

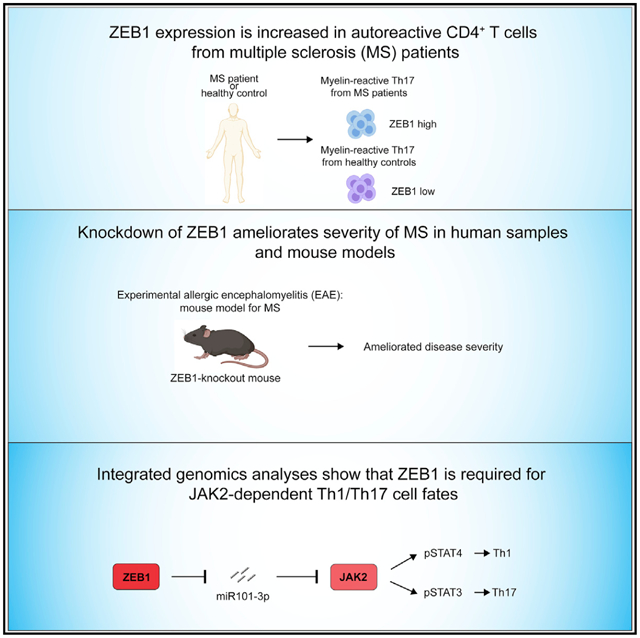
SUMMARY
Inappropriate CD4+ T helper (Th) differentiation can compromise host immunity or promote autoimmune disease. To identify disease-relevant regulators of T cell fate, we examined mutations that modify risk for multiple sclerosis (MS), a canonical organ-specific autoimmune disease. This analysis identified a role for Zinc finger E-box-binding homeobox (ZEB1). Deletion of ZEB1 protects against experimental autoimmune encephalitis (EAE), a mouse model of multiple sclerosis (MS). Mechanistically, ZEB1 in CD4+ T cells is required for pathogenic Th1 and Th17 differentiation. Genomic analyses of paired human and mouse expression data elucidated an unexpected role for ZEB1 in JAK-STAT signaling. ZEB1 inhibits miR-101-3p that represses JAK2 expression, STAT3/STAT4 phosphorylation, and subsequent expression of interleukin-17 (IL-17) and interferon gamma (IFN-γ). Underscoring its clinical relevance, ZEB1 and JAK2 downregulation decreases pathogenic cytokines expression in T cells from MS patients. Moreover, a Food and Drug Administration (FDA)-approved JAK2 inhibitor is effective in EAE. Collectively, these findings identify a conserved, potentially targetable mechanism regulating disease-relevant inflammation.
Graphical Abstract
In brief
Qian et al. show that ZEB1 is required for the development of the autoimmune disease multiple sclerosis (MS). ZEB1, a transcription factor, promotes JAK-STAT signaling during Th1/Th17 differentiation by repressing expression of a JAK2-targeting miRNA. ZEB1 and JAK2 are potentially clinically relevant therapeutic targets for MS.
INTRODUCTION
CD4+ Th cell differentiation is critical to mounting the proper cytokine response to a diverse array of foreign pathogens (Fang and Zhu, 2017). Inappropriate T helper (Th) cell differentiation promotes autoimmunity via the production of proinflammatory cytokines. Despite the central importance of Th cells to human immunity and health, our understanding of the mechanisms underlying CD4+ T cell fate remains incomplete.
JAK-STAT signaling pathways play pivotal but incompletely understood roles in determining CD4+ T cell fate (Villarino et al., 2017). In general, the JAK-STAT pathway is broadly similar across distinct T cell subtypes. Cytokines drive receptor tyrosine kinase phosphorylation, which in turn drive JAK activation and the subsequent phosphorylation of downstream STAT proteins. Dimerized phospho-STATs enter the nucleus and regulate transcription of genes that facilitate or inhibit T helper cell differentiation (Waldmann and Chen, 2017). Emerging data suggest that these processes are more complex than initially anticipated. Aside from cytokines, JAK-STATs can be modulated by cell-surface receptors and ubiquitin ligases (Cho et al., 2019; Meyer Zu Horste et al., 2018). Thus, regulation of this pathway, much as the broader regulation of Th cell differentiation, must be multi-layered.
Human genetics offers an unbiased approach to uncover novel regulators of disease processes. Multiple sclerosis (MS) is a canonical organ-specific autoimmune disease mediated by self-antigen specific Th1 and Th17 cells (Lee et al., 2017). Recently, a genome-wide association study (GWAS) implicated 200 common single nucleotide polymorphisms (SNPs) and 551 putative associated genes that increase or decrease the risk of MS (International Multiple Sclerosis Genetics Consortium, 2019b). For the most part, the mechanisms by which these genes promote or protect against MS remains unclear.
One gene, ZEB1, encodes a transcriptional repressor that binds to an E-box domain and recruits protein complexes (e.g., CtBP and HDAC proteins) that inhibit transcription (Manshouri et al., 2019). Recent reports suggest that ZEB1 plays a critical role in T cell development in the thymus (Arnold et al., 2012; Higashi et al., 1997; Zhang et al., 2020). Moreover, ZEB1 is required for the survival and maintenance of anti-viral memory CD8+ T cells (Guan et al., 2018). However, the role of ZEB1 in mature CD4+ T cells is not completely understood. Here, we find that ZEB1 promotes MS by positively regulating Th1 and Th17 differentiation in both human and mouse CD4+ T cells. Using a mouse model of MS,experimental autoimmune encephalomyelitis (EAE), we confirm that loss of ZEB1 prevents disease onset. Mechanistically, ZEB1 controls T cell differentiation across multiple lineages in both human and murine CD4+ T cells. It significantly promotes Th1 and Th17 and inhibits Th2 differentiation in both humans and mice in vitro. Systems level analyses of RNA sequencing (RNA-seq) data suggest that ZEB1 is necessary for the STAT3/STAT4 transcriptional programs required for Th1 and Th17 differentiation, respectively. This regulation occurs via its effects on a JAK2-targeting microRNA (miRNA), miR-101-3p. miR-101-3p is normally suppressed by ZEB1. Its expression is both necessary and sufficient to phenocopy the effects of ZEB1 loss on interferon-γ (IFN-γ) and interleukin-17 (IL-17) expression. Using MS patient samples, we find ZEB1 is required for IFN-γ and IL-17 expression in Th1, Th17, and Th1/Th17 cells. Last, fedratinib, a JAK2 inhibitor, shows efficacy in vivo in treating mouse models of MS.
RESULTS
Identifying Th1- and Th17-relevant genes that modulate MS risk
A recent GWAS study (47,429 MS patients and 68,374 controls) identified 200 SNPs and 551 putative genes that modify the risk for MS. These genes were hypothesized to mediate their effects in diverse cell types involved in MS inflammation (e.g., microglia and multiple immune cell types) (International Multiple Sclerosis Genetics Consortium, 2019b).
To find disease-relevant regulators of Th differentiation, we integrated the GWAS data with relevant gene expression data for relevant cell types. We first analyzed a recently published RNA-seq dataset of CD4+ T cells and CD14+ monocytes from 25 MS patients (International Multiple Sclerosis Genetics Consortium, 2019a). This analysis identified 87 MS-associated genes that were significantly enriched specifically in CD4+ T cells (log2Fold-Change > 2, adjust p value [padj] < 0.05) (Figures 1A, 1B and S1A; Table S1). Pathway analysis confirmed that this method enriched for genes that affect T cell biology and Th1-/Th17-related gene pathways (Enrichr; padj < 0.05) (Figure 1C).
Figure 1. ZEB1 mutations modulate risk for multiple sclerosis and experimental autoimmune encephalitis.
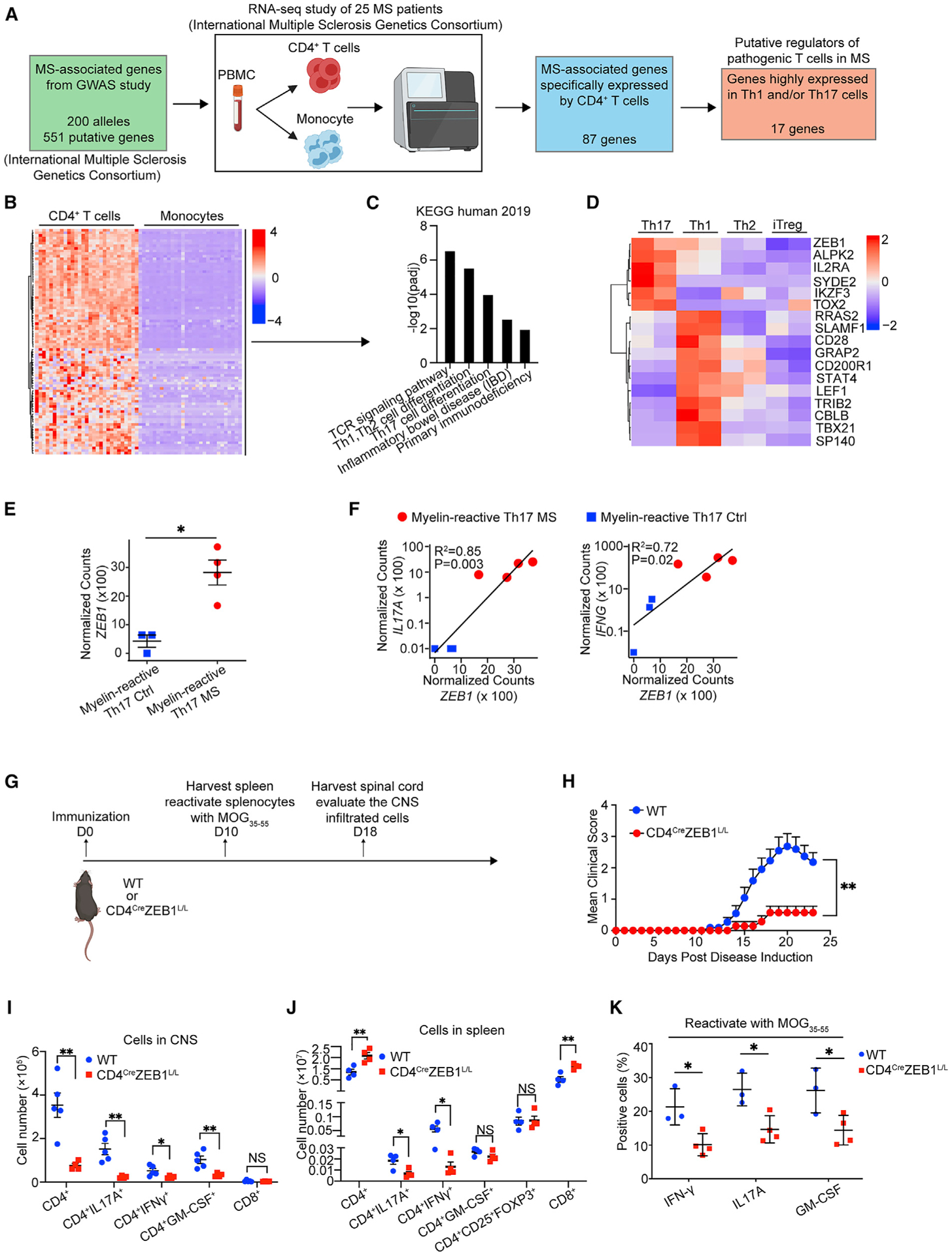
(A) Schematic to identify putative MS-relevant regulators of pathogenic Th1/Th17 differentiation.
(B) Heatmap of MS-associated genes expression in CD4+ T cells and monocytes from MS patients.
(C) Bar plot showing significantly enriched pathways in the genes in (B).
(D) Heatmap of genes highly expressed in Th1 and/or Th17 cells versus Th2 and Treg cells.
(E) Dot plot showing ZEB1 gene expression level in myelin-reactive Th17 cells from MS patients (Th17 MS) and healthy controls (Th17 Ctrl).
(F) Dot plot showing linear correlation of ZEB1 gene expression with IL-17A and IFNG gene expression in myelin-reactive Th17 cells from MS patients (Th17 MS) and healthy controls (Th17 Ctrl).
(G) Schematic displaying the experimental design for experimental autoimmune encephalomyelitis (EAE) model.
(H) Clinical scores of wild-type (WT, n = 11) and CD4CreZEB1L/L (n = 7) mice in EAE. Statistic difference is tested by nonparametric Mann-Whitney test.
(I) Flow cytometry evaluating the number of CD4+ cells, CD4+IL-17A+ cells, CD4+IFN-γ+ cells, CD4+GM-CSF+cells, and CD8+ cells in the CNS of CD4CreZEB1L/L (n = 4) and WT (n = 5) mice at 18 days post immunization.
(J) Flow cytometry evaluating the number of CD4+ cells, CD4+IL-17A+ cells, CD4+IFN-γ+ cells, CD4+GM-CSF+cells, CD4+CD25+FOXP3+ cells, and CD8+ cells in the spleen of CD4CreZEB1L/L (n = 4) and WT (n = 5) mice at 10 days post immunization.
(K) Flow cytometry analysis of cytokine expression in CD4+ T cells from reactivated splenocytes from CD4CreZEB1L/L and WT mice.
The data in (E) and (H)–(K) are presented as the mean ± SEM.
Statistical differences in (F) were tested using Pearson’s correlation coefficient test. All statistical differences in (E) and (I)–(K) were tested using unpaired Student’s t test (two-tailed). *p < 0.05, **p < 0.01.
To identify genes uniquely relevant for MS-relevant Th cells, we focused on genes significantly upregulated in Th17 or Th1 compared to Th2 and iTreg cells (padj < 0.05) (Figures 1D and S1B). This analysis identified 17 genes. This list was enriched for 7 genes (i.e., IKZF3, RRAS2, SLAMF1, CD28, STAT4, and TBX21) that have been functionally validated to be necessary for production of Th1 or Th17 cytokines in relevant human or mouse models. IL-2RA controls GM-CSF production in human Th cells and is associated with MS disease severity (Hartmann et al., 2014; Huang et al., 2014; Kwong et al., 2017; Lee et al., 2017; Linterman et al., 2014; Martínez-Riaño et al., 2019; Quintana et al., 2012).
Among the genes that are highly transcribed in both Th17 and Th1 cells (ZEB1, IL-2RA, and ALPK2) (Figure 1D), we focused on ZEB1 because orthogonal genomic studies suggest its fundamental importance to CD4+ T cell biology (Choi et al., 2015; Scott and Omilusik, 2019). In particular, a recent large-scale small interfering RNA (siRNA) screen suggested an as-yet undefined role for ZEB1 in Th17 cells (Yosef et al., 2013). Moreover, ZEB1 expression is significantly upregulated in myelin-reactive Th17 cells from MS patients compared to controls (Figures 1E and S1C) and is positive-correlated with the expression of IL-17A (R2 = 0.85, p = 0.003) and IFN-γ (R2 = 0.72, p = 0.02) (Figure 1F), respectively.
ZEB1 deficiency in T cells ameliorates EAE
The GWAS study predicts that ZEB1 modulates risk of developing MS. To test this, we subjected mice with conditional loss of ZEB1 specifically in T cells (CD4CreZEB1L/L) or wild-type (WT) controls to experimental autoimmune encephalitis, which recapitulates many aspects of MS (Ifergan et al., 2017) (Figure S2A).
