

Abstract
Background:
Activin receptor-like kinase 1 (ALK1) is an endothelial transmembrane serine threonine kinase receptor for BMP family ligands that plays a critical role in cardiovascular development and pathology. Loss-of-function mutations in the ALK1 gene cause type 2 hereditary hemorrhagic telangiectasia (HHT), a devastating disorder that leads to arteriovenous malformations (AVMs). Here we show that ALK1 controls endothelial cell polarization against the direction of blood flow and flow-induced endothelial migration from veins through capillaries into arterioles.
Methods:
Using Cre lines that recombine in different subsets of arterial, capillary-venous or endothelial tip cells, we showed that capillary-venous Alk1 deletion was sufficient to induce AVM formation in the postnatal retina.
Results:
ALK1 deletion impaired capillary-venous endothelial cell polarization against the direction of blood flow in vivo and in vitro. Mechanistically, ALK1 deficient cells exhibited increased integrin signaling interaction with VEGFR2, which enhanced downstream YAP/TAZ nuclear translocation. Pharmacological inhibition of integrin or YAP/TAZ signaling rescued flow migration coupling and prevented vascular malformations in Alk1 deficient mice.
Conclusions:
Our study reveals ALK1 as an essential driver of flow-induced endothelial cell migration and identifies loss of flow-migration coupling as a driver of AVM formation in HHT disease. Integrin-YAP/TAZ signaling blockers are new potential targets to prevent vascular malformations in HHT patients.
Keywords: HHT, arteriovenous malformations, mechanotransduction, Integrin, Hippo, BMP, VEGF
Introduction
Hereditary hemorrhagic telangiectasia (HHT) is an inherited autosomal dominant vascular disorder that causes arteriovenous malformations (AVMs) in more than 1.4 million people worldwide1. More than 90% of HHT cases are caused by heterozygous mutations in the endothelial surface receptors ENG (endoglin, mutated in HHT1) and ALK1 (ACVRL1, mutated in HHT2), and mutations in SMAD4 cause a combined juvenile polyposis-HHT syndrome that accounts for <5% of HHT cases2–5. ALK1 and ENG are receptors for TGF-β superfamily members BMP9 and BMP106, 7. Ligand binding activates ALK1/ENG receptor signaling to cytoplasmic SMAD1/5/8, which subsequently complex with SMAD4 and translocate into the nucleus to regulate gene expression8. Thus, known HHT mutations affect different components of an endothelial signaling pathway that prevents vessels from forming AVMs. A recent study has shown that somatic second-hits inactivating the remaining intact ALK1 or ENG allele occurred in the lesions, supporting that vascular malformations in HHT are caused by a two-hit mechanism9.
Whereas the genetics of AVM have been well studied, the underlying cellular and molecular principles are not fully understood, thus limiting the development of new treatment options. AVMs are direct connections between arteries and veins that lack an intermediate capillary bed2. AVMs in HHT patients appear most often in the skin, oral cavity, nasal, and gastrointestinal (GI) tract mucosa, lung, liver, and brain. Small AVMs in the skin and mucus membranes are called telangiectasias; rupture of these lesions leads to frequent epistaxis, GI bleeding, and anemia, all of which are major quality of life issues for HHT patients10. Larger AVMs in liver, lung, or brain may additionally cause life-threatening conditions such as high output heart failure and stroke11. We and others previously showed that pan-endothelial knockout of Alk1 using Alk1f/f Cdh5 CreERT2 in neonates led to AVMs in retina, brain and internal organs, indicating that endothelial ALK1 is necessary for proper vascular development12, 13. However, what types of ECs are responsible and how AVMs develop remain largely unknown.
Previous data from us and others have shown that BMP9/10-ALK1-ENG-SMAD4 signaling is enhanced by flow, and initiates a negative feedback signal that dampens flow-induced activation of AKT, thereby coordinating proper vascular remodeling12, 14–17. Mechanistically, blocking BMP9-ALK1-ENG signaling promotes endothelial phosphoinositide 3-kinase (PI3K)/AKT activation. ALK1 deficient ECs showed enhanced phosphorylation of the PI3K target AKT and vascular endothelial growth factor receptor 2 (VEGFR2)13, 18. Pharmacological VEGFR2 or PI3K inhibition prevented AVM formation in Alk1 deficient mice and decreased diameter of AVMs in Eng mutants19. Moreover, an increase in PI3K signaling has been recently confirmed in cutaneous telangiectasia biopsies of patients with HHT220, 21.
Here we investigated the origin of AVM-causing cells using novel Cre lines that delete Alk1 in subsets of ECs. In doing so, we observed that ECs in remodeling vessels move against the direction of blood flow, while maintaining vascular integrity. In response to the physical forces such as wall shear stress exerted by blood, ECs polarize their Golgi apparatus in front of the nucleus (front-rear polarity) and migrate against the blood flow from veins towards arteries. We further provide evidence that ALK1 contributes to flow-migration coupling via VEGFR2-integrin signaling and downstream YAP/TAZ nuclear translocation. Collectively, the data show that ALK1 controls flow-induced cell migration to prevent AVM formation and identify new targets with the potential to prevent vascular malformations in HHT patients.
Methods
The data and methods supporting this study’s findings are available from the corresponding author on request. Detailed methods are available in the Supplemental materials.
Mice
All animal experiments were performed under a protocol approved by Institutional Animal Care Use Committee of Yale University. The supplemental material contains a list of all mouse strains and protocols.
Statistical Analysis
All data are shown as mean ± standard error of the mean (SEM) and were analyzed using Student’s t-test, one-way ANOVA with Sidak’s multiple comparison test, and two-way ANOVA with Tukey’s multiple comparison test or Sidak’s multiple comparison test. All the statistical analyses were done using Prism 6 (GraphPad Software Inc., USA). ns: nonsignificant, p>0.05, * p < 0.05, ** p < 0.01, *** p < 0.001.
Results
Endothelial lineage tracing reveals flow-migration coupling in retinal vessels
To track the dynamics of endothelial flow-migration coupling in the mouse retina, we used three CreERT2 lines that recombine in subsets of ECs. These include Major Facilitator Superfamily Domain Containing 2a (MFSD2A), which recombines venous and capillary ECs but not arteries or tip cells in the brain vasculature22–24; Endothelial Cell-Specific Molecule 1 (ESM1), which recombines tip cells and their progeny25, 26; and the artery-specific BMX non-receptor tyrosine kinase (BMX)27 lines. We intercrossed these lines with mTmG reporter mice28 to lineage-trace GFP positive Mfsd2a, Esm1 and Bmx expressing ECs. Tamoxifen (Tx) was injected 12 h, 24 h and 48 h prior to sacrifice at P6 (Figure 1A–1I). At 12 h post injection, Mfsd2a-positive cells were absent from the tip cell position, but labeled veins and capillaries in the vascular plexus, as well as the distal pole of arterioles (Figure 1A). Esm1-positive cells were restricted to the tip position, while Bmx CreERT2 positive cells were located in the proximal part of retinal arterioles close to the optic nerve (Figure 1B and 1C and Figure IA in the Data Supplement). Hence the three CreERT2 lines labeled distinct and non-overlapping endothelial cell populations at this time point.
Figure 1. Retinal endothelial cell lineage tracing.
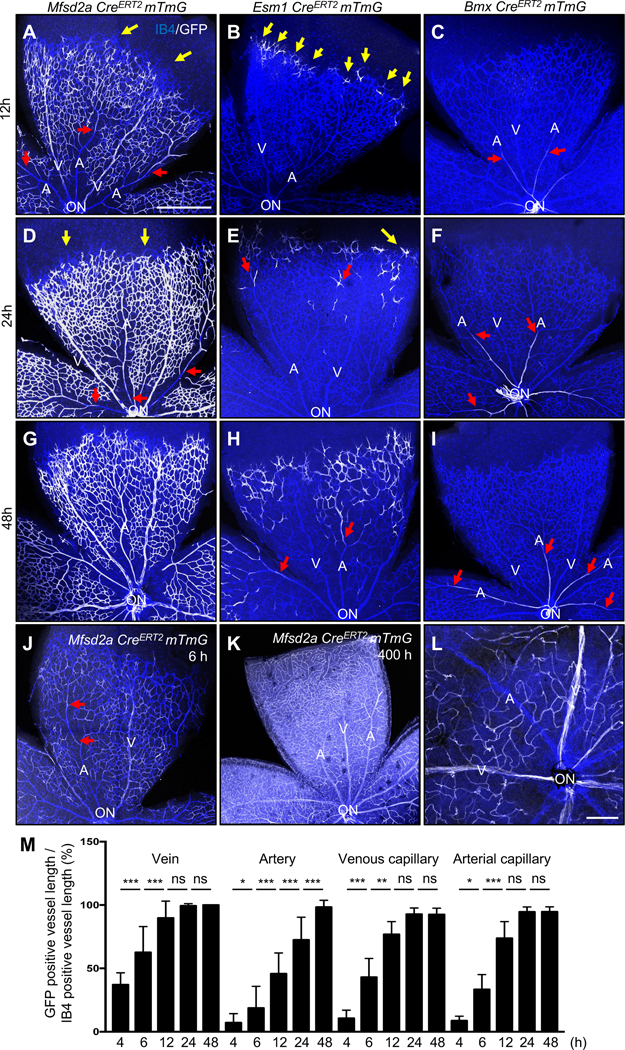
(A-I) P6 retina flat mount images labeled with IB4 (blue) and GFP (white) from Mfsd2a CreERT2 mTmG (A, D, G), Esm1 CreERT2 mTmG (B, E, H) and Bmx CreERT2 mTmG (C, F, I) mice injected with 100 μg tamoxifen (Tx) at P5.5 (12 h, A-C), P5 (for 24 h, D-F) and P4 (for 48 h, G-I) and dissected at P6. (J) 100 μg Tx was injected at P6 and dissected after 6 h in Mfsd2a CreERT2 mTmG mice. (K) 100 μg Tx was injected at P4 and dissected after 400 h (P21) (L) 2 mg/kg Tx was injected in P20 Mfsd2a CreERT2 mTmG mice and dissect at P21. Yellow arrows indicate tip cells and red arrows indicate location of GFP-expressing ECs in arteries. (M) Quantification of Mfsd2a CreERT2 mTmG GFP expressing vessel length over IB4 positive vessel length from optic nerve. n = 6–8 retinas per time point. P-value < 0.001, Error bars: SEM. *P-value < 0.05, **P-value < 0.01, ***P-value < 0.001, ns: nonsignificant, One-way ANOVA with Sidak’s multiple comparisons test. ON: optic nerve, V: vein, A: artery, Scale bars: 500 μm (A-K) and 50 μm (L).
24 h and 48 h after injection, Mfsd2a-positive cells were progressively colonized the arteries from the distal to the proximal part (Figure 1D and 1G). Esm1-positive cells were seen at the tip position and moving towards the distal parts of the arterioles at 24 h and 48 h after injection (Figure 1E and 1H), while Bmx-positive cells remained confined to the proximal arterioles (Figure 1F and 1I and Figure IA in the Data Supplement). Very few Mfsd2a-GFP positive cells were detected in arteries 4 h and 6 h post Tx injection (Figure IB in the Data Supplement and Figure 1J), while 400 h after P4 Tx injection, ie at P21, most of the retinal endothelium was GFP-positive (Figure 1K). By contrast, a single Tx injection at P20 labeled venous and capillary endothelium, but not arteries at P21 (Figure 1L), demonstrating that ECs of venous and capillary origin migrate against the direction of flow into neighboring arteries during vascular remodeling. To test the requirement of blood flow as a driver for the migration of the Mfsd2a-GFP positive cells, we injected Tx to P5 Mfsd2a CreERT2 mice for 12 h, then isolated the retinas and cultured them for another 12 h, which reduced arterial colonization by GFP-positive cells (Figure IC in the Data Supplement). To quantify displacement of Msfd2a-GFP positive cells, we measured the relative length of GFP-positive area in retinal arteries, veins and capillaries at different time points (Figure 1M). After 12 h, about 90% of venous and capillary vessel area was occupied by GFP-positive cells, while only 50% of the distal arterial vessel area was occupied by GFP-positive cells and this gradually increased over time until 48 h (Figure 1M), demonstrating quantifiable displacement of capillary and venous ECs towards arteries over time.
