakulendran T, Dow-dell E, Okur MN, Dementieva I, Sicheri F, Therrien M, Hancock JF, Ikura M, Koide S, O’Bryan JP Inhibition of RAS function through targeting an allosteric regulatory site. Nat. Chem. Biol 2017, 13, 62–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Wiechmann S; Maisonneuve P; Grebbin BM; Hoffmeister M; Kaulich M; Clevers H, Rajalingam K, Kurinov I, Farin HF; Sicheri F Conformation-specific inhibitors of activated Ras GTPases reveal limited Ras dependency of patient-derived cancer organoids. J. Biol. Chem 2020, 295, 4526–4540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Wu X; Upadhyaya P; Villalona-Calero MA; Briesewitz R; Pei D Inhibition of Ras–effector interactions by cyclic peptides. Medchemcomm 2013, 4, 378–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Upadhyaya P; Qian Z; Habir NAA; Pei D Direct Ras inhibitors identified from a structurally rigidified bicyclic peptide library. Tetrahedron 2014, 70, 7714–7720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Sakamoto K; Kamada Y; Sameshima T; Yaguchi M; Niida A; Sasaki S; Miwa M; Ohkubo S; Sakamoto J; Kamaura M; Cho N; Tani A KRas(G12D)-selective inhibitory peptides generated by random peptide T7 phage display technology. Biochem. Biophys. Res. Commun 2017, 484, 605–611. [DOI] [PubMed] [Google Scholar]
- (23).Zhang Z; Gao R; Hu Q; Peacock H; Peacock DM; Dai S; Shokat KM; Suga H GTP-state-selective cyclic peptide ligands of KRas(G12D) block its interaction with Raf. ACS Cent. Sci 2020, 6, 1753–1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Upadhyaya P; Qian Z; Selner NG; Clippinger SR; Wu Z; Briesewitz R; Pei D Inhibition of Ras signaling by blocking Ras-Effector interactions with cyclic peptides. Angew. Chem. Int. Ed 2015, 127, 7712–7716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Trinh TB; Upadhyaya P; Qian Z; Pei D Discovery of a direct Ras inhibitor by screening a combinatorial library of cell-permeable bicyclic peptides. ACS Comb. Sci 2016, 18, 75–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Sakamoto K; Masutani T; Hirokawa T Generation of KS-58 as the first KRas(G12D)-inhibitory peptide presenting anti-cancer activity in vivo. Sci. Rep 2020, 10, 21671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Patgiri A; Yadav KK; Arora PS; Bar-Sagi D An orthosteric inhibitor of the Ras-Sos interaction. Nat. Chem. Biol 2011, 7, 585–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Leshchiner ES; Parkhitko A; Bird GH; Luccarelli J; Bellairs JA; Escudero S; Opoku-Nsiah K; Godes M; Perrimon N; Walensky LD Direct inhibition of oncogenic KRAS by hydrocarbon-stapled SOS1 helices. Proc. Natl. Acad. Sci. U. S. A 2015, 112, 1761–1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Hong SH; Yoo DY; Conway L; Richards-Corke KC; Parker CG; Arora PS A Sos proteomimetic as a pan-Ras inhibitor. Proc. Natl. Acad. Sci. U. S. A 2021, 118, e2101027118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Vinogradov AA; Yin Y; Suga H Macrocyclic peptides as drug candidates: Recent progress and remaining challenges. J. Am. Chem. Soc 2019, 141, 4167–4181. [DOI] [PubMed] [Google Scholar]
- (31).Lam KS; Salmon SE; Hersh EM; Hruby VJ; Kazmierski WM; Knapp RJ A new type of synthetic peptide library for identifying ligand-binding activity. Nature 1991, 354, 82–84. [DOI] [PubMed] [Google Scholar]