We immunized WT and CD4CreZEB1L/L mice with myelin oligo-dendrocyte glycoprotein (MOG35–55) peptide to initiate EAE (Figure 1G) (Ifergan et al., 2016). Deletion of ZEB1 significantly delayed disease onset and decreased peak severity of EAE. Moreover, 43% of CD4CreZEB1L/L mice showed no signs of EAE (clinical score = 0) during the experiment (n ≥ 7 mice per group, p < 0.01) (Figure 1H). These results indicate that ZEB1 significantly reduced the risk of autoimmune demyelination.
Next, we examined the T cells in the central nervous system (CNS). At day 18 post-immunization, when the mice showed symptoms, the spinal cords from WT or CD4CreZEB1L/L mice were harvested and the infiltrated cells were evaluated. We found a significant and selective decrease in overall numbers of infiltrating CD4+ T cells in CD4CreZEB1L/L mice. Moreover, we found a significant reduction in CD4+ T cells producing IL-17A (6.6-fold), IFN-γ (2.3-fold), and GM-CSF (3.1-fold) from CD4CreZEB1L/L mice, which is consistent with the pathogenic roles of Th1 and Th17 cells in this model (Ifergan et al., 2017) (Figure 1I).
Next, we harvested the spleens of WT and CD4CreZEB1L/L mice and examined splenic T cells at day 10 post immunization (Figure 1G). The results showed that, relative to WT mice, CD4CreZEB1L/L mice had a slightly increased number of total CD4+ and CD8+ T cells (Figure 1J), suggesting that deletion of ZEB1 did not impair T cell viability. However, CD4CreZEB1L/L mice had significantly fewer IL-17A (2.4-fold reduction) and IFN-γ (4.8-fold reduction) producing CD4+ T cells in spleen (Figure 1J). In addition, we observed no difference of total cell number of Tregs (CD4+CD25+Foxp3+) and GM-CSF producing CD4+ T cells in spleens from WT and CD4CreZEB1L/L mice (Figure 1J). Ex vivo recall with MOG35–55 peptide showed decreased IFN-γ (2.1-fold reduction), IL-17A (1.8-fold reduction), and GM-CSF (1.8-fold reduction) cytokine production by splenocytes from CD4CreZEB1L/L mice (Figure 1K).
Mice with germline mutations in ZEB1 have fewer double negative (DN) T cells in thymus, suggesting a critical role in early thymic development (Zhang et al., 2020). We did not find any gross developmental defects with our CD4CreZEB1L/L model wherein Cre is induced during the double positive (DP) thymo-cyte stage. Nonetheless, to control for potential developmental effects induced by ZEB1 loss, we utilized a temporally controlled model (CD4CreErt2ZEB1L/L) (Figure S2B). We employed CD4CreErt2 mice as controls to control for the potential toxicity of Cre recombinase on CD4+ T cells (Higashi et al., 2009; Janbandhu et al., 2014; Schmidt-Supprian and Rajewsky, 2007).
Seven days post tamoxifen treatment, we immunized the CD4CreErt2 and CD4CreErt2ZEB1L/L mice with MOG35–55 peptide to initiate EAE (Figure S2C). Again, we found that, in CD4CreErt2 ZEB1L/L mice, ZEB1 deletion completely prevented (clinical score = 0) disease onset (n ≥ 6 mice per group, p < 0.0001) (Figure S2D). As before, in CNS, there was a significant decrease in overall numbers of CD4+ T cells and CD4+ T cells producing IL-17A (1.4-fold reduction), IFN-γ (1.7-fold reduction), and GM-CSF (1.9-fold reduction) (day 23 post immunization) (Figure S2E). In addition, we observed a modest decrease of IL-4 producing CD4+ T cells and Treg cells infiltration in CNS from CD4CreErt2 ZEB1L/L mice (Figure S2E).
ZEB1 is essential for Th1 and Th17 differentiation in mouse and human
We next queried ZEB1 expression level in T cells. In both humans and mice, ZEB1 levels were low in resting T cells but were dramatically induced by TCR stimulation (Figure S3A). Importantly, ZEB1 is appreciably expressed in both mouse and human Th17 cells and variably expressed in other in vitro differentiation conditions (Figures S3–S3I).
Next, we tested the effects of ZEB1 on mouse Th cells and iTreg in vitro differentiation. We isolated ZEB1-replete and ZEB1-deleted CD4+ naive T cells from WT and CD4CreZEB1L/L and differentiated the cells into Th cells (Th0, Th1, Th2, and Th17) and iTreg using previously described methods (Amsen et al., 2007; Benson et al., 2007; Ifergan et al., 2016) (Figure 2A). Consistent with the EAE model, ZEB1 deletion diminished IL-17A (2.1-fold reduction, n = 8 mice) (Figure 2B) and IFN-γ (1.5-fold reduction, n = 8 mice) (Figure 2C) expression from Th17 and Th1 cells respectively. However, ZEB1 loss increased IL-4 expression from mouse Th2 cells (1.6-fold increase, n = 4 mice, Figure 2D). ZEB1 had no effect on the numbers of iTregs (Figure 2E). The in vitro differentiation experiments using CD4CreErt2 and CD4CreErt2ZEB1L/L mice showed similar results (Figures S4A–S4E).
Figure 2. ZEB1 promotes Th1 and Th17 and inhibits Th2 differentiation in both humans and mice.
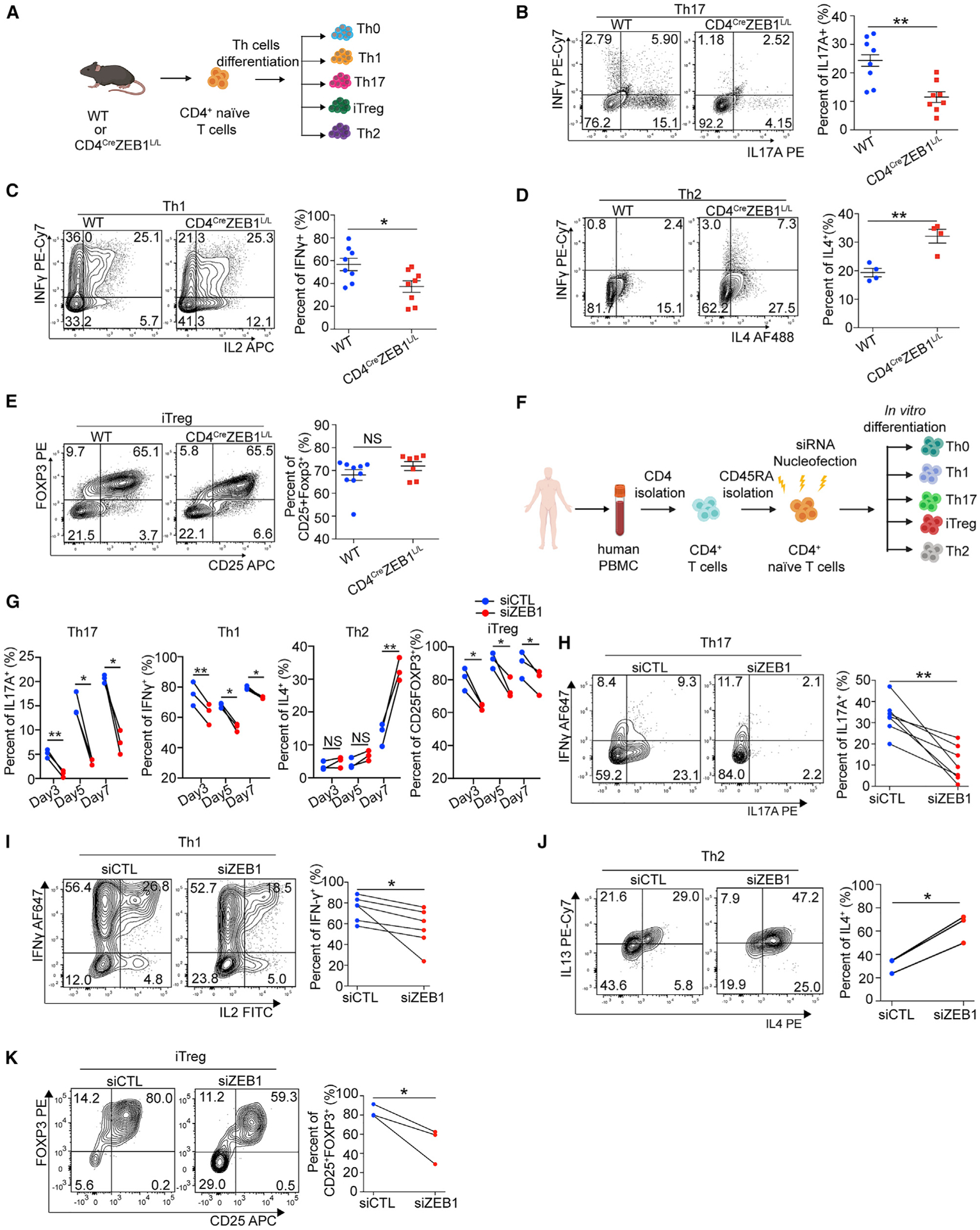
(A) Schematic for testing the role of ZEB1 in mouse Th cell in vitro differentiation.
(B–E) Flow cytometry evaluating Th cell differentiation of CD4+ naive T cells from WT or CD4CreZEB1L/L mice.
(F) Schematic of siRNA nucleofection and differentiation of naive CD4+ T cells under different polarizing conditions.
(G) Flow cytometry evaluating Th cell differentiation of human CD4+ naive T cells nucleofected with siZEB1 or siCTL at days 3, 5, and 7, respectively; n = 3 human donors.
(H–K) Flow cytometry evaluating Th cell differentiation of human CD4+ naive T cells nucleofected with siZEB1 or siCTL at the temporal peak of differentiation conditions.
Symbols in (B)–(E) represent individual mice. Symbols in (G)–(K) represent individual donors. Data in (B)–(E) are expressed as mean ± SEM. All statistical differences in (B)–(E) were tested using unpaired Student’s t test (two-tailed). All statistical differences in (G)–(K) were tested using paired Student’s t test (two-tailed). NS, not significant; *p < 0.05, **p < 0.01.
ZEB1 has previously been described in Jurkat cells to downregulate IL-2, a cytokine that inhibits Th17 differentiation (Quintana et al., 2012; Williams et al., 1991). In primary mouse CD4+ T cells, we did not detect an effect of ZEB1 on IL-2 expression, suggesting the effect of ZEB1 on Th17 cells is IL-2 independent (Figures S4F and S4G).
To determine if the effect is conserved, we performed analogous experiments on human CD4+ T cells. First, we utilized a SMARTpool siRNA system that consists of four siRNAs that target the same gene but have distinct sequences, minimizing off-target effects. These siRNA pools can be applied to human naive CD4+ T cells without prior TCR-stimulation (Figures 2F, S4H, and S4I). From this point on, we will refer to ZEB1-deficient CD4+ T cells (i.e., those transfected with siZEB1) as siZEB1. Isogenic ZEB1-replete CD4+ T cells from the same donor (i.e., those transfected with non-targeting scrambled siRNA controls) will be referred to as siCTL.
Following transfection with siZEB1 or siCTL, human naive CD4+ T cells were placed in different polarizing conditions (Figures 2F and S3D). For the siCTL cells, Th0, Th1, and iTreg differentiation reached peak rapidly (~70%–80% differentiated cell at day 3), whereas Th17 and Th2 differentiation was relatively delayed (5 and 7 days, respectively; n = 3 human donors) (Figures 2G and S4J).
As with mouse T cells, ZEB1 loss in human CD4+ T cells had no effect on proliferation (Figure S4K). However, ZEB1 loss inhibited human Th1, Th17, and increased Th2 differentiation. In contrast to mouse, it also decreased iTreg differentiation (n = 3 human donors) (Figure 2G). At the temporal peak of Th cell differentiation, siZEB1 significantly decreased IL-17A (3-fold reduction, n = 7 donors), IFN-γ (1.3-fold reduction, n = 6 donors), Foxp3 (1.6-fold reduction, n = 3 donors), and increased IL-4 (2.1-fold, n = 3 donors) expression in Th17, Th1, iTreg, and Th2 cells, respectively (Figures 2H–2K). As with mouse T cells, we found that ZEB1 loss did not increase IL-2 expression in human Th cells (Th0 [Figure S4L] and Th1 and Th17 cells [Figures S4M and S4N]), again suggesting that ZEB1-loss modulates human Th17 lineage commitment in an IL-2 independent manner. Collectively, both mouse and human data suggest that ZEB1 has a broad, conserved role in regulating CD4+ Th cell commitment with the strongest effect on Th17 cell differentiation.
ZEB1-deficiency leads to the loss of the canonical Th17 transcriptional program
Next, we performed single cell RNA-seq (scRNA-seq) to elucidate the effects of ZEB1 on cellular identity. For these experiments, we harvested human CD4+ T cells that were cultured under Th17 conditions for 5 days after nucleofection with either siZEB1 or siCTL. Analysis of these cohorts demonstrated that siZEB1 Th17 and siCTL Th17 cells clustered separately, suggesting a divergence in cell fate (Figure 3A). The siZEB1 cells were marked by upregulation of known target genes of the ZEB1 transcriptional repressor (EPCAM and CDH1) (Figure 3B). The siCTL cells were enriched for Th17 genes including Th17-associated transcription factors (RORC and RORA) and cell surface markers (IL-23R) (Figure 3B).
Figure 3. ZEB1 knockdown blocks global acquisition of the Th17 gene expression signature.