Alk1 deletion in capillary and venous ECs causes AVMs
To determine the origin of AVM forming cells in Alk1 mutants, we next intercrossed Mfsd2a, Esm1 and Bmx CreERT2 mice with Alk1f/f mTmG reporter mice. Tx was injected at P4 and mice were analyzed at P6 (Figure 2A). Efficient Alk1 deletion was verified in all three lines using immunostaining (Figure IIA–P in the Data Supplement). Interestingly, venous and capillary endothelial Alk1 deletion using the Mfsd2a CreERT2 driver line led to numerous AVMs in the retina (Figure 2B and 2E). By contrast, neither Alk1f/f Esm1 CreERT2 nor Alk1f/f Bmx CreERT2 mutants displayed any retinal AVMs (Figure 2C–2E). We analyzed the presence of retinal and brain AVMs by injection of latex dye into the left ventricle of P6 Alk1f/f Mfsd2a CreERT2 and control littermates (Figure 2F–2I). The latex dye does not cross the capillary beds and was retained within the arterial branches in Alk1f/f brain and retina (Figure 2F and 2G). In the Alk1f/f Mfsd2a CreERT2 mutants, the latex penetrated both the venous as well as the arterial branches via AVMs in the retina and brain (Figure 2H and 2I). To see whether Alk1f/f Esm1 CreERT2 and Alk1f/f Bmx CreERT2 could develop AVMs by longer-term exposure of Tx, Tx was injected at P1 and mice were analyzed at P6 (Figure IIQ in the Data Supplement). Neither Alk1f/f Esm1 CreERT2 nor Alk1f/f Bmx CreERT2 mutants exhibited any AVMs (Figure IIR and IIS in the Data Supplement).
Figure 2. Capillary-venous loss of ALK1 leads to AVMs.
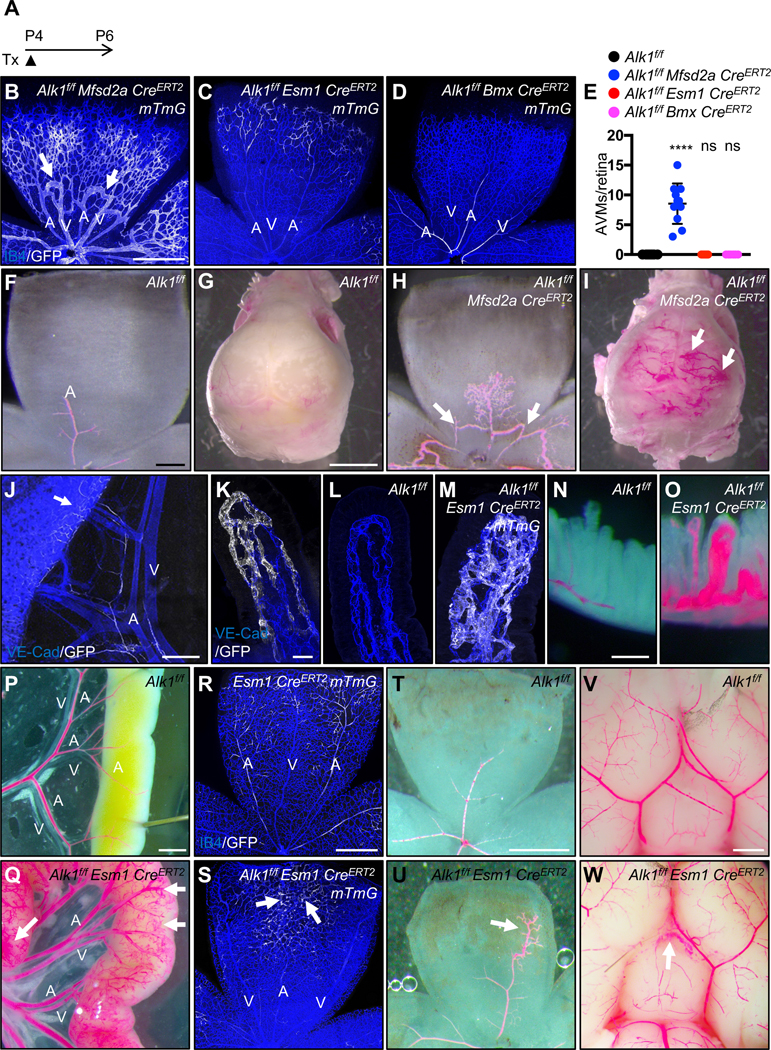
(A) Schematic representation of the experimental strategy used to delete Alk1 in mice (P4-P6). (B-D) P6 retina flat mount images labeled with IB4 (blue) and GFP (white) from Alk1f/f Mfsd2a CreERT2 mTmG (B), Alk1f/f Esm1 CreERT2 mTmG (C) and Alk1f/f Bmx CreERT2 mTmG pups (D) injected with 100 μg Tx at P4 and dissected at P6. White arrows indicate AVMs. (E) Quantification of AVM number. n = 6–11 mice per group. Error bars: SEM. **** P-value < 0.0001, ns: nonsignificant, One-way ANOVA with Sidak’s multiple comparisons test. (F-I) Vascular labeling with latex dye (red) of retinal and brain vessels in Alk1f/f (F and G) and Alk1f/f Mfsd2a CreERT2 (H and I) P6 pups. White arrows indicate AVMs. (J and K) GFP (white) and VE-Cad (blue) staining of mesentery and gastrointestinal (GI) tract (J) and lacteals (K) from P14 Esm1 CreERT2 mTmG. 100 μg Tx was injected at P4. An arrow indicates Esm1 positive capillary ECs (J). (L and M) VE-Cad (blue) and GFP (white) staining of jejunum lacteals from P14 Alk1f/f (L) and Alk1f/f Esm1 CreERT2 mTmG (M). (N-Q and T-W) 100 μg Tx was injected at P4 and dissected at P12. Vascular labeling with latex dye (red) of villi, GI tracts, retinas and brains in Alk1f/f (N,P,T and V) and Alk1f/f Esm1 CreERT2 (O, Q, U and W) P12 pups. (R and S) 100 μg Tx was injected at P4 and dissected at P12 (R and S). IB4 (blue) and GFP (white) staining of retinal flat mounts from Esm1 CreERT2 mTmG (R) and Alk1f/f Esm1 CreERT2 mTmG (S) P12 mice. An arrow indicates vascular malformations (Q, U and W). A: artery, V: vein, Scale bars: 500 μm (B-D), 200 μm (F and H), 2 mm (G and I). 400 μm (J), 25 μm (K-M), 200 μm (N and O), 1 mm (P-G and V-W), 500 μm (R-U), 200 μm.
Next, we examined the survival rate of these three mouse lines. Alk1f/f Mfsd2a CreERT2 mice died 5–6 days after gene deletion, most likely from ruptured brain AVMs, while Alk1f/f Bmx CreERT2 mice lived at least 50 days after gene deletion (Figure IIT in the Data Supplement). Interestingly, the Alk1f/f Esm1 CreERT2 mutants died 10–11 days after Tx injection (Figure IIT in the Data Supplement), suggesting they might develop AVMs in other tissues. Autopsy revealed massive intestinal hemorrhages in Alk1f/f Esm1 CreERT2 mice as a likely cause of death (Figure IIU in the Data Supplement). To define the Esm1 expression in intestines, Tx was injected at P4 and Esm1 CreERT2 mTmG mice were analyzed at P14. GFP-positive cells were found in scattered capillaries of the mesenteries, the intestinal wall and the intestinal villi (Figure 2J and 2K). We performed immunostaining of VE-Cadherin (VE-Cad) and GFP in P14 Alk1f/f and Alk1f/f Esm1 CreERT2 mTmG mice (Figure 2L and 2M). Alk1f/f Esm1 CreERT2 mTmG developed GFP-positive vascular malformations in capillaries of the intestinal villi (Figure 2M). Injection of latex dye confirmed the presence of AVMs in the intestinal villi and in the mesenteries (Figure 2N–2Q). To identify the presence of AVMs in other vascular beds, immunostaining and latex red dye injections were performed in P12 and P14 Esm1 CreERT2 mTmG, Alk1f/f and Alk1f/f Esm1 CreERT2 (mTmG) mice (Figure 2R–2W). Alk1f/f Esm1 CreERT2 mTmG developed GFP-positive vascular malformations in retinal capillaries, and migration of Alk1 mutant tip cell progeny into the arteries was perturbed (Figure 2R and 2S). Latex injection confirmed abnormal patterning of distal retinal arteries derived from the Esm1-positive tip cells (Figure 2T and 2U). The latex also revealed vascular malformations in the pial arteries of the brain in Alk1f/f Esm1 CreERT2 mice (Figure 2V and 2W), but full-blown AVMs were not observed in retina or brain. These data indicate that loss of ALK1 signaling in ESM1 expressing capillaries leads to intestinal vascular malformations.
Loss of ALK1 affects cell polarity and flow-migration coupling.
To test if flow-mediated EC polarization was altered in the absence of ALK1, we dissected retinas at P6 −48 h after Tx injection- and immunolabeled with IB4 to detect ECs, DAPI to label nuclei, the Golgi marker GOLPH4 and ALK1 (Figure 3A–3F). To analyze the orientation of the Golgi toward the flow direction in the retinal vessels, we measured the angles between the EC nuclei and the Golgi as well as the predicted blood flow vectors (Figure 3C’–3F’ and Figure 3G). In Alk1f/f retinas, ALK1 expressing arterial, venous and capillary ECs polarized against the direction of blood flow (Figure 3A, 3C, 3C’ and 3H). In contrast, ECs from Alk1f/f Mfsd2a CreERT2 retinas showed random Golgi distribution in veins, capillaries and AVMs (Figure 3B, 3E, 3E’, 3F, 3F’ and 3H). ECs in proximal arteries of Alk1f/f Mfsd2a CreERT2 retinas where Cre was not active maintained ALK1 expression and were polarized normally against the flow (Figure 3B, 3D, 3D’ and 3H). Quantification of polarization using a polarity index (PI), which ranges from 1 (strongly polarized) to 0 (random distribution) confirmed that Alk1f/f retinal ECs were strongly polarized against the direction of blood flow, while Alk1f/f Mfsd2a CreERT2 mutant ECs in capillaries, veins and AVMs displayed poor polarization against the direction of blood flow (Figure 3I). To determine whether the polarity defects preceded AVM development, Tx was injected at P4 and Alk1f/f Mfsd2a CreERT2 mice were analyzed after 24 h (P5) or 36 h (P5.5). Interestingly, AVMs started to appear at 24 h and were more pronounced at 36 h (Figure 3J and 3K). Analysis of cell polarity in P5 Alk1f/f Mfsd2a CreERT2 mutants and controls showed that venous and capillary ECs from Alk1f/f Mfsd2a CreERT2 retinas displayed poorly polarized Golgi distribution (Figure 3L and 3M), indicating that lack of flow-induced polarity preceded AVM formation and could be causally related to AVM development.
