

Abstract
The critical aspects of the corrosion of metal electrodes in cathodic reductions are covered. We discuss the involved mechanisms including alloying with alkali metals, cathodic etching in aqueous and aprotic media, and formation of metal hydrides and organometallics. Successful approaches that have been implemented to suppress cathodic corrosion are reviewed. We present several examples from electroorganic synthesis where the clever use of alloys instead of soft neat heavy metals and the application of protective cationic additives have allowed to successfully exploit these materials as cathodes. Because of the high overpotential for the hydrogen evolution reaction, such cathodes can contribute toward more sustainable green synthetic processes. The reported strategies expand the applications of organic electrosynthesis because a more negative regime is accessible within protic media and common metal poisons, e.g., sulfur-containing substrates, are compatible with these cathodes. The strongly diminished hydrogen evolution side reaction paves the way for more efficient reductive electroorganic conversions.
1. Introduction
1.1. General Background
The electrification of chemical transformations emerges as one of the most promising routes to promote sustainability without compromising economic competitiveness.1 The foundation of this approach is based on the use of electric current as a reagentless way to electrosynthesize the compounds of interest. Preferentially, in electrosynthesis, inexpensive and abundant renewable electricity is used both as an energy source and as a green reagent for reduction and oxidation reactions, which allows substituting conventional oxidizing or reducing agents for target reactions or avoiding hazardous reagents and reagent waste.2−8 Therefore, electrosynthesis will play a crucial part in decarbonizing the chemical industry and improving the sustainability of synthetic processes.9 If the electricity is affordable and the organic products generated exhibit enough added value, electrosynthesis will pay off.10
During the past two decades, anodic oxidations have received the lion’s share from the renaissance of organic electrosynthesis.11,12 However, cathodic electroconversions can also generate high value-added chemicals and help to mitigate the impact of chemical manufacturing on climate change. Cathodic conversions are of specific interest when biomass as a nonfossil feedstock is considered because deoxygenation processes are required.13
In cathodic electrosynthesis, electrons are fed to the catholyte by electrodes such as lead, tin, mercury, or carbon allotropes. The catholyte embodies the substrate(s), solvent, and commonly either a neutral, acidic, or basic supporting electrolyte for maintaining sufficient ionic conductivity. The simplest setup consists of two electrodes in an undivided cell under constant current control (Figure 1, left). More complicated setups can also be used. For example, instead of the current, the electrode potential can be controlled, which prolongs reaction time and requires an additional reference electrode and a more expensive and sophisticated power source. However, this setup can enhance the reaction’s selectivity. In addition, secondary and parallel reactions can be avoided by using a divided cell, where anolyte and catholyte are separated from each other by an ion-permeable membrane (i.e., Nafion) or a porous glass frit (Figure 1, right). This leads to a higher terminal voltage due to the additional ohmic resistance for the separator and an increased electrode distance when the same current density is used. For scalability reasons, only constant current modes are employed, and if the electrolysis is stopped in time, a practical setup is established providing good yields.14 In addition, cathodic corrosion appears mostly under electrosynthetic conditions wherein high current densities are involved and potential control is irrelevant.
Figure 1.

Undivided (left) and divided (right) beaker type cells. Reproduced with permission from ref (8). Copyright 2018 under CC-BY from Waldvogel et al.
Cathodic reductions are presently hindered by several issues that require attention before this technology can be broadly implemented in the chemical industry. One of the issues that needs to be urgently addressed is the fate of the cathodes, specifically their corrosion under reductive reaction conditions. This widely disregarded problem is especially crucial in the highly negative regime of reductive electroorganic synthesis, where heavy metals (e.g., lead, tin, mercury) with high overpotential for the hydrogen evolution reaction are commonly the best performing cathodes. Their deterioration through cathodic corrosion currently prevents the translation and implementation of the developed technologies for large-scale chemical manufacturing. The key challenges are the release of species with pronounced toxicity as an environmental and product-safety concern, and the effect of metal ions from the cathodic corrosion, e.g., Pb2+ on lowering the performance of cell separators such as Nafion.
1.2. Cathodic Electroorganic Synthesis
Reductions are fundamental in chemistry. Traditionally, substrates are reduced with dissolving metals (i.e., Na, K, Na/K, Li, Zn, Fe, Al, Ni), metal hydrides, or with hydrogen or hydrogen donors in the presence of a transition metal (TM) catalyst or a frustrated Lewis pair (FLP).15−19 Substrates can also be reduced electrochemically using inexpensive electrons ($0.006 mol–1) supplied directly by the cathode or indirectly using a mediator.20−25
In Scheme 1, the reaction conditions for three chemical (a–c) and one electrochemical (d) benzyl amine syntheses from benzamide are shown.26−29 In these cases, the thermochemical conversions necessitate, for example, excess of reducing agents, toxic reagents or solvents, or high pressure and temperature. In contrast, the electrochemical conditions are quite mild in comparison, and strikingly, the presence of strong acids is allowed, which is unattainable in reductions with dissolving metals, FLPs, or metal hydrides. The high tolerance to acids stems from the fact that common cathode materials (e.g., lead or mercury) have low exchange current densities for proton reduction (Table 1). As a consequence, substrates with relatively high negative reduction potentials can be reduced selectively in the presence of various proton donors (i.e., in water or alcohol solutions), even under acidic conditions.20 In addition to technical and economic benefits, electrosynthesis is aligned with the principles of green chemistry, if, for example, the amount of supporting electrolyte is minimized or recycled and the solvent is chosen wisely.30
Scheme 1. Selected Examples of Reduction of Benzamide to Benzyl Amine Using Transfer Hydrogenation (a), Heterogeneous Transition Metal Mediated Hydrogenation (b), Hydride Reduction (c), and Electrochemical Reduction (d).

Table 1. Exchange Current Densities of Several Cathodes Commonly Used in Cathodic Electroorganic Synthesis.
| cathode material | Pb | Sn | Hga | Cd | Cu | Pt (smooth) |
|---|---|---|---|---|---|---|
| exchange current density for hydrogen evolution: log(j0/ A m–2)b | –8.7 | –4.0 | –7.7 | –3.0 | –2.7 | 1.0 |
Banned in large quantities in most countries.
Measured in 1.0 M HCl at 20 °C; values from ref (31).
The low exchange current density for proton reduction is crucial but is not the only parameter that dictates the choice of a suitable cathode material for electroreductions.32 The cathode should also have a high electrical conductivity, and it should be inexpensive, easy to manufacture, easily machinable, mechanically durable, and chemically stable. The latter, viz, resistance to corrosion, is seldomly investigated or discussed in academic studies but is critical in the chemical industry as the downtime associated with the replacement of corroded cathodes translates into substantial operating expenses. Cathodic corrosion increases the overall cost of the synthesis but also complicates downstream processes and purification, especially if the electroreduction is at a late stage of the synthesis.33,34 The contamination of the reaction products with heavy metals from these cathodes can be particularly pronounced. Therefore, the high toxicity of the metallic and organometallic corrosion products overshadows the otherwise suitable properties of these metals. In addition, cathodic corrosion can induce morphological changes that can complicate the scenery in electroreductions. Some properties such as specific surface areas or surface compositions of the electrodes may change during the electrosynthesis, which can lead to rather large dissimilarities between nominal and absolute current densities as well as variations in overpotentials for the hydrogen evolution reaction (HER). This manifests in differences in performance between the pristine and used electrodes.35,36 Although some of these effects can be advantageous in some aspects, the cathodic corrosion is generally regarded as undesirable in electrosynthesis.
In this critical review, we first present a brief overview of the different mechanisms and reaction conditions that corrode the cathodes and, subsequently, discuss general concepts that can help to prevent the cathodic corrosion. Thereafter, we review reductive organic electrosynthetic processes that take advantage of these approaches to prevent the detrimental corrosion. We keep a strong focus on the lead cathodes as they are the most widely used cathodes in electroreductive industrial processes.25 For example, lead cathodes have been used in the reduction of pyridine to piperidine (Robinson Bros.),37 2-methylindole and tetrahydrocarbazole to 2-methylindolene and hexahydrocarbazole (L. B. Holliday, BASF),38,39 phthalic acid to dihydrophthalic acid (BASF),40 maleic to succinic acid (CECRI),41 acrylonitrile to adiponitrile (Monsanto, BASF),42 and anthranalic acid to o-aminobenzyl alcohol (BASF)43 (Scheme 2).
Scheme 2. Examples in Electroreductions with Lead Cathodes.

The different corrosion mechanisms and the parameters that affect the corrosion rate are common to various cathode materials. Therefore, in some parts, we also discuss the cathodic corrosion of electrode materials such as platinum, palladium, and gold. Because of their relatively high exchange current densities for proton reduction, they might not be as useful as cathodes in reductive organic electrosynthesis per se, but understanding the cathodic corrosion processes through them can prove very useful. In particular, platinum is commonly used as counter electrode in anodic oxidation reactions, and therefore its deterioration can have important consequences for the overall electrolysis.25,32 Finally, we suggest some simple guidelines for the organic electrosynthesis community to remain aware of cathodic corrosion and take it into account. As a final note, we do not review corrosion through anodic oxide formation that is followed by cathodic reduction of the metal oxides,44−46 cathodic corrosion of nanoparticles,47 or open-circuit corrosion.48 It is noteworthy that the latter can be an important aspect to consider when setting up electrosynthesis.
2. Cathodic Corrosion Processes
In cathodic corrosion, the cathode corrodes under negative polarization that is induced by an externally applied electric field. In contrast to anodic corrosion, which is a very well-known and understood process, the corrosion of the cathode is relatively counterintuitive. To put it in a very simple way, all the metals form cations but only few of them can form discrete anions (e.g., Au−, Pt2−) or anionic “Zintl-type” clusters (e.g., Pb94–, Bi42–, etc.).49−60 Furthermore, these metal anions or anionic clusters are extremely sensitive to oxygen and humidity. Consequently, they are not detected under ambient conditions. Therefore, the metallic form of a (post)transition metal is generally regarded as the most stable species that is immune to corrosion at cathodic potentials. In fact, some metals are even protected from the chemical corrosion by cathodic polarization.61 Yet, the cathodic corrosion has been documented for almost as long as electrochemistry itself. Already in 1808, Seebeck noticed that mercury cathodes react with ammonium carbonate,62,63 and in 1810, Sir Humphry Davy reported the corrosion of an arsenic cathode.64 Notably, the corrosion of lead and arsenic cathodes was studied by Reed in 1895.65 Haber and Bredig surveyed the early studies and reported their own studies on the cathodic corrosion of lead and other electrodes in 1898.66 Further studies were published in 1902 and 1903 by Haber and Sack.67−69 They proposed that, in the presence of alkali metal cations, cathodes form a bimetallic alloy that decomposes, causing cathodic corrosion.69 However, corrosion in acidic electrolytes with lead and bismuth cathodes was also observed, albeit to a lesser degree, and Haber consequently proposed a process involving the formation of metal hydrides in these cases.69 Later, Salzberg and co-workers studied the cathodic corrosion of lead and tin electrodes.70−72 They dismissed the alloying mechanism proposed by Haber and co-workers and suggested instead that the formation of metal hydrides is the major mechanism responsible for cathodic corrosion, including in the presence of alkali cations. Later, Kabanov argued in favor of the bimetallic corrosion pathway.73,74 Besides these two mechanisms, other proposed processes included cathodic etching,75,76 contact glow discharge,77 and anodic degradation of the cathode.78 In the following sections, we briefly discuss the main mechanisms that have been suggested to contribute to cathodic corrosion. We aim at providing a brief overview on the mechanisms but make no attempt to argue on their validity as, in most instances, the corrosion mechanism depends on the applied current densities and other experimental conditions such as electrolyte composition. Furthermore, several mechanisms may coexist under reaction conditions.
2.1. Alloying with Alkali Metals
Negative polarization of the cathode has been proposed to induce alloying with alkali cations.66−69,73,74 In the proposed mechanism, alkali cations (M+) from the electrolyte (Scheme 3a) form a bimetallic alloy with the cathode (b). The alloy is then oxidized and the alkali cation leaches from the cathode, yielding a sponge-like structure (c). Porous cathodes can then be pulverized due to the rapid decomposition of the alloy and evolution of hydrogen.69 Haber proposed this mechanism for the cathodic corrosions of several metals and alloys, for example, Pb, Sn, Bi, Sb, Hg, As, Tl, and Rose alloy electrodes.66
Scheme 3. Schematic Representation of Cathodic Corrosion through an Alloying-type Mechanism.

However, Salzberg’s experiments revealed some inconsistencies with this mechanism in the case of Pb.70 Specifically, he observed that the corrosion is faster for electrolytes with low concentrations of alkali ions, which is the exact opposite of the trend expected from thermodynamics when varying the chemical potential of the cations in solution. Similar reasoning was provided for tin cathodes.72 In the case of platinum cathodes, several convincing arguments were presented, suggesting that the corrosion is unlikely to follow the alloying mechanism.75,79 First, the cathodic corrosion started at relatively low negative potentials (−1.3 V vs NHE) that are insufficient to reduce Na+ to Na(0) as its E1/2 is −2.71 V (vs NHE).80 Furthermore, the corrosion was also observed in the presence of organic cations, i.e., conditions incompatible with alloying at least in a conventional sense.75 Moreover, instead of a spongy cathode, Yanson and Koper et al. observed the formation of etching pits, which further contrasts with the physical disintegration expected for the alloying mechanism.75 Last, decomposition of the alloyed cathode should be inhibited by high cathodic potentials but, in contrast, the corrosion of the cathode was assisted by it in their experiments.75 Conversely, Kabanov argued that the alloying mechanism prevails for Zn, Cd, Ag, Al, Pb, and Sb cathodes as, for example, potassium diffused through a zinc foil whereas cesium and tetramethylammonium did not.73,74 Moreover, a plot of the hydrogen evolution potential vs the logarithm of current density has been noted to change between −0.2 V and −0.3 V over time in the presence of sodium cations, which was attributed to the formation of a bimetallic alloy with a silver cathode.73 Brown has also reported the penetration of sodium into a lead cathode.81 Anawati, Frankel et al. explained their energy dispersive spectroscopy (EDS) results for a tin cathode exposed to K+ by the formation of either K or KSn bimetallic alloy.82 Although direct experimental evidence about the contribution of bimetallic alloys to cathodic corrosion is scarce, they have been discussed as plausible intermediates in many reports.36,78,82−84
2.2. Formation of Metal Hydrides
Several metals such as Cu,85 Pb,69−71,86 Sn,36,72,78,82,84,86,87 Bi,69,88,89 and Sb,83,90,91 can form (meta)stable hydrides that have been proposed as possible intermediates in their cathodic corrosion. In this mechanism, M–H bonds are formed at the interface between the electrode and aqueous solutions (Scheme 4)86,88 The metal hydrides can then be released from the cathode and either evaporate from the reaction mixture or react with an oxidizer (e.g., water) in the solution to produce hydrogen gas and metallic particles. These metal particles can then either remain suspended in the solution or redeposit back to the cathode.86 With lead and tin, dihydrides were proposed to form and then disproportionate to the metallic parent elements and the corresponding tetrahydrides (2MH2 → M(0) + MH4).72 However, it should be noted that some metal hydrides could form indirectly by oxidation of anionic intermediates during electrolysis.83 For example, the treatment of ZnSb alloy with dilute sulfuric acid was described to result in the formation of SbH3.92,93
Scheme 4. Mechanism of Cathodic Corrosion through Formation of Metal Hydrides.

Interestingly, PbH4 has remained largely elusive, but there has been at least one report on its formation from a lead cathode used in electrochemical hydride generation atomic absorption spectrometry.94 Furthermore, PbH4 has also been synthesized by other means.95,96 Huang, Li et al. reported the formation of lead and tin hydrides that subsequently decomposed to hydrogen gas and metal atoms, which later aggregated to form metallic clusters and hydrosols.86 Katsounaros et al. detected gaseous SnH4 originating from a tin cathode at more negative potentials than −2.4 V (vs Ag/AgCl) during the electrochemical reduction of nitrate to nitrogen.36 Salzberg and Mies were able to detect SnH4 evolving from the tin cathode as well, even though most of their corrosion products were colloidal in nature.72 Tin has also been observed to corrode from a two phase Al–Sn binary alloy cathode (0.4 wt % Sn).87 Upon cathodic corrosion, which was attributed to the formation of SnH4, empty pits were found where previously Sn phases resided. Chiacchiarelli, Frankel et al. studied in detail the tin cathodes corrosion mechanisms in aqueous KHCO3.78 Under some of the studied conditions, the weight loss of the cathode was likely associated to the formation of SnH4. Interestingly, they also observed deposits on the electrode that were suggested to be NaH and KH and noticed that the grain orientation affected the rate of the corrosion process (Figure 2). Later, Anawati, Frankel et al. studied the corrosion behavior of tin cathodes in KCl solutions.82 The cathode corroded probably because of SnH4 formation, and the deposits that were formed on the electrode were proposed to be either potassium metal or the bimetallic compound KSn. Under some conditions, SEM revealed that Sn particles were on the surface of the electrode after the electrolysis even though the electrode’s surface appearance did not change visually and SEM images did not reveal any corroded areas. Bismuth has been proposed to form hydrides as well,69,88,89 and intriguingly a correlation between corrosion and amount of produced hydrogen was observed in one study.89 Fascinatingly, the disintegration can be so vigorous that the corrosion products are ejected out of the solution and can be collected on a glass slide that is held in air above the cell.88 When the gas released by the cathodic process using an antimony electrode was passed through solid NaOH, deposits of Sb were found. It was believed to originate from SbH3.83 Furthermore, evolution of SbH3 has been observed from antimony cathodes at another occasion,90 and in another report, it was proposed as corrosive intermediate.91 Formation of Cu2O was observed when copper and 30% Cu–Ni cathodes were negatively polarized in aerated 0.5 M H2SO4 solutions.85 The authors suggested copper hydride as an intermediate which would subsequently oxidize to cuprite. A recent report by Hersbach, Koper et al. postulated ternary metal hydrides (AxMyHz, A = alkali or alkaline earth metal) as possible intermediates in the cathodic corrosion of Pt, Rh, and Au cathodes under aqueous alkaline conditions.97
Figure 2.

Scanning electron micrograph (left, a) and photograph (right, b) after polarization of a tin cathode at 175 mA cm–2 in 0.5 M KHCO3 at 500 and 3000 rpm, respectively. Reproduced with permission from ref (78). Copyright 2011, Springer Science Business Media BV.
Salzberg and co-workers showed that the corrosion of lead cathodes is the slowest in the presence of (NH4)2SO4 in the absence of an acid and, in the presence of an acid, the order of the rate in respect to cations is NH4+ < Cs+< K+∼ Na+ ∼ Rb+ < Li+.71 The corrosion is faster when lower concentration of supporting electrolyte is used.70,71 In the absence of any salt (i.e., in pure H2SO4 solutions), higher current densities were necessary for the corrosion.71 Furthermore, increasing the concentration of sulfuric acid reduced the disintegration of the lead cathode in the absence and presence of a constant amount of a conducting salt.71 With tin cathode, the current densities required for the corrosion were about an order of magnitude greater than with the lead cathode (i.e., ∼20 mA cm–2 and ∼500–600 mA cm–2 in 0.1 M Na2SO4 for Pb and Sn, respectively).71,72 Cations were found to increase the corrosion rate of tin cathodes in the order of H3O+ < NH4+ ≪ Li+< K+< Na+.72 Intriguingly, the addition of sulfuric acid decreased the corrosion rate in the presence of a constant amount of Na2SO4, and increasing the salt concentration at constant acidity increased the corrosion rates. In contrast, the corrosion rate decreased when the concentration of Na2SO4 and NaOH electrolytes were increased in the absence of acid.72 Furthermore, addition of KCl to Na2SO4 decreased the corrosion rate and addition of RbCl or CsCl completely stopped the disintegration of the tin cathode. The good performance of NH4+ with tin and lead cathodes was attributed to the ammonium reduction reaction that takes place at a lower potential than the hydride formation.71,72 In an another study performed with a tin cathode at −2.9 V (vs Ag/AgCl), the corrosion was observed to be 20 times slower when a small amount of NH4+ was added to the reaction mixture that contained K+ based electrolyte.36 Furthermore, the apparent rate constant of the reduction and the total electrolysis current increased experiment after experiment until it reached the third reaction, which was attributed to an increased specific surface area of the tin electrode caused by cathodic corrosion, something which could also be observed visually (Figure 3).
Figure 3.