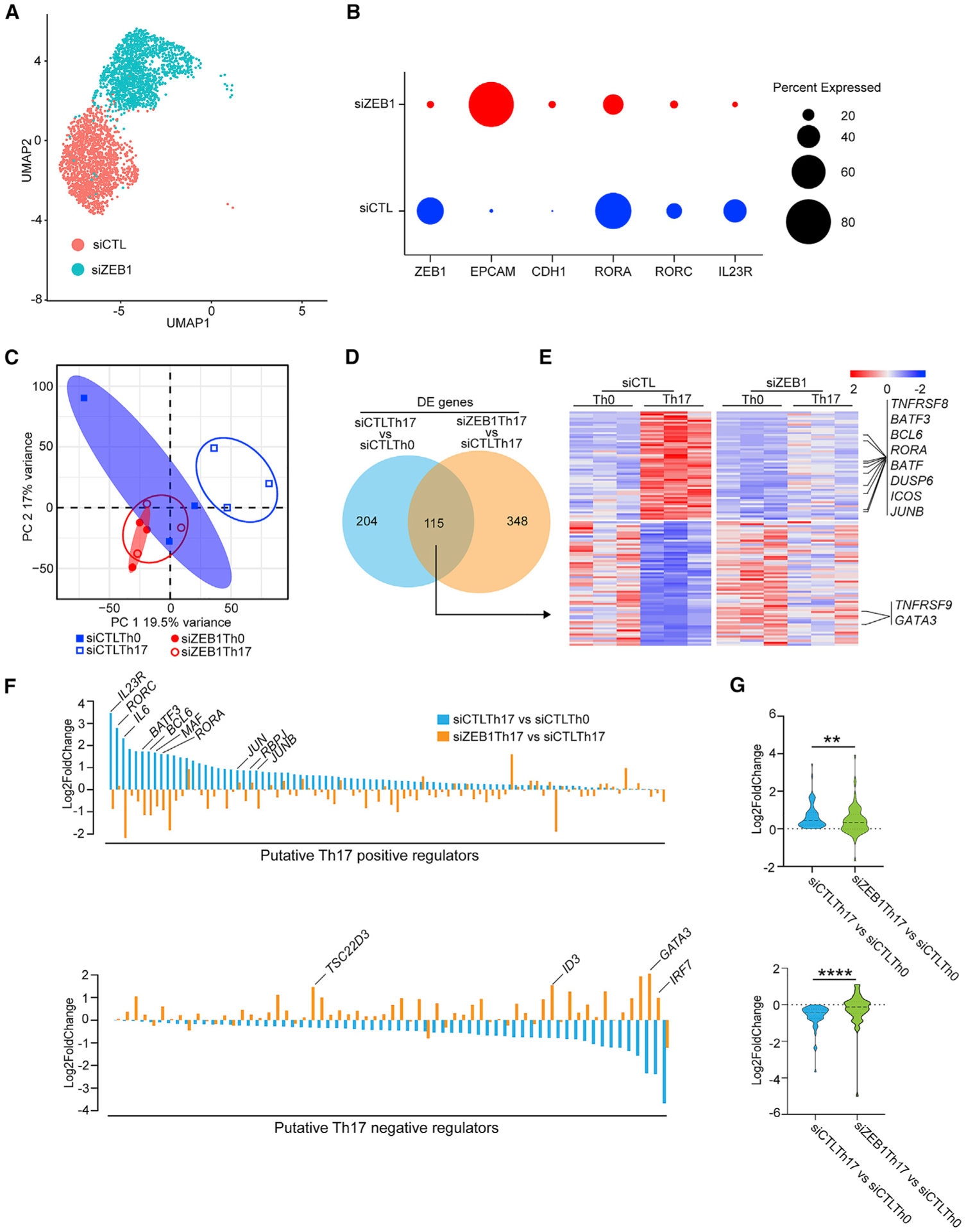
(A) Uniform manifold approximation and projection (UMAP) plot representing merged single-cell transcriptomes of siZEB1 Th17 (1699) and siCTL Th17 (1520) cells.
(B) Dot plot depicting selected scRNA-seq marker genes in siCTL and siZEB1 clusters. Dot size represents percentage of cells expressing the gene.
(C) PCA plot of the bulk transcriptomes of Th0 and Th17 cells in siZEB1 and siCTL cohorts (n = 3 independent experiments from 3 different healthy donors).
(D) Venn diagram displaying overlapped genes between differentially expressed (DE) genes in bulk RNA-seq of siCTL Th17 versus siCTL Th0 and siZEB1 Th17 versus siCTL Th17 cells.
(E) Heatmap of genes overlapped in (D).
(F) ZEB1-dependent changes in the expression of putative Th17 regulator genes.
(G) Violin plots of the expression changes between siCTL-treated Th0 cells and siCTL- or siZEB1-treated Th17 cells.
(A and B) Data are from scRNA-seq. (C–G) Data are from bulk RNA-seq.
p values in (D) were calculated using Wald test and adjusted for multiple-hypothesis testing using the Benjamini-Hochberg method. p values in (G) were calculated using paired Student’s t test (two-tailed). **p < 0.01, ****p < 0.0001.
We next generated bulk RNA-seq datasets on siZEB1 and siCTL CD4+ T cells cultured in Th17 (or control Th0) conditions because of its superior ability to detect and quantify gene transcripts (Chen et al., 2019). By principal component analysis (PCA), siCTL Th0 and siCTL Th17 cells clustered separately. These data suggest that there is a corresponding divergence in their cell fate during Th17 differentiation (Figure 3C). In contrast, siZEB1 Th0 and siZEB1 Th17 failed to cluster separately and instead clustered with the siCTL Th0 cells (Figure 3C). Collectively, these data suggest a global failure of siZEB1 Th17 cells to acquire the Th17 transcriptional program.
At the individual gene level, ZEB1 loss downregulated the expression of 54 genes normally upregulated by Th17 conditions (Figures 3D and 3E). These include canonical Th17 genes such as TNFRSF8, BATF, BATF3, BCL6, RORA, DUSP6, ICOS, and JUNB (padj < 0.05) (Figures 3D and 3E; Table S2). In contrast, ZEB1-loss significantly increased the expression of 61 genes normally suppressed by Th17 differentiation conditions (including negative regulators of Th17 differentiation TNFRSF9 and GATA3) (padj < 0.05) (Figures 3D and 3E; Table S2).
To explore this further, we assessed the effects of ZEB1-loss on the expression of an independently generated list of genes that putatively promote or inhibit Th17 differentiation (Yosef et al., 2013). We classified the genes in this list as putative positive (88 genes) or negative (63 genes) regulators of Th17 differentiation based on their relative expression in Th17 cells compared to Th0 controls (Figure 3F; Table S3).
Globally, siZEB1-treated cells showed a lower expression level of Th17 putative positive regulators (p < 0.01, upper panel) and a higher expression level of putative negative regulators (p < 0.0001, lower panel) (Figure 3G). Collectively, these results suggest that ZEB1 controls the expression of a broad range of Th17 regulators although the mechanisms were not clear.
ZEB1 deficiency downregulates STAT3 phosphorylation
Because the effects on Th17 differentiation are conserved across species, we hypothesized a common mechanism. We hypothesized ZEB1 targets would be similarly regulated by ZEB1 in both humans and mice (Figure 4A).
Figure 4. ZEB1 deficiency inhibits STAT3 phosphorylation during Th17 differentiation.
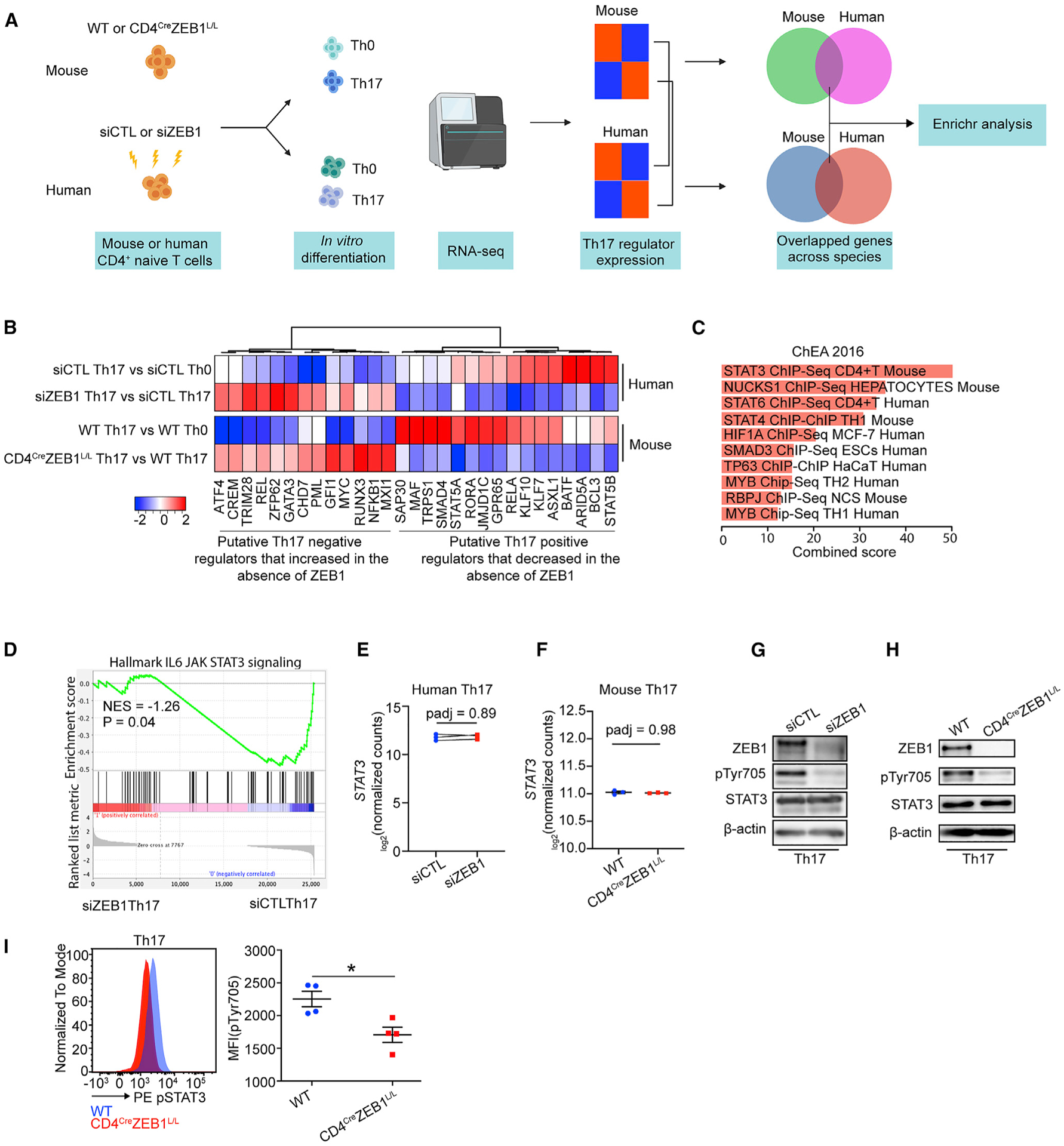
(A) Schematic of the experimental design to identify conserved effects of ZEB1 on both human and mouse Th17 transcriptional programs.
(B) Heatmap showing the expression changes of concordant ZEB1-dependent putative Th17 regulators in both human and mice.
(C) ChEA of conserved ZEB1-dependent Th17 regulator genes in (B) (p < 0.05).
(D) GSEA plot showing the effect of siZEB1 on JAK-STAT3 signaling in Th17 cells (p = 0.04). NES, normalized enrichment score.
(E and F) Transcription level of STAT3 in (E) human siCTL- and siZEB1-treated cells or (F) mouse WT and CD4CreZEB1L/L cells in Th17 condition.
(G and H) Western blots showing expression of total STAT3 and phospho-STAT3 (phosphorylation site: Tyr705) in human siCTL- or siZEB1-treated cells (G) and mouse WT or CD4CreZEB1L/L cells (H) under Th17 conditions.
(I) Flow cytometry results showing pTyr705 expression in mouse WT or CD4CreZEB1L/L cells under Th17 conditions.
A Representative result of at least two individual experiments is shown in (G) and (H). Symbols in (E) represent individual experiments. Symbols in (F) and (I) represents individual mice. The data in (F) and (I) are expressed as mean ± SEM. p values in (E) and (F) were calculated using Wald test and adjusted using the Benjamini-Hochberg method. p value in (I) was calculated using unpaired Student’s t test (two-tailed). *p < 0.05.
Thus, we generated RNA-seq data from naive CD4+ T cells from CD4creZEB1L/L or WT mice cultured in Th0 and Th17 differentiation conditions. In line with humans, naive CD4+ T cells from CD4creZEB1L/L mice broadly failed to acquire a Th17 transcriptional signature despite Th17 polarizing conditions (Figures S5A and S5B). Nonetheless, the transcription profiles of putative Th17 regulator genes between mice and humans were not identical (Figures S5C and S5D). Only 16 putative positive regulators (e.g., ARID5A, BATF, GPR65, RORA, and MAF) and 13 putative negative regulators (e.g., GFI1, GATA3, and ATF4) were similarly regulated across both species (Figure 4B; Table S4).
To identify pathways that regulate this conserved list of ZEB1-dependent Th17 regulators, we performed chromatin immuno-precipitation (ChIP) enrichment analysis (ChEA) (Figure 4C). This program examines cis-acting transcription factor (TF) binding sites that are found in gene sets more often than expected by chance (Lachmann et al., 2010). Unexpectedly, ZEB1 was not enriched in this gene set even though ZEB1 is itself a TF. Instead, the top hit was STAT3 (padj < 0.05) (Figure 4C). Additional hits in the top 10 included STAT4 and STAT6 which have similar consensus binding sites (Li et al., 2016). Other hits including SMAD3, HIF1A, and MYB either interact with STAT3 or are downstream targets of STAT3 (Dang et al., 2011; Levy and Lee, 2002; Yoon et al., 2015).
Because STAT3 is an important proximal positive regulator for Th17 differentiation, these data may explain the global failure of Th17 differentiation in the absence of ZEB1 (Tripathi et al., 2017). Thus, we performed gene set enrichment analysis (GSEA), which confirmed that STAT3 gene targets are significantly depleted in siZEB1 Th17 cells (p < 0.05) (Figure 4D) (Mootha et al., 2003; Subramanian et al., 2005).
To assess mechanisms, we first examined the effects of ZEB1 on STAT3 expression. However, we found that STAT3 expression was not affected by ZEB1 in either human or mouse Th17 cells (Figures 4E–4H). We then hypothesized that the effects of ZEB1 on STAT3 would be post-translational. Phosphorylation of STAT3 is critical for its dimerization, nuclear translocation, and activation of its downstream targets (Levy and Lee, 2002). By western blot, we observed that pSTAT3 (pTyr705) (but not total STAT3) was dramatically decreased in both ZEB1-deficient human (Figure 4G) and mouse Th17 cells (Figures 4H and 4I).
ZEB1 deficiency reduced JAK2 expression in Th17 and Th1 cells
To refine the mechanisms leading to pSTAT3 downregulation, we conducted parallel biochemical and RNA-seq time courses. First, we performed a time course western blot using antibodies that detected STAT3 phosphorylation sites induced by MAPK (pSTAT3 [pSer727]) or JAK (pSTAT3 [pTyr705]), respectively (Kopantzev et al., 2002; Lim and Cao, 2001). The results showed that from 12 h to 72 h, the expression of ZEB1 and expression of both pSTAT3 (pTyr705) and pSTAT3 (pSer727) increased in Th17 siCTL cells over time (Figure S6A). Interestingly, pSTAT3 (pTyr705), but not pSTAT3 (pSer727), was selectively decreased in siZEB1 cells. These results indicated that in human Th17 cells, ZEB1 is necessary for JAK-mediated phosphorylation of STAT3 (Figure S6A). The difference in pSTAT3 begins at 24 h (i.e., time of onset of ZEB1 expression in siCTL Th17 cells) and becomes more pronounced subsequently (Figure S6A). Therefore, ZEB1-dependent effects likely occur between 24 and 48 h, which was confirmed by a time course RNA-seq experiment (Figure S6B).
Time-lapse analyses identified 3 clusters of genes that were most dynamically altered over time between siCTL Th17 and siZEB1 Th17 cells (clusters 1–3, false discovery rate [FDR] <0.05) (Figure S6C; Table S5). Cluster 1 increased over time in siZEB1 cells (Figure S6C). This cluster was enriched for canonical ZEB1 targets with known ZEB1 binding sites near their promoters (i.e., CDH1, EPCAM, and LSR) (Figures S6D and S6E). Because ZEB1 is a transcriptional repressor, the increase in expression of ZEB1 targets in the absence of ZEB1 is an expected outcome.
In contrast, clusters 2 and 3 contain genes that were induced in siCTL Th17 but not in siZEB1 Th17 cells (Figure S6C). These two clusters contained multiple Th17 cytokines (i.e., IL-23R, IL-17A, IL17F, IL6, and IL-26) and positive regulators of Th17 differentiation (i.e., JUNB and RORA) (Figure S6D). They were also enriched for Th17-related pathways and JAK-STAT signaling pathways (Enrichr, padj < 0.05) (Figure S6F). However, there was no enrichment of ZEB1 or other transcription factor binding sites.