Figure 3. ALK1 controls cell polarization against the blood flow direction.
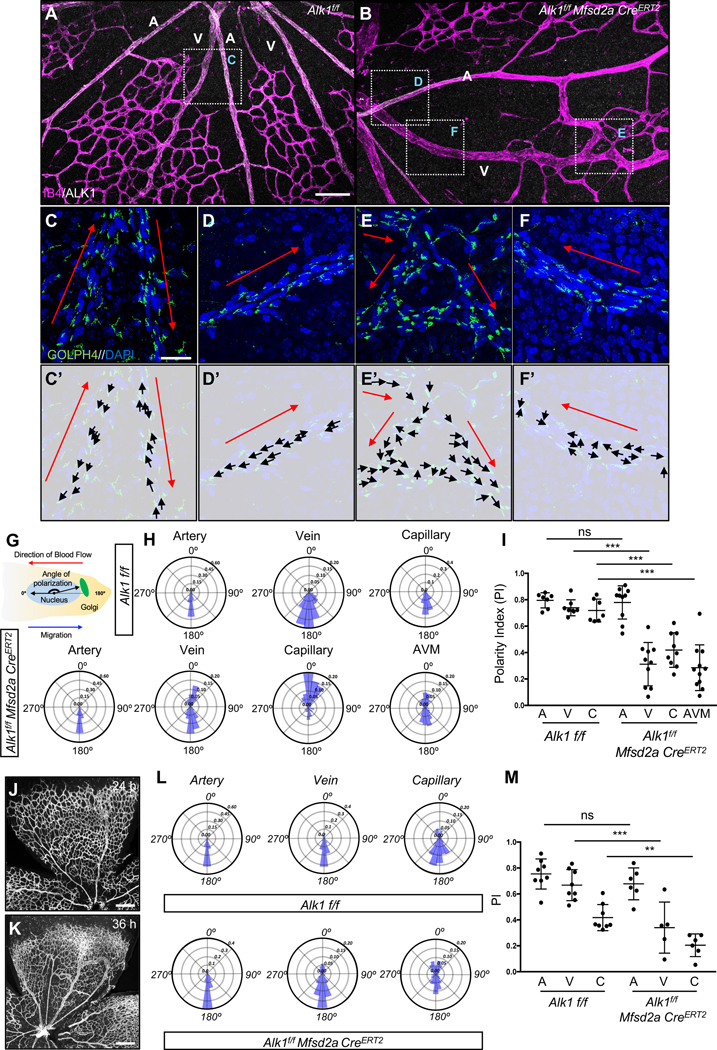
(A-B) IB4 (Magenta) and ALK1 (white) staining of retinal flat mounts from Alk1f/f (A) and Alk1f/f Mfsd2a CreERT2 (B) pups injected with 100 μg Tx at P4 and dissected at P6. (C-F) Higher magnification of insets in A and B. GOLPH4 (green) and DAPI (blue) staining of retina flat mounts. Red arrows indicate the blood flow direction. (C’-F’) Background images from Figure 2C–2F and corresponding polarity vectors (black arrows). (G) The polarity axis of each cell was defined as the angle between the direction of blood flow and the cell polarity axis, defined by a vector drawn from the center of the cell nucleus to the center of the Golgi apparatus. (H) Angular histograms showing the distribution of polarization angles of ECs in the artery, vein and capillaries from Alk1f/f and artery, vein, capillary and AVM from Alk1f/f Mfsd2a CreERT2 mouse retinas. n = 7–11 retinas. (I) polarity index (PI) box plots of ECs from artery, vein and capillary from Alk1f/f and artery, vein, capillary and AVM from Alk1f/f Mfsd2a CreERT2 P6 retinas. n = 7–11 retinas. (J and K) IB4 (gray) staining of retinal flat mounts from Alk1f/f Mfsd2a CreERT2 pups injected with 100 μg at P4 and dissected after 24 h (P5) (J) and 36 h (P5.5) (K). (L) Angular histograms showing the distribution of polarization angles of ECs in the artery, vein and capillary from Alk1f/f and Alk1f/f Mfsd2a CreERT2 P5 retinas at 24 h after Tx injection. (M) PI box plots of ECs from artery, vein and capillary from Alk1f/f and Alk1f/f Mfsd2a CreERT2 retinas at 24 h after Tx injection. n = 5–8 retinas/group. Error bars: SEM. **P-value < 0.01, ***P-value < 0.001, ns: nonsignificant, Two-way ANOVA with Tukey’s multiple comparisons test. Scale bars: 100 μm (A-B), 20 μm (C-F) and 500 μm (J-K)
To explore whether laminar flow affected the polarization of ALK1 mutant cells in vitro, we performed scratch wound assays with human umbilical vein endothelial cells (HUVECs) that were cultured in static conditions or subjected to laminar shear stress (15 dynes/cm2) and stained with a Golgi marker to determine cell polarity angles29. In static conditions, control siRNA transfected HUVECs were polarized towards the scratch areas in both the left and the right side of the wound (Figure 4A and 4C). Under laminar shear, the cells on the left side upstream of the scratch repolarize in the opposite direction to align against the flow (Figure 4E and 4G). By contrast, ALK1 deficient HUVECs showed random polarization in static conditions (Figure 4B and 4D). Most strikingly, they were unable to polarize against the direction of flow in the upstream scratch areas, and even the downstream polarization against the flow was impaired (Figure 4F and 4H). Polarity index calculation showed that flow significantly enhanced polarization of control siRNA transfected cells, and that ALK1 deletion prevented flow induced polarization (Figure 4I).
Figure 4. ALK1 controls EC polarization against the flow direction in vitro.

(A-B) Representative images of scratch wound assays after 18 h showing polarity angles of HUVECs transfected with Control (siCon) (A) or ALK1 (siALK1) (B) siRNAs under static conditions and immunolabeled with phalloidin(red), GM130 (green), and DAPI (blue). (E-F) Representative images of scratch wound assays showing polarity angles of siCon (E) or siALK1 (F) HUVECs with 18 h exposure to laminar shear stress (LSS) at 15 dynes/cm2. Left panels are upstream and right panels are downstream of flow. (C-D and G-H) Angular histograms showing polarization angles of siCon (C and G) or siALK1 ECs (D and H) at 18 h after scratch with (G-H) or without (C-D) LSS. Left is upstream and right is downstream of flow (G and H). (I) PI box plots of upstream (left) scratch areas from siCon or siALK1 transfected HUVECs at 18 h after with or without LSS. (C-I) n=6–8 images from 3 independent experiments. Error bars: SEM. *P-value < 0.05, **P-value < 0.01, ***P-value < 0.001, Two-way ANOVA with Sidak’s multiple comparison test. (J) Representative time lapse images of siCon or siALK1 HUVECs stably transduced with PH-AKT-mClover3 and plasma membrane targeting sequence of LCK-mRuby3. HUVEC monolayers in microfluidic chambers were exposed to 12 dynes/cm2 LSS under the microscope. 5 min (static) and 12 min (LSS) images were selected from the movies. The surface is color-coded by the value of PH-AKT intensity. (K) Local activation of PI3K was quantified by image analysis. PH-AKT intensity was normalized with average static intensity at each time point. 0 – 5 min : static and 5 – 24.5 min : LSS, n= 61, 41 cells from 3 independent experiments, Error bar : SEM. ***P-value < 0.001, Two-tailed unpaired t-test between siCon and siALK1 in average over the time. Scale bars : 50 μm (A-B and E-F), 20 μm (J).
Flow induces localization of phosphorylated AKT to the upstream edge of ECs30. Pleckstrin homology domain of AKT fused to GFP (PH-AKT-GFP) is a well-established biosensor of PI3K local activity which shows plasma membrane localized PH-AKT-GFP upon shear stress31. To examine whether ALK1 affected flow-induced PI3K localization, we performed live cell imaging. PH-AKT-mClover3 together with plasma membrane marker (LCK-mRuby3) were co-expressed as a biosensor and an internal control respectively. Control siRNA transfected HUVECs showed polarized activation of PI3K on their upstream edge within 5 minutes after flow, consistent with upstream Golgi polarization. By contrast, ALK1 knockdown significantly diminished this effect (Figure 4J, 4K and Movie I in the Data Supplement). This result indicated that ALK1 is required for flow-induced PI3K-AKT polarization.
Blockade of integrin prevents AVM formation.
Golgi orientation into scratch wounds and under flow is driven by integrin binding to ECM proteins and signaling to CDC4232, 33. Additionally, integrin activation and signaling is modulated by VEGFR2-PI3K signaling, which is altered following ALK1 deletion12, 13, 18,34. VEGFR2 interacts with integrins αvβ3 and α5β1 during vascularization35–37, prompting us to test if VEGFR2-integrin signaling was enhanced in ALK1 deficient ECs. Interestingly, immunolabeling with antibodies recognizing integrin β1 (ITGB1), α5 (ITGA5) and αv (ITGAV) showed increased ITGB1, ITGA5 and ITGAV expression in the AVM areas of P8 Alk1f/f Mfsd2a CreERT2 retinas when compared to wildtype controls (Figure 5A–5D and Figure IIIA–IIIC in the Data Supplement). We next tested if ALK1 knockdown affected VEGFR2 complex formation with integrins. VEGFR2 immunoprecipitation and immunoblotting for integrins showed that ALK1 deletion significantly enhanced VEGFR2 pulldown of ITGB1, ITGA5 and ITGAV, and that VEGFR2 knockdown abolished co-immunoprecipitation, without affecting total levels of ITGB1, ITGA5 and ITGAV in cell lysates (Figure 5E, 5F and Figure IIID in the Data Supplement). These results suggested that targeting integrin signaling with inhibitors could rescue AVM formation. To test this idea, we administered cilengitide, a small molecule inhibitor for integrin αvβ3 and αvβ5, or ATN161, a peptide inhibitor for integrin α5β1. Alk1 deletion was induced by Tx injection at P4, inhibitors were given intraperitoneally (i.p.) at 5 mg/kg at P4 and P5, and mice were analyzed at P6 (Figure 5G). Both cilengitide and ATN161 decreased AVM formation in Alk1f/f Mfsd2a CreERT2 or Alk1f/f Cdh5 CreERT2 mice (Figure 5H–5O). Immunostaining of Golgi markers showed that integrin inhibitors rescued polarization of Alk1 mutant cells against the direction of blood flow (Figure 5P–5T).
Figure 5. Integrin inhibition prevents AVM formation in Alk1 mutant retinas.