Tin cathode before electrolysis (left, a) and after extensive use (right, b). Reproduced with permission from ref (36). Copyright 2006 Elsevier Ltd.
The same research group later showed that the concentration of tin in solution after electrolysis at −1.8 V (vs Ag/AgCl) decreased from 79 to 38 mg L–1 when the NaCl electrolyte’s concentration was increased from 0.1 to 0.5 M.84 Interestingly, further increase did not limit the corrosion any further. In contrast, during the degradation study of tin cathodes in N2 atmosphere, Chiacchirarelli, Frankel et al. reported that decreasing the concentration of the KHCO3 supporting electrolyte or the current density decreases also the cathodic corrosion rate.78 Intriguingly, they also proposed an autoinhibitive corrosive process, where corrosion creates high local roughness that leads to local decrease of the current density which inhibits further corrosion of the cathode. Anawati, Franklin et al. showed that increasing the pH of the solution to make it slightly alkaline was beneficial in reducing the corrosion at potentials between −1.8 V and −2.2 V (vs SCE).82 The Pourbaix diagram of tin predicts that the formation of tin hydride is easier when the pH of the solution is low.36,82 Bismuth cathode was found to corrode at potentials between −2.8 and −1.8 V (vs Ag/AgCl) in 0.4 M NaHCO3 + 0.4 M Na2CO3 + 0.05 M NaNO3 supporting electrolyte system.89 Notably, the cathodic corrosion started to increase at around −2.3 V (vs Ag/AgCl).
2.3. Cathodic Etching
Cathodic etching is another mechanism that can corrode cathodes, and it has been documented in both aqueous (Scheme 5, up, a–c) and anhydrous aprotic solvents (Scheme 5, down, d–f) for a range of metals including Pt,75,79,80,97−116 Rh,75,97,98,101,102,117 Ir,101 Pd,98,101,106,107,118,119 Au,75,97,98,101,117 Ag,75,98,101 Cu,75,98,101,120 Re,98 Fe,98 Ni,75,98,101 Nb,75,98 Ru,75 Ti,98,121−123 V,121 W,121 Si,75,98 Nb,75 Ru,75 Al,98 Pb,56,57,124−131 Sn,125,127−133 Sb,83,91,129,134 Bi,128,129,135,136 Hg,62,125,128,137−149 Ga,129 and In.129 In this corrosive process, metal anions or anionic clusters are formed at the metallic cathode and stabilized by nonreducible cations from the electrolyte. The solvent environment plays an important role in the following steps. Under aprotic conditions, the anionic clusters can either remain at the electrode surface or leave it to suspend in the solution. Alternatively, ionometallic phases may form. In protic media, the metal anions or clusters are detached from the cathode. This is thought to be because a dynamic water-free layer with high pH forms at the solution–cathode interface.75 Thereafter, the free metal anions encounter oxidants (e.g., water) in the solution and quickly reoxidize to give metal atoms that agglomerate in the elemental state. Because this oxidative process takes place in the vicinity of the cathode, the agglomerates can be deposited back onto the electrode. This is corroborated by the fact that different crystal orientations in a gold cathode, which had dissimilar cathodic etching rates, had different surface concentrations of gold nanoparticles after electrolysis (Figure 4).75 Moreover, the agglomeration was also observed when discrete Bi42− anions were oxidized with alcohols to bismuth nanoparticles.58 In the case of post-transition metals, the anionic clusters may react with protic solvent and generate metal hydrides. For example, Yang, Li et al. suggested two alternative pathways for the generation of SbH3 in aqueous solutions.83 According to them, stibine can form either directly (Sb + 3H+ + 3e– → SbH3) or indirectly (Sb73– + nH2O → SbH3). This can complicate the analysis of corrosion mechanisms in protic conditions for these elements. Recently, the involvement of Zintl phases in the cathodic disintegration of Pb, Sn, and Sn50Pb50 cathodes was postulated in aqueous conditions.131
Scheme 5. Cathodic Etching in Aqueous Media (Up) or Aprotic Media (Down) and Subsequent Selected Plausible Reaction Steps.

Figure 4.

Gold cathode after operation at −10 V (vs RHE) in an aqueous electrolyte containing 1 M Na+ showing differing corrosion rates depending on crystal orientations (left, a), and different surface concentrations of gold nanoparticles (right, b). Reproduced, with permission from ref (75). Copyright 2011 Wiley-VCH.
2.3.1. Cathodic Etching in Anhydrous Aprotic Solvents
A cathodic etching mechanism has been proposed for anhydrous aprotic organic solvents wherein the formation of metal hydrides is unlikely. Kariv-Miller et al. studied extensively the cathodic corrosion in anhydrous DMF and diglyme solutions of Pb,124−129 Sn,125,127−129,132 Sb,129,134 Bi,128,129 Hg,125,128,137−144 Ga,129 In,129 Cr,129 and Pt129 cathodes at high negative potentials. In the presence of suitable tetraalkylammonium cations, corrosive products are formed at and by the cathode.124,126,127,129,132,134,138 Interestingly, when they introduced a ketone in the electrolysis cell, characteristic colors of ketyl radical anions were developed, indicating that the corrosion product retained a powerful anionic reductive character.126,127,129,134 The reduction of the ketone was later also monitored by EPR.126,127 Furthermore, when oxygen was brought into the electrolysis cell or when the potential of the cell was shifted to a more positive value, the tetraalkylammonium salts and the metallic elements were regenerated (Scheme 6).124−129,138 At one instance with Pb cathode, metallic particles were detected floating in the cell when oxygen was introduced in the cell.124 Also, the corresponding reaction between quaternary phosphonium and tertiary sulfonium supporting electrolytes with mercury cathode has been reported.145 The corrosion products, coined “tetraalkylammonium-metals”, have found use as mediators in the reductive electrosynthesis at very high negative potentials (Scheme 7).132,141,150−160 Furthermore, Medina-Ramos, Fenter et al. demonstrated that in the presence of imidazolium cations in acetonitrile, bismuth cathode undergoes structural changes that result in the partial dissolution of the cathode at negative potentials.135,136 In situ X-ray reflectivity measurements showed that the behavior of the cathode was mostly reversible, but excessive cycling enhanced the cathodic corrosion. Simonet and co-workers demonstrated that Pt cathodes corrode in dry DMF in the presence of tetraalkylammonium or alkali supporting electrolytes.104−109 It was noted that the corrosion products retained reductive anionic character, and they reacted with aromatic ketones and nitroaryl derivatives to produce the corresponding radical anions (Figure 5, right).104,106−109,113 Interestingly, EPR studies confirmed the existence of paramagnetic species when the corroded platinum cathode was dipped into the solution that contained either 9-fluorenone, 2,4-dinitrotoluene, or para-dinitrobenzene.107,109,113 After the electrolysis, the metallic state of platinum was regenerated by air, but this resulted in changes at surfaces of Pt cathodes (Figure 5, left and middle).104−109 The corrosion has also been studied by electrochemical atomic force microscopy with epitaxial Pt (100) thin layers deposited on MgO (100) in DMF.110−112 Here, the morphology was almost retained when the thin layer electrodes were regenerated electrochemically or by air exposure.110,111
Scheme 6. Chemically and Electrochemically Reversible Formation of “Zintl Type” Phase from Lead.

Scheme 7. Examples of Tetraalkylammonium Mediated Electroreductions with Mercury Cathodes.

Figure 5.

Platinum sheet prior to electrolysis (left, a), after cathodic polarization at −2.3 V (vs Ag/AgI) with 0.1 M CsI in DMF (middle, b), characteristic color of 2,4-dinitrotoluene radical anion after reduction by [Pt2–, Na+, NaI] (right, c). Reproduced with permission from ref (107). Copyright 2002 under CC BY-NC-ND 4.0 license.
Pd cathode was observed to corrode as well, and the corrosion resulted in modified surfaces in dry DMF in the presence of alkali or tetraalkylammonium supporting electrolytes (Figure 6).106,107,118,119
Figure 6.

Palladium before (left, a) and after cathodic polarization at −2.3 V (vs Ag/AgI) in 0.1 M Bu4NBr in DMF (right, b). Reproduced with permission from ref (118). Copyright 2001 Elsevier Ltd.
Empirical formulas of (R4N)n(Mm) have been determined for Pb, Sn, Sb, Bi, and Hg corrosion products by various techniques.124−127,138,140,142−144,146−149 Here, anionic clusters (i.e., Mmx–) hold the excess of electron density and the tetraalkylammonium cations stabilize these polymetallic anions.126 X-ray powder diffraction from isolated corrosion product with empirical formula of DMP(Pb5) showed that it has a face-centered cubic crystal structure. This indicates that crystallites of metallic Pb are part of its structure and, therefore, it does not consist of discrete molecular clusters.127 In the case of tin, it was suggested that the corrosion product with empirical formula TMA(Sn5) consists of negatively charged small particles or clusters (size less than 50 Å) arranged in a noncrystalline morphology.127,132 However, also discrete Zintl anions have been prepared from lead and tin cathodes in liquid ammonia or ethylenediamine.56,57,59 On the basis of in situ vibrational Raman spectroscopy and DFT calculations, Yang, Ji et al. demonstrated that the Zintl type phase of compounds are formed when Sn and Pb cathodes corrode in the presence of tetraalkylammonium cations and that these intermediates then either later oxidize or they undergo Hoffmann elimination to generate neutral metal particles.130 The Hoffmann elimination is supported by the detection of amine elimination products by GC/MS from the reaction mixture130 or from the thermal decomposition of the isolated corrosion products of lead and tin by mass spectrometry.126,127 Similar mechanism was also suggested to corrode Sn cathode in a 1-butyl-1-methylpyrrolidinium bis(trifluoromethylsulfonyl)imide ionic liquid133 and Sb cathodes in tetrabutylammonium hexafluorophospate or bromide supporting electrolytes in DMF or acetonitrile.83,91 Furthermore, Medina-Ramos, Fenter et al. proposed that bismuth thin-film cathode forms Bi/[Im]+y complexes when imidazolium cations are present in the supporting electrolyte.135,136 With platinum and palladium, the formation of different iono-metallic layers has been described.104,105,107−109,111,113,118,119
When Pb cathode was corroded in DMF in the presence of R4NPF6 (R = Me, Et, Bu) electrolytes, Me4NPF6 formed a black spongy material on the surface, Et4NPF6 yielded a spongy material as well as a black stream, and Bu4NPF6 produced only a black stream (Figure 7, right).130 Moreover, when the cathode was changed from lead to tin, Me4NPF6 produced a layer of gray and fluffy substance that eventually peeled off from the Sn cathode’s surface, and with Et4NPF6 and Bu4NPF6 supporting electrolytes, the Sn cathode directly decomposed into particles (Figure 7, left).130 In contrast, tin cathodes corrosion product prepared in DMPBF4 supporting electrolyte was stuck onto the electrode surface and could not be removed even by stirring the solution.127,132 Increasing the amount of tetramethylammonium or dimethyl pyrrolidinium based supporting electrolytes shifted the corrosion onset voltages toward more positive values for tin, bismuth, and lead cathodes in DMF.124,125,128 The order of corrosion onset potentials in respect to different cathode materials was found to be Sb (−1.92 V vs SCE; 0.05 M of supporting electrolyte) > Sn (−2.19 V vs SCE) > Bi (−2.27 V vs SCE) > Pb (−2.4 V vs SCE) > Hg (−2.59 V vs SCE) in 0.5 M DMPBF4.128,134 The density, melting point (for solid electrodes), and covalent radius follow the same order, which suggest that the strength of the metal lattice is an important parameter in the cathodic corrosion.128,134
Figure 7.

Time evolution of cathodic corrosion of tin (left, panel a) and lead (right, panel b) cathodes in DMF in 0.1 M solutions of tetramethylammonium (a), tetraethylammonium (b), and tetrabutylammonium (c) hexafluorophosphates. Reproduced with permission from ref (130). Copyright 2015 The Royal Society of Chemistry.
Cathodic corrosion onset voltages of antimony cathodes decreased in the order of Bu4NBF4 (−2.91 V vs SCE), DMPBF4 (−2.69 V vs SCE), and Me4NBF4 (−2.59 V vs SCE) in DMF.134 Interestingly, the corrosive currents with DMPBF4 and MeNBF4 were at least 10 times higher that when Bu4NBF4 was used. Furthermore, the use of Me4NBF4 and DMPBF4 lead to the formation of soluble corrosion product that transformed into a solid material after 14 h when the electrolysis was stopped. Interestingly, with Bu4NBF4, no preparative amount of solid product was formed. Matching this observation, Medina-Ramos, Fenter et al. showed that cathodic corrosion of a bismuth thin-film rotating ring-disk electrode was either attenuated or absent when tetrabutylammonium supporting electrolytes were employed, but when imidazolium cations in acetonitrile were used instead, the cathode corroded at potentials more negative than −1.25 V (vs Ag/AgCl).136 This difference in behavior of TBA and BMIM was attributed to the latter’s stronger binding to the bismuth cathode. In TBA, the positive charge is more sterically shielded in contrast to BMIM, which is why BMIM can penetrate and corrode the bismuth cathode better (Figure 8).
Figure 8.

ReaxFF snapshot highlighting the differences in penetration of BMIM (left, panel a) and TBA (right, panel b) into the bismuth cathode. Reproduced with permission from ref (136). Copyright 2018 American Chemical Society.
The different cations behave differently when platinum or palladium are polarized cathodically,104−107,109,118,119 and increasing the amount of supporting electrolyte also increases the corrosive currents for all electrolytes.104,105 In one example with palladium cathode, the corrosive currents decreased and the peak potentials became more negative in the order of Me4N+ > Et4N+ > Bu4N+ > Hex4N+ > Oct4N+ with halide counteranions in dry DMF (0.1 M).118,119 This order of the peak potentials was the same with Pt (100) thin layers.111 In the latter case, the bulkier the cation, more energetically demanding is its insertion into the platinum electrode and the formation of ionometallic phases.111 Interestingly, with palladium and platinum, the different anions on the tetrabutylammonium moiety affected the corrosion currents, and the order was reported to be ClO4– > BF4– > I– > PF6– = 0.107 With mercury cathode, correlation between more negative reduction potential and higher volume of the cation was uncovered.140
2.3.2. Cathodic Etching in Aqueous Media
Cathodic etching in aqueous alkaline conditions has been studied extensively because it is an appealing method to produce nanoparticles without ligands or exogenous reducing agents.75,76 The size and the shape of the nanoparticles can be controlled by adjusting the nature of the supporting electrolyte and the current density, which also consequently affects the corrosive processes.99,100 The etching of the metal cathodes is possible by either applying direct (DC) or alternate currents (AC). The corrosion mechanism is believed to be similar in both modes, but there is remarkable difference in the dispersion of the cathode to the solution. With DC, strongly alkaline conditions (e.g., >10 M NaOH) are required to disperse the cathode, whereas under milder alkaline conditions, nanoparticles form onto the electrode, which is evidenced by an increased surface area of these cathodes and SEM imaging (Figure 9).75,97,99 In contrast, AC waveforms disperse the cathode readily under softer conditions (e.g., 0.075–1 M NaOH).76,99 This behavior with AC stems mainly from the fact that during a positive phase the nanoparticles are partially oxidized, which facilitates their detachment from the electrodes.79
Figure 9.

Scanning electron micrographs of Pt electrodes corroded at −1 V (vs RHE) in 1 M NaOH (a,d), 5 M NaOH (b,e), and 10 M NaOH (c,f). Yellow markings in (d) feature triangular etching pits. Reproduced with permission from ref (97). Copyright 2018 under CC BY-NC-ND 4.0.
To date, pure Pt,75,76,79,80,97−103,114−116 Rh,75,76,97,98,101,102,117 Pd,98,101 Au,75,97,98,101,117 Ag,75,101 Ir,101 Cu,75,98,101,120 Re,98 Ru,75,101 Ir,101 Fe,98 Ni,75,98,101 Nb,75,98 Ti,98,121−123 V,121 W,121 Si,75,98 Sb,91 and Al98 electrodes have been reported to cathodically corrode through etching mechanism in aqueous media. Regarding alloys, Pt90Rh10,76,98 Pt70Rh30,76,98 Pt55Rh45,102 Pt20Rh80,76,98 Pt12Rh88,102 Pt80Ir20,76 Pt95Ru5,76 Pt50Ni50,76 AuCo,76 AuCu,76 and FeCo,76 Pt50Au50,101 Pd50Au50,101 AgxAu100–x (x = 10, 30, 50, 70, 90),101 Fe8.81Ti91.18,123 Cu6.28Ti93.72,123 and Sn42.82Ti57.18123 have been reported to corrode cathodically as well. In addition, metal oxides161 and in situ electrodeposited alloys such as PtBi, PtPb, PtSn, PdPb, and AuCu are also etched cathodically.161 Usually the corrosion products of these alloys retain the composition of the bulk electrode, which implies that clusters of metal anions dissolve directly into the electrolyte.76,98,123 However, later, Hersbach, Koper et al. discovered using various PtRh alloy cathodes that the corrosion behavior was dominated by platinum and the composition of the corrosion products were different at atomic level from the composition of the bulk alloy.102 Later, similar observation was made with PtAu and PdAu alloy cathodes.101
Much like in aprotic conditions, the mechanism in aqueous alkaline environment is proposed to proceed by the formation of cation-stabilized metastable metal anions.75,80 The presence of cations (alkali, alkaline earth, tetraalkylammonium, etc.) is crucial for the cathodic corrosion,75,79,98,99 and only a negligible amount of deterioration is observed in the presence of pure sulfuric acid in the case platinum and gold (Figure 10).75 In this process, cations are adsorbed to different crystallographic planes on different cathodes. For example, sodium cations adsorb preferentially to (100) sites of platinum, which leads to highly anisotropic etching of the cathode favoring the formation of the (100) terraces and steps and the removal of the (110) sites.80 In the corrosion of rhodium and gold, generation of (100) and (111) sites are more pronounced, respectively.117 Later studies showed that the site preference is favored at high NaOH concentrations and that 10 M KOH produced less (100) sites than 10 M NaOH with platinum.103 Interestingly, the pretreatment of the platinum electrode with flame-annealing also had an effect on the anisotropy of the corrosion.103 DFT calculations in vacuum indicated that the sodium is adsorbed more strongly to platinum than to rhodium and gold.117 Further DFT studies with explicit near-surface solvation confirmed that the sodium preferably binds to (100) type sites on platinum and showed that cations adsorb to different sites with various strengths depending on the metals (Figure 11).97 A detailed investigation on the cathodic corrosion at different monocrystalline Pt surfaces has also been reported.116 Furthermore, Hersebach, Koper et al. proposed that adsorbed hydrogen produced at the cathode can play a key role in the corrosion.97 Their hypothesis was based on DFT calculations on the hydrogen adsorption energies that were in good agreement with experimental onset potentials for the corrosion of the different cathodes. According to this postulate, ternary metal hydrides (AxMyHz; A = alkali or alkaline earth metal, M = transition metal, H = hydride) are formed and act as elusive intermediates in the cathodic etching.
Figure 10.

Scanning electron micrographs of Au cathode after cathodic treatment at −10 V (vs RHE) for 30 min in 1 M H2SO4 (left, a), and 1 M Na2SO4 (right, b). Reproduced with permission from ref (75). Copyright 2011 Wiley-VCH.
Figure 11.

Computational adsorption potentials of Li+, Na+, and K+ to Pt, Rh, and Au cathodes. Reproduced with permission from ref (97). Copyright 2018 under CC BY-NC-ND 4.0.
The rate of the cathodic etching under aqueous conditions depends on several properties on platinum cathodes. It was found that when increasing the concentration of NaOH electrolytes, the surface area of platinum cathode increased by 28%, 32%, and 74%, in 1, 5, and 10 M solutions, respectively.97 Interestingly, also the onset potential of the corrosion decreased from −1.4 V (vs NHE) to −1.3 V (vs NHE) for 1 or 5 M NaOH, and 10 M NaOH, respectively.97 Furthermore, the identity of the cation has also pronounced effect on the onset potential. For example, −1.3 V (vs NHE) was recorded in 1 M KOH, and −1.4 V (vs NHE) in 1 M NaOH and 1 M LiOH. When more negative potentials were applied (−1.0 V vs RHE), KOH was found to corrode the cathode the most, followed by NaOH and LiOH, which corroded the cathode the least.97 With rhodium, the onset potential order was similar to that with Pt, but in this case, Na+ corroded the cathode the most, followed by K+ and Li+.97 With gold, onset potentials were of −1.8 V and −1.7 V (vs NHE) for 5 M solutions of NaOH and KOH, respectively. Noteworthy, NaOH produced more surface area than KOH and LiOH.97 Interestingly, in 10 M NaOH, the onset potentials for the corrosion were −1.3 V (vs NHE) for rhodium and −1.6 V (vs NHE) for gold, respectively.117 The corrosion proceeds also in the presence of poly(vinylpyrrolidone) under ultrasonication.101
2.4. Formation of Organometallics
The synthesis of organometallic compounds from sacrificial metal cathodes has been studied extensively.162−166 In these reactions, transient radical species are generated through electron transfer from the cathode to the substrate (Scheme 8). The elusive intermediates can then react with the cathode to produce organometallic compounds. The preparative yields of organometallics are not always high, but conversely, the corrosion and the obtained compounds can pose significant problems.167 Because of the high reactivity of several cathodes (e.g., Scheme 8, right), it is of general interest to know which type of organic compounds can corrode common cathodes during the reductive electrosynthesis (Figure 12).
Scheme 8. Organometallic Corrosion of the Cathodes (left) and Rotating Disc Electrode after Electrolysis in DMF/Et4NClO4 (0.25 M) with 0.03 M EtI (right).

Reproduced with permission from ref (81). Copyright 1974 Elsevier BV.
Figure 12.

Selected classes of organic substrates and cathodes that can take part in cathodic corrosion through organometallic compound formation.
It has been reported on several occasions that at least Pb,29,81,162,163,168−184 Hg,167,169,176,180,185−192 Sb,168 As,168 Bi,168 Cd,170 Tl,162,176 and Sn163,166,168,170,176,177,183,193−198 corrode in the presence of various organic substrates. However, the organometallic products have not always been fully characterized, and in some cases, only their formation has been noted or proposed during reductive electrolysis. This might be due to the reactivity of these organometallic compounds and the fact that their decomposition is oftentimes accelerated by light.180 Interestingly, metallic parent elements and alkanes can be produced as decomposition products.180 In addition, gold and platinum can form covalent bonds with allyl moieties onto the surface of the cathode.199 These grafted layers can affect the performance of the cathode significantly by blocking further reactions.
Carbonyl compounds react with metallic cathodes to form alkyl organometallics in aqueous solutions (Figure 12, Scheme 9). High temperatures and acidic conditions seem to favor the formation of organometallics with lead and mercury cathodes.169 The formation of diisopropyl lead and mercury from acetone has been reported in numerous occasions,167,169,179,180 whereas diisobutyl lead and mercury have been obtained from methyl ethyl ketone167,169,185 and dimenthyl mercury from menthone.186 In addition, organomercury derivatives of phenylacetone, acetylbenzoyl, cyclopentanone, cyclohexanone, and methylcyclohexanones are known to form.167 Apart from the ketones, benzaldehyde can convert to dibenzylmercury, and lower temperatures favor its formation.187 In the electrocyclization of 2-nitrobenzaldehydes to 2,1-benzisoxazoles, Pb cathode has been described to function as a sacrificial electrode, which was evidenced by precipitation at the cathode.184 Moreover, electroreduction of levulinic to valeric acid corrodes at least lead, leaded bronze, tin, and cadmium cathodes.170
Scheme 9. Corrosive Formation of Selected Classes of Organometallic Compounds.