Interestingly, cluster 2 included a known regulator of STAT3 signaling, JAK2. JAK2 is a kinase that phosphorylates STAT3 in response to IL-23 and IL-6 in Th17 cells (Park et al., 2014; Teng et al., 2015). Other JAK family members were not affected by ZEB1 loss at the mRNA or protein level in human Th17 cells (Figures 5A and 5C). This effect was confirmed in human Th1 cells and mouse Th1 and Th17 cells (Figures 5A–5F).
Figure 5. ZEB1 promotes durable JAK2 expression in Th1 and Th17 cells.
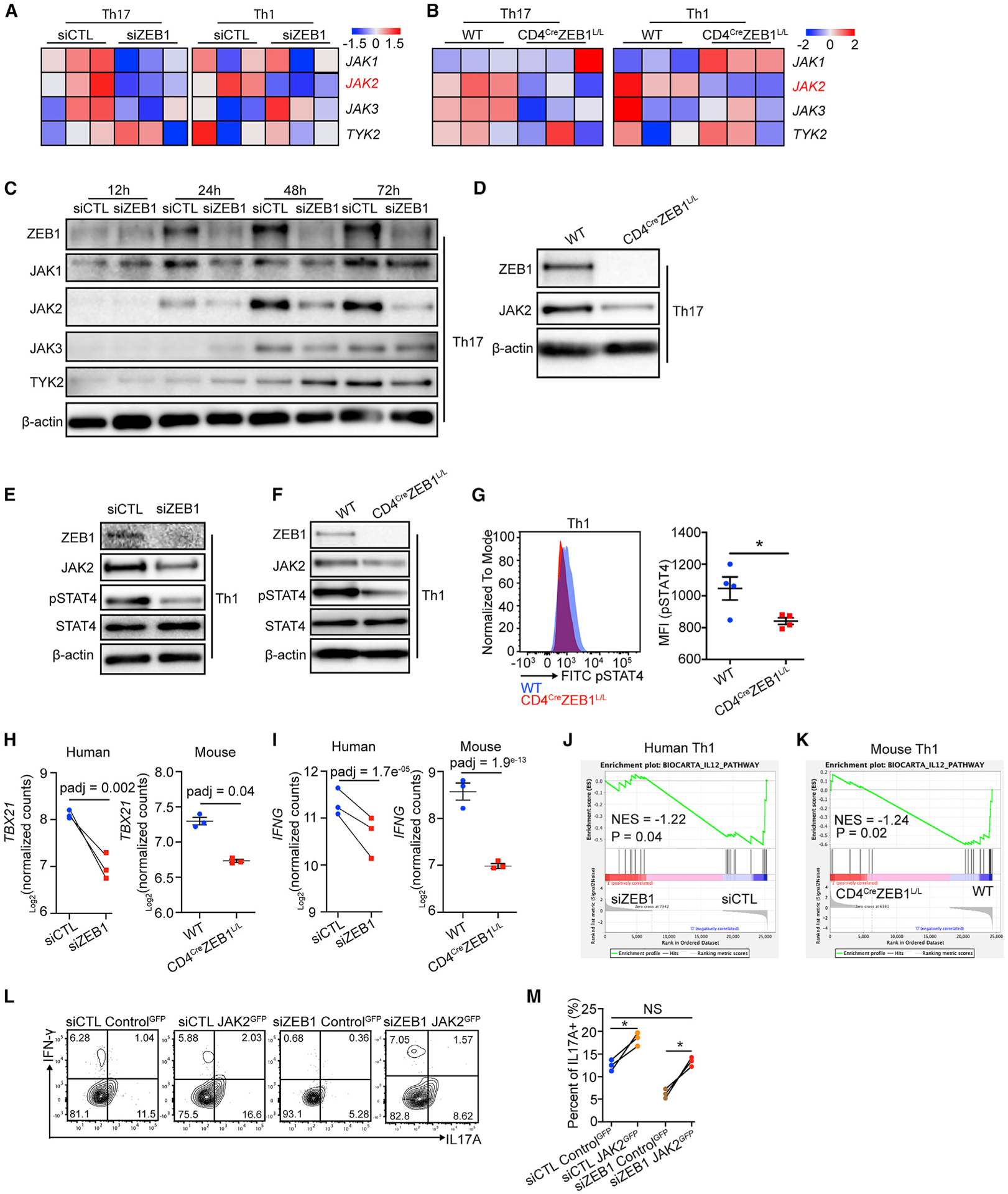
(A and B) Heatmap for JAK family gene expression in Th1 and Th17 conditions from siCTL and siZEB1 cohort (human, (A) or WT and CD4CreZEB1L/L mice (B).
(C) Western blot showing the expression of JAK family protein in human CD4+ T cells in Th17 conditions from siCTL and siZEB1 cohort at 12, 24, 48, and 72 h after nucleofection.
(D) Western blot showing JAK2 expression in Th17 cells from WT and CD4CreZEB1L/L mice.
(E and F) Western blot showing JAK2, total STAT4, and phosphorylated-STAT4 [pSTAT4 (pTyr693)] expression in Th1 cells from human siCTL and siZEB1 cohorts (E) or WT and CD4CreZEB1L/L mice (F).
(G) Flow cytometry showing pSTAT4 expression in Th1 cells from WT and CD4CreZEB1L/L mice.
(H and I) GSEA plot demonstrating the effect of ZEB1 loss on the expression of an IL12-STAT4 gene signature in (H) human and (I) mouse CD4+ Th1 cells. NES, normalized enrichment score.
(J and K) Transcription level of TBX21 and IFNG in (J) human siCTL- and siZEB1-treated cells or (K) mouse WT and CD4CreZEB1L/L cells in Th1 conditions.
(L and M) Flow cytometry evaluating JAK2 re-expression on Th17 differentiation in siZEB1 nucleofected CD4+ naive T cells. The contour plots (gated by GFP+) are representative 3 different human donors, summarized in (M).
Representative results from at least two individual experiments are shown in (C)–(F).
Statistical differences in (G) were determined using unpaired Student’s t test (two-tailed). Statistical differences in (M) were tested using paired Student’s t test (two-tailed). NS, not significant; *p < 0.05. p values in (H) and (I) were calculated using Wald test and adjusted using the Benjamini-Hochberg method.
In Th1 conditions, IL-12 activates JAK2, which is the kinase for STAT4, a transcription factor that regulates the expression of the Th1 master regulator T-BET and downstream cytokines such as IFN-γ. We therefore hypothesized an analogous mechanism by which ZEB1 affects Th1 cells. Indeed, we found a decrease of pSTAT4 (pTyr693) expression with ZEB1-loss in both human and mouse Th1 cells (Figures 5E–5G).
In line with this model, ZEB1 loss caused the downregulation of pSTAT4 targets in Th1 cells (TBX21 and IFNG) (Figures 5H and 5I). GSEA analysis confirmed that STAT4 gene signature expression requires ZEB1 in both human and mouse Th1 cells (Figures 5J and 5K).
We thus hypothesized that JAK2 loss would phenocopy the ZEB1 effects. As expected, siJAK2 reduced JAK2 levels and subsequent pSTAT3 (pTyr705) and pSTAT4 (pTyr693) in Th17 and Th1 conditions, respectively (Figure S6G). Consistent with our model, siJAK2, like siZEB1, significantly reduced IL-17A (fold change = 2.8, n = 3 human donors) and IFN-γ (fold change = 2.2, n = 3 human donors) expression in Th17 and Th1 cells, respectively (Figures S6H and S6I).
To determine if JAK2 expression is necessary for the ZEB1 effects, we co-transfected naive CD4+ T cells with a vector controlGFP or a vector expressing JAK2 cDNA and siRNAs (siCTL or siZEB1) into human CD4+ naive T cells before Th17 and Th1 polarization. The results showed that, compared to controlGFP (in both siCTL and siZEB1 cells), JAK2 re-expression restored IL-17A expression in siZEB1 cells (Figures 6L, 6M, S6J, and S6K) (n = 3 human donors). We found similar results for the JAK2 re-expression on IFN-γ expression in siZEB1 cells in Th1 differentiation (Figures S6L and S6M) (n = 3 human donors). These data confirmed that the reduction of IL-17A and IFN-γ in siZEB1 cells in Th17 and Th1 differentiation is due to decreased JAK2 expression.
Figure 6. ZEB1 promotes Th17 differentiation by repressing miR-101-3p that inhibits JAK2 expression.
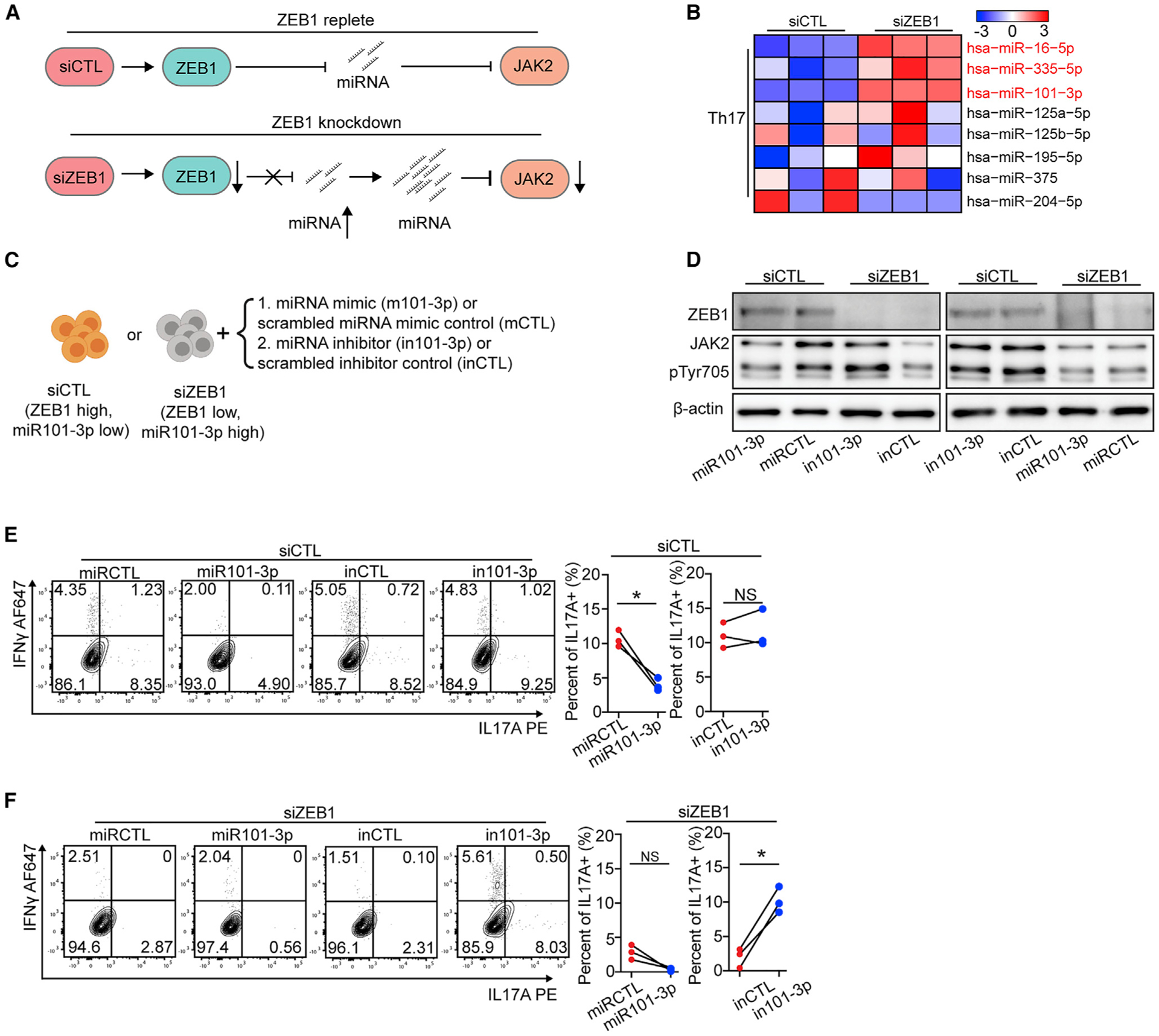
(A) Schematic displaying a model by which ZEB1 promotes JAK2 expression through repressing miRNA expression.
(B) Heatmap showing the expression of miRNAs, which putatively target JAK2, in Th17 cells from siCTL and siZEB1 cohorts (miRNAs with a padj < 0.1 were marked in red).
(C) Schematic of the experimental design to assess the necessity and sufficiency of siZEB1-dependent loss of JAK2 in Th17 differentiation condition.
(D) Western blots show that miR-101-3p is necessary and sufficient for siZEB1-dependent loss of JAK2 under Th17 differentiation conditions. Representative result from 3 independent experiments.
(E and F) Flow cytometry evaluating Th17 differentiation of human CD4+ naive T cells transfected with siCTL (E) or siZEB1 (F) and miRNA mimic (m101-3p or scrambled control, mCTL) or a hairpin inhibitor (in101-3p or scrambled control, inCTL). The contour plots are representative 3 different human donors, summarized in the graph at right.
All statistical differences in (E) and (F) were tested using paired Student’s t test (two-tailed). NS, not significant; *p < 0.05, **p < 0.01.
miR-101-3p is necessary and sufficient to phenocopy ZEB1 loss in human Th17 cells
The mechanism by which ZEB1 regulates JAK2 expression was obscure. First, ZEB1 is a transcriptional repressor whose loss is predicted to upregulate target genes (Spaderna et al., 2008). In addition, there is no ZEB1 binding site near the JAK2 promoter (2,000 bp up or downstream of the transcriptional start site [TSS]; ZEB1 binding site was evaluated by Find motif, IGV). Thus, we hypothesized that ZEB1’s effects on JAK2 expression were indirect. We therefore looked for a putative ZEB1 target, which would be upregulated after ZEB1 loss and could induce JAK2 mRNA downregulation. Based on the literature, a miRNA that controls JAK2 expression might be the ideal target (Figure 6A) (Ding et al., 2010; Navarro et al., 2009). To test this, we performed miRNA sequencing. The PCA analysis suggested that the miRNA transcriptomes clustered similarly to the mRNA transcriptomes (Figures S7A and S7B; Table S6).
To identify miRNAs that may target JAK2, we integrated our miRNA-seq data with miRTarBase that links miRNAs with target genes (Chou et al., 2018). This analysis identified 3 putative JAK2-targeting miRNAs that are upregulated in siZEB1 cells (padj < 0.1) (Figure 6B, Table S6). Among these miRNAs, miR-101-3p was the only one with a ZEB1 binding site near its promoter (71 bp downstream of TSS) (Figure S7C). Examination of the literature suggested that miR-101 was a viable candidate because there is a miR-101 binding site in the 3′-UTR of the JAK2 mRNA (Figure S7D). miR-101 expression was also shown in breast cancer cells to be sufficient to downregulate JAK2 expression (Wang et al., 2014). Moreover, miR-101-3p showed a decreased expression and protective effect in MS clinical data (Magner et al., 2016; Regev et al., 2017).