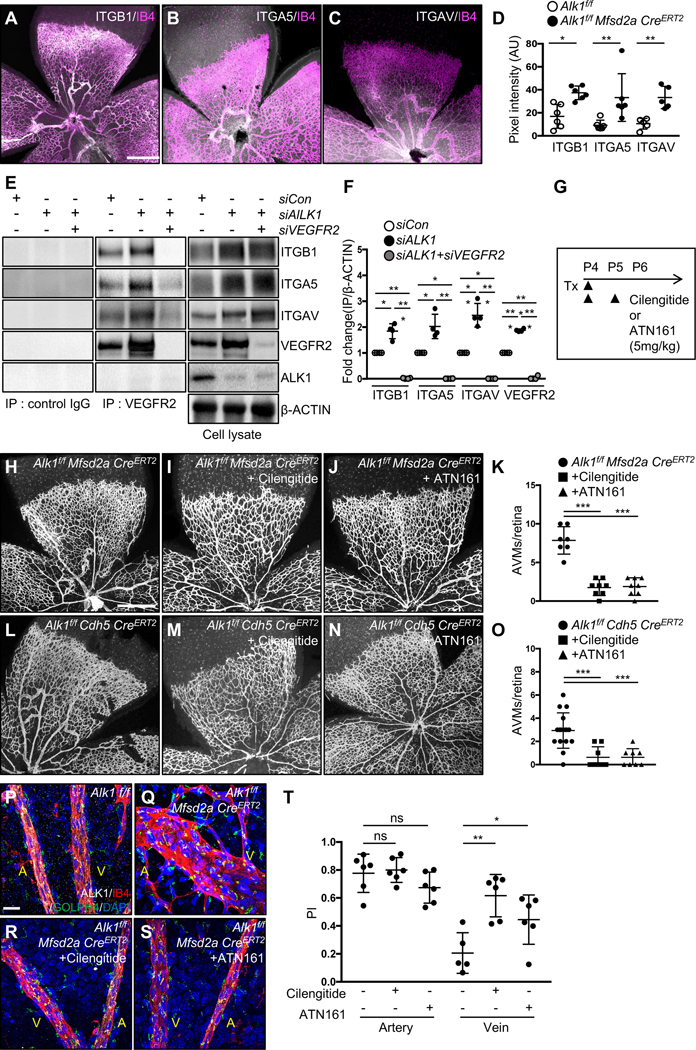
(A-C) IB4 (Magenta) and ITGB1(A, white), ITGA5 (B, white) or ITGAV (C, white) staining of retinal flat mounts from P8 Alk1f/f Mfsd2a CreERT2 pups. (D) Quantification of ITGB1, ITGA5 and ITGAV in P8 Alk1f/f Mfsd2a CreERT2 retinas. (E) VEGFR2 immunoprecipitation in siCon, siALK1 or siALK1+siVEGFR2 HUVECs and western blot analysis for ITGB1, ITGA5, ITGAV and ALK1. VEGFR2, ITGB1, ITGA5, ITGAV, ALK1 and β-ACTIN expression from the total cell lysates are shown as loading controls. Rabbit IgG was used as control. (F) Quantification of ITGB1, ITGA5 or ITGAV levels from immunoprecipitation normalized to β-ACTIN from total cell lysates. *P<0.05, **P<0.01, ***P-value < 0.001, One-way ANOVA with Sidak’s multiple comparisons test. (G) Experimental strategy to assess the effects of integrin inhibitors in Alk1 deleted retinas. Arrowheads indicate the time course of Tx (100 μg) and cilengitide (5mg/kg), ATN161 (5mg/kg) or vehicle administration. (H-J and L-N) IB4 staining of P6 retinal flat mounts from Alk1f/f Mfsd2a CreERT2 (H-J) or Alk1f/f CDH5 CreERT2 (L-N) injected with cilengitide (I and M) or ATN161 (J and N) at P4 and P5. (K and O) Quantification of the AVM number. Each dot represents one retina. n = 7–16 retinas per group. Error bars: SEM. ***P-value < 0.001, One-way ANOVA with Sidak’s multiple comparisons test. (P-S) IB4 (Magenta), ALK1 (white), GOLPH4 (green) and DAPI (blue) staining of retina flat mounts from Alk1f/f (P), Alk1f/f Mfsd2a CreERT2 (Q), Cilengitide (R) or ATN161 (S) injected Alk1f/f Mfsd2a CreERT2 pups. A: artery, V: vein, (T) PI box plots of ECs from artery and vein from Alk1f/f Mfsd2a CreERT2 injected cilengitide or ATN161 retinas. n=5–8 retinas/group. Error bars: SEM. *P-value < 0.05, **P-value < 0.01, ns: nonsignificant, One-way ANOVA with Sidak’s multiple comparisons test. Scale bars: 500 μm (A-C, G-I and K-M), 20 μm (P-S).
ALK1 controls Hippo pathway signaling.
Previous data reported an interaction between BMP9/ALK1 signaling and the YAP/TAZ pathway, and both of these pathways are regulated by blood flow in vivo38, 39. Moreover, integrins are potent regulators of YAP/TAZ activation in many systems including ECs40–42. To test whether laminar shear and ALK1 affected integrin and YAP/TAZ protein expression, HUVECs were transfected with control or ALK1 siRNA and cultured in static conditions or under laminar shear stress (15 dynes/cm2) for 18 h. Protein extracts from these cells were analyzed by western blot with antibodies against integrins, YAP or TAZ and expression levels were compared to β-ACTIN. Interestingly, ITGB1, ITGA5 and ITGAV as well as YAP and TAZ were all significantly increased in ALK1 deleted ECs when compared to control siRNA transfected cells, and their expression was further increased by laminar shear stress (Figure 6A and 6B). YAP and TAZ protein expression were also increased and appeared more nuclear in Alk1f/f Mfsd2a CreERT2 retina AVMs when compared to Alk1f/f control ECs (Figure 6C–6F). Immunostaining of HUVECs with YAP and TAZ antibodies revealed enhanced YAP and TAZ nuclear localization in ALK1 deficient cells, while YAP and TAZ were mainly located in the cytosol of control siRNA treated ECs (Figure 6G and 6H). To test whether other HHT pathway components ENG and SMAD4 also affected YAP/TAZ activity, we deleted ALK1, SMAD4, and ENG in HUVECs and immune-labeled for YAP and TAZ (Figure 7A). YAP and TAZ nuclear localization increased in ALK1, SMAD4, and ENG deleted ECs when compared to control siRNA transfected cells (Figure 7A and 7B). Treatment with the YAP/TAZ inhibitor Verteporfin (VP) blocked YAP/TAZ nuclear translocation in ALK1, SMAD4, and ENG depleted HUVECs (Figure 7A and 7B). ALK1 deletion also increased expression of the YAP/TAZ target cMYC43, 44 and enhanced cMYC nuclear localization. Nuclear cMYC expression was significantly reduced by VP treatment of ALK1 knockdown cells (Figure IVA–IVD in the Data Supplement). To examine VP activity in vivo, we administered VP (50 mg/kg, i.p.) into P4 and P5 Alk1f/f control mice (Figure 7C). VP injected control retinas developed blunted endothelial tip cells at the angiogenic front (Figure 7D), as reported in genetically Yap/Taz deficient endothelial mouse retinas38, 45, 46 indicating that the pharmacological inhibition was effective. Next, we injected VP into Alk1f/f Cdh5 CreERT2 or Alk1f/f Mfsd2a CreERT2 retinas, which led to a significant reduction of AVM formation and hemorrhage when compared to DMSO vehicle treated mutant retinas (Figure 7D–7F). VP treatment also rescued the polarization against the direction of blood flow (Figure 7G and 7H). These results demonstrated that ALK1 regulates Hippo pathway activation, and that VP-mediated inhibition of YAP/TAZ activity improved flow migration coupling and prevented AVMs in Alk1 mutant ECs.
Figure 6. ALK1 controls YAP/TAZ expression and localization.
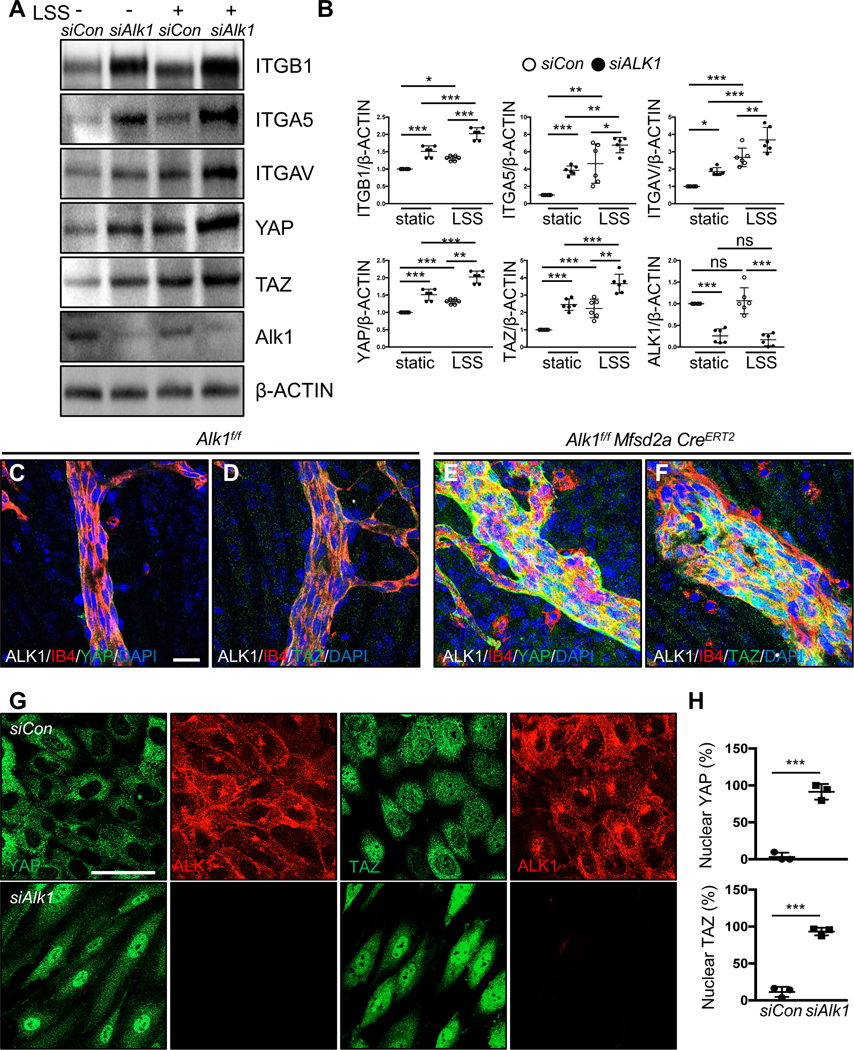
(A) Western blot analysis of HUVECs transfected with control and ALK1 siRNAs followed by 18 h exposure to LSS (15 dynes/cm2). (B) Quantification of ITGB1, ITGA5, ITGAV, YAP or TAZ levels normalized to β-ACTIN. *P<0.05, **P<0.01, ***P<0.001, Two-way ANOVA with Sidak’s multiple comparison test. (C-F) YAP and TAZ (green), ALK1 (gray), IB4 (red), DAPI (blue) staining of retinal flat mounts from P8 Alk1f/f (C-D) or Alk1f/f Mfsd2aCreERT2 (E-F) pups. A scale bar: 20 μm (C-F) (G) YAP or TAZ (green) and ALK1 (red) staining of siCon and siALK1 HUVECs. A scale bar: 50 μm. (H) Quantification of nuclear YAP and TAZ from siCon and siALK1 transfected HUVECs. ***P<0.001, n = 3 independent experiments. Two-tailed unpaired t-test between siCon and siALK1.
Figure 7. YAP/TAZ inhibition improves AVM formation in Alk1 mutant retinas.