In addition to isolated carbonyl compounds, also α,β-unsaturated systems form organometallics with different cathodes (Figure 12, Scheme 9). However, in these cases, the metal–carbon bond is formed between the carbon–carbon double bond of the α,β-unsaturated substrate and the metal. Tin cathode formed tetrakis(β-cyanoethyl)tin193 or hexakis(β-cyanoethyl)ditin,194 and tetrakis(β-cyanopropyl)tin195 compounds when it reacted with acrylonitrile or metacrylonitrile, respectively. Interestingly, in the case of acrylonitrile, alkaline or neutral solutions were necessary for the reaction, high temperature, and low current density increased the yields, and lead or mercury cathodes did not react at all.193 Furthermore, an unknown organometallic was formed in the reduction of 1-cyano-1,3-butadiene, and 2,4-pentadionic acid with tin cathode.166,195 With mercury, di(methylethylketyl)mercury and unknown organometallic compound was formed from methyl vinyl ketone188 and dimethylvinylcarbinol,189 respectively.
Formation of organometallics from substrates where sp3-carbon is attached to a suitable functional group (e.g., halogen, sulfate ester) are relatively well-known (Figure 12, Scheme 9). For example, electrosynthesis of tetraalkyl lead from primary alkyl bromides and iodides in an ethanol sodium hydroxide mixture was described in a patent already in 1922.171 Thereafter, many patents and academic studies described the electrosynthesis of tetraalkyl lead compounds. For example, synthesis has been described in the presence of casein in water,172 in nonhydroxylic solvents,168 nonhydroxylic solvent with a hydroxylic additive,173 acetonitrile,183 DMF,178 and in propylene carbonate,174,175 to name just a few examples. Interestingly, the synthesis of lesser alkylated congeners PbEt2, and PbEt3-Pb2Et6 has also been reported.174 To that end, these species can eventually disproportionate to metallic lead and PbEt4.81,174 Excitingly, Silversmith and Sloan described, that dialkyl sulfates (e.g., Et2SO4, 82%), benzyl dialkylphosphates, alkyl acetates (e.g., EtOAc, 10%), benzoates, trifluoroacetates, mesyls, tosylates, triflates, and allyl acetates can alkylate lead cathodes in addition to the more commonly known corrosive substrates of I (e.g., EtI, 72%), Br, and Cl (EtCl, 93%).168 Furthermore, tetraalkylammonium, trialkylsulfonium, trialkylselenonium, tetraphenylarsonium, and alkyltriphenylphosphonium cations could form organometallic derivates with lead as well but with a lower efficiency (Figure 12, Scheme 9). Formation of bis(2-cyanoethyl)lead from β-iodopropionitrile has also been reported.176 With mercury, the formation of dibenzyl mercuries from benzyl bromides,190 dicyclopropyl mercuries from the corresponding halo-derivatives,191 and bis(2-cyanoethyl)mercury from β-iodopropionitrile176 has been documented. Tin has been another extensively studied cathode, and it is known to react with alkyl bromides in acetonitrile in the presence of tetraalkylammonium salts,183 with alkyl bromides in a nonhydroxylic solvent,168 methyl iodide,196 alkyl iodides,198 alkyl chlorides,163 and β-iodopropionitrile.176 Interestingly, in acetonitrile–water solutions that contain tetraethylammonium bromide as supporting electrolyte, alkyl, phenyl, and allyl bromides form tetraalkyl, tetraphenyl, and tetraallyl tin, respectively.177 In this report, the author observed that tin organometallics converted into solid tetraethylammonium (alkyl)halostannates. This is important to realize when the supporting electrolyte is recycled or disposed.
Waldvogel and co-workers have reported that massive corrosion occurs when thiophene containing molecules are reduced at a lead cathode. The corrosion was attributed to the cathodic desulfurization process and organic lead corrosion.29 This research group also reported that the reduction of menthone oxime and its derivatives can corrode lead cathodes.181,182 Furthermore, the reduction of acetonitrile with a tin cathode produces tetramethyltin.197 In addition, diarylmercuries are obtained from diaryliodonium hydroxides.192 In adiponitrile synthesis, a relationship between oxygen produced at the anode and the corrosion of the lead cathode was described.33 Oxygen reduction reaction (ORR) has been reported to corrode zinc cathodes in an aqueous solution of NaCl (0.6 M) at more negative potentials than −1.075 V (vs SCE) (Figure 13).200 Deaerating the solution or lowering the potential to −1.3 V (vs SCE) decreased the corrosion notably. The beneficial effect of the latter was assigned to the reduction of ZnO to Zn. Oxygen also has detrimental effects on cathodes in acidic media at low negative potentials and, for example, cathodes such as Ag201 and Cu202−204 can corrode to some extent. With Cu cathodes, the initial rate of dissolution was high, but later the produced metal cations started to electrodeposit back onto the cathode and the corrosion reached a steady state.202,204 However, this type of corrosion could have important consequences when these cathode materials are used in flow cells and the solution is not deareated. There, the electrodeposition could be effectively inhibited, and, in addition, this type of corrosion-redeposit process could affect the Faradaic efficiency as well. Simonet and Jouikov reported that CO2 can form passivating carboxylate layers with Au, Ag, Ti, Pd, Pt, Rh, and Cu cathodes ([cathode]-CO2-M+) when reaction conditions involve CO2-saturated aprotic solvents and small-sized electrolyte cations (TMA+, Na+) at sufficiently negative potentials.205−209 Interestingly, anodic regeneration of the passivated Ag cathode resulted in the possible formation of Ag+.205 Moreover, copper cathode corroded under prolonged electrolysis at −1.8 V (vs Ag/AgCl), which could be observed visually (Figure 14).207
Figure 13.

Cathodic corrosion of Zn cathode in aqueous solution of NaCl (0.6 M) at −1.075 V (vs SCE) for 17 h (left, a), −1.3 V (vs SCE) for 7 h (middle, b), and −1.2 V (vs SCE) at deareated conditions for 7 h (right, c). Reproduced with permission from ref (200). Copyright 2018 Wiley-VCH.
Figure 14.

Copper cathode after electrolysis in CO2-saturated DMF (0.1 M TMABF4) at −1.8 V (vs Ag/AgCl). Reproduced with permission from ref (207). Copyright 2017 Elsevier BV.
Moreover, Ralph, Walsh et al. disclosed that lead cathodes form a lead chloride layer, mercury-plated copper cathodes slowly amalgamate, and titanium cathodes form hydrides when l-cystine is reduced to l-cysteine hydrochloride in a 2.0 M HCl solution (Scheme 10).34,210 This led to reduced current efficiencies with these cathodes.34 However, maintaining the potential more negative than −0.7 V (vs SCE) protected the lead cathode from the lead chloride formation, and lowering the potential to −1.5 V (vs SCE) cleaned the electrode through hydrogen evolution.211
Scheme 10. Different Corrosion Products of the Metal Cathodes in the Reduction of l-Cystine to l-Cysteine.

Silversmith and Sloan showed that when ethyl bromide corrodes the lead cathode, the lowest corrosion rates were achieved with triphenylmethyl bromide and sodium iodide as supporting electrolyte.168 In contrast, the use of lithium bromide or perchlorate or calcium bromide were the worst in terms of corrosion. Furthermore, out of several tested solvents, DMSO gave the lowest yield of tetraethyllead.168 Galli and Olivani observed the formation of PbEt4 from ethyl bromide in propylene carbonate when tetraalkylammonium, trialkylsulfonium, and pyridinium supporting electrolytes were used.175 Interestingly, when Li+, Ca2+, K+, and NH4+ cations were used as the supporting electrolytes instead, cathode weight did not decrease. The rate of the corrosion in respect to supporting electrolyte cation was reported to be Et4N+ ∼ Bu4N+ > EtMeS2+ ≫ Li+, Ca2+, K+, NH4+ = 0. However, in DMF, higher corrosion currents were obtained with NaClO4 rather than with Bu4NClO4 electrolyte with lead cathode in the presence of alkyl bromides or iodides.178 Interestingly, with a tin cathode, the current yields of the corrosion were in similar magnitude when Bu4NClO4 or NaClO4 were used with MeI in DMF.198 In addition, SnMe4 and PbMe4 were obtained in higher yields in Et4NBr than in Bu4NBr from tin and lead cathodes.163 Low temperature and current density decreased the corrosion of the tin cathode.163 Increasing the amount of tetraethylammonium bromide supporting electrolyte in acetonitrile–water solution also increased the corrosion rate of the tin cathode with methyl bromide.177 Less sloughing was observed with tetrapropylammonium and tetrabutylammonium bromide than tetraethylammonium supporting electrolytes.177 Grimshaw and Ramsey made some interesting observations when they studied the reduction of benzyl bromides with a mercury cathode.190 It was discovered that 4-H, 4-tBu, 4-Me, and 3,4-diCl substituted benzyl bromides gave dibenzylmercuries, whereas 3-Br and 4-NO2 gave the corresponding bibenzyl compounds (Scheme 11). Interestingly, the use of LiCl as a supporting electrolyte resulted in larger amounts of dibenzylmercury in comparison to tetrabutylammonium salts. Moreover, the yield of bis(3,4-dichlorophenyl)mercury decreased from 39% to 4.6% when the potential of the cell was changed from −0.94 to −1.30 V (vs Ag/AgCl), respectively.
Scheme 11. Substrate Specific Corrosion of Benzyl Bromides with Mercury Cathodes.

2.5. Short Summary on the Different Corrosive Mechanisms
As discussed in the previous sections, cathodic corrosion is a rather complex process as it can proceed through several mechanisms for each cathode. Many excellent studies have probed the different reaction conditions that affect the cathodic corrosion, but sometimes the results obtained contradict each other. Thus, the labyrinthine nature of the published data makes it hard to draw any general conclusions on the effect of different parameters on cathodic corrosion. One notable exception is in the cathodic etching type mechanisms, for which lowering the concentration of the supporting electrolyte was generally found to be beneficial to control the cathodic corrosion.97,104,105,124,125,128
From the point of view of organic electrosynthesis, the strategies for mitigating cathodic corrosion usually involve novel additives and new alloys. New mechanistic studies on cathodic corrosion could help in making both of these approaches more powerful by assisting in the building of a more thorough theoretical framework. For example, in the future, it would be valuable to know whether the etching in aprotic conditions is anisotropic like in aqueous environments and why some cathodes form metal hydrides relatively easily. Another interesting aspect would be to uncover the relative energies of the various corrosive pathways involved in the corrosion of metals.
3. Strategies for Inhibiting the Cathodic Corrosion
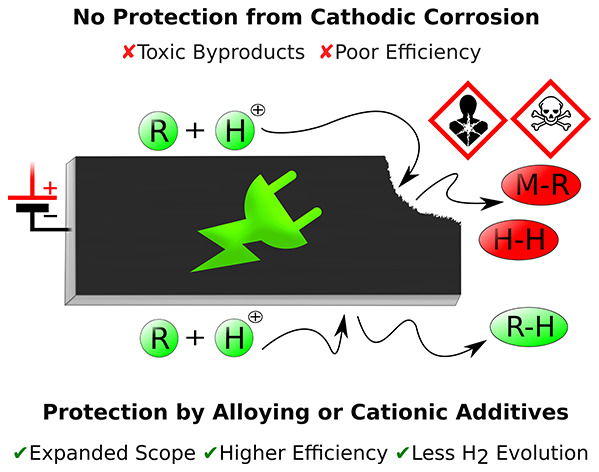
Two general strategies have emerged to reduce or inhibit the cathodic corrosion in reductive organic electrochemistry: cationic additives and alloying (Scheme 12). These two complementary strategies approach the same problem from two different perspectives. They either modify the electrical double layer (EDL) or the physicochemical properties of the cathodes. Both approaches were proven to be highly efficient, and generally, the reductive electrosynthesis proceeds with greater or the same activity and selectivity. Furthermore, in the best scenarios, the advantageous properties of the cathodes such as the high overpotential for hydrogen are enhanced. This has important implications for the Faradaic efficiency of electroreductions as the parasitic formation of dihydrogen can be suppressed by impressive extent, which improves the overall desirability of the reactions. Moreover, the tuning of the additives and alloying can improve other “secondary” properties such as extractability, machinability, softness, etc. Thus, both the corrosion resistance and the practical aspects may be improved at the same time. For example, leaded bronzes are significantly harder materials than the neat soft lead (Table 2). Consequently, larger electrodes can be manufactured from the former as the malleable nature of Pb can cause deformations under its own weight and, for instance, variations in electrode gap in flow cells. Furthermore, certain alloys such as leaded bronzes can be handled without gloves,212 which is not recommended with neat lead.
Scheme 12. General Strategies for Inhibiting the Cathodic Corrosion.

Table 2. Comparison of Selected Properties of Several Metals and Alloys Used Commonly in Reductive Organic Electrosynthesis.
| melting point (°C) | density (g/cm3) | electrical conductivity (MS/m) | tensile strength (N/mm2) | hardness (Brinell) | |
|---|---|---|---|---|---|
| Metal | |||||
| Cu22−25,221,222 | 1083 | 8.96 | 58.4 | 210 | 874 |
| Sn22−25,221,222 | 232 | 7.3 | 8.69 | 220 | 51 |
| Pb22−25,221,222 | 328 | 11.34 | 4.74 | 18 | 38 |
| Leaded Bronze (Material no.) | |||||
| CuSn10Pb10 (CC495K)35,220,223,224 | 762–928 | 9.0 | 6.0 | 290 | 85 |
| CuSn7Pb15 (CC496K)35,220,224−226 | 855–970 | 9.1 | 7.0 | 270 | 80 |
| CuSn5Pb20 (CC497K)35,220,224,227 | 855–950 | 9.1 | 7.0 | 230 | 60 |
3.1. Cationic Additives
When an electrode in an electrochemical cell is polarized, an electrical double layer will eventually form. This layer can be modified by introducing different concentrations of additives, and usually either inorganic or organic salts are used (e.g., as supporting electrolytes, see Figure 15). The additives can also have a profound effect on the cathodic corrosion. In reductive electrosynthesis, the cations have an articulate role in the degradation of the cathode, while the effect of the anion is usually minor but not always negligible. Essentially, the use of organic cations allows more freedom in fine-tuning the properties of the EDL in comparison to metal cations. Properties such as hydrophilicity, sterics, electron affinity, polarizability, etc., of the cation can be varied by changing the substituents and the central atom in the organic additive. For example, several organic tetraalkylammonium cations that have different molecular properties can decorate lead cathode’s surface as a dense layer. The modified EDL can then repel both protons and organic substrates, and consequently, the parasitic formation of hydrogen is inhibited and corrosive compounds (e.g., sulfur containing) can be used as substrates. In the latter case, a compact cationic layer bends the Fermi level and facilitates the tunneling of electrons into the bulk solution where they reduce the substrates.213,214 The resulting organic radical anions are then directly protonated by the protic electrolyte. In the former case, the Coulombic repulsion of the protons from the cationically decorated electrode surface prevents their discharging at the surface. The low interaction coefficient between protons and electrons promotes the reduction of the organic substrate. In addition, the metallic cathode is effectively shielded from the potentially corrosive substrates or intermediates. Applying this concept has proven highly effective, and, for example, Waldvogel and co-workers have demonstrated on several occasions that the cathodic corrosion of lead is inhibited when small amounts of (poly)cationic quaternary ammonium salts are added into the methanolic sulfuric acid electrolyte.29,181,182,215 To obtain a compact highly cationic layer, short alkyl moieties and dications with C3 spacer are much superior.
Figure 15.

Selected examples of different cationic additives used in reductive electroorganic synthesis.
3.2. Alloying
Alloying an electrosynthetically active base metal with different constituents or introducing a small amount of active metal into a base metal is an excellent approach to counter the cathodic corrosion. However, in contrast to the cationic additives, the physicochemical processes responsible for the increased resistance toward cathodic corrosion are not well-defined. However, some properties that have been described as beneficial for neat metals, such as the strength of the metal lattice,128,134 probably apply to alloys as well. Nevertheless, some highly useful details on the modified properties of alloyed electrodes have been described.216−218 According to Jacob and co-workers, the altered properties of the alloy electrodes can be divided into three different categories: (i) bifunctional mechanisms, (ii) delocalized electronic properties of the electrode, or (iii) local atomic configurations.218 In the first category, different components of the alloy catalyze different reaction steps or absorb different intermediates. From the point of view of the cathodic corrosion, the corrosive species might interact more readily with the other metal(s), whereas the substrate in turn would interact with the electrocatalytically active metal. In the second and third categories, the modified electronic band structure could lead to a smaller interaction toward the corrosive species or raise the energy barriers for the formation of corrosion intermediates. Furthermore, the local atomic configurations can be dynamic in nature, and they could be generated in operando.218 Elucidation of the dominant mechanisms in the corrosion resistance could lead to rational improvement of the alloys in the reductive electroorganic synthesis.
There are several lead alloys that are commercially available such as babbitt alloys, leaded coppers, lead-antimonies, Cerrosafe, Rose’s metal, Wood’s metal, leaded brasses, different solder alloys, and leaded bronzes. Some of the most studied alloys for reductive electrosynthesis are the leaded bronzes as they are inexpensive materials that are mechanically much more stable and machine workable than the soft pure lead. The ternary alloys, CuSn10Pb10, CuSn7Pb15, and CuSn5Pb20, contain different amounts of tin and lead alloyed with the copper base metal (Table 2). It is noteworthy that copper and lead do not form stable alloys due to the limited solubility of Pb in neat Cu (<0.09 atom % at 600 °C).219,220 Therefore, leaded copper alloys have to be formed as ternary systems, such as leaded bronzes, which were considered as uniform alloys. Intriguingly, it has been shown with femtosecond laser ablation/ionization mass spectrometry (LIMS) measurements in three dimensions that leaded bronzes are not homogeneous alloys (Figure 16).220 In fact, they contain micrometer size domains of lead-rich or pure lead phases mixed with a binary copper/tin alloy matrix. The highest segregation of lead phases and the highest inhomogeneity is seen in CuSn7Pb15 alloy, whereas CuSn5Pb20 represents the most homogeneous one.220 Furthermore, electrolysis for 3 h at −1.12 V (vs Ag/AgCl) or mechanical polishing can redistribute the lead phases on the surface. The latter is exemplified in Figure 17.212 In addition, with CuSn7Pb15 cathode, the inhomogeneity increased and the different phases became more separated after the reductive electrosynthesis.35 Interestingly, the redistribution has been noted to shift the HER and CO2 reduction reaction onset potentials for the leaded bronzes because these electroconversions occur at the surface. Consequently, the used and pristine alloys can show different performance in electrolysis/electrocatalytic reactions.35,212 It is noteworthy that the bulk composition of these electrodes should not be correlated with the amount of lead sites available in electrosynthesis. In fact, CuSn7Pb15, and not CuSn5Pb20, has the largest lead domains available for electrosynthetic reactions.35 Furthermore, after a mechanical polishing, the true surface composition of CuSn7Pb15 cathode was in fact Cu71Sn9Pb20.212
Figure 16.

EDS elemental mapping (a,c,e) and SEM pictures (b,d,f) from different leaded bronzes (left) and LIMS 3-D mapping from CuSn7Pb15 cathode (right), highlighting the heterogeneous nature of these materials. Reproduced with permission from ref (220). Copyright 2017 American Chemical Society.
Figure 17.
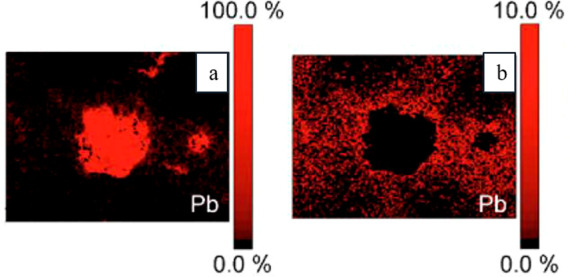
Scanning auger microscopy mapping of Pb from polished CuSn7Pb15, without cut off (left, a), and cut off at 10 at% (right, b) showing the lateral dispersion of the lead. Reproduced with permission from ref (212). Copyright 2019 Wiley-VCH.
Mercury is another electrode material with high hydrogen overpotential that can be used for organic reductive electrosynthesis. However, its liquid physical state, high toxicity, and unavailability in large quantities nowadays limits its use in reductive electrosynthesis. Nevertheless, mercury can form solid amalgams with many different metals, which can address some of the stated problems. For example, dental amalgam electrodes are similar to silver electrodes but with a higher hydrogen overvoltage.228−230 In another study, Pd, Au, Pt, and Cu amalgams behaved similarly than the pure Hg.231 Amalgam or mercury-plated electrodes have been used also in preparative organic electroreductive synthesis.34,42,153,210,231−236 For example, amalgamated copper, lead, and zinc were used to reduce vanillin to vanillyl alcohol,232 amalgamated lead and copper in the adiponitrile synthesis,42 amalgamated copper in the reduction of salicylic acid to salicylaldehyde,234 mercury-plated copper in the synthesis of l-cysteine hydrochloride,34,210 and amalgamated lead in the reduction of oxalic to glyoxylic acid (Scheme 13).235
Scheme 13. Examples for Amalgam or Mercury-Plated Cathodes in Organic Electroreductive Synthesis.