We then tested whether miR-101-3p is necessary and sufficient for the ZEB1 effect. Before Th17 and Th1 differentiation, we co-transfected a miRNA mimic (m101-3p or scrambled control, mCTL) or a hairpin inhibitor (in101-3p or scrambled control, inCTL) to miR-101-3p and siRNAs (siCTL or siZEB1) into human CD4+ naive T cells (Figure 6C). From the aforementioned results, we know that the ZEB1-replete siCTL cells expressed low level of miR101-3p. m101-3p decreased JAK2 expression, downstream STAT3 phosphorylation, and subsequent IL-17A expression (2.7-fold reduction, n = 3 human donors) in siCTL cells (Figures 6D and 6E). The hairpin inhibitor (in101-3p) binds the complimentary strand of the miR101-3p, inhibiting its ability to bind its mRNA target. In siCTL cells, nucleofection of in101-3p or inCTL showed no effect on JAK2 expression, STAT3 phosphorylation, or Th17 differentiation (Figures 6D and 6E).
In siZEB1 cells, the nucleofection of in101-3p restored JAK2 expression, STAT3 phosphorylation, and IL-17A expression (Figures 6D, 5.2-fold increase, n = 3 human donors, and 6F). We found similar effects of m101-3p and in101-3p on JAK2 expression, STAT4 phosphorylation, and IFN-γ production in siCTL and siZEB1 cells in Th1 differentiation (Figures S7E–S7G). Together, these results revealed that miR-101-3p is a potential negative regulator of Th17 and Th1 differentiation that is necessary and sufficient to phenocopy the ZEB1 effects on JAK2. Of note, the experiments in Figures 5L, 5M, and S6J–S6N employed a JAK2 cDNA that lacked its native 3′UTR and therefore its miR-101-3p binding sites.
ZEB1 and JAK2 are potential targets for the treatment of MS
Next, we tested whether the ZEB1-JAK2 axis may possibly serve as targets for MS treatment. Consistent with our findings in vitro, we observed a strong positive correlation between ZEB1 and JAK2 transcript expression in myelin-reactive Th17 cells (Cao et al., 2015) (Figure 7A). In addition, myelin-reactive Th17 from MS patients showed an enrichment of both ZEB1-associated Th17 signature genes (54 genes) (Table S2) and STAT3 target genes (Figures S7H and S7I). These results suggest that ZEB1 may be required not only for de novo Th17 differentiation but also for continued expression of functional cytokines in Th17 cells.
Figure 7. ZEB1 and JAK2 are potential targets for the treatment of MS.
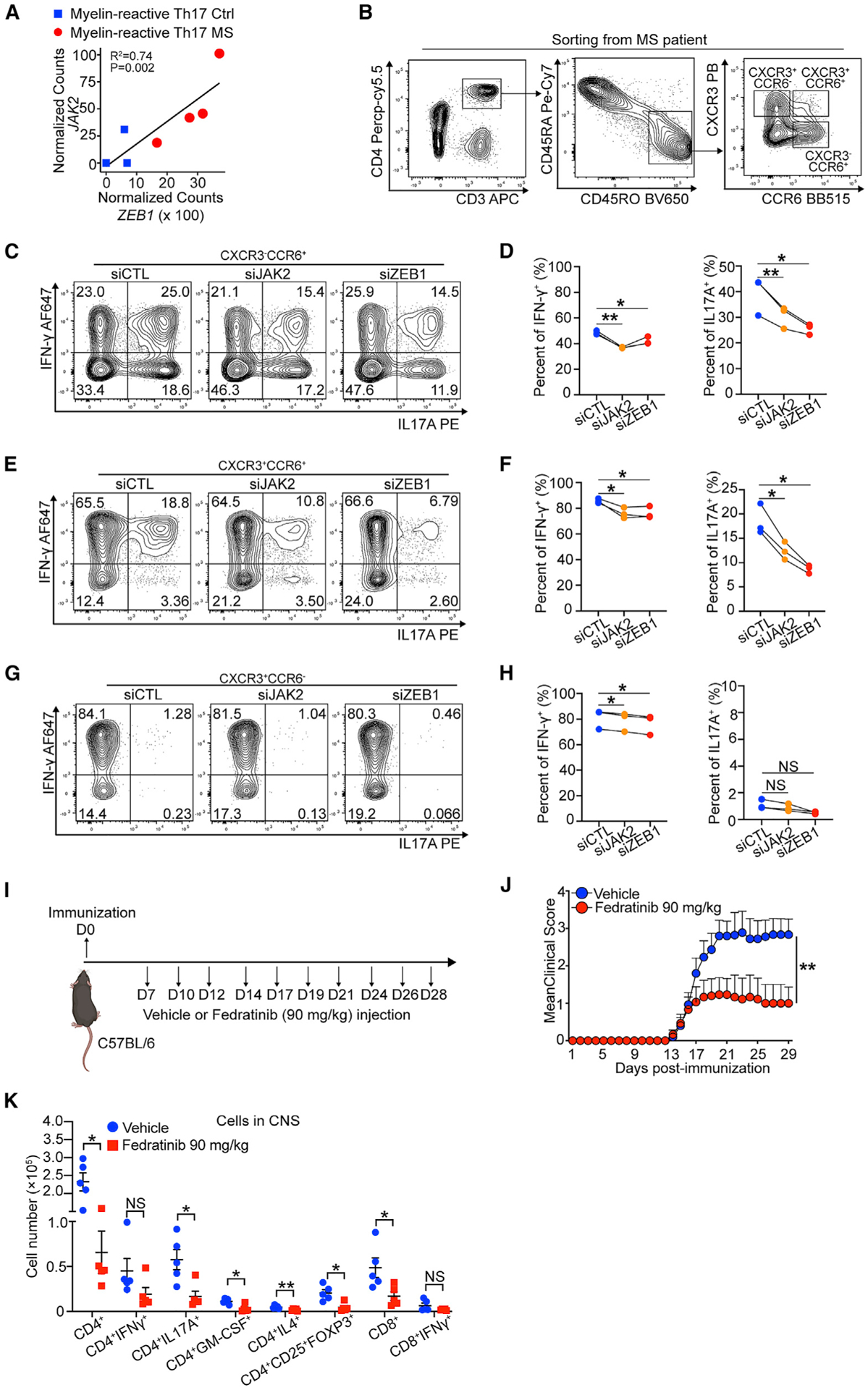
(A) Dot plot showing linear correlation of ZEB1 gene expression with JAK2 gene expression in myelin-reactive Th17 cells from MS patients (Th17 MS) and healthy controls (Th17 Ctrl).
(B) Contour plots showing sorting strategy for CD3+CD4+CD45RO+CCR6−CXCR3+ (Th1), CD3+CD4+CD45RO+CCR6+CXCR3+ (Th1/Th17), and CD3+CD4+CD45RO+CCR6+CXCR3− (Th17) cells from PBMC of MS patients (Silveira-Mattos et al., 2019).
(C–H) Flow cytometry evaluating IFN-γ and IL-17A expression in CCR6−CXCR3+ (Th1), CCR6+CXCR3+ (Th1/Th17), and CCR6+CXCR3− (Th17) transfected with siCTL, siJAK2, or siCTL. n = 3 different MS patients.
(I) Schematic displaying the experimental design for treating EAE using fedratinib.
(J) Clinical scores of C57BL/6 mice treated with vehicle or fedratinib (90 mg/kg) in EAE (n = 15 mice per group). Statistic difference is tested by nonparametric Mann-Whitney test.
(K) Flow cytometry evaluating the number of CD4+ cells, CD4+IFN-γ+ cells, CD4+IL-17A+ cells, CD4+GM-CSF+cells, CD4+IL4+ cells, CD4+CD25+FOXP3+ cells, CD8+ cells, and CD8+IFN-γ+ cells in the CNS of mice treated with vehicle or fedratinib at 29 days post immunization.
All statistical differences in (D), (F), (H), and (K) were tested using paired Student’s t test (two-tailed). NS, not significant; *p < 0.05, **p < 0.01.
To test our hypothesis, we sorted CD3+CD4+CD45RO+ CCR6−CXCR3+, CD3+CD4+CD45RO+CCR6+CXCR3−, and CD3+ CD4+CD45RO+CCR6+CXCR3− cells from 3 relapsing-remitting multiple sclerosis (RRMS) patient peripheral blood mononuclear cells (PBMCs) (Silveira-Mattos et al., 2019) (Figure 7B; Table S7). These cells represent Th1, Th17, and mixed Th1/Th17 cells, respectively. Then, we nucleofected the cells with siCTL, siZEB1, or siJAK2 ex vivo, respectively. We found that, comparing to siCTL, siZEB1 significantly decreased expression of IL-17A from CCR6+IL-17-producing cells (CXCR3−CCR6+ cells: 1.5-fold reduction, Figures 7C and 7D; CXCR3+CCR6+ cells: 2.1-fold reduction, Figures 7E and 7F). siJAK2 showed a similar effect (CXCR3−CCR6+ cells: 1.3-fold reduction, Figures 7C and 7D; CXCR3+CCR6+ cells: 1.5-fold reduction, Figures 7E and 7F). In all cell types, both siZEB1 and siJAK2 had significant but smaller effect on IFN-γ expression (Figures 7C–7H). Consistent with our previous results, western blots showed that both siZEB1 and siJAK2 again decreased JAK2 and decreased pSTAT3 (pTyr705)/pSTAT4 (pTyr693) expression in CXCR3+ and/or CCR6+ cells, respectively (Figure S7J).
Last, we tested the efficacy of inhibitors of this ZEB1-JAK2 pathway in an in vivo model. Because ZEB1 is not immediately targetable with drugs currently available in the clinic, we utilized fedratinib, a JAK2 inhibitor Food and Drug Administration (FDA)-approved for the treatment of adult patients with intermediate-risk or high-risk myelofibrosis (MF) (Mullally et al., 2020). We initiated the EAE model in WT mice. 7 days after immunization, we performed intraperitoneal (i.p.) treatment with 90 mg/kg fedratinib or vehicle controls (Figure 7I). At this time point, the antigen-specific T cells are primed, proliferating, and infiltrating the CNS (Saligrama et al., 2019; Shahi et al., 2019). We observed that fedratinib significantly reduced EAE disease severity (n = 15 mice per group, Figure 7J). Moreover, we found a significant decrease in overall numbers of CD4+ T cells and CD4+ T cells producing IL-17A (3.4-fold reduction) and GM-CSF (3.5-fold reduction) in the CNS. Other cell types that are less prevalent in the CNS, including IL-4 producing CD4+ T cells and Treg cells, were also significantly decreased (Figure 7K). Together, these results demonstrated JAK2 can be a potential target for MS treatment in vivo.
DISCUSSION
To better understand disease-relevant T helper differentiation, we studied multiple sclerosis, which is a debilitating organ-specific autoimmune disease propagated by pathogenic Th1/Th17 cells. Using an unbiased genetic approach, we identified ZEB1 as a regulator of MS-associated inflammation. Correspondingly, ZEB1 loss in T cells functionally ameliorates the development and the severity of EAE in a mouse model of MS.
Mechanistically, ZEB1 promotes both Th1 and Th17 differentiation but inhibits Th2 cytokine production in CD4+ T cells. Unexpectedly, ZEB1 does so by fine-tuning JAK-STAT signaling. JAK-STAT signaling is a broadly conserved pathway required for Th cell differentiation. Specificity is determined by the selective expression and activation of specific JAK or STAT isoforms (Villarino et al., 2017). Often, these JAK-STAT proteins activate competing transcriptional programs. For example, gain-of-function mutations in STAT1 in humans promote chronic fungal infections by inhibiting STAT3-dependent Th17 differentiation (Liu et al., 2011).
ZEB1, on the other hand, promotes both “competing” pro-inflammatory transcriptional programs. In CD4+ T cells, TCR engagement upregulates ZEB1 expression, which in turn downregulates a JAK2-targeting miRNA, miR101-3p. Therefore, in the presence of ZEB1, durable JAK2 expression enables phosphorylation of its pro-Th1 or pro-Th17 STAT targets in response to relevant cytokines in the microenvironment. Consistent with its role as a positive regulator of MS, the ZEB1 targets we identify here are dysregulated “appropriately” in MS. The ZEB1 target, miR-101-3p, is downregulated in the blood of patients with MS (Magner et al., 2016; Regev et al., 2017). Correspondingly, the miR-101-3p target, JAK2, is overexpressed in T cells isolated from MS patients (Conti et al., 2012).
ZEB1 thus modulates Th cell fate not by forcing cells to choose between Th1 and Th17 differentiation programs. Instead, it promotes both Th1 and Th17 differentiation at the expense of other anti-inflammatory transcriptional programs such as Th2. We thus hypothesize that ZEB1 acts as a differentiation checkpoint important for maintaining the healthy balance between pro-inflammatory Th cells required for defense against pathogenic microbes and overactive Th cells that promote autoimmune disease.
Because some autoimmune diseases, such as MS, are characterized by both Th1 and Th17 inflammation, the ZEB1-miR-101-3p-JAK2 signaling axis has potential therapeutic implications. Although small molecule non-specific pan-JAK inhibitors have shown efficacy in the treatment of autoimmune diseases, a JAK2-specific inhibitor may have improved therapeutic efficacy in MS (Banerjee et al., 2017). These data are supported by not only our data but also by studies with early-generation, non-specific JAK2 inhibitors, which were shown to ameliorate preclinical mouse models of MS (Constantin et al., 1999).
Last, our data provide insights into the role of naturally occurring ZEB1 mutations in another human disease. In Sezary syndrome, a T cell lymphoma, the tumor cells are mature CD4+ T cells of a Th2 phenotype. This Th2 tumor phenotype is thought to benefit the tumor by suppressing the host’s ability to mount pro-inflammatory and hence anti-lymphoma immune responses (Guenova et al., 2013). In the majority of patients (~65%), ZEB1 is inactivated by point mutations or copy number deletions (Choi et al., 2015; Park et al., 2017). Based on our data, we predict that ZEB1 loss may promote T cell lymphomagenesis by enforcing this pro-tumor, anti-inflammatory Th2 cell state and downregulating the expression of pro-inflammatory, anti-tumor Th1 or Th17 cytokines.
Collectively, our findings identify a mechanism regulating pathogenic Th differentiation in human disease and highlight the potential for ZEB1, miR-101-3p, and JAK2 as therapeutic targets for both T cell malignancies and organ-specific autoimmune diseases.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Jaehyuk Choi (jaehyuk.choi@northwestern.edu).
Materials availability
All unique/stable reagents generated in this study are available from the lead contact with a completed materials transfer agreement.
Data and code availability
Single-cell RNA-seq data, bulk RNA-seq data and microRNA-seq data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. Other data reported in this paper will be shared by the lead contact upon request. This paper does not report original code. All code and software used is open and freely available. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Relapsing-remitting multiple sclerosis patient samples
Fresh peripheral blood samples from de-identified RRMS patients were collected under the approvement by the Northwestern University Institutional Review Board. Their clinical characteristics are summarized in Table S7.
Mice
C57BL/6, CD4Cre and CD4CreErt2mice were purchased from Jackson Laboratory. ZEB1flox/flox (ZEB1L/L) mice were provided by Susan M. Kaech (NOMIS Center for Immunobiology and Microbial Pathogenesis, Salk Institute for Biological Studies, La Jolla, CA, USA) and were crossed to CD4Cre and CD4CreErt2 mice for generation of CD4-Cre+; ZEB1flox/flox (CD4CreZEB1L/L, ZEB1 knockout) mice, CD4-Cre−; ZEB1flox/flox (WT, wild-type) mice, CD4-CreErt2+; ZEB1flox/flox (CD4CreErt2ZEB1L/L, ZEB1 knockout) mice and CD4-CreErt2+; ZEB1+/+ (CD4CreErt2, wild-type) mice. All animal experiments were done with approved Northwestern University institutional animal care and use the committee protocols.