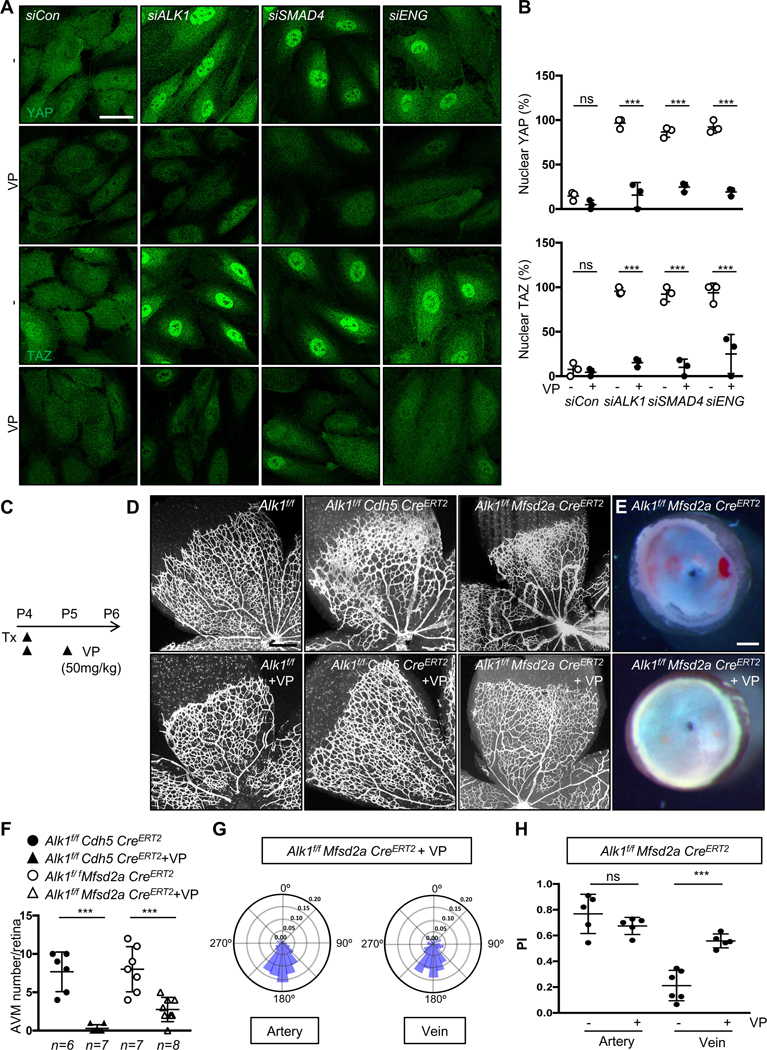
(A) YAP and TAZ staining of siCon, ALK1, SMAD4 or ENG siRNAs transfected HUVECs treated with DMSO or Verteporfin (VP, 5 μM) for 6 h. Nuclear YAP/TAZ localization in siALK1, siSMAD4 or siENG ECs is blocked by VP treatment. A scale bar: 50 μm. (B) Quantification of nuclear YAP and TAZ from siCon, siALK1,siSMAD4 and siENG transfected HUVECs. ***P<0.001, ns: nonsignificant, Two-way ANOVA with Tukey’s multiple comparisons test. (C) Experimental strategy to assess the effects of YAP/TAZ inhibition in EC specific Alk1 deleted vasculature. Arrowheads indicate the time course of Tx (100 μg) and VP (50mg/kg) or vehicle administration. (D) IB4 staining of P6 retinal flat mounts from VP injected Alk1f/f, Alk1f/f CDH5 CreERT2 or Alk1f/f Mfsd2a CreERT2 mice. (E) Stereomicroscopy images of vehicle or VP injected Alk1f/f Mfsd2a CreERT2 retinas. (F) Quantification of the AVM number/retina. Each dot represents one retina. n = 6–8 retinas per group. Error bars: SEM. ***P-value < 0.001, One-way ANOVA with Sidak’s multiple comparisons test. (G) Angular histograms showing polarization angles of artery and vein from Alk1f/f Mfsd2a CreERT2 with VP. (H) PI box plots of Alk1f/f Mfsd2a CreERT2 with vehicle or VP. n=5–6 retinas, Error bars: SEM, ***P-value < 0.001, ns: nonsignificant, Two-way ANOVA with Tukey’s multiple comparisons test. Scale bars : 50 μm (A), 500 μm (D), 300 μm (E)
VEGFR2, integrin and PI3K function upstream of YAP/TAZ in Alk1 mutants
To elucidate whether ALK1 modulation of the Hippo pathway was dependent on VEGFR2, integrins, and PI3K, we examined YAP/TAZ nuclear translocation by immunostaining. YAP/TAZ nuclear translocation was abolished by combined deletion of ALK1 and VEGFR2 (Figure 8A and 8B), by treating ALK1 deleted HUVECs with integrin inhibitors cilengitide or ATN161, and by treatment with the PI3K inhibitor wortmannin (Figure 8A, 8B and Figure VA–VC in the Data Supplement). Western blot analysis showed that combined deletion of ALK1 and VEGFR2 and/or ITGB1 reduced YAP/TAZ expression and phosphorylation of PI3K and AKT (Figure 8C and 8D), supporting that VEGFR2-integrin signaling through PI3K/AKT acts upstream of YAP/TAZ in ALK1 deficient HUVECs. Alk1 deletion in mouse brain ECs likewise revealed enhanced YAP/TAZ expression and enhanced PI3K/AKT phosphorylation, and this could be rescued by over-expression of ALK1 (Figure VIA and VIB in the Data Supplement). In vivo, cilengitide and ATN161 treatment of Alk1f/f Mfsd2a CreERT2 mice reduced YAP/TAZ expression when compared to Alk1f/f control retinas (Figure 8E), supporting that integrin signaling acted upstream of YAP/TAZ.
Figure 8. VEGFR2, integrin and PI3K function upstream of YAP/TAZ in Alk1 mutants.
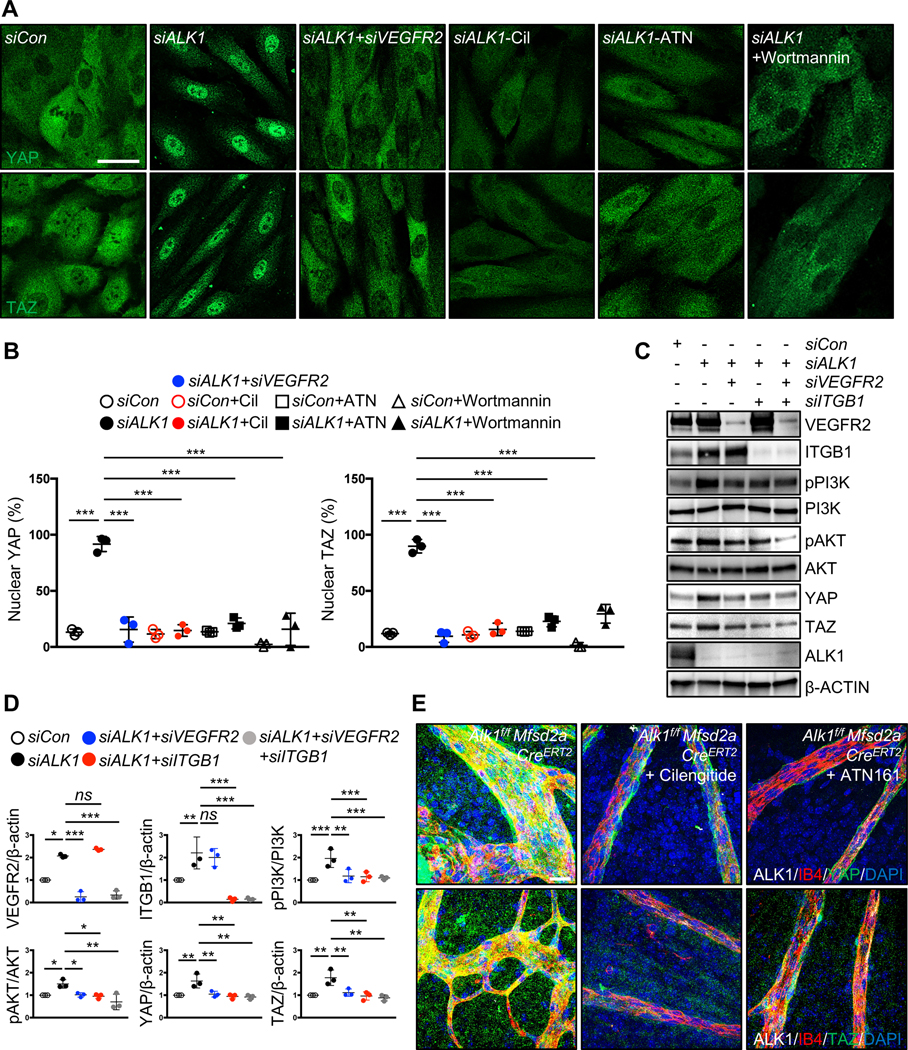
(A) YAP and TAZ staining for siCon, siALK1 and siALK1+siVEGFR2 transfected HUVECs and ALK1 siRNA transfected HUVECs treated with cilengitide (Cil, 5 μM), ATN161 (ATN, 5 μM) and wortmannin (100 mM) for 12 h. Nuclear YAP/TAZ localization in siALK1 ECs is blocked by siVEGFR2, cilengitide, ATN161 and wortmannin treatment. (B) Quantification of nuclear YAP and TAZ from for siCon, siALK1 and siALK1+siVEGFR2 transfected HUVECs and ALK1 siRNA transfected HUVECs treated with cilengitide, ATN16, wortmannin. ***P<0.001, n = 3 independent experiments. Error bars: SEM. ***P-value < 0.001, Two-way ANOVA with Tukey’s multiple comparisons test. (C) Western blot analysis of HUVECs transfected with control, ALK1, ALK1+VEGFR2, ALK1+ITGB1 or ALK1+VEGFR2+ITGB1 siRNAs. (D) Quantification of pPI3K/PI3K, pAKT/AKT and YAP or TAZ levels normalized to β-ACTIN. *P<0.05, **P<0.01, ***P<0.001, ns: nonsignificant, One-way ANOVA with Sidak’s multiple comparisons test. (E) YAP and TAZ (green), ALK1 (white), IB4 (red) and DAPI (blue) staining of retinal flat mounts from cilengitide or ATN161 injected Alk1f/f Mfsd2a CreERT2 P6 mice. Scale bars: 50 μm (A), 20 μm (E)
Discussion
This study shows that AVMs in Alk1 mutants originate from capillaries and veins and implicates integrin and Hippo pathway signaling in HHT. The data are consistent with a model whereby the presence of ALK1 suppresses integrin signaling interactions with VEGFR2, which limits PI3K activation and YAP/TAZ nuclear translocation. In the absence of ALK1, enhanced VEGFR2-integrin-PI3K signaling promotes YAP/TAZ nuclear translocation, and pharmacological inhibition of integrin or YAP/TAZ signaling prevents vascular malformations in Alk1 deficient mice (Figure VII in the Data Supplement), as does VEGFR2 or PI3K inhibition13, 20, 47, 48.
We found that Mfsd2a-positive capillary-venous cells migrated against the blood flow direction towards retinal arteries, highlighting endothelial flow-migration coupling as a critical process driving vascular remodeling29, 49, 50. The current concept suggests that blood flow attracts EC migration from low flow segments (veins and capillaries) towards high flow segments (arteries)49. Hence, disruption of flow-migration coupling and resulting accumulation of ECs in capillaries could cause capillary enlargement and thereby precipitate AVM formation. Consistent with this model, deletion of Alk1 in capillaries and veins using Mfsd2a CreERT2 led to disruption of Golgi polarization against the flow direction and caused retinal and cerebral AVMs. Mfsd2a is a brain-specific endothelial gene24, 51, hence our analysis of these mice was restricted to the brain and retina. In addition, a recent study showed that capillary/venous-specific deletion of the ALK1 co-receptor ENG using Engfl/fl Apj-CreERT2 mice induced retinal AVMs52, indicating that the ALK1-ENG complex is required in capillaries and veins to prevent AVM formation, and that defective flow-migration coupling is a hallmark of HHT. Whether venous or capillary ECs, or both, are involved in the vascular malformations, needs to be further investigated and will require generation of capillary or vein-specific Cre driver lines.