(a) Current effiency. (b) Superimposed alternating voltage 0–500 mV rms at 1.75 V (vs SCE); rms, root-mean-square.
However, in the latter synthesis, the amalgamation layer disappeared from the surface after 1 day of continuous operation.235 Furthermore, Ralph, Walsh et al. described that mercury-plated copper cathode amalgamated during the reductive electrosynthesis.34,210 Solid amalgams show some promise as they retain the high hydrogen overpotential of mercury, but the stability of these materials might be an issue to their use. We are not aware of any further studies, where, for example, the stability of the cathode or the amount of corroded mercury have been determined in reductive electrosynthesis.
4. Examples in Reductive Electroorganic Synthesis
4.1. Examples in Reductive Electroorganic Synthesis with Cationic Additives
Kulisch, Waldvogel et al. have studied the use of lead cathodes in the synthesis of optically pure menthyl amines from menthone oximes (Scheme 14, left). In this reaction, they observed that the lead cathodes corrode, which was attributed to the formation of PbSO4 and organometallic lead intermediates.181 Fascinatingly, a small quantity (0.5%) of a tetraalkylammonium salt in 2 M H2SO4 in methanol inhibited the corrosion but also lead to a tremendous improvement of the yield (Scheme 14, right). In this case, counter anions did not have any effect, and out of the several tested additives, triethylmethylammonium methylsulfate (MTES) proved to be best in terms of chemical and Faradaic yield. The use of additive allowed the complete prevention of the cathodic corrosion of the lead electrode in this electrosynthesis. Later, the substrate scope was broadened to other types of menthone derivatives as well.182 Similarly, the use of (poly)cationic additives efficiently inhibited the cathodic corrosion of the lead cathode. The most suitable additive in terms of yield and diastereomeric ratio depended on the substrate converted. For some, monocationic MTES was the most suitable additive, whereas for others, polycationic additive worked better. However, the additive strongly promoted the electroorganic reduction because the parasitic hydrogen evolution was almost inhibited.
Scheme 14. Deoxygenation of a Menthone Oxime to Menthyl Amine and Representative (Poly)cationic Additives Used to Control the Cathodic Corrosion (Left), Kinetic Traces (Right) from the Reduction Menthylamine (R = H) of (a) without Additive and (b) with MTES Additive (■, Menthylamines; ○, Menthylimine; ×, Menthone Oxime).

Kinetic traces reproduced with permission from ref (181). Copyright 2011 Wiley-VCH.
Later, Edinger, Waldvogel et al. studied in detail the use of (poly)cationic quaternary ammoniums to suppress cathodic corrosion in electroreductions with lead cathodes in the deoxygenation of amides to amines (Scheme 15).215 Interestingly, out of the several tested additives, N,N,N,N′,N′,N′-hexamethyl-1,3-propanediammonium methylsulfate proved to be the most suitable and current efficiencies of almost 100% were obtained. Noteworthy, all the tested (poly)cationic quaternary additives inhibited the cathode’s corrosion, which was attributed to the formation of PbSO4. Intriguingly, in the absence of an additive, the electrosynthesis was accompanied by significant hydrogen evolution and the surface of the electrode was roughened. The latter could be observed visually (Scheme 15, bottom right). In contrast, in the presence of cationic additive, the surface was smooth and homogeneous. ICP-MS measurement showed that <2.5 ppm levels of lead were present in the crude product and the recovered starting material when the additive was used in the electrosynthesis. Without the additive, the contamination of the crude product was several orders of magnitude higher accompanied by significant formation of PbSO4 precipitates.
Scheme 15. Deoxygenation of an Amide to Amine and Representative Polycationic Additives Used to Control the Cathodic Corrosion (Left), Experiment with Cationic Additive (Right, a) and without (Right, b).

Reproduced with permission from ref (215). Copyright 2014 Wiley-VCH.
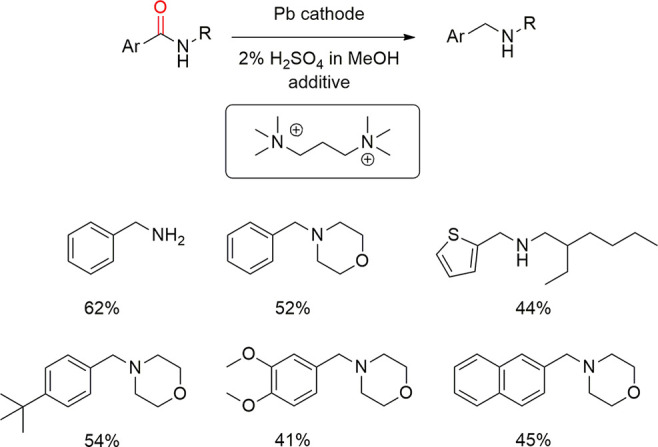
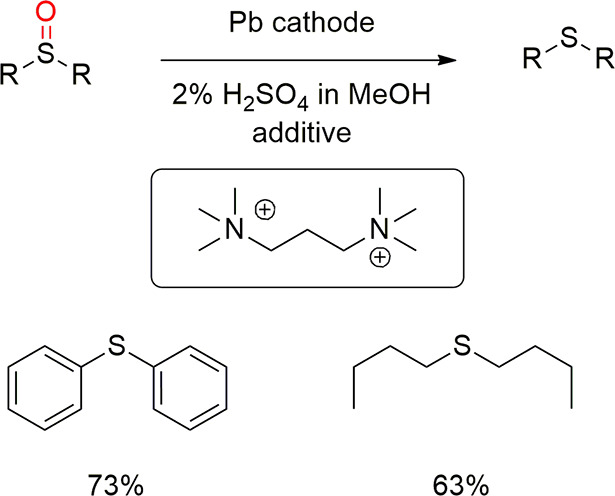
Later, Edinger, Waldvogel et al. broadened the scope of the amides significantly.29 Again, the use of N,N,N,N′,N′,N′-hexamethyl-1,3-propanediammonium methylsulfate allowed the synthesis of several deoxygenated amines from amides in the absence of cathodic corrosion (Scheme 16). Interestingly, when the amide was attached a thiophene functionality, the electroreduction in the absence of the additive caused a complete deterioration of the lead cathode, whereas the additive allowed to obtain the deoxygenated thiophene substrate in good yield. This was attributed to cathodic desulfurization processes and organic lead corrosion. In addition, two examples of electroreduction of sulfoxides to sulfides were also reported (Scheme 17).29 Interestingly, esters can also be deoxygenated with this protocol.237
Scheme 16. Deoxygenation of an Amide to Amine with Polycationic N,N,N,N′,N′,N′-Hexamethyl-1,3-propanediammonium Methylsulfate Additive.
Scheme 17. Deoxygenation of an Sulfoxides to Sulfide with Polycationic N,N,N,N′,N′,N′-Hexamethyl-1,3-propanediammonium Methylsulfate Additive.
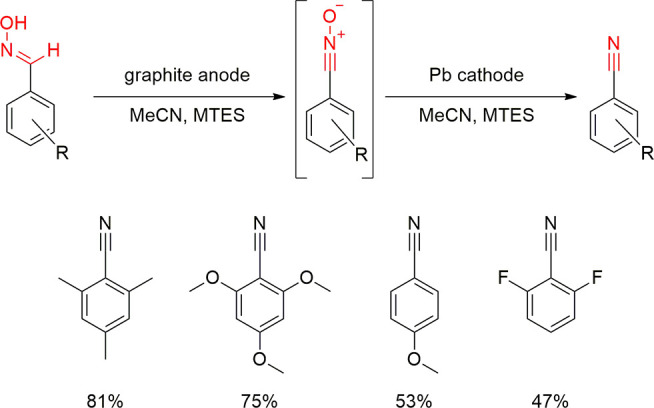
In consecutive paired electrolysis, a substrate is manipulated by subsequent redox processes. The research group of Moinet pioneered the technique and used it in the generation of nitrosobenzene derivatives from nitroarenes through hydroxylamines in several studies.238−246 After that, only a few examples of electrosyntheses have been published, wherein both electrodes are required for the same process.1,247,248 Hartmer and Waldvogel developed a domino oxidation–reduction sequence for the conversion of aldoximes to nitriles, where the first redox process is carried out on graphite anode and lead cathode is used in the second to generate the final compounds (Scheme 18).248 In this case, they utilized MTES, which is known from previous reports to protect the lead cathode from corrosion. Later this electrolysis was performed in flow as narrow gap flow electrolysis cells are of particular interest because they can offer a more energy efficient operation.10,249−251 In this case, the soft nature of lead turned out to be problematic, and consequently, the domino oxidation–reduction sequence of aldoximes to nitriles was performed at leaded bronzes in modular narrow gap flow cells.35
Scheme 18. Oxidation–Reduction Sequence for the Synthesis of Nitriles from Aldoximes in the Presence of MTES Additive.
Adiponitrile synthesis from acrylonitrile is one the most prominent examples of reductive electrosynthesis at an industrial scale. Annually more than 300 000 t of adiponitrile are produced via this route.252 The corrosion of the cathodes in this reaction is also well described. Nakagawa and Nagamori conducted the synthesis of adiponitrile in an undivided cell with lead or lead alloy PbM5-10 (M = Sb, Ag, Cu, Te) cathodes.33 The alloyed cathodes were described to exhibit improved mechanical strength and anticorrosion properties. Fascinatingly, when tributylethylammonium cations were added in concentrations between 0.02 and 0.08 M, the cathodic corrosion was reduced to below 1 mm/year, which was deemed acceptable (Scheme 19, right). The most beneficial combination was to use alkali metal and tributylethylammonium salts in conjunction. If only the former was used, the yield dropped and hydrogen evolution increased, and, if only the latter was added, the cell voltages were too high. The identity of the alkali salt and the tetraalkylammonium counteranion were not critical for the reaction. Interestingly, the tributylethylammonium additive outperformed ethyltripropylammonium and tetraethylammonium salts at reducing the cathodic corrosion. Moreover, researchers from BASF reported that the use of hexamethylene bis(dibutylethylammonium) phosphate (10.5 mmol/kg of aqueous electrolyte solution) was highly beneficial with bulk lead cathodes in this synthesis (Scheme 19, left).253 In this case, a corrosion rate of 0.35 mm/year was observed in comparison to a rate of 0.9 mm/year, which was obtained when tributylethylammonium phosphate was used at a higher concentration of 80.0 mmol/kg. However, when electrodeposited lead on a steel was used as cathode, the effect of the additives was similar: 0.25 mm/year and 0.21 mm/year corrosion rates were obtained with hexamethylene bis(dibutylethylammonium) and tributylethylammonium phosphates, respectively.
Scheme 19. The Effect of Hexamethylene Bis(dibutylethylammonium) and Tributylethylammonium Phosphate (Left), and the Concentration Effect of Tributylethylammonium Cation (Right) to the Cathodic Corrosion of Lead Cathodes in Adiponitrile Synthesis.

Right figure redrawn from ref (33).
4.2. Examples in Reductive Electroorganic Synthesis with Alloys
Researchers from BASF studied the use of electrochemically deposited lead and lead alloys on a circular steel disk in adiponitrile synthesis from acrylonitrile in the presence of 10.5 mmol/kg of hexamethylene bis(dibutylethylammonium) phosphate in the aqueous electrolyte (Table 3, entries 2–9).253 The binary and ternary alloys studied contained different amounts of copper, or copper and bismuth, selenium, or tellerium (entries 3–9). Impressive results were obtained. For example, massive Pb electrodes corroded at a rate of 0.35 mm/year (entry 1), whereas electrodeposited PbCu3.7 and PbCu1.3Se0.1 cathodes corroded only at a rate of 0.05 mm/year (entries 6 and 9). Fascinatingly, a longer lifetime of the cathode was obtained along with slightly better selectivity with PbCu1.3Se0.1 cathode (entry 9) in comparison to the massive Pb or electrodeposited Pb cathodes (Entries 1–2).
Table 3. Performance of Different Lead and Lead-alloy Cathodes in the Adiponitrile Synthesis.
| entrya |
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1b | 2c | 3c | 4c | 5c | 6c | 7c | 8c | 9c | |
| cathode | Pb | Pb | PbCu1.8 | PbCu0.8 | PbCu1.3 | PbCu3.7 | PbCu2.2Bi1.3 | PbCu1.3Te0.5 | PbCu1.3Se0.1 |
| corrosion (mg) | 0.2 | 0.14 | 0.03 | 0.09 | 0.04 | 0.03 | 0.045 | 0.05 | 0.03 |
| corrosion (mm/year) | 0.35 | 0.25 | 0.05 | 0.16 | 0.07 | 0.05 | 0.08 | 0.09 | 0.05 |
| time (h) | 90 | 90 | 200 | 209 | 96 | 90 | 96 | 96 | 96 |
| selectivity (%) | 90.3 | 90.4 | 90.9 | 91.4 | 90.4 | 88.8 | 90.0 | 90.9 | 90.9 |
Reaction conditions: undivided cell, steel anode, 20 A dm–2, 55 °C.
Bulk Pb cathode.
Electrodeposited on a circular steel plate (film thickness: 50 μm).
Gütz, Waldvogel et al. utilized leaded bronzes in a collaboration with Novartis Pharma in the cathodic mono- and didehalogenation reactions of 1,1-dibromo cyclopropane derivatives (Scheme 20).224 Interestingly, the use of a neat lead cathode with MTES additive led to almost 70 mol % of lead corrosion products with respect to the pyrrolidine precursor.226 In these reactions, leaded bronze cathodes in conjunction with the MTES additive proved to be extremely resistant to cathodic corrosion. ICP-MS measurements showed that only low levels of metallic residues (Pb 5 ppm, Cu 4 ppm, Sn 1 ppm) were in the crude electrolytic mixture. This was acceptable for pharmaceutical use, as the products obtained were intermediates and not the final synthesis product. Later on, the lab-scale electrosynthesis was scaled up to technically relevant submolar scale, and also a flow system was developed providing multimolar amounts of spirocyclopropane-proline.226 In this case, trace metal contents were less than 10 ppm, consisting mostly of copper. The selectivity for monohalo or completely dehalogenated product was controlled by the amount of applied charge as well as the electrolyte composition.
Scheme 20. Mono- or Didehalogenation of 1,1-Dibromo Cyclopropane Derivatives at CuSn7Pb15 Cathode.
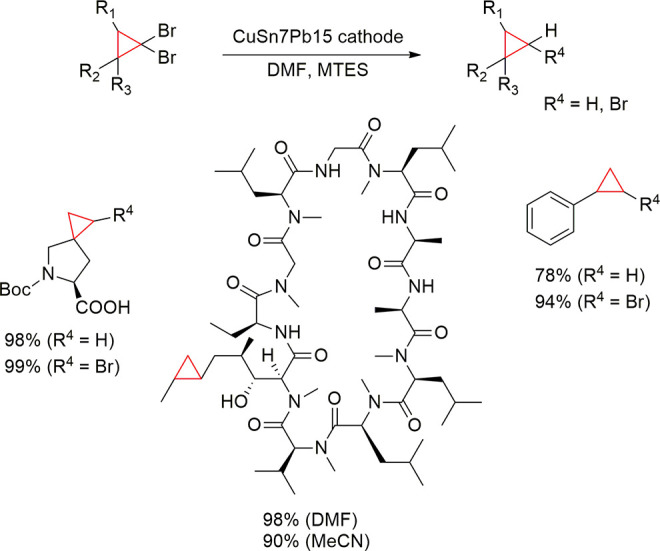
The leaded bronzes were shown also to be viable cathodes for the debromination of 2-amino-3,5-dibromopyridine, hydrodimerization of acrylonitrile, and domino oxidation–reduction of aldoximes to nitriles.35 Usually, with leaded bronze cathodes, similar or higher yields were obtained than with the neat lead cathode (Scheme 21). In the case of the selective dehalogenation of 2-amino-3,5-dibromopyridine, a slightly lower yield was achieved with the CuSn10Pb10 cathode, which was attributed to the decomposition of the substrate by Cu-rich domains at the cathode’s surface.
Scheme 21. Comparison of the Yields between Leaded Bronzes and Neat Lead Cathodes in Several Reductive Electrosyntheses.