Cell Lines
Jurkat cells were maintained in RPMI-1640 supplemented with 10% FCS and 1% Pen/Strep.
METHOD DETAILS
Tamoxifen induction
Tamoxifen (Cat# T5648, Sigma) was prepared by dissolving in the 5% ethanol-corn oil (Cat# C8267, Sigma) (v/v) for a final concentration of 20 mg/ml by shaking overnight at −37°C and stored at −20°C. Mice were given 1.5 mg tamoxifen solution (approximately 75 mg/kg) by i.p. injection daily for 5 consecutive days. One week after treatment ended, mice were processed for EAE induction or naive CD4+ T cell isolation.
EAE induction and activity
Eight to twelve weeks-old mice were injected subcutaneously with 100 μL of an emulsion containing 200 μg of Mycobacterium tuberculosis H37Ra (Cat# DF3114-33-8, Difco) and 200 μg of MOG35–55 (Cat# MOG3555-P2, Genemed Synthesis) distributed over three sites on the flank. On day 0 and 2 after immunization, 200 ng pertussis toxin (Cat# 180, List Biological Laboratories) were administered intraperitoneally (i.p.). Clinical signs of EAE were assessed daily according to the following scores: 0, no clinical sign of disease; 1, limp tail; 2, hind limp weakness; 3, partial hind limb paralysis; 4, complete hind limb paralysis; 5, hind and fore limb paralysis. Data are reported as the mean daily clinical score. For Fedratinib treatment, C57BL/6 mice were injected intraperitoneally with vehicle or 120 mg/Kg Fedratinib at day 7, 10, 12, 14, 17, 19, 21, 24, 26 post immunization.
Isolation of CNS leukocytes from EAE mice
CNS-immune cells were isolated by Percoll gradient centrifugation from homogenized brain and spinal cords combined as previously described (Ifergan et al., 2016). The number of cell subpopulations in the CNS were determined by multiplying the percentage of lineage marker–positive cells by the total number of mononuclear cells isolated from the CNS.
Ex vivo recall responses
Splenocytes were harvested from mice at the peak of disease, counted, and cultured in 96-well microtiter plates at a density of 106 cells/well in a total volume of 200 μL of R10 media (RPMI with 10% (volume/volume) fetal bovine serum (FBS), 2 mM L-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin). Cells were cultured at 37°C in the presence of MOG35–55 (Cat# MOG3555-P2, Genemed Synthesis; 20 μg/ml) or without any peptide for 72h.
Isolation of mouse CD4+ naive T cells and Th cells differentiation
Spleens were harvested from ZEB1-deficient or ZEB1-WT mice. Mouse CD4+ naive T cells were isolated from splenocytes by magnetic negative selection using the EasySep Mouse Naive CD4+ T Cell Isolation Kit (Cat #19765, STEMCELL). The CD4+ naive T cells were cultured in RPMI-1640 containing 10% Fetal Calf Serum (FCS), 50 mM 2-mercaptoethanol, 10 mM sodium pyruvate, and anti-CD3/CD28 Dynabeads (Cat #11452D, GIBCO) with following antibodies and cytokines for each T helper subset polarizing condition: Th0, 100 U/mL IL2 (Cat #200–02, PeproTech); Th1, 200 U/mL IL2, 10 ng/mL IL12 (Cat #210–12, PeproTech), 1 μg/mL anti-IL4 (Cat #554433, clone: 11B11, BD); Th2, 100 U/mL IL2, 12.5 ng/mL IL4 (Cat #214–14, PeproTech), 1 μg/mL anti-IFN-γ (Cat #554409, clone: XMG1.2, BD); Treg, 200 U/mL IL2, 25 ng/mL TGFβ (Cat #100–21, PeproTech), 1 μg/mL anti-IFN-γ; Th17, 10 ng/mL TGFβ, 50 ng/mL IL6 (Cat #216–16, PeproTech), 50 ng/mL IL23 (Cat #200–23, PeproTech), 1 μg/mL anti-IL4, 1 μg/mL anti-IFN-γ, 1 μg/mL anti-IL2 (Cat #554375, clone: S4B6, BD).
Isolation of human CD4+ naive T cells and Th cells differentiation
Fresh Leukopak samples from de-identified blood donors were purchased from AllCells, LLC. Peripheral blood mononuclear cells (PBMCs) were isolated by density gradient centrifugation using ISOLYMPH (Cat #759050, CTL). CD4+ T cells were isolated from PBMCs by magnetic positive selection using the Dynabeads Human CD4+ T Cell Isolation Kit (Cat #11331D, Invitrogen). CD4+ naive T cells were isolated from CD4+ T cells by magnetic positive selection using the Dynabeads pan mouse IgG (Cat #11041, Invitrogen) and Anti-CD45RA antibody (clone: HI100, Cat #14-0458-82, Invitrogen). The CD4+ naive T cells were cultured in RPMI-1640 containing 10% FCS, 50 mM 2-mercaptoethanol, 10 mM sodium pyruvate, and anti-CD3/CD28 Dynabeads (Cat #11161D, GIBCO) with following antibodies and cytokines for each T helper subset polarizing condition: Th0, 100 U/mL IL2 (Cat #200–02, PeproTech); Th1, 100 U/mL IL2, 10 ng/mL IL12 (Cat #200–12, PeproTech), 1 μg/mL anti-IL4 (Cat #16-7048-81, clone: MP4-25D2, eBioscience); Th2, 100 U/mL IL2, 12.5 ng/mL IL4 (Cat #200–04, PeproTech), 5 μg/mL anti-IFN-γ (Cat #14-7318-85, clone: NIB42, eBioscience); Treg, 100 U/mL IL2, 25 ng/mL TGFβ (Cat #100–21, PeproTech), 1 μg/mL anti-IFN-γ; Th17, 5 ng/mL TGFβ, 50 ng/mL IL-1β (Cat #200–01B, PeproTech, Rocky Hill, NJ), 50 ng/mL IL23 (Cat #200–23, PeproTech), 1 μg/mL anti-IL4 (Cat# 16-7048-81, clone: MP4-25D2, eBioscience), 1 μg/mL anti-IFN-γ (Cat #16-7318-81, clone: N1B42, eBioscience).
Sorting of pathogenic Th1 and Th17 cells from MS patients
Fresh peripheral blood samples from de-identified RRMS patients were collected under the approvement by the Northwestern University Institutional Review Board. Peripheral blood mononuclear cells (PBMCs) were isolated by density gradient centrifugation using ISOLYMPH (Cat #759050, CTL). The PBMCs were stained with Aqua LIVE/DEAD fixable staining reagents (Cat #L34957, Life Technologies), human FcR blocking reagent (Cat # 130-059-901, Miltenyi Biotec), APC anti-CD3 (clone OKT3, BioLegend), Percp-cy5.5 anti-CD4 (clone OKT4, BioLegend) PE-Cy7 anti-CD45RA (clone HI100, BioLegend), BV650 anti-CD45RO (clone UCHL1, BD Biosciences), Pacific Blue anti-CXCR3 (clone G025H7, BioLegend) and BB515 anti-CCR6 (clone 11A9, BD Biosciences). Then CD3+CD4+CD45RA−CD45RO+CCR6−CXCR3+ (Th1), CD3+CD4+CD45RA−CD45RO+CCR6+CXCR3+ (Th1/Th17) and CD3+CD4+ CD45RA−CD45RO+CCR6+CXCR3− (Th17) cells were sorted. After sorting, the cells were amplified by stimulated with anti-CD3/CD28 Dynabeads for 7 days.
siRNA and miRNA nucleofection
Human CD4+ naive T cells were nucleofected with 300 nM Accell SMARTpool siRNA, a mixture of 4 siRNAs against the same target: siCTL (Cat #D-001910-10-05, Dharmacon), siZEB1–1 (Cat #L-006564-01-0005, Smartpool OnTargetPlus), or siZEB1–2 (Cat #E006564-00-0005, Dharmacon) using Amaxa Human T cell Nucleofector kit (Cat #VPA-1002, Lonza) according to manufacturer’s manual. After nucleofection, cells were rest in RPMI-1640 containing 10% FCS for 6 hours before polarizing in Th differentiation conditions. For miRNA studies, 300 nM miR-101-3p mimics (Cat #C-300518-07-0002, Dharmacon) and microRNA Mimic Negative control (Cat #CN-001000-01-05, Dharmacon) or inhibitors (Cat #IH-300518-08-0002, Dharmacon) and microRNA hairpin inhibitor negative control (Cat #IN-001005-01-05, Dharmacon) were co-nucleofected with siCTL or siZEB1.
JAK2 re-expression
For JAK2 re-expression construct cloning, human JAK2 (Dharmacon) and copGFP DNA was cloned into pmaxCloning (Lonza) vector. Then, before Th1 and Th17 differentiation, human CD4+ naive T cells were co-transfected with 300 nM siRNAs (siCTL and siZEB1) and pmax-GFP (1 μg) or pmax-hJAK2-GFP (1 μg).
Flow Cytometry Analysis
All cells were first treated with human (Cat #130-059-901, Miltenyi Biotec) or mouse (Cat # 101301, BioLegend) Fc blockers. For surface markers, cells were then stained for 30 min at 4°C using the specified antibodies. To detect T lymphocytes cytokine expression, cells were activated for 6 h with 500 ng/ml ionomycin and 50 ng/ml phorbol 12-myristate 13-acetate 40 (PMA) in the presence bre-feldin A (Cat #420701, BioLegend) and Monensin solution (Cat #420601, BioLegend). Then, live-dead discrimination was performed using Violet LIVE/DEAD fixable staining reagents (Cat #L34963, Life Technologies) and the cells were fixed and permeabilized with a fixation and permeabilization kit (Cat #88-8824-00, eBioscience). Cells were acquired on BD LSRII or BD LSR Fortessa and analyzed using Flowjo version 10 software. Flow antibodies used for human included: CD3 (clone OKT3, BioLegend), CD4 (clone OKT4, BioLegend), CD45RA (clone HI100, BioLegend), IFN-γ AF647 (clone 4S.B3, BioLegend), IL-17A PE (clone N49–653, BD Biosciences), IL-2 FITC (clone MQ1–17H12, BioLegend), IL-13 PECy7 (clone 5ES10–5A2, BioLegend), IL-4 PE (clone 8D4–8, BD Biosciences), FOXP3 PE (clone 236A/E7, eBioscience), and CD25 APC (clone 4E3, eBioscience). For mouse staining, the following antibodies were used: CD3 (clone 17A2, BioLegend), CD4 (clone GK1.5, BioLegend), IFN-γ PECy7 (clone XMG1.2, BD Biosciences), IL-17A PE (clone TC11–18H10.1, BioLegend), IL-2 APC (clone JES6–5H4, BioLegend), IL-4 Alexa488 (clone 11B11, BD Bioscience), IL-13 PE (clone W17010B, BioLegend), FOXP3 PE (clone MF23, eBioscience), CD25 APC (PC61.5, eBioscience), pSTAT3 Y705 PE (clone 13A3–1, BioLegend), and pSTAT4 PE (38/p-Stat4, BD Biosciences).
Western blot analysis
Cells were harvested and lysed. Protein concentration was determined by BCA (Cat: #23227, Pierce) and 10 ug of protein were boiled in SDS containing Laemmli buffer. Protein was loaded on SDS-PAGE gel, and immunoblotting was performed using standard protocols. Antibodies were from cell signaling technology unless otherwise noted and included ZEB1 (clone E2G6Y), JAK2 (clone D2E12), STAT3 (clone 124H6), pSTAT3 Y705 (clone D3A7), pSTAT3 S727 (clone D8C2Z), STAT4 clone (C46B10), pSTAT4 Y693 (clone D2E4), JAK1 (clone D1T6W), JAK3 (clone D7B12), TYK2 (clone D4I5T), and β-actin (clone C4, Santa Cruz Biotechnology).
Single cell RNA-seq analysis
siCTLTh17 and siZEB1Th17 cells (1 × 106) were sorted using BD FACSMelody and stained with the following cell hashing antibodies; siCTLTh17 day5 (Cat #394663, Biolegend), siZEB1Th17 day 5 (Cat #394667, Biolegend). scRNA-seq was performed using 10x Genomics Single Cell 5′ v1 workflow and processed according to the manufacturer’s instructions. Alignment, filtering, barcode counting, and unique molecular identifier counting were performed using Cell Ranger v.2.1.0. Data were further analyzed using Seurat v.3.0 (Stuart et al., 2019). Briefly, cells with a percentage of mitochondrial genes below 5% were included. Cells with the number of detected genes lower than 200 were excluded. Cells were filtered for feature counts more than 1000 and less than 7000. Then data was normalized to unique molecular identifier count per million total counts and log-transformed. Then, 2000 variable genes were selected per dataset. Principal component analysis (PCA) was performed using variable genes. Clusters and UMAP plots were generated based on PCA dimensions.