Interestingly, deletion of Alk1 in Esm1-positive tip cells also led to defective flow-migration coupling and accumulation of the mutant cells in the vascular plexus ahead of the arteries, while control cells colonized the arterial tree. This underscores an important role of Alk1 in flow-migration coupling of retinal tip cells, but produced only mild retinal and brain vascular malformations when compared to pan-endothelial Alk1f/f Cdh5 CreERT2 and Alk1f/f Mfsd2a CreERT2 mice. One possible reason for the discrepant phenotypes is that disruption of cell polarity per se is not sufficient to induce AVMs. Another possibility is the differing flow environment: Esm1-positive tip cells migrate in a low-flow environment, whereas AVMs develop in high-flow regions of the retina, close to the optic nerve, and we and others have previously reported that blood flow potentiates ALK1-ENG-mediated shear stress sensing15, 26.
Quite strikingly, despite the lack of AVMs in retina and brain, the Alk1f/f Esm1 CreERT2 mice developed intestinal AVMs and succumbed to intestinal hemorrhage. Analysis of Esm1-driven GFP labeling revealed expression in capillary endothelium of the mesenteries, the gut wall and the intestinal villi, and GFP positive cells formed AVMs in those regions in Alk1f/f Esm1 CreERT2 mutant mice. Hence, capillary function of Alk1 was required to prevent intestinal AVM formation. Further analysis is required to assess whether flow-migration coupling also underlies intestinal vascular remodeling, but such studies will require endothelial specific fluorescent Golgi reporter mice to determine endothelial cell polarity.
Our in vitro data revealed that loss of ALK1 displayed disrupted endothelial Golgi orientation and polarization against the blood flow direction. Blocking blood flow in zebrafish alk1 mutants prevented AVM formation, directly demonstrating that blood flow induces AVM formation in the absence of ALK153. We and others have previously reported that BMP9/10-ALK1 signaling mechanistically links flow sensing and VEGFR2-PI3K/AKT pathway activation 12–15, 17, 20, 54. ALK1 signaling counteracted both flow and growth factor-induced AKT activation, and the absence of ALK1 overactivated PI3K/AKT signaling in AVMs 48, 55, 56. We extend these findings here by demonstrating that ALK1 is required for flow-induced PI3K-AKT polarization against the direction of blood flow. Besides enhanced VEGFR2/PI3K signaling, another study showed that loss of SMAD4 increased Angiopoietin2 and decreased TIE2 receptor expression57. Blocking Angiopoietin2 prevented AVM formation and normalized vessel diameters in endothelial Smad4 deficient mice, while TIE2 accumulated within the AVMs57, suggesting that increased TIE2 signaling could contribute to enhanced PI3K signaling in AVMs.
As new mechanistic findings, we report that integrins and YAP/TAZ signaling are involved in ALK1 signaling and AVM formation. Integrins are heterodimeric transmembrane receptors that are activated by flow and then bind to specific ECM proteins. RGD peptides or neutralizing antibodies against integrin α5β1 prevented laminar shear stress-induced increase in EC adhesion58–60. This correlates with our data that Alk1 mutant showed increased integrins in AVM regions and cilengitide and ATN161 improved AVMs in Alk1 mutant mice. Cilengitide is a selective αvβ3 and αvβ5 integrin inhibitor. Phase 3 trials using cilengitide for glioblastoma patients have failed to improve patient survival, however they were well tolerated61 and could be viable candidates for therapy in HHT patients.
Hippo-YAP/TAZ signaling regulates organ size, tissue regeneration and self-renewal as well as vascular development38, 39, 62–65. The core components of this pathway comprise a kinase cascade containing MST1/2 and LATS1/2. MST1/2 phosphorylates and activates LATS1/2, which then phosphorylate YAP/TAZ, causing their cytoplasmic sequestration, degradation and inactivation66, 67. When activated, YAP/TAZ shuffle from the cytosol to the nucleus where they interact with different transcription factors, of which TEAD transcription factors are the best characterized, to regulate expression of target genes involved in cell growth, survival, and migration68. Endothelial YAP/TAZ are important regulators of vascular development and function, and their activity is regulated by mechanical, metabolic and growth factor signals39, 45, 46, 62, 69–71. We found that ALK1 deletion in mice or in human ECs induced nuclear YAP/TAZ accumulation in a VEGFR2, PI3K and integrin-dependent manner, thereby linking YAP/TAZ signaling to HHT. The mechanistic details of Hippo pathway regulation by ALK1 remain to be determined. VEGFR2 signaling triggers integrin activation through PI3K72–74, alternatively PI3K can be activated downstream of VEGFR2 and integrins through outside-in signaling74–77. Because ITGB1 knockdown decreased PI3K activation in ALK1 knockdown cells, our data favor the latter model. Integrins are well known to activate YAP/TAZ through both mechanical and biochemical signaling pathways78, 79. Together, these results define a pathway in which loss of ALK1 activates VEGFR2-integrin and PI3K, which, in turn, activates YAP/TAZ. However, as nuclear YAP/TAZ accumulation alone is not reported to induce AVMs65, 80, we suggest that YAP/TAZ are necessary but not sufficient for AVM formation, and that VEGFR2, PI3K and integrins very likely contribute to lesion formation through additional effects on cell polarity and migration. Finally, YAP/TAZ overactivation suppressed BMP9-ALK1 signaling38, suggesting that negative feedback loops between these pathways occur during normal vascular development. We show that the YAP/TAZ inhibitor VP, which inhibits YAP/TAZ translocation to the nucleus81, 82 and blocks YAP-TEAD association by binding to YAP and changing its conformation83, rescued AVM formation in Alk1 mutant mice. VP photodynamic therapy is approved for the treatment of choroidal neovascularization due to age-related macular degeneration84 and both integrin inhibitors and verteporfin might be novel therapeutic options for HHT patients.
Supplementary Material
Clinical Perspective.
- What is New?
- Using Cre lines that delete the HHT causing Alk1 gene in different subsets of endothelial cells, we showed that Alk1 deletion in capillary and venous endothelium led to arteriovenous malformations (AVMs).
- Alk1 mutants displayed defective polarization against the direction of blood flow in capillary and venous endothelium, as well as increased integrin-VEGFR2 mediated PI3K activation of YAP/TAZ signaling.
- What are the clinical implications?
- Pharmacological integrin inhibition using cilengitide or ATN161, or YAP/TAZ inhibition using verteporfin prevented AVM formation in Alk1 mutant mice.
- Our study reveals integrin and YAP/TAZ as novel effectors of ALK1 signaling and AVM pathogenesis that could be targeted for AVM treatment in HHT patients.
Acknowledgements
We thank ATTRACT members Paul Oh, Lena Claesson-Welsh, Miguel Bernabeu and Holger Gerhardt for critical comments on the manuscript, Profs Bin Zhou (Shanghai Institute for Biological Sciences) and Ralf Adams (Max-Planck Institute, Munster, Germany) for mouse lines, and Dr. Jihoon Park for Python scripts to measure polarity and index.
Sources of Funding
This work was supported by grants from the Leducq Foundation (TNE ATTRACT, A.E, C.F) and NIH (P30 EY026878, R01EY025979 to A.E and R01 HL135582 to M.A.S). C.A.F was supported by European Research Council starting grant (679368) and the Fundação para a Ciência e a Tecnologia funding (CEECIND/02589/2018).
Non-standard Abbreviations and Acronyms
- AVMs
arteriovenous malformations
- GI
Gastrointestinal
- HHT
Hereditary hemorrhagic telangiectasia
- HUVECs
human umbilical vein endothelial cells
- i.p.
intraperitoneally
- LSS
laminar shear stress
- PI
Polarity index
- SEM
Standard error of the mean
- Tx
Tamoxifen
Footnotes
Disclosures
None.
References
- 1.Shovlin CL. Hereditary haemorrhagic telangiectasia: Pathophysiology, diagnosis and treatment. Blood Reviews. 2010;24:203–219. doi: 10.1016/j.blre.2010.07.001 [DOI] [PubMed] [Google Scholar]
- 2.McAllister KA, Grogg KM, Johnson DW, Gallione CJ, Baldwin MA, Jackson CE, Helmbold EA, Markel DS, McKinnon WC, Murrel J, et al. Endoglin, a TGF-β binding protein of endothelial cells, is the gene for hereditary haemorrhagic telangiectasia type 1. Nature Genetics. 1994;8:345–351. doi: 10.1038/ng1294-345 [DOI] [PubMed] [Google Scholar]
- 3.Johnson DW, Berg JN, Baldwin MA, Gallione CJ, Marondel I, Yoon SJ, Stenzel TT, Speer M, Pericak-Vance MA, Diamond A, et al. Mutations in the activin receptor–like kinase 1 gene in hereditary haemorrhagic telangiectasia type 2. Nature Genetics. 1996;13:189–195. doi: 10.1038/ng0696-189 [DOI] [PubMed] [Google Scholar]
- 4.Gallione CJ, Repetto GM, Legius E, Rustgi AK, Schelley SL, Tejpar S, Mitchell G, Drouin E, Westermann CJ and Marchuk DA. A combined syndrome of juvenile polyposis and hereditary haemorrhagic telangiectasia associated with mutations in MADH4 (SMAD4). Lancet. 2004;363:852–9. doi: 10.1016/s0140-6736(04)15732-2 [DOI] [PubMed] [Google Scholar]
- 5.Gallione CJ, Klaus DJ, Yeh EY, Stenzel TT, Xue Y, Anthony KB, McAllister KA, Baldwin MA, Berg JN, Lux A, et al. Mutation and expression analysis of the endoglin gene in Hereditary Hemorrhagic Telangiectasia reveals null alleles. Human Mutation. 1998;11:286–294. doi: 10.1002/(SICI)1098-1004(1998)11:4<286::AID-HUMU6>3.0.CO;2-B [DOI] [PubMed] [Google Scholar]
- 6.Roman BL and Hinck AP. ALK1 signaling in development and disease: new paradigms. Cellular and Molecular Life Sciences. 2017;74:4539–4560. doi: 10.1007/s00018-017-2636-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.David L, Mallet C, Mazerbourg S, Feige JJ and Bailly S. Identification of BMP9 and BMP10 as functional activators of the orphan activin receptor-like kinase 1 (ALK1) in endothelial cells. Blood. 2007;109:1953–61. doi: 10.1182/blood-2006-07-034124 [DOI] [PubMed] [Google Scholar]
- 8.Ruiz-Llorente L, Gallardo-Vara E, Rossi E, Smadja DM, Botella LM and Bernabeu C. Endoglin and alk1 as therapeutic targets for hereditary hemorrhagic telangiectasia. Expert Opinion on Therapeutic Targets. 2017;21:933–947. doi: 10.1080/14728222.2017.1365839 [DOI] [PubMed] [Google Scholar]