The cathodic conversion of N,O bonds is of actual interest and different electrode systems are employed.254 Wirtanen, Waldvogel et al. applied leaded bronze cathodes in the synthesis of 2H-2-(aryl)benzo[d]-1,2,3-triazoles and 2H-2-(aryl)benzo[d]-1,2,3-triazole N-oxides (Scheme 22).255 The leaded bronzes outperformed the lead as a cathode material, and among leaded bronzes, CuSn7Pb15 was found to be the most suitable cathode. In this case, no visible deterioration of the cathode was observed.
Scheme 22. Synthesis of 2H-2-(Aryl)benzo[d]-1,2,3-triazoles and 2H-2-(Aryl)benzo[d]-1,2,3-triazole N-Oxides with CuSn7Pb15 Cathode.
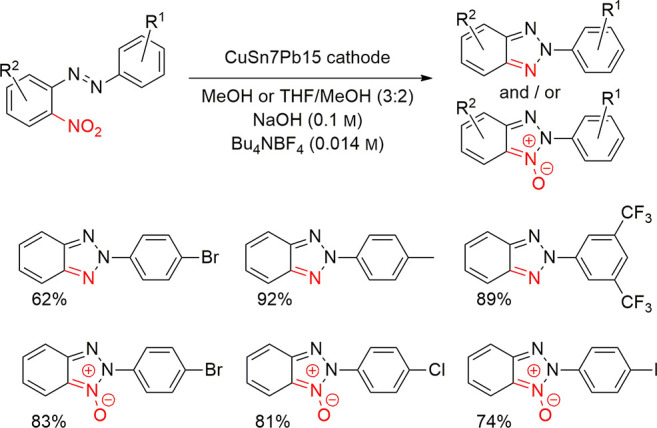
Strehl, Christoffers et al. studied the cathodic reduction of α-(ortho-iodophenyl)-β-oxoester cyclization (Scheme 23). In this reaction, the lead cathode gave a slightly lower yield than leaded bronze alloy CuSn17Pb.256
Scheme 23. Cathodic Reduction of α-(ortho-Iodophenyl)-β-oxoesters with CuSn17Pb Cathode.
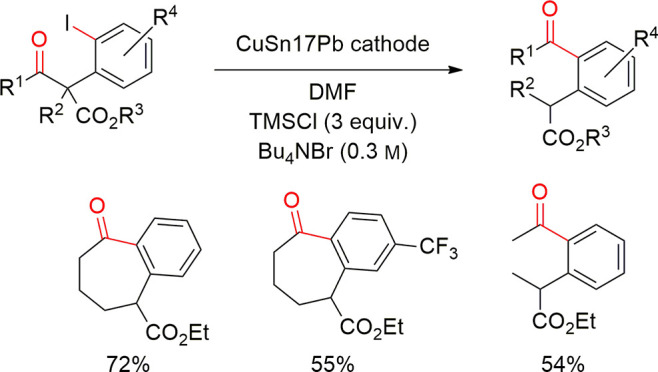
5. Summary
Acknowledging and addressing the issue of cathodic corrosion is urgent. At an industrial scale, cathodic corrosion can impede the adoption and implementation of electroorganic synthetic processes. It is noteworthy that it is at this scale that organic electrosynthesis would make the largest impact due to its highly sustainable nature and potential for decarbonizing the chemical industry, providing a climate-neutral solution for chemical conversions. Thus, broader perception and better awareness of the cathodic corrosion already at the lab-scale could accelerate the adoption of reductive organic electrosynthesis for technical processes. Some steps have been taken already for the better recognition of this type of corrosion. Recently, Hersbach and Koper emphasized this emerging issue of cathodic corrosion and proposed the adoption of updated Pourbaix diagrams as illustrated in Figure 18.257
Figure 18.
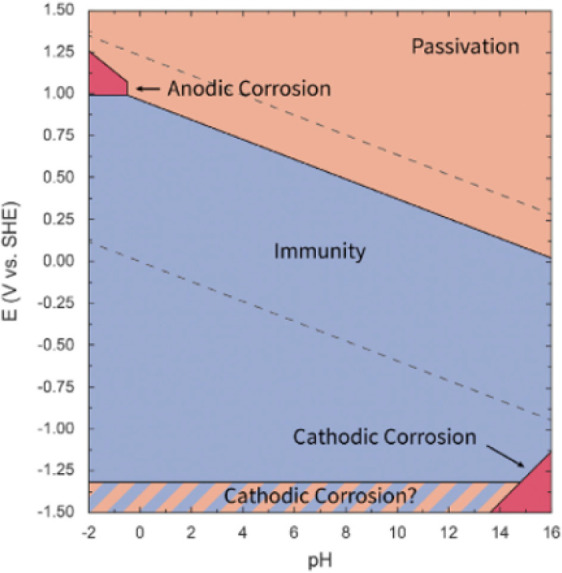
Updated Pourbaix diagram for platinum. Reproduced with permission from ref (257). Copyright 2002 under CC BY 4.0 license.
In the context of reductive organic electrosynthesis, we propose that some operationally simple measures should be undertaken more routinely. This is of particular significance when electrolyzing at high current density. Before and after the electrolysis, visual inspection and gravimetric analysis of the cathodes and determination of the metal in solution should allow assessing the magnitude of the cathodic corrosion. Although these techniques may at first seem to overlap, there are plausible scenarios where the full extent of the cathodic corrosion will be revealed only by the combination of these methods. For instance, deposit formation could counter the weight decrease caused by the cathodic corrosion, or the gaseous corrosion products might escape from the cell. Thus, in the former scenario, the corrosion would not be detected by gravimetric analysis, and in the latter scenario, measurements from the reaction mixture would not reveal the full extent of the corrosion. Regarding the use of alloys as cathodes, we strongly recommend documenting the used materials thoroughly in the supporting information by reporting the material number(s), supplier, composition, material properties, etc. This would result in an increased reproducibility and traceability especially for commercially available materials.14,258
6. Outlook
Although tremendous advances in the development of metal-free and high performance electrodes based on carbon (e.g., BDD) have been made,259−262 metal cathodes still offer unique features: the specific reactivity of cathodic surface (electrocatalysis), good thermal and electrical conductivity, easy machinability, and cost efficiency for electroorganic reductions at very negative potentials. Therefore, metal cathodes will be indispensable. The formation of ternary and higher alloys is currently not well investigated. The emerging concept of high entropy alloys (i.e., complex solid solutions or multiprincipal-component alloys) that contain five or more elements is a very promising area, and initial remarkable results, for example, in the reductions of oxygen, water, and carbon dioxide have been demonstrated.263−269 Interestingly, in one case, a high corrosion resistance was reported.265
Corrosion mitigation strategies will play an increasingly important role as electroorganic synthesis becomes widespread in large-scale chemical manufacturing as a unique technology to both decarbonize this industry and increase its sustainability by transitioning from petroleum to biomass as a feedstock. Electrosynthesis has already been highlighted for its unique advantages when integrated with other technologies in biorefineries.270 In particular, new synergies have been demonstrated when electrosynthesis is coupled with biocatalysis for the production of monomers from biomass.271−273 These synergies lower the number of unit operations needed for the overall transformation, open new routes for process intensification, and result in cost savings on the order of 30–50% compared to the corresponding thermocatalytic conversion.273,274 In addition to its intrinsic technological advantages compared to conventional thermocatalytic conversions, electrosynthesis was proposed to be well-suited for the distributed manufacturing of chemicals due to the modular nature of electrochemical reactors and their simple operation.270,275 Reductive electrosynthesis is already being considered for the distributed manufacturing of hydrogen (H2), ammonia (NH3), and hydrogen peroxide (H2O2) to overcome challenges with their transportation. Similar advantages exist when manufacturing chemicals from waste biomass. New technologies that could easily transform regional- and community-scale biomass into valuable chemicals using distributed chemical plants are therefore highly desired. These technologies would be particularly advantageous if using excess wind energy, as it represents an inexpensive, abundant (4 GW), and locally available source of renewable electricity in rural areas.276,277
Acknowledgments
The Carl Zeiss Stiftung is gratefully acknowledged for the perspective network ECHELON. Financial support by Deutsche Forschungsgemeinschaft (DFG: Wa1276/27-1 and Wa1276/31-1) is highly appreciated. Support by the focus area SusInnoScience (State Rhineland-Palatinate) is acknowledged. This material is based upon work supported in part by the National Science Foundation under grant numbers CBET-1512126 and IIP-1820147. This project has received funding from the European Union’s Horizon 2020 research and innovation program under the Marie Skłodowska-Curie grant agreement no. 844355 (T.W.).
Glossary
Abbreviations
- AC
alternating current
- AB
ammonium borane
- BDD
boron-doped diamond
- BMIM
1-butyl-3-metylimidazolium
- DC
direct current
- DFT
density functional theory
- DMF
dimethylformamide
- DMPBF4
1,1-dimethylpyrrolidinium tetrafluoroborate
- DMSO
dimethyl sulfoxide
- EDL
electrical double layer
- EDS
energy-dispersive X-ray spectroscopy
- EPR
electron paramagnetic resonance
- GC/MS
gas chromatography–mass spectrometry
- HER
hydrogen evolution reaction
- ICP-MS
inductively coupled plasma mass spectrometry
- LIMS
laser ablation/ionization mass spectrometry
- MS
mass spectrometry
- MTES
triethylmethylammonium methylsulfate
- NHE
normal hydrogen electrode
- ORR
oxygen reduction reaction
- ReaxFF
reactive force field
- RHE
reversible hydrogen electrode
- rms
root-mean-square
- SCE
saturated calomel electrode
- SEM
scanning electron microscope
- TBA
tetrabutylammonium
- TMA
tetramethylammonium
Biographies
Tom Wirtanen received his B.Sc. in Chemistry in 2010 and M.Sc. in Organic Chemistry in 2012 from the University of Helsinki. He visited in 2012 Prof. Dr. Jose A. Lopez-Sanchez at the University of Liverpool and finished his Ph.D. in 2018 at the University of Helsinki under supervision of Dr. Juho Helaja. From 2019, he has been working as a postdoctoral researcher at the Johannes Gutenberg University Mainz in the group of Prof. Dr. Siegfried R. Waldvogel focusing on reductive electrosynthesis.
Tobias Prenzel received his B.Sc. in Chemistry from Johannes Gutenberg University Mainz, working on oxidative coupling reactions with Mo(V) reagents in 2016, and his M.Sc., working on electroreductive cyclizations in 2019, supervised by Prof. Dr. Siegfried R. Waldvogel. He is currently focusing on the reductive electrochemical synthesis of heterocycles as a Ph.D. student in the Waldvogel group.
Jean-Philippe Tessonnier completed his Ph.D. at the University of Strasbourg in 2005 under the supervision of Drs. Marc-Jacques Ledoux and Cuong Pham-Huu. He then joined Prof. Robert Schlögl’s group at the Fritz Haber Institute of the Max Planck Society as a postdoctoral fellow and was promoted to project leader in 2008. He moved to the University of Delaware to work with Prof. Mark Barteau as a visiting researcher in 2010. He started his independent academic career in 2012 in the Department of Chemical and Biological Engineering at Iowa State University and was promoted to Associate Professor in 2018. He was awarded the Richard C. Seagrave Professorship in Chemical Engineering in 2020. His research interests span heterogeneous catalysis, organic electrosynthesis, and green process engineering for the synthesis of performance-advantaged chemicals and materials from biomass.
Siegfried R. Waldvogel studied chemistry in Konstanz and received his Ph.D. in 1996 from University of Bochum/Max Planck Institute for Coal Research with Prof. Dr. M. T. Reetz as supervisor. After Postdoctoral research at Scripps Research Institute in La Jolla, CA (Prof. Dr. J. Rebek, Jr.), he started his own research career in 1998 at University of Münster. After his professorship in 2004 at the University of Bonn, he became full professor of Organic Chemistry at Johannes Gutenberg University Mainz in 2010. His research interests are novel electro-organic transformations including biobased feedstocks from electrosynthetic screening to scale-up. In 2018, he cofounded ESy-Labs GmbH, which provides custom electrosynthesis and contract R&D.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
References
- Pollok D.; Waldvogel S. R. Electro-Organic Synthesis - a 21st Century Technique. Chem. Sci. 2020, 11, 12386–12400. 10.1039/D0SC01848A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiebe A.; Gieshoff T.; Möhle S.; Rodrigo E.; Zirbes M.; Waldvogel S. R. Electrifying Organic Synthesis. Angew. Chem., Int. Ed. 2018, 57, 5594–5619. 10.1002/anie.201711060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horn E. J.; Rosen B. R.; Baran P. S. Synthetic Organic Electrochemistry: An Enabling and Innately Sustainable Method. ACS Cent. Sci. 2016, 2, 302–308. 10.1021/acscentsci.6b00091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moeller K. D. Using Physical Organic Chemistry To Shape the Course of Electrochemical Reactions. Chem. Rev. 2018, 118, 4817–4833. 10.1021/acs.chemrev.7b00656. [DOI] [PubMed] [Google Scholar]
- Little R. D.; Moeller K. D. Introduction: Electrochemistry: Technology, Synthesis, Energy, and Materials. Chem. Rev. 2018, 118, 4483–4484. 10.1021/acs.chemrev.8b00197. [DOI] [PubMed] [Google Scholar]
- Fuchigami T.; Inagi S. Recent Advances in Electrochemical Systems for Selective Fluorination of Organic Compounds. Acc. Chem. Res. 2020, 53, 322–334. 10.1021/acs.accounts.9b00520. [DOI] [PubMed] [Google Scholar]
- Röckl J. L.; Pollok D.; Franke R.; Waldvogel S. R. A Decade of Electrochemical Dehydrogenative C,C-Coupling of Aryls. Acc. Chem. Res. 2020, 53, 45–61. 10.1021/acs.accounts.9b00511. [DOI] [PubMed] [Google Scholar]
- Waldvogel S. R.; Lips S.; Selt M.; Riehl B.; Kampf C. J. Electrochemical Arylation Reaction. Chem. Rev. 2018, 118, 6706–6765. 10.1021/acs.chemrev.8b00233. [DOI] [PubMed] [Google Scholar]
- Möhle S.; Zirbes M.; Rodrigo E.; Gieshoff T.; Wiebe A.; Waldvogel S. R. Modern Electrochemical Aspects for the Synthesis of Value-Added Organic Products. Angew. Chem., Int. Ed. 2018, 57, 6018–6041. 10.1002/anie.201712732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidler J.; Strugatchi J.; Gärtner T.; Waldvogel S. R. Does Electrifying Organic Synthesis Pay Off? The Energy Efficiency of Electro-organic Conversions. MRS Energy & Sustainability 2020, 7, 42. 10.1557/mre.2020.42. [DOI] [Google Scholar]
- Waldvogel S. R.; Janza B. Renaissance of Electrosynthetic Methods for the Construction of Complex Molecules. Angew. Chem., Int. Ed. 2014, 53, 7122–7123. 10.1002/anie.201405082. [DOI] [PubMed] [Google Scholar]
- Yan M.; Kawamata Y.; Baran P. S. Synthetic Organic Electrochemical Methods Since 2000: On the Verge of a Renaissance. Chem. Rev. 2017, 117, 13230–13319. 10.1021/acs.chemrev.7b00397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akhade S. A.; Singh N.; Gutiérrez O. Y.; Lopez-Ruiz J.; Wang H.; Holladay J. D.; Liu Y.; Karkamkar A.; Weber R. S.; Padmaperuma A. B.; et al. Electrocatalytic Hydrogenation of Biomass-Derived Organics: A Review. Chem. Rev. 2020, 120, 11370–11419. 10.1021/acs.chemrev.0c00158. [DOI] [PubMed] [Google Scholar]
- Beil S.; Pollok D.; Waldvogel S. R. Reproducibility in Electro-Organic Synthesis - Myths and Misunderstandings. Angew. Chem., Int. Ed. 2021, 60, 2–12. 10.1002/anie.202014544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frahm A. W.; Lehmann J.; Meise W.; Muth H.; Pachaly P.; Reimann E.; Rimek H.-J.; Sauerbier M.; Tinapp P.; Zymalkowski F.. Reduktion: Teil I. In Methoden der Organischen Chemie, Houben-Weyl, 4th ed.; Müller E., Bayer O., Eds.; Georg Thieme, 1980; pp 13–15, 563, 613. [Google Scholar]
- Bracht J.; Hajos A.; Hartter P.; Muth H.; Sauerbier M.. Reduktion: Teil II. In Methoden der Organischen Chemie, Houben-Weyl, 4th ed.; Müller E., Bayer O., Eds.; Georg Thieme, 1980; pp 1–3, 487. [Google Scholar]
- Lam J.; Szkop K. M.; Mosaferi E.; Stephan D. W. FLP Catalysis: Main Group Hydrogenations of Organic Unsaturated Substrates. Chem. Soc. Rev. 2019, 48, 3592–3612. 10.1039/C8CS00277K. [DOI] [PubMed] [Google Scholar]
- Stephan D. W.; Erker G. Frustrated Lewis Pair Chemistry: Development and Perspectives. Angew. Chem., Int. Ed. 2015, 54, 6400–6441. 10.1002/anie.201409800. [DOI] [PubMed] [Google Scholar]
- Stephan D. W. “Frustrated Lewis Pairs“: a Concept for New Reactivity and Catalysis. Org. Biomol. Chem. 2008, 6, 1535–1539. 10.1039/b802575b. [DOI] [PubMed] [Google Scholar]
- Utley J. H. P.; Little R. D.; Nielsen M. F.. Reductive Coupling. In Organic Electrochemistry, 4th ed.; Hammerich O., Lund H., Eds.; Taylor & Francis, 2000; pp 621–704. [Google Scholar]
- Heinze J.Aliphatic and Aromatic Hydrocarbons Reduction. In Organic Electrochemistry, 4th ed.; Hammerich O., Lund H., Eds.; Taylor & Francis, 2000; pp 861–889. [Google Scholar]
- Hammerich O.Reduction of Nitro Compounds and Related Substrates. In Organic Electrochemistry, 4th ed.; Hammerich O., Lund H., Eds.; Taylor & Francis, 2000; pp 1149–1200. [Google Scholar]
- Breinbauer R.; Peters M.. Reduction of Carboxylic Acids and Derivatives. In Organic Electrochemistry, 4th ed.; Hammerich O., Lund H., Eds.; Taylor & Francis, 2000; pp 1249–1265. [Google Scholar]
- Ludvík J.Reduction of Aldehydes, Ketones, and Azomethines. In Organic Electrochemistry, 4th ed.; Hammerich O., Lund H., Eds.; Taylor & Francis, 2000; pp 1201–1247. [Google Scholar]
- Steckhan E.Electrochemistry, 3. Organic Electrochemistry. In Ullmann’s Encyclopedia of Industrial Chemistry, 7th ed.; Elvers B., Ullmann F., Eds.; Wiley-VCH, 2011; pp 315–349. [Google Scholar]
- Broadbent H. S.; Bartley W. J. Rhenium Catalysts. VII. Rhenium(VI) Oxide. J. Org. Chem. 1963, 28, 2345–2347. 10.1021/jo01044a046. [DOI] [Google Scholar]
- Pan Y.; Luo Z.; Han J.; Xu X.; Chen C.; Zhao H.; Xu L.; Fan Q.; Xiao J. B(C6F5)3-Catalyzed Deoxygenative Reduction of Amides to Amines with Ammonia Borane. Adv. Synth. Catal. 2019, 361, 2301–2308. 10.1002/adsc.201801447. [DOI] [Google Scholar]
- Soai K.; Ookawa A. Mixed Solvents Containing Methanol as Useful Reaction Media for Unique Chemoselective Reductions with Lithium Borohydride. J. Org. Chem. 1986, 51, 4000–4005. 10.1021/jo00371a017. [DOI] [Google Scholar]
- Edinger C.; Waldvogel S. R. Electrochemical Deoxygenation of Aromatic Amides and Sulfoxides. Eur. J. Org. Chem. 2014, 2014, 5144–5148. 10.1002/ejoc.201402714. [DOI] [Google Scholar]
- Yuan Y.; Lei A. Is Electrosynthesis Always Green and Advantageous Compared to Traditional Methods?. Nat. Commun. 2020, 11, 802. 10.1038/s41467-020-14322-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieger P. H.Electrochemistry; Springer Netherlands, 1993. [Google Scholar]
- Couper A. M.; Pletcher D.; Walsh F. C. Electrode Materials for Electrosynthesis. Chem. Rev. 1990, 90, 837–865. 10.1021/cr00103a010. [DOI] [Google Scholar]
- Nakagawa K.; Nagamori Y.. Method for Producing Adiponitrile. U.S. Patent 4789442, Dec. 6, 1988.
- Ralph T. R.; Hitchman M. L.; Millington J. P.; Walsh F. C. The Importance of Batch Electrolysis Conditions during the Reduction of L-Cystine Hydrochloride. J. Electrochem. Soc. 2005, 152, D54–D63. 10.1149/1.1858831. [DOI] [Google Scholar]
- Gütz C.; Grimaudo V.; Holtkamp M.; Hartmer M.; Werra J.; Frensemeier L.; Kehl A.; Karst U.; Broekmann P.; Waldvogel S. R. Leaded Bronze: An Innovative Lead Substitute for Cathodic Electrosynthesis. ChemElectroChem 2018, 5, 247–252. 10.1002/celc.201701061. [DOI] [Google Scholar]