RNA-seq analysis
RNA from human T cells (5 × 105 cells, day 3 of differentiation) and mouse T cells (5 × 105 cells, day 4 of differentiation) was isolated with the RNeasy Micro kit (Cat: #74004, QIAGEN) according to the manufacturer’s protocol. RNA libraries were prepared using the QuantSeq 3′ mRNA-Seq Library Prep Kit FWD (Lexogen). Sequencing was performed on an Illumina Nextseq 2500 and generated 75 bp paired-end reads. Three biological replicates of each sample were sequenced. Reads were aligned using STAR (Dobin et al., 2013), gene specific transcripts were quantified using HT-Seq (Anders et al., 2015), and differentially expressed transcripts were identified using DE-Seq2 (Love et al., 2014). Time-course analyses were performed using maSigPro (Conesa et al., 2006). For Th17 regulator analysis, the list of Th17 regulators were culled from a published study (Tripathi et al., 2017). From the list, the genes upregulated in siCTL-treated cell in Th17 conditions versus Th0 controls were considered putative positive regulators. The genes downregulated in siCTL-Th17 conditions were considered putative negative regulators. P values were generated using default conditions. Volcano plots, PCA plots, and heatmaps were generated using R (https://cran.r-project.org). In the heatmaps, data was normalized and scaled per gene. Pathway enrichment analysis were performed through Enrichr or GSEA. All software employed default settings.
miRNA-seq analysis
Total RNA of human T cells transfected with siCTL or siZEB1 (5 × 105 cells, day 3 of Th17 differentiation) was isolated with the miRNeasy mini kit (Cat: #217004, QIAGEN) according to the manufacturer’s protocol. RNA libraries were prepared using the NEBNext Small RNA Library Prep Set for Illumina. 6% PAGE gel, followed by ethanol precipitation, was performed post library prep to purify small RNA library. Sequencing was performed on an Illumina Nextseq 2500 generating 75 bp paired-end reads. Three biological replicates of each sample were sequenced. miRNA-seq data was first trimmed with Cutadapt default parameters and then aligned with miRDeep2 and Bowtie (Friedländer et al., 2012; Langmead et al., 2009; Martin, 2011). miRNA expression quantification was performed with miRDeep2. Differentially expressed miRNAs were determined by DE-Seq2. PCA plots and heatmaps were generated using R (https://cran.r-project.org). All software employed default settings.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses were performed using GraphPad PRISM 9.0 (GraphPad software). Data are presented as the mean ± the standard error of the mean (SEM). Single comparisons of two means were analyzed by Student’s t test. For multiparametric data, two-way ANOVA with a Bonferroni post-test was used. Analyses of EAE scores were performed by nonparametric Mann-Whitney test. Only p values < 0.05 were considered significant.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| PE anti-mouse CD4 | BioLegend | Cat#100408; RRID:AB_312693 |
| PE/Cyanine7 anti-mouse/human CD44 | BioLegend | Cat#103030; RRID:AB_830787 |
| APC anti-mouse CD62L | BioLegend | Cat#104412; RRID:AB_313099 |
| Purified Rat Anti-Mouse IL-4 | BD Bioscience | Cat#554433; RRID:AB_395389 |
| PE-Cy7 Rat Anti-Mouse IFN-γ | BD Bioscience | Cat#557649; RRID:AB_396766 |
| PE anti-mouse IL-17A | BioLegend | Cat#506904; RRID:AB_315464 |
| APC anti-mouse IL-2 | BioLegend | Cat#503810; RRID:AB_315304 |
| Alexa Fluor® 488 anti-mouse IL-4 | BD Bioscience | Cat#557728; RRID:AB_396836 |
| PE anti-mouse IL-13 | BioLegend | Cat#159403; RRID:AB_2832569 |
| PE anti-FOXP3 Monoclonal | eBioscience | Cat#12-5773-82; RRID:AB_465936 |
| APC anti-CD25 Monoclonal Antibody | eBioscience | Cat#17-0251-82; RRID:AB_469366 |
| PE anti-STAT3 Phospho (Tyr705) Antibody | BioLegend | Cat#651004; RRID:AB_2571892 |
| PE Mouse Anti-Stat4 (pY693) | BD Bioscience | Cat#562073; RRID:AB_10895804 |
| Pacific Blue anti-human CD3 | BioLegend | Cat#317314; RRID:AB_571909 |
| PE anti-human CD4 | BioLegend | Cat#317410; RRID:AB_571955 |
| PerCP/Cyanine5.5 anti-human CD45RO | BioLegend | Cat#304222; RRID:AB_2174124 |
| PE/Cyanine7 anti-human CD45RA | BioLegend | Cat#304126; RRID:AB_10708879 |
| Alexa Fluor® 647 anti-human IFN-γ | BioLegend | Cat#502516; RRID:AB_493031 |
| PE Mouse anti-Human IL-17A | BD Bioscience | Cat#560486; RRID:AB_1645512 |
| FITC anti-human IL-2 | BioLegend | Cat#500304; RRID:AB_315091 |
| PE anti-human IL-13 | BioLegend | Cat#501903; RRID:AB_315198 |
| PE Mouse Anti-Human IL-4 | BD Bioscience | Cat#559333; RRID:AB_397230 |
| PE Mouse anti-Human FoxP3 | BD Bioscience | Cat#560852; RRID:AB_10563418 |
| APC anti-CD25 Monoclonal Antibody | eBioscience | Cat#17-0257-42; RRID:AB_11218671 |
| human FcR blocking reagent | Miltenyi Biotec | Cat#130-059-901; RRID:AB_2892112 |
| APC anti-human CD3 | BioLegend | Cat#317318; RRID:AB_1937212 |
| PerCP/Cyanine5.5 anti-human CD4 | BioLegend | Cat#317428; RRID:AB_1186122 |
| BV650 Mouse Anti-Human CD45RO | BD Bioscience | Cat#563749; RRID:AB_2744412 |
| Pacific Blue anti-human CD183 (CXCR3) | BioLegend | Cat#353724; RRID:AB_2561442 |
| BB515 Mouse Anti-Human CD196 (CCR6) | BD Bioscience | Cat#564479; RRID:AB_2738825 |
| ZEB1 (E2G6Y) XP® Rabbit mAb | Cell Signaling Technology | Cat#70512S; RRID:AB_2827862 |
| Jak2 (D2E12) XP® Rabbit mAb | Cell Signaling Technology | Cat#3230S; RRID:AB_2128522 |
| Stat3 (124H6) Mouse mAb | Cell Signaling Technology | Cat#9139S; RRID:AB_331757 |
| Phospho-Stat3 (Tyr705) (D3A7) XP® Rabbit mAb | Cell Signaling Technology | Cat#9145S; RRID:AB_2491009 |
| Phospho-Stat3 (Ser727) (D8C2Z) Rabbit mAb | Cell Signaling Technology | Cat#94994S; RRID:AB_2800239 |
| Stat4 (C46B10) Rabbit mAb | Cell Signaling Technology | Cat#2653S; RRID:AB_2255156 |
| Anti-p-Stat4 Antibody | Santa Cruz | Cat#sc-28296; RRID:AB_628294 |
| Purified Rat Anti-Mouse IFN-γ | BD Bioscience | Cat#554409; RRID:AB_398550 |
| Purified NA/LE Rat Anti-Mouse IL-2 | BD Bioscience | Cat#554375; RRID:AB_395355 |
| Pacific Blue anti-mouse CD3 | BioLegend | Cat#100214; RRID:AB_493645 |
| Bacterial and virus strains | ||
| DH5α | New England Biolabs | Cat#C2987I |
| Biological samples | ||
| Leukopak | ALLCELLS | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| RPMI 1640 Medium | GIBCO | Cat#21875034 |
| Recombinant human TGF-b1 | PeproTech | Cat#100-21 |
| Recombinant human IL-2 | PeproTech | Cat#200-02 |
| Recombinant human IL-4 | PeproTech | Cat#204-04 |
| Recombinant human IL-12 | PeproTech | Cat#200-12 |
| Recombinant human IL-1β | PeproTech | Cat#200-01B |
| Recombinant human IL-23 | PeproTech | Cat#200-23 |
| Recombinant mouse IL-12 | PeproTech | Cat#210-12 |
| Recombinant mouse IL-4 | PeproTech | Cat#214-14 |
| Recombinant mouse IL-6 | PeproTech | Cat#216-16 |
| Tamoxifen | Millipore Sigma | Cat#T5648 |
| Corn Oil | Millipore Sigma | Cat#C8267 |
| Mycobacterium tuberculosis H37Ra | Difco | Cat#DF3114-33-8 |
| MOG35–55 | Genemed Synthesis | Cat#MOG3555-P2 |
| Pertussis toxin | List Biological Laboratories | Cat#180 |
| Fedratinib | TargetMol | Cat#T1995-CUST |
| ISOLYMPH | CTL | Cat#759050 |
| CellTrace™ CFSE Cell Proliferation Kit | ThermoFisher | Cat#C34554 |
| LIVE/DEAD Fixable Violet Dead Cell Stain Kit | ThermoFisher | Cat#L34963 |
| LIVE/DEAD Fixable Aqua Dead Cell Stain Kit | ThermoFisher | Cat#L34957 |
| Critical commercial assays | ||
| Dynabeads CD4 Positive Isolation Kit | ThermoFisher | Cat#11331D |
| Dynabeads Pan Mouse IgG | ThermoFisher | Cat#11041 |
| Dynabeads Human T-Activator CD3/CD28 | ThermoFisher | Cat#11161D |
| EasySep Mouse Naive CD4+ T Cell Isolation Kit | STEMCELL | Cat#19765 |
| Dynabeads Mouse T-Activator CD3/CD28 | ThermoFisher | Cat#11452D |
| Amaxa Human T cell Nucleofector kit | Lonza | Cat# C34554 |
| RNeasy Micro kit | QIAGEN | Cat#74004 |
| QuantSeq 3′ mRNA-Seq Library Prep Kit FWD | Lexogen | Cat#015.96 |
| miRNeasy mini kit | QIAGEN | Cat#217004 |
| NEBNext Small RNA Library Prep Set for Illumina | NEB | E7330S |
| Deposited data | ||
| RNA-seq for Monocytes and CD4+ T cells of MS patients | International Multiple Sclerosis Genetics Consortium, 2019a | Dyrad: http://datadryad.org/stash/dataset/ doi:10.7272/Q6HQ3X3M |
| RNA-seq for mouse Th cells | Stubbington et al., 2015 | ArrayExpress: E-MTAB-2582 |
| RNA-seq | This paper | GEO: GSE179835 |
| single cell RNA-seq | This paper | GEO: GSE179835 |
| miRNA-seq | This paper | GEO: GSE179835 |
| Experimental models: Cell lines | ||
| Jurkat | ATCC | TIB-152 |
| Experimental models: Organisms/strains | ||
| C57BL/6 mice | Jackson | Cat#000664; RRID:IMSR_JAX:000664 |
| CD4CreErt2, C57BL/6 background | Jackson | Cat#022356; RRID:IMSR_JAX:022356 |
| CD4Cre, C57BL/6 background | Jackson | Cat#022071; RRID:IMSR_JAX:022071 |
| ZEB1fl/fl, C57BL/6 background | Dr. Susan M. Kaech | N/A |
| Oligonucleotides | ||
| siZEB1-1 | Dharmacon | Cat#L-006564-01-0005 |
| siZEB1-2 | Dhabormacon | Cat#E006564-00-0005 |
| miR-101-3p mimics | Dharmacon | Cat#C-300518-07-0002 |
| microRNA Mimic Negative control | Dharmacon | Cat#CN-001000-01-05 |
| miR-101-3p hairpin inhibitors | Dharmacon | Cat#IH-300518-08-0002 |
| microRNA hairpin inhibitor negative control | Dharmacon | Cat#IN-001005-01-05 |
| siCTL | Dharmacon | Cat#D-001910-10-05 |
| hJAK2-forward: ATGAAGTTTAAACAAGCTTGAATTCATGGGAATGGCCTGCCTTAC | IDT | N/A |
| hJAK2-reverse: CGGCCGCGCTAGCCTCGAGGGATCCTTAGCGAGATCCGGTGGAG | IDT | N/A |
| copGFP-forward: ATGAAGTTTAAACAAGCTTGAATTCGGGCAGAGCGCACATCGC | IDT | N/A |
| copGFP-reverse: CGGCCGCGCTAGCCTCGAGGGATCCTTAGCGAGATCCGGTGGAGC | IDT | N/A |
| Recombinant DNA | ||
| human JAK2 cDNA | Dharmacon | Cat#OHS6084-202638642 |
| pCDH-CMV-MCS-EF1α-copGFP | SBI | Cat#CD511B-1 |
| pmaxCloning Vector | Lonza | Cat#VDC-1040 |
| Software and algorithms | ||
| R (v3.6.0) | The R Foundation | https://www.r-project.org |
| ImageJ (Fiji) | NIH | https://imagej.nih.gov/ij/docs/guide/146-2.html; RRID: SCR_003070 |
| Flowjo | Flowjo LLC | https://www.flowjo.com/solutions/flowjo |
| STAR | GitHub | https://github.com/alexdobin/STAR |
| Samtools | MIT | https://github.com/samtools/samtools |
| DESeq2 | Bioconductor | http://bioconductor.org/packages/devel/bioc/html/DESeq2.html; RRID: SCR_015687 |
| Prism v9 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| Bowtie2 | GitHub | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
Highlights.
ZEB1 mutations modulate risk for multiple sclerosis (MS) in humans and in animal models
ZEB1 is required for Th1/Th17 and represses Th2 differentiation in both mice and humans
ZEB1 sustains JAK2 expression in Th1/Th17 cells by inhibiting JAK2-targeting miR-101-3p
Targeting ZEB1/JAK2 effectively reduces MS severity in in vitro and in vivo models
ACKNOWLEDGMENTS
We acknowledge the Northwestern University Research Computing Services, the Northwestern sequencing core, and Admera Health for their invaluable contributions. J.C. was supported in part by the NIH (K08-CA191019-01A1 and 1DP2AI136599-01), the Skin Cancer Foundation, the Leukemia Research Foundation, the Doris Duke Charitable Foundation, and the Damon Runyon Foundation (DRCRF CI-84-16, DDCF 2016095, and DDCF CRM Award). This work was supported by the Northwestern University Flow Cytometry Core Facility supported by Cancer Center Support Grant (NCI CA060553).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109602.
DECLARATION OF INTEREST
The authors declare no competing interests.
SUPPORTING CITATIONS
The following references appear in the Supplemental information: Agarwal et al. (2015); Georgakilas et al. (2016).