- 9.Snellings DA, Gallione CJ, Clark DS, Vozoris NT, Faughnan ME and Marchuk DA. Somatic Mutations in Vascular Malformations of Hereditary Hemorrhagic Telangiectasia Result in Bi-allelic Loss of ENG or ACVRL1. American Journal of Human Genetics. 2019;105:894–906. doi: 10.1016/j.ajhg.2019.09.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Govani FS and Shovlin CL. Hereditary haemorrhagic telangiectasia: A clinical and scientific review. European Journal of Human Genetics. 2009;17:860–871. doi: 10.1038/ejhg.2009.35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McDonald J, Bayrak-Toydemir P and Pyeritz RE. Hereditary hemorrhagic telangiectasia: An overview of diagnosis, management, and pathogenesis. Genetics in Medicine. 2011;13:607–616. doi: 10.1097/GIM.0b013e3182136d32 [DOI] [PubMed] [Google Scholar]
- 12.Tual-Chalot S, Mahmoud M, Allinson KR, Redgrave RE, Zhai Z, Oh SP, Fruttiger M and Arthur HM. Endothelial depletion of Acvrl1 in mice leads to arteriovenous malformations associated with reduced endoglin expression. PLoS One. 2014;9:e98646. doi: 10.1371/journal.pone.0098646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ola R, Dubrac A, Han J, Zhang F, Fang JS, Larrivée B, Lee M, Urarte AA, Kraehling JR, Genet G, et al. PI3 kinase inhibition improves vascular malformations in mouse models of hereditary haemorrhagic telangiectasia. Nature Communications. 2016;7:13650. doi: 10.1038/ncomms13650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ola R, Künzel Sandrine H, Zhang F, Genet G, Chakraborty R, Pibouin-Fragner L, Martin K, Sessa W, Dubrac A and Eichmann A. SMAD4 Prevents Flow Induced Arteriovenous Malformations by Inhibiting Casein Kinase 2. Circulation. 2018;138:2379–2394. doi: 10.1161/CIRCULATIONAHA.118.033842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Baeyens N, Larrivée B, Ola R, Hayward-Piatkowskyi B, Dubrac A, Huang B, Ross TD, Coon BG, Min E, Tsarfati M, et al. Defective fluid shear stress mechanotransduction mediates hereditary hemorrhagic telangiectasia. The Journal of Cell Biology. 2016;214:807. doi: 10.1083/jcb.201603106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Capasso TL, Li B, Volek HJ, Khalid W, Rochon ER, Anbalagan A, Herdman C, Yost HJ, Villanueva FS, Kim K, et al. BMP10-mediated ALK1 signaling is continuously required for vascular development and maintenance. Angiogenesis. 2020;23:203–220. doi: 10.1007/s10456-019-09701-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rochon ER, Menon PG and Roman BL. Alk1 controls arterial endothelial cell migration in lumenized vessels. Development. 2016;143:2593–602. doi: 10.1242/dev.135392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Han C, Choe S-w, Kim YH, Acharya AP, Keselowsky BG, Sorg BS, Lee Y-J and Oh SP. VEGF neutralization can prevent and normalize arteriovenous malformations in an animal model for hereditary hemorrhagic telangiectasia 2. Angiogenesis. 2014;17:823–830. doi: 10.1007/s10456-014-9436-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jin Y, Muhl L, Burmakin M, Wang Y, Duchez AC, Betsholtz C, Arthur HM and Jakobsson L. Endoglin prevents vascular malformation by regulating flow-induced cell migration and specification through VEGFR2 signalling. Nature Cell Biology. 2017;19:639–652. doi: 10.1038/ncb3534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Alsina-Sanchís E, García-Ibáñez Y, Figueiredo AM, Riera-Domingo C, Figueras A, Matias-Guiu X, Casanovas O, Botella LM, Pujana MA, Riera-Mestre A, et al. ALK1 Loss Results in Vascular Hyperplasia in Mice and Humans Through PI3K Activation. Arterioscler Thromb Vasc Biol. 2018;38:1216–1229. doi: 10.1161/atvbaha.118.310760 [DOI] [PubMed] [Google Scholar]
- 21.Iriarte A, Figueras A, Cerdà P, Mora JM, Jucglà A, Penín R, Viñals F and Riera-Mestre A. PI3K (Phosphatidylinositol 3-Kinase) Activation and Endothelial Cell Proliferation in Patients with Hemorrhagic Hereditary Telangiectasia Type 1. Cells. 2019;8. doi: 10.3390/cells8090971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pu W, Zhang H, Huang X, Tian X, He L, Wang Y, Zhang L, Liu Q, Li Y, Li Y, et al. Mfsd2a+ hepatocytes repopulate the liver during injury and regeneration. Nat Commun. 2016;7:13369. doi: 10.1038/ncomms13369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pu W, He L, Han X, Tian X, Li Y, Zhang H, Liu Q, Huang X, Zhang L, Wang QD, et al. Genetic Targeting of Organ-Specific Blood Vessels. Circ Res. 2018;123:86–99. doi: 10.1161/circresaha.118.312981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chow BW, Nuñez V, Kaplan L, Granger AJ, Bistrong K, Zucker HL, Kumar P, Sabatini BL and Gu C. Caveolae in CNS arterioles mediate neurovascular coupling. Nature. 2020;579:106–110. doi: 10.1038/s41586-020-2026-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pitulescu ME, Schmidt I, Giaimo BD, Antoine T, Berkenfeld F, Ferrante F, Park H, Ehling M, Biljes D, Rocha SF, et al. Dll4 and Notch signalling couples sprouting angiogenesis and artery formation. Nature Cell Biology. 2017;19:915. doi: 10.1038/ncb3555 [DOI] [PubMed] [Google Scholar]
- 26.Xu C, Hasan SS, Schmidt I, Rocha SF, Pitulescu ME, Bussmann J, Meyen D, Raz E, Adams RH and Siekmann AF. Arteries are formed by vein-derived endothelial tip cells. Nature Communications. 2014;5:5758. doi: 10.1038/ncomms6758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ehling M, Adams S, Benedito R and Adams RH. Notch controls retinal blood vessel maturation and quiescence. Development. 2013;140:3051. doi: 10.1242/dev.093351 [DOI] [PubMed] [Google Scholar]
- 28.Muzumdar MD, Tasic B, Miyamichi K, Li L and Luo L. A global double-fluorescent Cre reporter mouse. Genesis. 2007;45:593–605. doi: 10.1002/dvg.20335 [DOI] [PubMed] [Google Scholar]
- 29.Carvalho JR, Fortunato IC, Fonseca CG, Pezzarossa A, Barbacena P, Dominguez-Cejudo MA, Vasconcelos FF, Santos NC, Carvalho FA and Franco CA. Non-canonical Wnt signaling regulates junctional mechanocoupling during angiogenic collective cell migration. eLife. 2019;8:e45853. doi: 10.7554/eLife.45853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Melchior B and Frangos JA. Distinctive subcellular Akt-1 responses to shear stress in endothelial cells. Journal of Cellular Biochemistry. 2014;115:121–129. doi: 10.1002/jcb.24639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Várnai P and Balla T. Visualization of phosphoinositides that bind pleckstrin homology domains: Calcium- and agonist-induced dynamic changes and relationship to myo-[3H]inositol-labeled phosphoinositide pools. Journal of Cell Biology. 1998;143:501–510. doi: 10.1083/jcb.143.2.501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Etienne-Manneville S and Hall A. Integrin-mediated activation of Cdc42 controls cell polarity in migrating astrocytes through PKCzeta. Cell. 2001;106:489–98. doi: 10.1016/s0092-8674(01)00471-8 [DOI] [PubMed] [Google Scholar]
- 33.Tzima E, Kiosses WB, del Pozo MA and Schwartz MA. Localized cdc42 activation, detected using a novel assay, mediates microtubule organizing center positioning in endothelial cells in response to fluid shear stress. J Biol Chem. 2003;278:31020–3. doi: 10.1074/jbc.M301179200 [DOI] [PubMed] [Google Scholar]
- 34.Tzima E, Irani-Tehrani M, Kiosses WB, Dejana E, Schultz DA, Engelhardt B, Cao G, DeLisser H and Schwartz MA. A mechanosensory complex that mediates the endothelial cell response to fluid shear stress. Nature. 2005;437:426–31. doi: 10.1038/nature03952 [DOI] [PubMed] [Google Scholar]
- 35.Somanath PR, Malinin NL and Byzova TV. Cooperation between integrin ανβ3 and VEGFR2 in angiogenesis. Angiogenesis. 2009;12:177–185. doi: 10.1007/s10456-009-9141-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Simons M. An inside view: VEGF receptor trafficking and signaling. Physiology (Bethesda). 2012;27:213–22. doi: 10.1152/physiol.00016.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Serini G, Napione L, Arese M and Bussolino F. Besides adhesion: new perspectives of integrin functions in angiogenesis. Cardiovascular Research. 2008;78:213–222. doi: 10.1093/cvr/cvn045 [DOI] [PubMed] [Google Scholar]
- 38.Neto F, Klaus-Bergmann A, Ong YT, Alt S, Vion A-C, Szymborska A, Carvalho JR, Hollfinger I, Bartels-Klein E, Franco CA, et al. YAP and TAZ regulate adherens junction dynamics and endothelial cell distribution during vascular development. eLife. 2018;7:e31037. doi: 10.7554/eLife.31037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nakajima H, Yamamoto K, Agarwala S, Terai K, Fukui H, Fukuhara S, Ando K, Miyazaki T, Yokota Y, Schmelzer E, et al. Flow-Dependent Endothelial YAP Regulation Contributes to Vessel Maintenance. Developmental Cell. 2017;40:523–536.e6. doi: 10.1016/j.devcel.2017.02.019 [DOI] [PubMed] [Google Scholar]
- 40.Wang L, Luo JY, Li B, Tian XY, Chen LJ, Huang Y, Liu J, Deng D, Lau CW, Wan S, et al. Integrin-YAP/TAZ-JNK cascade mediates atheroprotective effect of unidirectional shear flow. Nature. 2016;540:579–582. doi: 10.1038/nature20602 [DOI] [PubMed] [Google Scholar]
- 41.Li B, He J, Lv H, Liu Y, Lv X, Zhang C, Zhu Y and Ai D. c-Abl regulates YAPY357 phosphorylation to activate endothelial atherogenic responses to disturbed flow. J Clin Invest. 2019;129:1167–1179. doi: 10.1172/jci122440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dupont S. Role of YAP/TAZ in cell-matrix adhesion-mediated signalling and mechanotransduction. Exp Cell Res. 2016;343:42–53. doi: 10.1016/j.yexcr.2015.10.034 [DOI] [PubMed] [Google Scholar]
- 43.Choi W, Kim J, Park J, Lee DH, Hwang D, Kim JH, Ashktorab H, Smoot D, Kim SY, Choi C, et al. YAP/TAZ Initiates Gastric Tumorigenesis via Upregulation of MYC. Cancer Res. 2018;78:3306–3320. doi: 10.1158/0008-5472.Can-17-3487 [DOI] [PubMed] [Google Scholar]