- Katsounaros I.; Ipsakis D.; Polatides C.; Kyriacou G. Efficient Electrochemical Reduction of Nitrate to Nitrogen on Tin Cathode at Very High Cathodic Potentials. Electrochim. Acta 2006, 52, 1329–1338. 10.1016/j.electacta.2006.07.034. [DOI] [Google Scholar]
- Parkers D. W.Improvements in the Electrolytic Manufacture of Piperidine. GB Patent 395741, Dec. 24, 1933.
- Hannebaum H.; Nohe H.; Müller H.-R.. Verfahren zur Herstellung Hydrierter Indole. DE Patent2403446 A1, Jul. 31, 1975.
- Nohe H.; Hannebaum H.. Verfahren zur Elektrochemischen Hydrierung von Indolen. DE Patent 2658951 A1, Jul. 6, 1978.
- Suter H.; Nohe H.; Beck F.; Hrubesch A.. Production of Cyclohexydiene Dicarboxylic Acids. U.S. Patent 3471381, Oct. 7, 1969.
- Vaghela S. S.; Ramachandraiah G.; Ghosh P. K.; Vasudevan D. Electrolytic Synthesis of Succinic Acid in a Flow Reactor with Solid Polymer Electrolyte Membrane. J. Appl. Electrochem. 2002, 32, 1189–1192. 10.1023/A:1021617918645. [DOI] [Google Scholar]
- Beck F.; Leitner H.. Production of Adiponitrile. U.S. Patent 3616320, Oct. 26, 1971.
- Coleman G. H.; Johnson H. L. o-Aminobenzyl Alcohol. Org. Synth. 1941, 21, 10. 10.15227/orgsyn.021.0010. [DOI] [Google Scholar]
- Huang W.; Chen S.; Zheng J.; Li Z. Facile Preparation of Pt Hydrosols by Dispersing Bulk Pt with Potential Perturbations. Electrochem. Commun. 2009, 11, 469–472. 10.1016/j.elecom.2008.12.021. [DOI] [Google Scholar]
- Huang W.; Wang M.; Zheng J.; Li Z. Facile Fabrication of Multifunctional Three-Dimensional Hierarchical Porous Gold Films via Surface Rebuilding. J. Phys. Chem. C 2009, 113, 1800–1805. 10.1021/jp8095693. [DOI] [Google Scholar]
- Liu J.; Huang W.; Chen S.; Hu S.; Li Z. Facile Electrochemical Dispersion of Bulk Rh into Hydrosols. Int. J. Electrochem. Sci. 2009, 4, 1302–1308. [Google Scholar]
- Vanrenterghem B.; Bele M.; Zepeda F. R.; Šala M.; Hodnik N.; Breugelmans T. Cutting the Gordian Knot of Electrodeposition via Controlled Cathodic Corrosion Enabling the Production of Supported Metal Nanoparticles Below 5 nm. Appl. Catal., B 2018, 226, 396–402. 10.1016/j.apcatb.2017.12.080. [DOI] [Google Scholar]
- Bernasconi E.; Genders D.; Lee J.; Longoni D.; Martin C. R.; Menon V.; Roletto J.; Sogli L.; Walker D.; Zappi G.; et al. Ceftibuten: Development of a Commercial Process Based on Cephalosporin C. Part II. Process for the Manufacture of 3-Exomethylene-7(R)-glutaroylaminocepham-4-carboxylic Acid 1(S)-Oxide. Org. Process Res. Dev. 2002, 6, 158–168. 10.1021/op0100706. [DOI] [Google Scholar]
- Schulz H.; Ritapal K.; Bronger W.; Klemm W. Über die Reaktion von Elementen der achten Nebengruppe mit Oxiden unedler Metalle im Wasserstoffstrom. Z. Anorg. Allg. Chem. 1968, 357, 299–313. 10.1002/zaac.19683570418. [DOI] [Google Scholar]
- Karpov A.; Nuss J.; Wedig U.; Jansen M. Covalently Bonded [Pt]− Chains in BaPt: Extension of the Zintl-Klemm Concept to Anionic Transition Metals?. J. Am. Chem. Soc. 2004, 126, 14123–14128. 10.1021/ja0401186. [DOI] [PubMed] [Google Scholar]
- Karpov A.; Nuss J.; Wedig U.; Jansen M. Cs2Pt: A Platinide(-II) Exhibiting Complete Charge Separation. Angew. Chem., Int. Ed. 2003, 42, 4818–4821. 10.1002/anie.200352314. [DOI] [PubMed] [Google Scholar]
- Karpov A.; Wedig U.; Dinnebier R. E.; Jansen M. Dibariumplatinide: (Ba2+)2Pt2-.2e– and Its Relation to the Alkaline-Earth-Metal Subnitrides. Angew. Chem., Int. Ed. 2005, 44, 770–773. 10.1002/anie.200462055. [DOI] [PubMed] [Google Scholar]
- Karpov A.; Konuma M.; Jansen M. An Experimental Proof for Negative Oxidation States of Platinum: ESCA-Measurements on Barium Platinides. Chem. Commun. 2006, 838–840. 10.1039/b514631c. [DOI] [PubMed] [Google Scholar]
- Iandelli A.; Palenzona A. The Phase Diagram of the Eu-Pt System and the Valency Behaviour of Europium and Ytterbium Intermetallics with Platinum. J. Less-Common Met. 1981, 80, P71–P82. 10.1016/0022-5088(81)90103-X. [DOI] [Google Scholar]
- Köhler J.; Chang J.-H.; Whangbo M.-H. Bonding and Oxidation State of a Transition Metal Atom Encapsulated in an Isolated Octahedral Cluster Cation of Main Group Elements: Synthesis, Crystal Structure, and Electronic Structure of Pt2In14Ga3O8F15 Containing Highly Positive 18-Electron Complex [PtIn6]10+ and Low-Valent In+ Ions. J. Am. Chem. Soc. 2005, 127, 2277–2284. 10.1021/ja044389s. [DOI] [PubMed] [Google Scholar]
- Chlistunoff J. B.; Lagowski J. J. Cathodic Generation and Oxidation of Lead Zintl Ion Pb94− in Potassium Iodide Solutions in Liquid Ammonia. J. Phys. Chem. B 1997, 101, 2867–2873. 10.1021/jp9630737. [DOI] [Google Scholar]
- Chlistunoff J. B.; Lagowski J. J. Surface Alloying of Lead as a Step in the Cathodic Generation of the Pb94- in KI and RbBr Liquid Ammonia Solutions. J. Phys. Chem. B 1998, 102, 5800–5809. 10.1021/jp980334z. [DOI] [Google Scholar]
- Lake S. M.; Lagowski J. J. Zintl Polyanions as Metal Nano Particle Precursors. Synth. React. Inorg., Met.-Org., Nano-Met. Chem. 2006, 36, 465–467. 10.1080/15533170600777903. [DOI] [Google Scholar]
- Stanley Pons B.; Santure D. J.; Craig Taylor R.; Rudolph R. W. Electrochemical Generation of the Naked Metal Anionic Clusters, Sn9-xPb4-x (x = 0 to 9). Electrochim. Acta 1981, 26, 365–366. 10.1016/0013-4686(81)85023-2. [DOI] [Google Scholar]
- Zintl E.; Kaiser H. Über die Fähigkeit der Elemente zur Bildung Negativer Ionen. Z. Anorg. Allg. Chem. 1933, 211, 113–131. 10.1002/zaac.19332110113. [DOI] [Google Scholar]
- Iversen P. E.; Lund H. Electrolytic Generation of Nucleophiles. Reductive Alkylation and Acylation of Disulfides. Acta Chem. Scand. 1974, 28b, 827–828. 10.3891/acta.chem.scand.28b-0827. [DOI] [Google Scholar]
- Gehlen A. F. D’une Letter de M. Gehlen à M. Descostils, sur Plusieurs Experiences Galvaniques.. Ann. Chim. 1808, 66, 191–193. [Google Scholar]
- Wisniak J. The History of Mercury - From Discovery to Incommodity. Rev. CENIC, Cienc. Quim. 2008, 39, 147–157. [Google Scholar]
- Davy H. II. II. The Bakerian Lecture for 1809. On Some New Electrochemical Researches, on Various Objects, Particularly the Metallic Bodies, from the Alkalies, and Earths, and on some Combinations of Hydrogene. Philos. Trans. R. Soc. London 1810, 100, 16–74. 10.1098/rstl.1810.0004. [DOI] [Google Scholar]
- Reed C. J. A Remarkable Electrolytic Phenomenon. J. Franklin Inst. 1895, 139, 283–286. 10.1016/0016-0032(85)90202-9. [DOI] [Google Scholar]
- Bredig G.; Haber F. Ueber Zerstäubung von Metallkathoden bei der Elektrolyse mit Gleichstrom. Ber. Dtsch. Chem. Ges. 1898, 31, 2741–2752. 10.1002/cber.18980310329. [DOI] [Google Scholar]
- Sack M. Über die Entstehung und Bedeutung von Natriumlegierungen bei der Kathodischen Polarisation. Z. Anorg. Chem. 1903, 34, 286–352. 10.1002/zaac.19030340130. [DOI] [Google Scholar]
- Haber F. Über Legierungspotentiale und Deckschichtenbildung; zugleich ein Nachtrag zu der Mitteilung über Kathoden-Auflockerung und Zerstäubung. Z. Elektrochem. Angew. Phys. Chem. 1902, 8, 541–552. 10.1002/bbpc.19020083202. [DOI] [Google Scholar]
- Haber F. The Phenomenon of the Formation of Metallic Dust From Cathodes. Trans. Am. Electrochem. Soc. 1902, 2, 189–196. [Google Scholar]
- Salzberg H. W. Cathodic Lead Disintegration and Hydride Formation. J. Electrochem. Soc. 1953, 100, 146–151. 10.1149/1.2781098. [DOI] [Google Scholar]
- Gastwirt L. W.; Salzberg H. W. Disintegration of Lead Cathodes, II. J. Electrochem. Soc. 1957, 104, 701–703. 10.1149/1.2428456. [DOI] [Google Scholar]
- Salzberg H. W.; Mies F. Cathodic Disintegration of Tin. J. Electrochem. Soc. 1958, 105, 64–66. 10.1149/1.2428763. [DOI] [Google Scholar]
- Kabanov B. N. Incorporation of Alkali Metals into Solid Cathodes. Electrochim. Acta 1968, 13, 19–25. 10.1016/0013-4686(68)80003-9. [DOI] [Google Scholar]
- Kabanov B. N.; Astakhov I. I.; Kiseleva I. G. Electrochemical Implantation of Alkali Metals. Russ. Chem. Rev. 1965, 34, 775–785. 10.1070/RC1965v034n10ABEH001562. [DOI] [Google Scholar]
- Yanson A. I.; Rodriguez P.; Garcia-Araez N.; Mom R. V.; Tichelaar F. D.; Koper M. T. M. Cathodic Corrosion: A Quick, Clean, and Versatile Method for the Synthesis of Metallic Nanoparticles. Angew. Chem., Int. Ed. 2011, 50, 6346–6350. 10.1002/anie.201100471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez P.; Tichelaar F. D.; Koper M. T. M.; Yanson A. I. Cathodic Corrosion as a Facile and Effective Method to Prepare Clean Metal Alloy Nanoparticles. J. Am. Chem. Soc. 2011, 133, 17626–17629. 10.1021/ja208264e. [DOI] [PubMed] [Google Scholar]
- Gangal U.; Srivastava M.; Sen Gupta S. K. Mechanism of the Breakdown of Normal Electrolysis and the Transition to Contact Glow Discharge Electrolysis. J. Electrochem. Soc. 2009, 156, F131–F136. 10.1149/1.3186023. [DOI] [Google Scholar]
- Chiacchiarelli L. M.; Zhai Y.; Frankel G. S.; Agarwal A. S.; Sridhar N. Cathodic Degradation Mechanisms of Pure Sn Electrocatalyst in a Nitrogen Atmosphere. J. Appl. Electrochem. 2012, 42, 21–29. 10.1007/s10800-011-0367-z. [DOI] [Google Scholar]
- Yanson Y. I.; Yanson A. I. Cathodic Corrosion. I. Mechanism of Corrosion via Formation of Metal Anions in Aqueous Medium. Low Temp. Phys. 2013, 39, 304–311. 10.1063/1.4795002. [DOI] [Google Scholar]
- Hersbach T. J. P.; Yanson A. I.; Koper M. T. M. Anisotropic Etching of Platinum Electrodes at the Onset of Cathodic Corrosion. Nat. Commun. 2016, 7, 12653. 10.1038/ncomms12653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown O. R.; Taylor K.; Thirsk H. R. The Reduction of Ethyl Iodide at a Lead Cathode. J. Electroanal. Chem. Interfacial Electrochem. 1974, 53, 261–269. 10.1016/S0022-0728(74)80139-7. [DOI] [Google Scholar]
- Anawati; Frankel G.S.; Agarwal A.; Sridhar N. Degradation and Deactivation of Sn Catalyst Used for CO2 Reduction as Function of Overpotential. Electrochim. Acta 2014, 133, 188–196. 10.1016/j.electacta.2014.04.057. [DOI] [Google Scholar]
- Yang Y.; Huang W.; Zheng J.; Li Z. Facile Preparation of Sb and Oxide-Coated Sb Nanoparticles via Cathodic Dispersion of Bulk Sb in Different Media. J. Solid State Electrochem. 2012, 16, 803–809. 10.1007/s10008-011-1432-6. [DOI] [Google Scholar]
- Katsounaros I.; Kyriacou G. Influence of the Concentration and the Nature of the Supporting Electrolyte on the Electrochemical Reduction of Nitrate on Tin Cathode. Electrochim. Acta 2007, 52, 6412–6420. 10.1016/j.electacta.2007.04.050. [DOI] [Google Scholar]
- Otsuka R.; Uda M. Cathodic Corrosion of Cu in H2SO4. Corros. Sci. 1969, 9, 703–705. 10.1016/S0010-938X(69)80101-0. [DOI] [Google Scholar]
- Huang W.; Fu L.; Yang Y.; Hu S.; Li C.; Li Z. Simultaneous Fabrications of Nanoparticles and 3D Porous Films of Sn or Pb from Pure Electrodes. Electrochem. Solid-State Lett. 2010, 13, K46–K48. 10.1149/1.3339058. [DOI] [Google Scholar]
- Kliškić M.; Radošević J.; Gudić S. Yield of Hydrogen During Cathodic Polarisation of Al-Sn Alloys. Electrochim. Acta 2003, 48, 4167–4174. 10.1016/S0013-4686(03)00602-9. [DOI] [Google Scholar]
- Chen X.; Chen S.; Huang W.; Zheng J.; Li Z. Facile Preparation of Bi Nanoparticles by Novel Cathodic Dispersion of Bulk Bismuth Electrodes. Electrochim. Acta 2009, 54, 7370–7373. 10.1016/j.electacta.2009.07.068. [DOI] [Google Scholar]
- Dortsiou M.; Kyriacou G. Electrochemical Reduction of Nitrate on Bismuth Cathodes. J. Electroanal. Chem. 2009, 630, 69–74. 10.1016/j.jelechem.2009.02.019. [DOI] [Google Scholar]
- Salzberg H. W.; Andreatch A. J. Evolution of Stibine at Antimony Cathodes. J. Electrochem. Soc. 1954, 101, 528–532. 10.1149/1.2781149. [DOI] [Google Scholar]
- Yang Y.; Yang X.; Zhang Y.; Hou H.; Jing M.; Zhu Y.; Fang L.; Chen Q.; Ji X. Cathodically Induced Antimony for Rechargeable Li-Ion and Na-Ion Batteries: The Influences of Hexagonal and Amorphous Phase. J. Power Sources 2015, 282, 358–367. 10.1016/j.jpowsour.2015.02.071. [DOI] [Google Scholar]
- Thompson L. LXVIII. On Antimoniuretted Hydrogen, with Some Remarks on Mr. Marsh’s Test for Arsenic. London, Edinburgh, and Dublin Philosophical Magazine and Journal of Science 1837, 10, 353–355. 10.1080/14786443708649170. [DOI] [Google Scholar]
- Stock A.; Doht W. Ueber die Darstellung des Antimonwasserstoffes. Ber. Dtsch. Chem. Ges. 1902, 35, 2270–2275. 10.1002/cber.190203502187. [DOI] [Google Scholar]
- Denkhaus E.; Beck F.; Bueschler P.; Gerhard R.; Golloch A. Electrolytic Hydride Generation Atomic Absorption Spectrometry for the Determination of Antimony, Arsenic, Selenium, and Tin—Mechanistic Aspects and Figures of Merit. Fresenius' J. Anal. Chem. 2001, 370, 735–743. 10.1007/s002160100718. [DOI] [PubMed] [Google Scholar]
- Wang X.; Andrews L. Infrared Spectra of Group 14 Hydrides in Solid Hydrogen: Experimental Observation of PbH4, Pb2H2, and Pb2H4. J. Am. Chem. Soc. 2003, 125, 6581–6587. 10.1021/ja029862l. [DOI] [PubMed] [Google Scholar]
- Li J.; Liu Y.; Lin T. Determination of Lead by Hydride Generation Atomic Absorption Spectrometry: Part 1. A New Medium for Generating Hydride. Anal. Chim. Acta 1990, 231, 151–155. 10.1016/S0003-2670(00)86412-0. [DOI] [Google Scholar]
- Hersbach T. J. P.; McCrum I. T.; Anastasiadou D.; Wever R.; Calle-Vallejo F.; Koper M. T. M. Alkali Metal Cation Effects in Structuring Pt, Rh, and Au Surfaces through Cathodic Corrosion. ACS Appl. Mater. Interfaces 2018, 10, 39363–39379. 10.1021/acsami.8b13883. [DOI] [PubMed] [Google Scholar]
- Yanson A. I.; Yanson Y. I. Cathodic corrosion. II. Properties of Nanoparticles Synthesized by Cathodic Corrosion. Low Temp. Phys. 2013, 39, 312–317. 10.1063/1.4795197. [DOI] [Google Scholar]
- Yanson A. I.; Antonov P. V.; Rodriguez P.; Koper M. Influence of the Electrolyte Concentration on the Size and Shape of Platinum Nanoparticles Synthesized by Cathodic Corrosion. Electrochim. Acta 2013, 112, 913–918. 10.1016/j.electacta.2013.01.056. [DOI] [Google Scholar]
- Yanson A. I.; Antonov P. V.; Yanson Y. I.; Koper M. Controlling the Size of Platinum Nanoparticles Prepared by Cathodic Corrosion. Electrochim. Acta 2013, 110, 796–800. 10.1016/j.electacta.2013.03.121. [DOI] [Google Scholar]
- Feng J.; Chen D.; Sediq A. S.; Romeijn S.; Tichelaar F. D.; Jiskoot W.; Yang J.; Koper M. T. M. Cathodic Corrosion of a Bulk Wire to Nonaggregated Functional Nanocrystals and Nanoalloys. ACS Appl. Mater. Interfaces 2018, 10, 9532–9540. 10.1021/acsami.7b18105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hersbach T. J. P.; Kortlever R.; Lehtimäki M.; Krtil P.; Koper M. T. M. Local Structure and Composition of PtRh Nanoparticles Produced Through Cathodic Corrosion. Phys. Chem. Chem. Phys. 2017, 19, 10301–10308. 10.1039/C7CP01059A. [DOI] [PubMed] [Google Scholar]
- Duca M.; Rodriguez P.; Yanson A. I.; Koper M. T. M. Selective Electrocatalysis on Platinum Nanoparticles with Preferential (100) Orientation Prepared by Cathodic Corrosion. Top. Catal. 2014, 57, 255–264. 10.1007/s11244-013-0180-5. [DOI] [Google Scholar]
- Simonet J.; Astier Y.; Dano C. On the Cathodic Behaviour of Tetraalkylammonium Cations at a Platinum Electrode. J. Electroanal. Chem. 1998, 451, 5–9. 10.1016/S0022-0728(98)00090-4. [DOI] [Google Scholar]
- Simonet J.; Labaume E.; Rault-Berthelot J. On the Cathodic Corrosion of Platinum in the Presence of Iodides in Dry Aprotic Solvents. Electrochem. Commun. 1999, 1, 252–256. 10.1016/S1388-2481(99)00050-8. [DOI] [Google Scholar]
- Simonet J. Electron Transfer at Platinum and Palladium Interfaces in Super-Dry Electrolytes. Generation of Iono-Metallic Layers. A Mini-Review. Electrochem. Commun. 2015, 53, 15–19. 10.1016/j.elecom.2015.01.018. [DOI] [Google Scholar]
- Cougnon C.; Simonet J. Cathodic Reactivity of Platinum and Palladium in Electrolytes in Superdry Conditions. Platinum Met. Rev. 2002, 46, 94–105. [Google Scholar]
- Simonet J. Cathodic Reactivity of Platinum Interface in the Presence of Tetramethylammonium Salts. A Pro-Base Cathode Material?. Electrochem. Commun. 2003, 5, 439–444. 10.1016/S1388-2481(03)00096-1. [DOI] [Google Scholar]
- Cougnon C. Cathodic Reactivity of Alkaline Metal Iodides Toward Platinum Bulk. The formation of New Reducing Phases. Electrochem. Commun. 2002, 4, 266–271. 10.1016/S1388-2481(02)00272-2. [DOI] [Google Scholar]
- Bergamini J.-F.; Ghilane J.; Guilloux-Viry M.; Hapiot P. In Situ EC-AFM Imaging of Cathodic Modifications of Platinum Surfaces Performed in Dimethylformamide. Electrochem. Commun. 2004, 6, 188–192. 10.1016/j.elecom.2003.11.012. [DOI] [Google Scholar]
- Ghilane J.; Guilloux-Viry M.; Lagrost C.; Hapiot P.; Simonet J. Cathodic Modifications of Platinum Surfaces in Organic Solvent: Reversibility and Cation Type Effects. J. Phys. Chem. B 2005, 109, 14925–14931. 10.1021/jp051846y. [DOI] [PubMed] [Google Scholar]
- Ghilane J.; Guilloux-Viry M.; Lagrost C.; Simonet J.; Hapiot P. Reactivity of Platinum Metal with Organic Radical Anions from Metal to Negative Oxidation States. J. Am. Chem. Soc. 2007, 129, 6654–6661. 10.1021/ja071483a. [DOI] [PubMed] [Google Scholar]
- Cougnon C.; Simonet J. Cathodic Immobilization of π-Acceptors such as Aromatic Ketones onto Platinum Interfaces Under Superdry Conditions. J. Electroanal. Chem. 2002, 531, 179–186. 10.1016/S0022-0728(02)01062-8. [DOI] [Google Scholar]
- Hersbach T. J. P.; Ye C.; Garcia A. C.; Koper M. T. M. Tailoring the Electrocatalytic Activity and Selectivity of Pt(111) through Cathodic Corrosion. ACS Catal. 2020, 10, 15104–15113. 10.1021/acscatal.0c04016. [DOI] [Google Scholar]
- Fichtner J.; Watzele S.; Garlyyev B.; Kluge R. M.; Haimerl F.; El-Sayed H. A.; Li W.-J.; Maillard F. M.; Dubau L.; Chattot R.; et al. Tailoring the Oxygen Reduction Activity of Pt Nanoparticles through Surface Defects: A Simple Top-Down Approach. ACS Catal. 2020, 10, 3131–3142. 10.1021/acscatal.9b04974. [DOI] [Google Scholar]