REFERENCES
- Agarwal V, Bell GW, Nam J-W, and Bartel DP (2015). Predicting effective microRNA target sites in mammalian mRNAs. eLife 4, e05005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amsen D, Antov A, Jankovic D, Sher A, Radtke F, Souabni A, Busslinger M, McCright B, Gridley T, and Flavell RA (2007). Direct regulation of Gata3 expression determines the T helper differentiation potential of Notch. Immunity 27, 89–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Pyl PT, and Huber W (2015). HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold CN, Pirie E, Dosenovic P, McInerney GM, Xia Y, Wang N, Li X, Siggs OM, Karlsson Hedestam GB, and Beutler B (2012). A forward genetic screen reveals roles for Nfkbid, Zeb1, and Ruvbl2 in humoral immunity. Proc. Natl. Acad. Sci. USA 109, 12286–12293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee S, Biehl A, Gadina M, Hasni S, and Schwartz DM (2017). JAK-STAT Signaling as a Target for Inflammatory and Autoimmune Diseases: Current and Future Prospects. Drugs 77, 521–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson MJ, Pino-Lagos K, Rosemblatt M, and Noelle RJ (2007). All-trans retinoic acid mediates enhanced T reg cell growth, differentiation, and gut homing in the face of high levels of co-stimulation. J. Exp. Med 204, 1765–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Y, Goods BA, Raddassi K, Nepom GT, Kwok WW, Love JC, and Hafler DA (2015). Functional inflammatory profiles distinguish myelin-reactive T cells from patients with multiple sclerosis. Sci, Transl, Med, 7, 287ra74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Ning B, and Shi T (2019). Single-Cell RNA-Seq Technologies and Related Computational Data Analysis. Front. Genet 10, 317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho JJ, Xu Z, Parthasarathy U, Drashansky TT, Helm EY, Zuniga AN, Lorentsen KJ, Mansouri S, Cho JY, Edelmann MJ, et al. (2019). Hectd3 promotes pathogenic Th17 lineage through Stat3 activation and Malt1 signaling in neuroinflammation. Nat. Commun 10, 701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi J, Goh G, Walradt T, Hong BS, Bunick CG, Chen K, Bjornson RD, Maman Y, Wang T, Tordoff J, et al. (2015). Genomic landscape of cutaneous T cell lymphoma. Nat. Genet 47, 1011–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou C-H, Shrestha S, Yang C-D, Chang N-W, Lin Y-L, Liao K-W, Huang W-C, Sun T-H, Tu S-J, Lee W-H, et al. (2018). miRTarBase update 2018: a resource for experimentally validated microRNA-target interactions. Nucleic Acids Res 46 (D1), D296–D302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conesa A, Nueda MJ, Ferrer A, and Talón M (2006). maSigPro: a method to identify significantly differential expression profiles in time-course microar-ray experiments. Bioinformatics 22, 1096–1102. [DOI] [PubMed] [Google Scholar]
- Constantin G, Laudanna C, Brocke S, and Butcher EC (1999). Inhibition of experimental autoimmune encephalomyelitis by a tyrosine kinase inhibitor. J. Immunol 162, 1144–1149. [PubMed] [Google Scholar]
- Conti L, De Palma R, Rolla S, Boselli D, Rodolico G, Kaur S, Silvennoinen O, Niccolai E, Amedei A, Ivaldi F, et al. (2012). Th17 cells in multiple sclerosis express higher levels of JAK2, which increases their surface expression of IFN-γR2. J. Immunol 188, 1011–1018. [DOI] [PubMed] [Google Scholar]
- Dang EV, Barbi J, Yang H-Y, Jinasena D, Yu H, Zheng Y, Bordman Z, Fu J, Kim Y, Yen H-R, et al. (2011). Control of T(H)17/T(reg) balance by hypoxia-inducible factor 1. Cell 146, 772–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding L, Xu Y, Zhang W, Deng Y, Si M, Du Y, Yao H, Liu X, Ke Y, Si J, and Zhou T (2010). MiR-375 frequently downregulated in gastric cancer inhibits cell proliferation by targeting JAK2. Cell Res 20, 784–793. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 5–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang D, and Zhu J (2017). Dynamic balance between master transcription factors determines the fates and functions of CD4 T cell and innate lymphoid cell subsets. J. Exp. Med 214, 1861–1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedländer MR, Mackowiak SD, Li N, Chen W, and Rajewsky N (2012). miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic. Acids Res 40, 37–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgakilas G, Vlachos IS, Zagganas K, Vergoulis T, Paraskevopoulou MD, Kanellos I, Tsanakas P, Dellis D, Fevgas A, Dalamagas T, and Hatzigeorgiou AG (2016). DIANA-miRGen v3.0: accurate characterization of microRNA promoters and their regulators. Nucleic Acids Res 44 (D1), D190–D195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan T, Dominguez CX, Amezquita RA, Laidlaw BJ, Cheng J, Henao-Mejia J, Williams A, Flavell RA, Lu J, and Kaech SM (2018). ZEB1, ZEB2, and the miR-200 family form a counterregulatory network to regulate CD8+ T cell fates. J. Exp. Med 215, 1153–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guenova E, Watanabe R, Teague JE, Desimone JA, Jiang Y, Dowlatshahi M, Schlapbach C, Schaekel K, Rook AH, Tawa M, et al. (2013). TH2 cytokines from malignant cells suppress TH1 responses and enforce a global TH2 bias in leukemic cutaneous T-cell lymphoma. Clin. Cancer Res 19, 3755–3763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann FJ, Khademi M, Aram J, Ammann S, Kockum I, Constantinescu C, Gran B, Piehl F, Olsson T, Codarri L, and Becher B (2014). Multiple sclerosis-associated IL2RA polymorphism controls GM-CSF production in human TH cells. Nat. Commun 5, 5056. [DOI] [PubMed] [Google Scholar]
- Higashi Y, Moribe H, Takagi T, Sekido R, Kawakami K, Kikutani H, and Kondoh H (1997). Impairment of T cell development in deltaEF1 mutant mice. J. Exp. Med 185, 1467–1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higashi AY, Ikawa T, Muramatsu M, Economides AN, Niwa A, Okuda T, Murphy AJ, Rojas J, Heike T, Nakahata T, et al. (2009). Direct hema-tological toxicity and illegitimate chromosomal recombination caused by the systemic activation of CreERT2. J. Immunol 182, 5633–5640. [DOI] [PubMed] [Google Scholar]
- Huang Y-H, Tsai K, Ma C, Vallance BA, Priatel JJ, and Tan R (2014). SLAM-SAP signaling promotes differentiation of IL-17-producing T cells and progression of experimental autoimmune encephalomyelitis. J. Immunol 193, 5841–5853. [DOI] [PubMed] [Google Scholar]
- Ifergan I, Chen S, Zhang B, and Miller SD (2016). Cutting Edge: MicroRNA-223 Regulates Myeloid Dendritic Cell-Driven Th17 Responses in Experimental Autoimmune Encephalomyelitis. J. Immunol 196, 1455–1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ifergan I, Davidson TS, Kebir H, Xu D, Palacios-Macapagal D, Cann J, Rodgers JM, Hunter ZN, Pittet CL, Beddow S, et al. (2017). Targeting the GM-CSF receptor for the treatment of CNS autoimmunity. J. Autoimmun 84, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International Multiple Sclerosis Genetics Consortium (2019a). A systems biology approach uncovers cell-specific gene regulatory effects of genetic associations in multiple sclerosis. Nat. Commun 10, 2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International Multiple Sclerosis Genetics Consortium (2019b). Multiple sclerosis genomic map implicates peripheral immune cells and microglia in susceptibility. Science 365, eaav7188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janbandhu VC, Moik D, and Fässler R (2014). Cre recombinase induces DNA damage and tetraploidy in the absence of loxP sites. Cell Cycle 13, 462–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopantzev Y, Heller M, Swaminathan N, and Rudikoff S (2002). IL-6 mediated activation of STAT3 bypasses Janus kinases in terminally differentiated B lineage cells. Oncogene 21, 6791–6800. [DOI] [PubMed] [Google Scholar]
- Kwong B, Rua R, Gao Y, Flickinger J Jr., Wang Y, Kruhlak MJ, Zhu J, Vivier E, McGavern DB, and Lazarevic V (2017). T-bet-dependent NKp46+ innate lymphoid cells regulate the onset of TH17-induced neuroinflammation. Nat. Immunol 18, 1117–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lachmann A, Xu H, Krishnan J, Berger SI, Mazloom AR, and Ma’ayan A (2010). ChEA: transcription factor regulation inferred from integrating genome-wide ChIP-X experiments. Bioinformatics 26, 2438–2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, and Salzberg SL (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee PW, Smith AJ, Yang Y, Selhorst AJ, Liu Y, Racke MK, and Lovett-Racke AE (2017). IL-23R-activated STAT3/STAT4 is essential for Th1/Th17-mediated CNS autoimmunity. JCI Insight 2, e91663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy DE, and Lee CK (2002). What does Stat3 do? J. Clin. Invest 109, 1143–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Rodriguez JP, Niu F, Pu M, Wang J, Hung L-W, Shao Q, Zhu Y, Ding W, Liu Y, et al. (2016). Structural basis for DNA recognition by STAT6. Proc. Natl. Acad. Sci. USA 113, 13015–13020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim CP, and Cao X (2001). Regulation of Stat3 activation by MEK kinase 1. J. Biol. Chem 276, 21004–21011. [DOI] [PubMed] [Google Scholar]
- Linterman MA, Denton AE, Divekar DP, Zvetkova I, Kane L, Ferreira C, Veldhoen M, Clare S, Dougan G, Espéli M, and Smith KG (2014). CD28 expression is required after T cell priming for helper T cell responses and protective immunity to infection. eLife 3, e03180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Okada S, Kong X-F, Kreins AY, Cypowyj S, Abhyankar A, Toubiana J, Itan Y, Audry M, Nitschke P, et al. (2011). Gain-of-function human STAT1 mutations impair IL-17 immunity and underlie chronic mucocutaneous candidiasis. J. Exp. Med 208, 1635–1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magner WJ, Weinstock-Guttman B, Rho M, Hojnacki D, Ghazi R, Ramanathan M, and Tomasi TB (2016). Dicer and microRNA expression in multiple sclerosis and response to interferon therapy. J. Neuroimmunol 292, 68–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manshouri R, Coyaud E, Kundu ST, Peng DH, Stratton SA, Alton K, Bajaj R, Fradette JJ, Minelli R, Peoples MD, et al. (2019). ZEB1/NuRD complex suppresses TBC1D2b to stimulate E-cadherin internalization and promote metastasis in lung cancer. Nat. Commun 10, 5125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M (2011). Cutadapt Removes Adapter Sequences From High-Throughput Sequencing Reads. EMBnet.journal 17, 10. [Google Scholar]
- Martínez-Riaño A, Bovolenta ER, Boccasavia VL, Ponomarenko J, Abia D, Oeste CL, Fresno M, van Santen HM, and Alarcon B (2019). RRAS2 shapes the TCR repertoire by setting the threshold for negative selection. J. Exp. Med 216, 2427–2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer Zu Horste G, Przybylski D, Schramm MA, Wang C, Schnell A, Lee Y, Sobel R, Regev A, and Kuchroo VK (2018). Fas Promotes T Helper 17 Cell Differentiation and Inhibits T Helper 1 Cell Development by Binding and Sequestering Transcription Factor STAT1. Immunity 48, 556–569.e7. [DOI] [PubMed] [Google Scholar]
- Mootha VK, Lindgren CM, Eriksson K-F, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstråle M, Laurila E, et al. (2003). PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet 34, 267–273. [DOI] [PubMed] [Google Scholar]
- Mullally A, Hood J, Harrison C, and Mesa R (2020). Fedratinib in myelofibrosis. Blood Adv 4, 1792–1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro A, Diaz T, Martinez A, Gaya A, Pons A, Gel B, Codony C, Ferrer G, Martinez C, Montserrat E, and Monzo M (2009). Regulation of JAK2 by miR-135a: prognostic impact in classic Hodgkin lymphoma. Blood 114, 2945–2951. [DOI] [PubMed] [Google Scholar]
- Park J-S, Lee J, Lim M-A, Kim E-K, Kim S-M, Ryu J-G, Lee JH, Kwok S-K, Park K-S, Kim H-Y, et al. (2014). JAK2-STAT3 blockade by AG490 suppresses autoimmune arthritis in mice via reciprocal regulation of regulatory T Cells and Th17 cells. J. Immunol 192, 4417–4424. [DOI] [PubMed] [Google Scholar]
- Park J, Yang J, Wenzel AT, Ramachandran A, Lee WJ, Daniels JC, Kim J, Martinez-Escala E, Amankulor N, Pro B, et al. (2017). Genomic analysis of 220 CTCLs identifies a novel recurrent gain-of-function alteration in RLTPR (p.Q575E). Blood 130, 1430–1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintana FJ, Jin H, Burns EJ, Nadeau M, Yeste A, Kumar D, Rangachari M, Zhu C, Xiao S, Seavitt J, et al. (2012). Aiolos promotes TH17 differentiation by directly silencing IL-2 expression. Nat. Immunol 13, 770–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regev K, Healy BC, Khalid F, Paul A, Chu R, Tauhid S, Tummala S, Diaz-Cruz C, Raheja R, Mazzola MA, et al. (2017). Association Between Serum MicroRNAs and Magnetic Resonance Imaging Measures of Multiple Sclerosis Severity. JAMA Neurol 74, 275–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saligrama N, Zhao F, Sikora MJ, Serratelli WS, Fernandes RA, Louis DM, Yao W, Ji X, Idoyaga J, Mahajan VB, et al. (2019). Opposing T cell responses in experimental autoimmune encephalomyelitis. Nature 572, 481–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt-Supprian M, and Rajewsky K (2007). Vagaries of conditional gene targeting. Nat. Immunol 8, 665–668. [DOI] [PubMed] [Google Scholar]
- Scott CL, and Omilusik KD (2019). ZEBs: Novel Players in Immune Cell Development and Function. Trends Immunol 40, 431–446. [DOI] [PubMed] [Google Scholar]
- Shahi SK, Freedman SN, Dahl RA, Karandikar NJ, and Mangalam AK (2019). Scoring disease in an animal model of multiple sclerosis using a novel infrared-based automated activity-monitoring system. Sci. Rep 9, 19194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silveira-Mattos PS, Narendran G, Akrami K, Fukutani KF, Anbalagan S, Nayak K, Subramanyam S, Subramani R, Vinhaes CL, Souza DO, et al. (2019). Differential expression of CXCR3 and CCR6 on CD4+ T-lymphocytes with distinct memory phenotypes characterizes tuberculosis-associated immune reconstitution inflammatory syndrome. Sci. Rep 9, 1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spaderna S, Schmalhofer O, Wahlbuhl M, Dimmler A, Bauer K, Sultan A, Hlubek F, Jung A, Strand D, Eger A, et al. (2008). The transcriptional repressor ZEB1 promotes metastasis and loss of cell polarity in cancer. Cancer Res 68, 537–544. [DOI] [PubMed] [Google Scholar]
- Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM, Hao Y, Stoeckius M, Smibert P, and Satija R (2019). Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stubbington MJ, Mahata B, Svensson V, Deonarine A, Nissen JK, Betz AG, and Teichmann SA (2015). An atlas of mouse CD4(+) T cell transcriptomes. Biol. Direct 10, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng MWL, Bowman EP, McElwee JJ, Smyth MJ, Casanova J-L, Cooper AM, and Cua DJ (2015). IL-12 and IL-23 cytokines: from discovery to targeted therapies for immune-mediated inflammatory diseases. Nat. Med 21, 719–729. [DOI] [PubMed] [Google Scholar]
- Tripathi SK, Chen Z, Larjo A, Kanduri K, Nousiainen K,Äijo T, Ricaño-Ponce I, Hrdlickova B, Tuomela S, Laajala E, et al. (2017). Genome-wide Analysis of STAT3-Mediated Transcription during Early Human Th17 Cell Differentiation. Cell Rep 19, 1888–1901. [DOI] [PubMed] [Google Scholar]
- Villarino AV, Kanno Y, and O’Shea JJ (2017). Mechanisms and consequences of Jak-STAT signaling in the immune system. Nat. Immunol 18, 374–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldmann TA, and Chen J (2017). Disorders of the JAK/STAT Pathway in T Cell Lymphoma Pathogenesis: Implications for Immunotherapy. Annu. Rev. Immunol 35, 533–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Li L, Guo R, Li X, Lu Y, Guan X, Gitau SC, Wang L, Xu C, Yang B, and Shan H (2014). miR-101 promotes breast cancer cell apoptosis by targeting Janus kinase 2. Cell. Physiol. Biochem 34, 413–422. [DOI] [PubMed] [Google Scholar]
- Williams TM, Moolten D, Burlein J, Romano J, Bhaerman R, Godillot A, Mellon M, Rauscher FJ, and Kant JA (1991). Identification of a zinc finger protein that inhibits IL-2 gene expression. Science 254, 1791–1794. [DOI] [PubMed] [Google Scholar]
- Yoon J-H, Sudo K, Kuroda M, Kato M, Lee I-K, Han JS, Nakae S, Imamura T, Kim J, Ju JH, et al. (2015). Phosphorylation status determines the opposing functions of Smad2/Smad3 as STAT3 cofactors in TH17 differentiation. Nat. Commun 6, 7600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yosef N, Shalek AK, Gaublomme JT, Jin H, Lee Y, Awasthi A, Wu C, Karwacz K, Xiao S, Jorgolli M, et al. (2013). Dynamic regulatory network controlling TH17 cell differentiation. Nature 496, 461–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Wencker M, Marliac Q, Berton A, Hasan U, Schneider R, Laubreton D, Cherrier DE, Mathieu A-L, Rey A, et al. (2020). Zeb1 represses TCR signaling, promotes the proliferation of T cell progenitors and is essential for NK1.1+ T cell development. Cell. Mol. Immunol Published online May 12, 2020. 10.1038/s41423-020-0459-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Single-cell RNA-seq data, bulk RNA-seq data and microRNA-seq data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. Other data reported in this paper will be shared by the lead contact upon request. This paper does not report original code. All code and software used is open and freely available. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.