- (32).Lian W; Upadhyaya P; Rhodes CA; Liu Y; Pei D Screening bicyclic peptide libraries for protein-protein interaction inhibitors: Discovery of a Tumor Necrosis Factor-α antagonist. J. Am. Chem. Soc 2013, 135, 11990–11995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Liu T; Qian Z; Xiao Q; Pei D High-throughput screening of one-bead-one-compound libraries: Identification of cyclic peptidyl inhibitors against calcineurin/NFAT interaction. ACS Comb. Sci 2011, 13, 537–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Degorce F; Card A; Soh S; Trinquet E; Knapik GP; Xie B HTRF: A technology tailored for drug discovery - a review of theoretical aspects and recent applications. Curr. Chem. Genomics 2009, 3, 22–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Butzler M; Worzella T; Hungriano M; Osorio F; Hanegraaff I; Cowan C Automated cytotoxicity profiling using CyBi®-FeliX instrument, cell health assay and thaw-and-use cells. Promega Corporation Website 2014, tpub_159.AccessedJune 10, 2021. https://www.promega.com/resources/pubhub/automated-profiling-using-the-cybi-felix-instrument-cell-health-assays-and-thawand-use-cells/ [Google Scholar]
- (36).Qian Z; Martyna A; Hard RL; Wang J; Appiah-Kubi G; Coss C; Phelps MA; Rossman JS; Pei D Discovery and mechanism of highly efficient cyclic cell-penetrating peptides. Biochemistry 2016, 55, 2601–2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Bery N; Rabbitts TH Bioluminescence Resonance Energy Transfer 2 (BRET2)‐based RAS biosensors to characterize RAS inhibitors. Curr. Protoc. Cell Biol 2019, 83, 1–22. [DOI] [PubMed] [Google Scholar]
- (38).Commisso C; Davidson S; Soydaner-Azeloglu R; Parker SJ; Kamphorst JJ; Hackett S; Grabocka E; Nofal M; Drebin JA; Thompson CB; Rabinowitz JD; Metallo CM; Vander Heiden MG; Bar-Sagi D Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature 2013, 497, 633–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Hunter JC; Manandhar A; Carrasco MA; Gurbani D; Gondi S; Westover KD Biochemical and structural analysis of common cancer-associated KRAS mutations. Mol. Cancer Res 2015, 13, 1325–35. [DOI] [PubMed] [Google Scholar]
- (40).Singh A; Settleman J Oncogenic KRas “addiction” and synthetic lethality. Cell Cycle 2009, 8, 2676–2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Sos ML; Fischer S; Ullrich R; Peifer M; Heuckmann J,M; Koker M; Heynck S; Stuckrath I; Weiss J; Fischer F; Michel K; Goel A; Regales L; Politi KA; Perera S; Getlik M; Heukamp LC; Ansen S, Zander T, Beroukhim R; Kashkar H; Shokat KM; Sellers WR; Rauh D; Orr C; Hoeflich KP; Friedman L; Wong K-K; Pao W; Thomas RK Identifying genotype-dependent efficacy of single and combined PI3K- and MAPK-pathway inhibition in cancer. Proc. Natl. Acad. Sci. U. S. A 2009, 106, 18351–18356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Zhong H; Sanchez C; Spitrzer D; Plambeck-Suess S; Gibbs J; Hawkins WG; Denardo D; Gao F; Pufahl RA; Lockhart AC; Xu M; Linehan D; Weber J; Wang-Gillam A Synergistic effects of concurrent blockade of PI3K and MEK pathways in pancreatic cancer preclinical models. PLoS ONE 2013, 8, e77243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Moghadamchargari Z; Huddleston J; Shirzadeh M; Zheng X; Clemmer DE; M Raushel F; Russell DH; Laganowsky A Intrinsic GTPase activity of K-RAS monitored by native mass spectrometry. Biochemistry 2019, 58, 3396–3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Buhrman G; De Serrano V; Mattos C Organic solvents order the dynamic switch II in Ras crystals. Structure 2003, 11, 747751. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.