- Arulmozhi N.; Hersbach T. J. P.; Koper M. T. M. Nanoscale Morphological Evolution of Monocrystalline Pt Surfaces During Cathodic Corrosion. Proc. Natl. Acad. Sci. U. S. A. 2020, 117, 32267–32277. 10.1073/pnas.2017086117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hersbach T. J. P.; Mints V. A.; Calle-Vallejo F.; Yanson A. I.; Koper M. T. M. Anisotropic Etching of Rhodium and Gold as the Onset of Nanoparticle Formation by Cathodic Corrosion. Faraday Discuss. 2016, 193, 207–222. 10.1039/C6FD00078A. [DOI] [PubMed] [Google Scholar]
- Cougnon C.; Simonet J. Are Tetraalkylammonium Cations Inserted into Palladium Cathodes? Formation of New Palladium Phases Involving Tetraalkylammonium Halides. J. Electroanal. Chem. 2001, 507, 226–233. 10.1016/S0022-0728(01)00431-4. [DOI] [Google Scholar]
- Cougnon C.; Simonet J. The Cathodic Insertion of Tetraalkylammonium Iodides into Palladium. Electrochem. Commun. 2001, 3, 209–214. 10.1016/S1388-2481(01)00135-7. [DOI] [Google Scholar]
- Najdovski I.; Selvakannan P. R.; O'Mullane A. P. Cathodic Corrosion of Cu Substrates as a Route to Nanostructured Cu/M (M = Ag, Au, Pd) Surfaces. ChemElectroChem 2015, 2, 106–111. 10.1002/celc.201402259. [DOI] [Google Scholar]
- Kromer M. L.; Monzó J.; Lawrence M. J.; Kolodziej A.; Gossage Z. T.; Simpson B. H.; Morandi S.; Yanson A.; Rodríguez-López J.; Rodríguez P. High-Throughput Preparation of Metal Oxide Nanocrystals by Cathodic Corrosion and Their Use as Active Photocatalysts. Langmuir 2017, 33, 13295–13302. 10.1021/acs.langmuir.7b02465. [DOI] [PubMed] [Google Scholar]
- Rodriguez P.; Plana D.; Fermin D. J.; Koper M. T. New Insights into the Catalytic Activity of Gold Nanoparticles for CO Oxidation in Electrochemical Media. J. Catal. 2014, 311, 182–189. 10.1016/j.jcat.2013.11.020. [DOI] [Google Scholar]
- Lawrence M. J.; Celorrio V.; Shi X.; Wang Q.; Yanson A.; Adkins N. J. E.; Gu M.; Rodríguez-López J.; Rodriguez P. Electrochemical Synthesis of Nanostructured Metal-Doped Titanates and Investigation of Their Activity as Oxygen Evolution Photoanodes. ACS Appl. Energy Mater. 2018, 1, 5233–5244. 10.1021/acsaem.8b00873. [DOI] [Google Scholar]
- Kariv-Miller E.; Lawin P. B. Tetraalkylammonium-Lead: Electrogeneration and Stoichiometry. J. Electroanal. Chem. Interfacial Electrochem. 1988, 247, 345–349. 10.1016/0022-0728(88)80156-6. [DOI] [Google Scholar]
- Lawin P. B.; Svetličić V.; Kariv-MilIer E. The Kinetics of Electrogeneration of (Dimethylpyrrolidinium)(Pb5). J. Electroanal. Chem. Interfacial Electrochem. 1989, 258, 357–368. 10.1016/0022-0728(89)85120-4. [DOI] [Google Scholar]
- Kariv-Miller E.; Christian P. D.; Svetlicic V. Ex Situ Structural Studies of a Tetraalkylammonium Lead Compound. Langmuir 1994, 10, 3338–3342. 10.1021/la00021a070. [DOI] [Google Scholar]
- Kariv-Miller E.; Christian P. D.; Svetlicic V. The First Cathodically Generated Tetraalkylammonium-Tin Compounds. Langmuir 1995, 11, 1817–1821. 10.1021/la00005a062. [DOI] [Google Scholar]
- Svetličič V.; Lawin P. B.; Kariv-Miller E. Reaction of Solid Cathodes with Tetraalkylammonium Electrolytes. J. Electroanal. Chem. Interfacial Electrochem. 1990, 284, 185–193. 10.1016/0022-0728(90)87071-Q. [DOI] [Google Scholar]
- Kariv-Miller E.; Lawin P. B.; Vajtner Z. The Reduction of Tetraalkylammonium Ions on Metal Electrodes. J. Electroanal. Chem. Interfacial Electrochem. 1985, 195, 435–438. 10.1016/0022-0728(85)80064-4. [DOI] [Google Scholar]
- Yang Y.; Qiao B.; Wu Z.; Ji X. Cathodic Corrosion: An Electrochemical Approach to Capture Zintl Compounds for Powder Materials. J. Mater. Chem. A 2015, 3, 5328–5336. 10.1039/C4TA05304D. [DOI] [Google Scholar]
- Pavesi D.; van de Poll R. C. J.; Krasovic J. L.; Figueiredo M.; Gruter G.-J. M.; Koper M. T. M.; Schouten K. J. P. Cathodic Disintegration as an Easily Scalable Method for the Production of Sn- and Pb-Based Catalysts for CO2 Reduction. ACS Sustainable Chem. Eng. 2020, 8, 15603–15610. 10.1021/acssuschemeng.0c04875. [DOI] [Google Scholar]
- Svetlicic V.; Gunderson E. G.; Kariv-Miller E.; Zutic V. Kinetics of Mediated Reductions by Cathodically Generated Dimethylpyrrolidinium-Tin Composites. The Case of Phenyl Bromides. Langmuir 1993, 9, 2210–2214. 10.1021/la00032a050. [DOI] [Google Scholar]
- Lu F.; Ji X.; Yang Y.; Deng W.; Banks C. E. Room Temperature Ionic Liquid Assisted Well-Dispersed Core-Shell Tin Nanoparticles Through Cathodic Corrosion. RSC Adv. 2013, 3, 18791–18793. 10.1039/c3ra43532f. [DOI] [Google Scholar]
- Fidler M. M.; Svetličić V.; Kariv-Miller E. An Electrochemical Study of Antimony Cathodes in Tetraalkylammonium Electrolyte Solutions. J. Electroanal. Chem. 1993, 360, 221–236. 10.1016/0022-0728(93)87015-N. [DOI] [Google Scholar]
- Medina-Ramos J.; Lee S. S.; Fister T. T.; Hubaud A. A.; Sacci R. L.; Mullins D. R.; DiMeglio J. L.; Pupillo R. C.; Velardo S. M.; Lutterman D. A.; et al. Structural Dynamics and Evolution of Bismuth Electrodes during Electrochemical Reduction of CO2 in Imidazolium-Based Ionic Liquid Solutions. ACS Catal. 2017, 7, 7285–7295. 10.1021/acscatal.7b01370. [DOI] [Google Scholar]
- Medina-Ramos J.; Zhang W.; Yoon K.; Bai P.; Chemburkar A.; Tang W.; Atifi A.; Lee S. S.; Fister T. T.; Ingram B. J.; et al. Cathodic Corrosion at the Bismuth-Ionic Liquid Electrolyte Interface under Conditions for CO2 Reduction. Chem. Mater. 2018, 30, 2362–2373. 10.1021/acs.chemmater.8b00050. [DOI] [Google Scholar]
- Kariv-Miller E.; Nanjundiah C. An Electrochemical Study of a Tetraalkylammonium Amalgam. J. Electroanal. Chem. Interfacial Electrochem. 1983, 147, 319–322. 10.1016/S0022-0728(83)80077-1. [DOI] [Google Scholar]
- Kariv-Miller E.; Svetličić V.; Christian P. D. N-Methylquinuclidinium-Mercury Compound: Electrodeposition and Catalysis. J. Electrochem. Soc. 1995, 142, 3386–3392. 10.1149/1.2049992. [DOI] [Google Scholar]
- Svetličić V.; Kariv-Miller E. The Growth of Ordered Films During the Cathodic Reduction of a Tetraalkylammonium Ion. J. Electroanal. Chem. Interfacial Electrochem. 1986, 209, 91–100. 10.1016/0022-0728(86)80188-7. [DOI] [Google Scholar]
- Ryan C. M.; Svetličić V.; Kariv-Miller E. Electrogenerated R4N(Hg)5 Films: Stoichiometry and Substituent Effects. J. Electroanal. Chem. Interfacial Electrochem. 1987, 219, 247–258. 10.1016/0022-0728(87)85043-X. [DOI] [Google Scholar]
- Kariv-Miller E.; Nanjundiah C.; Eaton J.; Swenson K. E. Dimethylpyrrolidinium Amalgam Formation and Catalysis of Organic Electroreductions. J. Electroanal. Chem. Interfacial Electrochem. 1984, 167, 141–155. 10.1016/0368-1874(84)87063-X. [DOI] [Google Scholar]
- Kariv-Miller E.; Svetličić V. Stoichiometry of a Tetraalkylammonium “Amalgam. J. Electroanal. Chem. Interfacial Electrochem. 1986, 205, 319–322. 10.1016/0022-0728(86)90243-3. [DOI] [Google Scholar]
- Kariv-Miller E.; Svetličić V.; Lawin P. B. Electrogenerated R4N(Hg)5 Films. J. Chem. Soc., Faraday Trans. 1 1987, 83, 1169–1177. 10.1039/f19878301169. [DOI] [Google Scholar]
- Ryan C. M.; Svetličić V.; Kariv-Miller E. The Kinetics of Cathodic Generation of R4N(Hg5). J. Chem. Soc., Faraday Trans. 1 1988, 84, 4023–4031. 10.1039/f19888404023. [DOI] [Google Scholar]
- Cottrell W. R. T.; Morris R. A. N. Quaternary Phosphonium and Tertiary Sulphonium Amalgams. Chem. Commun. (London) 1968, 409. 10.1039/c19680000409. [DOI] [Google Scholar]
- Brauer G.; Düsing G. Zur Kenntnis der Ammoniumamalgame. Z. Anorg. Allg. Chem. 1964, 328, 154–164. 10.1002/zaac.19643280306. [DOI] [Google Scholar]
- Garcia E.; Cowley A. H.; Bard A. J. Quaternary Ammonium Amalgams as Zintl Ion Salts and Their Use in the Synthesis of Novel Quaternary Ammonium Salts. J. Am. Chem. Soc. 1986, 108, 6082–6083. 10.1021/ja00279a090. [DOI] [PubMed] [Google Scholar]
- Littlehailes J. D.; Woodhall B. J. Quaternary Ammonium Amalgams. Chem. Commun. (London) 1967, 665–666. 10.1039/c19670000665. [DOI] [Google Scholar]
- Hoch C.; Simon A. Tetramethylammoniumamalgam, [N(CH3)4]Hg8. Z. Anorg. Allg. Chem. 2006, 632, 2288–2294. 10.1002/zaac.200600163. [DOI] [Google Scholar]
- Lawin P. B.; Hutson A. C.; Kariv-Miller E. Reduction of Organic Compounds at Lead Cathodes and Mediation by Dimethylpyrrolidinium Ion. J. Org. Chem. 1989, 54, 526–529. 10.1021/jo00264a004. [DOI] [Google Scholar]
- Misono A.; Osa T.; Yamagishi T.; Kodama T. Selective Electroreduction of the Benzene Nucleus. J. Electrochem. Soc. 1968, 115, 266–267. 10.1149/1.2411126. [DOI] [Google Scholar]
- Kariv-Miller E.; Vajtner Z. Electroreductive Dehalogenation of Fluorobenzenes. J. Org. Chem. 1985, 50, 1394–1399. 10.1021/jo00209a009. [DOI] [Google Scholar]
- Coleman J. P.; Wagenknecht J. H. Reduction of Benzene and Related Compounds in Aqueous Solution and Undivided Cells. J. Electrochem. Soc. 1981, 128, 322–326. 10.1149/1.2127411. [DOI] [Google Scholar]
- Swartz J. E.; Mahachi T. J.; Kariv-Miller E. Electrochemical Reduction of Ketones Mediated by (Dimethylpyrrolidinio)mercury. Reductive Cyclization of Unsaturated Ketones and Redox Catalysis Studies. J. Am. Chem. Soc. 1988, 110, 3622–3628. 10.1021/ja00219a042. [DOI] [Google Scholar]
- Fragoso-Luna L. M.; Frontana-Uribe B. A.; Cárdenas J. Deoxygenation of Aliphatic Acetate Derivatives Using Electrogenerated Organic Amalgams. Tetrahedron Lett. 2002, 43, 1151–1155. 10.1016/S0040-4039(01)02384-X. [DOI] [Google Scholar]
- Gunderson E. G.; Svetlicic V.; Kariv-Miller E. Mediated Reduction of Phenyl Bromides with Cathodically Generated Me2Py(Sn5). J. Electrochem. Soc. 1993, 140, 1842–1847. 10.1149/1.2220726. [DOI] [Google Scholar]
- Pacut R. I.; Kariv-Miller E. Birch-type Reductions in Aqueous Media. Benzo[b]thiophene and Diphenyl Ether. J. Org. Chem. 1986, 51, 3468–3470. 10.1021/jo00368a012. [DOI] [Google Scholar]
- Kariv-Miller E.; Mahachi T. J. Selective Cyclization and Pinacolization Directed by Tetraalkylammonium Ions. J. Org. Chem. 1986, 51, 1041–1045. 10.1021/jo00357a018. [DOI] [Google Scholar]
- Kariv-Miller E.; Pacut R. I.; Lehman G. K.. Organic Electroreductions at Very Negative Potentials. In Electrochemistry III; Steckhan E., Ed.; Springer: Berlin, Heidelberg, 1988; pp 97–130. [Google Scholar]
- Horner L.; Neumann H. Studien zum Vorgang der Wasserstoffübertragung, XII: Hydrierende Spaltung von Sulfonen mit Tetramethylammonium als Elektronenüberträger. Chem. Ber. 1965, 98, 1715–1721. 10.1002/cber.19650980606. [DOI] [Google Scholar]
- Lawrence M. J.; Kolodziej A.; Rodriguez P. Controllable Synthesis of Nanostructured Metal Oxide and Oxyhydroxide Materials via Electrochemical Methods. Curr. Opin. Electrochem. 2018, 10, 7–15. 10.1016/j.coelec.2018.03.014. [DOI] [Google Scholar]
- Tomilov A. P.; Brago I. N.. Electrochemical Synthesis of Organometallic Compounds. In Progress in Electrochemistry of Organic Compounds 1; Frumkin A. N., Érshler A. B., Eds.; Springer, 1971; pp 241–285. [Google Scholar]
- Ulery H. E. Electrosynthesis of Dialkyltin Derivatives. J. Electrochem. Soc. 1973, 120, 1493–1498. 10.1149/1.2403289. [DOI] [Google Scholar]
- Seyferth D. The Rise and Fall of Tetraethyllead. 2. Organometallics 2003, 22, 5154–5178. 10.1021/om030621b. [DOI] [Google Scholar]
- Lehmkuhl H. Preparative Scope of Organometallic Electrochemistry. Synthesis 1973, 1973, 377–396. 10.1055/s-1973-22221. [DOI] [Google Scholar]
- Tedoradze G. A. Electrochemical Synthesis of Organometallic Compounds. J. Organomet. Chem. 1975, 88, 1–36. 10.1016/S0022-328X(00)89328-6. [DOI] [Google Scholar]
- Arai T. The Electrolytic Reduction of Ketones at a Mercury Cathode.—Preparation of Organomercurie Compounds. Bull. Chem. Soc. Jpn. 1959, 32, 184–188. 10.1246/bcsj.32.184. [DOI] [Google Scholar]
- Silversmith E. F.; Sloan W. J. S.. Process for Preparing Organometal Compounds. U.S. Patent 3197392, Jul. 27, 1965.
- Tafel J. Eine Merkwürdige Bildungsweise von Quecksilberalkylen. Ber. Dtsch. Chem. Ges. 1906, 39, 3626–3631. 10.1002/cber.19060390419. [DOI] [Google Scholar]
- Bisselink R. J. M.; Crockatt M.; Zijlstra M.; Bakker I. J.; Goetheer E.; Slaghek T. M.; van Es D. S. Identification of More Benign Cathode Materials for the Electrochemical Reduction of Levulinic Acid to Valeric Acid. ChemElectroChem 2019, 6, 3285–3290. 10.1002/celc.201900734. [DOI] [Google Scholar]
- Calingaert G. H. F.Method for Producing Lead Compounds. U.S. Patent 1539297, May 26, 1925.
- Mead; Brian Method for Producing Lead Compounds. U.S. Patent 15667159, Dec. 29, 1925.
- Smeltz K. C.Electrolytic Process for Producing Tetraalkyl Lead Compounds. U.S. Patent 3392093, Apr. 30, 1968.
- Galli R. The Formation of Tetraethyllead by Electrochemical Reduction of Ethyl Bromide. J. Electroanal. Chem. Interfacial Electrochem. 1969, 22, 75–84. 10.1016/S0022-0728(69)80148-8. [DOI] [Google Scholar]
- Galli R.; Olivani F. The Effect of the Supporting Electrolyte on the Electroreduction of Ethyl Bromide. J. Electroanal. Chem. Interfacial Electrochem. 1970, 25, 331–339. 10.1016/S0022-0728(70)80349-7. [DOI] [Google Scholar]
- Tomilov A. P.; Smirnov Y. D.; Varshavskii S. L. Electrolytic Cyanoethylation of Lead, Tin, Mercury, and Thallium. Zh. Obshch. Khim. 1965, 35, 391–393. [Google Scholar]; Chem. Abstr. 1965, 63, 429619.
- Ulery H. E. Cathodic Synthesis of Tetraalkyltin Compounds. J. Electrochem. Soc. 1972, 119, 1474–1478. 10.1149/1.2404026. [DOI] [Google Scholar]
- Fleischmann M.; Pletcher D.; Vance C. J. The Reduction of Alkyl Halides at a Lead Cathode in Dimethylformamide. J. Electroanal. Chem. Interfacial Electrochem. 1971, 29, 325–334. 10.1016/S0022-0728(71)80094-3. [DOI] [Google Scholar]
- Tafel J. Ungesättigte Bleialkyle. Ber. Dtsch. Chem. Ges. 1911, 44, 323–337. 10.1002/cber.19110440150. [DOI] [Google Scholar]
- Sekine T.; Yamura A.; Sugino K. Mechanism of Hydrocarbon Formation in the Electrolytic Reduction of Acetone in Aqueous Sulfuric Acid. J. Electrochem. Soc. 1965, 112, 439–443. 10.1149/1.2423564. [DOI] [Google Scholar]
- Kulisch J.; Nieger M.; Stecker F.; Fischer A.; Waldvogel S. R. Efficient and Stereodivergent Electrochemical Synthesis of Optically Pure Menthylamines. Angew. Chem., Int. Ed. 2011, 50, 5564–5567. 10.1002/anie.201101330. [DOI] [PubMed] [Google Scholar]
- Edinger C.; Kulisch J.; Waldvogel S. R. Stereoselective Cathodic Synthesis of 8-substituted (1R,3R,4S)-Menthylamines. Beilstein J. Org. Chem. 2015, 11, 294–301. 10.3762/bjoc.11.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulery H. E. Polarographic Reduction of Alkyl Halides at a Stationary Lead Electrode. J. Electrochem. Soc. 1969, 116, 1201–1205. 10.1149/1.2412278. [DOI] [Google Scholar]
- Hyo Kim B.; Moo Jun Y.; Rack Choi Y.; Byung Lee D.; Baik W. Electrochemical Synthesis of 2,1-Benzisoxazoles by Controlled Potential Cathodic Electrolysis. Heterocycles 1998, 48, 749–754. 10.3987/COM-97-8077. [DOI] [Google Scholar]
- Haggerty C. J. The Electrolytic Reduction of Acetone at a Mercury Cathode. Trans. Am. Electrochem. Soc. 1929, 56, 421–427. [Google Scholar]
- Schall C.; Kirst W. Über die Kathodische Reduktion der Ketone am Beispiele des Menthons. Z. Elektrochem. Angew. Phys. Chem. 1923, 29, 537–546. [Google Scholar]
- Arai T.; Oguri T. Electrolytic Reduction of Benzaldehyde at a Mercury Cathode—Preparation of Dibenzyl Mercury. Bull. Chem. Soc. Jpn. 1960, 33, 1018a. 10.1246/bcsj.33.1018a. [DOI] [Google Scholar]
- Holleck L.; Marquarding D. Quecksilber-Alkylverbindungen durch Elektrolyse Konjugiert Ungesättigter Ketone. Naturwissenschaften 1962, 49, 468. 10.1007/BF00631894. [DOI] [Google Scholar]
- Lebedeva A. I. Electrolytic Hydrogenation of Dimethylethynylcarbinol and Dimethylvinylcarbinol. I. Effect of the Cathode Material. Zh. Obshch. Khim. 1948, 18, 1161–1167. [Google Scholar]; Chem. Abstr. 1949, 44, 4473.
- Grimshaw J.; Ramsey J. S. Electrochemical Reactions. Part III. The Reduction of Benzyl Bromides at a Mercury Cathode. J. Chem. Soc. B 1968, 60–62. 10.1039/j29680000060. [DOI] [Google Scholar]
- Mann C. K.; Webb J. L.; Walborsky H. M. Cyclopropanes. XX. Electrochemical Reduction of (+)-S-1-Bromo-1-methyl-2,2-diphenylcyclopropane. Tetrahedron Lett. 1966, 7, 2249–2255. 10.1016/S0040-4039(00)72409-9. [DOI] [Google Scholar]
- Azoo J. A.; Coll F. G.; Grimshaw J. Electrochemical Reactions. Part VII. Reduction of Diphenyliodonium Salts. J. Chem. Soc. C 1969, 2521–2522. 10.1039/j39690002521. [DOI] [Google Scholar]
- Tomilov A. P.; Kaabak L. V. Electrosynthesis of Tetrakis(2-cyanoethyl)tin. Zh. Prikl. Khim. 1959, 32, 2600–2601. [Google Scholar]; Chem. Abstr. 1960, 54, 37788.
- Brago I. N.; Kaabak L. V.; Tomilov A. P. Electrolytic Reduction of Acrylonitrile on a Tin Cathode. Zh. Vses. Khim. O-va. im. Mendeleeva 1967, 12, 472. [Google Scholar]; Chem. Abstr. 1967, 67, 504513.
- Kaabak L. V.; Tomilov A. P. Electrolytic Formation of Organotin Compounds and Their Chemical Properties. Zh. Obshch. Khim. 1963, 33, 2808–2810. [Google Scholar]; Chem. Abstr. 1964, 60, 31067.
- Brown O. R.; Gonzalez E. R.; Wright A. R. Cathodic Syntheses of Tin Alkyls—II. Reduction of Simple Alkyl Halides. Electrochim. Acta 1973, 18, 369–372. 10.1016/0013-4686(73)85006-6. [DOI] [Google Scholar]
- Fleischmann M.; Mengoli G.; Pletcher D. The Cathodic Reduction of Acetonitrile: A New Synthesis of Tin Tetramethyl. J. Electroanal. Chem. Interfacial Electrochem. 1973, 43, 308–310. 10.1016/S0022-0728(73)80501-7. [DOI] [Google Scholar]
- Fleischmann M.; Mengoli G.; Pletcher D. The Reduction of Simple Alkyl Iodides at Tin Cathodes in Dimethylformamide. Electrochim. Acta 1973, 18, 231–235. 10.1016/0013-4686(73)80018-0. [DOI] [Google Scholar]
- Simonet J. Free Allyl Radical: Catalytic Generation and Subsequent Immobilization onto Au, Pd, Pt, and Carbon Cathodes. Electrochem. Commun. 2011, 13, 1417–1419. 10.1016/j.elecom.2011.09.004. [DOI] [Google Scholar]