- 44.Yamaguchi H and Taouk GM. A Potential Role of YAP/TAZ in the Interplay Between Metastasis and Metabolic Alterations. Frontiers in Oncology. 2020;10. doi: 10.3389/fonc.2020.00928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sakabe M, Fan J, Odaka Y, Liu N, Hassan A, Duan X, Stump P, Byerly L, Donaldson M, Hao J, et al. YAP/TAZ-CDC42 signaling regulates vascular tip cell migration. Proc Natl Acad Sci U S A. 2017;114:10918–10923. doi: 10.1073/pnas.1704030114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kim J, Kim YH, Kim J, Park DY, Bae H, Lee DH, Kim KH, Hong SP, Jang SP, Kubota Y, et al. YAP/TAZ regulates sprouting angiogenesis and vascular barrier maturation. J Clin Invest. 2017;127:3441–3461. doi: 10.1172/jci93825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kim YH, Kim MJ, Choe SW, Sprecher D, Lee YJ and S PO. Selective effects of oral antiangiogenic tyrosine kinase inhibitors on an animal model of hereditary hemorrhagic telangiectasia. J Thromb Haemost. 2017;15:1095–1102. doi: 10.1111/jth.13683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ruiz S, Zhao H, Chandakkar P, Papoin J, Choi H, Nomura-Kitabayashi A, Patel R, Gillen M, Diao L, Chatterjee PK, et al. Correcting Smad1/5/8, mTOR, and VEGFR2 treats pathology in hereditary hemorrhagic telangiectasia models. Journal of Clinical Investigation. 2020;130:942–957. doi: 10.1172/JCI127425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Franco CA, Jones ML, Bernabeu MO, Geudens I, Mathivet T, Rosa A, Lopes FM, Lima AP, Ragab A, Collins RT, et al. Dynamic Endothelial Cell Rearrangements Drive Developmental Vessel Regression. PLOS Biology. 2015;13:e1002125. doi: 10.1371/journal.pbio.1002125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fonseca CG, Barbacena P and Franco CA. Endothelial cells on the move: dynamics in vascular morphogenesis and disease. Vasc Biol. 2020;2:H29–h43. doi: 10.1530/vb-20-0007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chow BW and Gu C. Gradual Suppression of Transcytosis Governs Functional Blood-Retinal Barrier Formation. Neuron. 2017;93:1325–1333.e3. doi: 10.1016/j.neuron.2017.02.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Singh E, Redgrave RE, Phillips HM and Arthur HM. Arterial endoglin does not protect against arteriovenous malformations. Angiogenesis. 2020;23:559–566. doi: 10.1007/s10456-020-09731-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Corti P, Young S, Chen CY, Patrick MJ, Rochon ER, Pekkan K and Roman BL. Interaction between alk1 and blood flow in the development of arteriovenous malformations. Development. 2011;138:1573–82. doi: 10.1242/dev.060467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tual-Chalot S, Garcia-Collado M, Redgrave RE, Singh E, Davison B, Park C, Lin H, Luli S, Jin Y, Wang Y, et al. Loss of endothelial endoglin promotes high-output heart failure through peripheral arteriovenous shunting driven by VEGF signaling. Circulation Research. 2020:243–257. doi: 10.1161/CIRCRESAHA.119.315974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Thalgott JH, Dos-Santos-Luis D, Hosman AE, Martin S, Lamandé N, Bracquart D, Srun S, Galaris G, De Boer HC, Tual-Chalot S, et al. Decreased expression of vascular endothelial growth factor receptor 1 contributes to the pathogenesis of hereditary hemorrhagic telangiectasia type 2. Circulation. 2018;138:2698–2712. doi: 10.1161/CIRCULATIONAHA.117.033062 [DOI] [PubMed] [Google Scholar]
- 56.Hwan Kim Y, Vu PN, Choe SW, Jeon CJ, Arthur HM, Vary CPH, Lee YJ and Oh SP. Overexpression of Activin Receptor-Like Kinase 1 in Endothelial Cells Suppresses Development of Arteriovenous Malformations in Mouse Models of Hereditary Hemorrhagic Telangiectasia. Circ Res. 2020;127:1122–1137. doi: 10.1161/circresaha.119.316267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Crist AM, Zhou X, Garai J, Lee AR, Thoele J, Ullmer C, Klein C, Zabaleta J and Meadows SM. Angiopoietin-2 Inhibition Rescues Arteriovenous Malformation in a Smad4 Hereditary Hemorrhagic Telangiectasia Mouse Model. Circulation. 2019;139:2049–2063. doi: 10.1161/CIRCULATIONAHA.118.036952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Xanthis I, Souilhol C, Serbanovic-Canic J, Roddie H, Kalli AC, Fragiadaki M, Wong R, Shah DR, Askari JA, Canham L, et al. β1 integrin is a sensor of blood flow direction. Journal of Cell Science. 2019;132(11). doi: 10.1242/JCS.229542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Urbich C, Walter DH, Zeiher AM and Dimmeler S. Laminar shear stress upregulates integrin expression role in endothelial cell adhesion and apoptosis. Circulation Research. 2000;87:683–689. doi: 10.1161/01.RES.87.8.683 [DOI] [PubMed] [Google Scholar]
- 60.Tzima E, Del Pozo MA, Shattil SJ, Chien S and Schwartz MA. Activation of integrins in endothelial cells by fluid shear stress mediates Rho-dependent cytoskeletal alignment. EMBO Journal. 2001;20:4639–4647. doi: 10.1093/emboj/20.17.4639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Stupp R, Hegi ME, Gorlia T, Erridge SC, Perry J, Hong YK, Aldape KD, Lhermitte B, Pietsch T, Grujicic D, et al. Cilengitide combined with standard treatment for patients with newly diagnosed glioblastoma with methylated MGMT promoter (CENTRIC EORTC 26071–22072 study): a multicentre, randomised, open-label, phase 3 trial. The Lancet Oncology. 2014;15:1100–1108. doi: 10.1016/S1470-2045(14)70379-1 [DOI] [PubMed] [Google Scholar]
- 62.Wang X, Freire Valls A, Schermann G, Shen Y, Moya IM, Castro L, Urban S, Solecki GM, Winkler F, Riedemann L, et al. YAP/TAZ Orchestrate VEGF Signaling during Developmental Angiogenesis. Developmental Cell. 2017;42:462–478.e7. doi: 10.1016/j.devcel.2017.08.002 [DOI] [PubMed] [Google Scholar]
- 63.Wang K-C, Yeh Y-T, Nguyen P, Limqueco E, Lopez J, Thorossian S, Guan K-L, Li Y-SJ and Chien S. Flow-dependent YAP/TAZ activities regulate endothelial phenotypes and atherosclerosis. Proceedings of the National Academy of Sciences. 2016;113:11525. doi: 10.1073/pnas.1613121113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kim J, Kim YH, Kim J, Park DY, Bae H, Lee D-H, Kim KH, Hong SP, Jang SP, Kubota Y, et al. YAP/TAZ regulates sprouting angiogenesis and vascular barrier maturation. The Journal of Clinical Investigation. 2017;127:3441–3461. doi: 10.1172/JCI93825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Boopathy GTK and Hong W. Role of Hippo Pathway-YAP/TAZ signaling in angiogenesis. Frontiers in Cell and Developmental Biology. 2019;10;7:49. doi: 10.3389/fcell.2019.00049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Halder G and Johnson RL. Hippo signaling: growth control and beyond. Development. 2011;138:9–22. doi: 10.1242/dev.045500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Piccolo S, Dupont S and Cordenonsi M. The biology of YAP/TAZ: hippo signaling and beyond. Physiol Rev. 2014;94:1287–312. doi: 10.1152/physrev.00005.2014 [DOI] [PubMed] [Google Scholar]
- 68.Zanconato F, Cordenonsi M and Piccolo S. YAP/TAZ at the Roots of Cancer. Cancer Cell. 2016;29:783–803. doi: 10.1016/j.ccell.2016.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wang KC, Yeh YT, Nguyen P, Limqueco E, Lopez J, Thorossian S, Guan KL, Li YJ and Chien S. Flow-dependent YAP/TAZ activities regulate endothelial phenotypes and atherosclerosis. Proc Natl Acad Sci U S A. 2016;113:11525–11530. doi: 10.1073/pnas.1613121113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Giampietro C, Disanza A, Bravi L, Barrios-Rodiles M, Corada M, Frittoli E, Savorani C, Lampugnani MG, Boggetti B, Niessen C, et al. The actin-binding protein EPS8 binds VE-cadherin and modulates YAP localization and signaling. J Cell Biol. 2015;211:1177–92. doi: 10.1083/jcb.201501089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sivaraj KK, Dharmalingam B, Mohanakrishnan V, Jeong HW, Kato K, Schröder S, Adams S, Koh GY and Adams RH. YAP1 and TAZ negatively control bone angiogenesis by limiting hypoxia-inducible factor signaling in endothelial cells. Elife. 2020;9:e50770 doi: 10.7554/eLife.50770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Byzova TV, Goldman CK, Pampori N, Thomas KA, Bett A, Shattil SJ and Plow EF. A mechanism for modulation of cellular responses to VEGF: activation of the integrins. Mol Cell. 2000;6:851–60. doi: [PubMed] [Google Scholar]
- 73.Kiosses WB, Shattil SJ, Pampori N and Schwartz MA. Rac recruits high-affinity integrin alphavbeta3 to lamellipodia in endothelial cell migration. Nat Cell Biol. 2001;3:316–20. doi: 10.1038/35060120 [DOI] [PubMed] [Google Scholar]
- 74.Shiojima I and Walsh K. Role of Akt signaling in vascular homeostasis and angiogenesis. Circ Res. 2002;90:1243–50. doi: 10.1161/01.res.0000022200.71892.9f [DOI] [PubMed] [Google Scholar]
- 75.Eliceiri BP. Integrin and growth factor receptor crosstalk. Circ Res. 2001;89:1104–10. doi: 10.1161/hh2401.101084 [DOI] [PubMed] [Google Scholar]
- 76.Frisch SM and Screaton RA. Anoikis mechanisms. Current Opinion in Cell Biology. 2001;13:555–562. doi: 10.1016/S0955-0674(00)00251-9 [DOI] [PubMed] [Google Scholar]
- 77.Park M-H, Kim AK, Manandhar S, Oh S-Y, Jang G-H, Kang L, Lee D-W, Hyeon DY, Lee S-H, Lee HE, et al. CCN1 interlinks integrin and hippo pathway to autoregulate tip cell activity. eLife. 2019;8:e46012. doi: 10.7554/eLife.46012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yun S, Hu R, Schwaemmle ME, Scherer AN, Zhuang Z, Koleske AJ, Pallas DC and Schwartz MA. Integrin α5β1 regulates PP2A complex assembly through PDE4D in atherosclerosis. J Clin Invest. 2019;129:4863–4874. doi: 10.1172/jci127692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kim N-G and Gumbiner BM. Adhesion to fibronectin regulates Hippo signaling via the FAK–Src–PI3K pathway. The Journal of Cell Biology. 2015;210:503. doi: 10.1083/jcb.201501025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Astone M, Lai JKH, Dupont S, Stainier DYR, Argenton F and Vettori A. Zebrafish mutants and TEAD reporters reveal essential functions for Yap and Taz in posterior cardinal vein development. Sci Rep. 2018;8:10189. doi: 10.1038/s41598-018-27657-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wang C, Zhu X, Feng W, Yu Y, Jeong K, Guo W, Lu Y and Mills GB. Verteporfin inhibits YAP function through up-regulating 14–3-3σ sequestering YAP in the cytoplasm. Am J Cancer Res. 2016;6:27–37. doi: [PMC free article] [PubMed] [Google Scholar]
- 82.Kato K, Diéguez-Hurtado R, Park DY, Hong SP, Kato-Azuma S, Adams S, Stehling M, Trappmann B, Wrana JL, Koh GY, et al. Pulmonary pericytes regulate lung morphogenesis. Nat Commun. 2018;9:2448. doi: 10.1038/s41467-018-04913-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Liu-Chittenden Y, Huang B, Shim JS, Chen Q, Lee SJ, Anders RA, Liu JO and Pan D. Genetic and pharmacological disruption of the TEAD-YAP complex suppresses the oncogenic activity of YAP. Genes Dev. 2012;26:1300–5. doi: 10.1101/gad.192856.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Messmer KJ and Abel SR. Verteporfin for Age-Related Macular Degeneration. Annals of Pharmacotherapy. 2001;35:1593–1598. doi: 10.1345/aph.10365 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.