- Prestat M.; Soares Costa J.; Lescop B.; Rioual S.; Holzer L.; Thierry D. Cathodic Corrosion of Zinc under Potentiostatic Conditions in NaCl Solutions. ChemElectroChem 2018, 5, 1203–1211. 10.1002/celc.201701325. [DOI] [Google Scholar]
- Volkova L. E.; Marshakov I. K.; Tutukina N. M.; Kreiser I. V. The Dissolution of Silver Cathodically Polarized in Acid Chloride Media. Prot. Met. 2006, 42, 140–143. 10.1134/S0033173206020068. [DOI] [Google Scholar]; Zashch. Met. 2006, 42, 154 - 157.10.1134/S0033173206020068
- Kreizer I. V.; Tutukina N. M.; Zartsyn I. D.; Marshakov I. K. The Dissolution of a Copper Cathode in Acidic Chloride Solutions. Prot. Met. 2002, 38, 226–232. 10.1023/A:1015609103529. [DOI] [Google Scholar]; Zashch. Met. 2002, 38, 262 - 268.
- Kreizer V.; Marshakov I. K.; Tutukina N. M.; Zartsyn I. D. The Effect of Oxygen on Copper Dissolution during Cathodic Polarization. Prot. Met. 2003, 39, 30–33. 10.1023/A:1021935023079. [DOI] [Google Scholar]; Zashch. Met 2003, 39, 35 - 39.
- Kreizer I. V.; Marshakov I. K.; Tutukina N. M.; Zartsyn I. D. Partial Reactions of Copper Dissolution under Cathodic Polarization in Acidic Media. Prot. Met. 2004, 40, 23–25. 10.1023/B:PROM.0000013107.65745.b0. [DOI] [Google Scholar]; Zashch. Met. 2004, 40, 28 - 30.
- Simonet J. Electrochemical Insertion of CO2 into Silver in a Large Extent. Electrochem. Commun. 2015, 58, 11–14. 10.1016/j.elecom.2015.05.011. [DOI] [Google Scholar]
- Jouikov V.; Simonet J. Cathodic Carboxylation of Gold in Thick {Au-CO2}n Layers. A model for Reversible Electrochemical Sequestration of CO2. Electrochem. Commun. 2015, 59, 40–42. 10.1016/j.elecom.2015.06.020. [DOI] [Google Scholar]
- Simonet J. Large-scale Cathodic Carboxylation of Copper Surfaces. Electrochem. Commun. 2017, 76, 67–70. 10.1016/j.elecom.2017.01.018. [DOI] [Google Scholar]
- Simonet J. Electrochemical Carboxylation of Titanium to Generate Versatile New Interfaces. Electrochem. Commun. 2018, 88, 67–70. 10.1016/j.elecom.2018.01.007. [DOI] [Google Scholar]
- Simonet J. A Simple Cathodic Process for Carboxylating Noble Metals and Generating New Versatile Electrode Interfaces. Electrochem. Commun. 2017, 85, 15–18. 10.1016/j.elecom.2017.09.020. [DOI] [Google Scholar]
- Ralph T. R.; Hitchman M. L.; Millington J. P.; Walsh F. C. The Electrochemistry of L-Cystine and L-Cysteine Part 2: Electrosynthesis of L-Cysteine at Solid Electrodes. J. Electroanal. Chem. 1994, 375, 17–27. 10.1016/0022-0728(94)03408-7. [DOI] [Google Scholar]
- Ralph T. R.; Hitchman M. L.; Millington J. P.; Walsh F. C. The Reduction of L-Cystine Hydrochloride at Lead Using Static and Rotating Disc Electrodes. J. Electroanal. Chem. 2005, 583, 260–272. 10.1016/j.jelechem.2005.06.009. [DOI] [Google Scholar]
- Gálvez-Vázquez M. D. J.; Moreno-García P.; Guo H.; Hou Y.; Dutta A.; Waldvogel S. R.; Broekmann P. Leaded Bronze Alloy as a Catalyst for the Electroreduction of CO2. ChemElectroChem 2019, 6, 2324–2330. 10.1002/celc.201900537. [DOI] [Google Scholar]
- Miller C.; Cuendet P.; Graetzel M. Adsorbed. Omega.-Hydroxy Thiol Monolayers on Gold Electrodes: Evidence for Electron Tunneling to Redox Species in Solution. J. Phys. Chem. 1991, 95, 877–886. 10.1021/j100155a072. [DOI] [Google Scholar]
- Schmickler W. A Theory of Resonance Tunnelling at Film-Covered Metal Electrodes. J. Electroanal. Chem. Interfacial Electrochem. 1977, 82, 65–80. 10.1016/S0022-0728(77)80250-7. [DOI] [Google Scholar]
- Edinger C.; Grimaudo V.; Broekmann P.; Waldvogel S. R. Stabilizing Lead Cathodes with Diammonium Salt Additives in the Deoxygenation of Aromatic Amides. ChemElectroChem 2014, 1, 1018–1022. 10.1002/celc.201402050. [DOI] [Google Scholar]
- Calle-Vallejo F.; Koper M. T. M.; Bandarenka A. S. Tailoring the Catalytic Activity of Electrodes with Monolayer Amounts of Foreign Metals. Chem. Soc. Rev. 2013, 42, 5210–5230. 10.1039/c3cs60026b. [DOI] [PubMed] [Google Scholar]
- Rodriguez J. Physical and Chemical Properties of Bimetallic Surfaces. Surf. Sci. Rep. 1996, 24, 223–287. 10.1016/0167-5729(96)00004-0. [DOI] [Google Scholar]
- Mueller J. E.; Krtil P.; Kibler L. A.; Jacob T. Bimetallic Alloys in Action: Dynamic Atomistic Motifs for Electrochemistry and Catalysis. Phys. Chem. Chem. Phys. 2014, 16, 15029–15042. 10.1039/C4CP01591F. [DOI] [PubMed] [Google Scholar]
- Chakrabarti D. J.; Laughlin D. E. The Cu-Pb (Copper-Lead) System. Bull. Alloy Phase Diagrams 1984, 5, 503–510. 10.1007/BF02872905. [DOI] [Google Scholar]
- Grimaudo V.; Moreno-García P.; Riedo A.; Meyer S.; Tulej M.; Neuland M. B.; Mohos M.; Gütz C.; Waldvogel S. R.; Wurz P.; et al. Toward Three-Dimensional Chemical Imaging of Ternary Cu-Sn-Pb Alloys Using Femtosecond Laser Ablation/Ionization Mass Spectrometry. Anal. Chem. 2017, 89, 1632–1641. 10.1021/acs.analchem.6b03738. [DOI] [PubMed] [Google Scholar]
- Shackelford J. F.; Alexander W.. CRC Materials Science and Engineering Handbook, 3rd ed.; CRC Press: Boca Raton, FL, 2001. [Google Scholar]
- Haynes W. M.CRC Handbook of Chemistry and Physics, 99th ed.; CRC Press by Taylor & Francis Group: Boca Raton, Fl, 2017. [Google Scholar]
- AS 1565 C93700; Copperalloys Ltd., 2021; https://www.copperalloys.net/alloys/as-1565-c93700 (accessed 2021-05-04).
- Gütz C.; Selt M.; Bänziger M.; Bucher C.; Römelt C.; Hecken N.; Gallou F.; Galvão T. R.; Waldvogel S. R. A Novel Cathode Material for Cathodic Dehalogenation of 1,1-Dibromo Cyclopropane Derivatives. Chem. - Eur. J. 2015, 21, 13878–13882. 10.1002/chem.201502064. [DOI] [PubMed] [Google Scholar]
- CuSn7Pb15-C; Copperalloys Ltd., 2021; https://www.copperalloys.net/alloys/cusn7pb15-c (accessed 2021-05-04).
- Gütz C.; Bänziger M.; Bucher C.; Galvão T. R.; Waldvogel S. R. Development and Scale-Up of the Electrochemical Dehalogenation for the Synthesis of a Key Intermediate for NS5A Inhibitors. Org. Process Res. Dev. 2015, 19, 1428–1433. 10.1021/acs.oprd.5b00272. [DOI] [Google Scholar]
- CuSn5Pb20-C; Copperalloys Ltd., 2021; https://www.copperalloys.net/alloys/cusn5pb20-c (accessed 2021-05-04).
- Mikkelsen Ø.; Schrøder K. H. Amalgam Electrodes for Electroanalysis. Electroanalysis 2003, 15, 679–687. 10.1002/elan.200390085. [DOI] [Google Scholar]
- Yosypchuk B.; Barek J. Analytical Applications of Solid and Paste Amalgam Electrodes. Crit. Rev. Anal. Chem. 2009, 39, 189–203. 10.1080/10408340903011838. [DOI] [Google Scholar]
- Danhel A.; Barek J. Amalgam Electrodes in Organic Electrochemistry. Curr. Org. Chem. 2011, 15, 2957–2969. 10.2174/138527211798357218. [DOI] [Google Scholar]
- Abbas S. A.; Kim S.-H.; Saleem H.; Ahn S.-H.; Jung K.-D. Preparation of Metal Amalgam Electrodes and Their Selective Electrocatalytic CO2 Reduction for Formate Production. Catalysts 2019, 9, 367. 10.3390/catal9040367. [DOI] [Google Scholar]
- Sharma P. L.; Gaur J. N. Electrolytic Reduction of Vanillin to Vanillyl Alcohol at Amalgamated Copper, Lead and Zinc Electrodes. J. Appl. Electrochem. 1981, 11, 173–176. 10.1007/BF00610977. [DOI] [Google Scholar]
- Ayyaswami A.; Krishnan V. Electroreduction of b-Furfuraldoxime at Mercury Cathode. Indian J. Chem. 1983, 22, 555–556. [Google Scholar]
- Vilambi N. R. K.; Chin D.-T. Reduction of Salicylic Acid to Salicylaldehyde with Modulated Alternating Voltage. J. Electrochem. Soc. 1987, 134, 3074–3077. 10.1149/1.2100342. [DOI] [Google Scholar]
- Gagyi-Pálffy E.; Prépostffy E.; Korányi G.. Electrosynthesis of Glyoxylic Acid. Period. Polytech. Chem. Eng., 198529, 95–101. [Google Scholar]
- Beck F. Electrosynthesis of Adiponitrile in Undivided Cells. J. Appl. Electrochem. 1972, 2, 59–69. 10.1007/BF00615193. [DOI] [Google Scholar]
- Waldvogel S. R.; Edinger C.. Process for Cathodic Deoxygenation of Amides and Esters in Solutions Containing Quaternary Ammonium Salts and Quaternary Phosphonium Salts. PCT Int. Appl.. WO 2013030316 A2, 20130307, Mar. 22, 2017.
- Lamoureux C.; Moinet C.; Tallec A. Cellules d‘Electrolyse a Circulation. Montages Particuliers et Applications a des Reactions Electrochimiques Consecutives et Opposees. Electrochim. Acta 1986, 31, 1–12. 10.1016/0013-4686(86)80053-6. [DOI] [Google Scholar]
- Lamoureux C.; Moinet C.; Tallec A. An electrolysis cell with close consecutive flow-through porous electrodes for particular organic electrosynthesis. J. Appl. Electrochem. 1986, 16, 819–824. 10.1007/BF01006527. [DOI] [Google Scholar]
- Frontana-Uribe B. A.; Moinet C. 2-Substituted Indazoles from Electrogenerated ortho-Nitrosobenzylamines. Tetrahedron 1998, 54, 3197–3206. 10.1016/S0040-4020(98)00058-1. [DOI] [Google Scholar]
- Moinet C.; Simonneaux G.; Autret M.; Hindre F.; Le Plouzennec M. Electroreduction of Porphyrin with a 4-Nithophenyl Group in the meso Position. Preparation of Nitroso and Amino Derivatives in a “Redox” Cell. Electrochim. Acta 1993, 38, 325–328. 10.1016/0013-4686(93)85147-Q. [DOI] [Google Scholar]
- Gault C.; Moinet C. Reaction of Electrogenerated 2-Nitrosobenzic Acids with Sulphinic Acids. A Convenient Route to N-Sulfonylbenzisoxazolones. Tetrahedron 1989, 45, 3429–3436. 10.1016/S0040-4020(01)81021-8. [DOI] [Google Scholar]
- Guennec N.; Moinet C. Etude Électrochimique des Cations (η5-Cyclopentadiényl fer η6-Nitrobénzene oú η6-Nitrotoluénes). Electrosynthèse des Dérivés Nitrosés Correspondants. J. Organomet. Chem. 1994, 465, 233–240. 10.1016/0022-328X(94)87060-8. [DOI] [Google Scholar]
- Guennec N.; Moinet C. Préparation de Cations η5-Cyclopentadienyl-η6-azobenzenes fer (1+). J. Organomet. Chem. 1995, 487, 177–185. 10.1016/0022-328X(94)05127-W. [DOI] [Google Scholar]
- Cristea C. V.; Moinet C.; Jitaru M.; Popescu I. C. Electrosynthesis of Nitroso Compounds from (1S, 2S)-2-Amino-l-(4-nitrophenyl)-propane-1,3-diol Derivatives. J. Appl. Electrochem. 2005, 35, 851–855. 10.1007/s10800-005-1320-9. [DOI] [Google Scholar]
- Frontana-Uribe B. A.; Moinet C.; Toupet L. N-Substituted 1-Aminoindoles from Electrogenerated N-Substituted 2-(ortho-Nitrosophenyl)ethylamines. Eur. J. Org. Chem. 1999, 1999, 419–430. . [DOI] [Google Scholar]
- Dong X.; Roeckl J. L.; Waldvogel S. R.; Morandi B. Merging Shuttle Reactions and Paired Electrolysis for Reversible Vicinal Dihalogenations. Science 2021, 371, 507–514. 10.1126/science.abf2974. [DOI] [PubMed] [Google Scholar]
- Hartmer M. F.; Waldvogel S. R. Electroorganic Synthesis of Nitriles via a Halogen-Free Domino Oxidation-Reduction Sequence. Chem. Commun. 2015, 51, 16346–16348. 10.1039/C5CC06437F. [DOI] [PubMed] [Google Scholar]
- Gleede B.; Selt M.; Gütz C.; Stenglein A.; Waldvogel S. R. Large, Highly Modular Narrow-Gap Electrolytic Flow Cell and Application in Dehydrogenative Cross-Coupling of Phenols. Org. Process Res. Dev. 2020, 24, 1916–1926. 10.1021/acs.oprd.9b00451. [DOI] [Google Scholar]
- Lipp A.; Selt M.; Ferenc D.; Schollmeyer D.; Waldvogel S. R.; Opatz T. Total Synthesis of (−)-Oxycodone via Anodic Aryl-Aryl Coupling. Org. Lett. 2019, 21, 1828–1831. 10.1021/acs.orglett.9b00419. [DOI] [PubMed] [Google Scholar]
- Gütz C.; Stenglein A.; Waldvogel S. R. Highly Modular Flow Cell for Electroorganic Synthesis. Org. Process Res. Dev. 2017, 21, 771–778. 10.1021/acs.oprd.7b00123. [DOI] [Google Scholar]
- Cardoso D. S. P.; Šljukić B.; Santos D. M. F.; Sequeira C. A. C. Organic Electrosynthesis: From Laboratorial Practice to Industrial Applications. Org. Process Res. Dev. 2017, 21, 1213–1226. 10.1021/acs.oprd.7b00004. [DOI] [Google Scholar]
- Sopher D.; Gieseler A.; Hibst H.; Harth K.; Jaeger P.. Electrode consisting of an Iron-Containing Core and a Lead-Containing Coating. U.S. Patent 5593557, Jan. 14, 1997.
- Wirtanen T.; Rodrigo E.; Waldvogel S. R. Recent Advances in the Electrochemical Reduction of Substrates Involving N-O Bonds. Adv. Synth. Catal. 2020, 362, 2088–2101. 10.1002/adsc.202000349. [DOI] [Google Scholar]
- Wirtanen T.; Rodrigo E.; Waldvogel S. R. Selective and Scalable Electrosynthesis of 2H-2-(Aryl)-benzod-1,2,3-triazoles and Their N-Oxides by Using Leaded Bronze Cathodes. Chem. - Eur. J. 2020, 26, 5592–5597. 10.1002/chem.201905874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strehl J.; Kahrs C.; Müller T.; Hilt G.; Christoffers J. Electrochemical-Induced Ring Transformation of Cyclic α-(ortho-Iodophenyl)-β-oxoesters. Chem. - Eur. J. 2020, 26, 3222–3225. 10.1002/chem.201905570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hersbach T. J.; Koper M. T. Cathodic Corrosion: 21st Century Insights into a 19th Century Phenomenon. Curr. Opin. Electrochem. 2021, 26, 100653. 10.1016/j.coelec.2020.100653. [DOI] [Google Scholar]
- Heard D. M.; Lennox A. J. J. Electrode Materials in Modern Organic Electrochemistry. Angew. Chem., Int. Ed. 2020, 59, 18866–18884. 10.1002/anie.202005745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang N.; Waldvogel S. R.; Jiang X. Electrochemistry of Carbon Dioxide on Carbon Electrodes. ACS Appl. Mater. Interfaces 2016, 8, 28357–28371. 10.1021/acsami.5b09825. [DOI] [PubMed] [Google Scholar]
- Lips S.; Waldvogel S. R. Use of Boron-Doped Diamond Electrodes in Electro-Organic Synthesis. ChemElectroChem 2019, 6, 1649–1660. 10.1002/celc.201801620. [DOI] [Google Scholar]
- Waldvogel S. R.; Mentizi S.; Kirste A. Boron-Doped Diamond Electrodes for Electroorganic Chemistry. Top. Curr. Chem. 2011, 320, 1–31. 10.1007/128_2011_125. [DOI] [PubMed] [Google Scholar]
- Waldvogel S. R.; Elsler B. Electrochemical Synthesis on Boron-Doped Diamond. Electrochim. Acta 2012, 82, 434–443. 10.1016/j.electacta.2012.03.173. [DOI] [Google Scholar]
- Batchelor T. A. A.; Löffler T.; Xiao B.; Krysiak O. A.; Strotkötter V.; Pedersen J. K.; Clausen C. M.; Savan A.; Li Y.; Schuhmann W.; et al. Complex Solid Solution Electrocatalyst Discovery by Computational Prediction and High-Throughput Experimentation. Angew. Chem., Int. Ed. 2021, 60, 6932–6937. 10.1002/anie.202014374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen J. K.; Batchelor T. A. A.; Bagger A.; Rossmeisl J. High-Entropy Alloys as Catalysts for the CO2 and CO Reduction Reactions. ACS Catal. 2020, 10, 2169–2176. 10.1021/acscatal.9b04343. [DOI] [Google Scholar]
- Zhang G.; Ming K.; Kang J.; Huang Q.; Zhang Z.; Zheng X.; Bi X. High Entropy Alloy as a Highly Active and Stable Electrocatalyst for Hydrogen Evolution Reaction. Electrochim. Acta 2018, 279, 19–23. 10.1016/j.electacta.2018.05.035. [DOI] [Google Scholar]
- Glasscott M. W.; Pendergast A. D.; Goines S.; Bishop A. R.; Hoang A. T.; Renault C.; Dick J. E. Electrosynthesis of High-Entropy Metallic Glass Nanoparticles for Designer, Multi-Functional Electrocatalysis. Nat. Commun. 2019, 10, 2650. 10.1038/s41467-019-10303-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X.; Si C.; Gao Y.; Frenzel J.; Sun J.; Eggeler G.; Zhang Z. Multi-Component Nanoporous Platinum-Ruthenium-Copper-Osmium-Iridium Alloy with Enhanced Electrocatalytic Activity Towards Methanol Oxidation and Oxygen Reduction. J. Power Sources 2015, 273, 324–332. 10.1016/j.jpowsour.2014.09.076. [DOI] [Google Scholar]
- Qiu H.-J.; Fang G.; Wen Y.; Liu P.; Xie G.; Liu X.; Sun S. Nanoporous High-Entropy Alloys for Highly Stable and Efficient Catalysts. J. Mater. Chem. A 2019, 7, 6499–6506. 10.1039/C9TA00505F. [DOI] [Google Scholar]
- Löffler T.; Meyer H.; Savan A.; Wilde P.; Garzón Manjón A.; Chen Y.-T.; Ventosa E.; Scheu C.; Ludwig A.; Schuhmann W. Discovery of a Multinary Noble Metal-Free Oxygen Reduction Catalyst. Adv. Energy Mater. 2018, 8, 1802269. 10.1002/aenm.201802269. [DOI] [Google Scholar]
- Harnisch F.; Urban C. Electrobiorefineries: Unlocking the Synergy of Electrochemical and Microbial Conversions. Angew. Chem., Int. Ed. 2018, 57, 10016–10023. 10.1002/anie.201711727. [DOI] [PubMed] [Google Scholar]
- Matthiesen J. E.; Carraher J. M.; Vasiliu M.; Dixon D. A.; Tessonnier J.-P. Electrochemical Conversion of Muconic Acid to Biobased Diacid Monomers. ACS Sustainable Chem. Eng. 2016, 4, 3575–3585. 10.1021/acssuschemeng.6b00679. [DOI] [Google Scholar]
- Suastegui M.; Matthiesen J. E.; Carraher J. M.; Hernandez N.; Rodriguez Quiroz N.; Okerlund A.; Cochran E. W.; Shao Z.; Tessonnier J.-P. Combining Metabolic Engineering and Electrocatalysis: Application to the Production of Polyamides from Sugar. Angew. Chem., Int. Ed. 2016, 55, 2368–2373. 10.1002/anie.201509653. [DOI] [PubMed] [Google Scholar]
- Matthiesen J. E.; Suástegui M.; Wu Y.; Viswanathan M.; Qu Y.; Cao M.; Rodriguez-Quiroz N.; Okerlund A.; Kraus G.; Raman D. R.; et al. Electrochemical Conversion of Biologically Produced Muconic Acid: Key Considerations for Scale-Up and Corresponding Technoeconomic Analysis. ACS Sustainable Chem. Eng. 2016, 4, 7098–7109. 10.1021/acssuschemeng.6b01981. [DOI] [Google Scholar]
- Bechthold I.; Bretz K.; Kabasci S.; Kopitzky R.; Springer A. Succinic Acid: A New Platform Chemical for Biobased Polymers from Renewable Resources. Chem. Eng. Technol. 2008, 31, 647–654. 10.1002/ceat.200800063. [DOI] [Google Scholar]
- Weber R. S.; Holladay J. E. Modularized Production of Value-Added Products and Fuels from Distributed Waste Carbon-Rich Feedstocks. Engineering 2018, 4, 330–335. 10.1016/j.eng.2018.05.012. [DOI] [Google Scholar]
- Chien A. A.; Yang F.; Zhang C. Characterizing Curtailed and Uneconomic Renewable Power in the Mid-Continent Independent System Operator. AIMS Energy 2018, 6, 376–401. 10.3934/energy.2018.2.376. [DOI] [Google Scholar]
- Bird L.; Lew D.; Milligan M.; Carlini E. M.; Estanqueiro A.; Flynn D.; Gomez-Lazaro E.; Holttinen H.; Menemenlis N.; Orths A.; et al. Wind and Solar Energy Curtailment: A Review of International Experience. Renewable Sustainable Energy Rev. 2016, 65, 577–586. 10.1016/j.rser.2016.06.082. [DOI] [Google Scholar]